Hypermelanoses
13
CONGENITAL HYPERMELANOSIS
LINEAR AND WHORLED NEVOID HYPERMELANOSIS
LINEAR AND WHORLED
NEVOID HYPERMELANOSIS
AT-A-GLANCE
■ Congenital diffuse streaky hyperpigmented macules in Blaschko lines.
■ Typically seen on the trunk and extremities.
■ No preceding inflammation or atrophy.
■ Onset in the first few weeks of life.
■ May fade with advancing age.
Linear and whorled nevoid hypermelanosis is a congenital condition causing diffuse streaky hyperpigmented macules along the lines of Blaschko.1
CLINICAL FEATURES
Linear and whorled nevoid hypermelanosis is characterized by widespread streaky hyperpigmented macules along the lines of Blaschko without preceding inflammation or atrophy in the first few weeks of life. Lesions are typically located on the trunk and limbs and do not cross the midline. Face, palms, soles, eyes, and mucous membranes are spared. Similar cases have been described under different descriptive names (zosteriform hyperpigmentation, zosteriform lentiginous nevus, zebra-like hyperpigmentation). Several cases have been reported with both hyperpigmentation and hypopigmentation. Pigmentary mosaicism is a useful term to encompass all these different phenotypes. Rarely, extracutaneous manifestations, such as developmental and growth retardation, facial and body asymmetry, ventricular septal defects, and pseudohermaphroditism, are observed. The frequency of extracutaneous manifestations is unknown, as these observations have been made in case reports only.
ETIOLOGY AND PATHOGENESIS
Most cases are sporadic but the presence of mosaicism has been confirmed in a few cases (mosaic trisomies 7, 14, 18, and 20, and X-chromosomal mosaicism) by chromosomal analysis.2
DIAGNOSIS
In addition to the typical clinical appearance, histologic examination reveals increased pigmentation of the basal layer and prominence or vacuolization of melanocytes. Pigment incontinence is usually absent.
DIFFERENTIAL DIAGNOSIS
Linear and whorled nevoid hypermelanosis should be differentiated from incontinentia pigmenti (IP) and epidermal nevus.
CLINICAL COURSE AND PROGNOSIS
Kalter and colleagues1 described typical onset in the first few weeks of life with progression during the initial years of life. The pigmentation may fade gradually with advancing age.
INCONTINENTIA PIGMENTI
INCONTINENTIA PIGMENTI
AT-A-GLANCE
■ X-linked dominant disorder resulting from mutation in the IKBKG (previously called NEMO) gene.
■ Lethal in male embryos.
■ Four clinical phases commencing at birth: vesicular, verrucous, hyperpigmented, and hypopigmented.
■ Congenital diffuse linear hyperpigmented macules in Blaschko lines.
IP, also known as Bloch-Sulzberger syndrome, was first described by Garrod and colleagues in
1906. It is an X-linked disorder primarily seen in females, which results in diffuse linear cutaneous hyperpigmentation.
CLINICAL FEATURES
Lesions usually proceed through 4 cutaneous stages, although the stages may sometimes overlap: (a) vesicular stage (from birth or shortly thereafter), which presents with multiple small and mediumsized vesicles following the lines of Blaschko;
13
(b) verrucous stage (between 2 and 8 weeks of age) consisting of wart-like plaques; (c) hyperpigmented stage (several months of age into adulthood); and (d) hypopigmented stage (from infancy onwards) (Fig. 77-1). The degree of hyperpigmentation varies among individuals but appears in streaks and whorls along Blaschko lines. Although it can appear on the limbs, it is usually pronounced on the trunk. The hypopigmented stage is characterized by linear, atrophic, hairless scars along Blaschko lines.
A
B
1352
ETIOLOGY AND PATHOGENESIS
IP is an X-linked, dominantly inherited disorder believed to be embryonically lethal in the majority of males. In most cases, IP is caused by a mutation in the IKBKG gene (previously called NEMO [nuclear factor κB (NF-κB) essential modulator]) on the X chromosome at Xq28.3,4 The gene encodes a regulatory component of the IKappaB kinase complex that activates the NF-κB pathway, which is required to protect against tumor necrosis factor-α–induced apoptosis.5
In females with IP, inactivation of 1 of the 2 X chromosomes through a process termed lyonization occurs during embryogenesis. Epidermal cells expressing the defective IKBKG gene give rise to typical skin lesions along the lines of Blaschko, reflecting the embryonic migration path of the affected keratinocytes. The cutaneous lesions in the vesicular stage represent the population of IKBKG-deficient cells that fail to activate NF-κB, leading to apoptosis. The number of IKBKG-deficient cells decreases secondary to apoptosis and is replaced by cells expressing the normal allele. Subsequently, the inflammatory and vesicular stage ends. The hyperproliferation in the second stage is likely a result of compensatory proliferation of normal IKBKG keratinocytes. Hyperpigmentation in the third stage results from incontinence of epidermal melanin pigment, which then migrates into the dermis and is engulfed by macrophages. Extracutaneous manifestations occur in a significant number of patients with IP. Ocular (30% to 70%), CNS (40%), dental, and skeletal anomalies are among the most common.
DIAGNOSIS
Histologically, the number of melanocytes appears to be normal, although a reduced number of melanocytes have been noted. The thin epidermis and absence or reduction of skin appendages in the dermis may contribute to the impression of hypopigmentation. Histologically, the areas of pigmentation show many melanin-laden melanophages, extensive deposits of melanin in the basal cell layer and dermis. Epidermal basal layer vacuolization and degeneration is also noted.
CLINICAL COURSE AND PROGNOSIS
Usually the hyperpigmentation fades gradually over several years, leaving hypopigmented skin (stage 4), which represents postinflammatory dermal scarring.
MANAGEMENT
A beneficial effect of topical steroids and topical tacrolimus in the vesicular stage has been reported.6
Hypopigmented and atrophic scarring may be amenable to noncultured epidermal cell grafting.7
DYSKERATOSIS CONGENITA
DYSKERATOSIS CONGENITA
AT-A-GLANCE
■ Congenital diffuse reticular hyperpigmentation with nail atrophy and leukoplakia.
■ Manifests in the first few years of life.
■ Extracutaneous manifestations are bone marrow failure and malignancies.
Dyskeratosis congenital (DKC) or Zinsser-Cole-Engman syndrome is characterized by cutaneous and hematologic manifestations.
CLINICAL FEATURES
Reticulate skin pigmentation especially on the neck and chest, nail atrophy in fingernails and toenails, and leukoplakia are seen in those with DKC. Extracutaneous manifestations include bone marrow failure (in more than 80% of cases) and malignancy in the second and third decades of life.
ETIOLOGY AND PATHOGENESIS
In all cases of DKC, the causative mutations are present in components of the telomerase complex. Rapidly dividing somatic cells express low but detectable levels of telomerase activity that slows telomere shortening occurring with each cycle of DNA replication, which eventually leads to cellular senescence (permanent loss of proliferative capacity). It is now thought that DKC is caused by defective telomere maintenance, which limits the proliferative capacity of hematopoietic and epithelial cells. Increased melanin synthesis occurs in senescent melanocytes, which is likely to account for the pigmentary changes noted in DKC. Critically short telomeres may force cells into “ replicative crisis,” at which time activation of an alternative “ALT” mechanism for lengthening telomeres in the absence of telomerase may lead to development of malignancies. The role of telomeres in cell biology (cellular aging) was actually first demonstrated through the finding of short telomeres in DKC.8
X-linked DKC is caused by mutations in the DKC1 gene located at Xq28, encoding for dyskerin. Females carrying 1 mutated allele are protected by expression of normal telomerase on the unaffected allele. In autosomal dominant DKC, the majority of cases are caused by mutations in TERC, the RNA component of the telomerase complex. TERT (telomerase reverse transcriptase) is affected much less often in autosomal dominant DKC. In the autosomal recessive form of DKC, mutations in telomerase-associated proteins such as NOP10, NHP2, and TINF2 are involved.9
13
DIAGNOSIS
Skin biopsy of hyperpigmented skin does not demonstrate specific changes. Epidermal atrophy and a chronic inflammatory cell infiltrate with numerous melanophages in the upper dermis are usually observed in histology.
DIFFERENTIAL DIAGNOSIS
DKC may be confused with Fanconi syndrome, which is characterized by short stature, hypoplastic or aplastic thumbs, and a reduced number of carpal bones. The patchy hyperpigmentation of the trunk, neck, groin, and axillary regions in Fanconi syndrome appears earlier than in DKC, that is, in the first few years of life.
CLINICAL COURSE AND PROGNOSIS
The autosomal dominant form has a better prognosis than other forms, possibly because of the presence of an unaffected allele with some preservation of telomerase activity. Usually, the hyperpigmentation fades gradually after several years.
NAEGELI-FRANCESCHETTI- JADASSOHN SYNDROME
NAEGELI-FRANCESCHETTI-
JADASSOHN SYNDROME
AT-A-GLANCE
■ Autosomal ectodermal dysplasias.
■ Reticulate hyperpigmentation on the neck and axillae.
■ Nail, teeth, and sweating abnormalities.
CLINICAL FEATURES
Reticulate hyperpigmentation is most prominent in neck and axillae. Palmoplantar diffuse keratoderma, absence of dermatoglyphs, nail and teeth changes, and heat intolerance owing to diminished or absent sweating are characteristic.10
ETIOLOGY AND PATHOGENESIS
The Naegeli-Franceschetti-Jadassohn syndrome and the related dermatopathia pigmentosa reticularis are autosomal dominant ectodermal dysplasias caused by mutations in the KRT14 gene.11 The KRT14 gene codes for keratin 14, which, together with keratin 5, forms intermediate keratin filaments. Keratin 14 is predominantly produced by the keratinocytes in the basal cell layer of the epidermis. Mutations in KRT14 gene lead to fragility of basal keratinocytes. It plays an important role during ontogenesis of dermatoglyphs and sweat glands.12
1353
13
Dermatopathia pigmentosa reticularis has been distinguished from Naegeli-Franceschetti-Jadassohn syndrome by lifelong persistence of skin hyperpigmentation, partial alopecia, and absence of dental anomalies. Both disorders have been mapped to 17q11.2-q21 and are considered to be allelic.11
HISTOLOGY
There are numerous melanophages in the upper dermis, next to a patchy epidermal hyperpigmentation. Eccrine glands histologically appear normal in number and structure.
DERMATOPATHIA PIGMENTOSA RETICULARIS
DERMATOPATHIA
PIGMENTOSA RETICULARIS
AT-A-GLANCE
■ Autosomal dominant.
■ Reticulate hyperpigmentation on the trunk.
■ Nail, sweating, and ocular abnormalities with nonscarring alopecia.
CLINICAL FEATURES
Reticulate hyperpigmentation on the trunk, palmoplantar keratoderma with punctiform accentuation, nail and ocular changes, noncicatricial alopecia, ichthyosis, hypohidrosis, widespread hyperkeratotic lesions, ainhum formation, severe periodontal disease, mechanic blister formation, and pigmentation of the oral mucosa are all seen in this condition.13
ETIOLOGY AND PATHOGENESIS
Dermatopathia pigmentosa reticularis is a rare autosomal dominant condition.
HISTOLOGY
Histologic examination shows pronounced pigmentary incontinence, liquefaction degeneration of the basal cell layer and hyalinization of dermal collagen.
DOWLING-DEGOS DISEASE
DOWLING-DEGOS DISEASE
AT-A-GLANCE
■ Autosomal dominant.
■ Reticulate hyperpigmentation in the flexures with acneiform perioral pits and comedo-like hyperkeratotic papules.
1354
CLINICAL FEATURES
Numerous small symmetrical brown-gray macules usually begin in the groin and axillae in the third or fourth decade of life and then spread to intergluteal and inframammary folds, neck, trunk, and arms. Comedo-like hyperkeratotic follicular papules on the neck and axilla and pitted perioral acneiform scars also appear.14
ETIOLOGY AND PATHOGENESIS
This rare autosomal dominant genodermatosis ( synonym: reticular pigmented anomaly of the flexures) is cause by mutations in the KRT5 gene on chromosome 12q13.13. Betz and colleagues demonstrated that haploinsufficiency in keratin 5 causes epithelial remodeling, melanosome mistargeting, and altered perinuclear organization of intermediate filaments.14 Keratin 5 and keratin 14 are 2 proteins that together form a keratin intermediate filament. It is hypothesized that keratin 5 haploinsufficiency causes an excess of keratin 14 that could be responsible for the pathology of Dowling- Degos disease by competing with transport adapters. Keratins could regulate the availability and positioning of AP-3 complexes in keratinocytes or alternatively could regulate the interaction of AP-3–dependent vesicles with motor proteins.
HISTOLOGY
The histology is very characteristic with filiform rete projections from both epidermis and follicular infundibulum with hyperpigmented tips, without an increase of melanocytes. The disorder has to be differentiated from acanthosis nigricans, which has a different histologic appearance.
CLINICAL COURSE AND PROGNOSIS
Progression of the hyperpigmentation is slow and usually is visible from early adult life, without sex predilection.
GALLI-GALLI DISEASE
GALLI-GALLI DISEASE
Patients with Galli-Galli disease show the diagnostic features of Dowling-Degos disease with the additional histopathologic finding of acantholysis in suprabasal epidermal layers. It is regarded as the acantholytic variant of Dowling-Degos disease. Mutations in the keratin 5 gene have been found in some patients.15
KITAMURA RETICULAR ACROPIGMENTATION
KITAMURA RETICULAR
ACROPIGMENTATION
AT-A-GLANCE
■ Autosomal dominant.
■ Reticulate hyperpigmentation in the flexures with acneiform perioral pits and comedo-like hyperkeratotic papules.
CLINICAL FEATURES
Kitamura reticular acropigmentation is characterized by angular and sharply demarcated reticulate, freckle-like hyperpigmented macules (but no hypopigmentation as seen in Dohi disorder [see section “Mixed Hypomelanosis and Hypermelanosis”]) beginning on the dorsa of the hands during the first decade of life and subsequently spreading to the rest of the body. The macules are slightly depressed; sometimes palmar pits can be observed. Most cases are from Japan.
ETIOLOGY AND PATHOGENESIS
Mutations in the ADAM10 gene (15q21.3). ADAM10 encodes for a zinc metalloprotease, a disintegrin, and a metalloprotease domain containing protein 10, which is involved in the shedding of skin substrates.16
HISTOLOGY
There is an increased number of melanocytes and melanogenic activity, and epidermal atrophy is present upon histologic examination.
DIFFERENTIAL DIAGNOSIS
Dowling-Degos disease, acropigmentation of Dohi, and dermatopathia pigmentosa reticularis are all in the differential diagnosis of Kitamura reticular acropigmentation.
HABER SYNDROME
HABER SYNDROME
Next to a reticulate pigmentation on trunk and axillae, patients with Haber disease also develop verruciformis papular lesions of the trunk and a distinct photosensitive rosacea-like facial erythema and telangiectasias, most commonly presenting in childhood.17
13
NEVUS OF OTA
NEVUS OF OTA
AT-A-GLANCE
■ Congenital circumscribed tan-brown hyperpigmentation with dermal melanocytosis.
■ Unilateral pigmentation in the area of the first and second branches of the trigeminal nerve.
■ Of those affected, 60% have scleral involvement.
■ Mostly occurs in Asian females.
Nevus of Ota (nevus fuscoceruleus ophthalmomaxillaris) was first described by Ota in 1939.18 It is characterized by blue-black or gray-brown dermal melanocytic pigmentation on the face.
EPIDEMIOLOGY
Even though nevus of Ota has been described in different skin types, it is most commonly seen in the Asian population with 0.6% of this population affected.19,20 Onset is usually noted at birth and in females, although it may be noted for the first time in early childhood or puberty.
CLINICAL FEATURES
Unilateral blue-black or gray-brown dermal melanocytic pigmentation is typically seen in areas innervated by the first and second branches of the trigeminal nerve.21 Nevus of Ota is now subclassified as mild (Type 1), moderate (Type 2), intensive (Type 3), and bilateral (Type 4). Of those affected, 60% have scleral pigmentation. Other sites of mucosal pigmentation include the conjunctiva and tympanic membrane (oculodermal melanocytosis) (Fig. 77-2). Malignant melanoma may rarely develop in a nevus of Ota. This necessitates careful followup of the lesion, especially if it occurs in white patients, in whom malignant degeneration seems to be more frequent. Malignant melanocytic tumors in association with nevus of Ota have been shown to arise in the chorioidea, brain, orbit, iris, ciliary body, and optic nerve. In addition, association with ipsilateral glaucoma and intracranial melanocytosis has been described.
ETIOLOGY AND PATHOGENESIS
Various triggers, including infection, trauma, ultraviolet light exposure, and hormonal influences, have been described.
DIAGNOSIS
The diagnosis is usually made clinically but may be confirmed on histology, which reveals evenly spread melanocytes throughout the entire dermis.
1355
13
A
B
DIFFERENTIAL DIAGNOSIS
Bilateral cases should be differentiated from Hori nevus, which is acquired, does not have mucosal involvement, and is less pigmented. Other dermal melanocytoses include nevus of Ito, Mongolian spots, and dermal melanocyte hamartomas (Table 77-1). Associated vascular malformations have been described in phakomatosis pigmentovascularis (port-wine stain type, Klippel-Trenaunay or Sturge- Weber syndrome).
CLINICAL COURSE AND PROGNOSIS
Nevus of Ota is persistent and does not usually undergo spontaneous regression.
1356
MANAGEMENT
Topical therapies are ineffective in the treatment of this dermal pigmentation. Even though cryotherapy and surgery have been used in the past, they should be avoided because of significant scarring. Laser surgery is the treatment of choice with quality-switched (QS) lasers including quality-switched ruby laser, QS alexandrite laser and QS neodymium:yttriumaluminum-garnet (Nd:YAG) laser having the most success in the treatment of this condition. Picosecond lasers also have been reported as effective in treating this condition.22
NEVUS OF ITO
NEVUS OF ITO
AT-A-GLANCE
■ Congenital circumscribed brown-tan hyperpigmentation with dermal melanocytosis.
■ Considered a variant of nevus of Ota.
■ Involvement of the acromioclavicular and deltoid region.
Nevus of Ito is a congenital dermal melanocytosis first described by Ito in 1954 as nevus fuscoceruleus acromiodeltoideus.23 It can be considered as a variant of nevus of Ota. Clinical, demographic, and histologic characteristics are similar to nevus of Ota and both lesions can occur simultaneously (see Table 77-1 and Fig. 77-3).
MONGOLIAN SPOTS
MONGOLIAN SPOTS
AT-A-GLANCE
■ Congenital circumscribed blue-tinged hyperpigmentation caused by dermal melanocytosis.
■ Mainly occurs in persons with skin of color.
■ Usually occurs in the sacral region.
EPIDEMIOLOGY
Mongolian spots are more common in the African, Asian, and Hispanic populations and are only rarely seen in whites.24 They occur in both sexes with a slight male predominance.
13
NEVUS OF OTA NEVUS OF ITO MONGOLIAN SPOT NEVUS OF HORI DERMAL MELANOCYTE HAMARTOMA
Epidemiology
■Mostly congenital
■Mostly congenital
■Sporadic (rare familial cases)
■Sporadic (rare familial cases)
■Asian and female predominance
■Asian and female predominance
■Blue to slate-gray mottled macular hyperpigmentation
■Blue to slate-gray mottled macular hyperpigmentation
Clinical presentation
Distribution
■Trigeminal nerve
■Acromioclavicular nerve
Histology
■Spindle-shaped melanocytes diffusely throughout the dermal layers; sometimes more band-like melanocytic proliferation and stromal fibrotic reaction
■Spindle-shaped melanocytes diffusely throughout the dermal layers; sometimes more band-like melanocytic proliferation and stromal fibrotic reaction
Therapy
■Quality-switched laser
■Quality-switched laser
■Cryotherapy
■Cryotherapy
■Surgery
■Surgery
■Rare malignant transformation
■No associated features of medical concern
■Rare malignant
■No associated features
Associated
Associated features
features
transformation
of medical concern
■Congenital
■Acquired
■Congenital
■Often familial
■Familial or sporadic
■Asian, African, and Hispanic populations with slight male predominance
■Asian and female predominance
■Uniform blue to slate-gray macular hyperpigmentation
■Brown-blue progressing to slate-gray mottled macular hyperpigmentation
■Mottled hyperpigmentation with small blue-gray macules in a diffuse pigmented patch
■Lower back and sacrum
■Especially malar region of the cheek (also forehead, upper eyelids, temple)
■Dermatomal distribution
■Spindle-shaped melanocytes diffusely throughout the dermal layers
■Dermal melanocytes in the upper and mid dermis
■Dermal melanocytes in the upper two-thirds of the dermis (including subpapillary layer)
■Usually spontaneous regression during childhood
■Qualityswitched laser in combination with bleaching cream and chemical peels
■None
■Possible association with inborn errors of metabolism
■No associated features of medical concern
■None
■Possible asso-
■No associ-
■None
ciation with inborn errors of metabolism
ated features of medical concern
CLINICAL FEATURES
Well-circumscribed, blue-tinged hyperpigmented macules in the sacral area are most commonly seen (Fig. 77-4). Mongolian spots also can be found in the gluteal and lumbar regions and on the thorax, abdomen, arms, legs, and shoulders. Several cases are described with extensive Mongolian spots involving large areas of the trunk and extremities that are associated with inborn errors of metabolism, such as lysosomal storage diseases, GM1- gangliosidosis and mucopolysaccharidosis.25
ETIOLOGY AND PATHOGENESIS
Histologically, these macules consist of spindle-shaped melanocytes in the lower dermis that have failed to migrate to the dermal–epidermal junction during fetal life.
1357
13
DIAGNOSIS
The diagnosis is usually made clinically, but may be confirmed on histology.
DIFFERENTIAL DIAGNOSIS
CLINICAL COURSE AND PROGNOSIS
In most cases, Mongolian spots spontaneously regress during childhood, but persistence into adulthood has been described.
MANAGEMENT
Spontaneous resolution usually means treatment can be avoided but laser treatment in childhood or adolescence can give favorable results, especially in sacral Mongolian spots.
1358
DERMAL MELANOCYTE HAMARTOMA
DERMAL MELANOCYTE
HAMARTOMA
AT-A-GLANCE
■ Congenital, circumscribed, blue-tinged hyperpigmentation with dermal melanocytosis.
■ Dermatomal patterns have been described.
Dermal melanocyte hamartoma is a distinctive form of congenital dermal melanocytosis, first described by Burkhart and colleagues in 1981.26 Gray-blue pigmentation, caused by melanocytes residing in the dermis, occurs in a dermatomal pattern.
FAMILIAL LENTIGINOSIS SYNDROMES
FAMILIAL LENTIGINOSIS
SYNDROMES
AT-A-GLANCE
■ Congenital conditions causing well-circumscribed pigmentation with lentiginosis.
■ Causes include Peutz-Jeghers syndrome, LEOPARD (lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness) syndrome, Carney complex, Bannayan-Ruvalcaba-Riley syndrome, and centrofacial lentiginosis.
Familial lentiginosis syndromes are characterized by the presence of lentigines—well-circumscribed brown macules (usually <5 mm in diameter), which have an increased number of melanocytes in the epidermis (epidermal melanocytic hypermelanosis) and an increased incidence of cardiovascular, endocrine, or GI neoplasias. Familial lentiginosis syndromes include Carney complex, Peutz-Jeghers syndrome (PJS), LEOPARD syndrome ( lentigines, electrocardiogram conduction defects, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness), arterial dissection and lentiginosis, Laugier-Hunziker syndrome, familial benign lentiginosis, Bannayan-Ruvalcaba-Riley syndrome (BRRS), centrofacial lentiginosis, and segmental and agminated lentiginosis.27 Genetic loci and gene mutations have been identified for Carney complex, PJS, and BRRS.
PEUTZ-JEGHERS SYNDROME
PJS was first described by Peutz (1921) and Jeghers (1949).28 It is a rare autosomal dominant genodermatosis with a predisposition for development of malignancies. More than half of all cases of PJS can be attributed to a mutation in STK11 (LKB1) on chromosome 19 (19q13.3),29 which is thought to act as a tumor-suppressor gene. Close surveillance of PJS patients for malignancies from a young age is warranted. Mucocutaneous pigmentation and intestinal hamartomatous polyposis are hallmarks of the condition (Fig. 77-5). The pigmentary lesions resemble those of Carney complex, with small hyperpigmented (brown-gray) macules typically appearing in childhood (not present at birth) on the lips and buccal mucosa; however, they may also involve the eyelids, hands, and feet. Quality-switched laser or intense pulsed light can be used to treat the pigmented lesions. The most common malignancies associated with PJS are GI (small intestine, colorectal, stomach, pancreas), but nongastrointestinal neoplasms, such as breast, cervix, and endocrine tumors (thyroid, testicular, ovarian), have been described.
LEOPARD SYNDROME
LEOPARD syndrome is a rare autosomal dominant condition caused by a heterozygous missense mutation in the PTPN11 gene, coding for the protein tyrosine phosphatase SHP-2 and situated on chromosome 12 (12q24.1). LEOPARD syndrome is allelic to Noonan syndrome and shares several clinical features with this disorder.30 The characteristic lentigines usually develop during childhood and in the first months of life (Fig. 77-6). Clinical diagnosis is primarily based on the typical facial features and the presence of hypertrophic cardiomyopathy and/or café-au-lait macules (CALMs). Recently, missense mutations in the RAF1 gene were found in 2 LEOPARD syndrome patients in whom no PTPN11 mutations could be discovered.
CARNEY COMPLEX
Carney complex is an autosomal dominant disorder first described by Carney and colleagues in 1985.31
Clinical components of Carney complex include spotty skin pigmentation, myxomas (heart, skin, breast), endocrine tumors (primary pigmented nodular adrenal disease [Cushing syndrome], testicular large-cell calcifying Sertoli cell tumor [sexual precocity], pituitary adenoma [acromegaly], thyroid tumors, ovarian cysts), and schwannomas.27 Multiple types of hyperpigmentation, such as lentigines, ephelides, blue nevi, junctional, dermal and compound nevi, and café-au-lait spots, have been described.32 The typical lesions consist
13
A
B
of spotty centrofacial pigmentation involving the vermilion border of the lips, the lacrimal caruncle, the conjunctival semilunar fold, and sometimes the sclera. Intraoral-pigmented spots are present in a limited number of cases. In at least half of the patients with Carney complex, a mutation in the gene encoding protein kinase A regulatory subunit 1A (PRKAR1A) mapped to 17q22-24 has been identified.33
1359
13
A
B
BANNAYAN-RILEY-RUVALCABA SYNDROME
BRRS is a second autosomal dominant hamartomatous polyposis syndrome. This designation replaces 3 previously described entities: (a) Riley-Smith,
1360
(b) Bannayan-Zonana, and (c) Ruvalcaba-Myhre- Smith syndromes.34 It is characterized by the classical triad of macrocephaly, genital lentiginosis, and intestinal polyposis. Besides the genital lentiginosis (sometimes presenting as larger CALMs), mucocutaneous manifestations include vascular malformations, lipomatosis, multiple acrochordons, and verrucous facial papules. More than 60% of BRRS patients show germline mutations in the PTEN (phosphatase and tensin homolog) gene on chromosome 10q23.3, a gene that is mutated in Cowden syndrome, which displays partial clinical overlap with BRRS34 Recently, the PTEN hamartoma tumor syndrome, encompassing 4 allelic disorders (Cowden syndrome, BRRS, Proteus syndrome, and Proteus-like syndrome), has been described.35
CENTROFACIAL LENTIGINOSIS
A horizontal band of lentigines across the central face is the hallmark of the autosomal dominant centrofacial lentiginosis syndrome. Other associated signs and symptoms include bone abnormalities, endocrine dysfunctions, neural tube defects, and mental retardation.36
FAMILIAL CAFÉ-AU-LAIT SYNDROMES
FAMILIAL CAFÉ-AU-LAIT
SYNDROMES
AT-A-GLANCE
■ Congenital circumscribed hypermelanosis with café-au-lait macules (CALMs) are seen in conditions like familial multiple CALMs (Legius syndrome), neurofibromatosis Type 1, McCune- Albright syndrome, Bloom syndrome, Watson syndrome, and Silver-Russel syndrome.
■ Well circumscribed hypermelanosis with CALMs.
CALMs consist of well-demarcated hyperpigmented patches of skin, varying in size from 0.5 cm to more than 20 cm. They are often present at birth or appear in the first few months of life. Between 0.3% and 18% of all newborns display isolated CALMs.37 Histologically, isolated CALMs show a normal number of melanocytes but increased epidermal melanin (epidermal melanotic hypermelanosis). Multiple CALMs are wellknown markers for several multisystem disorders.
LEGIUS SYNDROME
Legius syndrome is also known as familial multiple CALMs and is autosomal dominant with mutations in the SPRED1 gene (15q13.2). Multiple CALMs are present and axillary freckling and lipomas may be seen, but no neurofibromas are demonstrated. Macrocephaly and developmental delays are noted but are less severe than what is seen in neurofibromatosis Type 1.38,39
NEUROFIBROMATOSIS TYPE 1
Neurofibromatosis Type 1 (NF1) was first recognized by Friedrich von Recklinghausen in 1882 and is also called von Recklinghausen disease (see Chap. 135 for detailed discussions). NF1 is an autosomal dominant disease caused by a mutation in the NF1 gene that encodes the neurofibromin protein. The most important neurofibromin function involves downregulation of the Ras signal transduction pathway; consequently, it is considered a tumor-suppressor gene. NF1 has been considered a neurocristopathy and is characterized by a number of cutaneous and noncutaneous pigment cell-related manifestations such as CALMs, intertriginous freckling, and iris Lisch nodules. (Chap. 135 discusses the clinical diagnostic criteria for NF1.) The presence of 6 or more CALMs larger than 5 mm in greatest diameter in prepubertal individuals or larger than 15 mm after puberty is one of the hallmarks of the disease. NF1-associated CALMs, contrary to isolated CALMs, contain a significantly increased number of melanocytes in the epidermis. Intertriginous freckles, pathognomonic for NF1, also display increased numbers of epidermal melanocytes, which differentiates them from ordinary freckles (ephelides).
MCCUNE-ALBRIGHT SYNDROME
McCune-Albright syndrome was first described by McCune (1936) and Albright (1937) as a triad of poly/ monostotic fibrous dysplasia, CALMs, and hyperfunctioning endocrinopathies, including precocious puberty, hyperthyroidism, hypercortisolism, hypersomatotropism, and hypophosphatemic rickets.40 The CALMs are fewer in number and have more irregular borders than those seen in NF1. They are classically demarcated at the midline. McCune-Albright syndrome is caused by a postzygotic activating mutation of the α subunit of the cyclic adenosine monophosphate–regulating Gs protein. Consequently, a mosaic distribution of McCune-Albright syndrome cells bears the constitutively active adenylate cyclase.
BLOOM SYNDROME
Bloom syndrome is a rare, autosomal recessive, genetic-immunodeficiency-and-cancer-predisposition syndrome. It is characterized by a growth deficiency, unusual facies, CALMs, and a sun sensitivity with telangiectasia and erythema that can result in atrophy and scarring. Neoplasias that occur frequently in Bloom syndrome are acute leukemia, lymphoma, and squamous cell carcinoma.41 Bloom syndrome is caused by a DNA repair defect, which results in genomic instability, including a high frequency of sister-chromatid exchanges.42
The causative gene BLM has been mapped to 15q26.1 and encodes the BLM RecQ helicase homolog protein, a well-known member of the DNA helicase family, closely related to the helicases that is also defective in Werner syndrome and Rothmund-Thomson syndrome.43
13
WATSON SYNDROME
Watson syndrome is an autosomal dominant condition made up of pulmonary valvular stenosis, CALMs, and lower intelligence, that was first described by Watson in 1967.44 Later, the clinical phenotype of Watson syndrome was expanded with features overlapping with NF1 (macrocephaly, Lisch nodules, neurofibromas, short stature, intertriginous freckling), and it was shown to be allelic to NF1.45 Recent studies suggest an overlap of Watson syndrome with neurofibromatosis- Noonan syndrome.
SILVER-RUSSELL SYNDROME
Silver–Russell syndrome (SRS) is a clinically and genetically heterogeneous disorder first described by Silver (1953) and Russell (1954).46,47 Cardinal features of the disease are low birth weight, short stature resulting from intrauterine and postnatal growth retardation, and a small triangular face. Other associated symptoms include clinodactyly of the fifth finger, relative macrocephaly, and facial, limb, or body asymmetry. CALMs are a variable SRS feature. One or 2 CALMs are present in up to 20% of SRS patients. Many SRS cases are sporadic, but familial cases with different modes of inheritance have been described. Maternal uniparental disomy for chromosome 7 occurs in approximately 10% of patients, and several candidate regions for the SRS gene mutation have been described on this chromosome (7p11.2-p13, 7q31-qter, and 7q21). Other possible gene candidates could be situated on the long arm of chromosome 17 or on chromosome 11p15. Recently, methylation defects in the H19 imprinted domain chromosome 11p15 has been implicated in 35% to 65% of SRS cases.48
ACQUIRED HYPERMELANOSIS
ENDOCRINOPATHIES
ENDOCRINOPATHIES
AT-A-GLANCE
■ Diffuse, nonpatterned hyperpigmentation may be seen in conditions like Addison disease, Cushing syndrome, Nelson syndrome, pheochromocytoma, carcinoid syndrome, hyperthyroidism, acanthosis nigricans, and diabetes.
■ Circumscribed hyperpigmentation may be seen in conditions like acanthosis nigricans and maturational dyschromia
ADDISON DISEASE
Chapter 137 discusses endocrine diseases in detail. Addison disease is a clinical syndrome characterized by salt-wasting and diffuse cutaneous
1361
13
hyperpigmentation. In the developed world, Addison disease is usually autoimmune. It is associated with adrenal insufficiency with inadequate secretion of corticosteroid and androgenic hormones, leading to compensatory overproduction of adrenocorticotropic hormone (ACTH) by the pituitary gland. Hyperpigmentation is the most striking cutaneous sign of patients with chronic Addison disease and is the consequence of ACTH binding to the melanocortin-1 receptor. The hyperpigmentation occurs preferentially on sun-exposed areas (face, neck, hands), sites of trauma, scars, chronic pressure (knees, spine, knuckles, elbows, shoulders), in the palmar creases, nipples, areolae, axillae, perineum, and genitalia.
CUSHING SYNDROME
Chapter 137 discusses endocrine diseases in detail. Cushing syndrome is characterized by clinical signs and symptoms caused by chronic glucocorticoid excess. Various degrees of hyperpigmentation can be seen. It is usually most severe in patients with the ectopic ACTH syndrome. Patients present with generalized hyperpigmentation with accentuation in areas of sun exposure, chronic mild trauma, and pressure (shoulders, midriff, waist, elbows, knuckles, spine, knees), and on mucosal surfaces.
NELSON SYNDROME
Nelson syndrome comprises an enlarging pituitary tumor associated with elevated fasting plasma ACTH levels, hyperpigmentation, and neuroophthalmologic symptoms in patients with Cushing disease after bilateral adrenalectomy and inadequate hormonal replacement.49
PHEOCHROMOCYTOMA
Pheochromocytoma is a chromaffin cell tumor of the adrenal medulla with associated excessive production of catecholamines. Pallor of the face resulting from vasoconstriction may be observed. In contrast, addisonian-like hyperpigmentation has been reported and is probably caused by ectopic ACTH and melanocyte-stimulating hormone production by the tumor. Pigmentation rapidly fades after surgical treatment.50
CARCINOID SYNDROME
Diffuse hyperpigmentation resulting from melanocytestimulating hormone–producing tumors, such as gastric or thymic carcinoid tumors, have been described in carcinoid syndrome. Carcinoid syndrome also can be accompanied by a pellagra-like rash occurring on light-exposed skin. The rash is secondary to a tryptophan deficiency, as a large amount of dietary tryptophan is diverted to serotonin by the tumor. Treatment of the underlying tumor is critical in the management of affected patients.
1362
HYPERTHYROIDISM
Chapter 137 discusses endocrine diseases in detail. Thyrotoxicosis has multiple causes. The most common cause is Graves disease, characterized by circulating antibodies against thyroid-stimulating hormone receptors. The occurrence of hyperpigmentation in thyrotoxic patients has been estimated to be from 2% to as high as 40% in large series. The increased cutaneous pigmentation can be localized or generalized and is more common in those with dark skin. The distribution of hyperpigmentation is often similar to that in Addison disease with pigment deposition in the creases of the palms and soles. However, in contrast to Addison disease, involvement of the mucous membranes is uncommon and pigmentation of the nipples and genital skin is less striking. Hyperpigmentation associated with thyrotoxicosis is thought to be the result of an increased release of pituitary ACTH, compensating for accelerated cortisol degradation. The response of hyperpigmentation to therapy for the hyperthyroidism is reported to be poor.51 Autoimmune Graves disease has been reported in association with vitiligo.
DIABETES
Chapter 137 discusses diabetes in detail. Diabetic dermopathy is characterized by asymptomatic, irregular, light-brown, depressed patches on the anterior lower legs, often after mild trauma or the appearance of bullae. The pathogenesis is unknown.
ACANTHOSIS NIGRICANS
Acanthosis nigricans is characterized by thickened hyperpigmented velvety skin most often noted on the neck, axilla, popliteal and antecubital fossae, and inguinal folds (Fig. 77-7). It has, however, also been
described on the face and should be considered in the differential diagnosis of facial hyperpigmentation. When on the face, acanthosis nigricans presents as poorly demarcated hyperpigmentation with a predilection for the malar region inferior to the zygoma and the nasolabial folds, and has even been described on the supraalar creases.52 Recognizing this entity is becoming steadily more important as the rates of obesity and non–insulin-dependent diabetes increases. Acanthosis nigricans is discussed in more detail in Chap. 137.
MATURATIONAL DYSCHROMIA
Maturational dyschromia is a recently described entity noted in the African and Indian population in the fourth to fifth decades of life. It presents as dark-brown to black poorly demarcated areas of hyperpigmentation on the lateral forehead, temples, and zygoma. Histologic evaluation of hyperpigmented skin reveals mild to moderate proliferation of melanocytes with some reports of a papillomatous epidermis, suggesting a potential link or continuum with acanthosis nigricans.53 The etiology is still unclear and further research of this disorder is needed (Fig. 77-8).
NUTRITIONAL CONDITIONS
NUTRITIONAL CONDITIONS
Chapter 123 discusses nutritional disease in detail. Diffuse, nonpatterned pigmentary changes can be
13
secondary to nutritional conditions such as kwashiorkor, vitamin B12 deficiency, folic acid deficiency, or pellagra. The skin changes are reversible if the nutritional deficiency is corrected.
KWASHIORKOR
Kwashiorkor is the result of severe protein malnutrition in the presence of adequate caloric intake. It is a significant cause of death in children between 6 months and 5 years of age in developing countries, and is often associated with GI parasitoses. A child with kwashiorkor ceases to gain weight and becomes irritable and apathic, and develops edema, hepatomegaly, diarrhea, muscle wasting, and photophobia. Cutaneous manifestations are present in most cases. Skin changes usually begin with hypopigmentation of the face. In areas exposed to friction and pressure, such as knees, elbows, and buttocks, hyperkeratoses and scaling in association with hyperpigmentation develop, giving the skin a “flaky paint” appearance. Removing the scale leaves superficial erosions that turn into pale patches. Hair may become hypopigmented. Sometimes there are bands of hypopigmentation along the hair shafts (flag sign), a result of periods of worse malnutrition. Treatment of kwashiorkor consists of replacing protein sources. Changes in skin and hair are reversed by proper nutritional treatment.54
Kwashiorkor-like syndrome also has been described in cases of protein loss and malnutrition secondary to bowel disorders such as ulcerative colitis.
VITAMIN B12 DEFICIENCY
Cobalamin or vitamin B12 deficiency is caused by atrophic gastritis caused by Helicobacter pylori infection or an autoimmune process directed against gastric parietal cells, which lead to severe “intrinsic factor” deficiency (pernicious anemia). Blood examination reveals megaloblastic anemia and low levels of vitamin B12. Associated generalized hyperpigmentation of the skin and hypopigmentation/depigmentation or early graying of the hair have been reported. Skin histology shows increased melanin in the basal layer. Electron microscopy reveals a normal complement of melanosomes in melanocytes and surrounding keratinocytes. These findings suggest that an increase in melanin synthesis rather than a defect in melanin transport causes vitamin B12 deficiency–associated hyperpigmentation. Supplementation with vitamin B12 reverses the hyperpigmentation. Pernicious anemia is an autoimmune disorder and has been reported in association with vitiligo.55
FOLIC ACID DEFICIENCY
Folic acid deficiency associated with megaloblastic anemia has been reported to cause hyperpigmentation of the skin.
PELLAGRA
Pellagra is caused by a deficiency of niacin (vitamin B3) or its derivatives. The disease is rare and can be caused
1363
13
by chronic alcoholism, certain drugs (isoniazid, anticonvulsants), malabsorption secondary to inflammatory bowel disease, celiac disease, and carcinoid tumors. Pellagra is characterized by the clinical triad of dermatitis, dementia, and diarrhea. The first skin changes are erythema and edema following sun exposure, which fade with a dusky brown–red coloration on face, neck, and dorsal surfaces of the hands, arms, and feet. Later on the lesions become hyperpigmented, hyperkeratotic, and scaly with fissures and crusts (“goose-like”). Niacin supplementation causes a rapid clinical response.56
METABOLIC CONDITIONS
METABOLIC CONDITIONS
AT-A-GLANCE
■ Porphyria cutanea tarda and hemochromatosis may cause diffuse nonpatterned hyperpigmentation accentuated in photoexposed sites.
PORPHYRIA CUTANEA TARDA
Chapter 124 discusses the porphyrias in detail. Porphyria cutanea tarda is a metabolic disorder affecting the synthesis of heme. It is associated with diffuse brown hyperpigmentation, which is accentuated in photoexposed areas. In females, a melasma-like hyperpigmentation of the face may be observed.
HEMOCHROMATOSIS
Hemochromatosis is a disease of iron storage with a heterogeneous genetic basis that results in lowered levels of hepcidin. Approximately 10% of the population has a mutation in the hemochromatosis gene, with a lower prevalence noted in Africans and Asians compared with northern Europeans. This leads to increased intestinal absorption and deposition of iron in the liver, pancreas, and other organs, including the skin.57 Because hemochromatosis is now usually diagnosed early, hyperpigmentation is less frequently observed. Historically, hemochromatosis was diagnosed at an advanced stage with a classic triad of hyperpigmentation, diabetes mellitus (“bronze diabetes”), and hepatic cirrhosis. Darkening of the skin was present in 70% of the patients because of 2 different mechanisms: (a) hemosiderin deposition causing a diffuse, slate-gray color, and (b) increased epidermal melanin production. The pigmentation is usually generalized, but may be more pronounced in photoexposed areas, genitalia, and scars. Skin bronzing is reversible with phlebotomy, which remains the mainstay of treatment for this condition. Deferoxamine is most commonly used in secondary hemochromatosis.
1364
TUMORAL CONDITIONS
TUMORAL CONDITIONS
MAST CELL DISORDERS AND MELANOMA
Mast cell disorders and melanoma can lead to diffuse, nonpatterned hyperpigmentation and are discussed in Chaps. 42 and 116, respectively. Diffuse, generalized melanosis associated with advanced metastatic melanoma is a rare, although well-documented, condition. It is characterized by a slate bluish-gray to brown discoloration of the skin. Histology reveals melanin particles and melanin-containing histiocytes and dendritic cells in the dermis and subcutaneous fat. No melanoma cells can be detected in the skin, and there is no increase in epidermal melanin pigment or number of melanocytes. Melanosomes circulating in the blood have been detected, supporting the hypothesis that diffuse melanosis may result from tumor lysis with release of their organelles into the circulation and subsequent deposition in the skin.
PHYSICAL CAUSES
PHYSICAL CAUSES
AT-A-GLANCE
■ Ultraviolet, ionizing, and thermal radiation may cause diffuse nonpatterned hyperpigmentation in the area(s) affected.
■ Trauma may cause localized hyperpigmentation in areas of chronic friction.
ULTRAVIOLET RADIATION AND PIGMENTATION
Chapter 20 discusses pigmentation in detail. A major acute effect of ultraviolet radiation on normal human skin is tanning.
IONIZING RADIATION
Chapter 200 discusses radiotherapy in detail. Exposure of skin to ionizing radiation during accidents or after local fractionated radiotherapy can give rise to a cutaneous radiation syndrome characterized by fibrosis, keratosis, telangiectasias, and sharply demarcated lentiginous hyperpigmentation, resembling ultraviolet-induced lentigines ( radiation dermatitis). Small hypopigmented macules can be intermingled with zones of hyperpigmentation. Histology reveals altered melanin content in melanocytes and basal keratinocytes. Electron-beam therapy
has been reported to induce tan-like transient hyperpigmentation and transverse melanonychia when nails are exposed.58
THERMAL RADIATION
In superficial thermal burn injuries, when the melanocyte-bearing basal epidermis has not been destroyed, various degrees of hyperpigmentation result, depending on the patient’s skin color and time after the injury. Thermal injury resulting from laser therapy with intense, high-dose visible light also can give rise to hyperpigmentation, especially in dark-skinned patients. Cryotherapy, tissue destruction by application of cold, commonly causes (sometimes permanently) hypopigmentation in combination with peripheral hyperpigmentation in treated skin as a result of melanocyte injury.59
FRICTION MELANOSIS
Frictional melanosis in an acquired pigmentary disorder caused by repeated rubbing of the skin, especially over bony prominences. Marked increase in melanin and melanin incontinence is seen histologically.60 Diffuse hyperpigmentation is noted with some cases revealing mild lichenification.
TOXINS AND MEDICATIONS
TOXINS AND MEDICATIONS
AT-A-GLANCE
■ Toxins and medications are a common cause of acquired, diffuse, nonpatterned hyperpigmentation.
■ Fixed drug eruptions may cause well-demarcated brown-gray mucocutaneous macules.
■ Hyperpigmentation may be caused by direct deposition of the drug into the skin, deposition of melanin or melanin-drug complex in the dermis, or nonmelanin pigment produced by the drug.
■ Treatment involves cessation of the offending medication, and quality-switched and picosecond lasers.
Hyperpigmentation caused by toxic agents or medication accounts for 10% to 20% of all cases of acquired hyperpigmentation. CNS drugs, antineoplastic agents, antiinfectious drugs, antihypertensive medications, and hormones are most often the responsible agents (Table 77-2).
13
CLINICAL FEATURES
Clinical features vary with characteristic sites, patterns, and shades of discoloration noted with particular medications and toxins. Bluish-slate gray pigmentation is characteristic of dermal deposition of pigment. Pigmentation associated with melanin accumulation often worsens with sun exposure. A linear, sometimes flagellate hyperpigmentation can be observed in patients taking bleomycin or zidovudine. A diffuse hyperpigmentation on the palms and soles may be present in patients taking cyclophosphamide or doxorubicin. Diffuse slate-gray pigmentation also may be seen at the site of IV iron infusion (Fig. 77-9). Bleomycin and doxorubicin may produce localized hyperpigmentation around small joints. Estrogen-related hormonal substances and phenytoinlike medication may cause melasma-like pigmentation. Nail unit involvement can be observed with some medications, most frequently chemotherapeutic agents such as cyclophosphamide, zidovudine, psoralens, minocycline, antimalarials, and gold. Mucosal hyperpigmentation has been reported with cyclophosphamide, doxorubicin, zidovudine, minocycline, and some heavy metals. Amiodarone can produce blue-gray pigmentation in photoexposed areas from accumulation of a lipidlike substance in macrophages. Some of these patients display photosensitivity (Fig. 77-10). Chloroquine may give rise to a yellow-brown to bluish-gray pigmentation on the face, neck, lower extremities, and forearms after several years of intake, a result of deposition of a drug–melanin complex in the dermis. The nail unit and hard palate also may be involved. Exogenous ochronosis has been reported after chronic hydroquinone use (see section “Ochronosis”). Chlorpromazine and related phenothiazines can produce a bluish-gray cutaneous pigmentation that is worse in photoexposed areas with pigmentation of the conjunctivae. The risk of minocycline-induced pigmentation increases with prolonged therapy at doses above 100 mg daily.61 Three types of minocycline-induced pigmentation exist: Type 1 consists of blue-gray pigmentation in normal skin and areas of prior inflammation (often on the face) (Fig. 77-11); Type 2 is noted on the lower legs and forearms; and Type 3 is diffuse muddy brown photoexacerbated pigmentation. Pigmentation of the nails, sclerae, oral mucosa, thyroid, bones, and teeth also has been reported. Argyria patients present with a generalized grayishblue pigmentation caused by silver ingestion. The nails and the sclerae also may be involved. Although medication-induced pigmentation usually results in diffuse hyperpigmentation, fixed drug eruptions must be considered when well-demarcated mucocutaneous hyperpigmented macules are the presenting sign (Fig. 77-12). A preceding history of erythema and possible itching with pigmentation as the lesion resolves is classically seen after use of nonsteroidal antiinflammatory medications, certain antibiotics, and drugs containing codeine.
1365
13
TOPICAL TREATMENTS ANTIINFECTIOUS DRUGS CNS DRUGS ANTINEOPLASTIC AGENTS MISCELLANEOUS
Aminolevulinic acid Carmustine (BCNU) Bergamot Bimatoprost Carteolol Chlorhexidine Hydroquinone Imiquimod Latanoprost Nitrogen mustard Tretinoin Antihypertensive drugs
Amphotericin B Ceftriaxone Chloroquine Cidofovir Clofazimine Dapsone Demeclocycline Doxycycline Emtricitabine Enoxacin Foscarnet Ganciclovir Grepafloxacin Griseofulvin Hydroxychloroquine Indinavir Ketoconazole Linezolid Lomefloxacin Minocycline Ofloxacin Oxytetracycline Pyrimethamine Quinacrine Quinine Ribavirin Rifabutin Rifapentine Saquinavir Sertaconazole Smallpox vaccine Sparfloxacin Sulfadiazine Terbinafine Tetracycline Voriconazole Zidovudine
Amitriptyline Carbamazepine Chlorpromazine Citalopram Clomipramine Clonazepam Desipramine Diazepam Donepezil Eletriptan Fluoxetine Fluphenazine Fluvoxamine Haloperidol Imipramine Kava Loxapine Mephenytoin Mesoridazine Methamphetamine Molindone Olanzapine Paroxetine Perphenazine Phenytoin Pimozide Prochlorperazine Promazine Promethazine Risperidone Ropinirole Thioridazine Thiothixene Tiagabine Tolcapone Topiramate Trifluoperazine Venlafaxine Zaleplon
Aminolevulinic acid Carmustine (BCNU) Bergamot Bimatoprost Carteolol Chlorhexidine Hydroquinone Imiquimod Latanoprost Nitrogen mustard Tretinoin Antihypertensive drugs Acebutolol Betaxolol Bisoprolol Captopril Clonidine Diltiazem Esmolol Indapamide Labetalol Methyldopa Metoprolol Minoxidil Nisoldipine Propranolol Spironolactone Timolol Heavy metals Arsenic Bismuth Gold (compounds) Iron Lead Mercury Silver
Amphotericin B Ceftriaxone Chloroquine Cidofovir Clofazimine Dapsone Demeclocycline Doxycycline Emtricitabine Enoxacin Foscarnet Ganciclovir Grepafloxacin Griseofulvin Hydroxychloroquine Indinavir Ketoconazole Linezolid Lomefloxacin Minocycline Ofloxacin Oxytetracycline Pyrimethamine Quinacrine Quinine Ribavirin Rifabutin Rifapentine Saquinavir Sertaconazole Smallpox vaccine Sparfloxacin Sulfadiazine Terbinafine Tetracycline Voriconazole Zidovudine
Amitriptyline Carbamazepine Chlorpromazine Citalopram Clomipramine Clonazepam Desipramine Diazepam Donepezil Eletriptan Fluoxetine Fluphenazine Fluvoxamine Haloperidol Imipramine Kava Loxapine Mephenytoin Mesoridazine Methamphetamine Molindone Olanzapine Paroxetine Perphenazine Phenytoin Pimozide Prochlorperazine Promazine Promethazine Risperidone Ropinirole Thioridazine Thiothixene Tiagabine Tolcapone Topiramate Trifluoperazine Venlafaxine Zaleplon
Acebutolol Betaxolol Bisoprolol Captopril Clonidine Diltiazem Esmolol Indapamide Labetalol Methyldopa Metoprolol Minoxidil Nisoldipine Propranolol Spironolactone Timolol Heavy metals
Arsenic Bismuth Gold (compounds) Iron Lead Mercury Silver
ETIOLOGY AND PATHOGENESIS
In the majority of cases of medication-induced hyperpigmentation, the underlying pathogenic mechanism involves one of the following.
- Deposition of melanin in the dermis, usually in macrophages. Sometimes this melanin is complexed to the drug (drug–pigment complex, eg, hydroxychloroquine). Accumulation of melanin can occur after cutaneous inflammation (postinflammatory) and/or DNA damage (eg, carmustine). This type of hyperpigmentation is often increased by ultraviolet exposure and is usually more pronounced in photoexposed areas.
1366
Bevacizumab Bleomycin Busulfan Capecitabine Carboplatin Carmustine Cisplatin Cyclophosphamide Dactinomycin Daunorubicin Doxorubicin Epirubicin Estramustine Etoposide Floxuridine Fluorouracil Hydroxyurea Ifosfamide Irinotecan Mechlorethamine Mercaptopurine Methotrexate Mitomycin Mitotane Mitoxantrone Paclitaxel Pentostatin Procarbazine Thiotepa Vinblastine Vincristine Vinorelbine Hormones
Alitretinoin Amiodarone Azathioprine Cetirizine Cevimeline Cyclobenzaprine Cyclosporine Deferoxamine Dicumarol Dinoprostone Etodolac Glatiramer Heroin Interferon Isotretinoin Ketoprofen Leflunomide Levobupivacaine Lidocaine Methimazole Methoxsalen Methysergide Metoclopramide Niacin Nicotine Orphenadrine Pantoprazole Pentazocine Phenazopyridine Phenolphthalein Propylthiouracil Psoralens Quinidine Rabeprazole Riluzole Sulfasalazine Tacrolimus Toremifene Trioxsalen Vitamin A
Bevacizumab Bleomycin Busulfan Capecitabine Carboplatin Carmustine Cisplatin Cyclophosphamide Dactinomycin Daunorubicin Doxorubicin Epirubicin Estramustine Etoposide Floxuridine Fluorouracil Hydroxyurea Ifosfamide Irinotecan Mechlorethamine Mercaptopurine Methotrexate Mitomycin Mitotane Mitoxantrone Paclitaxel Pentostatin Procarbazine Thiotepa Vinblastine Vincristine Vinorelbine Hormones Chlorotrianisene Corticosteroids Diethylstilbestrol Estrogens Insulin Leuprolide Medroxyprogesterone Oral contraceptives Progestins Stanozolol
Alitretinoin Amiodarone Azathioprine Cetirizine Cevimeline Cyclobenzaprine Cyclosporine Deferoxamine Dicumarol Dinoprostone Etodolac Glatiramer Heroin Interferon Isotretinoin Ketoprofen Leflunomide Levobupivacaine Lidocaine Methimazole Methoxsalen Methysergide Metoclopramide Niacin Nicotine Orphenadrine Pantoprazole Pentazocine Phenazopyridine Phenolphthalein Propylthiouracil Psoralens Quinidine Rabeprazole Riluzole Sulfasalazine Tacrolimus Toremifene Trioxsalen Vitamin A
Chlorotrianisene Corticosteroids Diethylstilbestrol Estrogens Insulin Leuprolide Medroxyprogesterone Oral contraceptives Progestins Stanozolol
- Direct deposition of the medication in the skin (eg, carotene, heavy metals). Sometimes this type of pigmentation is accentuated in photoexposed areas, as ultraviolet light can induce a transformation in the deposited drug, which may then become more visible.
- Nonmelanin pigments synthesized or produced under the direct or indirect influence of the drug may cause cutaneous pigmentation.
DIAGNOSIS
The diagnosis may be made clinically if the characteristic appearance is seen and a temporal relationship
13
with exposure to a medication or toxin is noted. A skin biopsy also may be helpful in cases of medicationinduced pigmentation.
CLINICAL COURSE AND PROGNOSIS
Ceasing the offending medication is critical, but hyperpigmentation persists in many cases, sometimes for decades, even after discontinuation of the medication.
MANAGEMENT
Sun protection should be advised, especially in forms caused by melanin accumulation. Qualityswitched ruby, alexandrite, and Nd:YAG lasers have demonstrated favorable results in some cases (eg, amiodarone and minocycline-induced pigmentation), although multiple treatments spaced over many months is often necessary to achieve clinically notable results. Picosecond lasers have proven efficacious for medication-induced pigmentation in white skin, with fewer treatments over a shorter time frame achieving complete or near complete clearance of the pigmentation.62
1367
13
OCHRONOSIS
OCHRONOSIS
AT-A-GLANCE
■ May be congenital (autosomal recessive) or caused by the use of certain medications.
■ Results in asymptomatic diffuse nonpatterned hyperpigmentation.
■ More common in persons with skin of color.
■ Characteristic histologic appearance is yellowbrown banana-shaped globules in the dermis.
The term ochronosis is derived from the Greek word “ochre,” which refers to yellow discoloration. Endogenous and exogenous forms of the condition exist.
EXOGENOUS OCHRONOSIS Epidemiology: Although the majority of ochronosis cases have been reported in darker racial/ethnic groups in Africa, India, Thailand, China, and Singapore, it also has been described in other skin types. The exact worldwide incidence is unknown but assumed to be low.
Clinical Features: Asymptomatic blue-black and gray-black hyperpigmented macules are characteristic and can be found on the face (malar, temples, lower cheeks), posterolateral neck, back, and extensor skin of the extremities. Later stages include progressive hyperpigmented colloid milium (“caviar-like lesions), papulonodular lesions, and areas of scarring.63
No systemic involvement is noted with exogenous ochronosis.
Etiology and Pathogenesis: Exogenous ochronosis results from the use of certain medications, which form a homogentisic acid polymer-like substance during their metabolism. It has been most frequently reported in association with hydroquinone, usually at concentrations greater than 4%, and topicals, such as phenol or resorcinol, and quinine, an oral antimalarial. Exacerbating factors include unprotected ultraviolet light exposure, prolonged application of the offending medication on large surface areas, and use of topical resorcinol, phenol and mercury.
Diagnosis: Although early stages of exogenous ochronosis may appear similar to melasma, dermoscopy may be helpful in differentiating the two. Darkbrown globules and globular-like structures are seen on a diffuse brown background.64
Skin biopsy however, remains the gold standard for diagnosis of exogenous ochronosis.
1368
Histopathologically, there is a collection of yellowishbrown (ochronotic) banana-shaped globules in the papillary dermis. Homogenous, edematous dermal collagen also may be seen.
Differential Diagnosis: The early stages of ochronosis may be clinically indistinguishable from melasma, especially in Asians.
Clinical Course and Prognosis: Without cessation of the offending agent, exogenous ochronosis is progressive.
Management: Treatment is rarely helpful, but the offending drug should be stopped to prevent progression. Broad-spectrum sunscreen and sun avoidance are important. Topical retinoids, α-hydroxy acids, corticosteroids and physical therapies, such as chemical peels and quality-switched lasers, have all been reported in the treatment of exogenous ochronosis but there has been no universally efficacious treatment documented as of this writing. The following disorders can be associated with hyperpigmentation and are discussed in their respective chapters: systemic sclerosis (Chap. 63), pinta (Chap. 171), onchocerciasis (Chap. 177), phytophotodermatitis (Chap. 97), and flagellate pigmentation caused by bleomycin (Chap. 45).
SYSTEMIC SCLEROSIS
SYSTEMIC SCLEROSIS
Chapter 63 discusses systemic sclerosis in greater detail.
AT-A-GLANCE
■ Acquired diffuse hyperpigmentation, focal depigmentation over sites of friction, streaky hyperpigmentation, and reticulated pigmentation have all been described.
Different types of abnormal pigmentation have been described in systemic sclerosis. A diffuse, generalized hyperpigmentation similar to Addison’s disease but with normal levels of melanocyte-stimulating hormone can be observed in severe systemic sclerosis. Focal depigmentation with perifollicular hyperpigmentation may occur, especially on sites of friction (eg, shins, elbows, and dorsum of the hands). There can be localized hyperpigmentation and hypopigmentation. A streaky hyperpigmentation over blood vessels on a background of depigmentation also has been reported. Diffuse reticulated hyperpigmentation accentuated on the trunk has been reported in 1 case.65
INFECTIONS
INFECTIONS
CHIKUNGUNYA
AT-A-GLANCE
■ Acquired circumscribed or diffuse hyperpigmentation.
■ Caused by a mosquito-borne virus.
■ Persistent pigmentation with symptomatic relief being the only treatment option.
Chikungunya is a mosquito-borne virus first described in the 1950s in Tanzania, Africa.
Epidemiology: While first described in Africa, chikungunya has been noted in several dozen other countries in Africa, Asia, Europe, and the Americas. All age groups are affected.66
Clinical Features: Pigmentary changes are the most common cutaneous finding. Lentiginous central facial hyperpigmentation, diffuse hyperpigmentation of acrofacial lesions, and flagellate hyperpigmentation have all been described. Striking pigmentation of the nose also has been reported. A case of neonatal hyperpigmentation has been reported in a case of congenital chikungunya.67
A maculopapular eruption is noted at the onset of the illness and resolves over 7 to 10 days. Other mucocutaneous manifestations include vesiculobullous lesions, purpuric lesions, aphthous-like ulcers, and exacerbation of other primary dermatoses such as lichen planus and psoriasis. Systemic symptoms, such as fever, arthralgia, myalgia, and GI upset, are some of the extracutaneous manifestations.
Etiology and Pathogenesis: Aedes aegypti (tropics and subtropics) and Aedes albopictus (cooler regions of the world) have been implicated in large outbreaks of this mosquito-borne illness. The mechanism by which cutaneous hyperpigmentation occurs is not understood but various theories exist, including melanocyte phagocytosis of the invading pathogens68 followed by intraepidermal melanin dispersion or retention triggered by the virus.
Diagnosis: Lymphocytic vasculopathy and increased intraepidermal melanin is noted histologically. Melanophages are also noted and may account for the persistent cutaneous pigmentation.
Differential Diagnosis: Other viral illnesses and mosquito-borne illness fall into the wide differential diagnosis.
13
Clinical Course and Prognosis: Pigmentation often persists for many months.
Management: Symptom relief is the mainstay of treatment.
OTHER INFECTIONS
Erythematous, scaly papules termed pintids develop during the secondary stage of pinta. These initially red lesions can turn brown, slate-blue, black, or grayish.69
Chronic papular onchodermatitis is one of the skin manifestations of onchocerciasis. It is characterized by a severely pruritic maculopapular rash with hyperpigmented macules, most often occurring on shoulders, buttocks, and extremities (see Chap. 177).
PHYSIOLOGIC HYPERPIGMENTATION
PHYSIOLOGIC
HYPERPIGMENTATION
PIGMENTARY DEMARCATION LINES
AT-A-GLANCE
■ More common in persons with skin of color.
■ Linear demarcation with opposing hyperpigmentation and hypopigmentation.
■ Seen along “embryonic suture lines” in distinct patterns and areas of the face and limbs.
■ Can occur on the face, trunk, and limbs.
■ Usually become obvious in childhood.
Pigmentary demarcation lines (PDLs) are also known as Voigt or Futcher lines. They are abrupt changes from lighter to darker skin in a linear pattern mostly seen on the limbs, trunk, and face.
Epidemiology: PDLs are most common in persons with skin of color. PDLs often appear in childhood or puberty and persist throughout life.
Clinical Features: PDLs are classified based on location as follows:
■ Type A: This is the most common type and involves the lateral aspect of the upper anterior arms ( occasionally extending across the pectoral area);
■ Type B: Posterior medial lower limb;
■ Type C: Vertical hypopigmented line in sternal and parasternal areas;
■ Type D: Vertical line in spinal or paraspinal area;
1369
13
■ Type E: Hypopigmented oval areas, streaks, or bands on bilateral aspects of the chest, from the mid-third of clavicle to periareolar skin;
■ Type F: V-shaped hyperpigmented line between the malar prominence and the temple;
■ Type G: W-shaped hyperpigmented lines between the malar prominence and the temple; and
■ Type H: Linear hyperpigmentation from the angle of the mouth to the lateral aspect of the chin.
A pigmentary demarcation line anterior to the ear has not been described in the literature (at the time of this writing) but should now be added as a new pigmentary demarcation line “Type I” (Fig. 77-13).
Etiology and Pathogenesis: The cause of PDLs is unknown. The debate surrounding topography following lines of Blaschko or cutaneous innervation continues. Hormonal and genetic influences have been postulated with onset during pregnancy and variable inheritance patterns noted, including autosomal dominant inheritance patterns in some families.
Diagnosis: The diagnosis can usually be made clinically without need for biopsy.
Clinical Course and Prognosis: PDLs often appear in childhood or puberty and persist throughout life.
Management: Reassurance that these changes are a normal variant is needed. In addition, sun protection may diminish the difference between affected and unaffected skin.
1370
ACQUIRED IDIOPATHIC FACIAL PIGMENTATION
AT-A-GLANCE
■ Can be considered as another group of pigmentary demarcation lines.
■ More common in persons with skin of color.
■ Linear demarcation with opposing hyperpigmentation and hyperpigmentation.
■ Predominantly affects periorbital and perioral zones.
■ Usually become obvious in adulthood.
Acquired idiopathic facial pigmentation was described recently in the Indian population and can be considered another group of PDLs.70 The term acquired idiopathic facial pigmentation was coined after observing that some types of idiopathic facial pigmentation were nonnevoid, patterned, and bilateral with a darkbrown–gray color located around the eyes, lateral face, and chin.
Epidemiology: Onset is noted in early adulthood with occurrence in males and females in a 1:1 ratio. It often appears in adulthood and persists throughout life.
Clinical Features: Well-demarcated dark-brown– gray macular hyperpigmentation is noted on periorbital, zygomatic, and malar areas, root of the nose, and perioral and mandibular skin. The pigmentation is noted in a similar pattern to other nevoid conditions with patterns reflecting the normal patterns of embryologic spread of pigmentation on face. The absence of symptoms, scale, erythema, and lichenification should help differentiate it from other dermatoses and pigmentary disorders in the area.
Etiology and Pathogenesis: Although the cause is unknown, it is postulated this is a genetic disorder with exacerbation and accentuation resulting from physiologic factors such as ultraviolet light and aging.
Diagnosis: The diagnosis can usually be made clinically without need for biopsy.
Differential Diagnosis: Conditions like melasma and postinflammatory pigmentation may look similar to acquired idiopathic facial pigmentation and should be differentiated clinically and histologically.
Clinical Course and Prognosis: Acquired idiopathic facial pigmentation often appears in adulthood. It darkens with time and persists throughout life.
Management: Sun protection may help diminish the difference between affected and unaffected skin and occasionally topical hydroquinone can subtly lighten excessive pigmentation in the areas affected.
PERIORBITAL PIGMENTATION
AT-A-GLANCE
■ Primary dermatoses, such as lichen planus pigmentosus, medication-induced pigmentation, pigmented contact dermatitis, and postinflammatory hyperpigmentation, should be excluded.
■ Familial/racial/constitutional pigmentation is especially common in persons with skin of color.
Periorbital hyperpigmentation is also known as dark circles and is very common in certain racial ethnic types.
Epidemiology: Males and females, mainly with darker-skin types, are commonly affected. Onset is usually noted in young adults.
Clinical Features: Asymptomatic brown-black nonscaly and nonerythematous pigmentation is noted on the upper, lower, or both eyelids. Accentuation of skin creases is noted and visible vessels also may be seen (Fig. 77-14).
Etiology and Pathogenesis: The exact etiology of constitutional pigmentation is yet to be determined.
Diagnosis: A clinical diagnosis is made in most cases. Increased melanin in the basal epidermis and
13
melanophages in the upper dermis are seen histologically in those with constitutional pigmentation. Differential Diagnosis: Primary dermatoses, like lichen planus pigmentosus, medication-induced pigmentation, and eczema with lichenification, must be excluded.
Clinical Course and Prognosis: Constitutional pigmentation is usually progressive.
Management: Treatment of the underlying dermatosis, if present, is critical. Although topical therapy, peels, lasers, and autologous fat transplants have been reported to be useful, these studies don’t often specify the underlying cause of pigmentation, making it difficult to interpret the results.
MUCOSAL MELANOSIS
AT-A-GLANCE
■ Physiologic pigmentation in the oral cavity.
■ Macular pigmentation on the gingiva, oral mucosa, lips, and tongue.
■ More common in persons with skin of color.
Mucosal melanosis is common in persons with skin of color and should be differentiated from other causes of pigmentation in the oral cavity.71
Epidemiology: The limited literature available on this topic suggests the onset of pigmentation is gradual and likely to occur in middle-aged adults with richly pigmented skin.
Clinical Features: Diffuse and bilateral pigmentation may be noted but localized macular hyperpigmentation may be seen affecting one or more of the following locations: gingiva, buccal mucosa, lips, hard palate, and papillae of the tongue (Fig. 77-15). A study in China concluded that pigmented fungiform papillae
1371
13
of the tongue are relatively common in the Chinese population. Laugier-Hunziker syndrome is an acquired form of hyperpigmentation that presents as longitudinal melanonychia and pigmentation involving the oral mucosa, genitals, perianal region, fingers, and esophagus.
Etiology and Pathogenesis: The cause is unknown.
Diagnosis: The diagnosis can usually be made clinically without need for biopsy but if the diagnosis is in doubt, a biopsy can be helpful. Histologic examination demonstrates basal cell hypermelanosis, increased dermal melanophages, pigment incontinence, and normal melanocytes that in some instances are increased in number.
Differential Diagnosis: Many conditions can cause oral pigmentation. Drug-induced pigmentation, including fixed drug eruption, oral lichen planus, postinflammatory hyperpigmentation, dental amalgam, Addison disease, and malignancy, need to be excluded. Brown hyperpigmented macules in childhood should prompt investigation for genodermatoses such as Peutz-Jeghers, Bandler, Carney, and Cronkhite- Canada syndromes.
Clinical Course and Prognosis: Mucosal melanosis appears in childhood and persists without consequence throughout life.
Management: Reassurance is the most important component of management. No treatment is required.
LONGITUDINAL MELANONYCHIA
AT-A-GLANCE
■ Benign brown-black linear band of pigmentation of the nail plate.
■ More common in persons with skin of color.
■ Incidence increases with age.
Chapter 91 discusses longitudinal melanonychia in greater detail. Regular, thin hyperpigmented bands on multiple nails is characteristic of benign longitudinal pigmentation seen in darker individuals. Medications, trauma, and nevi in the nail apparatus may also cause longitudinal melanonychia and should be considered in the differential diagnosis. A single band located on the thumb or great toe, should prompt the clinician to diligently look for signs of melanoma clinically and dermoscopically. A biopsy also may be necessary to differentiate benign longitudinal melanonychia from melanoma.
1372
ACRAL HYPERPIGMENTED MACULES
AT-A-GLANCE
■ Benign brown well-demarcated macules on palms and soles.
■ More common in persons with skin of color.
■ Incidence increases with age.
Well-defined brown macules on the palms and soles reveal increased epidermal melanin histologically. Despite the parallel ridge pattern noted on dermoscopy, the number of lesions noted provides a clue to the benign nature of the lesions.72 It is important to exclude genodermatoses and drug-induced (especially chemotherapy) pigmentation that may present in a similar fashion.
ACQUIRED PATTERNED HYPERMELANOSIS
ACQUIRED PATTERNED
HYPERMELANOSIS
AT-A-GLANCE
■ Phytophotodermatitis, flagellate pigmentation caused by bleomycin, and flagellate mushroom dermatitis are all causes of patterned hyperpigmentation.
PHYTOPHOTODERMATITIS
Contact with plants containing phototoxic agents such as psoralens, with subsequent ultraviolet exposure, may lead to phytophotodermatitis, which is followed by patterned hyperpigmentation.
FLAGELLATE MUSHROOM DERMATITIS
Shiitake mushroom (Lentinula edodes) is typically grown and used in Asian cuisine but the Western world now consumes it in large quantities. Oral ingestion of raw or insufficiently cooked shiitake mushrooms is associated with a “flagellate mushroom dermatitis” that appears 12 hours to 5 days after consumption. While most reports have emerged from Japan, cases in Europe, the United States, and Canada also have been described.73 It is characterized by linear grouped, erythematous, and intensely pruritic papules that resembles bleomycininduced flagellate dermatitis. A pustular eruption also has been reported. The trunk and limbs are usually involved. The pathogenesis is not yet known, but it is hypothesized that a toxic reaction results from a thermolabile polysaccharide contained within the mushroom.
ERYTHEMA AB IGNE
ERYTHEMA AB IGNE
AT-A-GLANCE
■ Acquired diffuse reticulate pigmentation.
■ May also have epidermal atrophy, scaling, and hyperpigmentation.
■ Caused by chronic exposure to moderate heat.
■ Thyroid function should be checked.
Erythema ab igne is caused by frequent exposure to heat, usually infrared radiation, resulting in reticulate hyperpigmentation. Although it may occur anywhere on the body, it is usually localized to the thighs and lower legs.74
CLINICAL FEATURES
Tan–red-brown reticulate transient and blanchable erythema is noted acutely (usually about 3 weeks after the heat exposure) but fixed hyperpigmentation may be accompanied by epidermal atrophy, bullous lesions or hyperkeratosis, and scale in chronic cases. Common sites of involvement include the thighs, lower legs, abdomen, and back.
ETIOLOGY AND PATHOGENESIS
Erythema ab igne is caused by chronic exposure to moderate heat. It was seen in the past in people who frequently sat in front of open fires for warmth, but the introduction of central heating has seen a decline in this presentation. Nevertheless, it is seen after local application of heating pads, hot water bottles, or heating blankets (Fig. 77-16). Widespread cases secondary
13
to hot tub bathing in Japan also has been reported.45 Modern appliances that have been reported to elicit erythema ab igne are furniture with built-in heaters, car heaters, and laptop computers when rested on the thighs or legs. The reticulate nature of the cutaneous changes is thought to reflect the distribution of blood vessels. The heat is thought to damage superficial blood vessels in the epidermis, which, in turn, results in extravasation of red blood cells and hemosiderin, causing the cutaneous hyperpigmentation.
DIAGNOSIS
The diagnosis is usually made clinically. Thyroid function tests should be considered in patients with symptoms or signs that suggest hypothyroidism. If a skin biopsy is performed, histologic examination usually reveals vasodilation, extravasation of red blood cells, and deposition of melanin and hemosiderin in the dermis. Dermal edema and lymphohistiocytic dermal infiltration is also noted.75
DIFFERENTIAL DIAGNOSIS
Livedo reticularis and its various causes need to be considered, but the history often reveals chronic heat exposure in cases of erythema ab igne.
CLINICAL COURSE AND PROGNOSIS
The pigmentary changes usually resolve when the patient stops using the heat source on the skin, but depending on the chronicity of the exposure, pigmentation may persist for many months. Very rarely squamous cell carcinoma and Merkel cell carcinoma have been reported to arise within chronic lesions of erythema ab igne.
MANAGEMENT
Elimination of the heat source is critical, but topical retinoids and low-fluence quality-switched Nd:YAG laser also have been helpful in some persistent cases.76
PRURIGO PIGMENTOSA
PRURIGO PIGMENTOSA
AT-A-GLANCE
■ Acquired diffuse reticulate pigmentation on the trunk.
■ Most common in young females.
■ Initial phase of pruritic papules, vesicles, and papulovesicles is followed by asymptomatic hyperpigmentation.
■ Tetracyclines and dapsone are helpful for the inflammatory component of the condition.
1373
13
Prurigo pigmentosa is a rare condition, first described in Japan by Nagashima et al. in 1971.77 It most commonly affects the trunk and results in reticulated hyperpigmentation.
EPIDEMIOLOGY
While reports as of this writing have largely emerged from Japan, it is becoming increasingly documented in other parts of the world. It is most common in young females in their 20s with a female to male ratio of 4 to 6:1.78
CLINICAL FEATURES
Prurigo pigmentosa presents with intensely pruritic symmetrical urticarial, erythematous papules, papulovesicles, and vesicles that evolve into reticulated hyperpigmentation. The lesions develop on the upper back, chest, neck, and lumbosacral region. The papulovesicular lesions heal spontaneously over approximately 1 week, leaving nonpruritic reticular hyperpigmentation in its wake (Fig. 77-17). No systemic associations have been documented.
ETIOLOGY AND PATHOGENESIS
Environmental and metabolic factors have been suggested as causative agents, but the pathogenesis remains unknown.
DIAGNOSIS
The diagnosis of prurigo pigmentosa requires close clinicopathologic correlation. Histology of early lesions reveals a superficial neutrophilic perivascular and interstitial infiltrate. Later in the disease course, lesions demonstrate lichenoid inflammation with a predominance of lymphocytes. Spongiosis,
1374
necrotic keratinocytes, eosinophils, and vesiculation are also noted.79
DIFFERENTIAL DIAGNOSIS
Confluent reticulated papillomatosis (no preceding erythematous papules), pigmented contact dermatitis (responds to topical steroids), and Dowling-Degos disease (characteristic histologic findings) are the main differential diagnoses.
CLINICAL COURSE AND PROGNOSIS
The disease has a fluctuating course with exacerbations and recurrences.
MANAGEMENT
The eruption and pruritus respond well to minocycline or doxycycline 100-200 mg daily or dapsone 25 to 100 mg daily. The pigmentation resolves spontaneously.
NEVUS OF HORI
NEVUS OF HORI
AT-A-GLANCE
■ Acquired circumscribed hyperpigmentation in the form of symmetrical pinhead-sized macules on the malar region of the face.
■ Noted mainly in Asian populations.
■ Often coexists with other pigmentary conditions like melasma and flat seborrheic keratoses.
■ Best treated with quality-switched Nd:YAG lasers.
Nevus of Hori was first described in 1984 as acquired bilateral nevus of Ota–like macules (ABNOM) or acquired dermal melanocytosis.80
EPIDEMIOLOGY
It is a common condition noted in nearly 1% of the Asian population, particularly the Chinese and Japanese population. It is more common in females and appears in the third to fifth decades of life.
CLINICAL FEATURES
Nevus of Hori consists of blue-brown to slate-gray hyperpigmented macules ranging from pinhead size to a few millimeters in diameter (Fig. 77-18). Lesions are found on the face with predilection for the malar
region, although lesions on the temple and nose are not uncommon. Unlike nevus of Ota, nevus of Hori is bilateral and lacks ocular and mucosal membrane involvement.
ETIOLOGY AND PATHOGENESIS
Hori described 3 possible mechanisms for the pathogenesis of ABNOM: (a) “dropping” of epidermal melanocytes into the dermis, (b) migration of melanocytes from hair bulbs, and (c) reactivation of preexisting latent or immature dermal melanocytes. The third possibility has been suggested as being the most plausible mechanism with ultraviolet radiation and sex hormones as activating factors.
DIAGNOSIS
The diagnosis may require a biopsy, particularly if other pigmentary disorders, such as melasma, coexist in the affected area. Histology often reveals melanocytosis in the upper and mid dermis.
DIFFERENTIAL DIAGNOSIS
In many cases, ABNOM can occur simultaneously with other pigmentary abnormalities such as melasma, freckles, solar lentigines, and nevus of Ota.
CLINICAL COURSE AND PROGNOSIS
Nevus of Hori is usually gradually progressive.
MANAGEMENT
Although various lasers, such as ablative CO2, quality-switched 532-nm, quality-switched ruby, and fractional nonablative 2940-nm Erbium:YAG, have been used in isolation or in combination,81 qualityswitched lasers are the most commonly used form of treatment. Obtaining complete clearance is not always possible and multiple treatments are required. Quality-switched and picosecond lasers have demonstrated efficacy in lightening or clearing the lesions, but multiple treatments spaced many months apart are required.82,83
13
NEVUS OF BECKER
NEVUS OF BECKER
AT-A-GLANCE
■ Presents with acquired circumscribed hyperpigmentation.
■ Most commonly found on the trunk.
■ Usually associated with hypertrichosis.
■ Treatment with lasers can be attempted but current studies lack statistical significance.
This acquired hyperpigmented epidermal nevus was first described in 1949 (Fig. 77-19) and is also known as Becker hamartoma or Becker melanosis.
EPIDEMIOLOGY
Nevus of Becker appears to be androgen-dependent and is noted in approximately 0.5% of the population, usually becoming more prominent during adolescence. It is said to be more common in males but a newer study revealed equal incidence in male and female children.84
1375
13
CLINICAL FEATURES
This benign hamartoma may be cosmetically disfiguring and presents with a hyperpigmented macular or speckled area, most commonly in the scapular region, upper arms and chest, although it has been described in any area of the body. Hypertrichosis is common and often develops years after the hyperpigmentation is noted. The hairs are coarse and dark. Becker nevi are often first noted after an episode of intense sun exposure. Associated anomalies, such as ipsilateral breast hypoplasia, musculoskeletal abnormalities (eg, scoliosis, ipsilateral limb hypoplasia), maxillofacial abnormalities, and additional cutaneous hypoplasias, occur in the rare nevus of Becker syndrome.
ETIOLOGY AND PATHOGENESIS
Nevus of Becker is suggested to follow a paradominant inheritance pattern, which means it (almost) always occurs sporadically. The rare familial cases that have been described (especially in nevus of Becker syndrome) can be explained by a somatic mutation during embryogenesis, resulting in loss of heterozygosity and formation of a mutant cell population. This hypothesis is supported by the identification of chromosomal mosaicism in fibroblasts derived from a Becker nevus. The exact cause of the hyperpigmentation is unknown.
DIAGNOSIS
Epidermal acanthosis with variable hyperkeratosis and elongation of the rete ridges is noted on histology. Normal numbers of melanocytes, but increased levels of melanin in the basal epidermal layer are demonstrated. In the dermis, the number of arrector pili muscles is increased, making it difficult to differentiate from smooth muscle hamartomas.
DIFFERENTIAL DIAGNOSIS
Congenital hairy nevus, nevus of Ito, and CALMs are all potential differential diagnoses.
CLINICAL COURSE AND PROGNOSIS
Nevus of Becker may appear to darken during pregnancy and in summer. It persists throughout life.
MANAGEMENT
Treatment of nevus of Becker is challenging. If the lesion is small, surgical excision may be appropriate to consider. Although mechanical abrasion, cryotherapy, and argon and CO2 lasers have been used in the past for larger lesions, the high risk of scarring and dyspigmentation limit their use. Laser hair removal can give the illusion of lightening the lesion. Long-pulsed
1376
and picosecond alexandrite laser, as well as qualityswitched Nd:YAG, have been cited as useful in some studies, but the current evidence lacks statistical power and has inconsistent findings.85
CAFÉ-AU-LAIT MACULES
CAFÉ-AU-LAIT MACULES
AT-A-GLANCE
■ Acquired circumscribed hyperpigmentation.
■ May exist in isolation or as part of a genodermatosis.
■ Lasers may be used to lighten spots but relapses are common.
CALMs, also known as café-au-lait spots, may occur spontaneously or as part of a genodermatosis like NF1 and McCune-Albright syndrome.
EPIDEMIOLOGY
Up to one-third of the general population have CALMs. They are more frequent in darker Fitzpatrick skin types, but occur equally in males and females.86
CLINICAL FEATURES
Light-brown–tan–dark-brown well-demarcated homogenously hyperpigmented macules or patches can be noted anywhere on the body. The most commonly involved sites are the trunk, buttocks, and lower limbs (Fig. 77-20). When 6 or more CALMs are noted, investigation for underlying genodermatoses should be commenced.
ETIOLOGY AND PATHOGENESIS
A normal number of melanocytes produce an abnormal amount of melanin.
DIAGNOSIS
Histopathologic examination shows a normal to reduced number of sometimes hypertrophic melanocytes and increased melanin in the basal epidermal layer.
DIFFERENTIAL DIAGNOSIS
Nevus spilus, early nevus of Becker, and congenital nevus are all differential diagnoses.
CLINICAL COURSE AND PROGNOSIS
CALMs persist throughout life.
MANAGEMENT
For cosmetic reasons, quality-switched lasers can be used for treatment but relapses are common.
EPHELIDES
EPHELIDES
AT-A-GLANCE
■ Acquired circumscribed hyperpigmentation.
■ Autosomal dominant.
■ Appear on fair-skinned individuals.
EPIDEMIOLOGY
Ephelides appear in sun-exposed skin of fair-skinned individuals, often those with red or blond hair and Celtic ancestry. Males and females are equally affected.
CLINICAL FEATURES
Ephelides, or freckles, are asymptomatic, small, lightbrown macules that appear symmetrically on photoexposed sites.
13
ETIOLOGY AND PATHOGENESIS
Ephelides are inherited in an autosomal dominant pattern with a relationship noted to a mutation in the MCR-1 gene, with carriers demonstrating a significantly increased risk of developing these lesions.87
MCR-1 is the receptor for α-melanocyte-stimulating hormone, which activates the melanogenesis pathway via cyclic adenosine monophosphate. Decreased activity of this pathway promotes pheomelanin production.
DIAGNOSIS
Histopathologic examination shows a normal to reduced number of sometimes hypertrophic melanocytes but increased melanin in the basal epidermal layer.
DIFFERENTIAL DIAGNOSIS
Lentigines are the main differential diagnosis. The characteristic appearance in lighter-skin phototypes provides a clue to the diagnosis. The oral mucosa, palms, and soles are spared.
CLINICAL COURSE AND PROGNOSIS
They are more pronounced during spring and summer and fade during the winter. They appear in early childhood and often regress later in life.
MANAGEMENT
Photoprotection is important in those with light skin irrespective of the presence of ephelides but is especially important in individuals who wish to prevent darkening of the lesions. Topical depigmenting agents, such as hydroquinone, retinoids, α-hydroxy acids, and botanicals, may help to lighten pigmentation. Physical therapies, such as cryotherapy, are difficult to perform on very small lesions, so intense pulsed light or quality-switched lasers are preferred, but relapse should be expected.
ACROMELANOSIS PROGRESSIVA
ACROMELANOSIS
PROGRESSIVA
AT-A-GLANCE
■ Acquired circumscribed hyperpigmentation in newborns.
■ Usually involves acral and perineal areas.
1377
■ Spontaneously resolves.
13
EPIDEMIOLOGY
Very few cases have been described.
CLINICAL FEATURES
Diffuse brown hyperpigmentation is noted in newborns or during the first few weeks of life and is located on acral and or perineal areas. Progression to the trunk, oral mucosal involvement, and seizures were reported in one case.88
ETIOLOGY AND PATHOGENESIS
The cause is unknown.
DIAGNOSIS
Acromelanosis progressiva is a clinical diagnosis that becomes apparent over time, as the lesions fade. Histology reveals increased basal epidermal melanocytes.
DIFFERENTIAL DIAGNOSIS
Periungual hyperpigmentation, acropigmentation of Dohi, and reticulate acropigmentation of Kitamura are differential diagnoses to consider.
CLINICAL COURSE AND PROGNOSIS
Spontaneous resolution is noted.
MANAGEMENT
Reassurance is important as treatment is not required.
POSTINFLAMMATORY HYPERPIGMENTATION
POSTINFLAMMATORY
HYPERPIGMENTATION
AT-A-GLANCE
■ Acquired circumscribed hyperpigmentation in sites of prior inflammation or injury.
■ More common in persons with skin of color.
■ Hydroquinone remains the gold standard treatment.
■ Spontaneously resolves over a variable period of time.
Postinflammatory hyperpigmentation (PIH) is a common reactive melanosis caused by numerous preceding inflammatory (eg, acne, lichen planus, psoriasis) and cutaneous insults, such as drug and phototoxic
1378
reactions, infections, physical injury or trauma, and allergic reactions.
EPIDEMIOLOGY
PIH is far more common in darker-skin types ( Fitzpatrick Types III to VI) and can occur at any age. It is observed in a significant number of individuals of African ancestry who are suffering from acne.89
CLINICAL FEATURES
PIH consists of a macular hyperpigmentation at the site of inflammation (Fig. 77-21). A Wood lamp examination can determine the depth of the hyperpigmentation (see section “Melasma”). PIH after epidermal inflammation results in brown discoloration while inflammation in the dermis results in a gray-brown discoloration. The severity of PIH is proportional to the severity of the inflammation and degree of basement membrane disruption. Discovering the cause of the primary dermatosis should involve examination of the entire skin. Adverse effects on quality of life have been documented, especially in persons with skin of color, and should be addressed.
ETIOLOGY AND PATHOGENESIS
It is thought that inflammatory markers stimulate the melanocytes that drive PIH. Conditions causing epidermal inflammation cause an increase in epidermal keratinocytes, whereas dermal inflammation results in pigment incontinence into the dermis. The melanin is eventually cleared by macrophages. The cause for increased susceptibility of PIH in persons with skin of color has not been fully elucidated but is thought to relate to the increased amount of melanin in melanosomes.
DIAGNOSIS
Histologic features include pigment incontinence with accumulation of melanophages and increased melanin in epidermal layers.
DIFFERENTIAL DIAGNOSIS
When PIH is widespread on the trunk, it may look similar to urticaria pigmentosa but a positive Darier sign and the history of itching associated with the lesions should help to differentiate PIH from urticaria pigmentosa.
CLINICAL COURSE AND PROGNOSIS
The clinical course is variable and depends on the location of the inflammation or injury. It is more persistent in darker-skin types (Fitzpatrick Types III to VI) and after lichenoid inflammatory processes. Epidermal PIH often has spontaneous but slow fading.
A
B
13
MANAGEMENT
Management of PIH remains difficult. It is important, especially when treating patients with skin of color, to treat the primary dermatosis early to prevent PIH. The importance of photoprotection should not be underestimated. Pretreatment and posttreatment regimens are critical after physical therapies, especially for persons with skin of color, and may include sun protection, hydroquinone, hydroquinone alternatives, or topical corticosteroids.90
Topical hydroquinone remains the gold standard treatment for PIH (Table 77-3). A plethora of nonprescription cosmeceuticals containing licorice, soy, niacinamide, and various botanicals are available and numerous prescribed topicals, such as kojic acid, retinoids, and α-hydroxy acids may be used as monotherapy or in combination. Kligman’s formula and similar triple- therapy combinations containing hydroquinone, tretinoin, and a topical corticosteroid are also commonly prescribed. Physical therapies, including peels and lasers, should be used with caution given they can induce PIH if used too aggressively. Quality-switched and picosecond lasers have been described in isolation or as part of a broader treatment plan for PIH. Epidermal PIH is more amenable to topical therapy than dermal PIH.
MELASMA
MELASMA
AT-A-GLANCE
■ Acquired light-brown to dark-brown poorly circumscribed macules.
■ More common in persons with skin of color.
■ Malar skin, nose, forehead, and upper lip are commonly involved areas.
■ Hydroquinone remains the gold standard treatment.
■ Physical and oral therapies also can be used.
AGENTS MECHANISM OF ACTION
Hydroquinone Tyrosinase inhibitor
Retinoids Stimulate keratinocyte turnover, reduce melanosome transfer, tyrosinase inhibitor
Arbutin Tyrosinase inhibitor; inhibits melanosome maturation
Azelaic acid Tyrosinase inhibitor
Kojic acid Interacts with copper and inhibits tyrosinase
Tranexamic acid Plasmin inhibition; antioxidant
Tranexamic acid Plasmin inhibition; antioxidant
Adapted from Chaowattanapanit S, Silpa-archa N, Kohli I, et al. Postinflammatory hyperpigmentation: a comprehensive overview: treatment options and prevention. J Am Acad Dermatol. 2017;77(4):607-621.
1379
13
Melasma is one of the most common disorders of pigmentation and is sometimes referred to as chloasma or the mask of pregnancy.
EPIDEMIOLOGY
Melasma affects millions of people with a reported prevalence ranging from 9% in Hispanic populations in southern United States to 40% in Southeast Asians.91
It is cited as the most common pigmentary disorder in Indian women. It is mainly seen in premenopausal women, although men and postmenopausal women also may be affected.
CLINICAL FEATURES
Melasma typically presents with diffuse light-brown to dark-brown areas of pigmentation on the central face (Figs. 77-22 and 77-23). The malar and mandibular regions, forehead, chin, and upper lip are most commonly affected—centrofacial (63%: forehead, nose, chin, and upper lip); malar (21%: nose and cheeks); and mandibular (16%: ramus mandibulae) patterns are most common. Melasma spares the periorbital skin as well as the lips, neck, and ears. It may be seen less commonly on the forearms (also known as acquired brachial cutaneous dyschromatosis). The negative impact on quality of life has been documented repeatedly in the literature; consequently, symptoms of depression and anxiety need to be explored by the treating clinician.
ETIOLOGY AND PATHOGENESIS
The pathogenesis is not completely understood. Biologically active melanocytes, genetic and hormonal influences, and ultraviolet light exposure are known to be important. Specific precipitants, particularly oral contraceptives and estrogen replacement therapy, have been implicated in exacerbating the condition.
1380
Recently, visible light in individuals with skin of color and increased vascularity and vascular endothelial growth factor in the epidermis of lesional skin have been implicated in the pathogenesis of melasma.
DIAGNOSIS
A Wood lamp may help differentiate epidermal from dermal and mixed melasma in those with lighter skin types, although even Wood light–enhancing lesions, supposedly signifying epidermal melasma, have dermal melanin.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis may be broad depending on the area affected and the location of the pigment (epidermal, dermal, or mixed). It includes postinflammatory pigmentation, maturational dyschromia, medication-induced pigmentation, lichen planus pigmentosus, and possibly poikiloderma of Civatte.
CLINICAL COURSE AND PROGNOSIS
Despite lightening or resolution with treatment, melasma is a chronic and relapsing condition with exacerbations during pregnancy, sun or visible light exposure, and with certain hormonal treatments.
MANAGEMENT
Managing expectations and counseling on the recurrent and chronic nature of melasma is critical for patient satisfaction. Cessation of any obvious triggers is the first step in management. Photoprotection is central to management and should include sunavoidance, protective clothing, and regular application
of a broad-spectrum sunscreen with a physical blocker. Care should also be taken to protect from visible light in those with Fitzpatrick skin Types IV to VI. Epidermal pigmentation is known to be more responsive to topical therapy. Even though 4% hydroquinone remains the gold standard, various other topical agents have been used with variable success. Nonprescription creams containing soy, licorice, lignin peroxidase, and niacinamide are just some of the botanicals and vitamins said to help mild epidermal melasma. Preparations containing tretinoin, azelaic acid, and kojic acid are helpful when used for prolonged periods. Combining topical therapies is a useful strategy for epidermal melasma. Kligman’s formula is a popular combination of 5% hydroquinone, 0.1% tretinoin, and a mild topical corticosteroid. Another treatment, commonly referred to as triple-combination therapy, includes 4% hydroquinone, 0.05% tretinoin, and a topical corticosteroid. Both Kligman’s formula and triple-combination therapy have been deemed efficacious for epidermal melasma. Superficial chemical peels have proven efficacious and are useful when used as a treatment adjunct, but the cost and frequency of treatments must be considered. Furthermore, the risk of postinflammatory hyperpigmentation must be considered with appropriate pretreatment and posttreatment care, especially in individuals with skin of color. Laser therapy may be helpful in the treatment of melasma, but can also result in further unwanted hyperpigmentation. Various lasers, including intense pulsed light, erbium:YAG, and nonablative 1550-nm and 1927-nm fractional lasers have all been reported to be effective for melasma but low-fluence quality-switched Nd:YAG laser (also known as laser toning), is the most popular. This treatment modality has proved efficacious and safe when a few treatments are used in carefully selected cases. Recently, tranexamic acid has been proven to lighten epidermal and dermal melasma. It has been used in topical, intralesional, and oral forms. When used as an adjunct in low doses (500 to 700 mg daily) over a few months, it is efficacious and safe in treatment-resistant cases.92
PARTIAL UNILATERAL LENTIGINOSIS
PARTIAL UNILATERAL
LENTIGINOSIS
AT-A-GLANCE
■ Acquired circumscribed hyperpigmentation.
■ Appears at birth or in childhood.
■ May be a variant of mosaic neurofibromatosis Type 1.
■ Usually within 1 or more dermatomes.
Partial unilateral lentiginosis (PUL) is a rare pigmentary disorder also known as unilateral lentigines, lentiginous mosaicism, zosteriform lentiginous nevus, segmental lentiginosis, and agminated lentiginosis.
13
EPIDEMIOLOGY
Usually appears at birth or in childhood.
CLINICAL FEATURES
PUL is characterized by numerous lentigines with sharp margins. Lesions occur in a segmental pattern in the midline in 1 or more dermatomes.93
ETIOLOGY AND PATHOGENESIS
Its segmental pattern and presentation, which are accompanied by café-au-lait spots, Lisch nodules, or neurofibromas, suggests that PUL is a variant of mosaic NF1. Genetic studies will help to further elucidate this subject.
DIAGNOSIS
The characteristic history and clinical appearance are often diagnostic.
DIFFERENTIAL DIAGNOSIS
Mosaic NF1.
CLINICAL COURSE AND PROGNOSIS
PUL persists throughout life. No complications have been noted as a result of PUL.
MANAGEMENT
Quality-switched alexandrite and Nd:YAG lasers are reported to be treatment options.
RIEHL MELANOSIS
RIEHL MELANOSIS
AT-A-GLANCE
■ Acquired gray-brown-blue circumscribed reticulate hyperpigmentation.
■ Caused by topical contact sensitizers.
■ Mostly middle-aged women.
■ Located mainly on the face.
■ Lasers have been described as a treatment option.
Riehl melanosis, also known as female facial melanosis and pigmented contact dermatitis, was first described in 1917 by Riehl.
1381
13
EPIDEMIOLOGY
Riehl melanosis is mostly seen in middle-aged women, especially in darker-skin types, such as Latinos and Asians.
CLINICAL FEATURES
It is characterized by rapid onset of a reticular graybrown to almost black reticulate hyperpigmentation. The face (especially the forehead, zygomatic area, and temples) and the neck are principally involved, but the hands, forearms, and trunk also may be affected (Fig. 77-24). Inflammatory findings, such as erythema and pruritus, are usually absent although it may be observed in very early stages of the clinical course. Significant psychological stress may be reported by patients.
ETIOLOGY AND PATHOGENESIS
The pathogenesis is not understood fully understood but the hyperpigmentation has been postulated to be induced by repeated contact with threshold doses of a contact sensitizer such as fragrances, some pigments, and bactericides used in cosmetics and optical whiteners.
DIAGNOSIS
The main histopathologic feature is liquefactive degeneration of the basal layer of the epidermis, resulting in pigment incontinence in the dermis.
DIFFERENTIAL DIAGNOSIS
Poikiloderma of Civatte, erythrose peribuccal pigmentaire of Brocq, erythromelanosis follicularis faciei et colli, and medication-induced pigmentation are all differential diagnoses.
1382
CLINICAL COURSE AND PROGNOSIS
Riehl melanosis is progressive if the contact sensitizer is not avoided. It may take many years to lighten, but no systemic effects are noted.
MANAGEMENT
Unfortunately treatment is challenging. Cessation of the topical sensitizer is critical. Photoprotection and topical depigmenting agents (hydroquinone, α-hydroxy acids, tretinoin) have a limited effect on this dermal pigmentation, with more recent reports describing treatment with lasers and oral tranexamic acid. Intense pulsed-light therapy has been helpful in some cases, but carries the risk of postinflammatory hyperpigmentation. Low-fluence quality-switched Nd:YAG and “laser toning” have demonstrated moderate improvements in Asian skin and have even been used in combination with oral tranexamic acid and hydroquinone.94
CRONKHITE-CANADA SYNDROME
CRONKHITE-CANADA
SYNDROME
AT-A-GLANCE
■ Acquired generalized light-brown to dark-brown macules and patches.
■ Palms and soles often involved first.
■ Onychodystrophy and alopecia are also seen.
■ Nutritional supplementation and corticosteroids may help.
Cronkhite-Canada syndrome (CCS) is a very rare condition resulting in cutaneous and GI manifestations.
EPIDEMIOLOGY
Approximately 450 cases are reported in the literature. Adults of Asian (most commonly Japanese) and European extraction are usually affected, starting in the fourth decade of life.
CLINICAL FEATURES
Cronkhite-Canada syndrome initially presents with brown macules and patches noted on the palms and soles. This is followed by generalized pigmentary changes with onychodystrophy and alopecia.95
GI symptoms, such as abdominal pain, nausea, diarrhea, and loss of weight, are noted in addition to GI polyposis and occasional malignancies. Complications include, but are not limited to, protein and electrolyte
imbalance, anemia, and even portal vein thrombosis and glomerulonephritis. Cronkhite-Canada syndrome may be associated with other autoimmune conditions like systemic lupus erythematosus and rheumatoid arthritis.
ETIOLOGY AND PATHOGENESIS
An autoimmune process is suspected but the exact etiology remains elusive.
DIAGNOSIS
Clinicopathologic correlation is required to make the diagnosis. Histologic changes include increased epidermal melanin with dermal melanosis. Acanthosis and compact hyperkeratosis of the epidermis are also seen.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes other causes of congenital lentigines.
CLINICAL COURSE AND PROGNOSIS
Cronkhite-Canada syndrome is progressive and may cause significant morbidity and mortality because of the systemic manifestations of the condition.
MANAGEMENT
Immunosuppression with corticosteroids and nutritional supplements may be helpful.
ERYTHROSE PERIBUCCAL OF BROCQ
ERYTHROSE PERIBUCCAL
OF BROCQ
AT-A-GLANCE
■ Very rare dermatosis causing diffuse hyperpigmentation, erythema, and folliculocentric skin-colored papules.
■ Occurs mainly in women.
Erythrose peribuccal of Brocq is also known as erythrose pigmenta faciei, erythrosis pigmentosa peribuccalis, and melanosis perioralis et peribuccalis.
EPIDEMIOLOGY
Only a few cases, mainly in women, have been reported.
13
CLINICAL FEATURES
Erythema followed by diffuse light-brown to darkbrown pigmentation in the perioral region affecting the cutaneous upper and lower lip with sparing around the vermilion border is usually seen. In some cases, scale and loss of vellus hairs are noted.
ETIOLOGY AND PATHOGENESIS
The etiology is unknown, although ultraviolet light exposure and fragrances may play a role.
DIAGNOSIS
The diagnosis is usually made clinically. Histologic findings are nonspecific.
DIFFERENTIAL DIAGNOSIS
Melasma and PIH are the main differential diagnoses.
CLINICAL COURSE AND PROGNOSIS
Persistence and progression are the rule.
MANAGEMENT
Photoprotection and avoidance of triggers is recommended.
CUTANEOUS AMYLOIDOSIS
CUTANEOUS AMYLOIDOSIS
Macular amyloidosis presents as brown to gray-brown macules coalescing to plaques, mainly on the back (see Chap. 125).
ATROPHODERMA OF PASINI-PIERINI
ATROPHODERMA OF
PASINI-PIERINI
Chap. 70 discusses atrophic disorders of the skin in greater detail. The cutaneous lesions of atrophoderma of Pasini-Pierini can be described as single or multiple gray to violaceous-brown atrophic patches from 1 cm to more than 10 cm in diameter with slight depression and a characteristic “cliff-drop” border.
FIXED DRUG ERUPTION
FIXED DRUG ERUPTION
Fixed drug eruption can cause hyperpigmentation (see earlier in section “Toxins and Medications” and Chap. 45).
1383
13
ERYTHEMA DYSCHROMICUM PERSTANS
ERYTHEMA
DYSCHROMICUM
PERSTANS
AT-A-GLANCE
■ Acquired idiopathic dermal melanosis.
■ Blue-gray well-demarcated hyperpigmented macules and plaques.
■ Usually intermediate skin types (eg, Hispanic, Asian populations).
■ Seen in photoprotected sites.
■ Treatment is challenging and may require systemic therapy.
Erythema dyschromicum perstans (EDP), also known as ashy dermatosis, dermatosis cinecienta, and erythema chronicum figuratum melanodermicum, was first described by Ramirez in 1957.96 Patients with this eruption were labeled as los cenicientos, the ashen ones. The terms EDP, lichen planus pigmentosus, and idiopathic eruptive macular pigmentation have been used interchangeably and are often confused in the literature, even though they appear to be distinct clinical entities.
EPIDEMIOLOGY
EDP is mainly observed in females with intermediate skin types (eg, Hispanics and Asians). It is said to be a disease of young adults with most presenting between the second and third decades of life, although a recent review of Korean patients revealed onset in the third and fourth decades of life.
CLINICAL FEATURES
EDP presents with widespread asymptomatic welldemarcated blue-gray macules that are most commonly seen on photoprotected sites (Figs. 77-25 and 77-26). Lesions slowly expand over time and an erythematous border may be seen at the edge of the lesion in the acute stages of the condition. The erythematous border, especially in darker-skin types, may evolve into a hypopigmented border that accentuates the hyperpigmentation. Mucous membranes are not involved and no extracutaneous manifestations exist.
ETIOLOGY AND PATHOGENESIS
The etiology is unknown although human leukocyte antigen-D related (HLA-DR)–associated susceptibility has been described in a Mexican population.97
1384
DIAGNOSIS
The diagnosis usually requires clinicopathologic correlation. Histologically, early lesions reveal dermal edema and lichenoid inflammation, basal layer vacuolization and colloid bodies with a perivascular infiltrate. Later, melanin incontinence is noted in the deep dermis along with melanophages.
DIFFERENTIAL DIAGNOSIS
Lichen planus pigmentosus, idiopathic eruptive macular pigmentation, and PIH are the main differential diagnoses. Medication-induced pigmentation must also be excluded.
CLINICAL COURSE AND PROGNOSIS
The condition is chronic and very difficult to treat. There is a slow progression of the lesion over several years, usually without spontaneous regression.
MANAGEMENT
Topical therapies are not helpful for this acquired dermal disorder of pigmentation and improvement with systemic therapy is minimal. An 8- to 12-week course of dapsone 100 mg may lighten pigmentation and clofazimine 100 mg daily has demonstrated improvement in a small study.98 Unfortunately, even though many other treatments have been evaluated, none have resulted in appreciable lightening. Most recently, laser surgery has been studied by a group in the Netherlands, who concluded fractionated laser was unsuccessful for the treatment of EDP.99
LICHEN PLANUS PIGMENTOSUS
LICHEN PLANUS
PIGMENTOSUS
AT-A-GLANCE
■ Acquired idiopathic dermal melanosis.
■ Poorly demarcated brown to gray-brown hyperpigmented macules.
■ Usually seen with deeply pigmented skin (eg, South East Asians, Arabic).
■ Mostly in photoexposed sites.
■ Treatments provide only minimal improvement in pigmentation.
Lichen planus pigmentosus is considered a variant of lichen planus. Lichen planus is discussed in Chap. 32. It is often confused in the literature for EDP and idiopathic eruptive macular pigmentation.
13
EPIDEMIOLOGY
Lichen planus pigmentosus is mainly observed in middle-aged (third to fourth decade of life) individuals with more deeply pigmented skin (eg, South Asians, Southeast Asians, and the Arabic population).
CLINICAL FEATURES
Symmetrical brown to gray-brown poorly demarcated macules and patches are seen mainly in photoexposed sites such as the head (forehead and temples) and neck (Fig. 77-27). Lesions also may be seen in skin folds such as the axillae (known as lichen planus pigmentosus– inversus). Lesions may also rarely appear on mucous membranes. No extracutaneous manifestations exist.
ETIOLOGY AND PATHOGENESIS
The exact etiology is unknown but mustard and amla oils used in cooking and in hair care formulas, respectively, have been listed as possible causes in retrospective studies. Medication-induced pigmentation must also be excluded.
DIAGNOSIS
Biopsy is required and clinicopathologic correlation is critical. Epidermal atrophy, basal layer vacuolation with a perivascular lymphocytic infiltrate, and melanophages in the superficial dermis are noted histologically.
DIFFERENTIAL DIAGNOSIS
EDP, idiopathic eruptive macular pigmentation, and PIH are the main differential diagnoses.
CLINICAL COURSE AND PROGNOSIS
Some cases gradually resolve over many months, but these cases are the exceptions to the rule.
1385
13
MANAGEMENT
Photoprotection is critical. Topical tacrolimus (0.03% twice daily), topical and systemic corticosteroids, and topical vitamin A may be helpful in lichen planus pigmentosus.100 Treatment combinations and laser treatments also have been used with variable results. Recently, a prospective study reported low-dose oral isotretinoin (20 mg daily), used over 6 months in conjunction with sunscreen, caused stabilization and decrease in pigmentation, especially when used early in the course of disease.101
IDIOPATHIC ERUPTIVE MACULAR HYPERPIGMENTATION
IDIOPATHIC
ERUPTIVE MACULAR
HYPERPIGMENTATION
AT-A-GLANCE
■ Acquired idiopathic dermal melanosis.
■ Well-demarcated brown to gray hyperpigmented macules and plaques.
■ Usually smaller lesions than erythema dyschromicum perstans.
■ Usually appears in children and adolescents.
■ Usually appears on the face, trunk, and proximal extremities.
■ Treatments provide only a minimal improvement in pigmentation.
Idiopathic eruptive macular hyperpigmentation is a rare disorder of pigmentation, described initially by Degos and colleagues but first described in English in 1996.102,103
EPIDEMIOLOGY
Idiopathic eruptive macular hyperpigmentation is usually seen in children and adolescents.
CLINICAL FEATURES
Idiopathic eruptive macular hyperpigmentation is characterized by an eruption of asymptomatic, welldemarcated, nonscaly, brown macules and plaques (5 mm to several centimeters in diameter) involving the face, neck, trunk, and proximal extremities. No preceding inflammation or erythema is noted. A subset of patients have been noted to have thickened plaques (epidermal papillomatosis). No extracutaneous manifestations exist.
ETIOLOGY AND PATHOGENESIS
1386
The etiology is unknown.
DIAGNOSIS
Histopathologically, there is increased melanin in the basal epidermis and melanophages demonstrated in the dermis. The number of mast cells is normal. No basal layer change or lichenoid inflammation is seen.
DIFFERENTIAL DIAGNOSIS
EDP, lichen planus pigmentosus, and PIH are the main differential diagnoses. Drug-induced pigmentation must be excluded.
CLINICAL COURSE AND PROGNOSIS
Lesions spontaneously disappear over several months to years without scarring.
MANAGEMENT
Spontaneous resolution means that in most situations, treatment is not needed. Topical lightening agents like retinoids, α-hydroxy acids, and hydroquinone are possible treatments.
MIXED HYPOMELANOSIS AND HYPERMELANOSIS
MIXED HYPOMELANOSIS
AND HYPERMELANOSIS
AT-A-GLANCE
Causes of mixed hypomelanosis and hypermelanosis include:
■ Dyschromatosis hereditaria universalis.
■ Dyschromatosis symmetrica hereditaria.
■ Reticulate acropigmentation of Dohi.
■ Familial progressive hyperpigmentation with (Westerhof syndrome) or without hypopigmentation.
■ Vagabond leucoderma.
DYSCHROMATOSIS HEREDITARIA UNIVERSALIS
Dyschromatosis hereditaria universalis is a rare autosomal dominant disorder (mutation in the ABCB6 gene on chromosome 12q21-q23) that usually presents in infancy or early childhood in Japanese families and is characterized by pinpoint to pea-sized hypopigmented and hyperpigmented macules, distributed in a reticulated pattern over the trunk, abdomen, and limbs, usually sparing the face and palmoplantar surfaces.104
RETICULATE ACROPIGMENTATION OF DOHI
Reticulate acropigmentation of Dohi is a localized form of dyschromatosis universalis hereditaria, also called dyschromatosis symmetrica hereditaria. It is characterized by small, symmetric, hyperpigmented and hypopigmented macules on the dorsal hands and feet, and is mainly seen in young children in South American and Asian families.104
FAMILIAL PROGRESSIVE HYPERPIGMENTATION WITH (WESTERHOF SYNDROME) OR WITHOUT HYPOPIGMENTATION
Familial progressive hyperpigmentation with or without hypopigmentation is an autosomal dominant condition caused by a heterozygous mutation in the KIT ligand gene (KITLG) on chromosome 12q22. It presents at birth or early infancy as diffuse hyperpigmentation and may demonstrate café-au-lait macules and larger hypopigmented ash-leaf macules on the face, neck, trunk, and limbs. The case series of Westerhof syndrome in 1978 described some family members with retarded growth and mental retardation.105
VAGABOND LEUKODERMA
Vagabond leukoderma is a condition found in persons living in poor hygienic conditions. Many abuse alcohol, live on an inadequate diet, and are infested with lice and/or scabies. Diffuse light-brown hyperpigmentation is present at the shoulder and waist girdle, and the neck and back are dotted with depigmented macules. The condition improves upon institution of a healthier lifestyle and likely represents a coexistence of many disorders
ACKNOWLEDGMENTS
We would like to acknowledge the contribution of the authors of this chapter in the previous edition: Hilde Lapeere, Barbara Boone, Sofie De Schepper, Evelien Verhaeghe, Mireille Van Gele, Katia Ongenae, Nanja Van Geel, Jo Lambert, and Lieve Brochez.
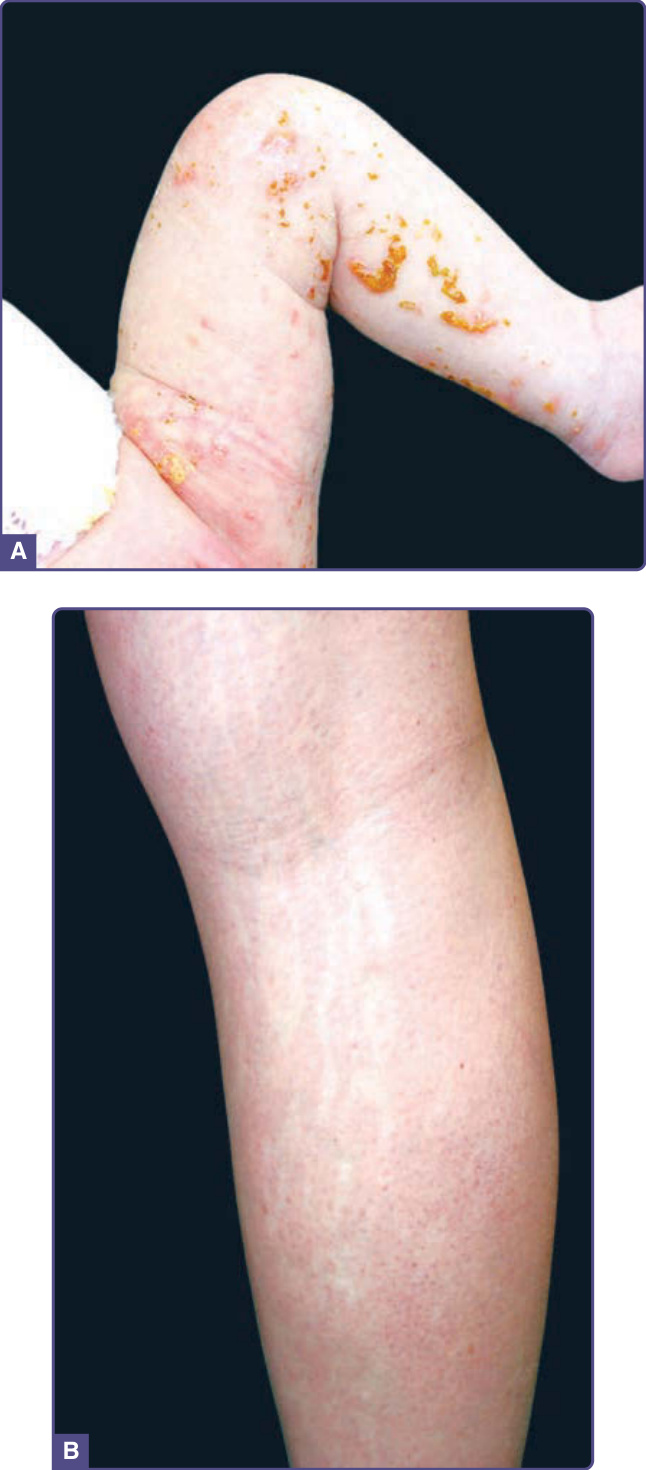
Figure 77-1 Incontinentia pigmenti in a mother and her baby. A, Verrucous lesions in a 2-week-old baby. B, Hypopigmented atrophic lesions following Blaschko lines.
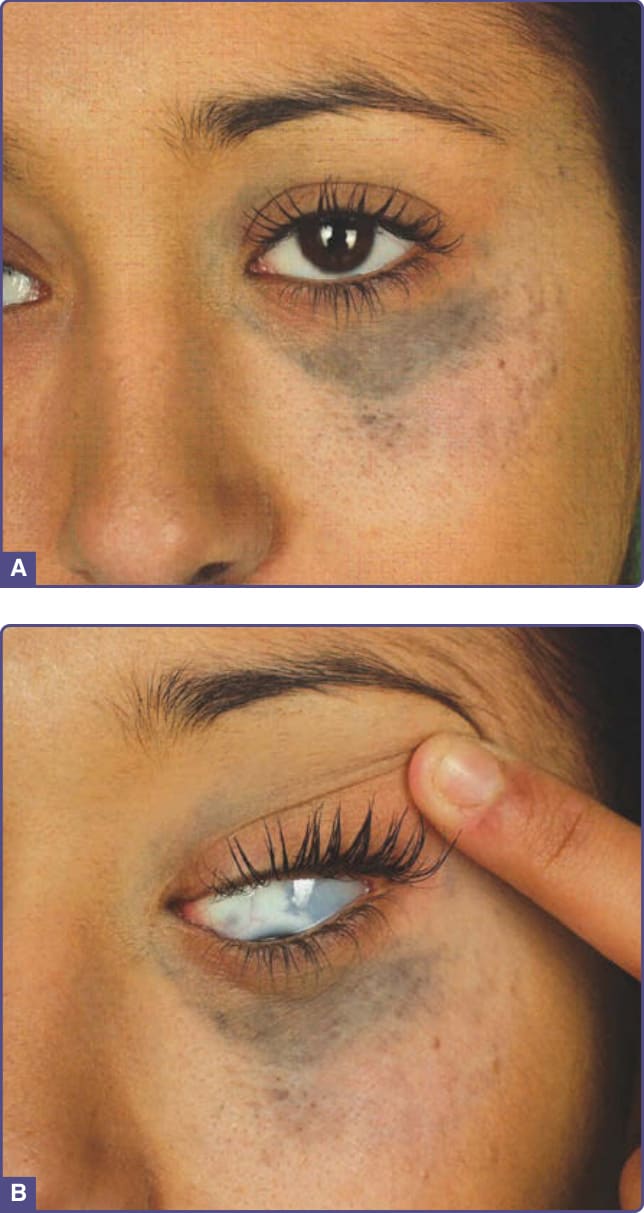
Figure 77-2 Nevus of Ota in a 20-year-old woman. A, Hyperpigmentation around the orbit. B, The hyperpigmentation extends into the sclera.
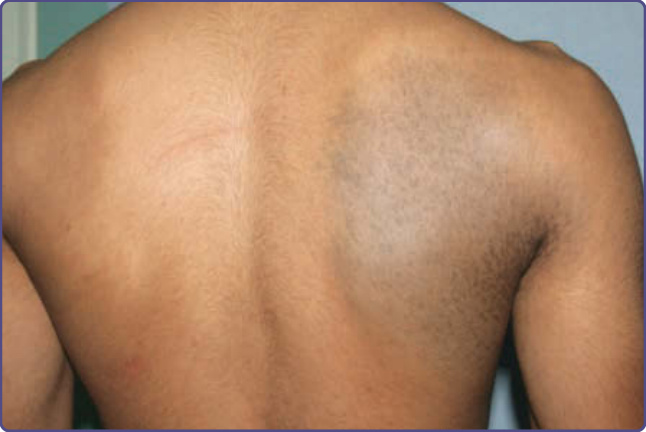
Figure 77-3 Nevus of Ito on the back of a Middle Eastern adolescent.
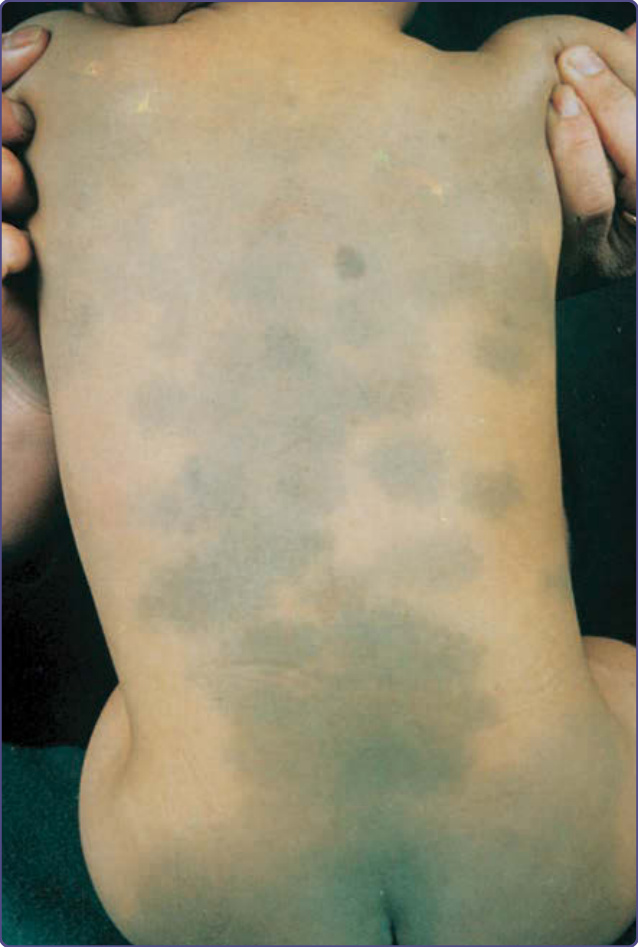
Figure 77-4 Classic Mongolian spots in the lumbosacral region and aberrant or extrasacral Mongolian spots on the back.
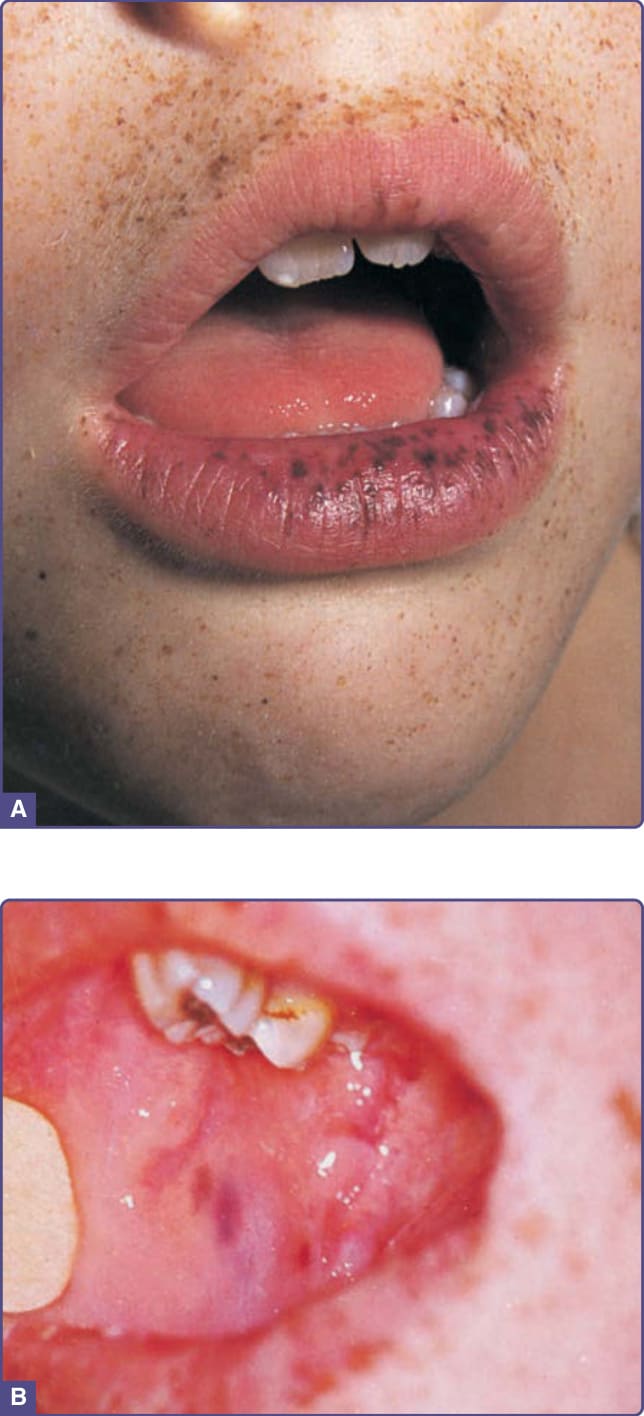
Figure 77-5 Peutz-Jeghers syndrome. A, Lentigines, which are dark-brown to gray-blue, appear on the lips, around the mouth, and on the fingers. Lip macules may, over time, disappear. B, Macules of the buccal mucosa are blue to blue-black and are pathognomonic; unlike lip lesions, these do not tend to disappear with time.
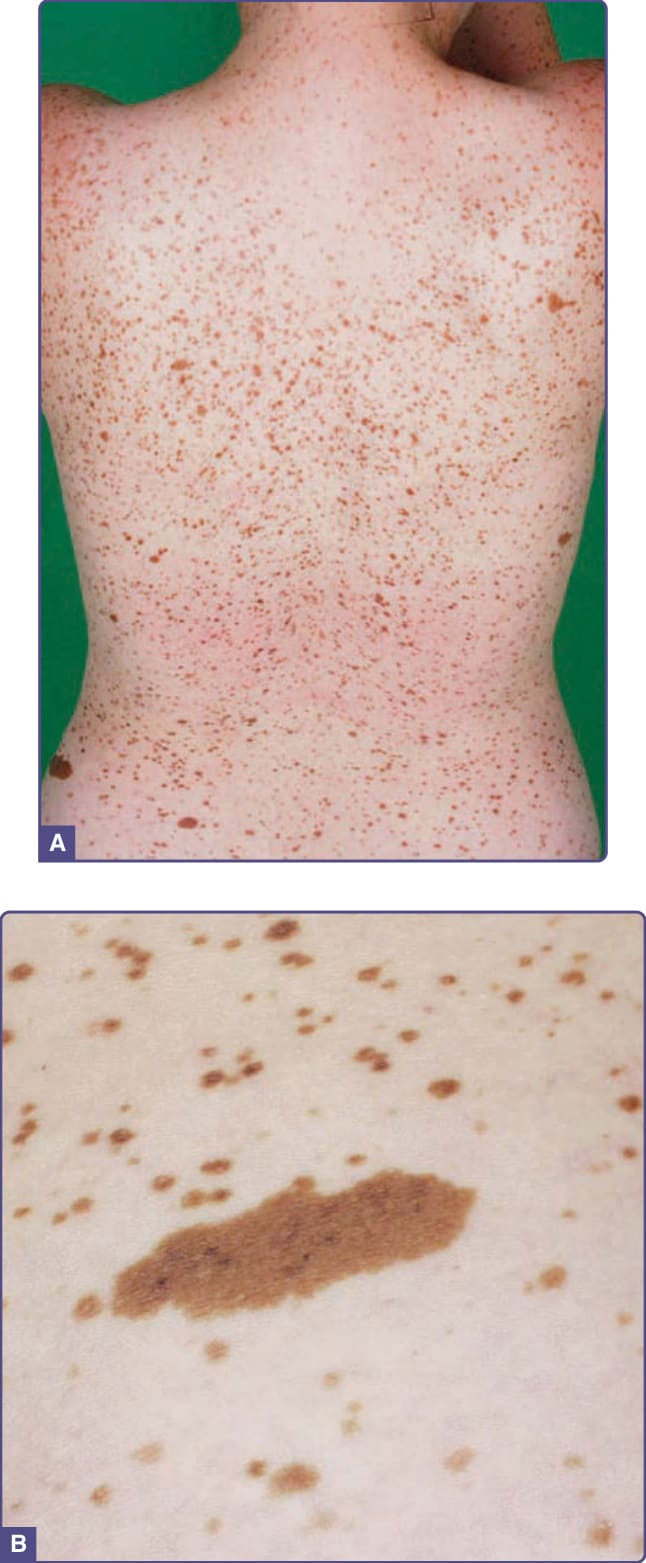
Figure 77-6 LEOPARD (lentigines, electrocardiogram conduction defects, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness) syndrome. A and B, A 27-year-old woman with LEOPARD syndrome. Note the characteristic widespread lentigines and several café-au-lait macules.
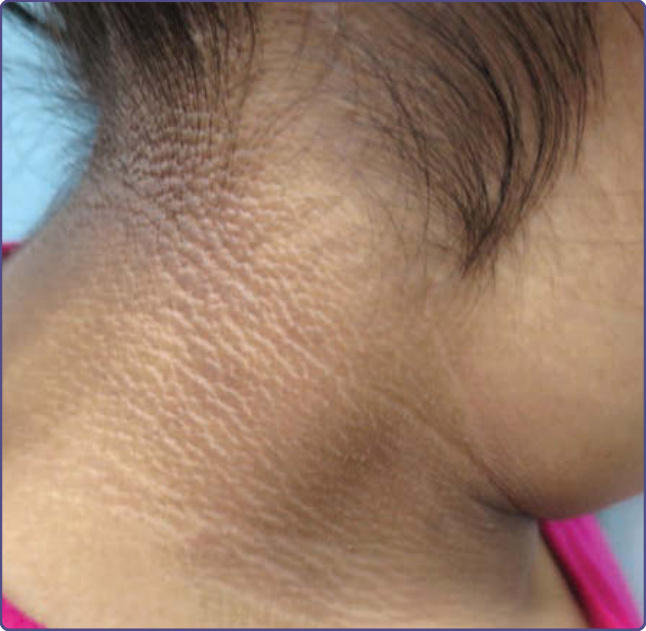
Figure 77-7 Acanthosis nigricans on the neck of a young girl of Filipino origin.
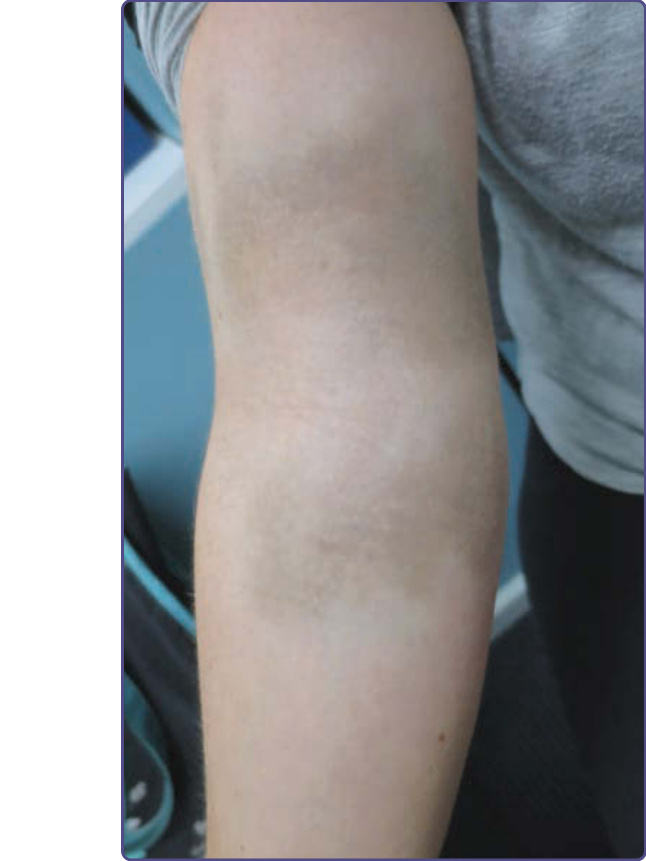
Figure 77-9 Diffuse gray pigmentation secondary to IV iron infusion in the arm of a white woman.
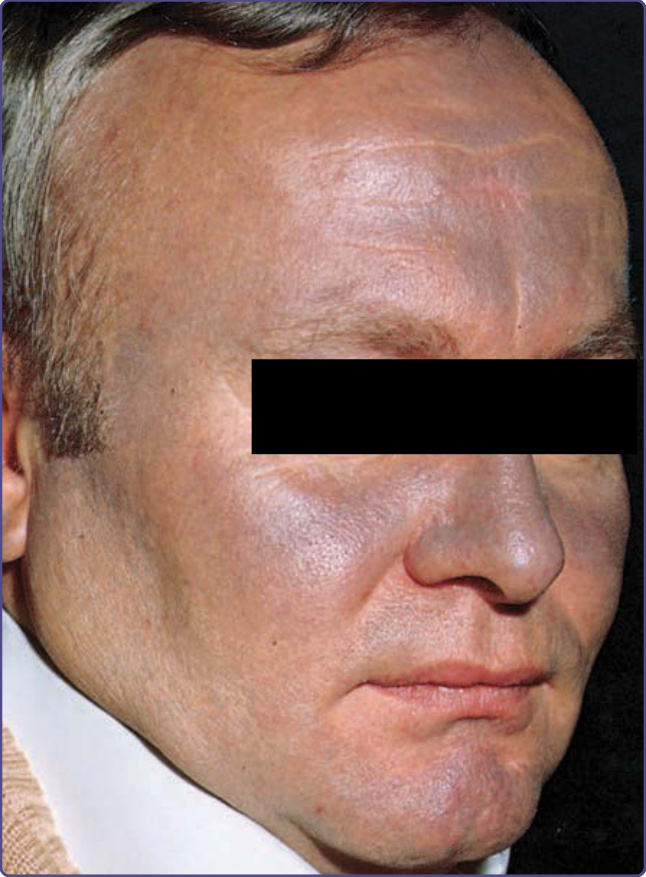
Figure 77-10 Amiodarone hyperpigmentation. This patient exhibits a striking amiodarone-induced, slate-gray pigmentation of the face. The blue color (ceruloderma) is a result of the deposition of a brown pigment in the dermis, contained in macrophages and endothelial cells.
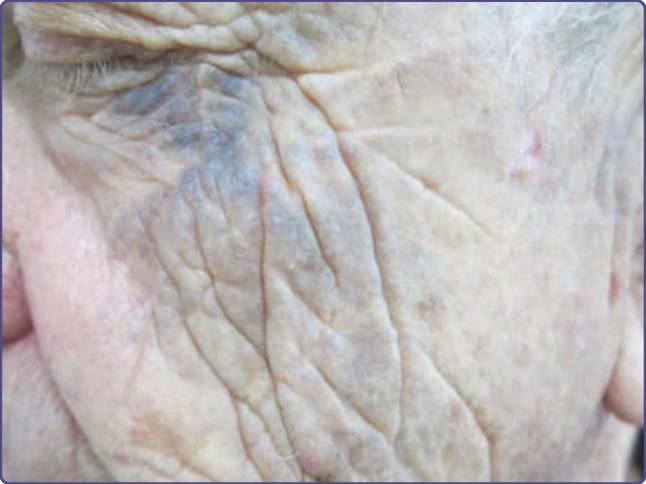
Figure 77-11 Slate-gray minocycline-induced pigmentation on the cheek of a white male.
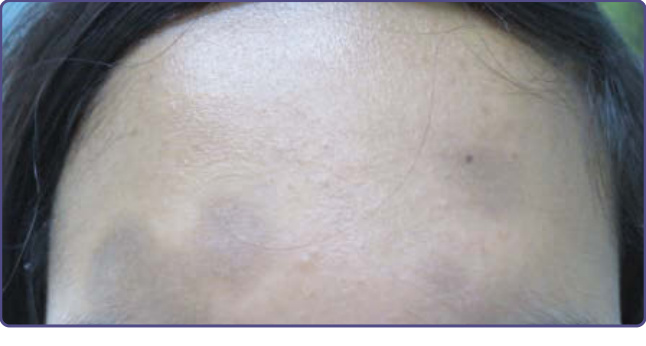
Figure 77-12 Fixed drug eruption on the face secondary to nonsteroidal antiinflammatory medication in a young Sri Lankan girl.
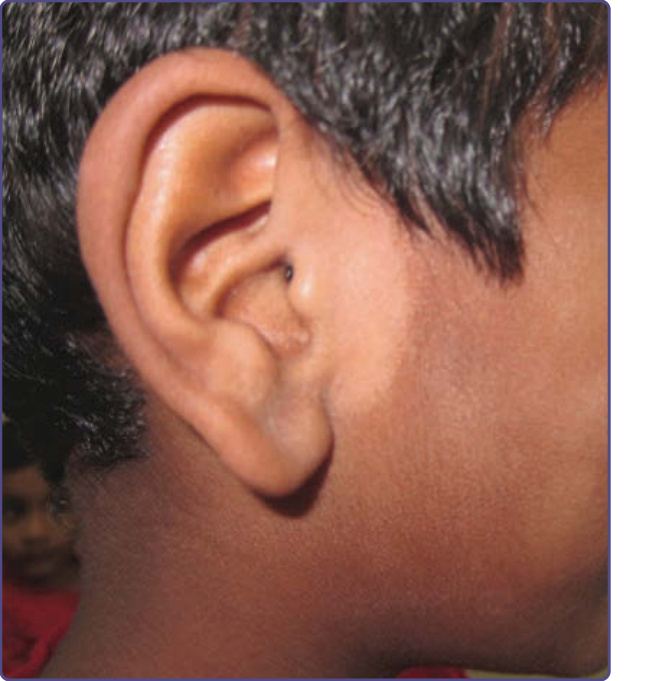
Figure 77-13 Pigmentary demarcation line anterior to the ear in a boy of Sri Lankan descent.
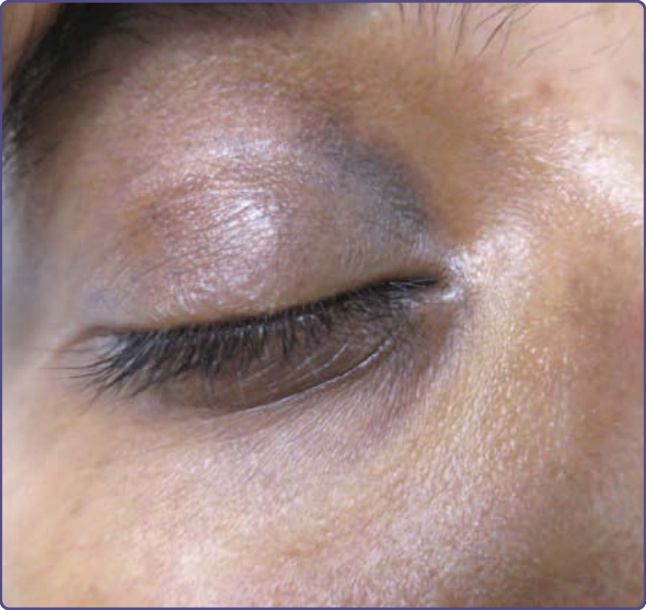
Figure 77-14 Periorbital pigmentation in an Indian lady.
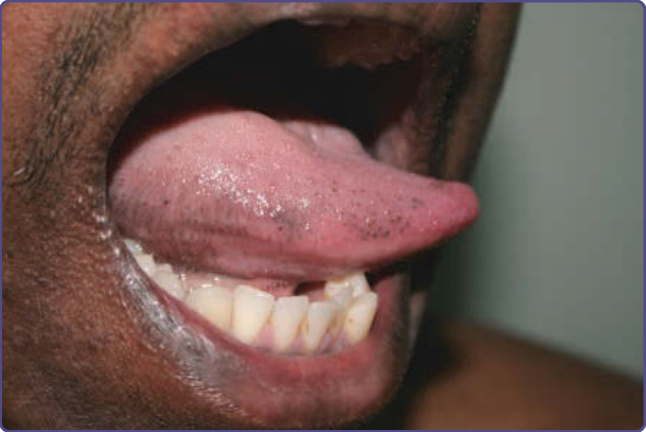
Figure 77-15 Benign pigmentation of the papillae of the tongue in an Indian man.
Figure 77-16 Reticular hyperpigmentation (erythema ab igne) on the abdomen resulting from repeated applications of a heating pad over several years.
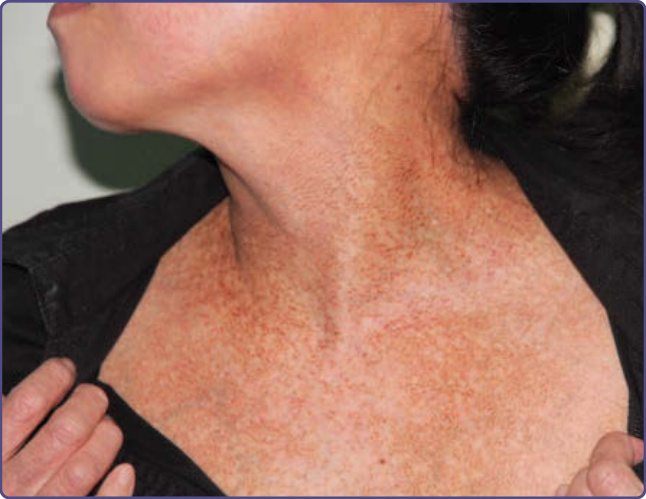
Figure 77-17 Reticulate pigmentation seen in prurigo pigmentosa in a Japanese woman.
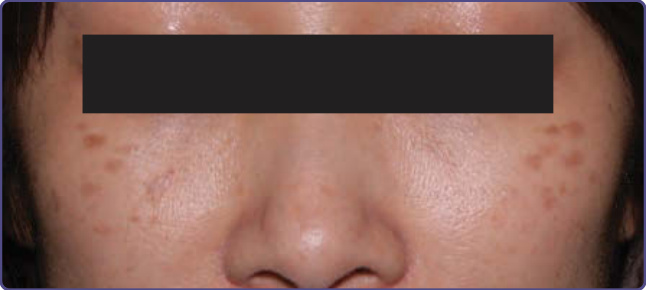
Figure 77-18 Nevus of Hori.
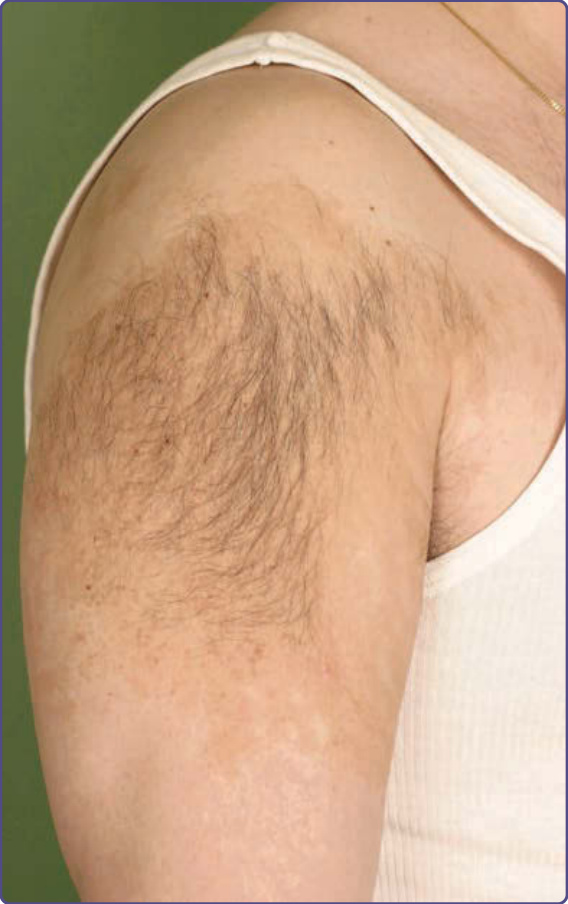
Figure 77-19 Nevus of Becker on the upper arm and shoulder of a young man. Note the typical localization and marked hypertrichosis.
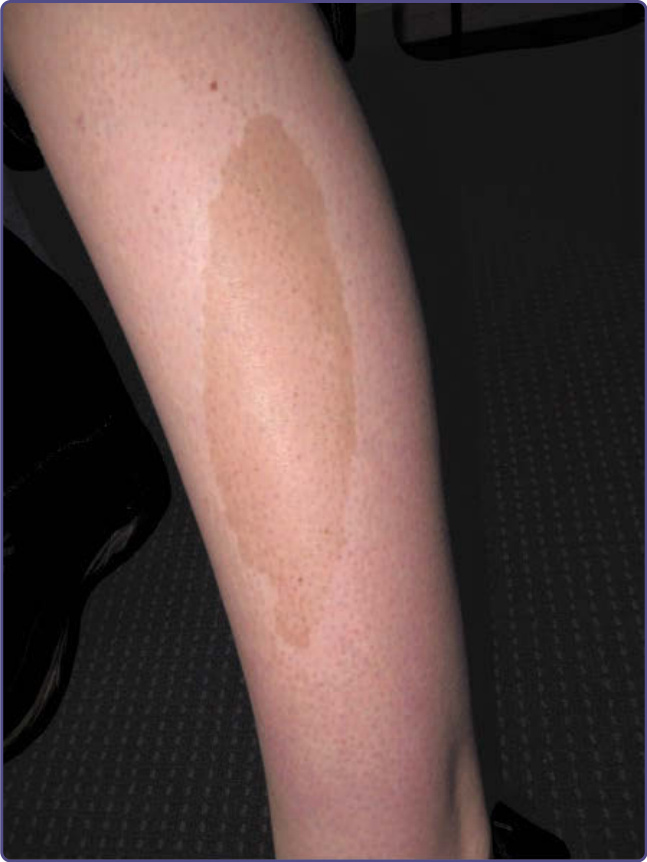
Figure 77-20 Café-au-lait macule on the lower leg of a young girl.
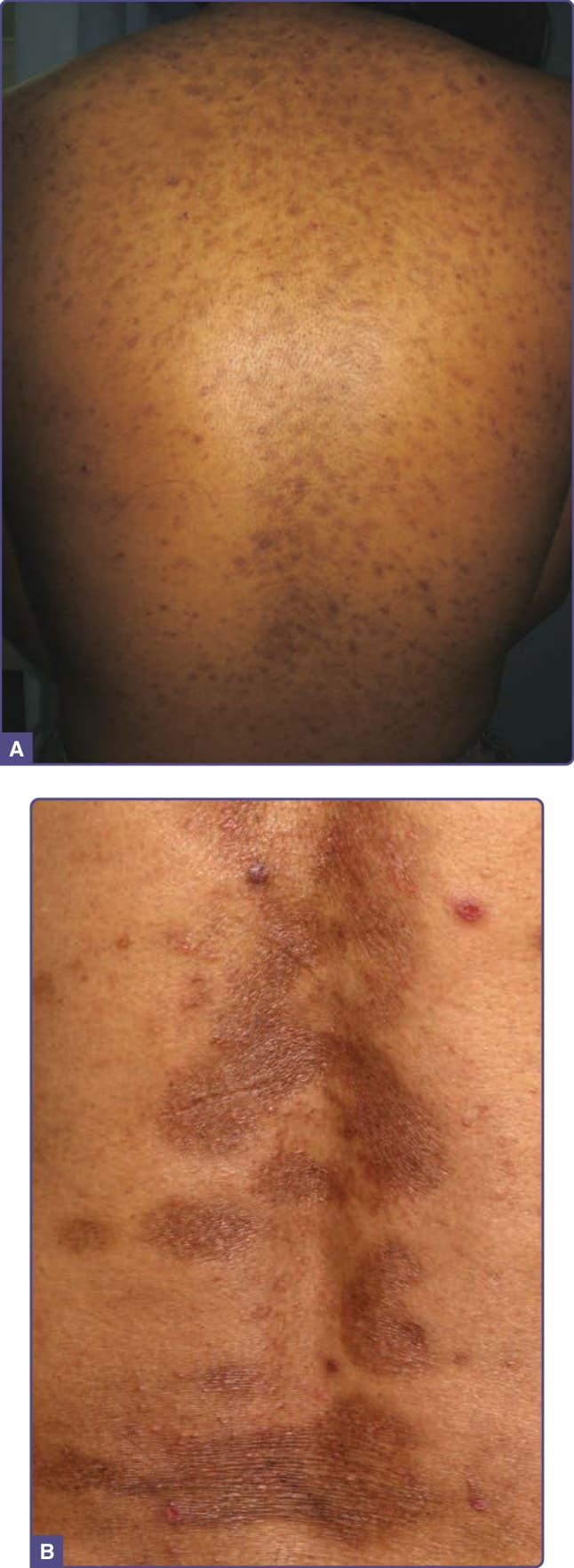
Figure 77-21 A, Postinflammatory hyperpigmentation secondary to acne on the back of a young African girl. B, Postinflammatory hyperpigmentation secondary to psoriasis on the back of a Chinese man.
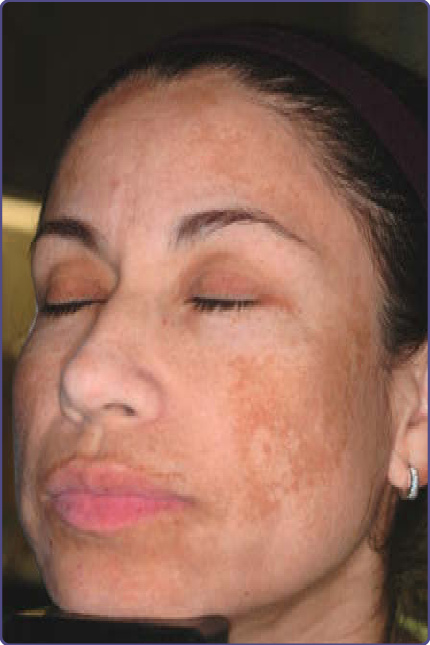
Figure 77-23 Hyperpigmentation of the forehead, cheek, upper lip, and chin caused by melasma in a Latino female.
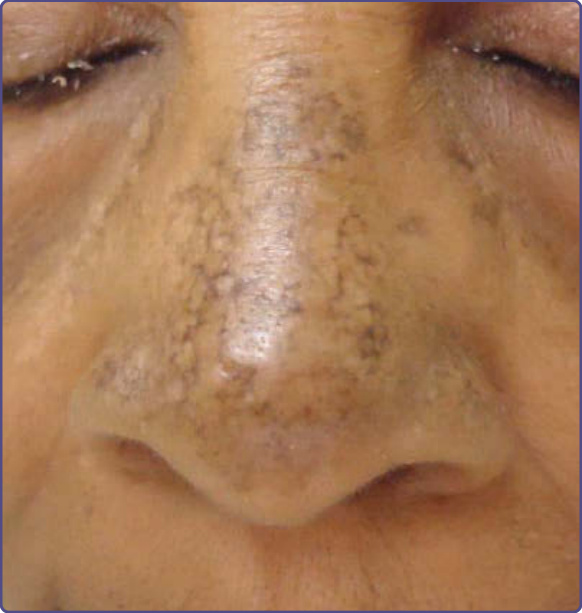
Figure 77-24 Riehl melanosis on the face.
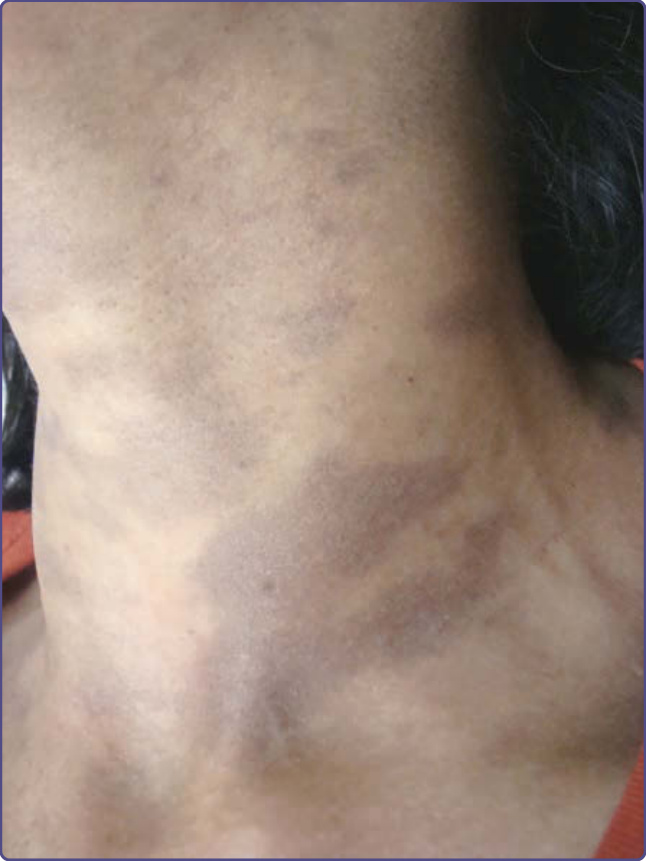
Figure 77-25 Acquired dermal melanosis–erythema dyschromicum perstans/on the neck of a Hispanic woman.
Figure 77-26 Erythema dyschromicum perstans on the trunk. (Used with permission of Juan Carlos Mendez, Department of Dermatology, UANL. Monterrey, México.)
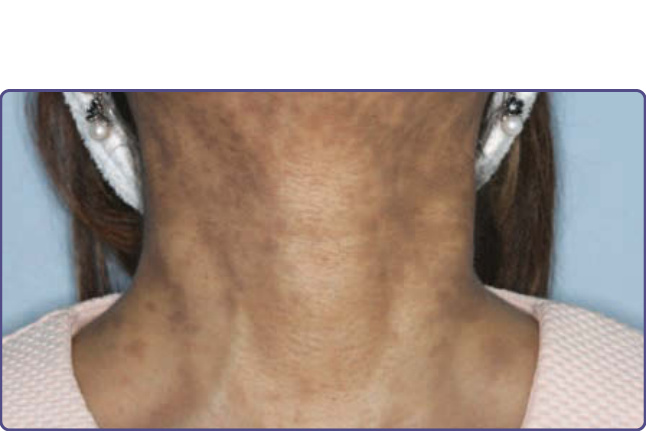
Figure 77-27 Acquired dermal melanosis–erythema dyschromicum perstans/lichen planus pigmentosus on the neck of a Sri Lankan lady.
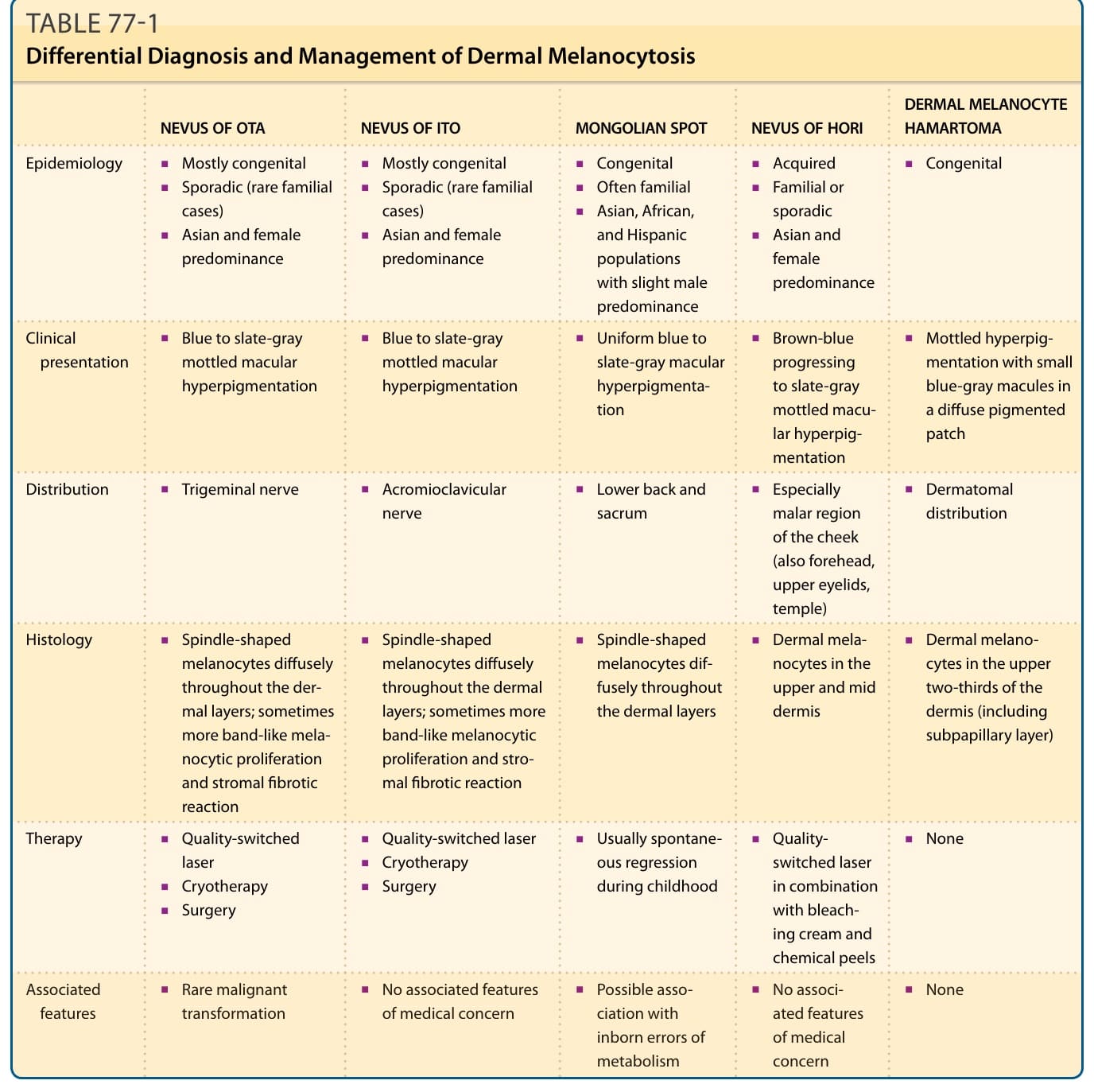
TABLE 77-1 Differential Diagnosis and Management of Dermal Melanocytosis
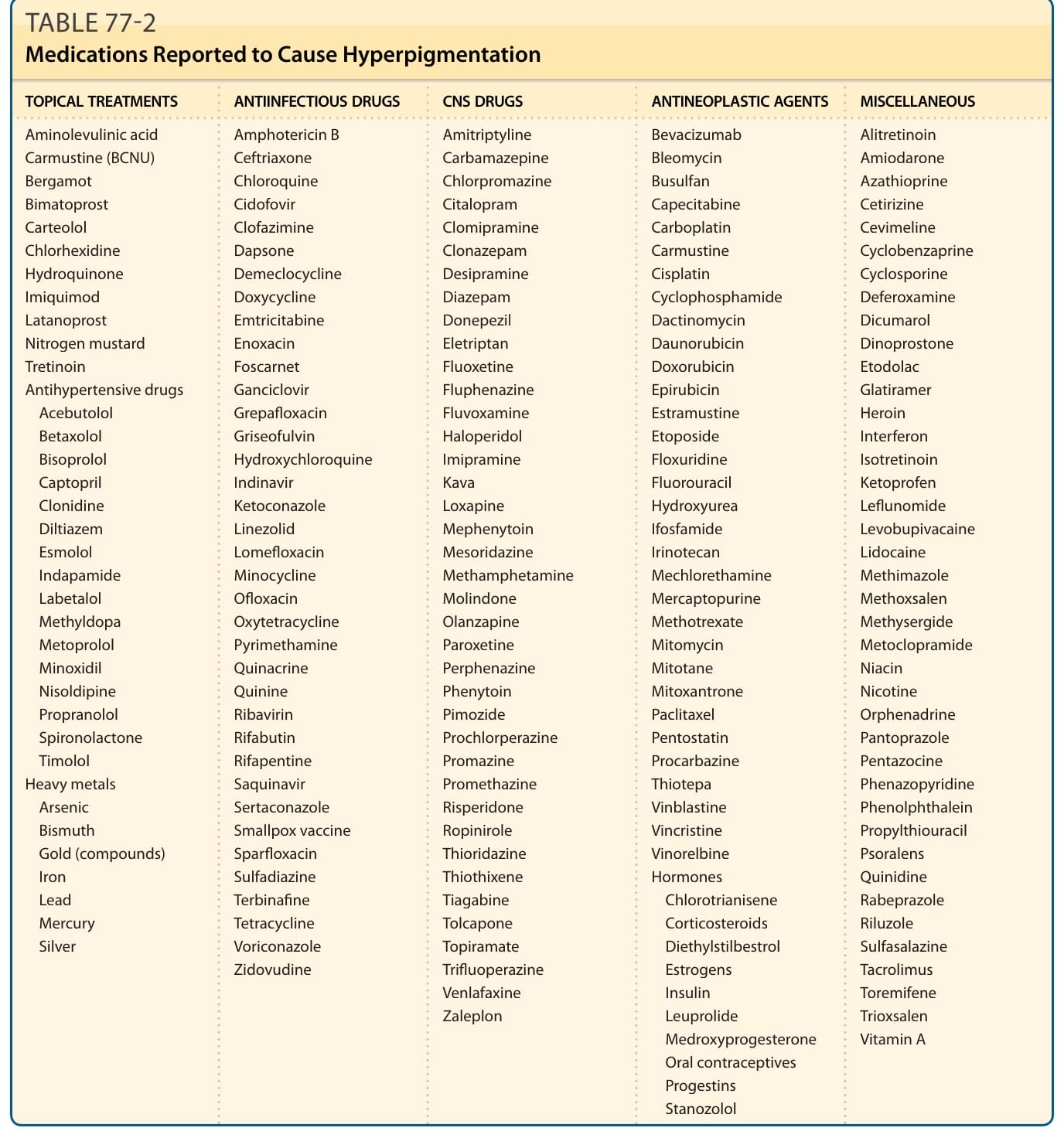
TABLE 77-2 Medications Reported to Cause Hyperpigmentation
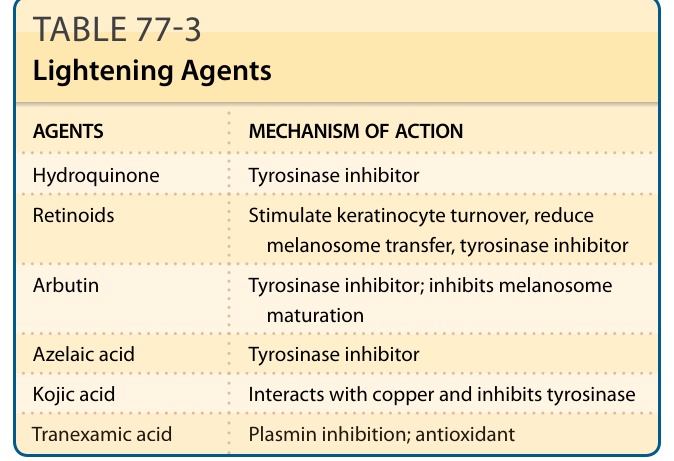
TABLE 77-3 Lightening Agents