全身性硬化症 (Systemic Sclerosis) 精華筆記
定義與流行病學
- 硬皮症(scleroderma,全身性硬化症 [systemic sclerosis, SSc])是一種多系統自體免疫疾病,特徵為自體免疫過程、血管內皮細胞損傷 (vascular endothelial cell injury)、發炎 (inflammation) 與廣泛纖維母細胞活化 (activation of fibroblasts)。
- 最常受累器官:皮膚、食道、肺、心臟與腎臟。
- 女性較常受累,女男比 3:1 至 14:1;發病年齡 30 至 50 歲,男性發病較早。
- 為罕見病,但在所有自體免疫風濕性疾病中具最高的疾病特異性死亡率 (case-specific mortality);輕症常未被診斷。
分類與亞型
- 幾乎總是存在的特徵:雷諾現象 (Raynaud phenomenon, RP) 與皮膚硬化;皮膚硬化的範圍界定主要亞群。
- 分類標準:1980 年 ACR 標準(敏感度 97%、特異度 98%)已較舊;新版 ACR/EULAR 標準採評分系統,納入異常甲褶微血管、指尖病灶與自體抗體,可早期診斷。1988 年 LeRoy 提出局限型 vs 瀰漫型分類,至今仍廣泛使用。
- 瀰漫型皮膚 SSc (diffuse cutaneous SSc):進行性、RP 通常在皮膚增厚發病後 1 年內出現;軀幹、顏面、上臂、大腿快速受累;常見抗 Scl-70(抗拓樸異構酶-I antitopoisomerase-I)或抗 RNA 聚合酶 III 抗體;較易出現肺纖維化、心臟受累與硬皮症腎危象。
- 局限型皮膚 SSc (limited cutaneous SSc):長期既存 RP 病史;皮膚變化局限於膝、肘遠端(含顏面);50% 至 70% 表現抗著絲點抗體 (anticentromere antibodies),常合併孤立性肺動脈高壓 (isolated PAH)。傳統縮寫 CREST(calcinosis、RP、esophageal dysmotility、sclerodactyly、telangiectasias)即指此型。
- 早期(未分化)SSc:RP 陽性加至少 1 項額外特徵,尚未符合 ACR 標準。
- 無硬皮的 SSc (SSc sine scleroderma):極少數(1.5%),有血管、免疫與器官纖維化特徵但無皮膚硬化。
- SSc 重疊症候群 (overlap syndrome):兼具 SSc 與另一自體免疫風濕病特徵;多呈高效價抗 U1RNP、抗 nRNP、抗纖維蛋白原或抗 PmScl 抗體。包含混合性結締組織病 (MCTD,高效價抗 U1RNP,預後較佳、對抗發炎治療反應良好) 與肌炎重疊型 (Pm-Scl 抗體、技工手 mechanic hands、早發皮下鈣化)。
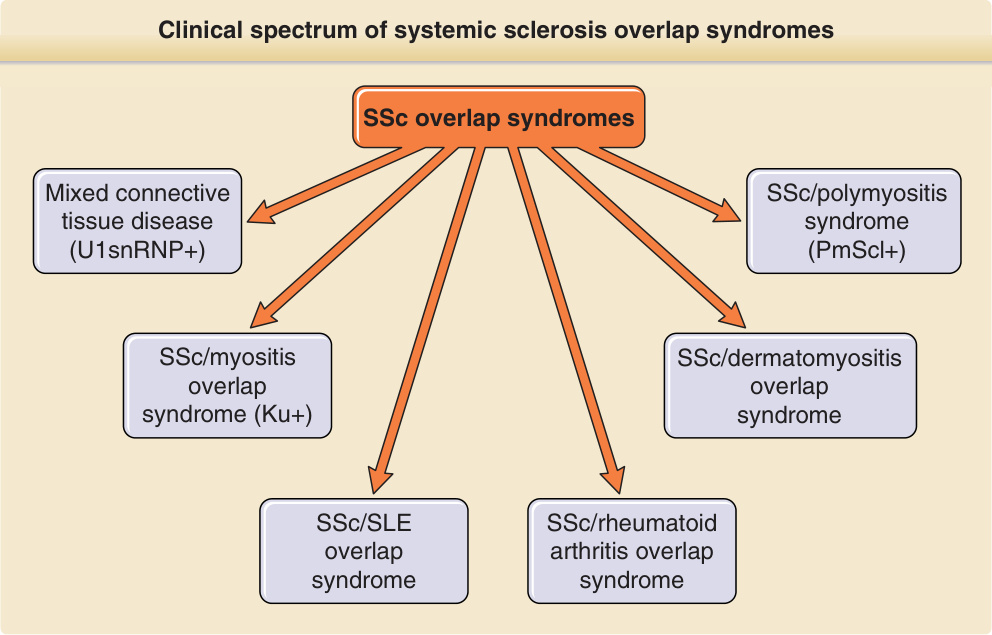
圖 63-3:SSc 重疊症候群的臨床譜。
器官表現
- 雷諾現象 (RP):見於 >90% 病人,常為本病首發、可早於本病數年;為手指/腳趾小動脈反覆血管痙攣 (vasospasm),由寒冷或情緒壓力誘發;典型三相性(蒼白/缺血 → 反應性充血 → 發紺)。
- 皮膚受累(主要特徵 cardinal feature):最先見於手指與手;先非凹陷性水腫(腫脹手指 puffy fingers),後硬化與皮膚增厚(指端硬化 sclerodactyly)。顏面:毛細血管擴張 (telangiectasias)、鳥喙狀鼻、口裂縮小 (microstomia)、口周放射狀皺褶、面具樣外觀。其他:皮膚鈣質沉著症 (calcinosis cutis,好發壓力點)、鹽和胡椒狀色素變化、少汗/無汗。
- 指端潰瘍 (digital ulceration):約 50% 病人某時點出現,為結構性血管疾病主要外部特徵;併發症含嚴重指端缺血、甲溝炎、感染、壞疽、骨髓炎、指腹喪失或截肢。
- 心肺:最常為纖維化與肺動脈高壓 (PAH)。PAH 目前是 SSc 最常見的疾病相關死亡原因,最典型為局限型合併孤立性 PAH。心臟亦可因纖維化或發炎性心肌炎受累,致心律不整、傳導阻滯與心臟功能不全。
- 胃腸道 (>60%,最常見內臟受累):食道為主(吞嚥困難、逆流性胃灼熱);下食道括約肌減弱增食道炎風險,可致消化性狹窄、瘻管、巴瑞特食道(可進展為腺癌);胃排空延遲、胃竇血管擴張(須內視鏡偵測);腸道可見假性阻塞、吸收不良、腹瀉、便秘、營養不良。
- 腎臟:硬皮症腎危象 (SRC) 見於 5% 至 10% 病人,突發顯著高血壓(>140/90 mm Hg,或收縮壓/舒張壓升高 ≥30/≥20 mm Hg),伴血清肌酸酐上升、蛋白尿、血尿、血小板減少或溶血,繼以急性腎衰竭;多發生於病程前 12 個月。應避免腎毒性藥物與高劑量普賴鬆龍 (prednisolone, >7.5 mg/day)。
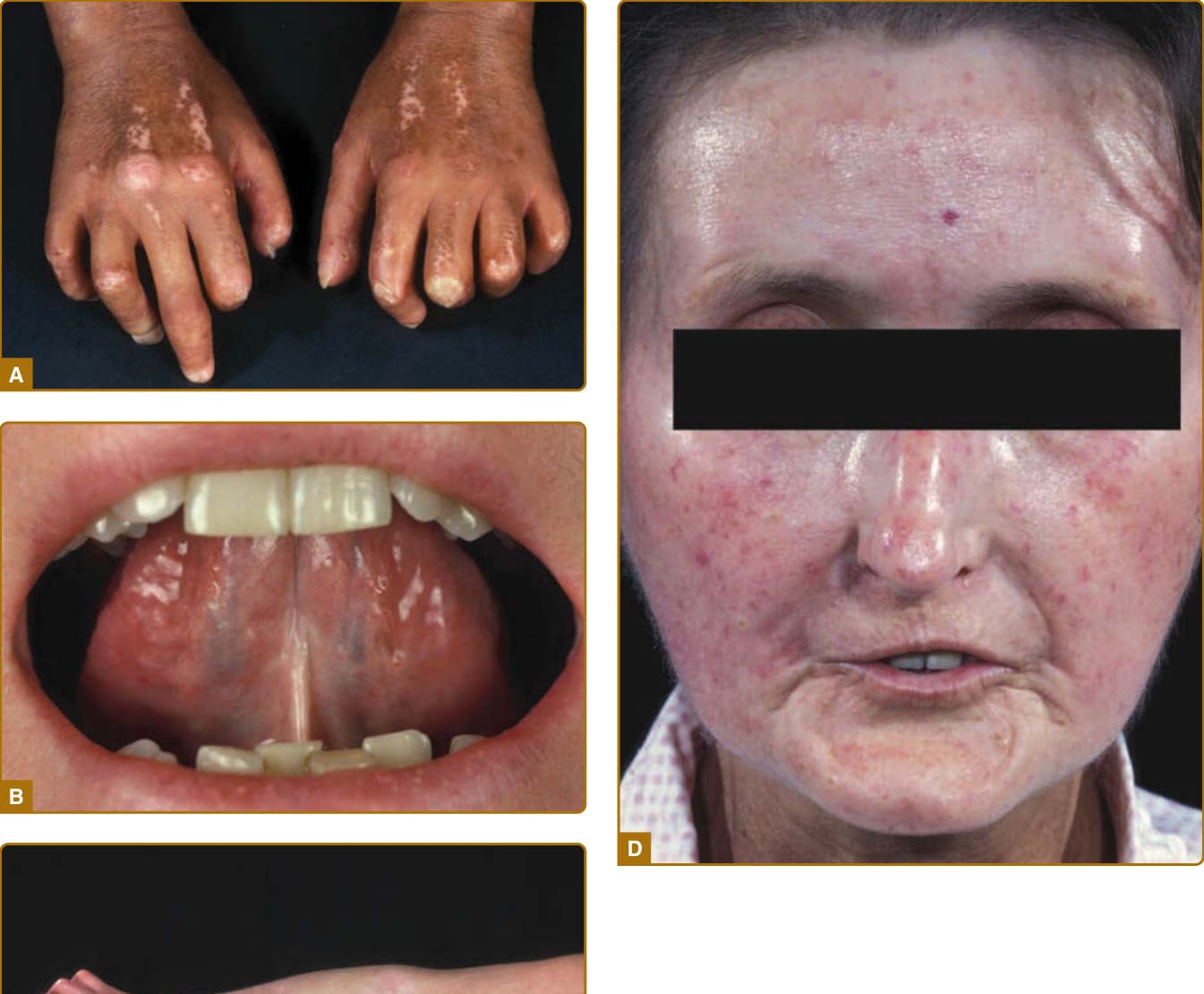
圖 63-1:瀰漫型皮膚 SSc 的廣泛皮膚受累(指端硬化、攣縮、口裂縮小、典型顏面相貌)。
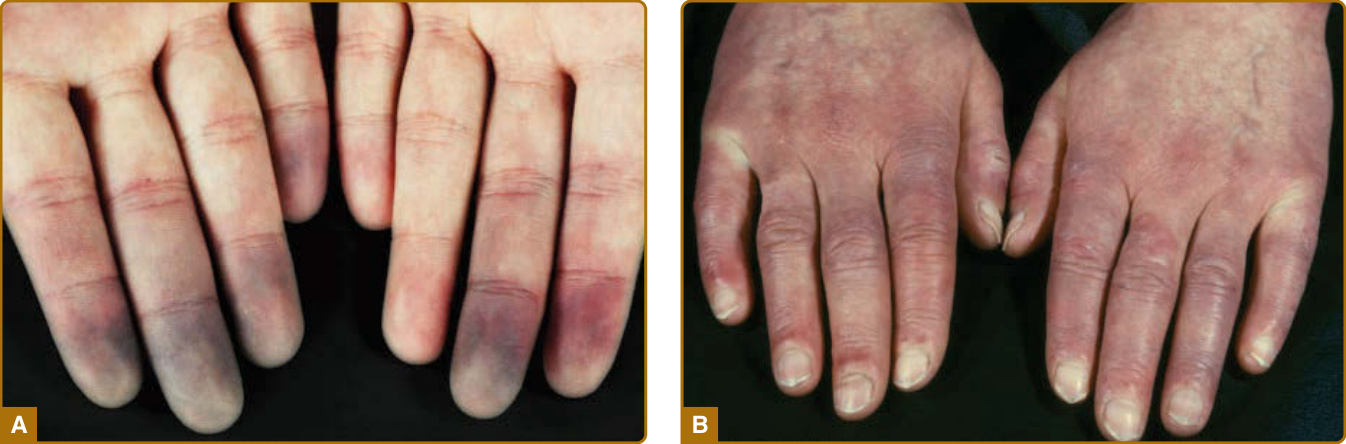
圖 63-2:早期疾病臨床特徵(雷諾現象、腫脹手指)。
致病機轉
- 三大面向:發炎、血管病變、纖維化,涉及內皮細胞、上皮細胞、纖維母細胞與淋巴細胞交互作用。
- 遺傳因子:一等親相對風險高 13 倍;自體抗體圖譜有強遺傳決定性,受 MHC 單倍型限制(如 HLA-DRB1∗1302/HLA-DQB1∗0604/0605 與抗纖維蛋白原相關);表觀遺傳機轉 (epigenetic mechanisms) 為重要額外因子。
- 環境因子:類硬皮症症候群與溶劑(氯乙烯、苯、甲苯、環氧樹脂)、藥物(博來黴素 bleomycin、卡比多巴、潘他唑新等)相關;矽肺症男性發展 SSc 風險約為未暴露者 190 倍。博來黴素引起的變化呈劑量依賴、停藥可逆。整體而言化學暴露僅佔一小部分。
- 血管病變 (vasculopathy):早期事件,可能為原發;特徵為血管收縮、內膜/外膜增生、發炎與血栓;血管舒張性與收縮性介質失衡導致組織缺氧;產生 PAH、SRC 與指端血管病變。
- 免疫事件:早期浸潤以單核球譜系為主,後 T 淋巴細胞(CD4+、寡克隆擴增、Th2 表型)佔主導;B 細胞透過分泌 IL-6 與 TGF-β 誘導 ECM 生成並產生自體抗體。部分以 RNA 聚合酶抗體為特徵者與惡性腫瘤相關。
- 纖維化 (fibrosis):關鍵為纖維母細胞誘導為活化的肌纖維母細胞 (activated myofibroblasts);關鍵纖維化性細胞激素含 TGF-β(核心角色)、結締組織生長因子、PDGF 與內皮素-1。

圖 63-5:SSc 的致病機轉示意圖。
組織病理學
- 真皮下三分之二與皮下纖維小梁纖維化,源自細胞外基質 (ECM) 過度沉積,最顯著為第 I 型與第 III 型膠原蛋白。
- 早期細胞期:膠原束蒼白、均質、平行皮面、腫脹,伴血管周圍淋巴細胞浸潤;晚期纖維化期:毛囊皮脂腺單位與外分泌腺消失、膠原束緊密堆積、表皮腳 (rete ridges) 消失、皮膚漸無血管化。
診斷
- RP/血管:甲褶微血管鏡檢查 (nailfold capillaroscopy) 為非侵入、最有用的診斷與預後工具,可分早期/活動期/晚期模式;加雷射都卜勒灌流影像。
- 皮膚硬化:改良式 Rodnan 皮膚評分 (modified Rodnan skin score),評估 17 個部位、分第 1~3 級(輕/中/重);輔以 20-MHz 超音波、MRI、皮褶測量計等。
- 心肺:至少每年追蹤肺功能檢查、心臟超音波、6 分鐘步行測試與 HRCT。肺一氧化碳瀰散能力(DLCO ≤75%)受損為肺纖維化與 PAH 的早期標記。右心導管術為 PAH 黃金標準:PAH 定義為靜息平均肺動脈壓 ≥25 mm Hg 且肺微血管楔壓 ≤15 mm Hg。
- 胃腸道:上消化道內視鏡(食道炎)、閃爍掃描或 24 小時 pH 測壓(蠕動)。
- 腎臟:定期血壓監測、尿液分析微電泳、肌酸酐清除率。
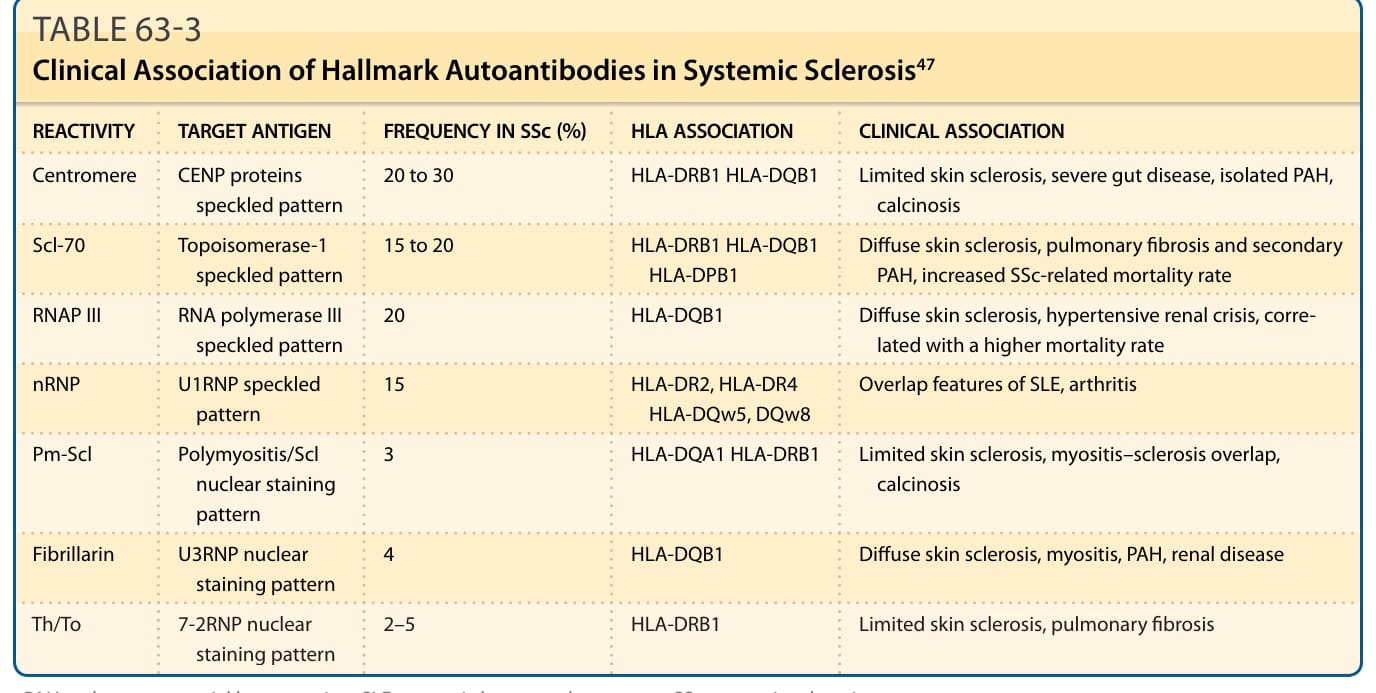
表 63-3:SSc 標誌性自體抗體的臨床關聯。
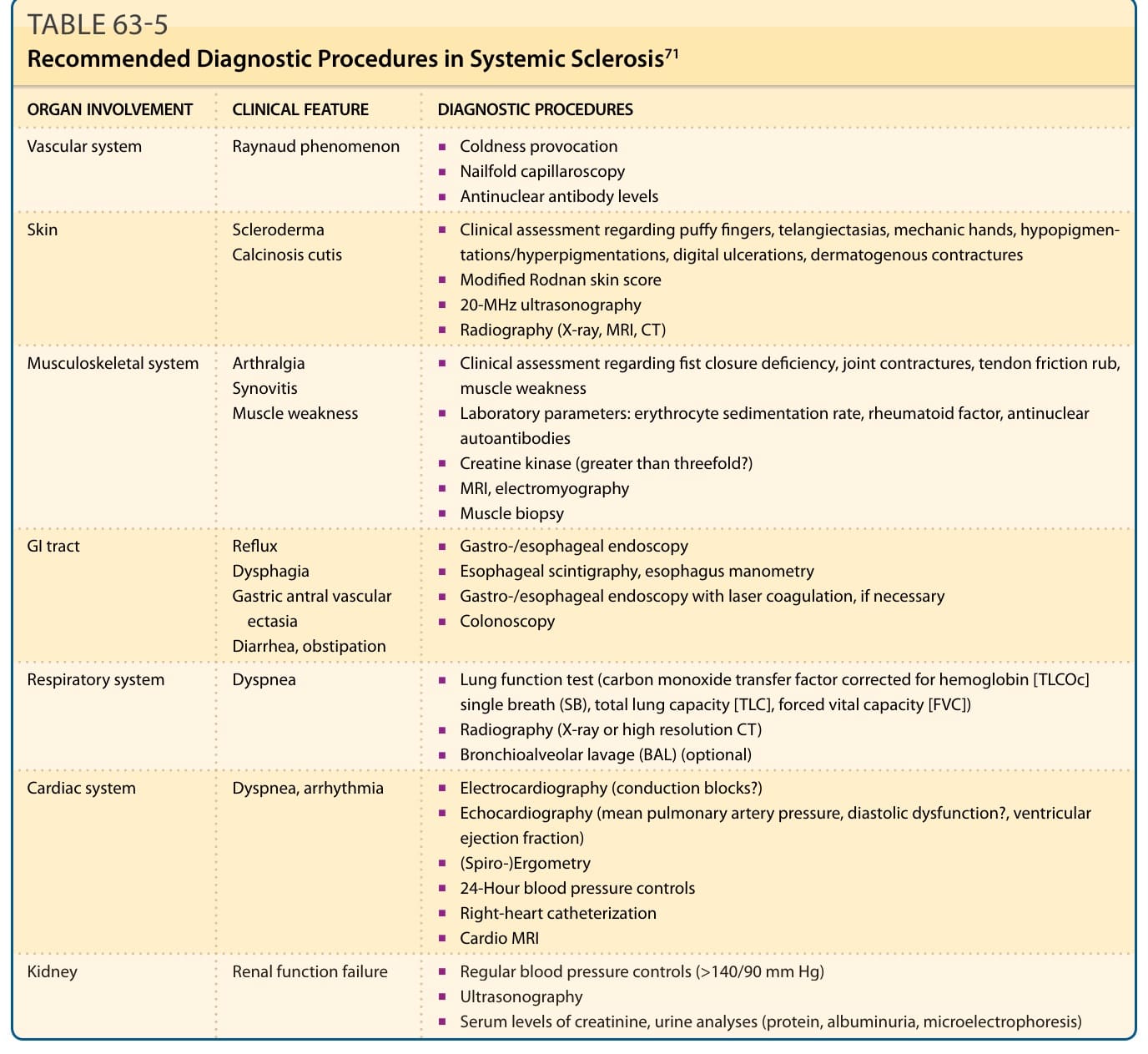
表 63-5:SSc 的建議診斷程序。
鑑別診斷
- SSc 診斷為臨床性,尚無正式診斷標準。須排除:局限性(局部性)硬皮症、嗜酸性筋膜炎、硬皮樣遺傳性皮膚病、慢性萎縮性肢端皮膚炎、環境誘發類硬皮症症候群、成人硬腫病 (scleroderma adultorum Buschke)、糖尿病性硬皮病、硬化性黏液水腫 (scleromyxedema)、腎源性纖維化皮膚病、遲發性皮膚紫質症、移植物抗宿主病、惡性腫瘤中的類硬皮症病灶等。
臨床病程與預後
- 取決於亞群。局限型:RP 早於其他器官表現多年,皮膚纖維化局限肢端,主要併發症為指端潰瘍與肺高壓。
- 瀰漫型:纖維化早發、快速擴散,肺(肺纖維化)、心、腎表現早出現並常決定預後;初期快速惡化,後期活性可降低、硬化皮膚可變軟、攣縮可減輕。
- 早期偵測與多學科管理 (multidisciplinary management) 可大幅改善預後。
處置
疾病修飾治療
- 一般性免疫抑制可改善皮膚受累與間質性肺疾病;最佳證據為環磷醯胺 (cyclophosphamide),黴酚酸酯 (mycophenolate mofetil, MMF) 已證實與口服環磷醯胺同等有效。
- 標準免疫抑制失敗時,利妥昔單抗 (rituximab) 可改善經選擇病人的病程;自體造血幹細胞移植用於某些經選擇病人。
- 抗纖維化治療仍具挑戰,目前尚無已證實的抗纖維化藥物(抗 TGF-β 抗體於早期研究有改善)。
指端血管病變
- 減少雷諾發作:避免尼古丁、擬交感神經藥物、情緒壓力與寒冷;保暖、暖手器、石蠟浴、減少手指創傷。
- 一線:鈣離子通道阻斷劑 (calcium channel blockers) 與血管收縮素 II 受體拮抗劑 (angiotensin II receptor antagonists)。
- 非腸道前列環素衍生物(尤其伊洛前列素 iloprost)有助癒合指端潰瘍,為嚴重指端缺血治療主軸;抗血小板藥(阿斯匹靈、氯吡格雷)亦用。
- 波生坦 (bosentan,口服雙重特異性內皮素受體拮抗劑) 在 2 大型試驗顯著減少新發指端潰瘍,但對已確立潰瘍癒合無正面效果。第 5 型磷酸二酯酶抑制劑(西地那非 sildenafil、他達拉非 tadalafil)亦用但缺前瞻試驗數據。
- 外科:指端微動脈鬆解術;腰椎交感神經切除術(下肢 RP/潰瘍)。
皮膚受累
- 物理治療與規律運動為關鍵;淋巴引流、局部類固醇、鈣調神經磷酸酶抑制劑、保濕乳霜。
- 全身:免疫抑制劑、短期全身性類固醇、光照治療(紫外線 A1、PUVA);UVA1 似能抑制纖維化與發炎。
- 乾癢皮膚:局部皮質類固醇、大麻素致效劑、辣椒素、潤膚劑、光照治療。
- 甲胺喋呤 (methotrexate) 於 2 個隨機試驗改善早期瀰漫型皮膚評分;環磷醯胺亦改善皮膚硬化;MMF 為平衡療效與副作用的選項;蛋白激酶抑制劑(如伊馬替尼 imatinib)結果參差且耐受性不佳。
心肺
- PAH(功能受限達 NYHA 第 III 級):多以口服內皮素受體拮抗劑(波生坦、安立生坦 ambrisentan)或第 5 型磷酸二酯酶抑制劑(西地那非、他達拉非)治療;進展則組合或加非腸道前列環素,伊洛前列素有吸入式給藥。
- SSc 肺纖維化 (SSc-PF):環磷醯胺於 2 個隨機對照試驗顯示適度扣除安慰劑後益處,為多數中心治療嚴重或進行性者之選;MMF 與環磷醯胺穩定肺功能療效相當且副作用較佳。波生坦於大型研究未優於安慰劑。
硬皮症腎危象 (SRC)
- 約三分之二就診病例需腎臟替代療法,約半數最終可停止透析(恢復可發生於腎危象後 >24 個月,故腎移植決定應延後)。
- 最關鍵:迅速辨識並治療顯著高血壓、啟動血管收縮素轉化酶抑制劑 (ACE inhibitor);為醫療急症。引入 ACE 抑制劑前,已確立 SRC 的 12 個月死亡率 >90%。
- 食道症狀:質子幫浦抑制劑與增加下食道括約肌張力藥(如多潘立酮 domperidone);小腸細菌過度生長用廣效抗生素。
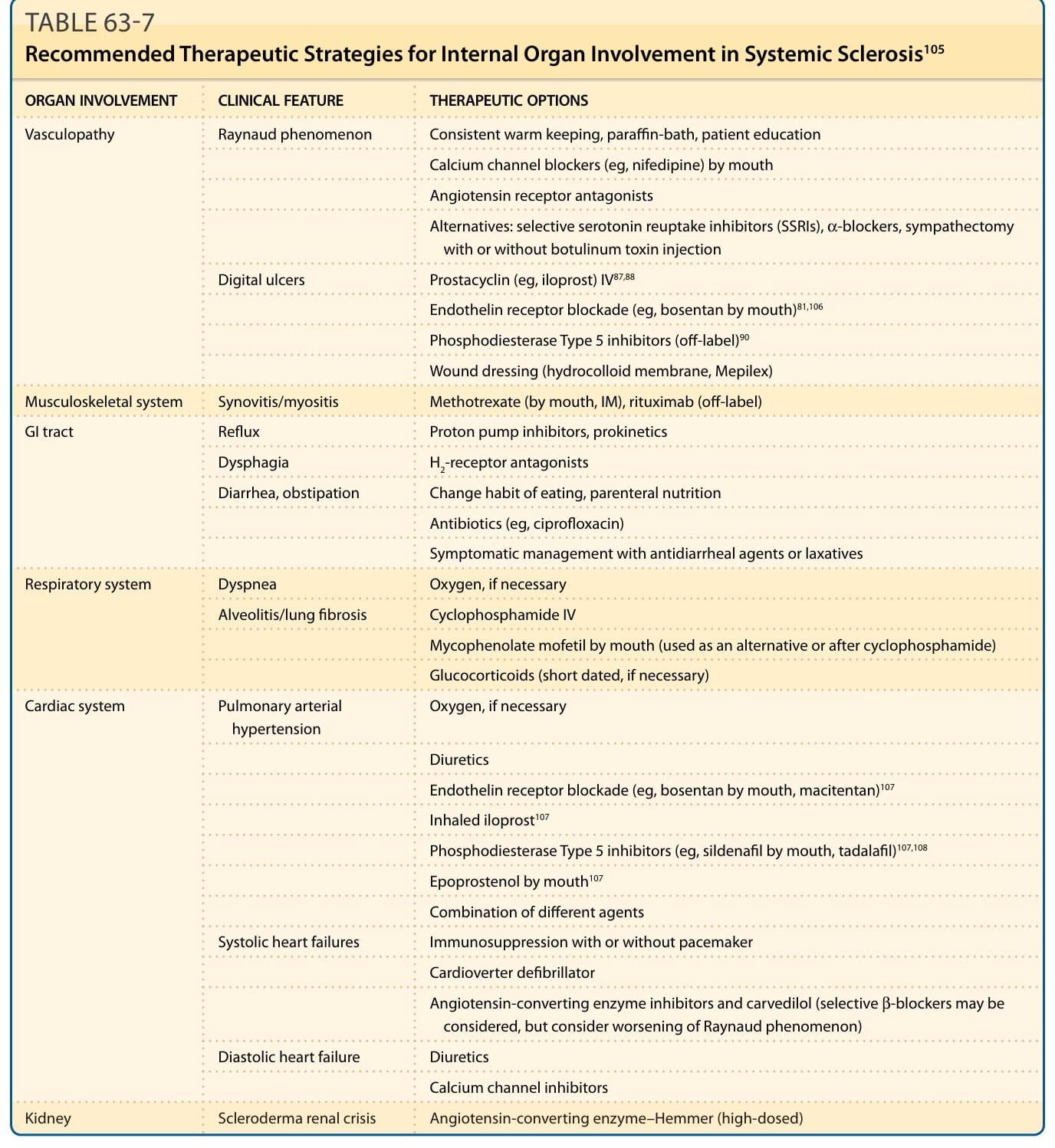
表 63-7:SSc 內臟器官受累的建議治療策略。