硬腫病與硬化黏液水腫 (Scleredema and Scleromyxedema)
總論
- 以黏液 (mucin) 增加、膠原蛋白 (collagen) 過量沉積或纖維細胞 (fibrocyte) 增生為特徵的皮膚病,分別稱黏液沉積症 (mucinoses)、硬化性疾病 (sclerosing disorders)、纖維化性疾病 (fibrosing disorders),皆可視為各種「纖維病變 (fibropathy)」。
- 硬腫病 (scleredema) = 過量黏液+膠原蛋白沉積;硬化黏液水腫 (scleromyxedema) = 過量黏液沉積+纖維細胞增生。
- 纖維母細胞 (fibroblast) 源自骨髓 CD34 陽性造血前驅細胞,主要負責沉積膠原蛋白、彈性蛋白 (elastin) 與基質。誘發纖維細胞過度產生黏液/膠原或增生的刺激因子廣泛(感染、發炎、糖尿病、藥物、重金屬如釓 gadolinium、基因突變、毒素、細胞激素、副蛋白等),確切機轉仍未充分闡明。
硬腫病 (Scleredema)
- 定義:以真皮黏液沉積症 (dermal mucinosis) 與輕度硬化 (mild sclerosis) 為特徵的急性皮膚硬結 (induration);由 Buschke 於 1902 年描述,又稱 Buschke 硬腫病、scleredema adultorum(兒童/青少年亦可發病,故後者為誤導性名稱)。
- 疾病關聯:
- 青少年感染後型 (postinfectious scleredema of youth)。
- 成年糖尿病相關型 (diabetes mellitus–associated)。
- 副蛋白血症相關型 (paraproteinemia-associated)。
- 罕見:副甲狀腺功能亢進、結締組織疾病、HIV 感染。
- 流行病學:罕見,男女相同。超過半數發生於兒童/青少年期,最常見於上呼吸道感染之後(鏈球菌 Streptococcal 最常見)。成年糖尿病相關型為第二常見表現。
- 致病機轉:第 1 型膠原蛋白 (Type 1 collagen) 與玻尿酸鹽 (hyaluronate) 為增加的主要纖維母細胞產物;確切機轉不明。血清因子/免疫球蛋白可能刺激纖維母細胞(副蛋白血症病人血清在體外 in vitro 可增加培養纖維母細胞的膠原蛋白產生)。
- 臨床表現:頸部、肩部、上背部皮膚急性發作的非凹陷性硬結 (nonpitting induration),可侵犯顏面與手臂;皮膚光滑、呈蠟樣 (waxy)、毛囊開口明顯,可呈「橘皮樣 (peau d’orange)」。觸診較視診更易察覺;受累與未受累皮膚交界融合難辨。罕見侵犯食道、骨髓、神經、肝臟、唾液腺。可有關節/舌/眼活動受限、肌肉無力或觸痛。
- 組織病理:低倍鏡下呈非漸細的「方形 (square)」外觀,真皮比例顯著增加;外泌汗腺單位 (eccrine units) 減少或位置升高;纖維母細胞數量與形態正常;膠原束略增厚並被細微黏液沉積分隔;黏液染色(阿爾辛藍 Alcian blue、膠體鐵 colloidal iron)辨識黏液。
- 預後/病程:感染後型通常 1 至 2 年內消退;糖尿病相關型遷延但血糖控制改善可好轉;免疫球蛋白病變相關 (gammopathy-associated) 型較慢性、可能對多種療法有抗性。
- 治療:抗生素不影響感染後型病程;治療相關疾病(改善血糖控制、治療副蛋白血症)可能改善;紫外線 A1 光療 (UVA-1) 與補骨脂素加紫外線 A 光療 (psoralen plus UVA) 療效不一;甲胺喋呤 (methotrexate) 無益;曾對局部放射治療/電子束照射有反應;單株免疫球蛋白病變相關型曾對體外光分離術 (extracorporeal photopheresis) 與靜脈注射免疫球蛋白有反應。
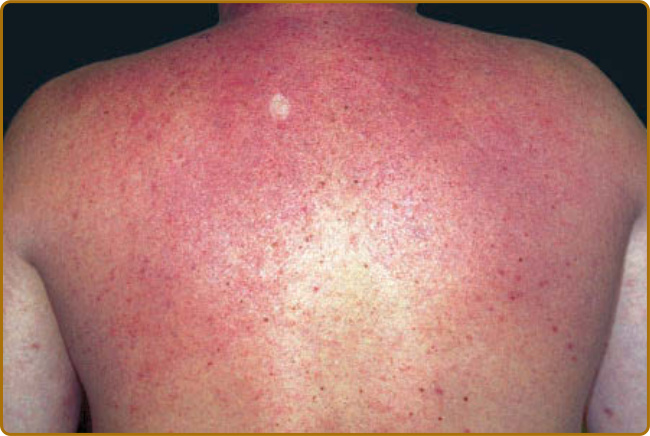
圖 67-1:硬腫病。背部皮膚蠟樣、非凹陷性硬結。
硬化黏液水腫 (Scleromyxedema)
- 定義:以真皮纖維化 (dermal fibrosis) 與黏液沉積症為特徵、甲狀腺功能正常的慢性、進行性病況;通常與副蛋白血症(典型為免疫球蛋白 G-kappa)相關。
- 臨床變異型:全身性融合性苔癬樣疹(即 scleromyxedema)、散在性丘疹疹(苔癬樣黏液水腫 lichen myxedematosus)、嬰兒/成人型丘疹性黏液沉積症、自癒性、肢端持續性、結節型、苔癬樣斑塊、蕁麻疹樣斑塊等。
- 流行病學:罕見,男女相同,中年至晚年發病。
- 致病機轉:不明。多數病人有單株副蛋白血症,典型為意義未明的單株免疫球蛋白病變 (MGUS),罕見符合多發性骨髓瘤標準。可見過量玻尿酸鹽產生與纖維母細胞增生,但缺乏單株蛋白直接作用於纖維母細胞的證據;間接機轉包括循環細胞激素、發炎介質與遷移自血液的纖維母細胞前驅細胞譜系。
- 臨床表現:全身性苔癬樣疹由四肢/軀幹散布的眾多細小(1 至 3 mm)丘疹組成;scleromyxedema 以融合性苔癬樣斑塊表現,可有紅斑或過度色素沉著。多數侵犯顏面致「牛樣面容 (bovine facies)」;軀幹四肢受累常致柔韌度與活動範圍下降。多數可辨識意義未明單株副蛋白血症,通常為 IgG-λ 型(局部變異型較少見);進展為多發性骨髓瘤罕見。可合併肌肉無力、攣縮、限制性肺病、肺高壓、食道蠕動障礙及神經學疾病(癲癇發作、腕隧道症候群、周邊神經病變、精神病等)。依定義甲狀腺功能正常。
- 組織病理:表淺至中真皮黏液沉積混雜纖維母細胞增生;Rongioletti 等人於 44 位中 10 位見上皮樣組織球間質增生、呈環狀肉芽腫樣 (granuloma annulare-like) 型態;發炎甚少顯著。與腎源性全身性纖維化 (nephrogenic systemic fibrosis) 組織學高度相似,但脂肪小葉間隔 (pannicular septae) 為區分重點:後者一致受累、scleromyxedema 從不受累;且 scleromyxedema 局限真皮上半部。
- 預後/病程:局部「自癒性 (self-healing)」型多自限;但部分局部型曾合併內臟侵犯或進展(非典型丘疹性黏液沉積症),建議追蹤。全身/系統性 scleromyxedema 慢性進行性、預後不良;呼吸衰竭、腦部疾病與感染常導致逐漸衰退與死亡。
- 治療:美法侖 (Melphalan) 數十年來使用、療效不一。據報告具療效者:糖皮質素 (glucocorticoids)、靜脈注射免疫球蛋白、沙利竇邁 (thalidomide)、體外光分離術、干擾素-α (interferon-α)、合併化學治療、補骨脂素加紫外線 A。10 位以靜脈注射免疫球蛋白治療者有 8 位達部分至完全緩解。自體周邊血液幹細胞移植 (autologous peripheral blood stem cell transplantation) 已使重度病人獲顯著緩解。
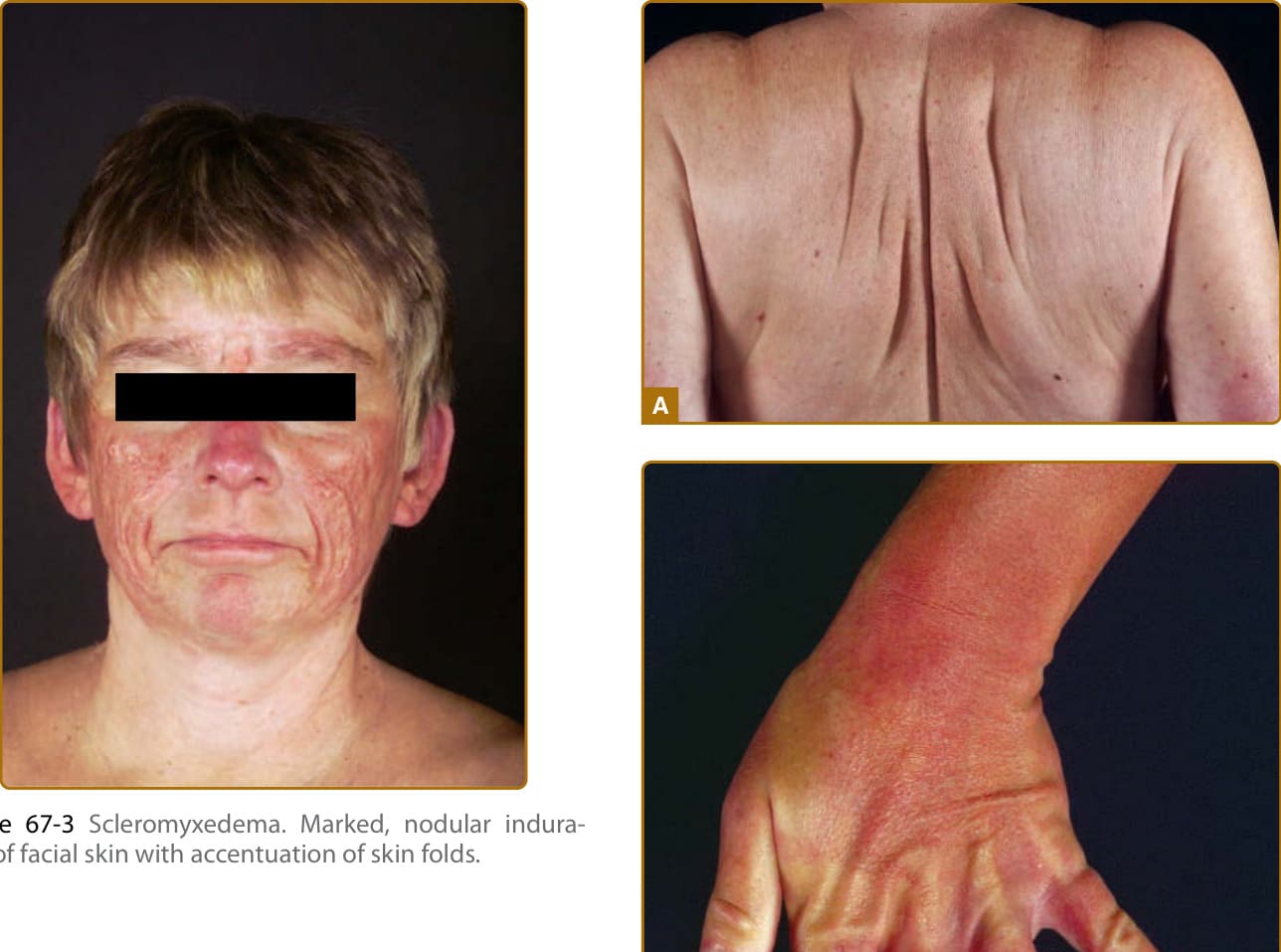
圖 67-3:硬化黏液水腫。顏面結節性硬結,皮膚皺褶加深(牛樣面容)。
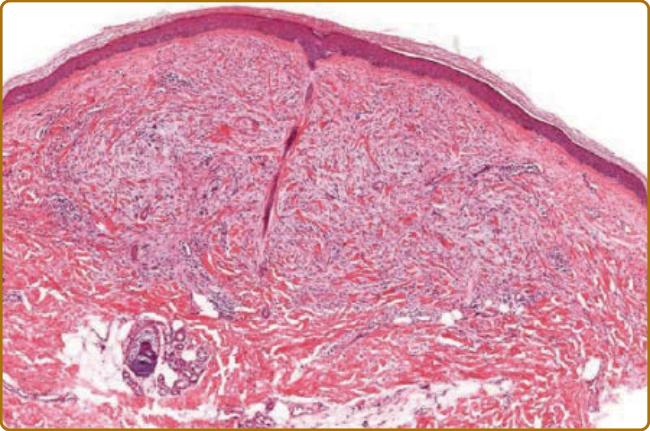
圖 67-5:硬化黏液水腫。表淺至中真皮纖維增生與黏液增加。