色素過度沉著症 (Hypermelanoses)
一、先天性色素過度沉著 (Congenital Hypermelanosis)
線狀渦輪狀痣樣色素過度沉著 (Linear and Whorled Nevoid Hypermelanosis)
- 先天性、沿 Blaschko 線分布的瀰漫條紋狀色素過度沉著斑,出生後最初幾週發病,無先前發炎或萎縮。
- 病灶位於軀幹與四肢、不跨越中線;臉、掌蹠、眼、黏膜不受侵犯。
- 屬色素鑲嵌現象 (pigmentary mosaicism);多數偶發,少數證實染色體鑲嵌 (三染色體 7/14/18/20、X 染色體)。
- 組織學:基底層色素增加、黑色素細胞增生或空泡化,通常無色素失禁。鑑別:色素失禁症、表皮痣。
- 病程:可隨年齡逐漸消退。
色素失禁症 (Incontinentia Pigmenti, IP)
- X 連鎖顯性,由 Xq28 之 IKBKG (舊稱 NEMO) 基因突變引起,男性胚胎多致死,主要見於女性 (Bloch-Sulzberger 症候群)。
- 四個皮膚階段(可重疊):(a) 水疱期(出生時)、(b) 疣狀期(2–8 週)、(c) 色素過度沉著期(數月至成年,沿 Blaschko 線條紋渦輪狀,軀幹最明顯)、(d) 色素減退期(線狀萎縮無毛瘢痕)。
- 機轉:女性 X 染色體 lyonization;IKBKG 缺陷細胞無法活化 NF-κB 而凋亡;第三期色素來自表皮黑色素失禁入真皮被巨噬細胞吞噬。
- 皮膚外:眼部(30%–70%)、中樞神經(40%)、牙齒與骨骼異常。
- 治療:水疱期可用局部類固醇與局部 tacrolimus;色素減退萎縮瘢痕可考慮非培養表皮細胞移植。
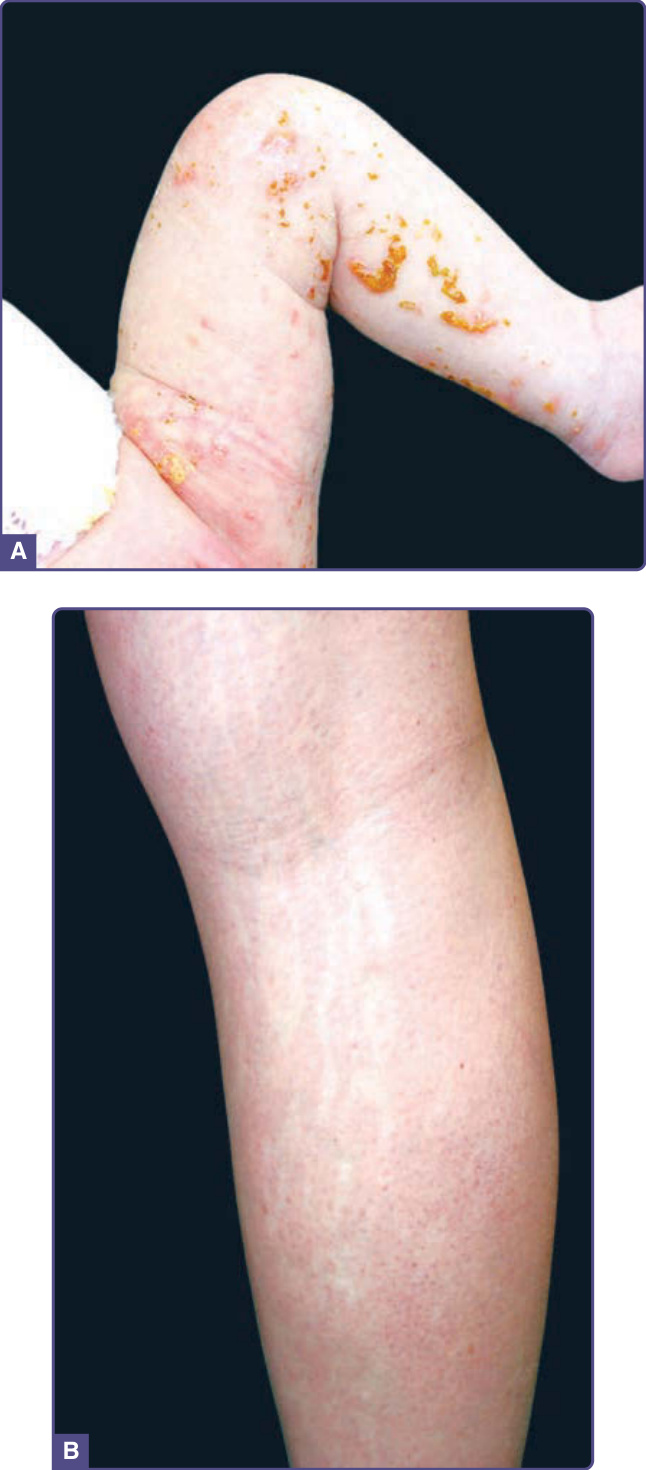
圖 77-1:母親與嬰兒的色素失禁症 — A 疣狀病灶、B 沿 Blaschko 線的色素減退萎縮病灶。
先天性角化不良 (Dyskeratosis Congenita, DKC)
- 三聯:網狀色素過度沉著(頸、胸)+ 甲萎縮 + 白斑(leukoplakia);出生後最初幾年顯現。
- 皮膚外:骨髓衰竭(>80%)、第二與第三個十年出現惡性腫瘤。
- 機轉:端粒酶複合體成分突變致端粒維持缺陷;X 連鎖型 DKC1(dyskerin)、自體顯性多為 TERC(較少 TERT)、自體隱性涉 NOP10/NHP2/TINF2。
- 鑑別:Fanconi 症候群(身材矮小、拇指發育不全、腕骨減少,色素沉著出現更早)。預後:自體顯性型較佳。
其他網狀色素疾病
- Naegeli-Franceschetti-Jadassohn 症候群:自體顯性外胚層發育不良,KRT14 突變(定位 17q11.2-q21);頸與腋窩網狀色素沉著、掌蹠角皮症、皮膚紋理缺失、甲與牙齒異常、出汗減少致耐熱不良。
- 網狀色素性皮膚病 (Dermatopathia Pigmentosa Reticularis):與上者等位;軀幹網狀色素沉著、非瘢痕性禿髮、無牙齒異常、色素沉著終生持續。
- Dowling-Degos 病:自體顯性,KRT5 突變(12q13.13);屈側網狀色素沉著、粉刺樣角化過度丘疹、口周痤瘡樣凹陷瘢痕;組織學見絲狀表皮突帶色素尖端、黑色素細胞不增加(須與黑色棘皮症區別)。
- Galli-Galli 病:Dowling-Degos 之棘層鬆解(acantholysis)變異型,部分有 KRT5 突變。
- Kitamura 網狀肢端色素沉著:自體顯性,ADAM10 突變(15q21.3);手背稜角狀網狀雀斑樣斑,第一個十年起,多見於日本;黑色素細胞與黑色素生成活性增加。
- Haber 症候群:軀幹/腋窩網狀色素沉著 + 疣狀丘疹 + 光敏酒糟樣顏面紅斑與毛細血管擴張。
真皮黑色素細胞增多症 (Dermal Melanocytoses)
- 太田母斑 (Nevus of Ota):先天性、單側,位於三叉神經第一與第二分支區的藍黑/灰棕色真皮黑色素細胞色素沉著;60% 有鞏膜侵犯(眼皮膚黑色素細胞增多症),多見亞洲女性(該族群 0.6%);分輕/中/重/雙側四型。罕見惡性黑色素瘤(白人較頻),須追蹤。治療:雷射為首選(品質開關紅寶石、紫翠玉、Nd:YAG;皮秒雷射亦有效);局部療法無效,應避免冷凍與手術(瘢痕)。
- 伊藤母斑 (Nevus of Ito):太田母斑變異型,侵犯肩鎖與三角肌區。
- 蒙古斑 (Mongolian Spots):薦骨區帶藍色斑,多見有色皮膚,略以男性為主;廣泛型可伴先天性代謝異常(溶酶體儲積病、GM1-神經節苷脂貯積症、黏多醣貯積症);多於兒童期自發消退,必要時雷射。
- 真皮黑色素細胞錯構瘤:皮節分布的灰藍色色素沉著。
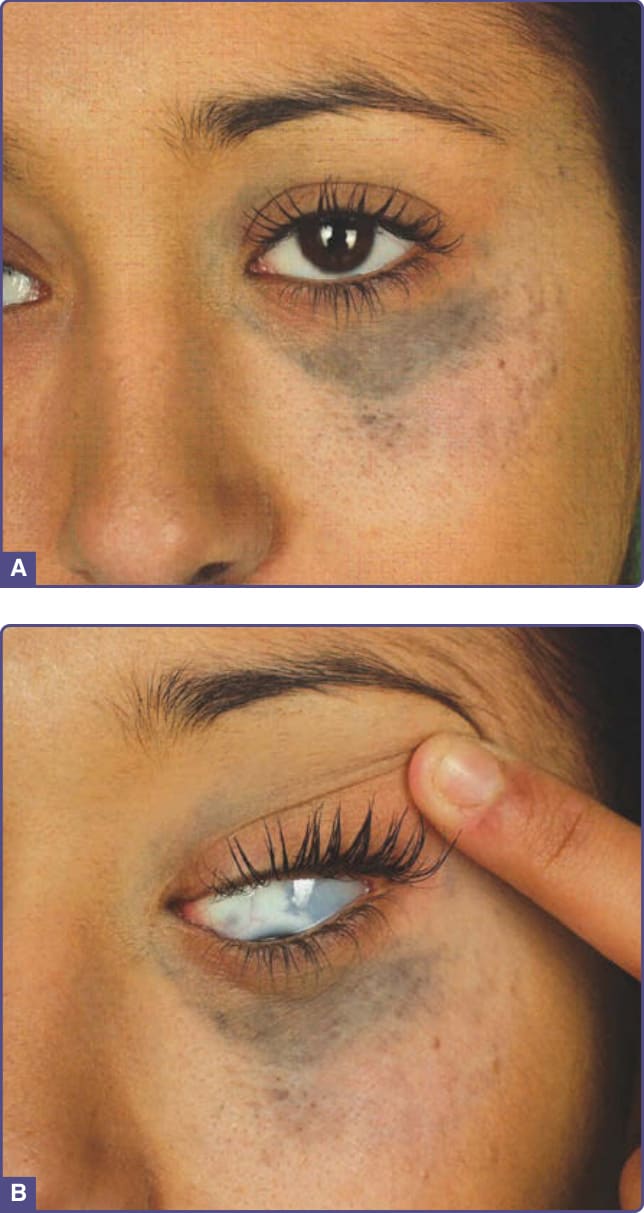
圖 77-2:太田母斑 — 眼眶周圍色素沉著延伸入鞏膜。
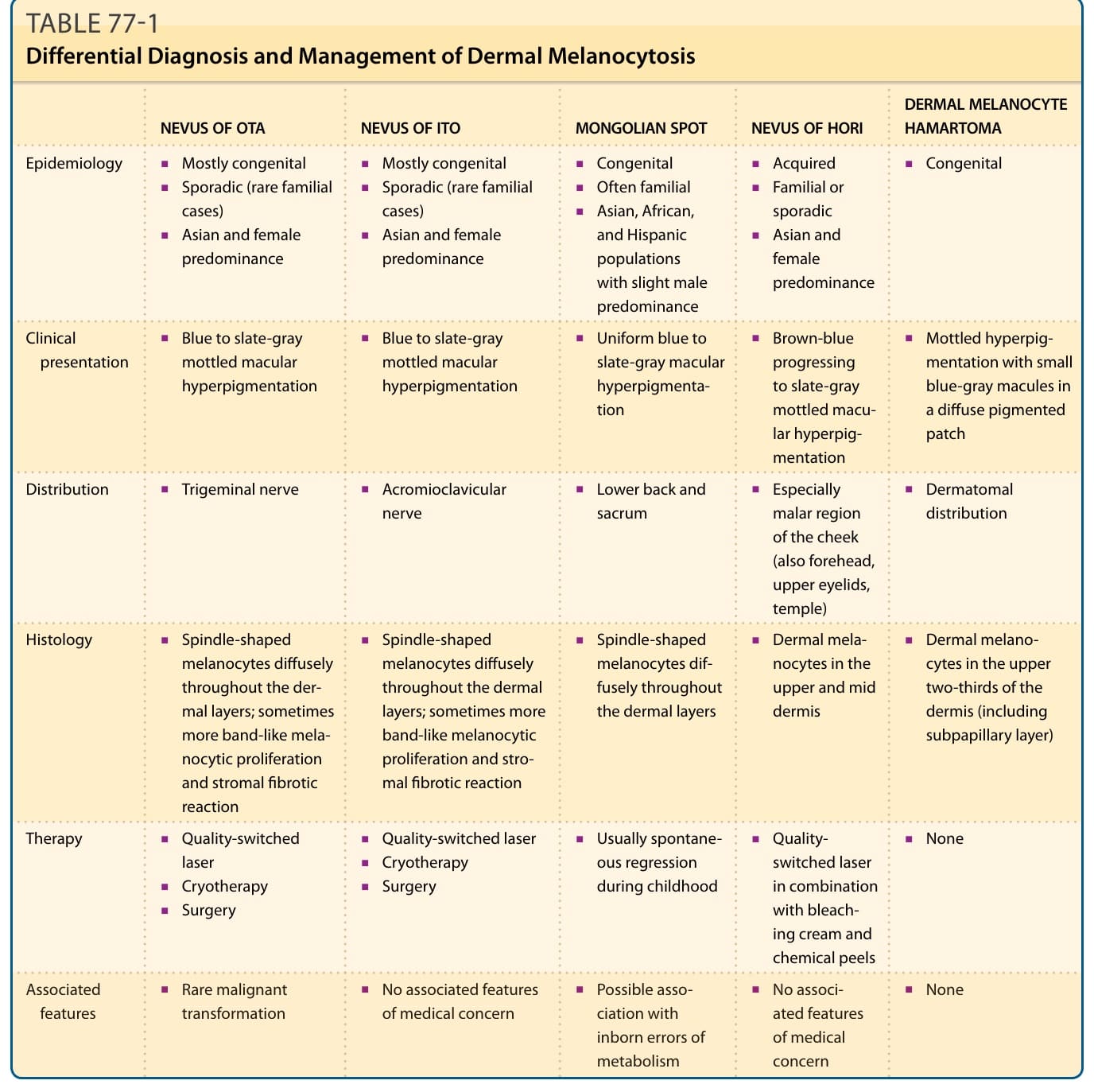
表 77-1:真皮黑色素細胞增多症的鑑別診斷與治療(太田/伊藤/蒙古斑/Hori 痣/真皮黑色素細胞錯構瘤之流行病學、臨床、分布、組織學、治療對照)。
家族性小痣病症候群 (Familial Lentiginosis Syndromes)
- 小痣(lentigines,通常 <5 mm)為表皮黑色素細胞增多,伴心血管/內分泌/胃腸腫瘤風險增加。
- Peutz-Jeghers 症候群 (PJS):自體顯性,STK11/LKB1(19q13.3)突變;嘴唇與頰黏膜小型棕灰色斑(兒童期出現)+ 腸道錯構瘤性息肉病;胃腸惡性腫瘤風險高,需自年輕時監測;色素斑可用品質開關雷射或強脈衝光。
- LEOPARD 症候群:自體顯性,PTPN11(SHP-2,12q24.1)突變(與 Noonan 等位;少數 RAF1);診斷依顏面特徵 + 肥厚型心肌病與/或 CALMs。
- Carney 複合症:自體顯性,PRKAR1A(17q22-24);斑點狀中顏面色素沉著(唇紅緣、淚阜、結膜)+ 黏液瘤 + 內分泌腫瘤。
- Bannayan-Riley-Ruvalcaba 症候群 (BRRS):自體顯性,>60% 有 PTEN(10q23.3)生殖系突變;三聯:巨頭畸形 + 生殖器小痣病 + 腸道息肉病(屬 PTEN 錯構瘤腫瘤症候群)。
- 中顏面小痣病:中央顏面水平小痣帶 + 骨骼/內分泌/神經管缺損。
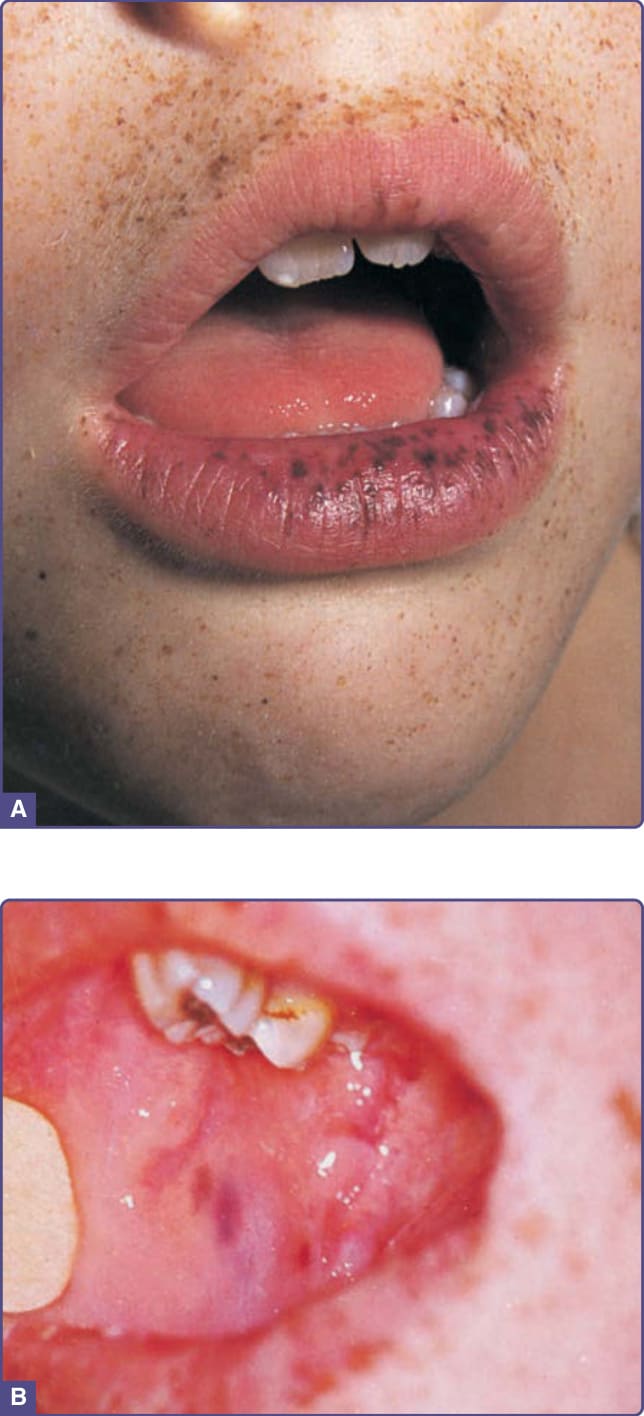
圖 77-5:Peutz-Jeghers 症候群 — 唇、口周與手指小痣;頰黏膜藍黑色斑具特異性病徵且不隨時間消失。
家族性咖啡牛奶斑症候群 (Familial Café-au-Lait Syndromes)
- CALMs:界線分明色素斑(0.5 至 >20 cm),常出生時或出生後數月出現;孤立性 CALMs 黑色素細胞數正常但表皮黑色素增加;6 個以上 CALMs 應啟動潛在疾病調查。
- Legius(SPRED1, 15q13.2)、第 1 型神經纖維瘤病 NF1(NF1 基因,神經纖維瘤蛋白下調 Ras;CALMs 含黑色素細胞增多、間擦部位雀斑為特異性病徵)、McCune-Albright(Gsα 合子後活化突變,鑲嵌;纖維性骨發育不良 + CALMs 邊界不規則中線分界 + 內分泌亢進)、Bloom(BLM, 15q26.1, DNA 修復缺陷)、Watson(與 NF1 等位)、Silver-Russell(11p15 印記/7 號染色體;矮小三角臉)。
二、後天性色素過度沉著 (Acquired Hypermelanosis)
內分泌病變 (Endocrinopathies)
- Addison 病:失鹽 + 瀰漫色素過度沉著,因腎上腺功能不全致 ACTH 代償過產、結合黑皮質素-1 受體;偏好日曬區、創傷/瘢痕/受壓處、掌紋、乳頭、腋窩、會陰生殖器。
- Cushing 症候群:異位性 ACTH 症候群時最嚴重的全身性色素沉著。
- Nelson 症候群:雙側腎上腺切除後垂體瘤增大、ACTH 升高、色素沉著。
- 嗜鉻細胞瘤:可致類 Addison 樣色素沉著(異位 ACTH/MSH),術後迅速消退。
- 類癌症候群:MSH 產生致瀰漫色素沉著 + 色胺酸缺乏之糙皮病樣疹。
- 甲狀腺功能亢進:多 Graves 病;色素沉著分布類似 Addison 但黏膜少受侵,對治療反應不佳。
- 黑色棘皮症:增厚絨毛狀色素沉著皮膚,見頸、腋、膕窩、肘前窩、鼠蹊;肥胖與非胰島素依賴型糖尿病相關。
- 成熟性色素異常:第四至五個十年非洲與印度族群,外側前額/顳部/顴部深棕黑色界線不清色素沉著。
營養性疾病 (Nutritional)
- 缺乏矯正後皮膚變化可逆。惡性營養不良症(kwashiorkor):嚴重蛋白質營養不良,「剝落油漆」外觀、毛髮旗幟徵;補充蛋白可逆轉。維生素 B12 缺乏:全身色素過度沉著 + 毛髮色素減退,基底層黑色素增加(黑色素合成增加而非運輸缺陷),補充可逆。葉酸缺乏、糙皮病(pellagra):菸鹼酸缺乏,皮膚炎-失智-腹瀉三聯,光照處色素沉著,補充菸鹼酸快速反應。
代謝性疾病 (Metabolic)
- 遲發性皮膚紫質症:光照區加重的瀰漫棕色色素沉著,女性可見黑斑樣色素沉著。
- 血色素沉著症:鐵儲積、鐵調素降低;經典三聯(色素沉著 + 「青銅色糖尿病」+ 肝硬化),70% 皮膚變黑(含鐵血黃素沉積 + 表皮黑色素生成增加);放血(phlebotomy)為主要治療,次發型用 deferoxamine。
腫瘤性疾病
- 肥大細胞疾病與黑色素瘤可致瀰漫色素沉著。晚期轉移性黑色素瘤的瀰漫黑色素沉著症:皮膚板岩藍灰至棕色,真皮含黑色素組織細胞/樹突細胞,表皮黑色素細胞不增加,可能源於腫瘤溶解。
物理性病因 (Physical Causes)
- 紫外線:急性曬黑。游離輻射:皮膚輻射症候群(纖維化、角化症、小痣樣色素沉著);電子束治療可致暫時色素沉著與橫向黑甲。熱輻射:淺層燒傷與強雷射熱損傷致色素沉著(深膚色尤甚);冷凍治療可致色素減退伴周邊色素過度沉著。摩擦性黑色素沉著:骨突起處反覆摩擦,黑色素增加與失禁。
毒素與藥物 (Toxins and Medications)
- 佔後天色素過度沉著 10%–20%;最常見:中樞神經藥、抗腫瘤藥、抗感染藥、抗高血壓藥、荷爾蒙。
- 臨床線索:藍板岩灰色 = 真皮色素沉積;與黑色素相關者日曬惡化。bleomycin/zidovudine 致鞭痕狀;cyclophosphamide/doxorubicin 致掌蹠色素沉著;amiodarone(脂質樣物質)致光照區藍灰色;chloroquine(藥物-黑色素複合體)致黃棕至藍灰色;chlorpromazine 致藍灰色;minocycline 風險隨每日劑量高於 100 mg 之延長治療而增加,分三型;銀質沉著病(argyria)致全身灰藍色;界線分明黏膜皮膚斑須考慮固定性藥疹。
- 機轉:(1) 黑色素 ± 藥物複合體沉積真皮巨噬細胞;(2) 藥物直接沉積(carotene、重金屬);(3) 藥物所致非黑色素色素。
- 治療:停藥(色素常仍持續數年至數十年)、防曬;品質開關紅寶石/紫翠玉/Nd:YAG 與皮秒雷射有效(常需多次)。
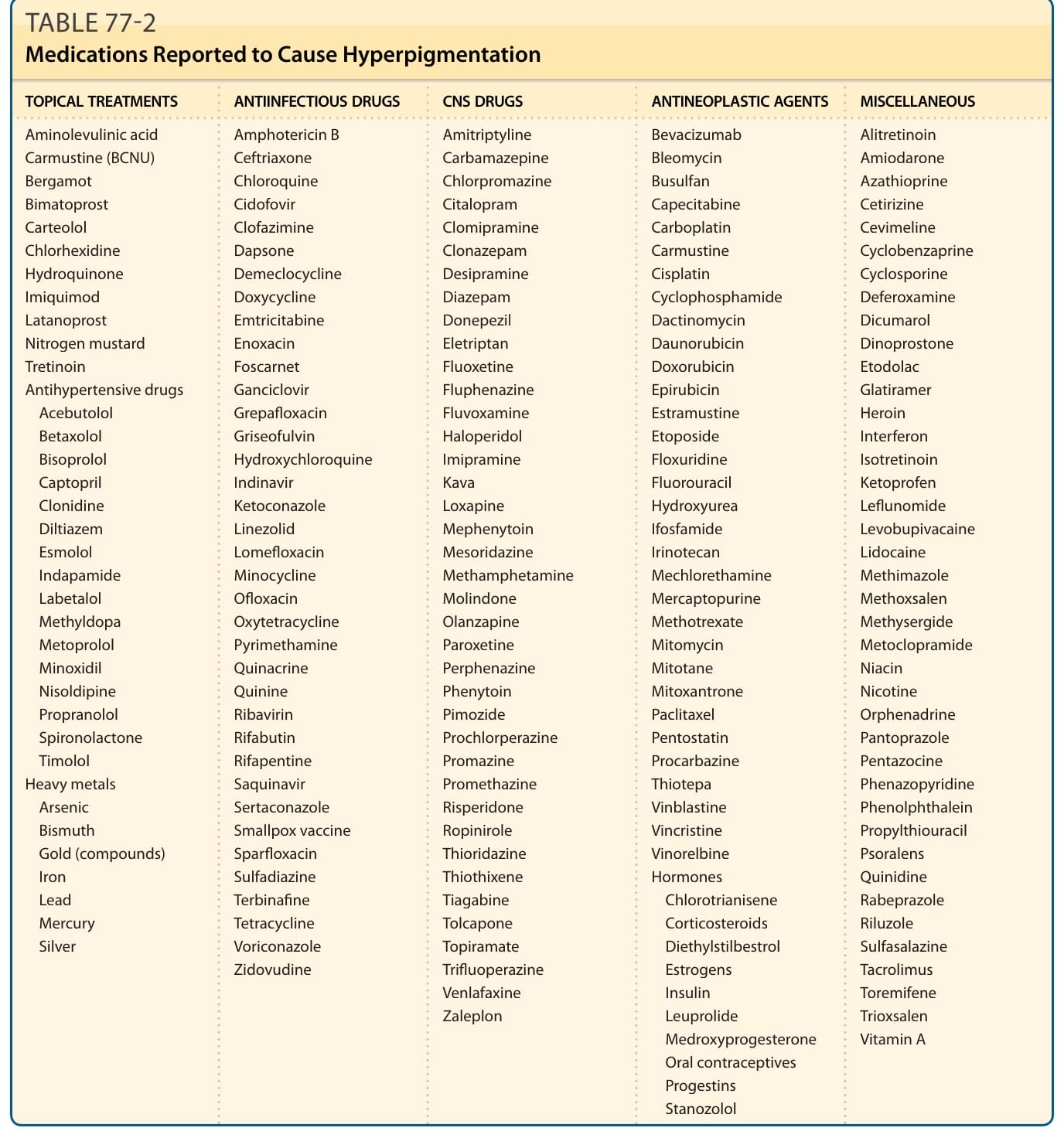
表 77-2:曾被報導造成色素過度沉著的藥物(局部治療、抗高血壓、重金屬、抗感染、中樞神經、抗腫瘤與荷爾蒙等分類)。
褐黃病 (Ochronosis)
- 可內源性(自體隱性)或外源性(藥物);無症狀瀰漫色素沉著,有色皮膚較常見。組織學特徵:真皮中黃棕色香蕉形小球。
- 外源性褐黃病:藍黑/灰黑色斑(顏面顴顳下臉頰、後外側頸、背、四肢伸側),晚期見膠樣粟丘疹「魚子醬樣」病灶;最常與 hydroquinone(常 >4%) 相關,亦見 phenol、resorcinol、quinine;皮膚鏡有助於與黑斑區別,但皮膚切片為診斷黃金標準;治療鮮少有效,須停藥、嚴格防曬。
全身性硬化症、感染
- 全身性硬化症:類 Addison 但 MSH 正常的瀰漫色素沉著;摩擦部位局灶脫色伴毛囊周圍色素沉著。
- 屈公病 (Chikungunya):蚊媒病毒(埃及斑蚊、白線斑蚊);中央顏面小痣樣與鞭痕狀色素沉著,鼻部顯著;色素常持續數月,症狀緩解為主要治療。品他病(品他疹)、蟠尾絲蟲病亦可致色素沉著。
三、生理性色素過度沉著 (Physiologic Hyperpigmentation)
- 色素分界線(Voigt/Futcher 線):有色皮膚常見,沿胚胎縫合線,按位置分 A–H 型(本書新增耳前「I 型」);兒童期出現終生持續;治療為保證 + 防曬。
- 後天性特發性顏面色素沉著:PDLs 另一群組,眼周/顴/口周深棕灰色斑,成年期出現。
- 眼周色素沉著(黑眼圈)、黏膜黑色素沉著(牙齦/頰黏膜/唇/舌,須與藥物、扁平苔癬、Addison、惡性等區別;Laugier-Hunziker 症候群)、縱向黑甲(拇指/大趾單一帶須排除黑色素瘤)、肢端色素過度沉著斑(掌蹠良性棕斑,皮膚鏡平行脊型態)。
四、後天性型態化色素過度沉著 (Acquired Patterned Hypermelanosis)
- 植物日光性皮膚炎(psoralens + 紫外線)、鞭痕狀香菇皮膚炎(生食/未熟香菇,熱不穩定多醣毒性反應)。
- 火激紅斑 (Erythema Ab Igne):慢性中度熱(紅外線)致網狀色素過度沉著,多見大腿小腿(電熱墊/熱水袋/筆電);急性可壓退網狀紅斑、慢性固定色素沉著;須查甲狀腺功能;罕見可發生鱗狀細胞癌/梅克爾細胞癌;治療為去除熱源(± 局部類視黃醇、低能量 QS Nd:YAG)。
- 色素性癢疹 (Prurigo Pigmentosa):年輕女性(女:男 4–6:1),軀幹搔癢性丘疹/水疱演變為網狀色素沉著;治療:minocycline 或 doxycycline 100-200 mg daily,或 dapsone 25 to 100 mg daily,色素自發消退。
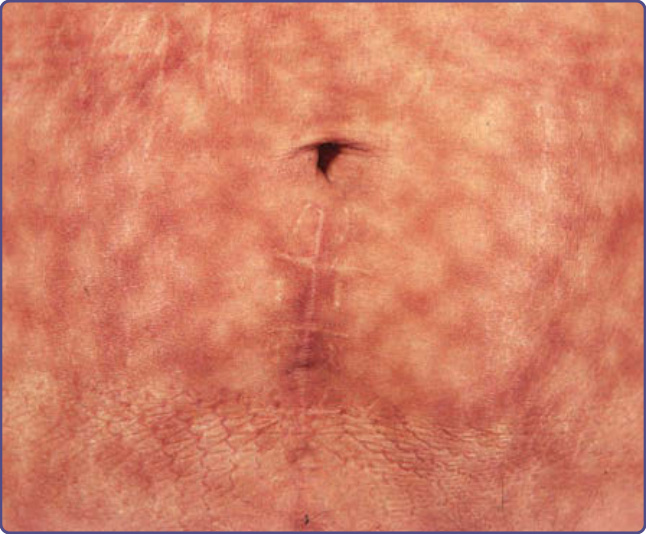
圖 77-16:腹部因多年使用電熱墊所致的火激紅斑網狀色素過度沉著。
五、後天性局限性色素過度沉著(代表性疾病)
- Hori 痣 (ABNOM):後天性雙側顴部對稱針頭大小藍棕至板岩灰色斑,亞洲女性(近 1%),第三至五個十年;與太田母斑不同為雙側、無眼/黏膜侵犯;治療以 QS Nd:YAG 最佳(需多次,皮秒亦有效)。
- Becker 痣:雄激素依賴(約 0.5% 人口),青春期顯著,肩胛/上臂/胸部色素斑伴多毛症;半顯性遺傳;治療具挑戰性,雷射證據不足。
- 咖啡牛奶斑 (CALMs):可孤立或為遺傳性皮膚病一部分;6 個以上應調查;QS 雷射治療但復發常見。
- 雀斑 (Ephelides):自體顯性、與 MCR-1 突變相關,淺膚色凱爾特血統日曬處;夏增冬退;防曬、淡化劑、IPL/QS 雷射(預期復發)。
- 進行性肢端黑色素沉著:新生兒肢端/會陰色素沉著,自發消退。
發炎後色素過度沉著 (Postinflammatory Hyperpigmentation, PIH)
- 常見反應性黑色素沉著,有色皮膚(Fitzpatrick III–VI)遠為常見;表皮型棕色、真皮型灰棕色;嚴重度與發炎及基底膜破壞程度成正比;Wood 燈判定深度。
- 機轉:發炎標記刺激黑色素細胞;表皮發炎致角質細胞黑色素增加,真皮發炎致色素失禁,終由巨噬細胞清除。
- 病程:可變,深膚色與苔癬樣過程後較持久;表皮 PIH 多緩慢自發消退。
- 治療:及早治原發病、防曬;局部 hydroquinone 為黃金標準;麴酸、類視黃醇、α-羥基酸、Kligman 配方/三重療法;換膚與雷射須謹慎(過度可誘發 PIH);表皮型較適合局部療法。
黑斑 (Melasma)
- 最常見色素疾病之一(黃褐斑/妊娠面斑);盛行率 9%(美國南部西班牙裔)至 40%(東南亞);主要停經前女性。
- 臨床:中央顏面對稱淺至深棕色界線不清色素沉著;型態分中顏面型(63%)、顴部型(21%)、下頷型(16%);不侵犯眼周、唇、頸、耳。
- 機轉:生物活性黑色素細胞、遺傳與荷爾蒙、紫外線(口服避孕藥/雌激素加重);近期關注可見光與血管/VEGF 增加。
- 治療:管理期望、停觸發因素;防曬為核心(深膚色須防可見光);4% hydroquinone 為黃金標準;Kligman 配方(5% hydroquinone、0.1% tretinoin + 溫和類固醇)、三重組合療法(4% hydroquinone、0.05% tretinoin + 類固醇);tretinoin/azelaic acid/kojic acid;淺層換膚;低能量 QS Nd:YAG「雷射調色」最受歡迎;tranexamic acid 低劑量(500 to 700 mg daily)輔助於治療抗性病例有效。
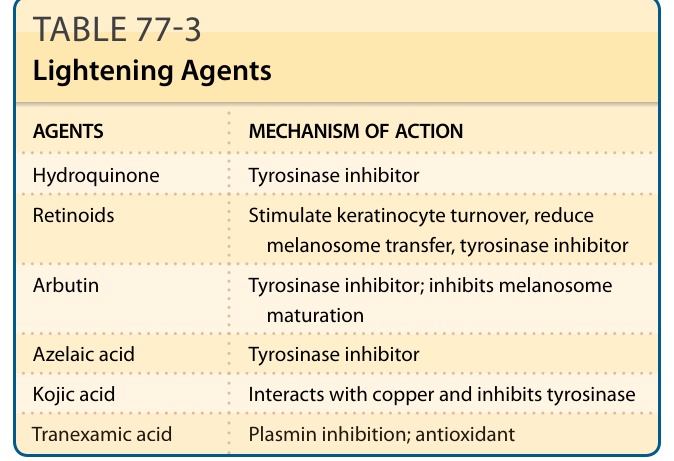
表 77-3:淡化製劑(hydroquinone、類視黃醇、arbutin、azelaic acid、kojic acid、tranexamic acid 及其作用機制)。
其他局限/網狀色素疾病
- 部分單側小痣病 (PUL):節段性小痣,可能為鑲嵌型 NF1 變異;QS 紫翠玉與 Nd:YAG 雷射。
- Riehl 黑色素沉著(色素性接觸性皮膚炎):接觸致敏劑致顏面灰棕至近黑色網狀色素沉著,中年女性;停用致敏劑、防曬、雷射、口服 tranexamic acid。
- Cronkhite-Canada 症候群:全身棕色斑(掌蹠先受侵)+ 甲營養不良 + 禿髮 + 胃腸息肉病;懷疑自體免疫;皮質類固醇 + 營養補充。
- Brocq 口周紅變:口周瀰漫棕色色素沉著,唇紅緣周圍不受侵;防曬與避觸發因素。
- 皮膚澱粉樣變性(斑狀)、Pasini-Pierini 萎縮性皮膚病(灰紫棕色萎縮斑、「懸崖落差」邊緣)、固定性藥疹。
後天性真皮黑色素沉著(灰皮病譜系)
- 持久性色素異常性紅斑 (EDP / 灰皮病):中間膚質女性(西班牙裔、亞洲),光防護部位廣泛藍灰色斑,急性期紅斑性邊界;局部療法無效,dapsone 100 mg(8–12 週)或 clofazimine 100 mg daily 可能淡化,分段雷射不成功。
- 色素性扁平苔癬:扁平苔癬變異型,深色皮膚中年,光照部位棕至灰棕色界線不清斑(腋窩型稱 inversus);芥末油/印度醋栗油可能相關;防曬、局部 tacrolimus(0.03% twice daily)、皮質類固醇,低劑量口服 isotretinoin(20 mg daily)6 個月併防曬可穩定。
- 特發性爆發性斑狀色素過度沉著:兒童青少年,顏面/軀幹/近端四肢界線分明棕色斑塊,無先前發炎;數月至數年自發消退,常不需治療。
六、混合性色素減退與色素過度沉著 (Mixed Hypomelanosis and Hypermelanosis)
- 遺傳性泛發性色素異常症:自體顯性,ABCB6(12q21-q23);軀幹/四肢網狀色素減退與過度沉著斑,日本家族。
- Dohi 網狀肢端色素沉著(遺傳性對稱性色素異常症):手足背小型對稱色素過度沉著與減退斑,南美與亞洲幼兒。
- 伴(Westerhof 症候群)或不伴色素減退的家族性進行性色素過度沉著:自體顯性,KITLG(12q22);瀰漫色素沉著 ± 灰葉斑。
- 流浪者白斑:不良衛生條件,肩與腰帶瀰漫色素沉著、頸背脫色斑,改善生活方式後好轉。