色素過度沉著症 (Hypermelanoses)
PART 13
黑色素細胞疾病 (Melanocytic Disorders)
先天性色素過度沉著 (CONGENITAL HYPERMELANOSIS)
線狀渦輪狀痣樣色素過度沉著 (Linear and Whorled Nevoid Hypermelanosis)
重點一覽 (AT-A-GLANCE)
■ 沿 Blaschko 線分布的先天性瀰漫條紋狀色素過度沉著斑 (macules)。
■ 典型見於軀幹與四肢。
■ 不伴隨先前的發炎或萎縮。
■ 出生後最初幾週發病。
■ 可能隨年齡增長而消退。
線狀渦輪狀痣樣色素過度沉著 (linear and whorled nevoid hypermelanosis) 是一種先天性疾病,造成沿 Blaschko 線分布的瀰漫條紋狀色素過度沉著斑。
臨床特徵 (CLINICAL FEATURES)
線狀渦輪狀痣樣色素過度沉著的特徵是在出生後最初幾週出現沿 Blaschko 線分布、不伴隨先前發炎或萎縮的廣泛條紋狀色素過度沉著斑。病灶通常位於軀幹與四肢,且不跨越中線。臉部、手掌、足底、眼睛與黏膜不受侵犯。類似的病例曾以不同的描述性名稱被報導 (帶狀色素過度沉著 zosteriform hyperpigmentation、帶狀小痣樣痣 zosteriform lentiginous nevus、斑馬樣色素過度沉著 zebra-like hyperpigmentation)。已有數個病例同時呈現色素過度沉著與色素減退。色素鑲嵌現象 (pigmentary mosaicism) 是一個能涵蓋所有這些不同表現型的有用名詞。罕見地,可觀察到皮膚外的表現,例如發育與生長遲緩、顏面與身體不對稱、心室中隔缺損 (ventricular septal defects) 以及假兩性畸形 (pseudohermaphroditism)。皮膚外表現的發生頻率未知,因為這些觀察僅見於病例報告。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
大多數病例為偶發性,但少數病例已透過染色體分析證實鑲嵌現象的存在 (鑲嵌型三染色體 7、14、18 與 20,以及 X 染色體鑲嵌現象)。
診斷 (DIAGNOSIS)
除典型臨床外觀外,組織學檢查顯示基底層色素增加,以及黑色素細胞 (melanocytes) 增生或空泡化 (vacuolization)。通常不見色素失禁 (pigment incontinence)。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
線狀渦輪狀痣樣色素過度沉著應與色素失禁症 (incontinentia pigmenti, IP) 及表皮痣 (epidermal nevus) 區別。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
Kalter 與其同事描述其典型在出生後最初幾週發病,並於最初幾年內進展。色素沉著可能隨年齡增長而逐漸消退。
色素失禁症 (Incontinentia Pigmenti)
重點一覽 (AT-A-GLANCE)
■ 一種 X 連鎖顯性疾病,源於 IKBKG (先前稱為 NEMO) 基因突變。
■ 在男性胚胎中致死。
■ 從出生開始有四個臨床階段:水疱期 (vesicular)、疣狀期 (verrucous)、色素過度沉著期 (hyperpigmented) 與色素減退期 (hypopigmented)。
■ 沿 Blaschko 線分布的先天性瀰漫線狀色素過度沉著斑。
色素失禁症 (IP),又稱 Bloch-Sulzberger 症候群,最早由 Garrod 與其同事於 1906 年描述。它是一種主要見於女性的 X 連鎖疾病,造成瀰漫線狀的皮膚色素過度沉著。
臨床特徵 (CLINICAL FEATURES)
病灶通常經歷四個皮膚階段,儘管各階段有時可能重疊:(a) 水疱期 (從出生時或出生後不久開始),表現為沿 Blaschko 線分布的多個小型與中型水疱;(b) 疣狀期 (約 2 至 8 週齡之間),由疣狀斑塊 (wart-like plaques) 構成;(c) 色素過度沉著期 (數月齡至成年);以及 (d) 色素減退期 (從嬰兒期開始) (圖 77-1)。色素過度沉著的程度因人而異,但沿 Blaschko 線呈條紋與渦輪狀出現。雖然它可出現於四肢,但通常以軀幹上最為明顯。色素減退期的特徵是沿 Blaschko 線分布的線狀、萎縮性、無毛的瘢痕。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
IP 是一種 X 連鎖、顯性遺傳的疾病,被認為在大多數男性中為胚胎致死。在大多數病例中,IP 是由位於 X 染色體 Xq28 上的 IKBKG 基因 (先前稱為 NEMO [nuclear factor κB (NF-κB) essential modulator,核因子 κB 必需調節因子]) 突變所引起。該基因編碼 IKappaB 激酶複合體的一個調節成分,此複合體可活化 NF-κB 路徑,而後者是保護細胞免於腫瘤壞死因子-α (tumor necrosis factor-α) 誘導之凋亡 (apoptosis) 所必需的。
在患有 IP 的女性中,兩條 X 染色體中的一條會在胚胎發育期間經由一種稱為 lyonization (萊昂化) 的過程而失活。表達缺陷 IKBKG 基因的表皮細胞會沿 Blaschko 線形成典型的皮膚病灶,反映出受影響角質細胞 (keratinocytes) 的胚胎遷移路徑。水疱期的皮膚病灶代表那群無法活化 NF-κB 的 IKBKG 缺陷細胞,導致凋亡。IKBKG 缺陷細胞的數量因凋亡而減少,並被表達正常等位基因的細胞所取代。隨後,發炎與水疱階段結束。第二階段的過度增生很可能是正常 IKBKG 角質細胞代償性增生的結果。第三階段的色素過度沉著源於表皮黑色素 (epidermal melanin) 的失禁,這些色素隨後遷移進入真皮並被巨噬細胞 (macrophages) 吞噬。皮膚外表現出現於相當數量的 IP 病人。眼部 (30% 至 70%)、中樞神經系統 (40%)、牙齒與骨骼異常是其中最常見者。
診斷 (DIAGNOSIS)
組織學上,黑色素細胞的數量看似正常,但也曾觀察到黑色素細胞數量減少。較薄的表皮以及真皮中皮膚附屬器的缺失或減少,可能造成色素減退的印象。在組織學上,色素沉著的區域顯示許多載滿黑色素的噬黑色素細胞 (melanophages)、基底細胞層與真皮中廣泛的黑色素沉積。亦可見表皮基底層的空泡化與退化。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
色素過度沉著通常會在數年間逐漸消退,留下色素減退的皮膚 (第 4 期),代表發炎後的真皮瘢痕。
治療 (MANAGEMENT)
已有報導指出局部類固醇 (topical steroids) 與局部 tacrolimus 在水疱期具有有益效果。
色素減退與萎縮性瘢痕可能適合以非培養表皮細胞移植 (noncultured epidermal cell grafting) 治療。
先天性角化不良 (Dyskeratosis Congenita)
重點一覽 (AT-A-GLANCE)
■ 先天性瀰漫網狀色素過度沉著,伴隨甲萎縮與白斑 (leukoplakia)。
■ 在出生後最初幾年顯現。
■ 皮膚外表現為骨髓衰竭與惡性腫瘤。
先天性角化不良 (dyskeratosis congenita, DKC) 或 Zinsser-Cole-Engman 症候群,以皮膚與血液學表現為特徵。
臨床特徵 (CLINICAL FEATURES)
DKC 患者可見特別位於頸部與胸部的網狀皮膚色素沉著、手指甲與腳趾甲的甲萎縮,以及白斑。皮膚外表現包括骨髓衰竭 (見於超過 80% 的病例) 以及在生命第二與第三個十年出現的惡性腫瘤。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在所有 DKC 病例中,致病性突變都存在於端粒酶複合體 (telomerase complex) 的組成成分中。快速分裂的體細胞表達低但可偵測到的端粒酶活性,可減緩每一輪 DNA 複製所發生的端粒縮短,而端粒縮短最終會導致細胞老化 (senescence,永久喪失增殖能力)。現在認為 DKC 是由端粒維持缺陷所引起,此缺陷限制了造血與上皮細胞的增殖能力。老化的黑色素細胞中會發生黑色素合成增加,這很可能解釋了 DKC 中所觀察到的色素變化。臨界短的端粒可能迫使細胞進入「複製危機 (replicative crisis)」,此時在缺乏端粒酶的情況下,活化一種延長端粒的替代性「ALT」機制,可能導致惡性腫瘤的發生。端粒在細胞生物學 (細胞老化) 中的角色,實際上最早正是透過在 DKC 中發現短端粒而得到證實。
X 連鎖型 DKC 是由位於 Xq28 的 DKC1 基因突變所引起,該基因編碼 dyskerin。攜帶一個突變等位基因的女性,因未受影響等位基因表達正常的端粒酶而受到保護。在自體顯性 DKC 中,大多數病例是由 TERC (端粒酶複合體的 RNA 成分) 突變所引起。TERT (telomerase reverse transcriptase,端粒酶逆轉錄酶) 在自體顯性 DKC 中受影響的頻率低得多。在自體隱性型 DKC 中,則涉及端粒酶相關蛋白如 NOP10、NHP2 與 TINF2 的突變。
診斷 (DIAGNOSIS)
色素過度沉著皮膚的切片並不顯示特異性變化。組織學上通常可觀察到表皮萎縮,以及上真皮中帶有眾多噬黑色素細胞的慢性發炎細胞浸潤。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
DKC 可能與 Fanconi 症候群混淆,後者的特徵是身材矮小、拇指發育不全或缺如,以及腕骨數量減少。Fanconi 症候群中軀幹、頸部、鼠蹊部與腋窩區域的斑駁色素過度沉著出現得比 DKC 更早,亦即在出生後最初幾年。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
自體顯性型的預後優於其他型別,可能是因為存在一個未受影響的等位基因,保留了部分端粒酶活性。通常色素過度沉著會在數年後逐漸消退。
Naegeli-Franceschetti-Jadassohn 症候群 (Naegeli-Franceschetti-Jadassohn Syndrome)
重點一覽 (AT-A-GLANCE)
■ 自體外胚層發育不良 (ectodermal dysplasias)。
■ 頸部與腋窩的網狀色素過度沉著。
■ 甲、牙齒與出汗異常。
臨床特徵 (CLINICAL FEATURES)
網狀色素過度沉著在頸部與腋窩最為明顯。掌蹠瀰漫性角皮症 (palmoplantar diffuse keratoderma)、皮膚紋理 (dermatoglyphs) 缺失、甲與牙齒變化,以及因出汗減少或缺如所致的耐熱不良,均為其特徵。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
Naegeli-Franceschetti-Jadassohn 症候群與相關的網狀色素性皮膚病 (dermatopathia pigmentosa reticularis) 是自體顯性外胚層發育不良,由 KRT14 基因突變所引起。KRT14 基因編碼角蛋白 14 (keratin 14),它與角蛋白 5 (keratin 5) 一同形成中間角蛋白絲 (intermediate keratin filaments)。角蛋白 14 主要由表皮基底細胞層的角質細胞所產生。KRT14 基因突變導致基底角質細胞的脆弱性。它在皮膚紋理與汗腺的個體發生 (ontogenesis) 期間扮演重要角色。
網狀色素性皮膚病與 Naegeli-Franceschetti-Jadassohn 症候群的區別在於皮膚色素過度沉著終生持續、部分禿髮,以及無牙齒異常。這兩種疾病都已被定位到 17q11.2-q21,並被認為是等位的 (allelic)。
組織學 (HISTOLOGY)
上真皮中有眾多噬黑色素細胞,緊鄰斑駁的表皮色素過度沉著。外分泌腺 (eccrine glands) 在組織學上的數量與結構看似正常。
網狀色素性皮膚病 (Dermatopathia Pigmentosa Reticularis)
重點一覽 (AT-A-GLANCE)
■ 自體顯性。
■ 軀幹的網狀色素過度沉著。
■ 甲、出汗與眼部異常,伴隨非瘢痕性禿髮。
臨床特徵 (CLINICAL FEATURES)
此病可見軀幹的網狀色素過度沉著、伴點狀加重的掌蹠角皮症、甲與眼部變化、非瘢痕性 (noncicatricial) 禿髮、魚鱗癬 (ichthyosis)、少汗症 (hypohidrosis)、廣泛的角化過度病灶、指趾自截 (ainhum) 形成、嚴重的牙周病、機械性水疱形成,以及口腔黏膜色素沉著。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
網狀色素性皮膚病是一種罕見的自體顯性疾病。
組織學 (HISTOLOGY)
組織學檢查顯示明顯的色素失禁、基底細胞層的液化退化,以及真皮膠原的玻璃樣變 (hyalinization)。
Dowling-Degos 病 (Dowling-Degos Disease)
重點一覽 (AT-A-GLANCE)
■ 自體顯性。
■ 屈側的網狀色素過度沉著,伴隨痤瘡樣口周凹陷,以及粉刺樣 (comedo-like) 角化過度丘疹。
臨床特徵 (CLINICAL FEATURES)
眾多細小的對稱性棕灰色斑通常於生命第三或第四個十年開始於鼠蹊部與腋窩,隨後擴散至臀間褶、乳房下褶、頸部、軀幹與手臂。頸部與腋窩也會出現粉刺樣角化過度的毛囊性丘疹,以及凹陷的口周痤瘡樣瘢痕。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
這種罕見的自體顯性遺傳性皮膚病 (同義詞:屈側網狀色素異常 reticular pigmented anomaly of the flexures) 是由位於 12q13.13 染色體上 KRT5 基因的突變所引起。Betz 與其同事證實,角蛋白 5 的單倍體不足 (haploinsufficiency) 會造成上皮重塑、黑色素小體 (melanosome) 錯誤定位,以及中間絲核周組織的改變。角蛋白 5 與角蛋白 14 是共同形成角蛋白中間絲的兩種蛋白。假設認為角蛋白 5 單倍體不足會造成角蛋白 14 過量,後者可能藉由與運輸接合蛋白 (transport adapters) 競爭而導致 Dowling-Degos 病的病理。角蛋白可能調節角質細胞中 AP-3 複合體的可用性與定位,或者可能調節 AP-3 依賴性囊泡與動力蛋白 (motor proteins) 的相互作用。
組織學 (HISTOLOGY)
其組織學非常具特徵性,可見來自表皮與毛囊漏斗部的絲狀表皮突 (filiform rete projections),且帶有色素過度沉著的尖端,黑色素細胞並未增加。本病須與黑色棘皮症 (acanthosis nigricans) 區別,後者具有不同的組織學外觀。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
色素過度沉著的進展緩慢,通常自成年早期開始可見,無性別偏好。
Galli-Galli 病 (Galli-Galli Disease)
Galli-Galli 病的病人呈現 Dowling-Degos 病的診斷性特徵,並額外具有表皮基底上層棘層鬆解 (acantholysis) 的組織病理學發現。它被視為 Dowling-Degos 病的棘層鬆解變異型。在某些病人中已發現角蛋白 5 基因的突變。
Kitamura 網狀肢端色素沉著 (Kitamura Reticular Acropigmentation)
重點一覽 (AT-A-GLANCE)
■ 自體顯性。
■ 屈側的網狀色素過度沉著,伴隨痤瘡樣口周凹陷,以及粉刺樣角化過度丘疹。
臨床特徵 (CLINICAL FEATURES)
Kitamura 網狀肢端色素沉著的特徵是有稜角且界線分明的網狀、雀斑樣色素過度沉著斑 (但無 Dohi 疾病所見的色素減退 [見「混合性色素減退與色素過度沉著」一節]),於生命第一個十年開始於手背,隨後擴散至身體其餘部位。這些斑略呈凹陷;有時可觀察到掌部凹陷。大多數病例來自日本。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
ADAM10 基因 (15q21.3) 的突變。ADAM10 編碼一種鋅金屬蛋白酶、一種解離素 (disintegrin),以及一種含金屬蛋白酶結構域的蛋白 10,其參與皮膚受質的脫落 (shedding)。
組織學 (HISTOLOGY)
組織學檢查可見黑色素細胞數量與黑色素生成活性 (melanogenic activity) 增加,並存在表皮萎縮。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
Dowling-Degos 病、Dohi 肢端色素沉著 (acropigmentation of Dohi),以及網狀色素性皮膚病,都在 Kitamura 網狀肢端色素沉著的鑑別診斷之列。
Haber 症候群 (Haber Syndrome)
除了軀幹與腋窩的網狀色素沉著外,Haber 病的病人還會發展出軀幹的疣狀丘疹病灶,以及一種獨特的光敏性、酒糟樣 (rosacea-like) 顏面紅斑與毛細血管擴張,最常於兒童期表現。
太田母斑 (Nevus of Ota)
重點一覽 (AT-A-GLANCE)
■ 先天性局限性黃褐色色素過度沉著,伴隨真皮黑色素細胞增多症 (dermal melanocytosis)。
■ 位於三叉神經第一與第二分支區域的單側色素沉著。
■ 受影響者中有 60% 有鞏膜侵犯。
■ 主要發生於亞洲女性。
太田母斑 (nevus of Ota,眼上頜青褐色痣 nevus fuscoceruleus ophthalmomaxillaris) 最早由 Ota 於 1939 年描述。其特徵是顏面上呈藍黑色或灰棕色的真皮黑色素細胞性色素沉著。
流行病學 (EPIDEMIOLOGY)
儘管太田母斑已在不同膚質中被描述,但它最常見於亞洲族群,該族群中有 0.6% 的人受影響。發病通常見於出生時與女性,但也可能在兒童早期或青春期首次被注意到。
臨床特徵 (CLINICAL FEATURES)
單側的藍黑色或灰棕色真皮黑色素細胞性色素沉著,典型見於三叉神經第一與第二分支所支配的區域。太田母斑現今被進一步分類為輕度 (第 1 型)、中度 (第 2 型)、重度 (第 3 型) 與雙側 (第 4 型)。受影響者中有 60% 有鞏膜色素沉著。其他黏膜色素沉著的部位包括結膜與鼓膜 (眼皮膚黑色素細胞增多症 oculodermal melanocytosis) (圖 77-2)。惡性黑色素瘤罕見地可能在太田母斑中發生。這使得對病灶進行仔細追蹤成為必要,尤其當它發生於白人病人時,後者的惡性退變似乎較為頻繁。與太田母斑相關的惡性黑色素細胞性腫瘤已被證實會出現在脈絡膜、腦、眼眶、虹膜、睫狀體與視神經。此外,亦有與同側青光眼及顱內黑色素細胞增多症相關的描述。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
已描述各種觸發因素,包括感染、創傷、紫外線曝曬與荷爾蒙影響。
診斷 (DIAGNOSIS)
診斷通常以臨床做出,但可由組織學確認,後者顯示黑色素細胞均勻分布於整個真皮中。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
雙側病例應與 Hori 痣 (Hori nevus) 區別,後者為後天獲得性、無黏膜侵犯且色素較淺。其他真皮黑色素細胞增多症包括伊藤母斑 (nevus of Ito)、蒙古斑 (Mongolian spots) 與真皮黑色素細胞錯構瘤 (dermal melanocyte hamartomas) (表 77-1)。在斑痣性血管畸形 (phakomatosis pigmentovascularis,葡萄酒色斑型、Klippel-Trenaunay 或 Sturge-Weber 症候群) 中已描述相關的血管畸形。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
太田母斑為持續性,通常不會自發性消退。
治療 (MANAGEMENT)
局部療法對這種真皮色素沉著的治療無效。儘管過去曾使用冷凍治療與手術,但因會造成顯著瘢痕而應避免。雷射手術是首選治療,其中以品質開關 (quality-switched, QS) 雷射最為成功,包括品質開關紅寶石雷射、QS 紫翠玉 (alexandrite) 雷射,以及 QS 銣:釔鋁石榴石 (neodymium:yttrium-aluminum-garnet, Nd:YAG) 雷射。皮秒雷射 (picosecond lasers) 亦曾被報導對此病的治療有效。
伊藤母斑 (Nevus of Ito)
重點一覽 (AT-A-GLANCE)
■ 先天性局限性棕褐色色素過度沉著,伴隨真皮黑色素細胞增多症。
■ 被視為太田母斑的變異型。
■ 侵犯肩鎖 (acromioclavicular) 與三角肌 (deltoid) 區域。
伊藤母斑 (nevus of Ito) 是一種先天性真皮黑色素細胞增多症,最早由 Ito 於 1954 年描述為肩峰三角肌青褐色痣 (nevus fuscoceruleus acromiodeltoideus)。它可被視為太田母斑的變異型。其臨床、人口統計與組織學特徵與太田母斑相似,且兩種病灶可同時發生 (見表 77-1 與圖 77-3)。
蒙古斑 (Mongolian Spots)
重點一覽 (AT-A-GLANCE)
■ 由真皮黑色素細胞增多症所引起的先天性局限性帶藍色色素過度沉著。
■ 主要發生於有色皮膚 (skin of color) 的人。
■ 通常發生於薦骨 (sacral) 區域。
流行病學 (EPIDEMIOLOGY)
蒙古斑在非洲、亞洲與西班牙裔族群中較為常見,而在白人中僅罕見。它發生於兩性,並略以男性為主。
表 77-1:真皮黑色素細胞增多症 (Dermal Melanocytosis) 的鑑別診斷與治療
| 項目 | 太田母斑 (NEVUS OF OTA) | 伊藤母斑 (NEVUS OF ITO) | 蒙古斑 (MONGOLIAN SPOT) | Hori 痣 (NEVUS OF HORI) | 真皮黑色素細胞錯構瘤 (DERMAL MELANOCYTE HAMARTOMA) |
|---|---|---|---|---|---|
| 流行病學 (Epidemiology) | 多為先天性;亞洲與女性為主 | 多為先天性;亞洲與女性為主 | 偶發性 (罕見家族性病例);亞洲、非洲與西班牙裔族群,略以男性為主 | 後天獲得性;常為家族性;亞洲與女性為主 | 偶發性 (罕見家族性病例);家族性或偶發性 |
| 臨床表現 (Clinical presentation) | 藍至板岩灰色斑駁狀斑性色素過度沉著 | 藍至板岩灰色斑駁狀斑性色素過度沉著 | 均勻的藍至板岩灰色斑性色素過度沉著 | 由棕藍色進展為板岩灰色的斑駁狀斑性色素過度沉著 | 斑駁狀色素過度沉著,於瀰漫性色素斑片中可見小型藍灰色斑 |
| 分布 (Distribution) | 三叉神經 (trigeminal nerve) | 肩鎖神經 (acromioclavicular nerve) | 下背部與薦骨 | 尤其臉頰顴部 (malar region),亦見於前額、上眼瞼、顳部 | 皮節分布 (dermatomal distribution) |
| 組織學 (Histology) | 紡錘形黑色素細胞瀰漫分布於各真皮層;有時呈較帶狀的黑色素細胞增生與基質纖維化反應 | 紡錘形黑色素細胞瀰漫分布於各真皮層;有時呈較帶狀的黑色素細胞增生與基質纖維化反應 | 紡錘形黑色素細胞瀰漫分布於各真皮層 | 真皮黑色素細胞位於上真皮與中真皮 | 真皮黑色素細胞位於真皮上三分之二 (包括乳頭下層) |
| 治療 (Therapy) | 品質開關雷射;冷凍治療;手術 | 品質開關雷射;冷凍治療;手術 | 通常於兒童期自發性消退 | 品質開關雷射併用漂白霜與化學換膚 | 無 |
| 相關特徵 (Associated features) | 罕見惡性轉變 | 無醫學上需顧慮的相關特徵 | 可能與先天性代謝異常 (inborn errors of metabolism) 相關 | 無醫學上需顧慮的相關特徵 | 無 |
臨床特徵 (CLINICAL FEATURES)
最常見的是位於薦骨區域、界線分明、帶藍色的色素過度沉著斑 (圖 77-4)。蒙古斑也可見於臀部與腰部區域,以及胸部、腹部、手臂、腿部與肩部。已描述數個廣泛蒙古斑的病例,侵犯軀幹與四肢的大片區域,並與先天性代謝異常相關,例如溶酶體儲積病 (lysosomal storage diseases)、GM1-神經節苷脂貯積症 (GM1-gangliosidosis) 與黏多醣貯積症 (mucopolysaccharidosis)。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
組織學上,這些斑由位於下真皮、在胎兒期間未能遷移至真皮-表皮交界處的紡錘形黑色素細胞所構成。
診斷 (DIAGNOSIS)
診斷通常以臨床做出,但可由組織學確認。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
在大多數病例中,蒙古斑會在兒童期自發性消退,但也曾描述持續至成年的情況。
治療 (MANAGEMENT)
自發性消退通常意味著可避免治療,但在兒童期或青春期進行雷射治療可獲得良好結果,尤其是薦骨部蒙古斑。
真皮黑色素細胞錯構瘤 (Dermal Melanocyte Hamartoma)
重點一覽 (AT-A-GLANCE)
■ 先天性、局限性、帶藍色的色素過度沉著,伴隨真皮黑色素細胞增多症。
■ 已描述皮節型態 (dermatomal patterns)。
真皮黑色素細胞錯構瘤是一種特殊型態的先天性真皮黑色素細胞增多症,最早由 Burkhart 與其同事於 1981 年描述。由位於真皮中的黑色素細胞所引起的灰藍色色素沉著,以皮節型態出現。
家族性小痣病症候群 (Familial Lentiginosis Syndromes)
重點一覽 (AT-A-GLANCE)
■ 造成界線分明的色素沉著並伴隨小痣病 (lentiginosis) 的先天性疾病。
■ 病因包括 Peutz-Jeghers 症候群、LEOPARD (小痣 lentigines、心電圖異常 electrocardiographic abnormalities、眼距過寬 ocular hypertelorism、肺動脈狹窄 pulmonary stenosis、生殖器異常 abnormalities of genitalia、生長遲緩 retardation of growth,以及耳聾 deafness) 症候群、Carney 複合症、Bannayan-Ruvalcaba-Riley 症候群,以及中顏面小痣病 (centrofacial lentiginosis)。
家族性小痣病症候群的特徵是存在小痣 (lentigines)——界線分明的棕色斑 (通常直徑 <5 mm),這些病灶在表皮中具有增多數量的黑色素細胞 (表皮黑色素細胞性色素過度沉著 epidermal melanocytic hypermelanosis),並有心血管、內分泌或胃腸道腫瘤的發生率增加。家族性小痣病症候群包括 Carney 複合症、Peutz-Jeghers 症候群 (PJS)、LEOPARD 症候群 (小痣、心電圖傳導缺陷、眼距過寬、肺動脈狹窄、生殖器異常、生長遲緩,以及感覺神經性耳聾)、動脈剝離與小痣病、Laugier-Hunziker 症候群、家族性良性小痣病、Bannayan-Ruvalcaba-Riley 症候群 (BRRS)、中顏面小痣病,以及節段性與聚集性小痣病 (segmental and agminated lentiginosis)。Carney 複合症、PJS 與 BRRS 的基因座與基因突變已被鑑定。
Peutz-Jeghers 症候群 (Peutz-Jeghers Syndrome)
PJS 最早由 Peutz (1921) 與 Jeghers (1949) 描述。它是一種罕見的自體顯性遺傳性皮膚病,具有發展惡性腫瘤的傾向。超過半數的 PJS 病例可歸因於位於第 19 號染色體 (19q13.3) 上 STK11 (LKB1) 的突變,該基因被認為作為腫瘤抑制基因 (tumor-suppressor gene)。對 PJS 病人自年輕時起密切監測惡性腫瘤是有必要的。黏膜皮膚色素沉著與腸道錯構瘤性息肉病 (intestinal hamartomatous polyposis) 是本病的標誌 (圖 77-5)。色素病灶類似 Carney 複合症者,為小型的色素過度沉著 (棕灰色) 斑,典型於兒童期出現 (出生時不存在) 於嘴唇與頰黏膜;但它們也可能侵犯眼瞼、手與足。品質開關雷射或強脈衝光 (intense pulsed light) 可用於治療色素病灶。與 PJS 相關最常見的惡性腫瘤為胃腸道者 (小腸、結直腸、胃、胰臟),但也曾描述非胃腸道腫瘤,例如乳房、子宮頸與內分泌腫瘤 (甲狀腺、睪丸、卵巢)。
LEOPARD 症候群 (LEOPARD Syndrome)
LEOPARD 症候群是一種罕見的自體顯性疾病,由 PTPN11 基因的雜合錯義突變 (heterozygous missense mutation) 所引起,該基因編碼蛋白酪胺酸磷酸酶 SHP-2,並位於第 12 號染色體 (12q24.1)。LEOPARD 症候群與 Noonan 症候群為等位 (allelic),並與此疾病共有數項臨床特徵。特徵性的小痣通常於兒童期與生命最初幾個月發展 (圖 77-6)。臨床診斷主要基於典型的顏面特徵,以及肥厚型心肌病 (hypertrophic cardiomyopathy) 與/或咖啡牛奶斑 (café-au-lait macules, CALMs) 的存在。近期在 2 名無法發現 PTPN11 突變的 LEOPARD 症候群病人中,發現了 RAF1 基因的錯義突變。
Carney 複合症 (Carney Complex)
Carney 複合症是一種自體顯性疾病,最早由 Carney 與其同事於 1985 年描述。
Carney 複合症的臨床組成包括斑點狀皮膚色素沉著、黏液瘤 (myxomas,心臟、皮膚、乳房)、內分泌腫瘤 (原發性色素性結節性腎上腺疾病 [Cushing 症候群]、睪丸大細胞鈣化型 Sertoli 細胞瘤 [性早熟]、垂體腺瘤 [肢端肥大症]、甲狀腺腫瘤、卵巢囊腫),以及神經鞘瘤 (schwannomas)。已描述多種型態的色素過度沉著,例如小痣、雀斑 (ephelides)、藍痣 (blue nevi)、交界痣、真皮痣與複合痣,以及咖啡牛奶斑。典型病灶由斑點狀的中顏面色素沉著構成,侵犯嘴唇的唇紅緣 (vermilion border)、淚阜 (lacrimal caruncle)、結膜半月皺襞 (conjunctival semilunar fold),有時還有鞏膜。口內色素斑點存在於有限數量的病例中。在至少半數的 Carney 複合症病人中,已鑑定出編碼蛋白激酶 A 調節亞單位 1A (PRKAR1A) 的基因突變,該基因定位於 17q22-24。
Bannayan-Riley-Ruvalcaba 症候群 (Bannayan-Riley-Ruvalcaba Syndrome)
BRRS 是第二種自體顯性錯構瘤性息肉病症候群。此命名取代了三個先前描述的疾病:(a) Riley-Smith、(b) Bannayan-Zonana,以及 (c) Ruvalcaba-Myhre-Smith 症候群。其特徵是巨頭畸形 (macrocephaly)、生殖器小痣病 (genital lentiginosis) 與腸道息肉病的經典三聯徵。除生殖器小痣病 (有時表現為較大的 CALMs) 外,黏膜皮膚表現包括血管畸形、脂肪瘤病 (lipomatosis)、多發性軟纖維瘤 (acrochordons),以及疣狀顏面丘疹。超過 60% 的 BRRS 病人在第 10 號染色體 10q23.3 上的 PTEN (phosphatase and tensin homolog) 基因顯示生殖系突變 (germline mutations),該基因在 Cowden 症候群中發生突變,而後者與 BRRS 有部分臨床重疊。近期已描述 PTEN 錯構瘤腫瘤症候群 (PTEN hamartoma tumor syndrome),涵蓋 4 種等位疾病 (Cowden 症候群、BRRS、Proteus 症候群與類 Proteus 症候群)。
中顏面小痣病 (Centrofacial Lentiginosis)
橫跨中央顏面的一條水平小痣帶,是自體顯性中顏面小痣病症候群的標誌。其他相關的徵象與症狀包括骨骼異常、內分泌功能障礙、神經管缺損 (neural tube defects),以及智能遲緩。
家族性咖啡牛奶斑症候群 (Familial Café-au-Lait Syndromes)
重點一覽 (AT-A-GLANCE)
■ 在諸如家族性多發性 CALMs (Legius 症候群)、第 1 型神經纖維瘤病 (neurofibromatosis Type 1)、McCune-Albright 症候群、Bloom 症候群、Watson 症候群與 Silver-Russell 症候群等疾病中,可見伴隨咖啡牛奶斑 (CALMs) 的先天性局限性色素過度沉著。
■ 伴隨 CALMs 的界線分明色素過度沉著。
CALMs 由界線分明的色素過度沉著皮膚斑片構成,大小從 0.5 cm 至超過 20 cm 不等。它們常於出生時即存在或在出生後最初幾個月出現。所有新生兒中有 0.3% 至 18% 呈現孤立性 CALMs。組織學上,孤立性 CALMs 顯示正常數量的黑色素細胞但表皮黑色素增加 (表皮黑色素性色素過度沉著 epidermal melanotic hypermelanosis)。多發性 CALMs 是數種多系統疾病眾所周知的標記。
Legius 症候群 (Legius Syndrome)
Legius 症候群又稱家族性多發性 CALMs,為自體顯性,具有 SPRED1 基因 (15q13.2) 的突變。多發性 CALMs 存在,並可能見到腋窩雀斑 (axillary freckling) 與脂肪瘤,但不出現神經纖維瘤 (neurofibromas)。可見巨頭畸形與發育遲緩,但較第 1 型神經纖維瘤病所見者輕微。
第 1 型神經纖維瘤病 (Neurofibromatosis Type 1)
第 1 型神經纖維瘤病 (NF1) 最早由 Friedrich von Recklinghausen 於 1882 年認識,亦稱 von Recklinghausen 病 (詳細討論見第 135 章)。NF1 是一種自體顯性疾病,由編碼神經纖維瘤蛋白 (neurofibromin) 的 NF1 基因突變所引起。神經纖維瘤蛋白最重要的功能涉及對 Ras 訊號傳導路徑的下調 (downregulation);因此,它被視為腫瘤抑制基因。NF1 已被視為一種神經嵴病 (neurocristopathy),其特徵是若干與色素細胞相關的皮膚與非皮膚表現,例如 CALMs、間擦部位雀斑 (intertriginous freckling),以及虹膜 Lisch 結節 (iris Lisch nodules)。(第 135 章討論 NF1 的臨床診斷準則。) 在青春期前個體中存在 6 個或更多最大直徑大於 5 mm 的 CALMs,或在青春期後存在大於 15 mm 者,是本病的標誌之一。與孤立性 CALMs 相反,NF1 相關的 CALMs 在表皮中含有顯著增多數量的黑色素細胞。間擦部位雀斑為 NF1 的特異性病徵 (pathognomonic),同樣呈現增多數量的表皮黑色素細胞,這使其與普通雀斑 (ephelides) 區別。
McCune-Albright 症候群 (McCune-Albright Syndrome)
McCune-Albright 症候群最早由 McCune (1936) 與 Albright (1937) 描述為多發/單發性纖維性骨發育不良 (poly/monostotic fibrous dysplasia)、CALMs,以及功能亢進性內分泌病變的三聯徵,後者包括性早熟、甲狀腺功能亢進、皮質醇增多症、生長激素增多症 (hypersomatotropism),以及低磷血症性佝僂病。其 CALMs 數量較少,且邊界比 NF1 所見者更不規則。它們典型以中線為界。McCune-Albright 症候群是由調控環腺苷單磷酸 (cyclic adenosine monophosphate) 的 Gs 蛋白 α 次單位之合子後活化突變 (postzygotic activating mutation) 所引起。因此,帶有組成型活化腺苷酸環化酶 (constitutively active adenylate cyclase) 的 McCune-Albright 症候群細胞呈鑲嵌分布。
Bloom 症候群 (Bloom Syndrome)
Bloom 症候群是一種罕見的自體隱性遺傳性免疫缺陷與癌症易感性症候群。其特徵是生長缺陷、不尋常的顏面、CALMs,以及伴隨毛細血管擴張與紅斑的日光敏感性,後者可導致萎縮與瘢痕。Bloom 症候群中頻繁發生的腫瘤為急性白血病、淋巴瘤與鱗狀細胞癌。Bloom 症候群是由一種 DNA 修復缺陷所引起,導致基因組不穩定,包括姊妹染色分體交換 (sister-chromatid exchanges) 的高頻率。
致病基因 BLM 已被定位到 15q26.1,編碼 BLM RecQ helicase 同源蛋白,這是 DNA 解旋酶家族中眾所周知的成員,與在 Werner 症候群及 Rothmund-Thomson 症候群中同樣有缺陷的解旋酶密切相關。
Watson 症候群 (Watson Syndrome)
Watson 症候群是一種自體顯性疾病,由肺動脈瓣狹窄、CALMs 與智能較低所構成,最早由 Watson 於 1967 年描述。後來,Watson 症候群的臨床表現型擴展至包含與 NF1 重疊的特徵 (巨頭畸形、Lisch 結節、神經纖維瘤、身材矮小、間擦部位雀斑),並被證實與 NF1 為等位。近期的研究顯示 Watson 症候群與神經纖維瘤病-Noonan 症候群有重疊。
Silver-Russell 症候群 (Silver-Russell Syndrome)
Silver-Russell 症候群 (SRS) 是一種臨床與遺傳上異質性的疾病,最早由 Silver (1953) 與 Russell (1954) 描述。本病的主要特徵為低出生體重、因子宮內與出生後生長遲緩所致的身材矮小,以及一張小而呈三角形的臉。其他相關症狀包括第五指的指彎曲 (clinodactyly)、相對巨頭畸形,以及顏面、肢體或身體不對稱。CALMs 是 SRS 的一項可變特徵。多達 20% 的 SRS 病人存在 1 或 2 個 CALMs。許多 SRS 病例為偶發性,但也曾描述具有不同遺傳模式的家族性病例。約 10% 的病人發生第 7 號染色體的母系單親二體 (maternal uniparental disomy),並已在此染色體上描述數個 SRS 基因突變的候選區域 (7p11.2-p13、7q31-qter 與 7q21)。其他可能的候選基因可能位於第 17 號染色體長臂或第 11 號染色體 11p15。近期,第 11p15 號染色體 H19 印記區域 (imprinted domain) 的甲基化缺陷被認為與 35% 至 65% 的 SRS 病例有關。
後天性色素過度沉著 (ACQUIRED HYPERMELANOSIS)
內分泌病變 (Endocrinopathies)
重點一覽 (AT-A-GLANCE)
■ 在諸如 Addison 病、Cushing 症候群、Nelson 症候群、嗜鉻細胞瘤 (pheochromocytoma)、類癌症候群 (carcinoid syndrome)、甲狀腺功能亢進、黑色棘皮症與糖尿病等疾病中,可見瀰漫、無型態 (nonpatterned) 的色素過度沉著。
■ 在諸如黑色棘皮症與成熟性色素異常 (maturational dyschromia) 等疾病中,可見局限性色素過度沉著。
Addison 病 (Addison Disease)
第 137 章詳細討論內分泌疾病。Addison 病是一種臨床症候群,以失鹽 (salt-wasting) 與瀰漫性皮膚色素過度沉著為特徵。在已開發世界中,Addison 病通常為自體免疫性。它與腎上腺功能不全 (adrenal insufficiency) 相關,後者伴隨皮質類固醇與雄激素荷爾蒙分泌不足,導致垂體代償性過度產生促腎上腺皮質激素 (adrenocorticotropic hormone, ACTH)。色素過度沉著是慢性 Addison 病病人最顯著的皮膚徵象,是 ACTH 結合至黑皮質素-1 受體 (melanocortin-1 receptor) 的結果。色素過度沉著優先發生於日光曝曬區域 (臉、頸、手)、創傷部位、瘢痕、慢性受壓處 (膝、脊柱、指關節、肘、肩),以及掌紋、乳頭、乳暈、腋窩、會陰與生殖器。
Cushing 症候群 (Cushing Syndrome)
第 137 章詳細討論內分泌疾病。Cushing 症候群的特徵是由慢性糖皮質素過量所引起的臨床徵象與症狀。可見各種程度的色素過度沉著。它通常在異位性 ACTH 症候群 (ectopic ACTH syndrome) 的病人中最為嚴重。病人呈現全身性色素過度沉著,並於日光曝曬、慢性輕度創傷與受壓區域 (肩、腹部、腰、肘、指關節、脊柱、膝),以及黏膜表面加重。
Nelson 症候群 (Nelson Syndrome)
Nelson 症候群包含一個逐漸增大的垂體腫瘤,伴隨空腹血漿 ACTH 濃度升高、色素過度沉著,以及神經眼科症狀,見於患有 Cushing 病、在雙側腎上腺切除術後且荷爾蒙替代不足的病人。
嗜鉻細胞瘤 (Pheochromocytoma)
嗜鉻細胞瘤是腎上腺髓質的嗜鉻細胞腫瘤,伴隨兒茶酚胺 (catecholamines) 的過度產生。可觀察到因血管收縮所致的顏面蒼白。相反地,亦曾報導類 Addison 病樣的色素過度沉著,這可能是由腫瘤產生異位性 ACTH 與黑色素細胞刺激素 (melanocyte-stimulating hormone) 所引起。色素沉著在手術治療後迅速消退。
類癌症候群 (Carcinoid Syndrome)
在類癌症候群中曾描述由產生黑色素細胞刺激素的腫瘤 (例如胃或胸腺類癌腫瘤) 所致的瀰漫性色素過度沉著。類癌症候群也可伴隨發生於光照皮膚的糙皮病樣 (pellagra-like) 皮疹。此皮疹繼發於色胺酸 (tryptophan) 缺乏,因為大量的膳食色胺酸被腫瘤轉向合成血清素 (serotonin)。治療潛在腫瘤對受影響病人的處置至關重要。
甲狀腺功能亢進 (Hyperthyroidism)
第 137 章詳細討論內分泌疾病。甲狀腺毒症 (thyrotoxicosis) 有多種病因。最常見的原因是 Graves 病,其特徵是循環中存在對抗甲狀腺刺激素受體 (thyroid-stimulating hormone receptors) 的抗體。在大型病例系列中,甲狀腺毒症病人發生色素過度沉著的比率估計從 2% 高至 40%。增加的皮膚色素沉著可為局限性或全身性,且在深色皮膚者中較為常見。色素過度沉著的分布常類似 Addison 病,色素沉積於掌蹠的皺褶處。然而,與 Addison 病相反,黏膜的侵犯不常見,且乳頭與生殖器皮膚的色素沉著較不顯著。與甲狀腺毒症相關的色素過度沉著被認為是垂體 ACTH 釋放增加的結果,以代償加速的皮質醇降解。據報導,色素過度沉著對甲狀腺功能亢進治療的反應不佳。自體免疫性 Graves 病曾被報導與白斑症 (vitiligo) 相關。
糖尿病 (Diabetes)
第 137 章詳細討論糖尿病。糖尿病性皮膚病 (diabetic dermopathy) 的特徵是位於小腿前側、無症狀、不規則、淺棕色的凹陷斑片,常於輕度創傷或水疱出現後發生。其致病機轉未明。
黑色棘皮症 (Acanthosis Nigricans)
黑色棘皮症的特徵是增厚的色素過度沉著、絨毛狀皮膚,最常見於頸部、腋窩、膕窩與肘前窩,以及鼠蹊褶 (圖 77-7)。然而,它也曾被描述於顏面,並應納入顏面色素過度沉著的鑑別診斷。當位於顏面時,黑色棘皮症表現為界線不清的色素過度沉著,偏好顴骨下方的顴部區域與鼻唇溝,甚至曾被描述於鼻翼上溝。隨著肥胖與非胰島素依賴型糖尿病的比率增加,認識此疾病正變得日益重要。黑色棘皮症在第 137 章有更詳細的討論。
成熟性色素異常 (Maturational Dyschromia)
成熟性色素異常是一種近期描述的疾病,見於非洲與印度族群中生命第四至第五個十年的人。它表現為位於外側前額、顳部與顴部、深棕色至黑色、界線不清的色素過度沉著區域。色素過度沉著皮膚的組織學評估顯示黑色素細胞輕度至中度增生,部分報告指出有乳頭瘤狀 (papillomatous) 表皮,暗示其與黑色棘皮症可能有關聯或連續性。其病因仍不明,需要對此疾病進行進一步研究 (圖 77-8)。
營養性疾病 (Nutritional Conditions)
第 123 章詳細討論營養性疾病。瀰漫、無型態的色素變化可繼發於營養性疾病,例如惡性營養不良症 (kwashiorkor)、維生素 B12 缺乏、葉酸缺乏,或糙皮病 (pellagra)。若營養缺乏得到矯正,皮膚變化是可逆的。
惡性營養不良症 (Kwashiorkor)
惡性營養不良症是在熱量攝取充足情況下嚴重蛋白質營養不良的結果。它是開發中國家 6 個月至 5 歲兒童重要的死亡原因,且常與胃腸道寄生蟲病相關。患有惡性營養不良症的兒童會停止增重,並變得易怒與淡漠,並發展出水腫、肝腫大、腹瀉、肌肉萎縮與畏光。大多數病例存在皮膚表現。皮膚變化通常始於顏面的色素減退。在暴露於摩擦與壓力的區域,例如膝、肘與臀部,會發展出伴隨色素過度沉著的角化過度與脫屑,使皮膚呈現「剝落油漆 (flaky paint)」外觀。移除鱗屑後留下淺表糜爛,並轉變為蒼白斑片。毛髮可能變得色素減退。有時沿髮幹會出現色素減退帶 (旗幟徵 flag sign),這是營養不良較嚴重時期的結果。惡性營養不良症的治療在於補充蛋白質來源。皮膚與毛髮的變化可經由適當的營養治療而逆轉。
惡性營養不良症樣症候群也曾被描述於繼發於腸道疾病 (如潰瘍性結腸炎) 的蛋白質流失與營養不良病例中。
維生素 B12 缺乏 (Vitamin B12 Deficiency)
鈷胺素 (cobalamin) 或維生素 B12 缺乏是由幽門螺旋桿菌 (Helicobacter pylori) 感染所致的萎縮性胃炎,或針對胃壁細胞的自體免疫過程所引起,後者導致嚴重的「內因子 (intrinsic factor)」缺乏 (惡性貧血 pernicious anemia)。血液檢查顯示巨球性貧血 (megaloblastic anemia) 與低濃度的維生素 B12。曾報導相關的全身性皮膚色素過度沉著,以及毛髮的色素減退/脫色或早期變灰。皮膚組織學顯示基底層黑色素增加。電子顯微鏡顯示黑色素細胞與周圍角質細胞中黑色素小體的數量正常。這些發現暗示,造成維生素 B12 缺乏相關色素過度沉著的是黑色素合成增加,而非黑色素運輸缺陷。補充維生素 B12 可逆轉色素過度沉著。惡性貧血是一種自體免疫疾病,曾被報導與白斑症相關。
葉酸缺乏 (Folic Acid Deficiency)
與巨球性貧血相關的葉酸缺乏曾被報導會造成皮膚色素過度沉著。
糙皮病 (Pellagra)
糙皮病是由菸鹼酸 (niacin,維生素 B3) 或其衍生物缺乏所引起。本病罕見,可由慢性酒精中毒、某些藥物 (isoniazid、抗痙攣藥)、繼發於發炎性腸道疾病的吸收不良、乳糜瀉 (celiac disease),以及類癌腫瘤所引起。糙皮病的特徵是皮膚炎 (dermatitis)、失智 (dementia) 與腹瀉 (diarrhea) 的臨床三聯徵。最初的皮膚變化是日光曝曬後的紅斑與水腫,這些變化會以暗棕紅色消退於顏面、頸部,以及手、手臂與足的背面。之後病灶會變得色素過度沉著、角化過度與脫屑,並伴有龜裂與結痂 (「鵝皮樣 goose-like」)。補充菸鹼酸可造成快速的臨床反應。
代謝性疾病 (Metabolic Conditions)
重點一覽 (AT-A-GLANCE)
■ 遲發性皮膚紫質症 (porphyria cutanea tarda) 與血色素沉著症 (hemochromatosis) 可造成瀰漫、無型態的色素過度沉著,並於光照部位加重。
遲發性皮膚紫質症 (Porphyria Cutanea Tarda)
第 124 章詳細討論紫質症 (porphyrias)。遲發性皮膚紫質症是一種影響血基質 (heme) 合成的代謝疾病。它與瀰漫性棕色色素過度沉著相關,後者於光照區域加重。在女性中,可觀察到顏面的黑斑樣 (melasma-like) 色素過度沉著。
血色素沉著症 (Hemochromatosis)
血色素沉著症是一種鐵儲積疾病,具有異質性的遺傳基礎,導致鐵調素 (hepcidin) 濃度降低。約 10% 的人口在血色素沉著症基因上有突變,與北歐人相比,非洲人與亞洲人的盛行率較低。這導致腸道鐵吸收增加,並沉積於肝臟、胰臟與其他器官,包括皮膚。由於血色素沉著症現今通常早期即可診斷,色素過度沉著較不常見。在歷史上,血色素沉著症在晚期才被診斷,具有色素過度沉著、糖尿病 (「青銅色糖尿病 bronze diabetes」) 與肝硬化的經典三聯徵。70% 的病人出現皮膚變黑,原因有兩種不同機轉:(a) 含鐵血黃素 (hemosiderin) 沉積造成瀰漫、板岩灰色的顏色,以及 (b) 表皮黑色素生成增加。色素沉著通常為全身性,但可能在光照區域、生殖器與瘢痕處更為明顯。皮膚的青銅色變化可經由放血 (phlebotomy) 逆轉,後者仍是此病治療的主要方式。Deferoxamine 最常用於次發性血色素沉著症。
腫瘤性疾病 (Tumoral Conditions)
肥大細胞疾病與黑色素瘤 (Mast Cell Disorders and Melanoma)
肥大細胞疾病與黑色素瘤可導致瀰漫、無型態的色素過度沉著,分別在第 42 與第 116 章討論。與晚期轉移性黑色素瘤相關的瀰漫、全身性黑色素沉著症 (melanosis) 是一種罕見但有充分記錄的疾病。其特徵是皮膚呈板岩藍灰色至棕色的變色。組織學顯示真皮與皮下脂肪中有黑色素顆粒,以及含黑色素的組織細胞 (histiocytes) 與樹突細胞 (dendritic cells)。皮膚中無法偵測到黑色素瘤細胞,且表皮黑色素或黑色素細胞數量並無增加。已偵測到循環於血液中的黑色素小體,支持瀰漫性黑色素沉著症可能源於腫瘤溶解 (tumor lysis)、其胞器釋放進入循環並隨後沉積於皮膚的假說。
物理性病因 (Physical Causes)
重點一覽 (AT-A-GLANCE)
■ 紫外線、游離輻射 (ionizing radiation) 與熱輻射可在受影響區域造成瀰漫、無型態的色素過度沉著。
■ 創傷可在慢性摩擦區域造成局限性色素過度沉著。
紫外線輻射與色素沉著 (Ultraviolet Radiation and Pigmentation)
第 20 章詳細討論色素沉著。紫外線輻射對正常人類皮膚的一個主要急性效應是曬黑 (tanning)。
游離輻射 (Ionizing Radiation)
第 200 章詳細討論放射治療。皮膚在意外事故期間或局部分次放射治療後暴露於游離輻射,可引發皮膚輻射症候群 (cutaneous radiation syndrome),其特徵是纖維化、角化症、毛細血管擴張,以及界線分明的小痣樣色素過度沉著,類似紫外線誘導的小痣 (放射性皮膚炎 radiation dermatitis)。小型色素減退斑可與色素過度沉著區交織。組織學顯示黑色素細胞與基底角質細胞中黑色素含量的改變。已有報導指出電子束治療 (electron-beam therapy) 在指甲暴露時會誘發曬黑樣的暫時性色素過度沉著與橫向黑甲 (transverse melanonychia)。
熱輻射 (Thermal Radiation)
在淺層熱燒傷損傷中,當帶有黑色素細胞的基底表皮未被破壞時,會產生各種程度的色素過度沉著,取決於病人的膚色與損傷後的時間。由強烈、高劑量可見光雷射治療所致的熱損傷,也可造成色素過度沉著,尤其在深色皮膚病人中。冷凍治療——藉由施加寒冷造成組織破壞——常因黑色素細胞損傷而在治療皮膚中造成 (有時為永久性的) 色素減退,併周邊色素過度沉著。
摩擦性黑色素沉著 (Friction Melanosis)
摩擦性黑色素沉著是一種後天性色素疾病,由皮膚反覆摩擦所引起,尤其在骨突起處之上。組織學上可見黑色素明顯增加與黑色素失禁。可見瀰漫性色素過度沉著,部分病例呈現輕度苔癬化。
毒素與藥物 (Toxins and Medications)
重點一覽 (AT-A-GLANCE)
■ 毒素與藥物是後天性、瀰漫、無型態色素過度沉著的常見原因。
■ 固定性藥疹 (fixed drug eruptions) 可造成界線分明的棕灰色黏膜皮膚斑。
■ 色素過度沉著可由藥物直接沉積於皮膚、黑色素或黑色素-藥物複合體沉積於真皮,或由藥物所產生的非黑色素色素所引起。
■ 治療涉及停用致病藥物,以及品質開關與皮秒雷射。
由毒性製劑或藥物引起的色素過度沉著佔所有後天性色素過度沉著病例的 10% 至 20%。中樞神經系統藥物、抗腫瘤製劑、抗感染藥物、抗高血壓藥物與荷爾蒙最常是責任製劑 (表 77-2)。
臨床特徵 (CLINICAL FEATURES)
臨床特徵隨特定藥物與毒素所引起的特徵性部位、型態與變色色調而異。藍板岩灰色色素沉著是色素真皮沉積的特徵。與黑色素堆積相關的色素沉著常隨日光曝曬而惡化。服用 bleomycin 或 zidovudine 的病人可觀察到線狀、有時為鞭痕狀 (flagellate) 的色素過度沉著。服用 cyclophosphamide 或 doxorubicin 的病人可能出現掌蹠的瀰漫性色素過度沉著。靜脈鐵劑輸注部位也可見瀰漫板岩灰色色素沉著 (圖 77-9)。Bleomycin 與 doxorubicin 可在小關節周圍產生局限性色素過度沉著。雌激素相關的荷爾蒙物質與 phenytoin 類藥物可造成黑斑樣色素沉著。某些藥物可觀察到甲單位 (nail unit) 的侵犯,最常見的是化療製劑,例如 cyclophosphamide、zidovudine、psoralens、minocycline、抗瘧藥與金 (gold)。Cyclophosphamide、doxorubicin、zidovudine、minocycline 與某些重金屬曾被報導造成黏膜色素過度沉著。Amiodarone 可因脂質樣物質堆積於巨噬細胞中,而在光照區域產生藍灰色色素沉著。其中一些病人呈現光敏性 (圖 77-10)。Chloroquine 在服用數年後可引起顏面、頸部、下肢與前臂的黃棕色至藍灰色色素沉著,這是藥物-黑色素複合體沉積於真皮的結果。甲單位與硬腭也可能受侵犯。已有報導指出慢性使用 hydroquinone 後發生外源性褐黃病 (exogenous ochronosis) (見「褐黃病」一節)。Chlorpromazine 與相關的吩噻嗪類 (phenothiazines) 可產生藍灰色皮膚色素沉著,於光照區域更為嚴重,並伴隨結膜的色素沉著。Minocycline 誘導色素沉著的風險隨著每日劑量高於 100 mg 的延長治療而增加。Minocycline 誘導的色素沉著存在三種型別:第 1 型由正常皮膚與先前發炎區域 (常於顏面) 的藍灰色色素沉著構成 (圖 77-11);第 2 型見於小腿與前臂;第 3 型為瀰漫、泥棕色、光照加重的色素沉著。亦曾報導指甲、鞏膜、口腔黏膜、甲狀腺、骨骼與牙齒的色素沉著。銀質沉著病 (argyria) 病人因攝入銀而呈現全身性灰藍色色素沉著。指甲與鞏膜也可能受侵犯。儘管藥物誘導的色素沉著通常造成瀰漫性色素過度沉著,但當界線分明的黏膜皮膚色素過度沉著斑為呈現徵象時,必須考慮固定性藥疹 (圖 77-12)。在使用非類固醇抗發炎藥物、某些抗生素,以及含可待因 (codeine) 的藥物後,典型可見先有紅斑並可能伴隨搔癢、隨病灶消退而留下色素沉著的病史。
表 77-2:曾被報導造成色素過度沉著的藥物 (Medications Reported to Cause Hyperpigmentation)
| 局部治療 (TOPICAL TREATMENTS)/抗高血壓藥物/重金屬 | 抗感染藥物 (ANTIINFECTIOUS DRUGS) | 中樞神經系統藥物 (CNS DRUGS) | 抗腫瘤製劑 (ANTINEOPLASTIC AGENTS) | 其他 (MISCELLANEOUS) |
|---|---|---|---|---|
| 局部治療: Aminolevulinic acid、Carmustine (BCNU)、Bergamot、Bimatoprost、Carteolol、Chlorhexidine、Hydroquinone、Imiquimod、Latanoprost、Nitrogen mustard、Tretinoin。抗高血壓藥物: Acebutolol、Betaxolol、Bisoprolol、Captopril、Clonidine、Diltiazem、Esmolol、Indapamide、Labetalol、Methyldopa、Metoprolol、Minoxidil、Nisoldipine、Propranolol、Spironolactone、Timolol。重金屬 (Heavy metals): Arsenic、Bismuth、Gold (compounds)、Iron、Lead、Mercury、Silver | Amphotericin B、Ceftriaxone、Chloroquine、Cidofovir、Clofazimine、Dapsone、Demeclocycline、Doxycycline、Emtricitabine、Enoxacin、Foscarnet、Ganciclovir、Grepafloxacin、Griseofulvin、Hydroxychloroquine、Indinavir、Ketoconazole、Linezolid、Lomefloxacin、Minocycline、Ofloxacin、Oxytetracycline、Pyrimethamine、Quinacrine、Quinine、Ribavirin、Rifabutin、Rifapentine、Saquinavir、Sertaconazole、天花疫苗 (Smallpox vaccine)、Sparfloxacin、Sulfadiazine、Terbinafine、Tetracycline、Voriconazole、Zidovudine | Amitriptyline、Carbamazepine、Chlorpromazine、Citalopram、Clomipramine、Clonazepam、Desipramine、Diazepam、Donepezil、Eletriptan、Fluoxetine、Fluphenazine、Fluvoxamine、Haloperidol、Imipramine、Kava、Loxapine、Mephenytoin、Mesoridazine、Methamphetamine、Molindone、Olanzapine、Paroxetine、Perphenazine、Phenytoin、Pimozide、Prochlorperazine、Promazine、Promethazine、Risperidone、Ropinirole、Thioridazine、Thiothixene、Tiagabine、Tolcapone、Topiramate、Trifluoperazine、Venlafaxine、Zaleplon | Bevacizumab、Bleomycin、Busulfan、Capecitabine、Carboplatin、Carmustine、Cisplatin、Cyclophosphamide、Dactinomycin、Daunorubicin、Doxorubicin、Epirubicin、Estramustine、Etoposide、Floxuridine、Fluorouracil、Hydroxyurea、Ifosfamide、Irinotecan、Mechlorethamine、Mercaptopurine、Methotrexate、Mitomycin、Mitotane、Mitoxantrone、Paclitaxel、Pentostatin、Procarbazine、Thiotepa、Vinblastine、Vincristine、Vinorelbine。荷爾蒙 (Hormones): Chlorotrianisene、Corticosteroids、Diethylstilbestrol、Estrogens、Insulin、Leuprolide、Medroxyprogesterone、口服避孕藥 (Oral contraceptives)、Progestins、Stanozolol | Alitretinoin、Amiodarone、Azathioprine、Cetirizine、Cevimeline、Cyclobenzaprine、Cyclosporine、Deferoxamine、Dicumarol、Dinoprostone、Etodolac、Glatiramer、Heroin、Interferon、Isotretinoin、Ketoprofen、Leflunomide、Levobupivacaine、Lidocaine、Methimazole、Methoxsalen、Methysergide、Metoclopramide、Niacin、Nicotine、Orphenadrine、Pantoprazole、Pentazocine、Phenazopyridine、Phenolphthalein、Propylthiouracil、Psoralens、Quinidine、Rabeprazole、Riluzole、Sulfasalazine、Tacrolimus、Toremifene、Trioxsalen、Vitamin A |
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在大多數藥物誘導色素過度沉著的病例中,其潛在致病機轉涉及以下之一。
-
黑色素沉積於真皮,通常位於巨噬細胞內。有時這種黑色素與藥物形成複合體 (藥物-色素複合體 drug-pigment complex,例如 hydroxychloroquine)。黑色素的堆積可發生於皮膚發炎後 (發炎後) 與/或 DNA 損傷後 (例如 carmustine)。此型色素過度沉著常因紫外線曝曬而增加,且通常於光照區域更為明顯。
-
藥物直接沉積於皮膚 (例如胡蘿蔔素 carotene、重金屬)。有時此型色素沉著於光照區域加重,因為紫外線可誘導沉積藥物發生轉化,使其可能變得更明顯。
-
在藥物直接或間接影響下所合成或產生的非黑色素色素,可能造成皮膚色素沉著。
診斷 (DIAGNOSIS)
若見到特徵性外觀,並注意到與藥物或毒素暴露的時間關係,即可在臨床上做出診斷。在藥物誘導色素沉著的病例中,皮膚切片也可能有所幫助。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
停用致病藥物至關重要,但在許多病例中,即使在停藥後,色素過度沉著仍持續存在,有時長達數十年。
治療 (MANAGEMENT)
應建議防曬,尤其是在由黑色素堆積引起的型別中。品質開關紅寶石、紫翠玉與 Nd:YAG 雷射在某些病例 (例如 amiodarone 與 minocycline 誘導的色素沉著) 中已展現良好結果,儘管常需要在數月間進行多次治療才能達到臨床上顯著的結果。皮秒雷射已被證實對白皮膚中藥物誘導的色素沉著有效,在較短的時間範圍內以較少的治療次數即可達到色素沉著的完全或近乎完全清除。
褐黃病 (Ochronosis)
重點一覽 (AT-A-GLANCE)
■ 可為先天性 (自體隱性) 或由某些藥物的使用所引起。
■ 造成無症狀、瀰漫、無型態的色素過度沉著。
■ 在有色皮膚的人中較為常見。
■ 特徵性的組織學外觀是真皮中黃棕色香蕉形小球 (banana-shaped globules)。
褐黃病 (ochronosis) 一詞源自希臘文「ochre」,意指黃色變色。本病存在內源性與外源性兩種型態。
外源性褐黃病 (EXOGENOUS OCHRONOSIS)
流行病學:儘管大多數褐黃病病例報導於非洲、印度、泰國、中國與新加坡的較深色種族/族裔群體,但它也曾被描述於其他膚質。確切的全球發生率未知,但假定為低。
臨床特徵:無症狀的藍黑色與灰黑色色素過度沉著斑為其特徵,可見於顏面 (顴部、顳部、下臉頰)、後外側頸部、背部,以及四肢的伸側皮膚。較晚期包括進行性色素過度沉著的膠樣粟丘疹 (colloid milium,「魚子醬樣 caviar-like」病灶)、丘疹結節性病灶,以及瘢痕區域。
外源性褐黃病不見全身性侵犯。
病因與致病機轉:外源性褐黃病源於某些藥物的使用,這些藥物在代謝過程中形成一種類似尿黑酸聚合物 (homogentisic acid polymer-like) 的物質。它最常被報導與 hydroquinone 相關,通常於濃度大於 4% 時,以及局部製劑如 phenol 或 resorcinol,與 quinine (一種口服抗瘧藥) 相關。加重因素包括無防護的紫外線曝曬、致病藥物在大面積區域上的長期塗抹,以及局部 resorcinol、phenol 與汞 (mercury) 的使用。
診斷:儘管外源性褐黃病的早期階段外觀可能類似黑斑 (melasma),皮膚鏡 (dermoscopy) 可能有助於區別二者。在瀰漫性棕色背景上可見深棕色小球與小球樣結構。
然而,皮膚切片仍是外源性褐黃病診斷的黃金標準。
組織病理學上,乳頭真皮中有一群黃棕色 (褐黃) 的香蕉形小球。亦可見均質、水腫的真皮膠原。
鑑別診斷:褐黃病的早期階段在臨床上可能與黑斑無法區別,尤其在亞洲人中。
臨床病程與預後:若不停用致病製劑,外源性褐黃病為進行性。
治療:治療鮮少有幫助,但應停用致病藥物以防止進展。廣效防曬與避免日光至關重要。局部類視黃醇 (retinoids)、α-羥基酸 (α-hydroxy acids)、皮質類固醇,以及物理療法 (如化學換膚與品質開關雷射),都曾被報導用於外源性褐黃病的治療,但截至撰寫本文時,尚無記錄到普遍有效的治療。以下疾病可與色素過度沉著相關,並在其各自的章節中討論:全身性硬化症 (systemic sclerosis,第 63 章)、品他病 (pinta,第 171 章)、蟠尾絲蟲病 (onchocerciasis,第 177 章)、植物日光性皮膚炎 (phytophotodermatitis,第 97 章),以及由 bleomycin 引起的鞭痕狀色素沉著 (第 45 章)。
全身性硬化症 (Systemic Sclerosis)
第 63 章更詳細討論全身性硬化症。
重點一覽 (AT-A-GLANCE)
■ 後天性瀰漫色素過度沉著、摩擦部位之上的局灶性脫色、條紋狀色素過度沉著,以及網狀色素沉著,均曾被描述。
在全身性硬化症中曾描述不同型態的異常色素沉著。在嚴重的全身性硬化症中,可觀察到類似 Addison 病但黑色素細胞刺激素濃度正常的瀰漫、全身性色素過度沉著。可能發生局灶性脫色併毛囊周圍色素過度沉著,尤其在摩擦部位 (例如脛部、肘部與手背)。可有局限性的色素過度沉著與色素減退。亦曾報導在脫色背景上沿血管分布的條紋狀色素過度沉著。有 1 例曾報導以軀幹加重的瀰漫網狀色素過度沉著。
感染 (Infections)
屈公病 (Chikungunya)
重點一覽 (AT-A-GLANCE)
■ 後天性局限性或瀰漫性色素過度沉著。
■ 由一種蚊媒病毒所引起。
■ 持續性色素沉著,症狀緩解是唯一的治療選項。
屈公病 (chikungunya) 是一種蚊媒病毒,最早於 1950 年代在非洲坦尚尼亞描述。
流行病學:雖然最早在非洲描述,屈公病已在非洲、亞洲、歐洲與美洲的數十個其他國家被注意到。所有年齡層均受影響。
臨床特徵:色素變化是最常見的皮膚發現。小痣樣的中央顏面色素過度沉著、肢端顏面病灶的瀰漫性色素過度沉著,以及鞭痕狀色素過度沉著,均曾被描述。亦曾報導鼻部顯著的色素沉著。曾在一例先天性屈公病的病例中報導新生兒色素過度沉著。
在疾病發作時可見斑丘疹 (maculopapular eruption),並於 7 至 10 天內消退。其他黏膜皮膚表現包括水疱大疱性病灶、紫斑性病灶、口瘡樣潰瘍,以及其他原發性皮膚病 (如扁平苔癬與乾癬) 的惡化。全身性症狀,如發燒、關節痛、肌肉痛與胃腸不適,是其中一些皮膚外表現。
病因與致病機轉:埃及斑蚊 (Aedes aegypti,熱帶與亞熱帶) 與白線斑蚊 (Aedes albopictus,世界較涼爽地區) 已被認為與這種蚊媒疾病的大規模爆發有關。皮膚色素過度沉著發生的機制尚未明瞭,但存在各種理論,包括黑色素細胞對入侵病原體的吞噬作用,隨後由病毒觸發的表皮內黑色素分散或滯留。
診斷:組織學上可見淋巴球性血管病變 (lymphocytic vasculopathy) 與表皮內黑色素增加。亦可見噬黑色素細胞,這可能解釋持續性的皮膚色素沉著。
鑑別診斷:其他病毒性疾病與蚊媒疾病落在廣泛的鑑別診斷之中。
臨床病程與預後:色素沉著常持續數月之久。
治療:症狀緩解是治療的主要方式。
其他感染 (Other Infections)
在品他病 (pinta) 的次發階段會發展出紅斑性、脫屑性丘疹,稱為品他疹 (pintids)。這些最初為紅色的病灶可轉變為棕色、板岩藍色、黑色或灰色。
慢性丘疹性蟠尾絲蟲皮膚炎 (chronic papular onchodermatitis) 是蟠尾絲蟲病的皮膚表現之一。其特徵是嚴重搔癢的斑丘疹,伴隨色素過度沉著斑,最常發生於肩部、臀部與四肢 (見第 177 章)。
生理性色素過度沉著 (PHYSIOLOGIC HYPERPIGMENTATION)
色素分界線 (Pigmentary Demarcation Lines)
重點一覽 (AT-A-GLANCE)
■ 在有色皮膚的人中較為常見。
■ 線狀分界,伴隨相對的色素過度沉著與色素減退。
■ 沿「胚胎縫合線 (embryonic suture lines)」見於顏面與四肢的獨特型態與區域。
■ 可發生於顏面、軀幹與四肢。
■ 通常於兒童期變得明顯。
色素分界線 (pigmentary demarcation lines, PDLs) 又稱 Voigt 線或 Futcher 線。它們是從較淺至較深皮膚的突然改變,呈線狀型態,最常見於四肢、軀幹與顏面。
流行病學:PDLs 在有色皮膚的人中最為常見。PDLs 常於兒童期或青春期出現,並終生持續。
臨床特徵:PDLs 依位置分類如下:
■ A 型:這是最常見的型別,侵犯上臂前部的外側 (偶爾跨越胸肌區域);
■ B 型:後內側下肢;
■ C 型:胸骨與胸骨旁區域的垂直色素減退線;
■ D 型:脊柱或脊柱旁區域的垂直線;
■ E 型:雙側胸部的色素減退橢圓形區域、條紋或帶狀,從鎖骨中三分之一至乳暈周圍皮膚;
■ F 型:顴突與顳部之間的 V 形色素過度沉著線;
■ G 型:顴突與顳部之間的 W 形色素過度沉著線;以及
■ H 型:從口角至下巴外側的線狀色素過度沉著。
耳前的色素分界線在文獻中尚未被描述 (截至撰寫本文時),但現今應將其新增為新的色素分界線「I 型」(圖 77-13)。
病因與致病機轉:PDLs 的原因未知。關於其拓撲學是遵循 Blaschko 線或皮膚神經支配的爭論仍在持續。已有人提出荷爾蒙與遺傳的影響,並注意到於懷孕期間發病以及可變的遺傳模式,包括某些家族中的自體顯性遺傳模式。
診斷:診斷通常可在臨床上做出,無需切片。
臨床病程與預後:PDLs 常於兒童期或青春期出現,並終生持續。
治療:需要向病人保證這些變化是正常變異。此外,防曬可能減少受影響與未受影響皮膚之間的差異。
後天性特發性顏面色素沉著 (Acquired Idiopathic Facial Pigmentation)
重點一覽 (AT-A-GLANCE)
■ 可被視為色素分界線的另一個群組。
■ 在有色皮膚的人中較為常見。
■ 線狀分界,伴隨相對的色素過度沉著與色素過度沉著。
■ 主要影響眼周與口周區域。
■ 通常於成年期變得明顯。
後天性特發性顏面色素沉著近期在印度族群中被描述,可被視為 PDLs 的另一個群組。「後天性特發性顏面色素沉著」一詞,是在觀察到某些型態的特發性顏面色素沉著為非痣樣、有型態且雙側、呈深棕灰色並位於眼睛周圍、外側顏面與下巴後所創。
流行病學:發病見於成年早期,男性與女性以 1:1 的比例發生。它常於成年期出現並終生持續。
臨床特徵:界線分明的深棕灰色斑性色素過度沉著見於眼周、顴弓 (zygomatic) 與顴部 (malar) 區域、鼻根、口周與下頷皮膚。色素沉著的型態與其他痣樣疾病相似,其型態反映顏面色素胚胎擴散的正常型態。無症狀、無鱗屑、無紅斑與無苔癬化,應有助於將其與該區域的其他皮膚病及色素疾病區別。
病因與致病機轉:儘管原因未知,據推測這是一種遺傳疾病,因生理因素 (如紫外線與老化) 而加重與加強。
診斷:診斷通常可在臨床上做出,無需切片。
鑑別診斷:諸如黑斑與發炎後色素沉著等疾病可能看起來類似後天性特發性顏面色素沉著,應在臨床與組織學上加以區別。
臨床病程與預後:後天性特發性顏面色素沉著常於成年期出現。它隨時間變深並終生持續。
治療:防曬可能有助於減少受影響與未受影響皮膚之間的差異,偶爾局部 hydroquinone 可微妙地淡化受影響區域的過度色素沉著。
眼周色素沉著 (Periorbital Pigmentation)
重點一覽 (AT-A-GLANCE)
■ 應排除原發性皮膚病,例如色素性扁平苔癬 (lichen planus pigmentosus)、藥物誘導的色素沉著、色素性接觸性皮膚炎 (pigmented contact dermatitis),以及發炎後色素過度沉著。
■ 家族性/種族性/體質性色素沉著在有色皮膚的人中尤其常見。
眼周色素過度沉著又稱黑眼圈 (dark circles),在某些種族族裔型別中非常常見。
流行病學:男性與女性,主要為較深膚質者,常受影響。發病通常見於年輕成人。
臨床特徵:無症狀的棕黑色、無鱗屑、無紅斑色素沉著見於上眼瞼、下眼瞼或兩者。可見皮膚皺褶的加重,也可能見到可見的血管 (圖 77-14)。
病因與致病機轉:體質性色素沉著的確切病因尚待確定。
診斷:大多數病例做出臨床診斷。在體質性色素沉著者中,組織學上可見基底表皮黑色素增加與上真皮的噬黑色素細胞。
鑑別診斷:必須排除原發性皮膚病,如色素性扁平苔癬、藥物誘導的色素沉著,以及伴苔癬化的濕疹。
臨床病程與預後:體質性色素沉著通常為進行性。
治療:若存在潛在皮膚病,治療之至關重要。儘管局部療法、換膚、雷射與自體脂肪移植曾被報導有用,但這些研究常未指明色素沉著的潛在原因,使結果難以解讀。
黏膜黑色素沉著 (Mucosal Melanosis)
重點一覽 (AT-A-GLANCE)
■ 口腔的生理性色素沉著。
■ 牙齦、口腔黏膜、嘴唇與舌頭的斑性色素沉著。
■ 在有色皮膚的人中較為常見。
黏膜黑色素沉著在有色皮膚的人中常見,應與口腔內色素沉著的其他原因區別。
流行病學:關於此主題可獲得的有限文獻顯示,色素沉著的發病是漸進的,且很可能發生於膚色濃郁的中年成人。
臨床特徵:可見瀰漫且雙側的色素沉著,但也可能見到局限性的斑性色素過度沉著,侵犯以下一個或多個位置:牙齦、頰黏膜、嘴唇、硬腭,以及舌乳頭 (圖 77-15)。一項中國的研究結論指出,舌頭的色素性蕈狀乳頭 (pigmented fungiform papillae) 在中國族群中相對常見。Laugier-Hunziker 症候群是一種後天性色素過度沉著型態,表現為縱向黑甲 (longitudinal melanonychia),以及侵犯口腔黏膜、生殖器、肛周區域、手指與食道的色素沉著。
病因與致病機轉:原因未知。
診斷:診斷通常可在臨床上做出,無需切片,但若診斷有疑慮,切片可能有所幫助。組織學檢查顯示基底細胞色素過度沉著、真皮噬黑色素細胞增加、色素失禁,以及正常的黑色素細胞 (在某些情況下數量增加)。
鑑別診斷:許多疾病可造成口腔色素沉著。需排除藥物誘導的色素沉著 (包括固定性藥疹)、口腔扁平苔癬、發炎後色素過度沉著、牙科汞合金 (dental amalgam)、Addison 病,以及惡性腫瘤。兒童期的棕色色素過度沉著斑應促使對遺傳性皮膚病 (如 Peutz-Jeghers、Bandler、Carney 與 Cronkhite-Canada 症候群) 進行調查。
臨床病程與預後:黏膜黑色素沉著於兒童期出現,並終生持續而無不良後果。
治療:保證是處置中最重要的部分。不需要治療。
縱向黑甲 (Longitudinal Melanonychia)
重點一覽 (AT-A-GLANCE)
■ 甲板的良性棕黑色線狀色素帶。
■ 在有色皮膚的人中較為常見。
■ 發生率隨年齡增加。
第 91 章更詳細討論縱向黑甲。多個指甲上規則、細的色素過度沉著帶,是較深膚色個體所見良性縱向色素沉著的特徵。藥物、創傷與甲器 (nail apparatus) 內的痣也可能造成縱向黑甲,應納入鑑別診斷。位於拇指或大腳趾的單一色素帶,應促使臨床醫師在臨床與皮膚鏡下勤勉地尋找黑色素瘤的徵象。切片可能也是區別良性縱向黑甲與黑色素瘤所必需的。
肢端色素過度沉著斑 (Acral Hyperpigmented Macules)
重點一覽 (AT-A-GLANCE)
■ 手掌與足底界線分明的良性棕色斑。
■ 在有色皮膚的人中較為常見。
■ 發生率隨年齡增加。
手掌與足底界線分明的棕色斑,組織學上顯示表皮黑色素增加。儘管皮膚鏡下可見平行脊型態 (parallel ridge pattern),但所見病灶的數量提供了其良性本質的線索。重要的是排除可能以類似方式呈現的遺傳性皮膚病與藥物誘導 (尤其是化療) 的色素沉著。
後天性型態化色素過度沉著 (ACQUIRED PATTERNED HYPERMELANOSIS)
重點一覽 (AT-A-GLANCE)
■ 植物日光性皮膚炎、由 bleomycin 引起的鞭痕狀色素沉著,以及鞭痕狀香菇皮膚炎 (flagellate mushroom dermatitis),都是型態化色素過度沉著的原因。
植物日光性皮膚炎 (Phytophotodermatitis)
接觸含有光毒性製劑 (如 psoralens) 的植物,隨後暴露於紫外線,可導致植物日光性皮膚炎,繼之以型態化色素過度沉著。
鞭痕狀香菇皮膚炎 (Flagellate Mushroom Dermatitis)
香菇 (shiitake mushroom,Lentinula edodes) 通常種植並用於亞洲料理,但西方世界現今也大量食用。生食或烹煮不足的香菇之口服攝取,與一種「鞭痕狀香菇皮膚炎」相關,後者於食用後 12 小時至 5 天出現。雖然大多數報告來自日本,但歐洲、美國與加拿大的病例也曾被描述。其特徵是線狀成群、紅斑性、劇烈搔癢的丘疹,類似 bleomycin 誘導的鞭痕狀皮膚炎。亦曾報導膿疱性皮疹。軀幹與四肢通常受侵犯。其致病機轉尚不明,但據推測是由香菇內含的一種熱不穩定多醣 (thermolabile polysaccharide) 所致的毒性反應。
火激紅斑 (Erythema Ab Igne)
重點一覽 (AT-A-GLANCE)
■ 後天性瀰漫網狀色素沉著。
■ 也可能有表皮萎縮、脫屑與色素過度沉著。
■ 由慢性暴露於中度熱所引起。
■ 應檢查甲狀腺功能。
火激紅斑 (erythema ab igne) 是由頻繁暴露於熱 (通常為紅外線輻射) 所引起,造成網狀色素過度沉著。雖然它可發生於身體任何部位,但通常局限於大腿與小腿。
臨床特徵 (CLINICAL FEATURES)
急性期可見黃紅棕色網狀、暫時性且可被壓退 (blanchable) 的紅斑 (通常在熱暴露後約 3 週),但在慢性病例中,固定的色素過度沉著可能伴隨表皮萎縮、大疱性病灶或角化過度與鱗屑。常見的侵犯部位包括大腿、小腿、腹部與背部。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
火激紅斑是由慢性暴露於中度熱所引起。過去常見於為取暖而頻繁坐在明火前的人,但中央暖氣的引入已使此種表現減少。儘管如此,它仍見於局部使用電熱墊、熱水袋或電熱毯後 (圖 77-16)。日本亦曾報導繼發於泡熱水澡 (hot tub bathing) 的廣泛病例。曾被報導引發火激紅斑的現代器具有:內建加熱器的家具、汽車暖氣,以及置於大腿或腿上的筆記型電腦。皮膚變化的網狀本質被認為反映了血管的分布。熱被認為會損傷表皮中的淺表血管,後者進而導致紅血球與含鐵血黃素的外滲,造成皮膚色素過度沉著。
診斷 (DIAGNOSIS)
診斷通常以臨床做出。對於有暗示甲狀腺功能低下症狀或徵象的病人,應考慮甲狀腺功能檢查。若進行皮膚切片,組織學檢查通常顯示血管擴張、紅血球外滲,以及黑色素與含鐵血黃素沉積於真皮。亦可見真皮水腫與淋巴組織細胞性 (lymphohistiocytic) 真皮浸潤。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
需考慮網狀青斑 (livedo reticularis) 及其各種原因,但在火激紅斑的病例中,病史常顯示慢性熱暴露。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
色素變化通常在病人停止對皮膚使用熱源後消退,但取決於暴露的慢性程度,色素沉著可能持續數月之久。非常罕見地,曾報導鱗狀細胞癌與梅克爾細胞癌 (Merkel cell carcinoma) 在火激紅斑的慢性病灶內發生。
治療 (MANAGEMENT)
消除熱源至關重要,但局部類視黃醇與低能量品質開關 Nd:YAG 雷射在某些持續性病例中也有所幫助。
色素性癢疹 (Prurigo Pigmentosa)
重點一覽 (AT-A-GLANCE)
■ 軀幹的後天性瀰漫網狀色素沉著。
■ 最常見於年輕女性。
■ 搔癢性丘疹、水疱與丘疹水疱的初始階段,繼之以無症狀的色素過度沉著。
■ 四環素類 (tetracyclines) 與 dapsone 對本病的發炎成分有所幫助。
色素性癢疹 (prurigo pigmentosa) 是一種罕見疾病,最早由 Nagashima 等人於 1971 年在日本描述。它最常侵犯軀幹,並造成網狀色素過度沉著。
流行病學 (EPIDEMIOLOGY)
雖然截至撰寫本文時的報告大多來自日本,但它在世界其他地區正越來越多被記錄。它最常見於 20 多歲的年輕女性,女性對男性的比例為 4 至 6:1。
臨床特徵 (CLINICAL FEATURES)
色素性癢疹表現為劇烈搔癢、對稱的蕁麻疹樣、紅斑性丘疹、丘疹水疱與水疱,這些病灶演變為網狀色素過度沉著。病灶發展於上背部、胸部、頸部與腰薦區域。丘疹水疱性病灶在約 1 週內自發性癒合,留下非搔癢性的網狀色素過度沉著 (圖 77-17)。尚未記錄到任何全身性關聯。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
已有人提出環境與代謝因素作為致病製劑,但致病機轉仍未明。
診斷 (DIAGNOSIS)
色素性癢疹的診斷需要密切的臨床病理相關性。早期病灶的組織學顯示淺表的嗜中性球血管周圍與間質浸潤。在疾病病程稍後,病灶呈現以淋巴球為主的苔癬樣 (lichenoid) 發炎。亦可見海綿狀水腫 (spongiosis)、壞死的角質細胞、嗜酸性球與水疱形成。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
融合性網狀乳頭瘤病 (confluent reticulated papillomatosis,無先前的紅斑性丘疹)、色素性接觸性皮膚炎 (對局部類固醇有反應),以及 Dowling-Degos 病 (特徵性的組織學發現),是主要的鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
本病具有波動性病程,伴隨惡化與復發。
治療 (MANAGEMENT)
皮疹與搔癢對 minocycline 或 doxycycline 100-200 mg daily,或 dapsone 25 to 100 mg daily 反應良好。色素沉著會自發性消退。
Hori 痣 (Nevus of Hori)
重點一覽 (AT-A-GLANCE)
■ 後天性局限性色素過度沉著,呈位於顏面顴部區域的對稱針頭大小斑形式。
■ 主要見於亞洲族群。
■ 常與其他色素疾病 (如黑斑與扁平脂漏性角化症 flat seborrheic keratoses) 並存。
■ 以品質開關 Nd:YAG 雷射治療效果最佳。
Hori 痣最早於 1984 年描述為後天性雙側太田母斑樣斑 (acquired bilateral nevus of Ota–like macules, ABNOM) 或後天性真皮黑色素細胞增多症。
流行病學 (EPIDEMIOLOGY)
它是一種常見疾病,見於近 1% 的亞洲族群,尤其是中國與日本族群。它在女性中較常見,並出現於生命第三至第五個十年。
臨床特徵 (CLINICAL FEATURES)
Hori 痣由藍棕色至板岩灰色的色素過度沉著斑構成,大小從針頭大小至數毫米直徑不等 (圖 77-18)。病灶見於顏面,偏好顴部區域,儘管顳部與鼻部的病灶也不少見。與太田母斑不同,Hori 痣為雙側性,且不見眼部與黏膜的侵犯。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
Hori 描述了 ABNOM 致病機轉的 3 種可能機制:(a) 表皮黑色素細胞「掉落」進入真皮,(b) 黑色素細胞自毛球 (hair bulbs) 遷移,以及 (c) 預先存在的潛伏或未成熟真皮黑色素細胞的再活化。第三種可能性被認為是最合理的機制,以紫外線輻射與性荷爾蒙作為活化因素。
診斷 (DIAGNOSIS)
診斷可能需要切片,尤其當其他色素疾病 (如黑斑) 在受影響區域並存時。組織學常顯示上真皮與中真皮的黑色素細胞增多症。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
在許多病例中,ABNOM 可與其他色素異常 (如黑斑、雀斑、日光性小痣 solar lentigines,以及太田母斑) 同時發生。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
Hori 痣通常為漸進性進展。
治療 (MANAGEMENT)
儘管各種雷射 (如剝離性 CO2、品質開關 532-nm、品質開關紅寶石,以及分段非剝離性 2940-nm 鉺:YAG [Erbium:YAG]) 曾被單獨或併用使用,品質開關雷射是最常使用的治療形式。要獲得完全清除並非總是可能,且需要多次治療。品質開關與皮秒雷射已展現淡化或清除病灶的療效,但需要間隔數月的多次治療。
Becker 痣 (Nevus of Becker)
重點一覽 (AT-A-GLANCE)
■ 以後天性局限性色素過度沉著表現。
■ 最常見於軀幹。
■ 通常與多毛症 (hypertrichosis) 相關。
■ 可嘗試以雷射治療,但目前的研究缺乏統計顯著性。
這種後天性色素過度沉著的表皮痣最早於 1949 年描述 (圖 77-19),亦稱 Becker 錯構瘤或 Becker 黑色素沉著。
流行病學 (EPIDEMIOLOGY)
Becker 痣似乎是雄激素依賴性的 (androgen-dependent),見於約 0.5% 的人口,通常於青春期變得更顯著。據說在男性中較常見,但一項較新的研究顯示在男孩與女孩中發生率相等。
臨床特徵 (CLINICAL FEATURES)
這種良性錯構瘤可能造成外觀缺陷,並以色素過度沉著的斑性或斑點狀區域表現,最常見於肩胛區域、上臂與胸部,儘管它已被描述於身體任何區域。多毛症常見,且常於色素過度沉著被注意到後數年才發展。這些毛髮粗糙且深色。Becker 痣常於一次強烈日光曝曬後首次被注意到。相關異常,如同側乳房發育不全、肌肉骨骼異常 (例如脊柱側彎、同側肢體發育不全)、上頜顏面異常,以及其他皮膚發育不全,發生於罕見的 Becker 痣症候群中。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
Becker 痣被認為遵循半顯性遺傳模式 (paradominant inheritance pattern),這意味著它 (幾乎) 總是偶發性發生。曾被描述的罕見家族性病例 (尤其在 Becker 痣症候群中) 可由胚胎發育期間的體細胞突變來解釋,導致雜合性喪失 (loss of heterozygosity) 與突變細胞群的形成。此假說由在源自 Becker 痣的纖維母細胞中鑑定出染色體鑲嵌現象所支持。色素過度沉著的確切原因未知。
診斷 (DIAGNOSIS)
組織學上可見表皮棘層肥厚 (acanthosis)、伴隨可變的角化過度,以及表皮突 (rete ridges) 的延長。可見正常數量的黑色素細胞,但基底表皮層的黑色素含量增加。在真皮中,立毛肌 (arrector pili muscles) 的數量增加,使其難以與平滑肌錯構瘤 (smooth muscle hamartomas) 區別。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
先天性多毛痣 (congenital hairy nevus)、伊藤母斑與 CALMs 都是潛在的鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
Becker 痣可能在懷孕期間與夏季顯得變深。它終生持續。
治療 (MANAGEMENT)
Becker 痣的治療具挑戰性。若病灶小,手術切除可能是適當的考量。儘管機械磨皮 (mechanical abrasion)、冷凍治療,以及氬 (argon) 與 CO2 雷射過去曾用於較大的病灶,但瘢痕與色素異常的高風險限制了它們的使用。雷射除毛可給予淡化病灶的錯覺。長脈衝與皮秒紫翠玉雷射,以及品質開關 Nd:YAG,曾在某些研究中被引述為有用,但目前的證據缺乏統計力,且有不一致的發現。
咖啡牛奶斑 (Café-au-Lait Macules)
重點一覽 (AT-A-GLANCE)
■ 後天性局限性色素過度沉著。
■ 可孤立存在或作為遺傳性皮膚病的一部分。
■ 雷射可用於淡化斑點,但復發常見。
CALMs,又稱咖啡牛奶斑,可自發性發生或作為遺傳性皮膚病 (如 NF1 與 McCune-Albright 症候群) 的一部分。
流行病學 (EPIDEMIOLOGY)
多達三分之一的一般人口有 CALMs。它們在較深的 Fitzpatrick 膚質中較常見,但在男性與女性中發生率相等。
臨床特徵 (CLINICAL FEATURES)
淺棕至棕褐至深棕色、界線分明、均質色素過度沉著的斑或斑片,可見於身體任何部位。最常侵犯的部位是軀幹、臀部與下肢 (圖 77-20)。當注意到 6 個或更多 CALMs 時,應開始對潛在的遺傳性皮膚病進行調查。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
正常數量的黑色素細胞產生異常量的黑色素。
診斷 (DIAGNOSIS)
組織病理學檢查顯示正常至減少數量的、有時為肥大的黑色素細胞,以及基底表皮層的黑色素增加。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
斑痣 (nevus spilus)、早期 Becker 痣,以及先天性痣,都是鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
CALMs 終生持續。
治療 (MANAGEMENT)
基於美容原因,品質開關雷射可用於治療,但復發常見。
雀斑 (Ephelides)
重點一覽 (AT-A-GLANCE)
■ 後天性局限性色素過度沉著。
■ 自體顯性。
■ 出現於膚色白皙的個體。
流行病學 (EPIDEMIOLOGY)
雀斑出現於膚色白皙個體的日光曝曬皮膚,常為紅髮或金髮且具凱爾特 (Celtic) 血統者。男性與女性受影響的程度相等。
臨床特徵 (CLINICAL FEATURES)
雀斑 (ephelides,或 freckles) 是無症狀、小型、淺棕色的斑,對稱出現於光照部位。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
雀斑以自體顯性模式遺傳,並注意到與 MCR-1 基因突變的關聯,攜帶者顯示發展這些病灶的風險顯著增加。
MCR-1 是 α-黑色素細胞刺激素 (α-melanocyte-stimulating hormone) 的受體,後者經由環腺苷單磷酸活化黑色素生成路徑。此路徑活性降低會促進褐黑素 (pheomelanin) 的產生。
診斷 (DIAGNOSIS)
組織病理學檢查顯示正常至減少數量的、有時為肥大的黑色素細胞,但基底表皮層的黑色素增加。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
小痣 (lentigines) 是主要的鑑別診斷。較淺膚色表型中的特徵性外觀提供了診斷的線索。口腔黏膜、手掌與足底不受侵犯。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
它們在春季與夏季更為明顯,並於冬季消退。它們出現於兒童早期,並常於生命較晚期消退。
治療 (MANAGEMENT)
無論是否存在雀斑,對淺膚色者而言防曬都很重要,但對希望防止病灶變深的個體尤其重要。局部去色素製劑,如 hydroquinone、類視黃醇、α-羥基酸與植物萃取物 (botanicals),可能有助於淡化色素沉著。物理療法 (如冷凍治療) 在非常小的病灶上難以施行,因此偏好強脈衝光或品質開關雷射,但應預期會復發。
進行性肢端黑色素沉著 (Acromelanosis Progressiva)
重點一覽 (AT-A-GLANCE)
■ 新生兒的後天性局限性色素過度沉著。
■ 通常侵犯肢端與會陰區域。
■ 自發性消退。
流行病學 (EPIDEMIOLOGY)
僅有極少數病例被描述。
臨床特徵 (CLINICAL FEATURES)
瀰漫性棕色色素過度沉著見於新生兒或生命最初幾週,位於肢端與/或會陰區域。一例曾報導進展至軀幹、口腔黏膜侵犯與癲癇發作。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
原因未知。
診斷 (DIAGNOSIS)
進行性肢端黑色素沉著是一種隨時間 (病灶消退時) 變得明顯的臨床診斷。組織學顯示基底表皮黑色素細胞增加。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
甲周色素過度沉著 (periungual hyperpigmentation)、Dohi 肢端色素沉著,以及 Kitamura 網狀肢端色素沉著,是需考慮的鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
可見自發性消退。
治療 (MANAGEMENT)
由於不需要治療,保證很重要。
發炎後色素過度沉著 (Postinflammatory Hyperpigmentation)
重點一覽 (AT-A-GLANCE)
■ 先前發炎或損傷部位的後天性局限性色素過度沉著。
■ 在有色皮膚的人中較為常見。
■ Hydroquinone 仍是黃金標準治療。
■ 在可變的時間段內自發性消退。
發炎後色素過度沉著 (postinflammatory hyperpigmentation, PIH) 是一種常見的反應性黑色素沉著 (reactive melanosis),由眾多先前的發炎 (例如痤瘡、扁平苔癬、乾癬) 與皮膚傷害 (如藥物與光毒性反應、感染、物理損傷或創傷,以及過敏反應) 所引起。
流行病學 (EPIDEMIOLOGY)
PIH 在較深膚質 (Fitzpatrick 第 III 至 VI 型) 中遠為常見,且可發生於任何年齡。它見於相當數量罹患痤瘡的非洲血統個體。
臨床特徵 (CLINICAL FEATURES)
PIH 由發炎部位的斑性色素過度沉著構成 (圖 77-21)。Wood 燈 (Wood lamp) 檢查可判定色素過度沉著的深度 (見「黑斑」一節)。表皮發炎後的 PIH 造成棕色變色,而真皮的發炎則造成灰棕色變色。PIH 的嚴重度與發炎的嚴重度及基底膜破壞的程度成正比。發現原發性皮膚病的原因應涉及對整個皮膚的檢查。已記錄到對生活品質的不良影響,尤其在有色皮膚的人中,並應加以處理。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
據認為發炎標記 (inflammatory markers) 刺激驅動 PIH 的黑色素細胞。造成表皮發炎的疾病導致表皮角質細胞增加,而真皮發炎則導致色素失禁進入真皮。黑色素最終由巨噬細胞清除。有色皮膚的人對 PIH 易感性增加的原因尚未完全闡明,但據認為與黑色素小體中黑色素的量增加有關。
診斷 (DIAGNOSIS)
組織學特徵包括色素失禁併噬黑色素細胞的堆積,以及表皮層黑色素增加。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
當 PIH 廣泛分布於軀幹時,它可能看起來類似色素性蕁麻疹 (urticaria pigmentosa),但陽性的 Darier 徵 (Darier sign) 以及與病灶相關的搔癢病史,應有助於將 PIH 與色素性蕁麻疹區別。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
臨床病程可變,並取決於發炎或損傷的位置。它在較深膚質 (Fitzpatrick 第 III 至 VI 型) 中,以及在苔癬樣發炎過程後較為持久。表皮 PIH 常自發性但緩慢地消退。
治療 (MANAGEMENT)
PIH 的處置仍然困難。重要的是,尤其在治療有色皮膚的病人時,要及早治療原發性皮膚病以預防 PIH。防曬的重要性不應被低估。在物理療法後,治療前與治療後的方案至關重要,尤其對有色皮膚的人,可能包括防曬、hydroquinone、hydroquinone 替代品,或局部皮質類固醇。
局部 hydroquinone 仍是 PIH 的黃金標準治療 (表 77-3)。市面上有大量含甘草 (licorice)、大豆 (soy)、菸鹼醯胺 (niacinamide) 與各種植物萃取物的非處方藥妝品 (cosmeceuticals),許多處方局部製劑,如麴酸 (kojic acid)、類視黃醇與 α-羥基酸,可作為單一療法或併用。含 hydroquinone、tretinoin 與局部皮質類固醇的 Kligman 配方及類似的三重療法 (triple-therapy) 組合,也常被處方。包括換膚與雷射在內的物理療法應謹慎使用,因為若使用過於積極,它們可能誘發 PIH。品質開關與皮秒雷射曾被描述單獨使用,或作為 PIH 更廣泛治療計畫的一部分。表皮 PIH 比真皮 PIH 更適合局部療法。
黑斑 (Melasma)
重點一覽 (AT-A-GLANCE)
■ 後天性淺棕至深棕色、界線不清的斑。
■ 在有色皮膚的人中較為常見。
■ 顴部皮膚、鼻、前額與上唇是常侵犯的區域。
■ Hydroquinone 仍是黃金標準治療。
■ 物理與口服療法也可使用。
表 77-3:淡化製劑 (Lightening Agents)
| 製劑 (AGENTS) | 作用機制 (MECHANISM OF ACTION) |
|---|---|
| Hydroquinone | 酪胺酸酶抑制劑 (Tyrosinase inhibitor) |
| 類視黃醇 (Retinoids) | 刺激角質細胞更新、減少黑色素小體轉移、酪胺酸酶抑制劑 |
| Arbutin | 酪胺酸酶抑制劑;抑制黑色素小體成熟 |
| Azelaic acid | 酪胺酸酶抑制劑 |
| Kojic acid | 與銅交互作用並抑制酪胺酸酶 |
| Tranexamic acid | 抑制纖維蛋白溶酶 (plasmin);抗氧化劑 |
改編自 Chaowattanapanit S, Silpa-archa N, Kohli I, et al. Postinflammatory hyperpigmentation: a comprehensive overview: treatment options and prevention. J Am Acad Dermatol. 2017;77(4):607-621.
黑斑 (melasma) 是最常見的色素疾病之一,有時被稱為黃褐斑 (chloasma) 或妊娠面斑 (mask of pregnancy)。
流行病學 (EPIDEMIOLOGY)
黑斑影響數百萬人,報導的盛行率從美國南部西班牙裔族群的 9% 至東南亞人的 40% 不等。
它被引述為印度女性中最常見的色素疾病。它主要見於停經前女性,儘管男性與停經後女性也可能受影響。
臨床特徵 (CLINICAL FEATURES)
黑斑典型表現為中央顏面瀰漫的淺棕至深棕色色素沉著區域 (圖 77-22 與 77-23)。顴部與下頷區域、前額、下巴與上唇最常受影響——中顏面型 (63%:前額、鼻、下巴與上唇);顴部型 (21%:鼻與臉頰);以及下頷型 (16%:下頜支 ramus mandibulae) 是最常見的型態。黑斑不侵犯眼周皮膚以及嘴唇、頸部與耳朵。它較少見於前臂 (亦稱後天性臂部皮膚色素異常 acquired brachial cutaneous dyschromatosis)。文獻中已反覆記錄到對生活品質的負面影響;因此,治療臨床醫師需要探詢憂鬱與焦慮的症狀。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
致病機轉未完全瞭解。生物活性黑色素細胞、遺傳與荷爾蒙影響,以及紫外線曝曬,已知是重要的。特定的誘發因素,尤其是口服避孕藥與雌激素替代療法,已被認為與本病的惡化有關。
近期,有色皮膚個體中的可見光,以及病灶皮膚表皮中血管增生與血管內皮生長因子 (vascular endothelial growth factor) 的增加,已被認為與黑斑的致病機轉有關。
診斷 (DIAGNOSIS)
Wood 燈可能有助於在較淺膚質者中區別表皮型與真皮型及混合型黑斑,儘管即使是 Wood 光增強的病灶 (據稱代表表皮型黑斑) 也有真皮黑色素。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷可能很廣泛,取決於受影響的區域與色素的位置 (表皮、真皮或混合)。它包括發炎後色素沉著、成熟性色素異常、藥物誘導的色素沉著、色素性扁平苔癬,以及可能的 Civatte 皮膚異色症 (poikiloderma of Civatte)。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
儘管經治療後淡化或消退,黑斑仍是一種慢性與復發性疾病,於懷孕期間、日光或可見光曝曬,以及某些荷爾蒙治療下會惡化。
治療 (MANAGEMENT)
管理期望並就黑斑復發與慢性的本質進行諮商,對病人的滿意度至關重要。停止任何明顯的觸發因素是處置的第一步。防曬是處置的核心,應包括避免日光、防護衣物,以及規律塗抹含物理性遮蔽劑的廣效防曬乳。對 Fitzpatrick 膚質第 IV 至 VI 型者,也應注意防護可見光。已知表皮色素沉著對局部療法較有反應。儘管 4% hydroquinone 仍是黃金標準,各種其他局部製劑已被使用,成效不一。含大豆、甘草、木質素過氧化物酶 (lignin peroxidase) 與菸鹼醯胺的非處方乳霜,只是據稱有助於輕度表皮型黑斑的一些植物萃取物與維生素。含 tretinoin、azelaic acid 與 kojic acid 的製劑於長期使用時有所幫助。併用局部療法是表皮型黑斑的有用策略。Kligman 配方是一種廣受歡迎的組合,含 5% hydroquinone、0.1% tretinoin 與一種溫和的局部皮質類固醇。另一種常被稱為三重組合療法 (triple-combination therapy) 的治療,包括 4% hydroquinone、0.05% tretinoin 與一種局部皮質類固醇。Kligman 配方與三重組合療法都被認為對表皮型黑斑有效。淺層化學換膚已被證實有效,並在作為治療輔助時有用,但必須考量治療的成本與頻率。此外,必須考量發炎後色素過度沉著的風險,並進行適當的治療前與治療後照護,尤其在有色皮膚的個體中。雷射治療可能有助於黑斑的治療,但也可能造成進一步不想要的色素過度沉著。各種雷射,包括強脈衝光、鉺:YAG,以及非剝離性 1550-nm 與 1927-nm 分段雷射,都曾被報導對黑斑有效,但低能量品質開關 Nd:YAG 雷射 (亦稱雷射調色 laser toning) 是最受歡迎的。此治療方式在謹慎選擇的病例中使用少數幾次治療時,已被證實有效且安全。近期,tranexamic acid 已被證實可淡化表皮型與真皮型黑斑。它曾以局部、病灶內與口服形式使用。當在數月間以低劑量 (500 to 700 mg daily) 作為輔助使用時,它在治療抗性病例中有效且安全。
部分單側小痣病 (Partial Unilateral Lentiginosis)
重點一覽 (AT-A-GLANCE)
■ 後天性局限性色素過度沉著。
■ 出生時或兒童期出現。
■ 可能是鑲嵌型第 1 型神經纖維瘤病的變異型。
■ 通常在 1 個或多個皮節內。
部分單側小痣病 (partial unilateral lentiginosis, PUL) 是一種罕見的色素疾病,亦稱單側小痣、小痣鑲嵌現象 (lentiginous mosaicism)、帶狀小痣樣痣、節段性小痣病,以及聚集性小痣病。
流行病學 (EPIDEMIOLOGY)
通常於出生時或兒童期出現。
臨床特徵 (CLINICAL FEATURES)
PUL 的特徵是眾多具銳利邊緣的小痣。病灶以節段型態出現於中線,位於 1 個或多個皮節內。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
其節段型態與表現,伴隨咖啡牛奶斑、Lisch 結節或神經纖維瘤,暗示 PUL 是鑲嵌型 NF1 的變異型。基因研究將有助於進一步闡明此主題。
診斷 (DIAGNOSIS)
特徵性的病史與臨床外觀常具診斷性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑲嵌型 NF1。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
PUL 終生持續。尚未注意到 PUL 造成的併發症。
治療 (MANAGEMENT)
品質開關紫翠玉與 Nd:YAG 雷射被報導為治療選項。
Riehl 黑色素沉著 (Riehl Melanosis)
重點一覽 (AT-A-GLANCE)
■ 後天性灰棕藍色局限性網狀色素過度沉著。
■ 由局部接觸致敏劑 (contact sensitizers) 所引起。
■ 多為中年女性。
■ 主要位於顏面。
■ 雷射曾被描述為治療選項。
Riehl 黑色素沉著,又稱女性顏面黑色素沉著與色素性接觸性皮膚炎,最早於 1917 年由 Riehl 描述。
流行病學 (EPIDEMIOLOGY)
Riehl 黑色素沉著多見於中年女性,尤其在較深膚質者中,如拉丁裔與亞洲人。
臨床特徵 (CLINICAL FEATURES)
其特徵是快速發病的網狀灰棕色至近乎黑色的網狀色素過度沉著。顏面 (尤其是前額、顴弓區域與顳部) 與頸部主要受侵犯,但手、前臂與軀幹也可能受影響 (圖 77-24)。發炎發現,如紅斑與搔癢,通常不存在,儘管在臨床病程的非常早期階段可能觀察到。病人可能報告顯著的心理壓力。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
致病機轉未完全瞭解,但據推測色素過度沉著是由反覆接觸閾值劑量的接觸致敏劑所誘導,例如化妝品與光學增白劑中使用的香料、某些色素與殺菌劑。
診斷 (DIAGNOSIS)
主要的組織病理學特徵是表皮基底層的液化退化,導致真皮的色素失禁。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
Civatte 皮膚異色症、Brocq 口周紅變色素沉著 (erythrose peribuccal pigmentaire of Brocq)、顏面頸部毛囊性紅黑色素沉著 (erythromelanosis follicularis faciei et colli),以及藥物誘導的色素沉著,都是鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
若不避免接觸致敏劑,Riehl 黑色素沉著為進行性。它可能需要許多年才能淡化,但不見全身性效應。
治療 (MANAGEMENT)
不幸地,治療具挑戰性。停用局部致敏劑至關重要。防曬與局部去色素製劑 (hydroquinone、α-羥基酸、tretinoin) 對這種真皮色素沉著的效果有限,較近期的報告描述以雷射與口服 tranexamic acid 治療。強脈衝光治療在某些病例中有所幫助,但帶有發炎後色素過度沉著的風險。低能量品質開關 Nd:YAG 與「雷射調色」在亞洲皮膚中已展現中度的改善,甚至曾與口服 tranexamic acid 及 hydroquinone 併用。
Cronkhite-Canada 症候群 (Cronkhite-Canada Syndrome)
重點一覽 (AT-A-GLANCE)
■ 後天性全身性淺棕至深棕色斑與斑片。
■ 手掌與足底常首先受侵犯。
■ 也可見甲營養不良 (onychodystrophy) 與禿髮。
■ 營養補充與皮質類固醇可能有幫助。
Cronkhite-Canada 症候群 (Cronkhite-Canada syndrome, CCS) 是一種非常罕見的疾病,造成皮膚與胃腸道表現。
流行病學 (EPIDEMIOLOGY)
文獻中報導約 450 個病例。亞洲 (最常為日本) 與歐洲血統的成人通常受影響,始於生命第四個十年。
臨床特徵 (CLINICAL FEATURES)
Cronkhite-Canada 症候群最初表現為注意到於手掌與足底的棕色斑與斑片。繼之以全身性色素變化,伴隨甲營養不良與禿髮。
胃腸道症狀,如腹痛、噁心、腹瀉與體重減輕,除胃腸道息肉病與偶發的惡性腫瘤外亦被注意到。併發症包括但不限於蛋白質與電解質失衡、貧血,甚至門靜脈血栓 (portal vein thrombosis) 與腎絲球腎炎。Cronkhite-Canada 症候群可能與其他自體免疫疾病 (如全身性紅斑性狼瘡與類風濕性關節炎) 相關。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
懷疑為自體免疫過程,但確切病因仍難以捉摸。
診斷 (DIAGNOSIS)
需要臨床病理相關性以做出診斷。組織學變化包括表皮黑色素增加併真皮黑色素沉著。亦可見表皮的棘層肥厚與緻密角化過度。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
鑑別診斷包括先天性小痣的其他原因。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
Cronkhite-Canada 症候群為進行性,並可能因本病的全身性表現而造成顯著的罹病率與死亡率。
治療 (MANAGEMENT)
以皮質類固醇進行免疫抑制與營養補充可能有幫助。
Brocq 口周紅變 (Erythrose Peribuccal of Brocq)
重點一覽 (AT-A-GLANCE)
■ 非常罕見的皮膚病,造成瀰漫性色素過度沉著、紅斑,以及毛囊中心性 (folliculocentric) 膚色丘疹。
■ 主要發生於女性。
Brocq 口周紅變亦稱顏面紅變色素沉著 (erythrose pigmenta faciei)、口周色素性紅變 (erythrosis pigmentosa peribuccalis),以及口周黑色素沉著 (melanosis perioralis et peribuccalis)。
流行病學 (EPIDEMIOLOGY)
僅有少數病例 (主要為女性) 被報導。
臨床特徵 (CLINICAL FEATURES)
通常可見先有紅斑,繼之以口周區域瀰漫的淺棕至深棕色色素沉著,侵犯皮膚的上唇與下唇,唇紅緣周圍不受侵犯。在某些病例中,可見鱗屑與絨毛 (vellus hairs) 的喪失。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
病因未知,儘管紫外線曝曬與香料可能扮演某種角色。
診斷 (DIAGNOSIS)
診斷通常以臨床做出。組織學發現為非特異性。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
黑斑與 PIH 是主要的鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
持續與進展是常態。
治療 (MANAGEMENT)
建議防曬與避免觸發因素。
皮膚澱粉樣變性 (Cutaneous Amyloidosis)
斑狀澱粉樣變性 (macular amyloidosis) 表現為棕至灰棕色斑,融合成斑塊,主要位於背部 (見第 125 章)。
Pasini-Pierini 萎縮性皮膚病 (Atrophoderma of Pasini-Pierini)
第 70 章更詳細討論皮膚的萎縮性疾病。Pasini-Pierini 萎縮性皮膚病的皮膚病灶可被描述為單一或多發的灰至紫棕色萎縮性斑片,直徑從 1 cm 至超過 10 cm,伴隨輕微凹陷與特徵性的「懸崖落差 (cliff-drop)」邊緣。
固定性藥疹 (Fixed Drug Eruption)
固定性藥疹可造成色素過度沉著 (見前文「毒素與藥物」一節與第 45 章)。
持久性色素異常性紅斑 (Erythema Dyschromicum Perstans)
重點一覽 (AT-A-GLANCE)
■ 後天性特發性真皮黑色素沉著。
■ 藍灰色界線分明的色素過度沉著斑與斑塊。
■ 通常為中間膚質 (例如西班牙裔、亞洲族群)。
■ 見於光防護部位 (photoprotected sites)。
■ 治療具挑戰性,可能需要全身性療法。
持久性色素異常性紅斑 (erythema dyschromicum perstans, EDP),又稱灰皮病 (ashy dermatosis)、灰色皮膚病 (dermatosis cinecienta),以及黑皮病性慢性圖形紅斑 (erythema chronicum figuratum melanodermicum),最早由 Ramirez 於 1957 年描述。患有此皮疹的病人被標記為 los cenicientos,即「灰燼之人」。EDP、色素性扁平苔癬與特發性爆發性斑狀色素沉著 (idiopathic eruptive macular pigmentation) 這些名詞曾被交替使用,並在文獻中常被混淆,儘管它們似乎是不同的臨床疾病。
流行病學 (EPIDEMIOLOGY)
EDP 主要見於中間膚質的女性 (例如西班牙裔與亞洲人)。據說它是年輕成人的疾病,大多數於生命第二與第三個十年之間表現,儘管一項近期對韓國病人的回顧顯示其於第三與第四個十年發病。
臨床特徵 (CLINICAL FEATURES)
EDP 表現為廣泛的無症狀、界線分明的藍灰色斑,最常見於光防護部位 (圖 77-25 與 77-26)。病灶隨時間緩慢擴大,在本病的急性階段,病灶邊緣可見紅斑性邊界。紅斑性邊界,尤其在較深膚質者中,可能演變為一個色素減退的邊界,使色素過度沉著更為突出。黏膜不受侵犯,且不存在皮膚外表現。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
病因未知,儘管在墨西哥族群中已描述與人類白血球抗原-D 相關 (human leukocyte antigen-D related, HLA-DR) 的易感性。
診斷 (DIAGNOSIS)
診斷通常需要臨床病理相關性。組織學上,早期病灶顯示真皮水腫與苔癬樣發炎、基底層空泡化,以及伴隨血管周圍浸潤的膠樣小體 (colloid bodies)。稍後,深真皮中可見黑色素失禁併噬黑色素細胞。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
色素性扁平苔癬、特發性爆發性斑狀色素沉著,以及 PIH,是主要的鑑別診斷。亦須排除藥物誘導的色素沉著。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
本病為慢性且非常難治療。病灶在數年間緩慢進展,通常不會自發性消退。
治療 (MANAGEMENT)
局部療法對這種後天性真皮色素疾病無幫助,全身性療法的改善也很微小。為期 8 至 12 週的 dapsone 100 mg 療程可能淡化色素沉著,clofazimine 100 mg daily 在一項小型研究中已展現改善。不幸地,儘管已評估許多其他治療,但無一導致明顯的淡化。最近,雷射手術已由荷蘭的一個團隊研究,他們的結論是分段雷射 (fractionated laser) 對 EDP 的治療不成功。
色素性扁平苔癬 (Lichen Planus Pigmentosus)
重點一覽 (AT-A-GLANCE)
■ 後天性特發性真皮黑色素沉著。
■ 界線不清的棕至灰棕色色素過度沉著斑。
■ 通常見於深度色素沉著的皮膚 (例如東南亞人、阿拉伯人)。
■ 多見於光照部位。
■ 治療僅提供色素沉著的微小改善。
色素性扁平苔癬被視為扁平苔癬的一種變異型。扁平苔癬在第 32 章討論。它在文獻中常被誤認為 EDP 與特發性爆發性斑狀色素沉著。
流行病學 (EPIDEMIOLOGY)
色素性扁平苔癬主要見於具有較深度色素沉著皮膚 (例如南亞人、東南亞人與阿拉伯族群) 的中年 (生命第三至第四個十年) 個體。
臨床特徵 (CLINICAL FEATURES)
對稱的棕至灰棕色、界線不清的斑與斑片,主要見於光照部位,如頭部 (前額與顳部) 與頸部 (圖 77-27)。病灶也可見於皮膚褶處,如腋窩 (稱為反向色素性扁平苔癬 lichen planus pigmentosus–inversus)。病灶罕見地也可能出現於黏膜。不存在皮膚外表現。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
確切病因未知,但分別用於烹飪與護髮配方的芥末油 (mustard oil) 與印度醋栗油 (amla oil),在回溯性研究中被列為可能的原因。亦須排除藥物誘導的色素沉著。
診斷 (DIAGNOSIS)
需要切片,且臨床病理相關性至關重要。組織學上可見表皮萎縮、基底層空泡化併血管周圍淋巴球浸潤,以及淺真皮的噬黑色素細胞。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
EDP、特發性爆發性斑狀色素沉著,以及 PIH,是主要的鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
某些病例在數月間逐漸消退,但這些病例是常態的例外。
治療 (MANAGEMENT)
防曬至關重要。局部 tacrolimus (0.03% twice daily)、局部與全身性皮質類固醇,以及局部維生素 A,可能對色素性扁平苔癬有幫助。治療組合與雷射治療也曾被使用,結果不一。近期,一項前瞻性研究報導,低劑量口服 isotretinoin (20 mg daily),於 6 個月間併用防曬乳使用,造成色素沉著的穩定與減少,尤其在病程早期使用時。
特發性爆發性斑狀色素過度沉著 (Idiopathic Eruptive Macular Hyperpigmentation)
重點一覽 (AT-A-GLANCE)
■ 後天性特發性真皮黑色素沉著。
■ 界線分明的棕至灰色色素過度沉著斑與斑塊。
■ 病灶通常比持久性色素異常性紅斑為小。
■ 通常出現於兒童與青少年。
■ 通常出現於顏面、軀幹與近端四肢。
■ 治療僅提供色素沉著的微小改善。
特發性爆發性斑狀色素過度沉著是一種罕見的色素疾病,最初由 Degos 與其同事描述,但首次以英文描述是在 1996 年。
流行病學 (EPIDEMIOLOGY)
特發性爆發性斑狀色素過度沉著通常見於兒童與青少年。
臨床特徵 (CLINICAL FEATURES)
特發性爆發性斑狀色素過度沉著的特徵是無症狀、界線分明、無鱗屑的棕色斑與斑塊 (直徑 5 mm 至數公分) 的皮疹,侵犯顏面、頸部、軀幹與近端四肢。不見先前的發炎或紅斑。已注意到一部分病人具有增厚的斑塊 (表皮乳頭瘤病 epidermal papillomatosis)。不存在皮膚外表現。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
病因未知。
診斷 (DIAGNOSIS)
組織病理學上,基底表皮的黑色素增加,並在真皮中可見噬黑色素細胞。肥大細胞的數量正常。不見基底層變化或苔癬樣發炎。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
EDP、色素性扁平苔癬,以及 PIH,是主要的鑑別診斷。須排除藥物誘導的色素沉著。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
病灶在數月至數年間自發性消失,不留瘢痕。
治療 (MANAGEMENT)
自發性消退意味著在大多數情況下不需要治療。局部淡化製劑,如類視黃醇、α-羥基酸與 hydroquinone,是可能的治療。
混合性色素減退與色素過度沉著 (Mixed Hypomelanosis and Hypermelanosis)
重點一覽 (AT-A-GLANCE)
混合性色素減退與色素過度沉著的原因包括:
■ 遺傳性泛發性色素異常症 (dyschromatosis hereditaria universalis)。
■ 遺傳性對稱性色素異常症 (dyschromatosis symmetrica hereditaria)。
■ Dohi 網狀肢端色素沉著 (reticulate acropigmentation of Dohi)。
■ 伴 (Westerhof 症候群) 或不伴色素減退的家族性進行性色素過度沉著。
■ 流浪者白斑 (vagabond leucoderma)。
遺傳性泛發性色素異常症 (Dyschromatosis Hereditaria Universalis)
遺傳性泛發性色素異常症是一種罕見的自體顯性疾病 (第 12 號染色體 12q21-q23 上 ABCB6 基因的突變),通常於日本家族的嬰兒期或兒童早期表現,其特徵是針尖至豌豆大小的色素減退與色素過度沉著斑,以網狀型態分布於軀幹、腹部與四肢,通常不侵犯顏面與掌蹠表面。
Dohi 網狀肢端色素沉著 (Reticulate Acropigmentation of Dohi)
Dohi 網狀肢端色素沉著是遺傳性泛發性色素異常症的一種局限型態,亦稱遺傳性對稱性色素異常症。其特徵是手與足背面小型、對稱的色素過度沉著與色素減退斑,主要見於南美與亞洲家族的年幼兒童。
伴 (Westerhof 症候群) 或不伴色素減退的家族性進行性色素過度沉著 (Familial Progressive Hyperpigmentation with [Westerhof Syndrome] or without Hypopigmentation)
伴或不伴色素減退的家族性進行性色素過度沉著是一種自體顯性疾病,由第 12 號染色體 12q22 上 KIT 配體基因 (KIT ligand gene, KITLG) 的雜合突變所引起。它於出生時或嬰兒早期表現為瀰漫性色素過度沉著,並可能呈現咖啡牛奶斑,以及位於顏面、頸部、軀幹與四肢、較大的色素減退灰葉斑 (ash-leaf macules)。1978 年的 Westerhof 症候群病例系列描述了某些家族成員伴有生長遲緩與智能遲緩。
流浪者白斑 (Vagabond Leukoderma)
流浪者白斑是一種見於生活在不良衛生條件下的人的疾病。許多人酗酒、飲食不足,並感染蝨子與/或疥蟲。瀰漫性淺棕色色素過度沉著存在於肩部與腰帶處,頸部與背部則點綴著脫色斑。本病在採行較健康的生活方式後改善,並很可能代表許多疾病的共存。
致謝 (ACKNOWLEDGMENTS)
我們在此感謝本章前一版作者的貢獻:Hilde Lapeere、Barbara Boone、Sofie De Schepper、Evelien Verhaeghe、Mireille Van Gele、Katia Ongenae、Nanja Van Geel、Jo Lambert,以及 Lieve Brochez。
圖片 (FIGURES)
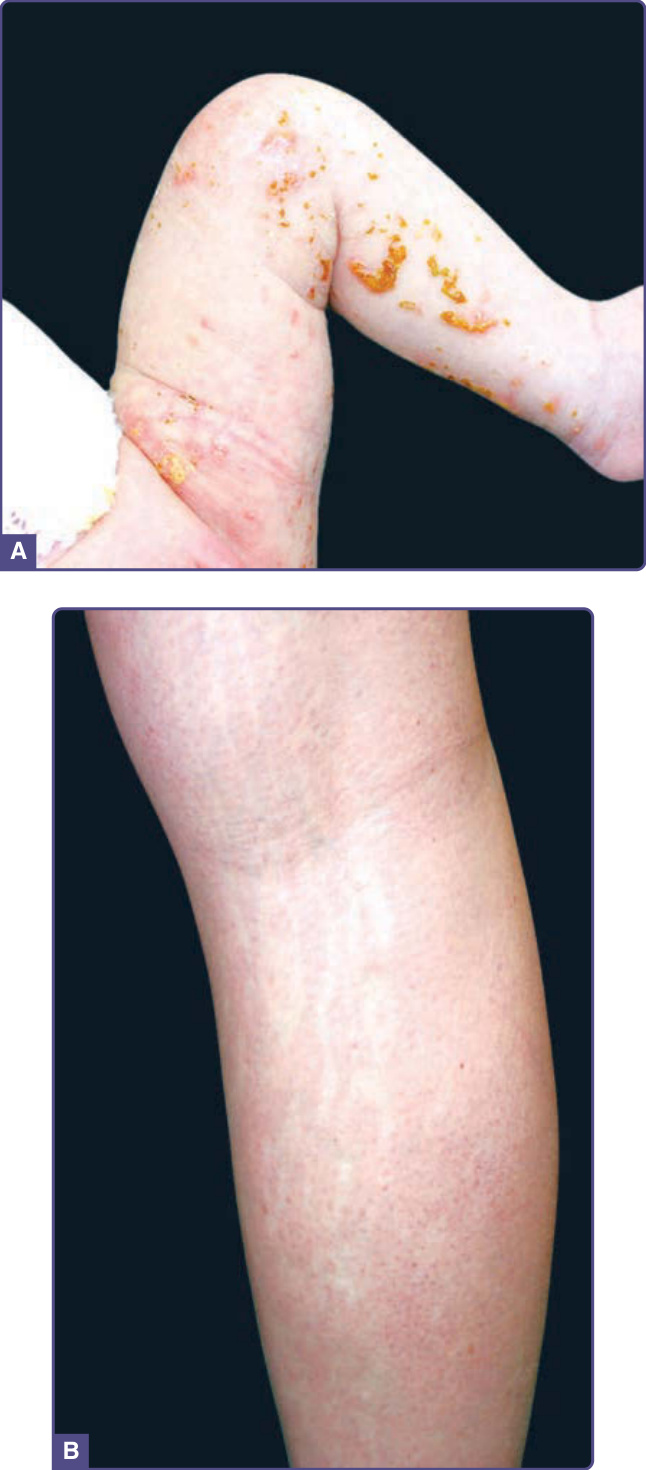
圖 77-1:一位母親與她的嬰兒的色素失禁症 (Incontinentia pigmenti)。A,一名 2 週大嬰兒的疣狀病灶。B,沿 Blaschko 線分布的色素減退萎縮性病灶。
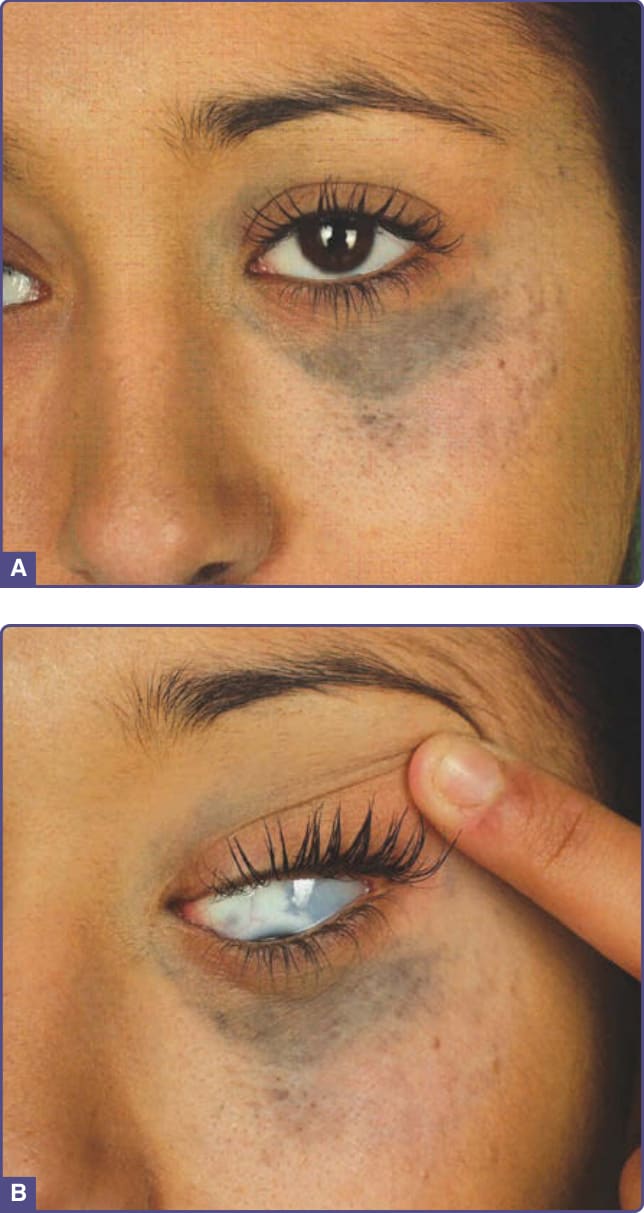
圖 77-2:一名 20 歲女性的太田母斑 (Nevus of Ota)。A,眼眶周圍的色素過度沉著。B,色素過度沉著延伸進入鞏膜。
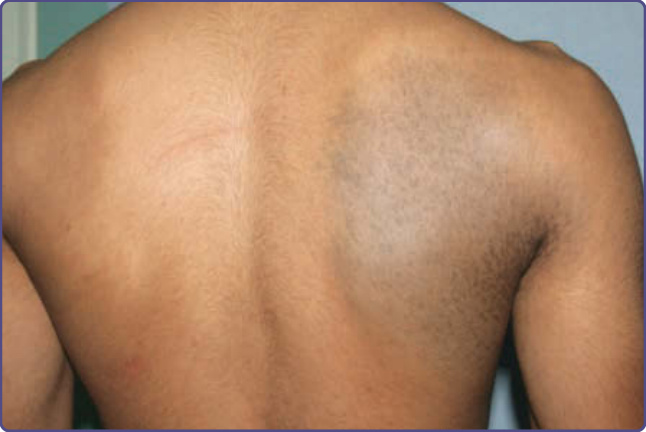
圖 77-3:一名中東青少年背部的伊藤母斑 (Nevus of Ito)。
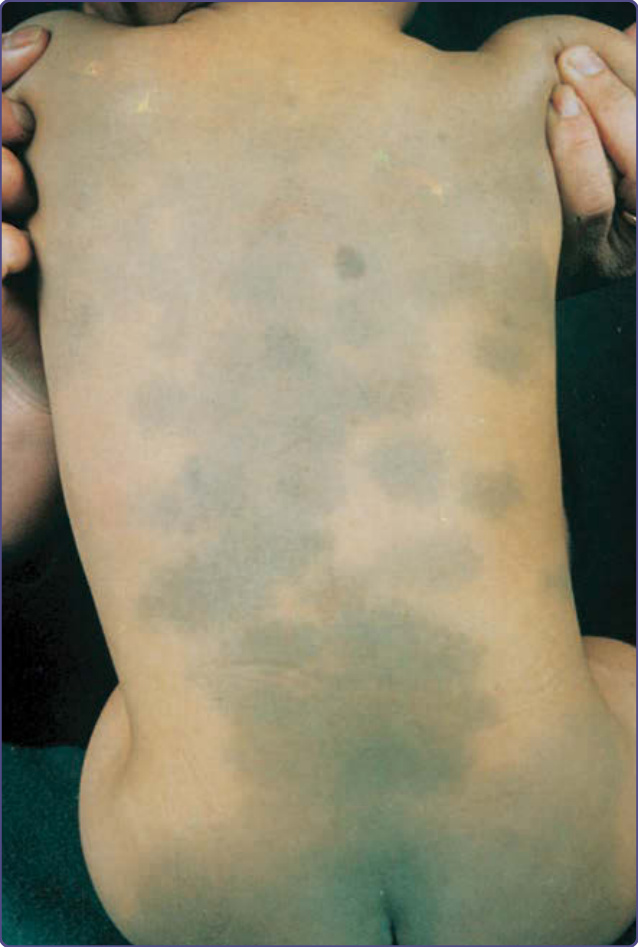
圖 77-4:腰薦區域的典型蒙古斑 (Mongolian spots),以及背部的異位或薦外蒙古斑。
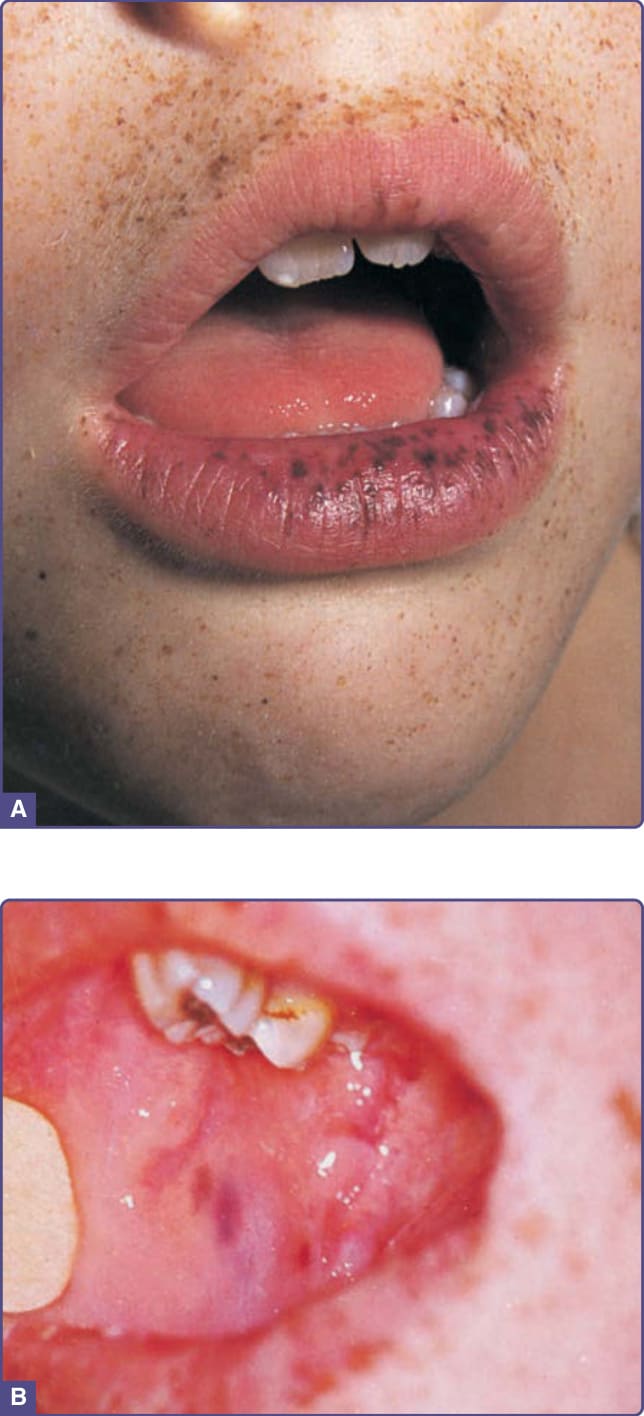
圖 77-5:Peutz-Jeghers 症候群。A,呈深棕色至灰藍色的小痣 (lentigines) 出現於嘴唇、嘴巴周圍與手指上。唇部斑可能隨時間消失。B,頰黏膜的斑為藍至藍黑色且具特異性病徵 (pathognomonic);與唇部病灶不同,這些病灶不傾向隨時間消失。
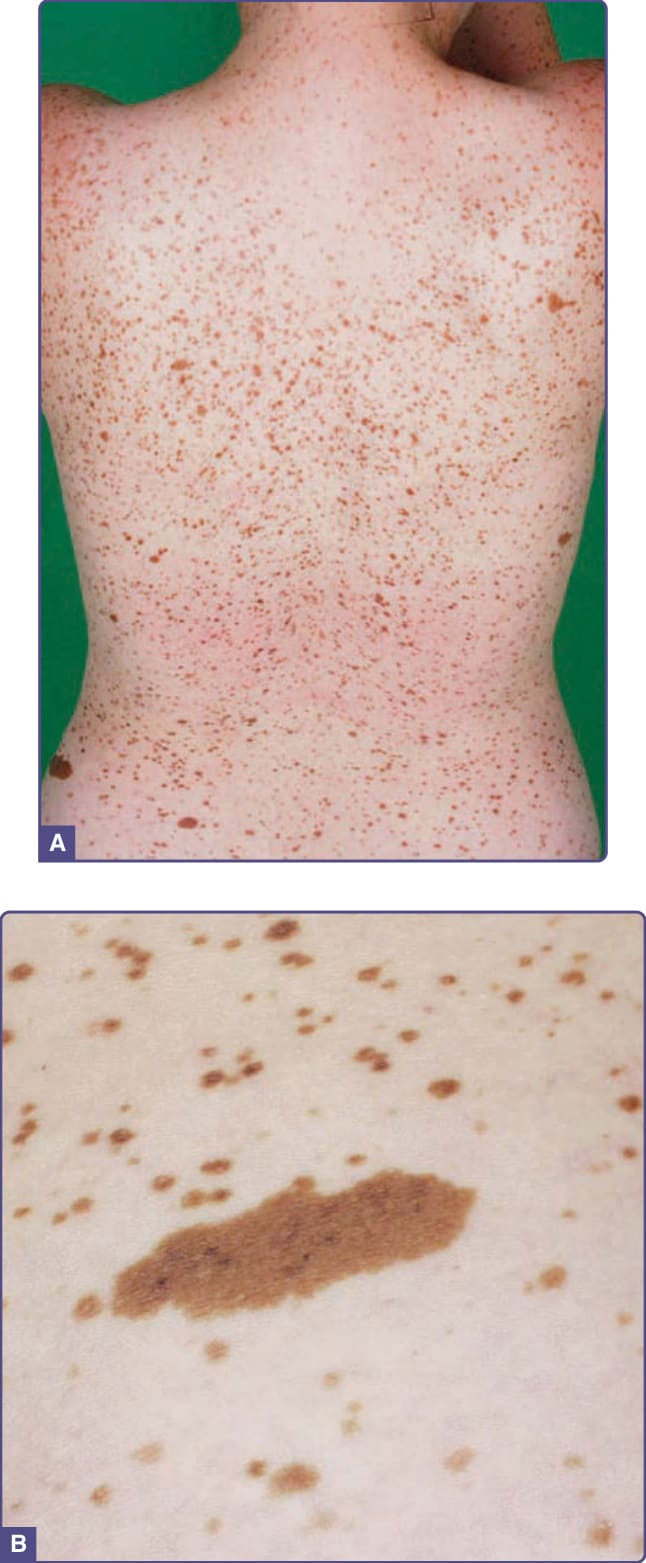
圖 77-6:LEOPARD (小痣 lentigines、心電圖傳導缺陷 electrocardiogram conduction defects、眼距過寬 ocular hypertelorism、肺動脈狹窄 pulmonary stenosis、生殖器異常 abnormalities of genitalia、生長遲緩 retardation of growth,以及感覺神經性耳聾 sensorineural deafness) 症候群。A 與 B,一名患有 LEOPARD 症候群的 27 歲女性。注意其特徵性的廣泛小痣與數個咖啡牛奶斑 (café-au-lait macules)。
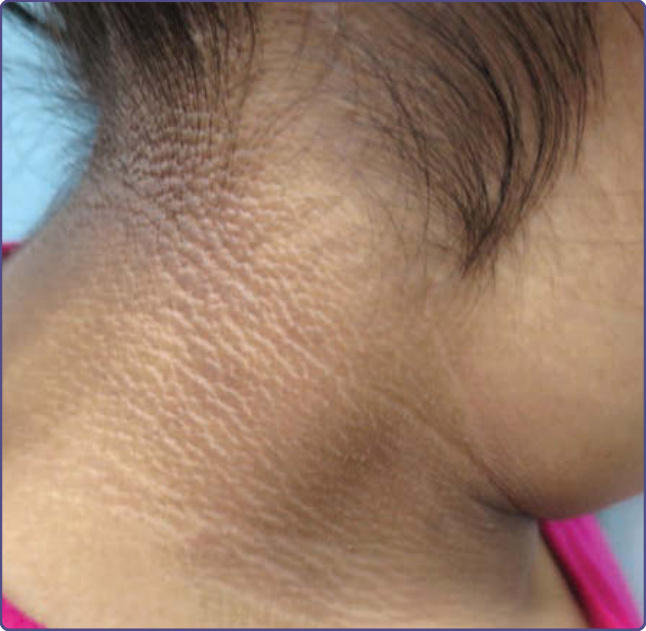
圖 77-7:一名菲律賓裔年輕女孩頸部的黑色棘皮症 (Acanthosis nigricans)。
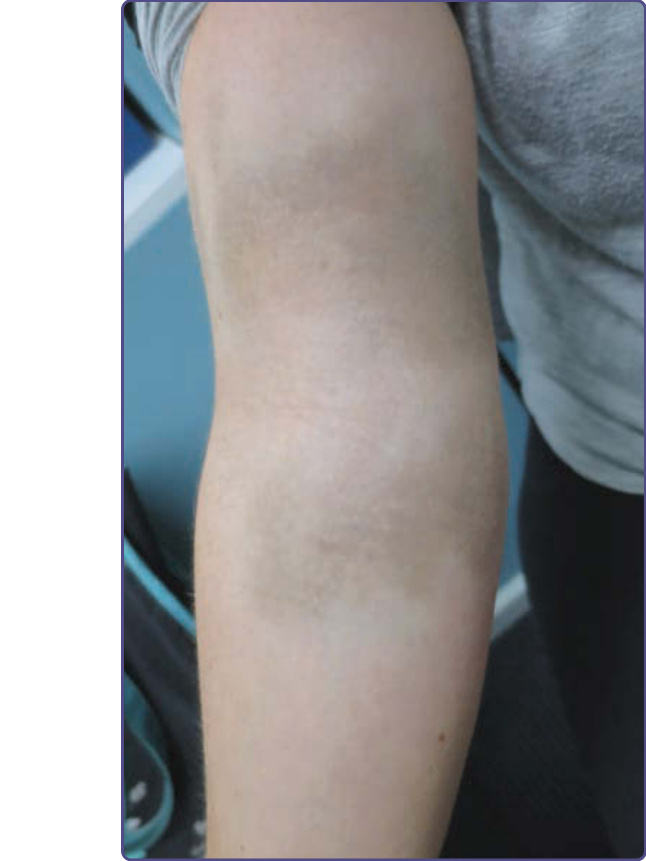
圖 77-9:一名白人女性手臂繼發於靜脈鐵劑輸注的瀰漫性灰色色素沉著。
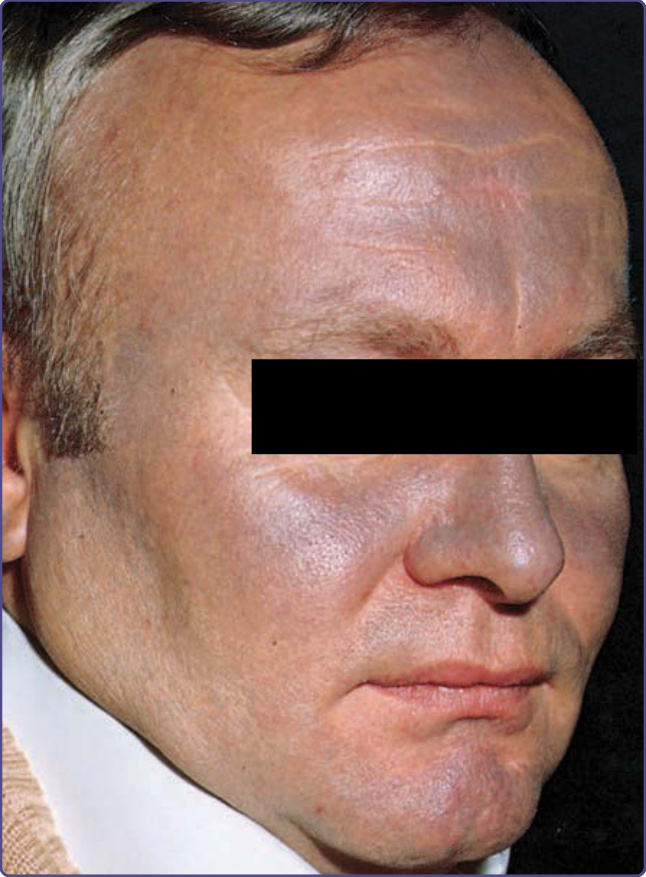
圖 77-10:Amiodarone 色素過度沉著。此病人呈現顯著的 amiodarone 誘導之顏面板岩灰色色素沉著。藍色 (青皮病 ceruloderma) 是棕色色素沉積於真皮、含於巨噬細胞與內皮細胞中的結果。
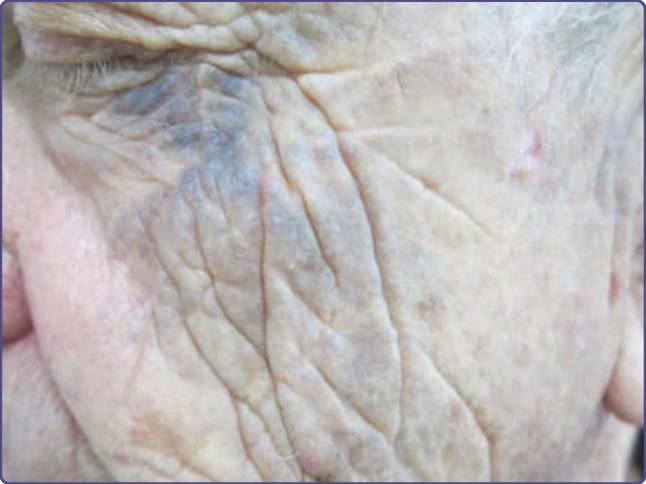
圖 77-11:一名白人男性臉頰上的板岩灰色 minocycline 誘導色素沉著。
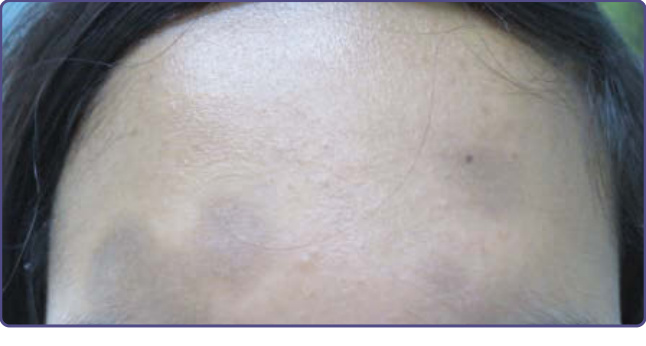
圖 77-12:一名斯里蘭卡年輕女孩顏面繼發於非類固醇抗發炎藥物的固定性藥疹 (Fixed drug eruption)。
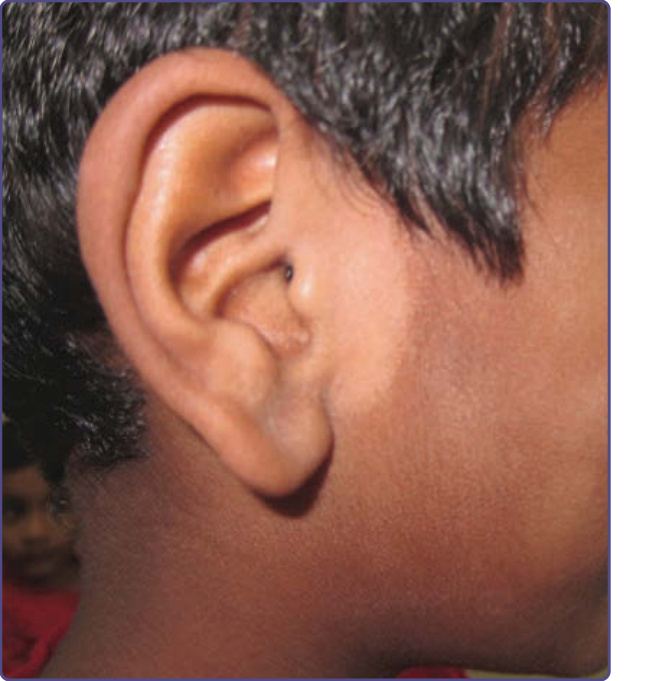
圖 77-13:一名斯里蘭卡裔男孩耳前的色素分界線 (Pigmentary demarcation line)。
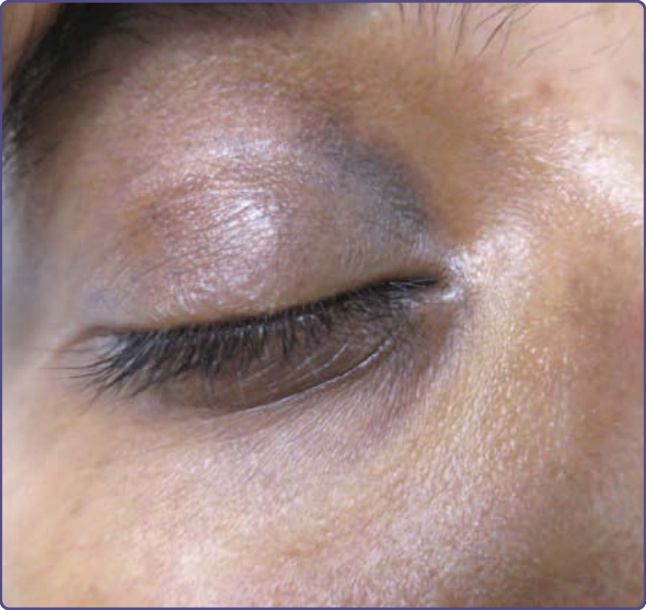
圖 77-14:一名印度女士的眼周色素沉著 (Periorbital pigmentation)。
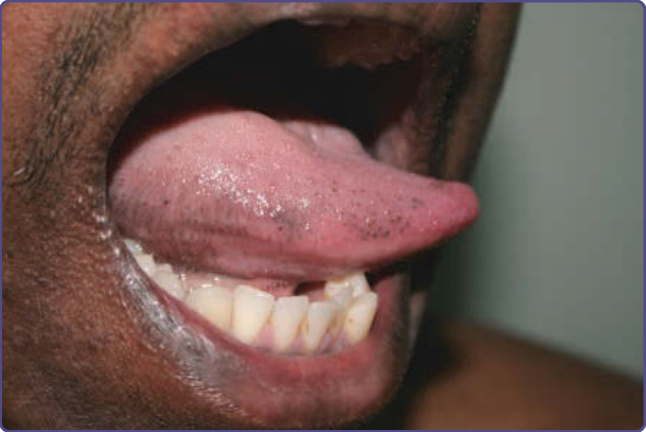
圖 77-15:一名印度男性舌乳頭的良性色素沉著。
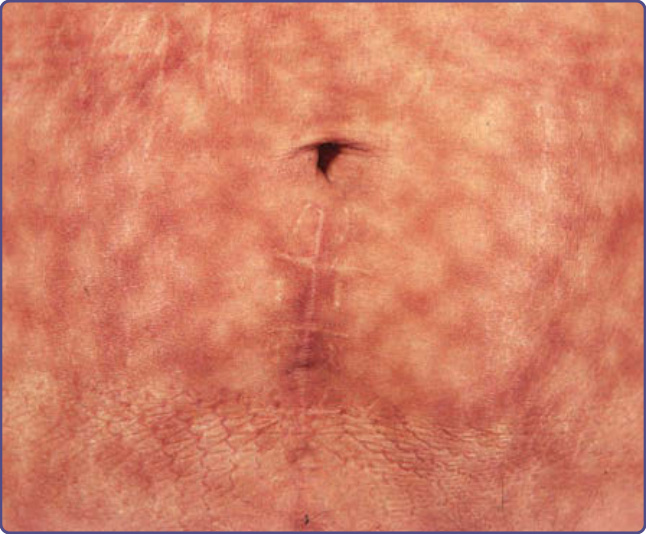
圖 77-16:腹部因多年反覆使用電熱墊所致的網狀色素過度沉著 (火激紅斑 erythema ab igne)。
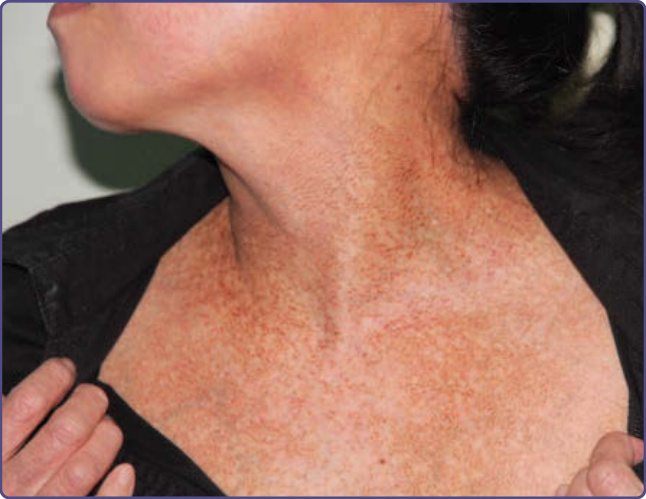
圖 77-17:一名日本女性色素性癢疹 (prurigo pigmentosa) 所見的網狀色素沉著。
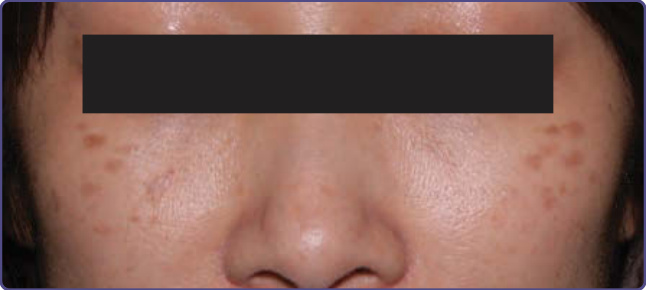
圖 77-18:Hori 痣 (Nevus of Hori)。
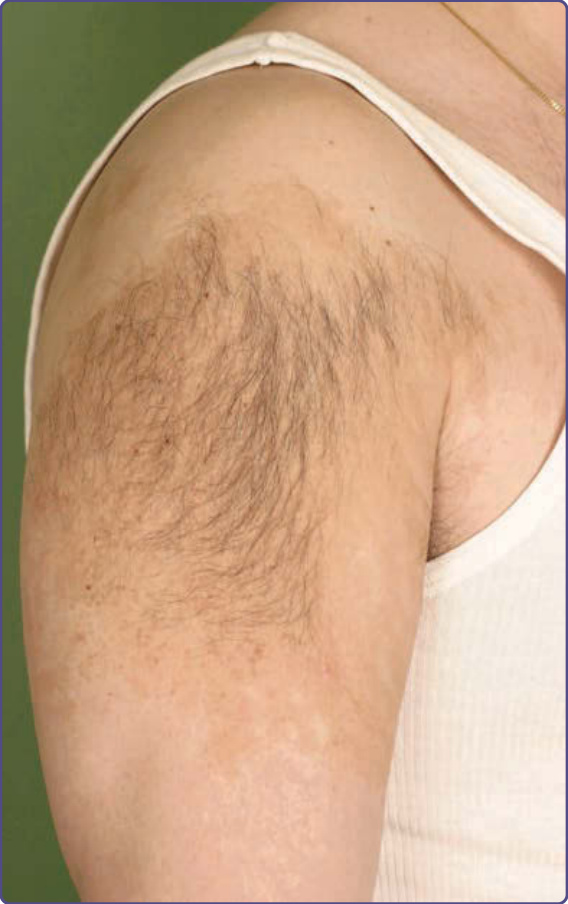
圖 77-19:一名年輕男性上臂與肩部的 Becker 痣 (Nevus of Becker)。注意其典型的位置與明顯的多毛症。
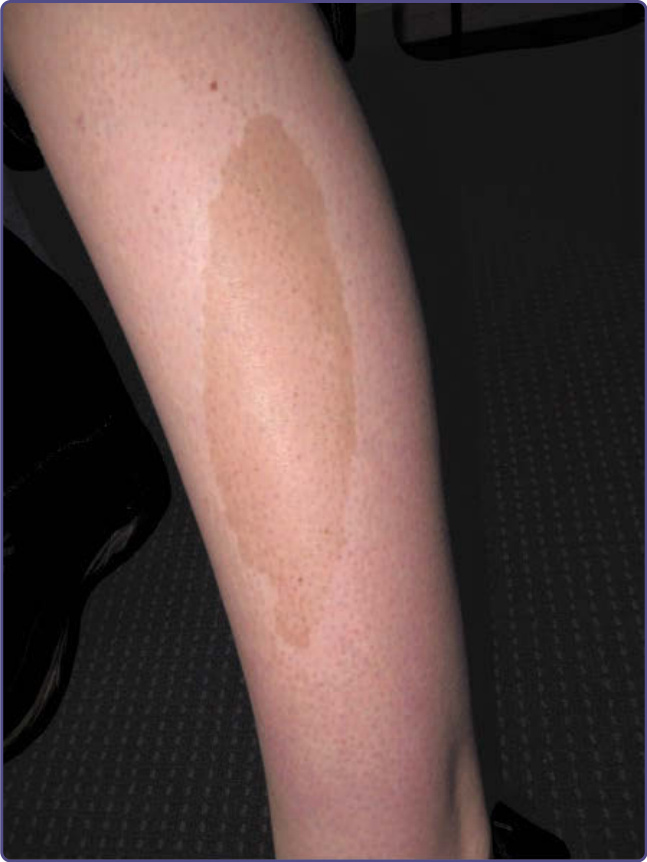
圖 77-20:一名年輕女孩小腿上的咖啡牛奶斑 (Café-au-lait macule)。
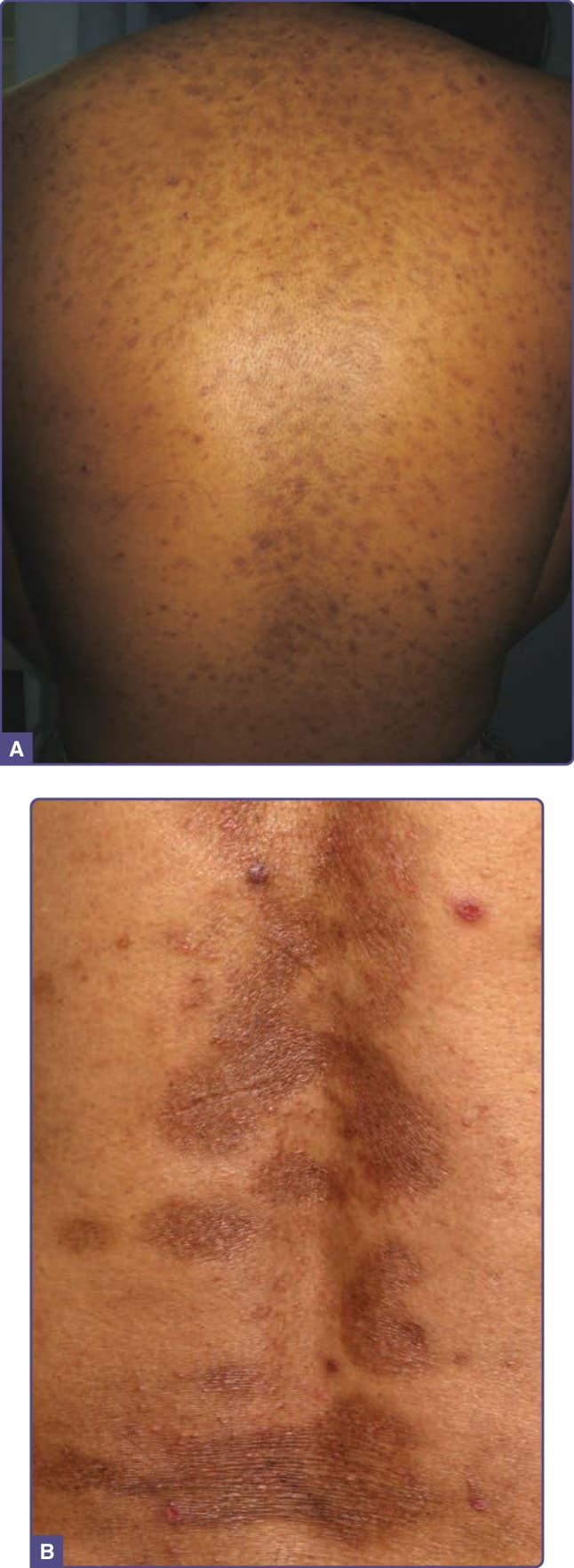
圖 77-21:A,一名非洲年輕女孩背部繼發於痤瘡的發炎後色素過度沉著 (Postinflammatory hyperpigmentation)。B,一名中國男性背部繼發於乾癬的發炎後色素過度沉著。
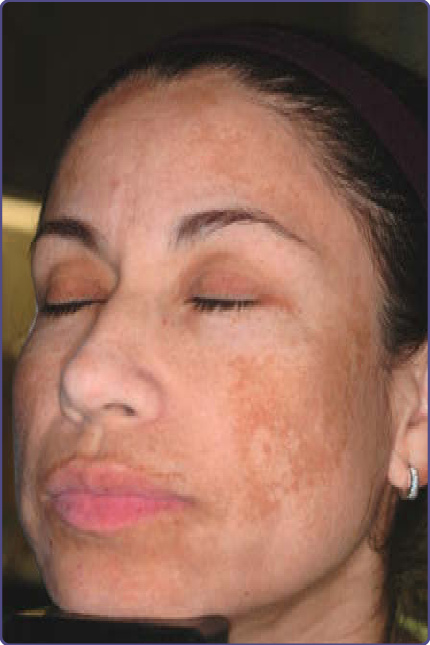
圖 77-23:一名拉丁裔女性因黑斑 (melasma) 所致的前額、臉頰、上唇與下巴色素過度沉著。
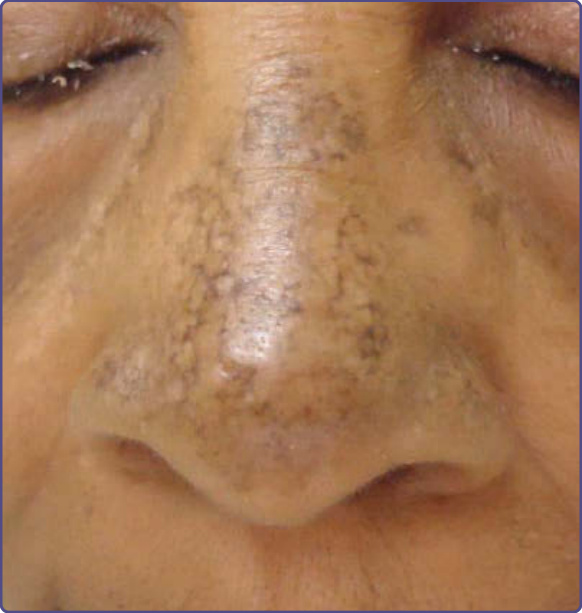
圖 77-24:顏面的 Riehl 黑色素沉著 (Riehl melanosis)。
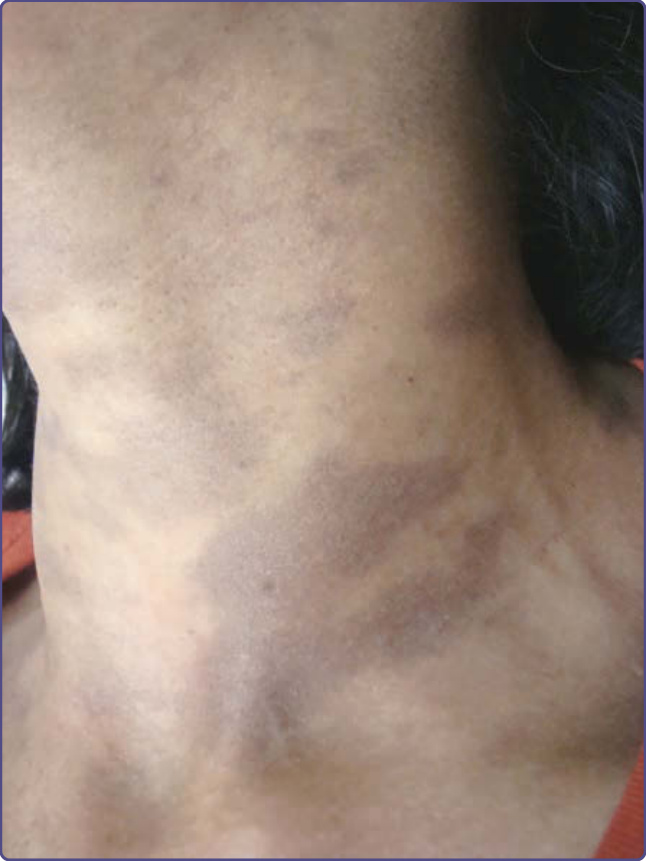
圖 77-25:一名西班牙裔女性頸部的後天性真皮黑色素沉著—持久性色素異常性紅斑 (erythema dyschromicum perstans)。
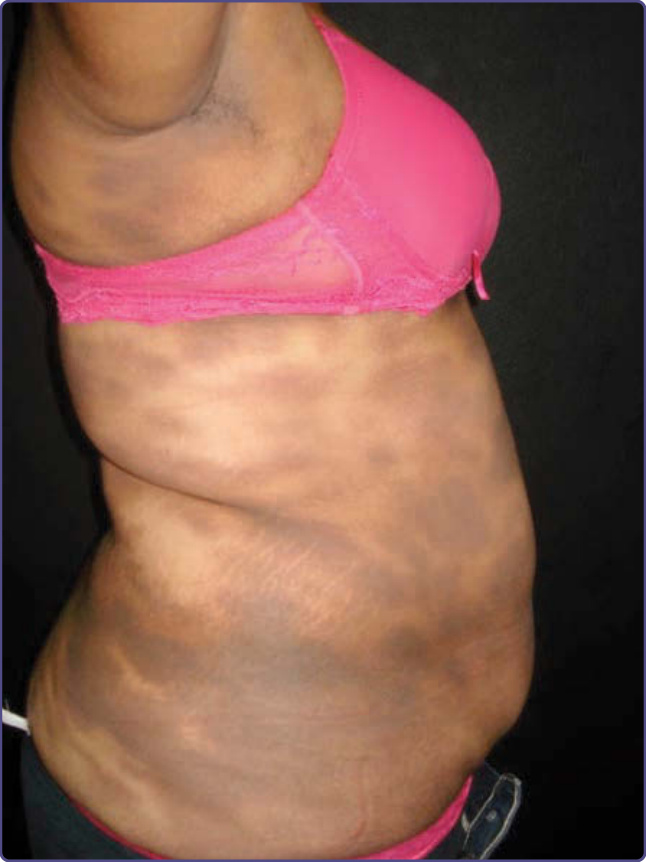
圖 77-26:軀幹上的持久性色素異常性紅斑 (Erythema dyschromicum perstans)。(經 Juan Carlos Mendez 許可使用,皮膚科,UANL,墨西哥蒙特雷。)
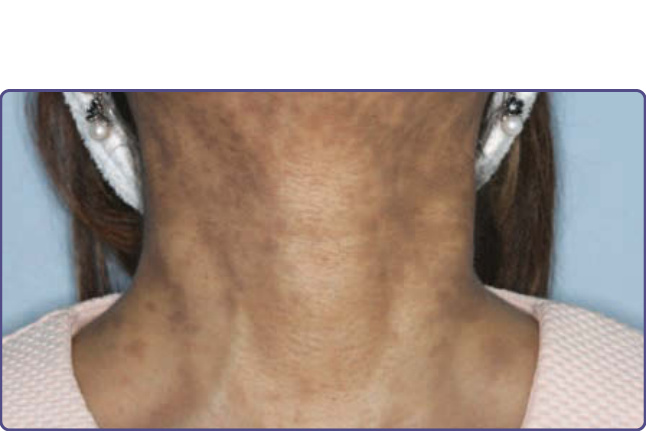
圖 77-27:一名斯里蘭卡女士頸部的後天性真皮黑色素沉著—持久性色素異常性紅斑/色素性扁平苔癬 (lichen planus pigmentosus)。
表格 (TABLES)
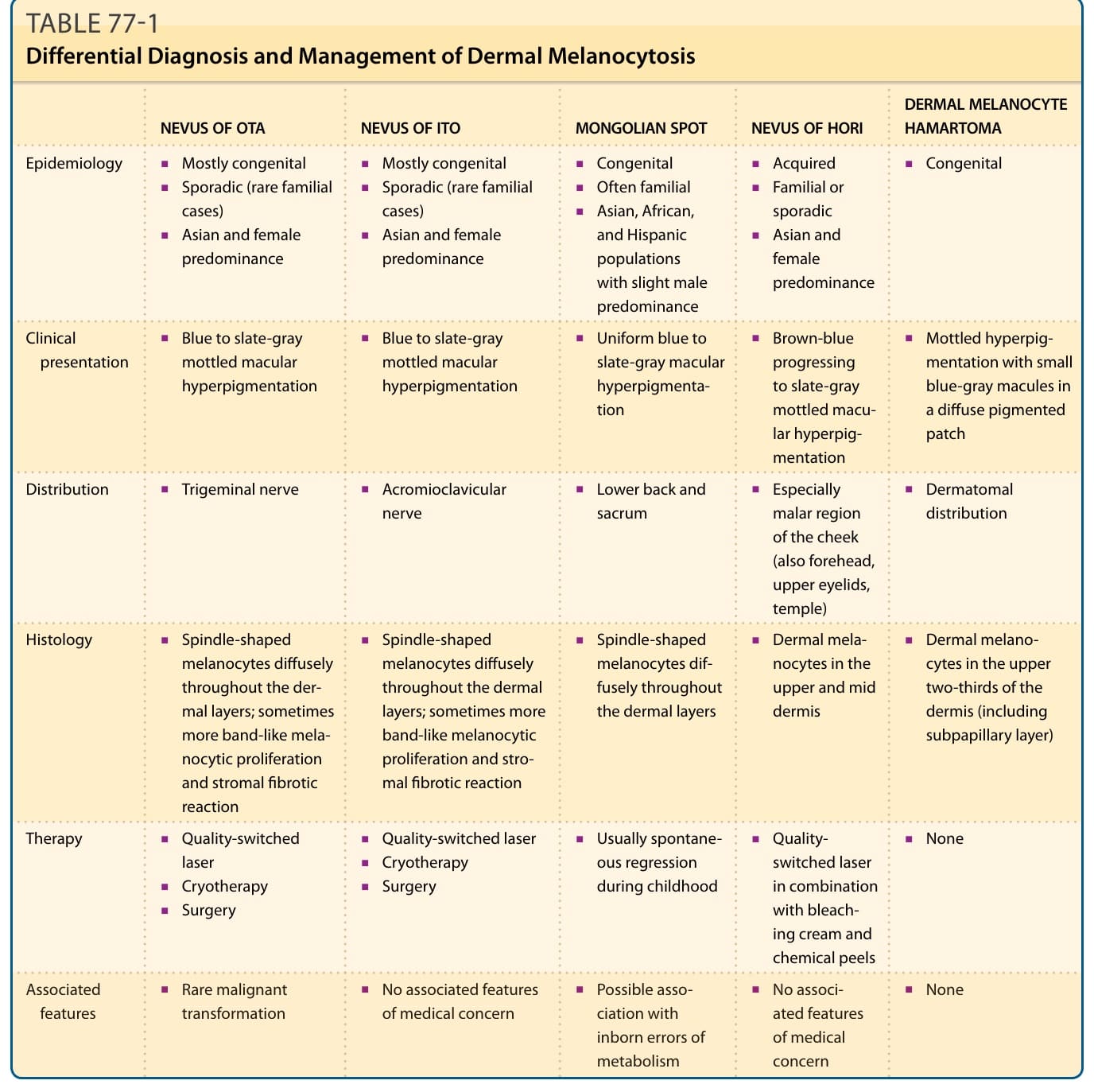
表 77-1:真皮黑色素細胞增多症 (Dermal Melanocytosis) 的鑑別診斷與治療 (Differential Diagnosis and Management of Dermal Melanocytosis)
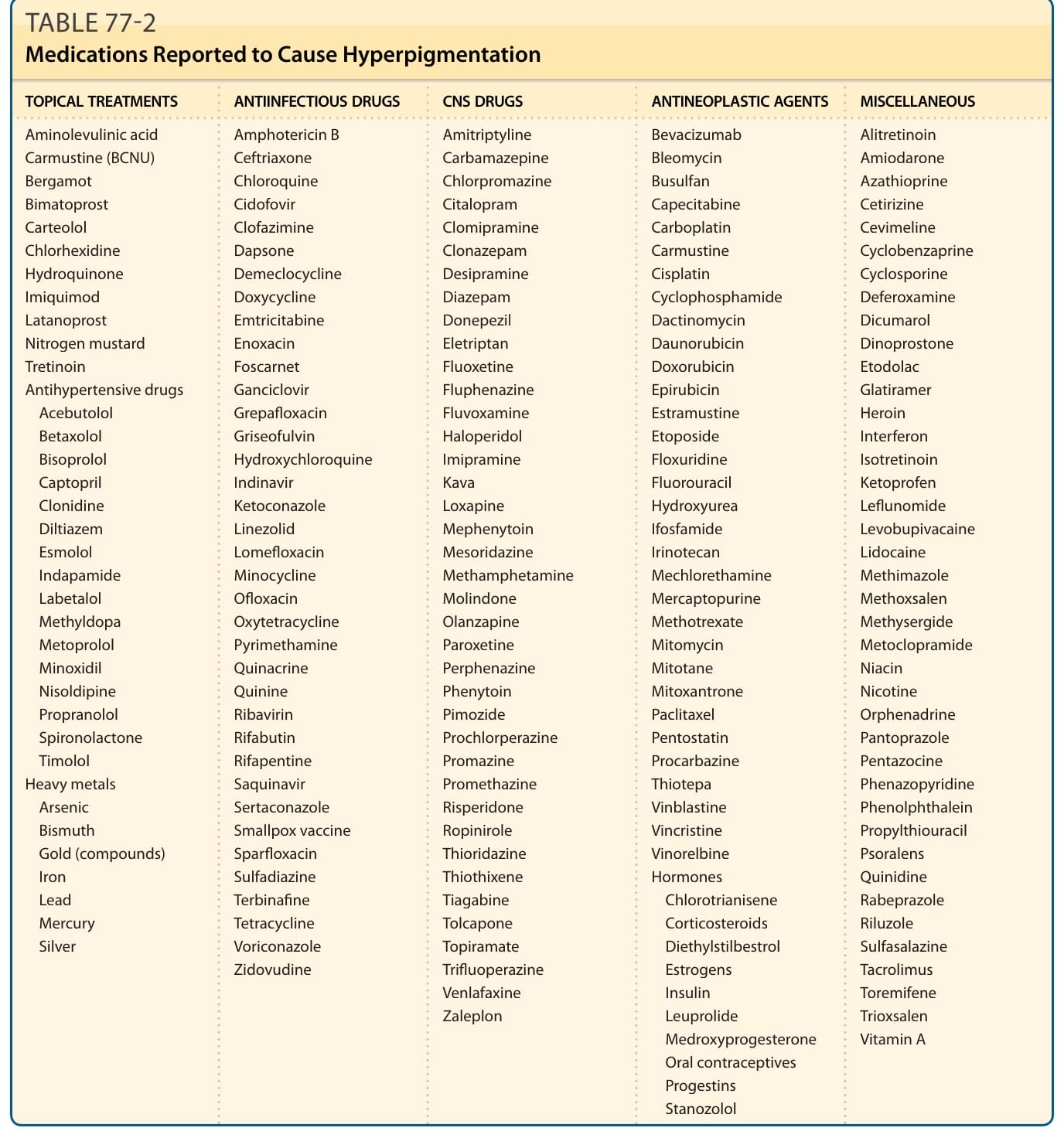
表 77-2:曾被報導造成色素過度沉著的藥物 (Medications Reported to Cause Hyperpigmentation)
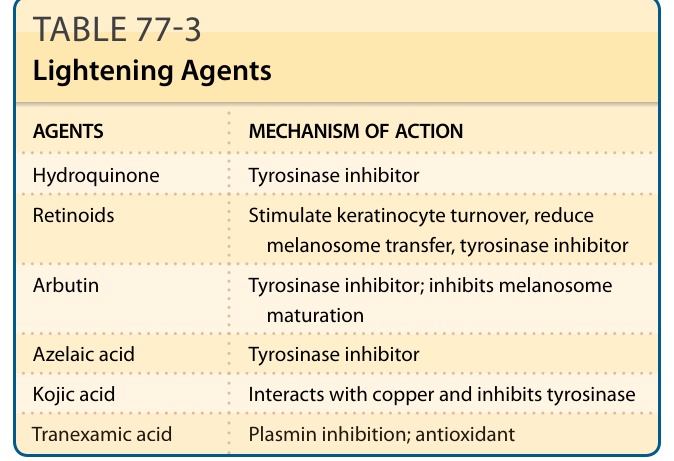
表 77-3:淡化製劑 (Lightening Agents)