Scleredema and Scleromyxedema
10
Skin disorders that are characterized by increased mucin content, excessive collagen deposition, or fibrocyte hyperplasia are designated as mucinoses, sclerosing disorders, or fibrosing disorders, respectively. As the fibrocyte (fibroblast) is the cellular component of their pathogenesis, these conditions are aptly viewed as various forms of “fibropathy.” In some conditions, one fibrocyte product predominates, such as excessive mucin production in pretibial myxedema or collagen deposition in scleroderma. In others conditions there is a combination of excessive mucin and collagen deposition (scleredema) or excessive mucin deposition and fibrocyte hyperplasia (scleromyxedema and nephrogenic systemic fibrosis). Hence, the nosology of some of these conditions does not accurately describe what is observed histopathologically. For example, “fibromyxedema” more accurately depicts the histopathogenesis of scleromyxedema, as scleromyxedema implies a combination of sclerosis and mucinosis, which more accurately describes scleredema. Fibroblasts are derived from CD34-positive hematopoietic precursors that originate in the bone marrow and populate the skin during development and wound repair. The chief role of the skin fibroblast is
SCLEREDEMA
AT-A-GLANCE
■ Acute skin induration characterized by dermal mucinosis and mild sclerosis.
■ Disease associations:
■ Postinfectious scleredema of youth.
■ Diabetes mellitus–associated scleredema of adulthood.
■ Paraproteinemia-associated scleredema.
■ Rare associations: hyperparathyroidism, connective tissue disease, HIV infection.
■ Histopathology: pandermal thickening with broad collagen bundles separated by mucin deposits, chiefly hyaluronate.
HISTORICAL ASPECTS
HISTORICAL ASPECTS
Scleredema was described by Buschke in 19021 and is also designated as scleredema of Buschke and scleredema adultorum, although the latter is a misleading term because children or adolescents can develop the condition.
to deposit collagen, elastin, and ground substance, the major constituents of the dermis and pannicular septae and the substrate to which the epidermis and skin appendages attach. Stimuli that cause fibrocytes to overproduce mucin or collagen, or to proliferate are protean and broad ranging. Infectious triggers, inflammatory states, diabetes mellitus, drugs, heavy metals (eg, gadolinium), genetic mutations, and toxins (eg, tainted rapeseed oil and l-tryptophan in eosinophilia myalgia syndrome), as well as specific mediators, such as eicosenoids, growth factors, cytokines, chemokines, immunoglobulins-paraproteins, and other circulating factors, have been implicated, but the precise mechanisms that result in the varied forms of fibrocyte pathology (fibropathy) are poorly characterized. Some of these stimuli may recruit bone-marrow derived fibrocyte precursors to the skin; others may act directly on fibrocytes already residing in the skin. This chapter focuses on the distinctive conditions of scleredema and scleromyxedema (Table 67-1.). Other diseases relevant to cutaneous fibrocytes are discussed separately in other chapters and are listed in Table 67-2.
■ Clinical course:
■ Postinfectious: usually resolves in 1 to 2 years.
■ Diabetes mellitus–associated: may improve with better control of diabetes, but a prolonged course is expected.
■ Paraprotein associated: often more chronic although remissions observed with specific treatments.
■ Treatment: treat underlying disease; UVA-1; psoralen plus ultraviolet A light.
EPIDEMIOLOGY
EPIDEMIOLOGY
Scleredema is rare, although the precise incidence and prevalence are unknown. Men and women are affected with equal frequency. More than half of cases of scleredema occur in childhood or adolescence, most commonly after an upper respiratory tract infection.2
10
CONDITION EPIDEMIOLOGY ASSOCIATED CONDITIONS CLINICAL MANIFESTATIONS HISTOLOGIC FINDINGS COURSE/ PROGNOSIS TREATMENT OPTIONS
Scleredema
Childhood or adolescence Adulthood
Postinfectious
Acute onset, nonpitting neck induration; shoulders, upper back, face, and arms may be affected. Affected skin feels smooth and waxy.
Diabetes mellitus Paraproteinemia Also hyperparathyroidism, connective tissue disease, HIV
Adulthood
Scleromyxedema
Mid to late
“Squared” appearance on low-power microscopy; more dermis in affected skin compared to adjacent unaffected skin; decreased number/higher placement of eccrine glands; mucin stains (Alcian blue, colloidal iron) can identify mucin deposits
Abates in 1-2 years Protracted course Chronic, resistant to therapies
Treatment of underlying condition (except for postinfectious scleredema); UVA-1 phototherapy may be effective
Paraproteinemia Generalized lichenoid
Scleromyxedema (also, lichen myxedematosus)
Mid to late adulthood, males and females equally affected
Superficial to mid-
Chronic,
Treatment of
Paraproteinemia
Generalized lichenoid eruption has minute papules on extremities and trunk Confluent, lichenoid plaques
(also, lichen myxedematosus)
adulthood, males and females equally affected
Superficial to mid- dermal fibroplasia and mucin deposition
Chronic, progressive course
Treatment of underlying cause
eruption has minute papules on extremities and trunk Confluent, lichenoid
plaques
Streptococcal infection is most commonly identified, but other infectious agents also have been implicated. Disease development in adulthood in association with adult-onset diabetes mellitus is the second most common presentation of scleredema. Uncommonly, scleredema occurs in association with paraproteinemia or multiple myeloma, and this association possibly is becoming more common as other causes diminish in frequency with better treatment and management of underlying disease.
PATHOGENESIS
PATHOGENESIS
Type 1 collagen and hyaluronate appear to be the major fibroblast products that are increased in scleredema-affected skin.3-5 The precise mechanism(s) for increased collagen and glycosaminoglycan production in scleredema is not known. Stimulation of fibroblasts by serum factors or immunoglobulin may be related to the pathogenesis of scleredema, particularly cases associated with infectious agents or a paraproteinemia. Serum from a patient with scleredema associated with paraproteinemia was able to increase collagen production by cultured fibroblasts in vitro.6
However, the factor(s) involved has not been elucidated. Other soluble circulating cytokines and small molecule mediators likely also play critical roles.
CLINICAL FINDINGS
CLINICAL FINDINGS
The clinical findings of scleredema are distinctive. An acute onset of nonpitting induration of neck,
1164
dermal fibroplasia and mucin deposition
progressive course
underlying cause
shoulders, and upper back skin may be followed by involvement of the face and arms. Characteristically, the affected skin appears smooth and waxy, with tense dermal induration and prominent follicular ostia, at times imparting a “peau d’orange” appearance (Fig. 67-1). In general, however, skin changes are better felt on palpation than seen. The skin of the upper trunk (especially the back) is a favored site for scleredema, but more widespread involvement may be observed. The affected and unaffected skin blend imperceptibly. Scleredema involving the esophagus, bone marrow, nerve, liver, or salivary glands has been described rarely.7,8 However, investigations to identify internal organ involvement by scleredema are infrequently pursued. Patients may report symptoms of restricted motion of joints, the tongue, or eyes, as well as weak or tender muscles.
HISTOPATHOLOGY
HISTOPATHOLOGY
Punch biopsies of affected skin reveal a nontapered (square) appearance on low power. The proportion of dermis in dramatically increased in comparison to adjacent nonaffected skin (Fig. 67-2A). A decreased number or higher placement of eccrine units may be appreciated. Fibroblasts are normal in number and morphology. The collagen bundles are slightly thickened and separated from each other by subtle deposits of mucin. Stains for mucin (Alcian blue, colloidal iron) are often used to identify the mucin deposits (Fig. 67-2B).
CHAPTER
Excessive Collagen Production by Fibroblasts—Sclerosing Disorders
■Scleroderma
63 64 129 124 40, 63 63
■Morphea
■Sclerodermoid graft-versus-host disease
■Sclerodermoid porphyria cutanea tarda
■Eosinophilic fasciitis
■Sclerodermoid drug reactions (bleomycin, epoxy resin, pentazocine)
■Lipodermatosclerosis (sclerosing panniculitis)
73, 148
■Collagenoma
136
Fibroblast Hyperplasia—Fibrosing Disorders
■Fibromatosis (Dupuytren contracture, Peyronie disease) 72
Excessive Collagen and Mucin Production by Fibroblasts— Scleromucinoses
■Scleredema 67
Fibroblast Hyperplasia and Excessive Mucin Production by Fibroblasts—Fibromucinoses
■Lichen myxedematosus and scleromyxedema
67
■Nephrogenic systemic fibrosis/ nephrogenic fibrosing dermopathy
133
■Toxic oil syndrome
64 40
■Eosinophilia-myalgia syndrome
Fibroblast Hyperplasia and Excessive Collagen Production by Fibroblasts—Fibrosclerosing Disorders
■Juvenile hyaline fibromatosis
■Gingival fibromatosis 72 72
Excessive Mucin Production by Fibroblasts—Mucinoses
■Pretibial myxedema
■Pretibial myxedema
137 137 137 62
137 137 137 62
■Generalized myxedema
■Generalized myxedema
■Localized myxedema
■Localized myxedema
■Reticular erythematous mucinosis and plaque-like mucinosis
■Reticular erythematous mucinosis and
plaque-like mucinosis
■Connective tissue disease
■Connective tissue disease
61, 62, 63, 66, 68, 69 146
61, 62, 63, 66, 68, 69 146
■Degos disease
■Degos disease
10
A
B
1165
10
PROGNOSIS AND CLINICAL COURSE
PROGNOSIS AND
CLINICAL COURSE
Postinfectious scleredema usually abates in 1 to 2 years. Scleredema associated with adult-onset diabetes tends to be protracted, although some patients appear to improve with better glucose control. Gammopathy-associated scleredema is more chronic and can be resistant to many therapies.
TREATMENT
TREATMENT
Antibiotics do not appear to affect the course of postinfectious scleredema. Treatment of the associated disease (eg, improved glucose control or treatment of paraproteinemia) may lead to improvement. Ultraviolet A1 phototherapy has been variably effective in some patients,9-11 as has psoralen plus ultraviolet A phototherapy.11,12 Treatment with methotrexate did not provide benefit in a series of cases of scleredema. Scleredema has responded to local radiotherapy or electron-beam irradiation. Scleredema associated with monoclonal gammopathy has responded to extracorporeal photopheresis and intravenous immunoglobulin.13
SCLEROMYXEDEMA
AT-A-GLANCE
AT A GLANCE
■ Chronic, progressive condition characterized by dermal fibrosis and mucinosis and normal thyroid function.
■ Usually associated with paraproteinemia (typically immunoglobulin G-kappa).
■ Clinical variants
■ Generalized, confluent lichenoid eruption (scleromyxedema).
■ Discrete papular (rarely nodular) eruption on the trunk or extremities (lichen myxedematosus).
■ Papular mucinosis of infancy.
■ Papular mucinosis of adulthood.
■ Self-healing papular mucinosis.
■ Acral persistent papular mucinosis.
■ Nodular variant.
■ Localized or generalized lichenoid plaques (but distinct from plaque-like mucinosis/reticulated erythematous mucinosis).
■ Urticarial plaques
■ Histopathology: superficial to mid-dermal fibroplasia and mucin deposition.
1166
■ Clinical course: chronic progressive course and poor outcome in generalized, systemic cases; localized and self-healing forms have a better prognosis.
■ Treatment: correct paraproteinemia: through clonal plasma cell targeted therapies, intravenous immune globulin, autologous stem-cell transplant.
EPIDEMIOLOGY
EPIDEMIOLOGY
Lichen myxedematosus/scleromyxedema is a rare disease afflicting men and women with equal frequency and symptom onset in mid to later life.14-16
PATHOGENESIS
PATHOGENESIS
The pathogenesis of scleromyxedema is unknown. Most patients with scleromyxedema have a monoclonal paraproteinemia, typically designated as monoclonal gammopathy of undetermined significance. Rare patients meet criteria for multiple myeloma. Excessive hyaluronate production and fibroblast proliferation are observed, but support for a direct effect of the monoclonal protein on fibroblasts is lacking.17,18 Indirect mechanisms appear to include circulating cytokines, inflammatory mediators, and fibroblast precursor cell lineages that migrate from the blood, take up residence in the dermis and, in more extensive cases, in other tissues, and synthesize mucin.
CLINICAL FINDINGS
CLINICAL FINDINGS
Several distinct clinical presentations, and hence nosologic designations have been recognized. Although there is considerable clinical overlap and patients may show progression from limited to widespread involvement, it is useful to distinguish localized from generalized disease as well as patients that present with discrete papules from those with confluent plaques. The generalized lichenoid eruption consists of numerous minute (1 to 3 mm) papules scattered on the extremities and the trunk. Scleromyxedema presents with confluent lichenoid plaques. Individual lesions and plaques may exhibit marked erythema or hyperpigmentation. The face is involved in most cases, resulting in significant deformity, “bovine facies” (Fig. 67-3). The trunk and extremities are usually affected (Fig. 67-4A) and often results in decreased flexibility and range of motion in the involved areas (Fig. 67-4B). A monoclonal paraproteinemia of undetermined significance, usually immunoglobulin G λ (IgG-λ) type
is identified in most patients. Paraproteinemia is less common in localized variants. Progression to multiple myeloma occurs rarely. Muscle weakness, contractures, restrictive lung disease, upper airway involvement, pulmonary hypertension, esophageal dysmotility, and neurologic disorders (seizures, motor impairment, carpal tunnel syndrome, depression, memory loss, aphasia, peripheral neuropathy, and psychosis) have been reported in association with scleromyxedema. Thyroid function is normal by definition.
HISTOPATHOLOGY
HISTOPATHOLOGY
The classical histopathologic findings in scleromyxedema consist of superficial to mid-dermal mucin deposition with admixed fibroblast proliferation (Fig. 67-5). Recently, Rongioletti et al reported an interstitial proliferation of epithelioid histiocytes imparting a granuloma annulare-like pattern in 10 of 44 patients studied.19 A mild perivascular and interstitial T-cell infiltrate is also seen, but inflammation is rarely a prominent feature. Occasional multinucleated histiocytes with or without elastophagocytosis may be identified in biopsies of scleromyxedema. A highly unusual histologic presentation with a marked dermal granulomatous infiltrate was observed in association with fibromucinous deposition in a 56-year-old male with scleromyxedema.20
Moreover, scleromyxedema is histologically distinct from other mucinoses as well as sclerosing and
10
A
B
1167
10
fibrosing conditions. Nephrogenic systemic fibrosis (previously designated nephrogenic fibrosing dermopathy) shows close histologic resemblance to scleromyxedema. However, the pannicular septae are the focus for histologic distinction: they are consistently involved in nephrogenic systemic fibrosis and never involved in scleromyxedema. Moreover, scleromyxedema tends to be restricted to the upper half of the dermis and nephrogenic systemic fibrosis becomes more prominent in the lower dermis and then extends to pannicular septae.
PROGNOSIS AND CLINICAL COURSE
PROGNOSIS AND CLINICAL
COURSE
Localized, “self-healing” variants are usually confined to the skin and are self-limiting. However, prospective followup and caution is advised as some cases fitting criteria for localized disease have been associated with internal organ involvement or progression to more generalized disease (so-called atypical papular mucinosis). Scleromyxedema usually follows a chronic and progressive clinical course with more systemic organ and tissue involvement, and a poor outcome is expected. Respiratory failure, cerebral disease, and infection usually lead to a gradual decline and death.
TREATMENT
TREATMENT
Melphalan had been used for decades to treat scleromyxedema with variable efficacy.21-23 Other therapies directed toward quantitatively reducing or ameliorating the effects of the paraproteinemia have shown variable results. Agents or therapies with reported efficacy include: glucocorticoids,24 intravenous immunoglobulin,25 thalidomide,26 extracorporeal photophoresis,27 interferon-α,28 combination chemotherapy,29 and psoralen and ultraviolet A.30 Partial to complete remission was achieved in 8 of 10 patients treated with intravenous immunoglobulin.31 Autologous peripheral blood stem cell transplantation has resulted in dramatic remission of scleromyxedema in patients with severe disease.32
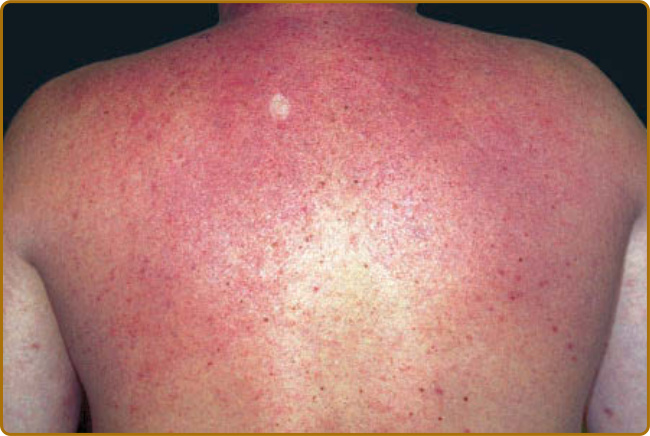
Figure 67-1 Scleredema. Waxy, nonpitting induration of back skin.
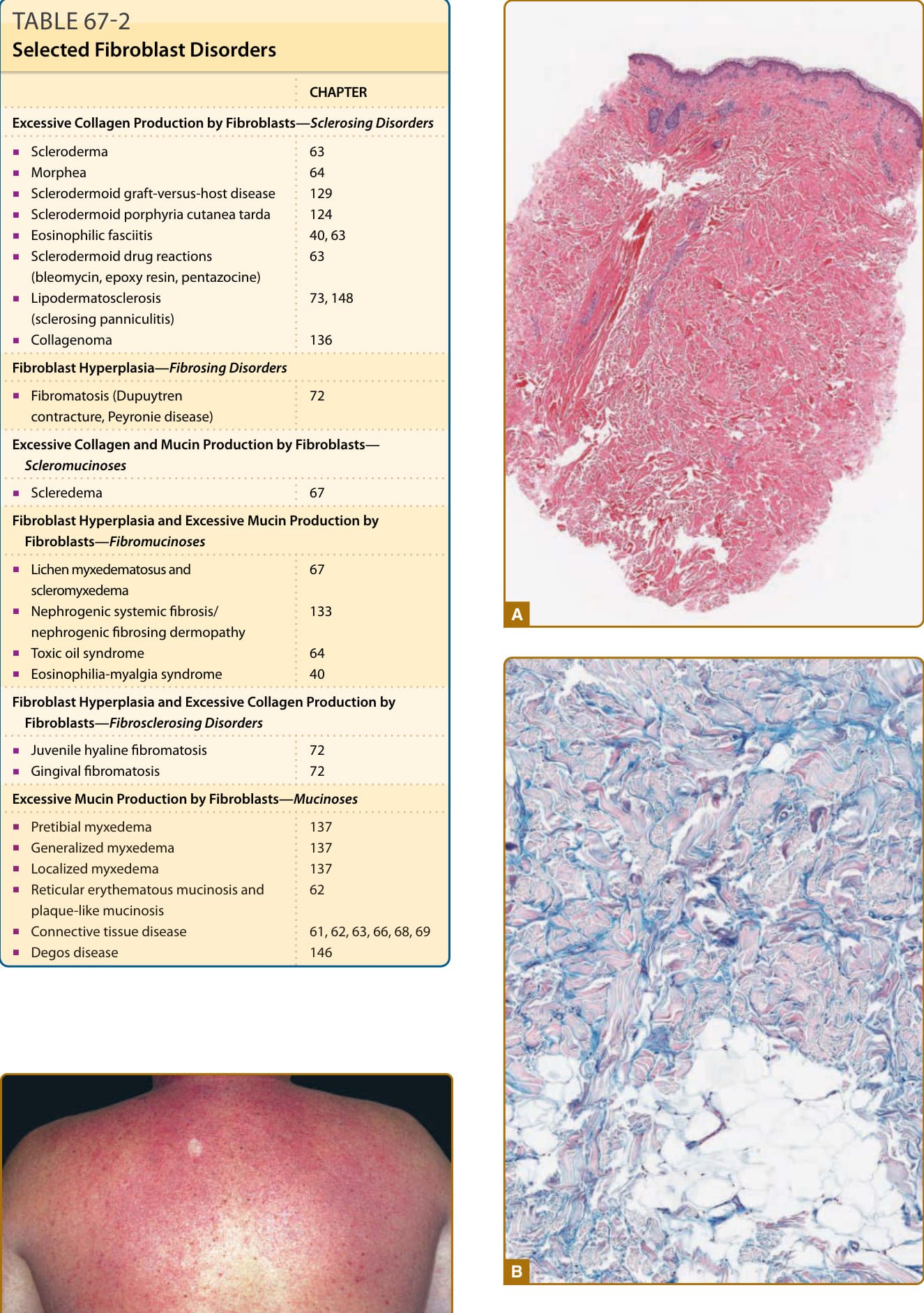
Figure 67-2 A, Scleredema. Thickened dermis characterized by enlarged collagen bundles separated by clear spaces (×15 original magnification). B, Scleredema. Mucin deposition identified in clear spaces by acid mucopolysaccharide stain (×80 original magnification).
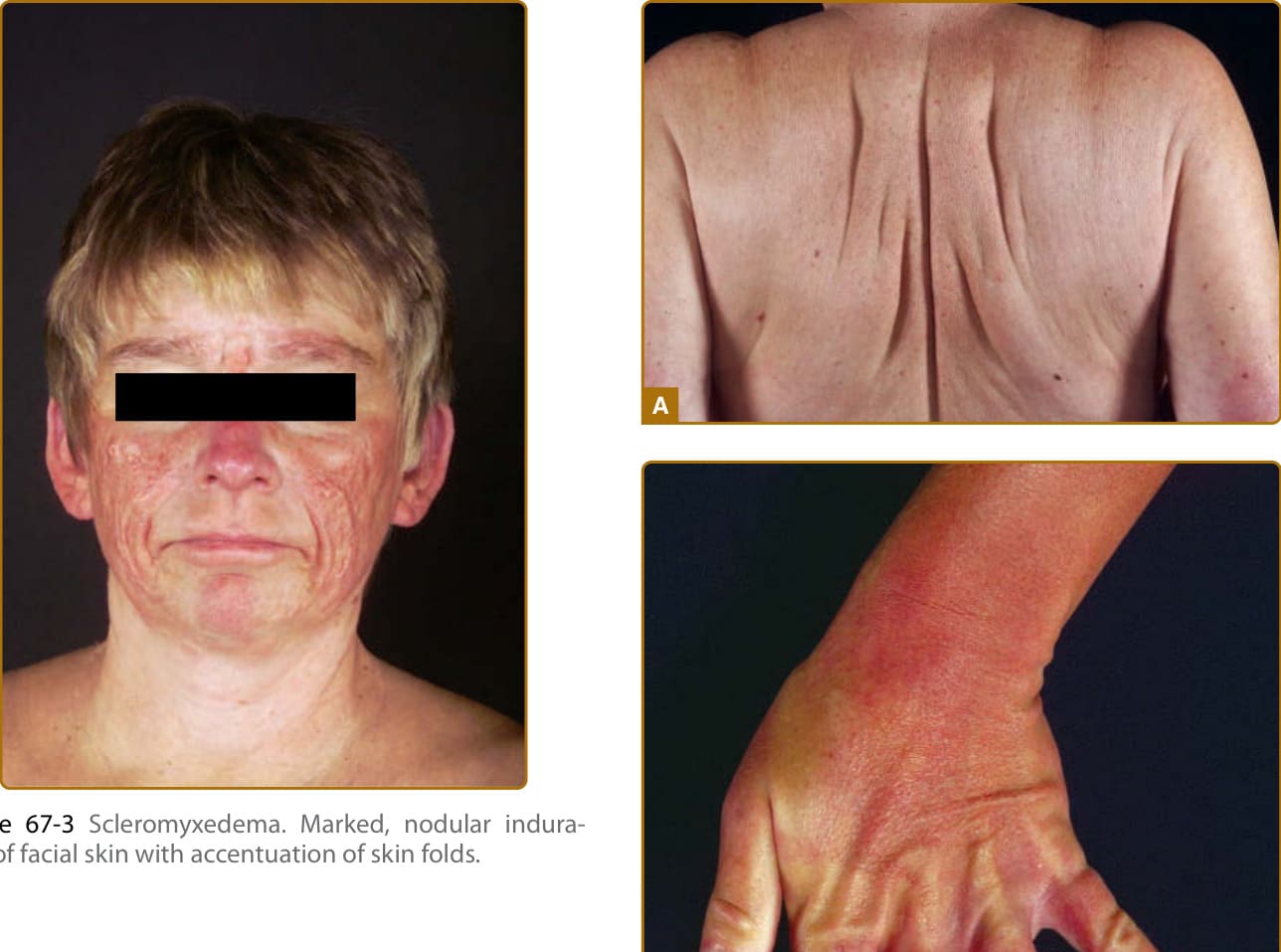
Figure 67-3 Scleromyxedema. Marked, nodular induration of facial skin with accentuation of skin folds.
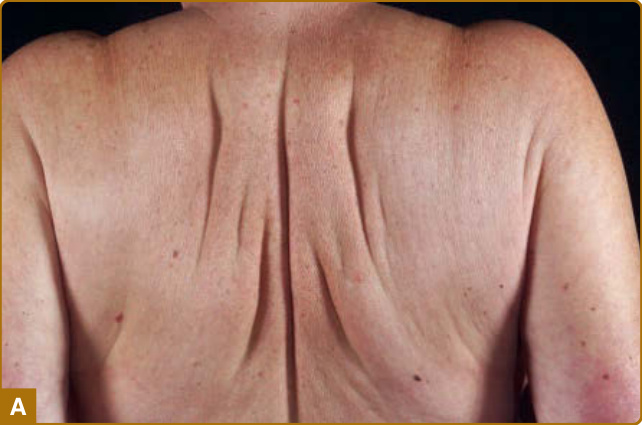
Figure 67-4 A, Scleromyxedema. Thickened bound-down skin of the back. B, Scleromyxedema. Confluent lichenoid papules and indurated skin of the hand and wrist.
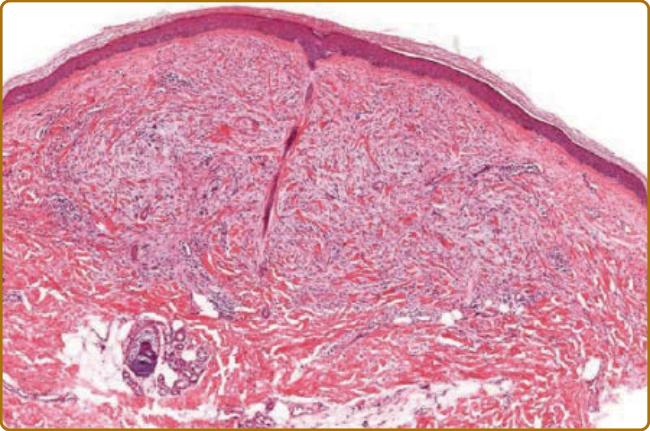
Figure 67-5 Scleromyxedema. Superficial to middermal fibroplasia and increased mucin (×40 original magnification).
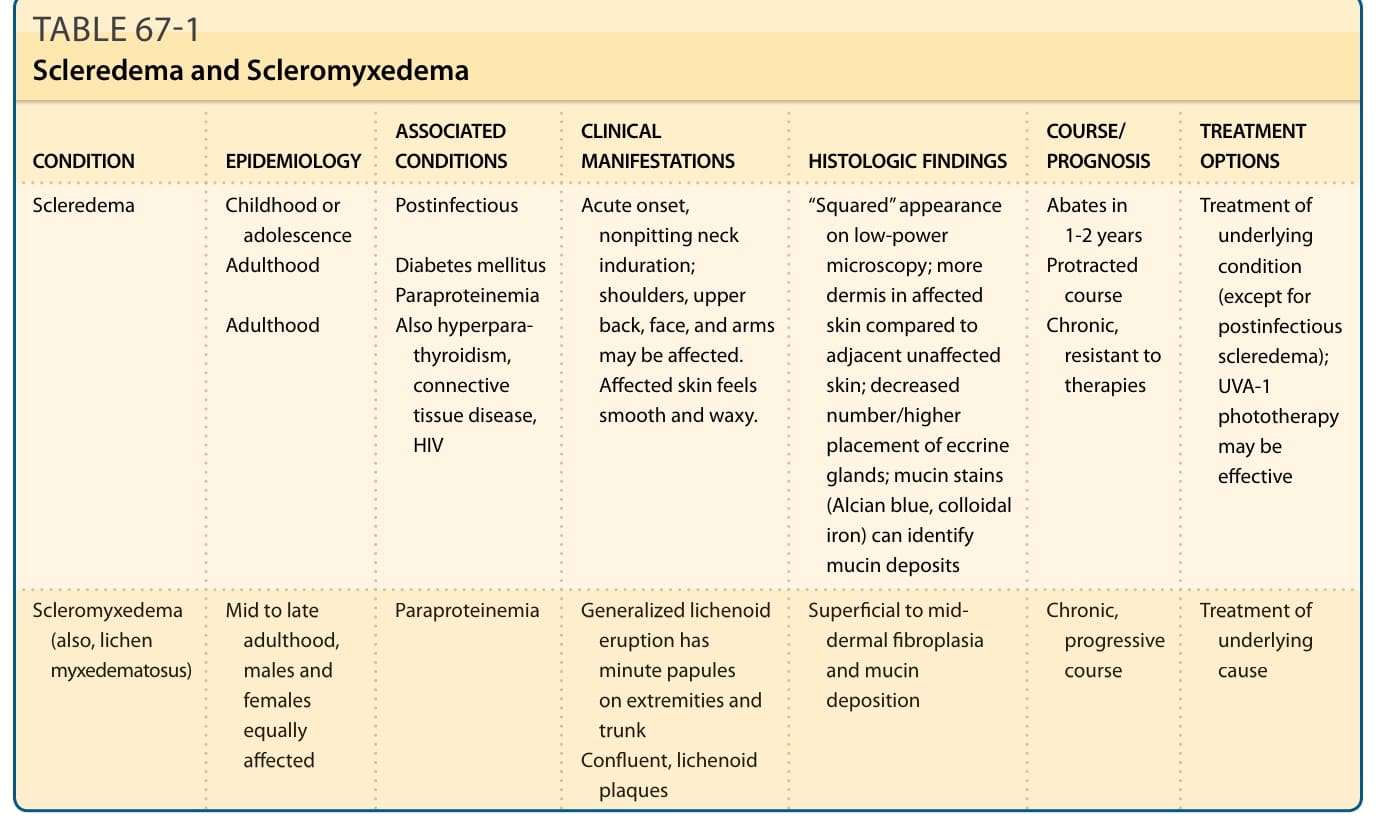
TABLE 67-1 Scleredema and Scleromyxedema
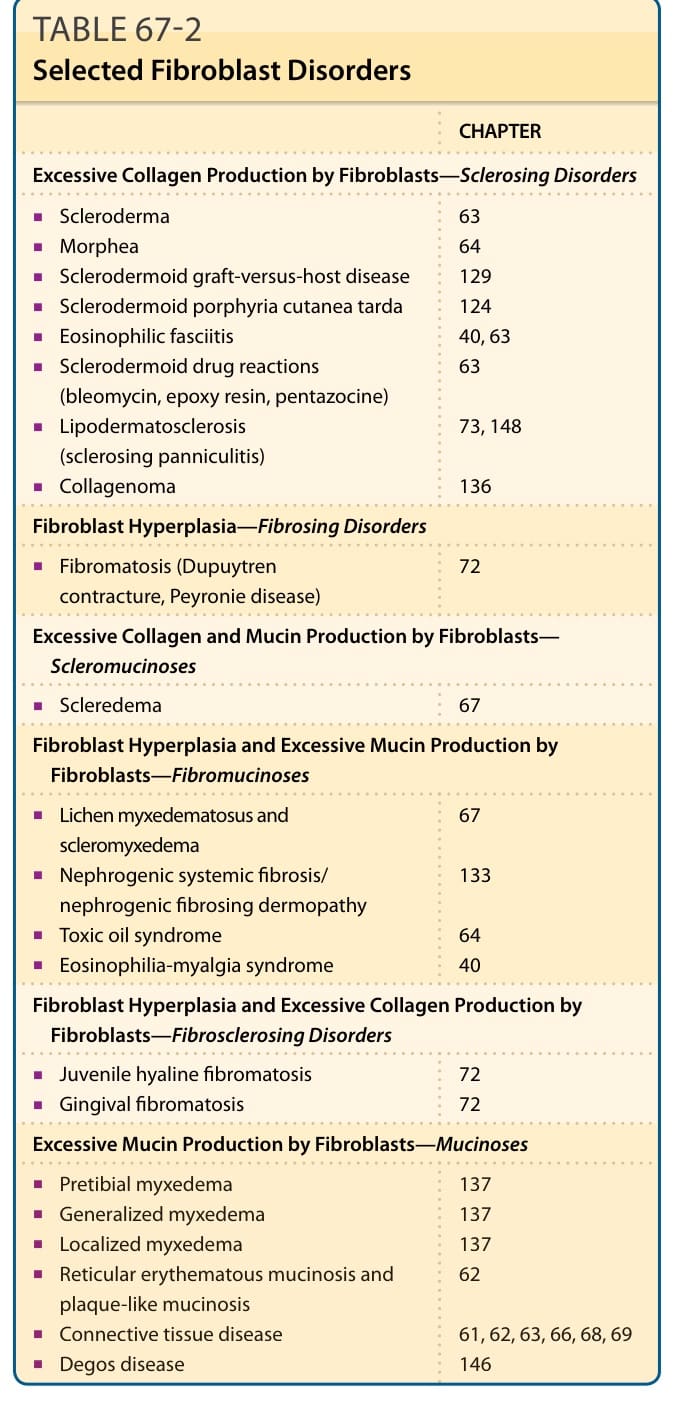
TABLE 67-2 Selected Fibroblast Disorders