Systemic Sclerosis
10
AT-A-GLANCE
■ Scleroderma (systemic sclerosis [SSc]) is a multisystemic autoimmune disease characterized by vasculopathy, inflammation, and fibrosis of the skin and many other organs.
■ Differential diagnosis of SSc includes severe forms of localized scleroderma, as well as many other scleroderma-like conditions.
■ Raynaud phenomenon, circulating autoantibodies, and skin sclerosis are almost always present and are important for the early diagnosis.
■ Patients with SSc are classified into 2 major subtypes depending on the extent of skin sclerosis (diffuse cutaneous systemic sclerosis and limited cutaneous systemic sclerosis).
■ Patients with an overlap syndrome, including mixed connective tissue disease, are characterized by additional clinical features of other rheumatic diseases.
■ Involvement of internal organs (digestive tract, lung, kidney, and heart) can lead to severe dysfunction and determines the prognosis.
■ The heterogeneity and clinical course of SSc and SSc overlap syndromes require the urgent need of interdisciplinary collaborations and regular followup visits.
■ Although the disease is still not curable, there have been substantial advances in developing new therapeutic approaches and treating organ-based complications based on a better understanding of the pathophysiology.
DEFINITIONS
Scleroderma (systemic sclerosis [SSc]) is a multisystem disease, characterized by autoimmunologic processes, vascular endothelial cell injury, inflammation, and an extensive activation of fibroblasts. There is a large individual variability in the extent of skin and organ involvement, as well as in disease progression and prognosis. Skin, esophagus, lung, heart, and kidneys are the most frequently affected organs.
EPIDEMIOLOGY
Women are more frequently affected by SSc, with a female-to-male ratio between 3:1 and 14:1.1-4 The age of disease onset ranges between 30 and 50 years.1
However, male patients have earlier onset than female patients. Blacks with SSc are frequently younger than whites. SSc is a rare disease; however, incidence rates increased from 0.6 to 16 patients per million inhabitants and prevalence rates rose from 2 to 233 patients per million inhabitants per year,2-5 depending on methodologic differences in case definition and ascertainment as well as the investigated time period. It can be assumed that these numbers represent an underestimation as patients with mild disease remain often undiagnosed. SSc has the highest case-specific mortality of any of the autoimmune rheumatic diseases, but it varies individually, depending on racial or ethnic differences, presence and severity of organ involvement, SSc subsets, age at diagnosis, and gender differences. Although not curable, there have been substantial advances in treatment options for organ-based complications of SSc.
CLINICAL FEATURES OF SYSTEMIC SCLEROSIS
SSc usually starts with a Raynaud phenomenon, which can precede the disease for many years. The clinical manifestations depend to a large extent on the subset and stage of disease. The clinical features of established SSc are diverse with severe fibrosis of the skin and all additional cutaneous manifestations. These include hardening of the skin, development of contractures, digital ulcerations and calcifications. They also reflect the multiple patterns of internal organ involvement and the consequences of progression of the underlying pathologic processes of vasculopathy, inflammation, and fibrosis. Particular consideration must be given to the hallmark complications of hypertensive scleroderma renal crisis (SRC), pulmonary arterial hypertension (PAH), pulmonary fibrosis (PF), and GI dysmotility.
CLASSIFICATION AND DEFINITION OF DIFFERENT SSc SUBSETS
CLASSIFICATION AND
DEFINITION OF DIFFERENT
SS
c
SUBSETS
The heterogeneity of SSc arises from the range of disease manifestations that vary in extent and severity of organ involvement between patients. However, some
clinical features that are almost always present are Raynaud phenomenon (RP) and skin sclerosis. The extent of skin sclerosis defines each major disease subset, each of which has particular clinical characteristics, although there are also common features to each. In 1980, the American College of Rheumatology (ACR) published preliminary classification criteria for SSc for patients with established disease,6 which criteria showed 97% sensitivity and 98% specificity for SSc. According to the criteria, the diagnosis is proven, if either 1 major criterion or at least 2 minor criteria are found. The major criterion are scleroderma proximal to the metacarpophalangeal or metatarsophalangeal joints; the minor criteria include sclerodactyly, digital ulcerations and/or pitting digital scars, and bibasilar PF. Although these criteria have been used for many years they do not allow the inclusion of patients with early SSc and some patients with limited cutaneous systemic sclerosis. As a result, new ACR/European League Against Rheumatism criteria were developed, which are based on a score system and consider several additional criteria such as abnormal nailfold capillaries, fingertip lesions, and autoantibodies. These new criteria now allow early diagnosis of patients and include those patients in clinical trials before extensive fibrosis develops.7
In 1988, a descriptive subclassification of limited versus diffuse SSc was introduced by LeRoy,8 which was primarily associated with the extent of cutaneous involvement. This classification has been widely accepted and used in clinical practice. In 2001, LeRoy and Medsger9 published amended criteria, with the additional presence of autoantibodies and nailfold capillaroscopic alterations. Furthermore, these criteria include a separate group of patients with early onset of SSc, with minimal skin thickening. It was mandatory that patients with early (limited) SSc have evidence of RP plus scleroderma-specific autoantibodies and/or nailfold capillaroscopic manifestations.10,11 Although there are several other classifications published, for example, by Nadashkevich and colleagues and by Maricq and Valter,10,12
the initial LeRoy classification is still widely used in daily clinical practice. Diffuse cutaneous SSc is defined as a progressive form of SSc with an early onset of RP, usually within 1 year of onset of skin thickening. This subset is characterized by rapid skin involvement of trunk, face, upper arms, and thighs, showing very frequently, anti–scleroderma 70 (antitopoisomerase-I) or anti-RNA polymerase III antibodies.8 Furthermore, there is a higher propensity to develop PF, cardiac involvement, and SRC (Fig. 63-1). Limited cutaneous SSc is characterized by a long preexisting history of RP and skin changes of the extremities distal to the knee and elbow joints, including facial skin.8 This SSc-subset variant often (50% to 70% of cases) presents with anticentromere antibodies and is frequently associated with isolated PAH. The traditional acronym CREST (calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasias) is assigned to the limited form of SSc (Fig. 63-2).
10
Patients suffering from early SSc, also known as undifferentiated SSc, are defined by positive RP and at least 1 additional feature of SSc (positive nailfold capillary alterations, puffy fingers, pulmonary hypertension) and/or detectable scleroderma-associated autoantibodies without fulfilling the ACR criteria.1,13,14
A very small proportion of cases (1.5%) develop vascular (RP and/or PAH), immunologic (most commonly anticentromere antibodies), and organ-based fibrotic features of SSc, but do not show skin sclerosis.15
Patients suffering from this subset are classified as SSc sine scleroderma. Patients with features of scleroderma together with those of another autoimmune rheumatic disease are designated SSc overlap syndrome (Fig. 63-3 and Table 63-1). SSc overlap syndrome is defined as a disease occurring with clinical aspects of SSc (according to the ACR criteria) or main symptoms of SSc simultaneously with those of other connective tissue diseases or other autoimmune diseases, such as dermatomyositis, Sjögren syndrome, systemic lupus erythematosus, vasculitis, and polyarthritis. These patients present mostly high titers of anti-U1RNP, anti-nRNP, antifibrillarin, or anti-PmScl antibodies.16
This group of patients includes well-defined patients suffering from mixed connective tissue disease (MCTD), characterized by high titers of circulating anti-U1RNP antibodies (Table 63-2). There is still an ongoing discussion, whether MCTD represents a distinct disease entity or may be an early form of another connective tissue disease. These MCTD patients have varying clinical features with symptoms of systemic lupus erythematosus or rheumatoid arthritis with Raynaud syndrome, and later developing sclerodermatous lesions. They have swollen fingers and puffy hands. Non-Raynaud symptoms are skin sclerosis at acral regions and internal manifestations that occur later. There are often intense inflammatory symptoms with heavy arthralgia. MCTD patients can develop pericarditis, pleuritis, and pulmonary hypertension. However, there is a good response to antiinflammatory/anti-immune therapy and the prognosis is clearly better than in patients with classic scleroderma.17,18
Another already well-defined subset within the overlap syndromes are patients with sclerodermatous lesions, who also suffer from an intense myositis. These patients are usually characterized by specific Pm-Scl autoantibodies, have typical mechanic hands, and develop early intense subcutaneous calcifications. Similar to MCTD patients, they respond well to an early antiinflammatory treatment (eg, methotrexate/ glucocorticosteroids).19
Other overlap syndromes include patients with Raynaud and lupus erythematosus or rheumatoid arthritis symptoms, who later develop sclerodermatous lesions. Many of these patients have specific circulating autoantibodies that probably represent distinct disease entities. However further studies using molecular markers are still required to clarify their disease identity within the spectrum of scleroderma-related diseases.19
1087
10
A
B
C
A B
D
1088
10
Clinical spectrum of systemic sclerosis overlap syndromes
SSc overlap syndromes
Mixed connective tissue disease (U1snRNP+)
SSc/myositis overlap syndrome (Ku+)
SSc/SLE overlap syndrome
SSc/polymyositis syndrome (PmScl+)
SSc/dermatomyositis overlap syndrome
SSc/rheumatoid arthritis overlap syndrome
In addition, the frequency and timing of different visceral manifestations of SSc differs between major subsets. However, there is some overlap between subsets in terms of organ-based disease and the extent and severity of skin sclerosis. In all patients, the extent and severity of skin sclerosis can be assessed by the modified Rodnan skin score. Skin score at baseline correlates with disease severity and outcome in diffuse cutaneous SSc. Thickening and fibrosis of the skin as one of the first recognized phenomenon in SSc still forms the basis of most classification criteria and proposed subsets of this disease spectrum. It also has to be mentioned that another classification of SSc-related diseases, which is completely based on autoantibodies, has been proposed. There is evidence from association studies that this may be clinically meaningful, as indicated in Table 63-3. Moreover, genetic association analysis using a candidate gene approach has demonstrated an association between serologically based subsets of SSc that are stronger than with SSc overall. The significance of this is uncertain, and it should be noted that a genetic basis for
■Raynaud phenomenon
■Raynaud phenomenon
■Sclerodactyly
■Sclerodactyly
■Mechanic hands
■Mechanic hands
■Myositis
■Myositis
■Pulmonary involvement
■Pulmonary involvement
■Calcinosis cutis
■Calcinosis cutis
■Serositis
■Serositis
■PmScl autoantibodies
■PmScl autoantibodies
autoantibody reactivity has been well described, leading to the suggestion that the serologic subsets may be more genetically homogeneous than unselected SSc cases or clinically defined subsets of SSc. Large-scale studies in multinational patient cohorts using molecular and clinical markers are probably required to revise the current classification system of this heterogeneous spectrum of diseases.
ORGAN MANIFESTATION
ORGAN MANIFESTATION
RAYNAUD PHENOMENON
Distinctive for this disease is the initial onset of RP, which appears in more than 90% of SSc patients (see Fig. 63-2).1 It is defined by recurrent attacks of vasospasm of small digital arterioles/arteries at fingers and toes, usually caused by cold and/or other stimuli, for example, emotional stress. RP clinically appears
■Raynaud phenomenon
■Raynaud phenomenon
■Puffy fingers/hands
■Puffy fingers/hands
■Sclerodactyly
■Sclerodactyly
■Oesophageal involvement
■Oesophageal involvement
■Pulmonary hypertension/interstitial lung disease
■Pulmonary hypertension/interstitial lung disease
■Myositis
■Myositis
■Arthritis and arthralgia
■Arthritis and arthralgia
■Serositis
■Serositis
■Anemia/lymphopenia
■Anemia/lymphopenia
1089
■High titers of U1RNP antibodies
■High titers of U1RNP antibodies
10
REACTIVITY TARGET ANTIGEN FREQUENCY IN SSc (%) HLA ASSOCIATION CLINICAL ASSOCIATION
Centromere CENP proteins speckled pattern 20 to 30 HLA-DRB1 HLA-DQB1 Limited skin sclerosis, severe gut disease, isolated PAH, calcinosis
Scl-70 Topoisomerase-1 speckled pattern 15 to 20 HLA-DRB1 HLA-DQB1 HLA-DPB1 Diffuse skin sclerosis, pulmonary fibrosis and secondary PAH, increased SSc-related mortality rate
RNAP III RNA polymerase III speckled pattern 20 HLA-DQB1 Diffuse skin sclerosis, hypertensive renal crisis, correlated with a higher mortality rate
nRNP U1RNP speckled pattern 15 HLA-DR2, HLA-DR4 HLA-DQw5, DQw8 Overlap features of SLE, arthritis
Pm-Scl Polymyositis/Scl nuclear staining pattern
3 HLA-DQA1 HLA-DRB1 Limited skin sclerosis, myositis–sclerosis overlap, calcinosis
Fibrillarin U3RNP nuclear staining pattern 4 HLA-DQB1 Diffuse skin sclerosis, myositis, PAH, renal disease
Th/To 7-2RNP nuclear
2–5 HLA-DRB1 Limited skin sclerosis, pulmonary fibrosis
Th/To 7-2RNP nuclear staining pattern 2–5 HLA-DRB1 Limited skin sclerosis, pulmonary fibrosis
staining pattern
PAH, pulmonary arterial hypertension; SLE, systemic lupus erythematosus; SSc, systemic sclerosis.
suddenly and is clearly restricted and is accompanied by painful pallor/ischemia of single or several digits/ toes, followed by reactive hyperemia after reheating at the end of a RP attack, in some cases cyanosis (triphasic RP) also ensues (see Fig. 63-2).
SKIN INVOLVEMENT
Skin involvement is a cardinal feature of SSc and usually appears first in the fingers and hands. Within time, patients develop nonpitting edema of the fingers (puffy fingers), hands, and extremities, followed by an increasing induration and skin thickening (sclerodactyly) (see Figs. 63-1 and 63-2). Depending on the localization of skin thickening, restricted mobility of joints (dermatogenous contractures), and/or restricted breath excursion may be present. Typical facial features include telangiectasias, a beak-shaped nose, and reduced mouth aperture (microstomia). The typical facial appearance of SSc patients is characterized by a radial furrowing around the mouth, no expression, a stiff and mask-like facial appearance, and sclerosis of the frenulum. Besides cosmetic/aesthetic problems, this causes considerable difficulties regarding eating and oral hygiene (see Fig. 63-1). The abnormal deposition of cutaneous and/or subcutaneous calcium (calcinosis cutis), usually occurs over pressure points (acral, joints) (Fig. 63-4). Calcinosis cutis next to joints is Thibierge-Weissenbach syndrome. Further skin manifestations include hypopigmented and hyperpigmented (salt-and-pepper) skin (see Fig. 63-1), and loss of hair follicles and sweat glands (hypohidrosis/anhydrosis).20
Approximately 50% of patients with SSc are affected by digital ulceration associated with vasculopathy at some point in their disease. This is the major external feature of structural vessel disease, probably
1090
attributable to thickened intima and lumen-occluded vessels. Tender and painful pitting scars are very frequent and, on occasion, progress to ulcers. These occur on the finger- or toe-tips, over the extensor surfaces of the joints as a result of microtrauma or in association with the abovementioned calcinosis cutis. Digital ulcers are associated with strong, local pain and a major impact on quality of life regarding all-day functions (eg, dressing, eating). Other complications include critical digital ischemia, paronychia, infections, gangrene, osteomyelitis, and finger pulp loss or amputation.
CARDIOPULMONARY MANIFESTATIONS
There are different ways that the cardiopulmonary system may be involved, most often appearing as fibrosis and PAH. The differentiation between these manifestations is often clinically difficult because of similar overlapping clinical features, such as dyspnea, nonproductive cough, disturbed diffusion capacity, and cyanoses. PAH is currently the most common cause of disease-related death in SSc.21 It occurs in both limited and diffuse cutaneous subsets, although the most typical cases are those of limited SSc associated with isolated PAH. This condition has substantial similarities to idiopathic PAH. Thus, 2 patterns of disease occur in SSc. Most cases have PAH, but there are some patients with late-stage extensive interstitial lung fibrosis in SSc that develop a true secondary pulmonary hypertension.22 Besides the right-heart worsening caused by PAH, the heart could also be involved by diffuse or focal fibrosis or from inflammatory myocarditis. This may lead to diastolic or systolic dysfunction, as well as a restricted contractibility of the myocardium. These patients clinically
A
C D
10
B
present cardiac arrhythmia, paroxysmal tachycardia, incomplete or complete right-heart blocks, and heart insufficiency.23
GI INVOLVEMENT
GI involvement is the most common internal organ involvement in patients suffering from both limited and diffuse SSc (>60%).1 Many parts of the GI tract may be impaired, affecting motility, digestion, absorption, and excretion.24
Esophageal involvement includes symptoms like dysphagia, heartburn resulting from reflux, nausea, and/or vomiting. A weakened lower esophageal sphincter and impaired peristalsis increase the risk for esophagitis. If untreated, this could lead to peptic esophagitis, gastric/esophageal ulcerations, peptic stricture formations, and fistulae. Chronic gastroesophageal reflux can be complicated after a time by a higher risk for Barrett esophagus, which may progress into an adenocarcinoma. Possible gastric manifestations include atrophy of mucous membrane–associated ulcerations and delayed gastric emptying. Gastric antral vascular ectasia is also an important complication in some SSc patients and needs to be detected by endoscopy because it can lead to severe, often not recognized, bleeding.25
SSc can also affect the intestine, and includes atonic dilation, constrictions, malabsorption, pseudoobstruction, diarrhea, constipation, fecal incontinence, and severe malnutrition.
KIDNEY INVOLVEMENT
SRC appears in 5% to 10% of SSc patients, and may cause an abrupt onset of significant systemic hypertension (>140/90 mm Hg, or a rise in systolic/diastolic blood pressure ≥30/≥20 mm Hg), together with an increase in serum creatinine, proteinuria, hematuria, thrombocytopenia, or hemolysis followed by an acute renal failure.26 Studies suggest that a chronic vasculopathy with reduced glomerular filtration rate is frequent. In addition, there is evidence of an increase in fibrillar collagen deposition within the renal interstitium in SSc. Many cases occur within the first 12 months of disease, and in up to 25% of patients with SRC, the diagnosis of SSc is made at the time of the renal presentation. End-organ damage can result in encephalopathy with generalized seizures or flash pulmonary edema. Microangiopathic anemia is common, and sometimes disseminated, intravascular coagulation develops. Nephrotoxic drugs and high-dose prednisolone (>7.5 mg/day) should be avoided in patients suffering from SSc.27
1091
10
ETIOLOGY AND PATHOGENESIS
The pathogenesis of this complex autoimmune disease involves multiple cell types (endothelial cells, epithelial cells, fibroblasts, and lymphocytic cells) interacting through a variety of mechanisms that are
dependent on their microenvironment and several key mediators. Major facets of the disease include inflammation, vasculature, and the activation of connective tissueproducing cells (Fig. 63-5). The clinical heterogeneity of SSc makes it likely that distinct pathogenetic mechanisms predominate in particular patients or subsets of disease. Similarly, the key pathways are not necessarily
The pathophysiology of scleroderma
Injury
EC damage
Inflammation
IL-4/ IL-13 Innate immune cells
Cytokines CTGH PDGF Growth factors TGF-β
Profibrotic macrophages
TGF-β
RELM-β
Endothelial cells
TGF-β
Circulating precursors
Resting fibroblast
T cells B cells Autoantibodies
TGF-βR
PDG-R
Mechanical tension
ET-R
Activated myofibroblast
Adipocytes
Growth factors Lysyt hydroxylase Other fibrogenic cytokines
Fibrillin, collagens, FN, COMP, etc.
Stiff fibrous matrix
Genetic predisposition
1092
the same at different stages of SSc. Although a genetic component to etiopathogenesis is likely and evidence supports genetic factors determining severity and susceptibility, there are also strong arguments, supporting environmental and chemical factors as triggers for the disease.28
GENETIC FACTORS
GENETIC FACTORS
The best evidence for a genetic contribution to SSc and related diseases comes from studies that report familial clustering and from the limited twin studies that have been undertaken. Although the absolute risk for familial-occurring SSc is relative low, the relative risk for first-degree relatives is 13-fold higher compared to the normal population.29 Several studies suggest that a positive family history for SSc is the strongest risk factor, but ethnicity also contributes.29 Assassi and colleagues suggest that members of SSc-affected families tend to show concordant sclerodermaspecific autoantibodies.30 Further support is provided from genetic association studies with candidate gene approaches. Most success has been observed in genetic analysis of individual components of the disease such as autoantibody profiles. These appear to have a strong genetic determinant, and this may underlie the apparent mutual exclusivity of the SSc hallmark reactivities. It has been demonstrated that the ability to mount an immune response to a particular SSc-associated antigen is restricted by major histocompatibility complex haplotype. Several studies suggest an association of HLA-DRB1∗1302 and HLA-DQB1∗0604/0605 haplotypes with antifibrillarin-positive patients,31 while HLA-SRB1∗0301 occurs in patients with anti–Pm-Scl antibodies.32 Observation from a large number of studies examining genetic markers has identified a number of candidate genes (eg, anemia-inducing factor [AIF]-1, cluster of differentiation [CD] 19, CD22, CD86, cytotoxic T-lymphocyte antigen [CTLA]-4, CCL-2, CCL-5, chemokine ligand [CXCL]-8, chemokine-related receptor [CXCR]-2, interleukin [IL]-1α, IL-1β, IL-2, IL-10, IL-13, macrophage migration inhibitory factor (MIF), protein tyrosine phosphatase non-receptor 22 (PTPN22) tumor necrosis factor [TNF]-α).29,33 Most of the more recent genome-wide association studies have identified loci relevant for the innate immune system; some of these associations are already rather robust and might lead to new therapeutic approaches. Others reflect the altered connective tissue response. However, as with other complex diseases, in very many instances, it has not always possible to replicate initially promising data. Studies of genetically homogeneous populations have been especially informative, including those of the Choctaw Nation of Native Americans. However, it is of interest that some of the associations are very plausible in terms of molecular pathogenesis. It is likely that epistasis and the effect of multiple modifier genes confound simple genetic association studies in SSc, just as in other complex diseases.34 There is increasing evidence that epigenetic
10
mechanisms by modulating chromatin structure and gene expression of cytokines/growth factors critical for the activation of the immune response and/or the fibrotic reaction are important additional factors contributing to the development of scleroderma.
ENVIRONMENTAL FACTORS
ENVIRONMENTAL FACTORS
Scleroderma-like syndromes have been reported in association with numerous environmental toxins and drugs. These agents include solvents (vinyl chloride, benzene, toluene, epoxy resins), drugs (bleomycin, carbidopa, pentazocine, cocaine, docetaxel, metaphenylenediamine), and miscellaneous substances.35
SSc was reported to occur in underground coal and gold miners. In male patients with silicosis who were older than 40 years of age, the likelihood of developing SSc was approximately 190 times greater than in males not exposed to silica, and 50 times greater than in males without silicosis but exposed to silica dust.36 The role of silicone gel implants and other silicone products in the development of scleroderma has been questioned.37
However, most epidemiologic studies have failed to show a significant association. An unusual form of scleroderma characterized by RP, morphea-like skin changes, capillary abnormalities of the nailfold (similar to those in SSc), osteolysis of the distal phalanges, and hepatic and PF may occur in workers exposed to polyvinyl chloride. Bleomycin also produces PF, RP, and cutaneous changes indistinguishable from those of SSc.37 The development of these changes appears to be dose-dependent and is reversible on discontinuation of the drug. Collectively, chemical exposures account for a small fraction of scleroderma-like diseases. Large epidemiologic studies have not yet revealed a significant role for toxins and drugs in scleroderma.
HISTOPATHOLOGY
HISTOPATHOLOGY
The histopathology of SSc shows fibrosis of the lower two-thirds of the dermis and the subcutaneous fibrous trabeculae, because of excessive deposition of extracellular matrix (ECM) proteins, most notably collagen Types I and III (Fig. 63-6).28
Panniculitis and mucoid edema also may be prominent features in the early stages, whereby subcutaneous fat is replaced by a fibrous connective tissue. It is possible to differentiate histologically an early cellular stage on the one hand and a later fibrotic stage on the other hand. In the early stages, the dermis presents pathologically collagen bundles within the reticular dermis, and appear pale, homogenous, running parallel to the skin surface, and swollen, and there is often a perivascular lymphocytic infiltrate. These inflammatory cell infiltrates are localized between the collagen bundles but mainly around the vessels, and can also spread into subcutaneous fat tissue. The infiltrate can also entrap sweat glands. The epidermis often becomes
1093
10
atrophic in the overlying areas. Vessels of all sizes may be involved in SSc. In the early stages, there may only be dilation of capillaries, then endothelial proliferation and complete occlusion of vessels occur. With the progression of scleroderma, the involved skin becomes more avascular and inflammation decreases. In later stages, pilosebaceous units and eccrine glands disappear, collagen bundles appear to be packed closely, and there may be an effacement of the rete ridges.20
VASCULOPATHY
VASCULOPATHY
Vasculopathy in SSc is an early event and is based on inappropriate vascular remodeling and repair processes. It involves the microcirculation and arterioles and is very likely a primary event in the pathogenetic processes of the disease. Vascular abnormalities are characterized by vasoconstriction, adventitial and intimal proliferation, inflammation, and thrombosis.28 The earliest signs of vascular dysfunction are represented by enhanced vascular permeability with an imbalance between vasodilatory (nitric oxide, prostacyclin, calcitonin gene-related peptide) and vasoconstrictive mediators (endothelin-1, angiotensin II, α2- adrenoreceptors). Consequently, the impaired blood flow leads to tissue hypoxia, which induces strong expression of vascular endothelial growth factor and its receptors, associated with a defect of vasculogenesis. However, inflammatory cytokines like TNF-α may stimulate or inhibit angiogenesis depending on the duration of the stimulus.38
1094
In addition to these functional abnormalities, intravascular and structural changes contribute to overt RP, and in the course of time to progressive reduction of vessels and blood flow. This pattern of obliterative vasculopathy may clinically manifest in all vessels of virtually all organs. Early lesions in the microcirculation because of structural damage are initially seen in the nailfold capillaries and as vasospastic responses in RP. Furthermore, vascular changes, that is, overgrowth of the endothelium and deposition of scar tissue, produce some of the major complications of SSc, including PAH, SRC, and digital vasculopathy.
IMMUNE EVENTS
IMMUNE EVENTS
There are early inflammatory changes in the skin and lung of patients with SSc. The first inflammatory infiltrates in lesional skin are predominantly cells of the monocyte lineage (T cells, macrophages, B cells, and mast cells).39 There are several lines of evidence for the crucial role of the innate immune system in SSc. This is underlined by the association of interferon regulatory factor-5 variants with scleroderma.40,41 Several studies also demonstrate the role of macrophages as important contributors of cytokines, which influence the fibrotic response.42
Later, T lymphocytes predominate and are detectable in both the circulation and affected organs. These T cells are predominantly CD4+, bear markers of activation, exhibit oligoclonal expansion, which suggests an antigen-driven proliferation, and show a predominant T-helper 2 phenotype.43,44 Consequently, increased serum levels of T-helper 2 cell–derived cytokines (IL-2, IL-4, IL-10, IL-13, and IL-17) have been observed in scleroderma patients.45,46
In addition to T cells, B cells are also found in involved skin. Several studies suggest that B cells are able to induce ECM production through secretion of IL-6 and transforming growth factor-β (TGF-β) and are involved in the production of autoantibodies. Several of these autoantibodies are associated with defined subsets of the disease and are important diagnostic markers (see Table 63-3).47 The potential role of autoantibodies in pathogenesis is a fascinating and exciting area. The majority of SSc cases have circulating antibodies. These include a number of hallmark reactivities, as well as autoantibodies, that occur in other autoimmune rheumatic diseases (eg, anticyclic citrullinated peptide, rheumatoid factor) but also antibodies that may have functional significance, as they are directed against cell-surface antigens (eg, antiendothelial cell antibodies, antifibrillin antibodies, anti–platelet-derived growth factor [PDGF] receptor antibodies) (Table 63-4).48-50 However, functional impact of these antibodies remains an area of investigation. There is growing evidence of functional significance for antiendothelial cell autoantibodies and for antifibroblast-reacting antibodies. Reports also suggest the presence of antifibrillin autoantibodies and stimulatory autoantibodies reacting with PDGF
10
FUNCTIONAL ANTIBODY FREQUENCY IN SYSTEMIC SCLEROSIS CLINICAL ASSOCIATION
Anti–platelet-derived growth factor receptor 33% to 100%
■Seem to induce skin fibrosis as a result of activation of fibroblasts into myofibroblasts and fibroblast-like cells
■First functional antibodies discovered in systemic sclerosis (SSc)
Antiendothelial cell antibodies 44% to 84%
■Mediates endothelial cell damage and activation of fibroblasts resulting from stimulation of proinflammatory and fibrotic cytokines
■Associated with severe organ manifestation
■Associated with perivascular, vascular (digital ulcers [DUs]) and lung involvement (pulmonary arterial hypertension [PAH])
■Also found in other rheumatic diseases
Antifibroblast antibodies 26% to 58%
■Associated with Scl-70 antibodies and the prevalence of interstitial lung disease and PAH
■Increased prevalence in diffuse cutaneous SSc compared to limited cutaneous SSc
Antifibrillin-1 >50%
■Activates fibroblasts by stimulation of the release of transforming growth factor-ββ
49% to 52%
■Inhibits the degradation of extracellular matrix proteins, because of an inhibition of MMP collagenase activity
Anti–matrix metalloproteinase (MMP) 1, anti-MMP3
■Correlates with the extent of fibrosis (skin, lung, kidney)
82% to 83%
■Simultaneous presence has been described in SSc patients (cross-reactivity)
82% to 83% ■Simultaneous presence has been described in SSc patients (cross-reactivity)
Angiotensin II Type 1
Angiotensin II Type 1 receptor and endothelin Type A receptor
■Associated with early and severe disease, PAH, DUs, renal crisis, diffuse cutaneous SSc, and lung fibrosis
■Associated with early and severe disease, PAH, DUs, renal crisis, diffuse cutaneous SSc, and
receptor and endothelin Type A receptor
lung fibrosis
receptors.48-50 Microchimerism and graft-versus-host disease mechanisms have been suggested in some cases, although the relatively high frequency of microchimerism in healthy individuals or other disease states suggests that this may be contributory rather than causal if it has a role in SSc. Careful clinical studies have identified a subset of SSc patients (characterized by RNA polymerase antibodies), who develop the disease in association with the occurrence of malignancies.51-53 This led to the hypothesis that fibrosis may represent an immune response against tumor antigens and initiated a discussion on the relationship of autoimmunity and malignancy in general. Further studies are required to clarify this issue.
FIBROSIS
FIBROSIS
SSc is a multisystem fibrotic disease. The initial inflammation and hypoxia induces in fibroblasts the production of several proteins that are involved in ECM remodeling as, for example, thrombospondin-1, fibronectin-1, lysylhydroxylase-2, and TGF-β–induced proteins.54 At the same time there is a disturbed balance between synthesis and degradation mechanisms leading to the excess of ECM in specialized organs, which is then responsible for much of the morbidity and mortality of the disease. The key event in the development of fibrosis is the induction of fibroblasts into activated myofibroblasts. Alternatively, also other cell types (eg, circulating precursor cells, endothelial cells, and epithelial cells) can be converted into myofibroblasts. The initiation of this process includes a number of key cytokines and growth factors that may
represent logical therapeutic targets. These include fibrogenic cytokines such as TGF-β, connective tissue growth factor, PDGF, and endothelin-1.28,55-57 Especially TGF-β has been shown to play a central role,58 which is also underscored by extensive expression profiling studies using skin biopsies from scleroderma patients in different stages of the diseases.59 This has already led to therapeutic approaches using antibodies against TGF-β in early clinical studies.60
Myofibroblasts are characterized by a high contractility, ECM production, and cytokine release. This function together with altered biophysical properties of the resulting connective tissue lead to persistent activation of fibroblasts with an excessive deposition of ECM components. However, crucial for understanding the mechanisms of this disease is the close connection between autoimmunity, vasculopathy, and fibrosis. This was recently demonstrated in a mouse model characterized by downregulation of the transcription factors Friend leukemia integration 1 (Fli1) and Kruppel-like factor 5 (KLF5), which develop a scleroderma like disease with the production of autoantibodies.61
DIAGNOSIS
RAYNAUD PHENOMENON
RAYNAUD PHENOMENON
Patients presenting only with RP should be studied for capillary alterations as well as autoantibody status. All these are predictors for the development of SSc, which together make the diagnosis of SSc rather likely.
1095
10
ORGAN INVOLVEMENT CLINICAL FEATURE DIAGNOSTIC PROCEDURES
Vascular system Raynaud phenomenon
■Coldness provocation
■Nailfold capillaroscopy
■Antinuclear antibody levels
■Clinical assessment regarding puffy fingers, telangiectasias, mechanic hands, hypopigmentations/hyperpigmentations, digital ulcerations, dermatogenous contractures
Skin Scleroderma Calcinosis cutis
■Modified Rodnan skin score
■20-MHz ultrasonography
■Radiography (X-ray, MRI, CT)
■Clinical assessment regarding fist closure deficiency, joint contractures, tendon friction rub, muscle weakness
Musculoskeletal system Arthralgia Synovitis Muscle weakness
■Laboratory parameters: erythrocyte sedimentation rate, rheumatoid factor, antinuclear autoantibodies
■Creatine kinase (greater than threefold?)
■MRI, electromyography
■Muscle biopsy
■Gastro-/esophageal endoscopy
GI tract Reflux Dysphagia Gastric antral vascular ectasia Diarrhea, obstipation
■Esophageal scintigraphy, esophagus manometry
■Gastro-/esophageal endoscopy with laser coagulation, if necessary
■Colonoscopy
Respiratory system Dyspnea
■Lung function test (carbon monoxide transfer factor corrected for hemoglobin [TLCOc] single breath (SB), total lung capacity [TLC], forced vital capacity [FVC])
■Radiography (X-ray or high resolution CT)
■Bronchioalveolar lavage (BAL) (optional)
Cardiac system Dyspnea, arrhythmia
■Electrocardiography (conduction blocks?)
■Echocardiography (mean pulmonary artery pressure, diastolic dysfunction?, ventricular ejection fraction)
■(Spiro-)Ergometry
■24-Hour blood pressure controls
■Right-heart catheterization
■Cardio MRI
Kidney Renal function failure
■Regular blood pressure controls (>140/90 mm Hg)
Kidney Renal function failure ■Regular blood pressure controls (>140/90 mm Hg)
■Ultrasonography
■Ultrasonography
■Serum levels of creatinine, urine analyses (protein, albuminuria, microelectrophoresis)
■Serum levels of creatinine, urine analyses (protein, albuminuria, microelectrophoresis)
To identify and visualize vascular cutaneous alterations caused by SSc, nailfold capillaroscopy is a noninvasive, simple, and one of the most useful diagnostic and prognostic methods (Table 63-5). Furthermore, it is a useful tool to categorize capillary changes into early, active, and late patterns. Laser Doppler perfusion imaging is also a noninvasive microvascular imaging technique able to provide maps of the cutaneous blood flow.62
SKIN SCLEROSIS
SKIN SCLEROSIS
Skin involvement should be evaluated using the modified Rodnan Skin Score. Usually, 17 sites are assessed and skin thickness is categorized to grade 1, 2, or 3, corresponding to mild, medium, and severe, according to palpation of the skin by a trained examiner (Fig. 63-7). Newer techniques for calculating skin thickening also have been evaluated. In addition
1096
to the modified Rodnan skin score,63 the 20-MHz ultrasonography,64 MRI,65 and plicometer66 methods are useful for assessing skin thickening (recommended diagnostic procedures are listed in Table 63-5). Further physical procedures to monitor skin fibrosis are the durometer,67 cutometer,68 and elastometer.69 In addition to these noninvasive methods, skin biopsy with histologic evaluation of the dermal skin thickness is an appropriate but invasive method. This method enables the characterization of the inflammatory infiltrates.
CARDIOPULMONARY INVOLVEMENT
CARDIOPULMONARY
INVOLVEMENT
Individuals with SSc and cardiopulmonary symptoms should be followed up at least annually using pulmonary function tests, echocardiography, a 6-minute walk test, and high-resolution CT (HRCT).70 Pulmonary
10
Modified Rodnan skin score
Face:
Upper arm ri:
Forearm ri:
Hand ri:
Fingers ri:
Thigh ri:
Lower leg ri:
Foot ri:
Sum of scores:
Upper arm le:
Trunk/thorax:
Abdomen:
Forearm le:
Hand le:
Fingers le:
Thigh le:
Lower leg le:
Foot le:
function tests are the most important techniques to determine possible cardiopulmonary involvement, because of impaired diffusion capacity of the lung for carbon monoxide (DLCO ≤75%) being an early marker of both lung fibrosis and PAH.71
To determine the presence of interstitial lung involvement, that is, subpleural localized line opacities, ground-glass opacities, and subpleural cysts with honeycomb formations, HRCT and/or thoracic radiography should be used. Followup should also include transthoracic Doppler echocardiography, a noninvasive procedure, that can indicate a hypertrophy with or without enlargement of the right ventricle, paradoxical motion of the interventricular septum, tricuspid valve insufficiency, and pericardial effusion. Right-heart catheterization is indeed the gold standard, but is an invasive diagnostic procedure to determine PAH. PAH is defined as a mean pulmonary artery pressure of ≥25 mm Hg at rest together with a pulmonary capillary wedge pressure of ≤15 mm Hg as determined by right-heart catheterization.70,72,73
Cardiac MRI is also a potential strategy for assessing myocardiac involvement in SSc. Besides imaging procedures, there is also some promise for the use of
N-terminal brain natriuretic peptide to detect right ventricular impairment (see Table 63-5). Early detection of cardiac involvement is crucial to prevent and to allow early treatment of cardiomyopathy and severe cardiac arrhythmias.70
GI INVOLVEMENT
GI INVOLVEMENT
The presence of esophagitis can be determined by upper GI endoscopy with histologic evaluations. Impaired motility of the esophagus can usually be diagnosed by scintigraphic evaluation following a radiolabeled meal or 24-hour pH manometry (see Table 63-5).71
KIDNEY INVOLVEMENT
KIDNEY INVOLVEMENT
Early diagnosis is the key role in improving the outcome of SRC using regular blood pressure monitoring, urine analysis microelectrophoresis, and determination of creatinine clearance (see Table 63-5).26
1097
10
DIFFERENTIAL DIAGNOSIS
The diagnosis of SSc is clinical. Although there are criteria that were developed to facilitate the distinction of SSc from other connective tissue diseases, no formal diagnostic criteria have been developed. However, determination of the correct subset of the disease, including the SSc overlap syndromes, is required to enable judging of prognosis and involvement of certain organs and for determining the therapeutic approach. There are several differential diagnoses that imitate scleroderma: circumscript (localized) scleroderma; eosinophilic fasciitis; sclerodermiform genodermatoses; acrodermatitis chronica atrophicans; sclerodermalike syndromes induced by environmental factors; scleroderma adultorum Buschke; scleroderma diabeticorum; scleromyxedema; nephrogenic fibrosing dermopathy; porphyria cutanea tarda; graft-versus-host disease; and scleroderma-like lesions in malignancies. These certainly have to be excluded. Table 63-6 outlines the differential diagnosis of SSc.
CLINICAL COURSE AND PROGNOSIS
The development of the disease depends very much on the specific subset. Patients with the limited form develop RP already many years prior to the onset of other organ manifestations. Skin fibrosis remains localized to the acral areas and the main complications are the development of digital ulcerations and pulmonary hypertension. In the diffuse form, however, fibrosis occurs early and together with inflammation, joint pain and shows a rapid spreading to almost all parts of the integument. In these patients, manifestations of the lung (lung fibrosis), heart, and kidney occur early in the disease course and often determine the prognosis. There is still a high number of deaths associated with this disease subset. Several studies indicate that the diffuse cutaneous SSc patients show a rapid
Differential Diagnosis
Differential Diagnosis
■Circumscript (localized) scleroderma (morphea)
■Circumscript (localized) scleroderma (morphea)
■Eosinophilic fasciitis
■Eosinophilic fasciitis
■Lichen sclerosus et atrophicans
■Lichen sclerosus et atrophicans
■Sclerodermiform genodermatoses (eg, progeria, acrogeria)
■Sclerodermiform genodermatoses (eg, progeria, acrogeria)
■Sclerodermiform acrodermatitis chronica atrophicans
■Sclerodermiform acrodermatitis chronica atrophicans
■Scleroderma adultorum Buschke
■Scleroderma adultorum Buschke
■Scleroderma diabeticorum
■Scleroderma diabeticorum
■Scleroderma amyloidosus
■Scleroderma amyloidosus
■Scleromyxedema
■Scleromyxedema
■Mixed connective tissue disease (MCTD)
■Mixed connective tissue disease (MCTD)
■Nephrogenic fibrosing dermopathy
■Nephrogenic fibrosing dermopathy
■Sclerodermiform porphyria cutanea tarda
■Sclerodermiform porphyria cutanea tarda
■Sclerodermiform chronic graft-versus-host disease
■Sclerodermiform chronic graft-versus-host disease
1098
■Eosinophilia-myalgia syndrome
■Eosinophilia-myalgia syndrome
worsening of the disease in the initial years. In later years, the activity of the disease is reduced and symptoms can improve. Surprisingly, the sclerotic skin also can become softer and contractures can be diminished. Although SSc is still a life-threatening disease, a multidisciplinary management of the patients with early detection and treatment of complications may lead to a much-improved prognosis during the patient’s life.
MANAGEMENT
DISEASE-MODIFYING TREATMENT
DISEASE-MODIFYING
TREATMENT
Three facets of SSc are potentially amenable to therapeutic modulation, which raises the possibility of true disease-modifying treatment. At present, vascular therapies and immunomodulation have the widest range of candidate therapies. Tables 63-7 and 63-8 summarize these approaches. General immunosuppression can be of benefit by improving skin involvement and interstitial lung disease. The best evidence is available for cyclophosphamide but more recently mycophenolate mofetil (MMF) has been shown to be as effective as oral cyclophosphamide and is used by many centers.74,75 There is also evidence that rituximab can lead to improvement of the disease course in a select group of patients, if standard immunosuppression has failed.76 Efficiency of immunosuppression in general is demonstrated by trials from the United States and Europe using high-intensity immunosuppression with autologous hemopoietic stem cell transplantation in some selected patients.77-80 However, side effects always have to be considered (Table 63-9). Antifibrotic treatment remains still a challenge, although during the last few years a number of new approaches have been generated mainly based on the better understanding of the underlying mechanisms. A newer study using a new antibody against TGF-β led to an improvement of the severity of skin involvement and a reduction of the expression of several TGF-β–dependent genes.60 Some encouragement is also provided by newer clinical trials of idiopathic PF. However, at present there is no proven antifibrotic agent. Figure 63-8 is a simplified schematic for integrating putative disease-modifying therapy with programs of screening and surveillance that permit timely intervention in SSc with organ-based strategies that currently form the basis of the majority of SSc therapeutics. Possibilities for targeted disease modifying therapy depend on the availability of therapeutic agents and a clear understanding of their role in pathogenesis. There has been much more success in the field of organ-based therapeutics in SSc, which already had a high impact especially on the quality of life of many patients. Early detection of these organ-specific complications is required to enable early intervention.
10
ORGAN INVOLVEMENT CLINICAL FEATURE THERAPEUTIC OPTIONS
Vasculopathy Raynaud phenomenon Consistent warm keeping, paraffin-bath, patient education
Calcium channel blockers (eg, nifedipine) by mouth
Angiotensin receptor antagonists
Alternatives: selective serotonin reuptake inhibitors (SSRIs), α-blockers, sympathectomy with or without botulinum toxin injection
Digital ulcers Prostacyclin (eg, iloprost) IV87,88
Endothelin receptor blockade (eg, bosentan by mouth)81,106
Phosphodiesterase Type 5 inhibitors (off-label)90
Wound dressing (hydrocolloid membrane, Mepilex)
Musculoskeletal system Synovitis/myositis Methotrexate (by mouth, IM), rituximab (off-label)
GI tract Reflux Proton pump inhibitors, prokinetics
Dysphagia H2-receptor antagonists
Diarrhea, obstipation Change habit of eating, parenteral nutrition
Antibiotics (eg, ciprofloxacin)
Symptomatic management with antidiarrheal agents or laxatives
Respiratory system Dyspnea Oxygen, if necessary
Alveolitis/lung fibrosis Cyclophosphamide IV
Mycophenolate mofetil by mouth (used as an alternative or after cyclophosphamide)
Glucocorticoids (short dated, if necessary)
Cardiac system Pulmonary arterial hypertension Oxygen, if necessary
Diuretics
Endothelin receptor blockade (eg, bosentan by mouth, macitentan)107
Inhaled iloprost107
Phosphodiesterase Type 5 inhibitors (eg, sildenafil by mouth, tadalafil)107,108
Epoprostenol by mouth107
Combination of different agents
Systolic heart failures Immunosuppression with or without pacemaker
Cardioverter defibrillator
Angiotensin-converting enzyme inhibitors and carvedilol (selective β-blockers may be considered, but consider worsening of Raynaud phenomenon)
Diastolic heart failure Diuretics
Calcium channel inhibitors
Kidney Scleroderma renal crisis Angiotensin-converting enzyme–Hemmer (high-dosed)
Kidney Scleroderma renal crisis Angiotensin-converting enzyme–Hemmer (high-dosed)
DIGITAL VASCULOPATHY AND ITS COMPLICATIONS
DIGITAL VASCULOPATHY
AND ITS COMPLICATIONS
Simple but important recommendations for reducing the frequency of Raynaud attacks include reducing vasoconstriction by avoiding precipitating factors like nicotine, sympathomimetics, emotional stress and coldness, and instead to have good home heating, thick and airtight clothes, thermochemical or microwaveable hand warmers, electrically heated gloves, soles, or infrared hyperthermy, regular paraffin wax bath treatments, and minimizing finger trauma.
Therapy requires a close interaction between several medical disciplines applying topical and systemic therapies. Current local management of digital ulcers includes a combination of nonpharmacologic care, antibiotics (in case of infection), analgesia, and individually applied wound dressings, if necessary. Potential pharmacologic treatment requires optimal therapy for RP, including agents that have the potential for vascular remodeling and/or dilation, such as calcium channel blockers and angiotensin II receptor antagonists,81,82 which should be considered first-line therapy. The results have been contradictory with other pharmacologic treatment options, such as diltiazem and angiotensin-converting enzyme inhibitors.83-86
1099
10
CLINICAL FEATURE THERAPEUTIC OPTIONS
Skin hardening
■Lymphatic drainage
■Physiotherapy
■Topical treatment with steroids or calcineurin inhibitors
■Systemic treatment with steroids (short dated) and/or immunosuppressants
■Phototherapy (psoralen and ultraviolet A, ultraviolet A1, extracorporeal photochemotherapy)
■Topical treatment with steroids, capsaicin
Dryness and itching
■Cannabinoid agonists
■Emollients
■Phototherapy (psoralen and ultraviolet A, ultraviolet A1)
■Systemic treatment with antihistamines or gabapentin
Digital ulcerations
■IV iloprost
■Bosentan by mouth
■Hydrocolloid dressings
■Skin substitutes
■Physical therapy
Calcifications
■Bisphosphonate by mouth
■Local corticosteroid injection
■Laser therapy
■Surgery
Telangiectases
■Laser therapy
■Camouflage
■Bleaching agents, camouflage, sunscreens
■Bleaching agents, camouflage, sunscreens
Hyperpigmentation
Hyperpigmentation and hypopigmentation
■Salicylic acid and chemical peelings
■Salicylic acid and chemical peelings
and hypopigmentation
■Hydroquinone, retinoids, corticosteroids
■Hydroquinone, retinoids, corticosteroids
Parenteral prostacyclin derivatives, in particular iloprost, are widely used, and help to heal digital ulcers and may prevent recurrent lesions. Prostacyclin derivatives by IV infusion are the mainstay of therapy for critical digital ischemia.87,88 Antiplatelet agents, such as aspirin and clopidogrel, are also used, especially in critical digital ischemia. There has been enthusiasm about therapies that are effective for PAH in digital vasculopathy. Thus, in 2
■Recommended in poor-prognosis diffuse cutaneous systemic sclerosis (SSc)
■Patients should not have severe organ manifestations, which render this option highly toxic
Pro Contra
■Improved long-term survival
■10% Transplant-related mortality
■Improved long-term
■10% Transplant-related mortality
■Patients with cardiopulmonary disease have to be excluded
■Patients with cardiopulmonary
survival
■Improved event-free survival
■Improved event-free
disease have to be excluded
1100
survival
Algorithm summarizing current approach to management of systemic sclerosis
Systemic slerosis
lSSc dSSc Overlap SSc
Therapy: Manage according to severity and activity of overlap features - arthritis, myositis, lupus
Vascular Therapy: Vascular Immunosupressive Antifibrotic
Identification and treatment of organ-based complications
large, controlled trials, bosentan, an oral dual-specificity endothelin receptor antagonist, was shown to significantly reduce the number of new digital ulcers, compared with placebo.89 However, no positive effect on healing of established ulcers was demonstrated. Other agents, such as phosphodiesterase Type 5 inhibitors sildenafil and tadalafil, also have been used for treatment of RP and digital ulcers, but prospective clinical trial data are not available.90
Surgical treatments include digital microarteriolysis, which can benefit single fingers with refractory ulcers. Whenever possible, surgical amputation of digits is avoided, and prolonged treatment with parenteral prostacyclin in combination with phosphodiesterase Type 5 inhibitors and potent analgesia may help with this. Lumbar sympathectomy may be helpful for lower-limb RP or ulceration. Generally, a temporary procedure is performed initially to determine the likely benefit from a definitive sympathectomy. In cases of critical digital ischemia, antiplatelet therapies are often given, with anecdotal reports of the benefit of clopidogrel in preventing digital infarction (see Table 63-7).
SKIN INVOLVEMENT
SKIN INVOLVEMENT
Key elements in the management of skin manifestations of SSc are physical therapy and regular exercise
to maintain circulation, joint mobility, and muscle strength, all aimed at improving the quality of life of SSc patients. Skin affected by scleroderma tends to be very dry, taut, and susceptible to trauma. Skin hardening can be improved by physical therapy and exercise, lymphatic drainage, topical treatment with steroids, calcineurin inhibitors, and moisturizing crèmes. Systemic therapies include immunosuppressive drugs, systemic steroids (for just a short time), and phototherapy (ultraviolet A1 or psoralen and ultraviolet A). Ultraviolet A1 phototherpay appears to inhibit fibrotic and inflammatory processes and reduce the amount of sclerotic skin.91
Dry and itching skin should be treated topically with corticosteroids, cannabinoid agonists, capsaicin, emollients, and phototherapy (see above). Local steroid injections and laser or surgical therapies could also be tried for the treatment of calcinosis cutis. Laser therapies or noninvasive methods like camouflage have been used for telangiectases. Bleaching agents, salicylic acids, and chemical peels, as well as camouflage, retinoids, and corticosteroids, may have the potential to improve hyperpigmentation or hypopigmentations (see Table 63-8). Two randomized clinical trials have shown that methotrexate improves the skin score in early diffuse SSc, while positive effects on other organ manifestations have not been established.92,93 On the other hand, in two randomized clinical trials, cyclophosphamide improved skin sclerosis.94,95 For balancing efficacy and side effects, MMF might be an interesting option to influence skin fibrosis.74 Protein kinase inhibitors (eg, imatinib) have been used, but as of this writing, controlled clinical trials have been mixed and suggest poor tolerability.96,97
CARDIOPULMONARY MANIFESTATIONS
CARDIOPULMONARY
MANIFESTATIONS
It is increasingly appreciated that a group of patients with some lung fibrosis have predominantly PAH and that this group may respond to standard PAH therapies. There have been substantial advances in the treatment of PAH over the past decade. Most cases are treated with oral agents, either an endothelin receptor antagonist (eg, bosentan, ambrisentan) or a phosphodiesterase 5 inhibitor (eg, sildenafil, tadalafil), once the PAH is significantly functionally limited (New York Heart Association class III). Later, if progression takes place, a combination of oral treatment or introduction of parenteral prostacyclin is used, by either the IV or subcutaneous route. Inhaled delivery systems for iloprost are available. Although PAH is probably responsible for more deaths than lung fibrosis in SSc, lung fibrosis remains an important complication. Treatment of SSc-PF remains challenging.98 Adding to a substantial body of uncontrolled or retrospective data suggesting benefit for cyclophosphamide in SSc-PF, the results of 2 randomized, double-blind, placebo-controlled trials have been
10
reported. Both show a modest placebo-subtracted benefit for cyclophosphamide.99-101 For change in forced vital capacity (percent predicted), this was statistically significant for the Scleroderma Lung Study comparing oral cyclophosphamide with placebo, showing a strong trend (p = 0.06) in the trial of IV cyclophosphamide followed by oral azathioprine. At present, most centers use cyclophosphamide as treatment for severe or progressive SSc-PF, defining the extent and severity by pulmonary function tests and HRCT. The extent of disease by HRCT and a history of progressive restrictive abnormality on pulmonary function tests is the best predictor of future decline in lung function and is generally used to make decisions about therapy. Newer clinical trials have shown equal efficacy in the treatment of both MMF and cyclophosphamide for stabilizing lung function of patients with scleroderma and ILD.74 MMF therapy is associated with a stability of lung function for up to 36 months, with a better side-effect profile than in patients treated with azathioprine.102 Other therapies that are in use include carbocysteine and low-dose corticosteroids. The place of other immunosuppressive strategies remains uncertain and requires evaluation in prospective multicenter clinical trials. It is noteworthy that despite there being a strong theoretic rationale for using the endothelin receptor antagonist bosentan as a therapy for lung fibrosis, bosentan was not superior to placebo in a recent large multicenter study of SSc-PF cases. Cardiac involvement from SSc is also an important contributor to mortality but remains one of the least-well understood and poorly recognized of the internal organ complications of SSc. A large number of studies confirm that radionuclide imaging, electrophysiologic, and functional abnormalities are frequent in SSc, but the significance of these findings is uncertain. Hemodynamically significant cardiac involvement occurs in up to 10% of cases of diffuse cutaneous SSc. An inflammatory component of myocarditis may respond to immunosuppressive treatment, and so an operational approach to management of cardiac scleroderma. Although this is not yet based on sufficient reliable data, it could form a basis for prospective evaluation of the significance of impaired left ventricular ejection fraction and elevated circulating troponin levels in SSc. There have been many advances in SSc, including a better appreciation of the diversity of the condition, improved understanding of the underlying pathologic mechanisms, and major progress in treating organbased complications. This includes the accumulation of robust clinical trial data that demonstrate effectiveness or lack of benefit of individual therapies and in validation of measures of disease assessment.103
GI INVOLVEMENT
GI INVOLVEMENT
Involvement of the GI tract occurs frequently in SSc. Esophageal symptoms can respond very well to proton pump inhibitors and agents that increase lower esophageal sphincter tone such as domperidone, although high-dose treatment may be associated with an increased risk of cardiac arrhythmia. Midgut
1101
10
involvement takes many forms. Pseudoobstruction requires conservative management initially, but subsequently may require parenteral nutritional supplementation. Small intestinal bacterial overgrowth can be treated using broad-spectrum antibiotics, and pancreatic insufficiency may require enzyme supplements. Large bowel involvement is a major challenge. Anorectal incontinence sometimes responds well to an implanted sacral nerve stimulator or to less-elaborate approaches, such as bioplastic injection to increase the internal anal sphincter bulk. Associated rectal prolapse may require additional surgical intervention. Chronic constipation, sometimes with overflow diarrhea, is a common problem. An adjustment to diet and judicious use of stimulating, softening, or bulking aperients is recommended, but an individualized approach with substantial patient involvement is generally the most successful approach. On occasion, defunctioning colostomy is needed, but this is only appropriate in a very limited number of cases.
SCLERODERMA RENAL CRISIS
SCLERODERMA
RENAL CRISIS
Overall, approximately two-thirds of the cases of SRC that present to a specialist center require renal replacement therapy. Of these, approximately one-half of cases eventually recover sufficiently to discontinue dialysis. This can occur over 24 months after the renal crisis, and so decisions about renal transplantation should be postponed depending on the outcome. The possibility of late recovery distinguishes SRC from other causes of end-stage renal failure. These outcomes are possible through the use of angiotensin-converting enzyme inhibitors as routine therapy for SRC. Before their availability, the mortality from established SRC was greater than 90% at 12 months. The most critical aspect of management of SRC is prompt identification and treatment of significant hypertension in the context of scleroderma, with initiation of angiotensin-converting enzyme inhibitors. This is a medical emergency and any features of renal impairment or end-organ damage should prompt hospitalization.104
OTHER SUPPORTIVE PROCEDURES
OTHER SUPPORTIVE
PROCEDURES
All these organ-specific therapeutic approaches have to be supported by several general measures to help the patients. These general measures include recommendations for keeping the home and the body warm, and for optimizing nutritional status. Paraffin waxing and physical therapy has to be provided. Patients need to be taught to deal with the complications of daily life
1102
DIFFUSE CUTANEOUS SYSTEMIC SCLEROSIS LIMITED CUTANEOUS SYSTEMIC SCLEROSIS
UNITED KINGDOM (n = 741) GERMANY (n = 1190)
UNITED KINGDOM (n = 1505)
CLINICAL FEATURES GERMANY (n = 780)
Raynaud phenomenon 95.3% 97% 96.3% 99%
Skin Hardening 95.9% 100% 90% 90%
Digital ulcerations 36% 28% 24.3% 13%
PAH 20.2% 12% 14% 15%
Lung fibrosis 62.9% 38% 26.7% 16%
GI tract involvement 65.2% 90% 60.7% 90%
Heart involvement 20% 3% 9.9% 1%
Kidney involvement 15.9% 19% 9.7% 3%
Musculoskeletal involvement
48.8% 45% 38.8% 35%
Musculo-
48.8% 45% 38.8% 35%
skeletal involvement
and to recognize early those symptoms that indicate disease progression and new organ involvement.
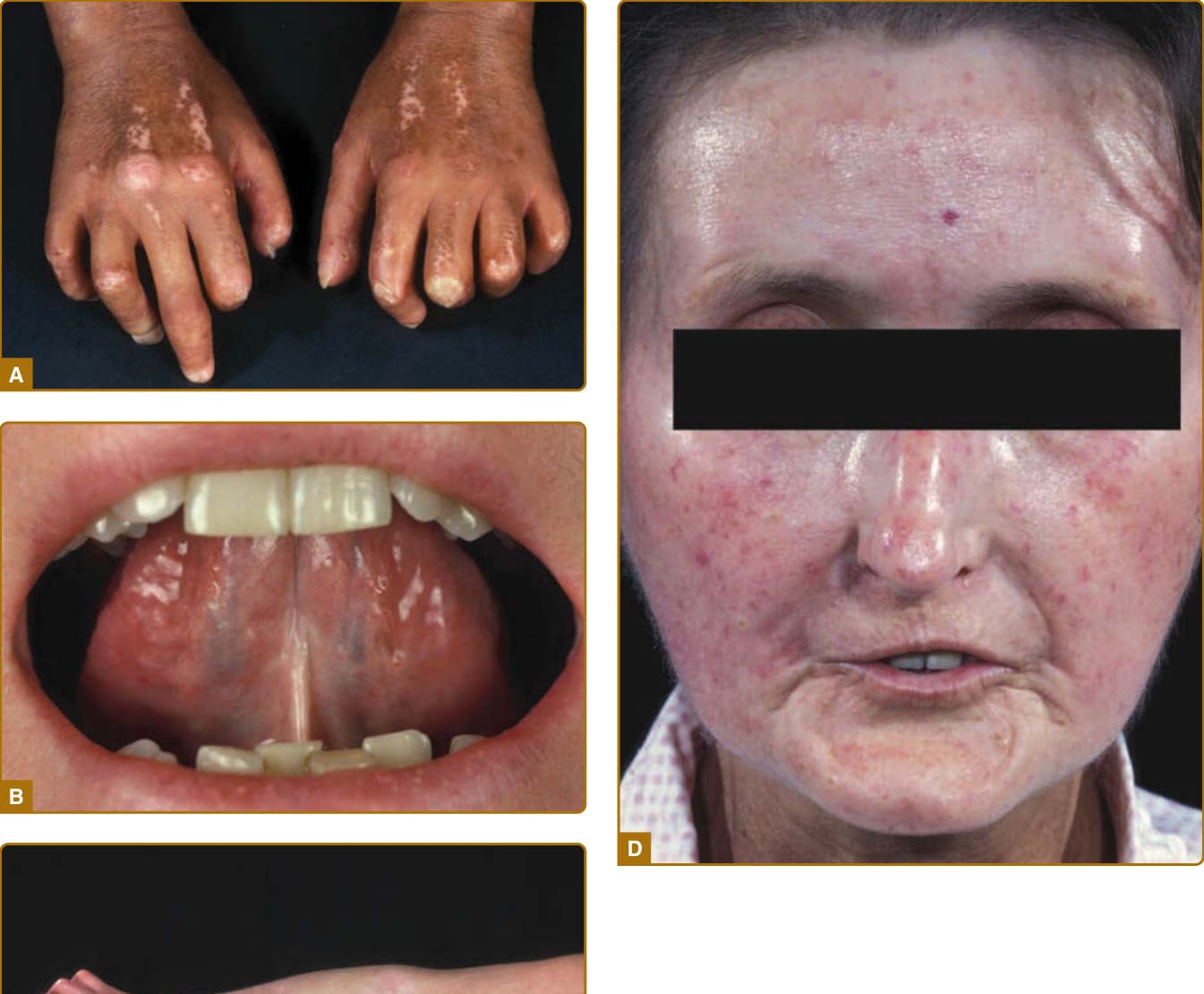
Figure 63-1 Extensive skin involvement in patients with diffuse cutaneous systemic sclerosis. A, Sclerodactyly with dermatogenous contractures (restricted mobility of digital joints) and salt-and-pepper hyperpigmentations and hypopigmentations. B, Microstomia (radial furrowing around the mouth) with frenulum sclerosis. C, Skin thickening proximal of the metacarpophalangeal joints. D, Typical scleroderma facial physiognomy with hypermimia, microstomia, telangiectasias, and a beaked nose.
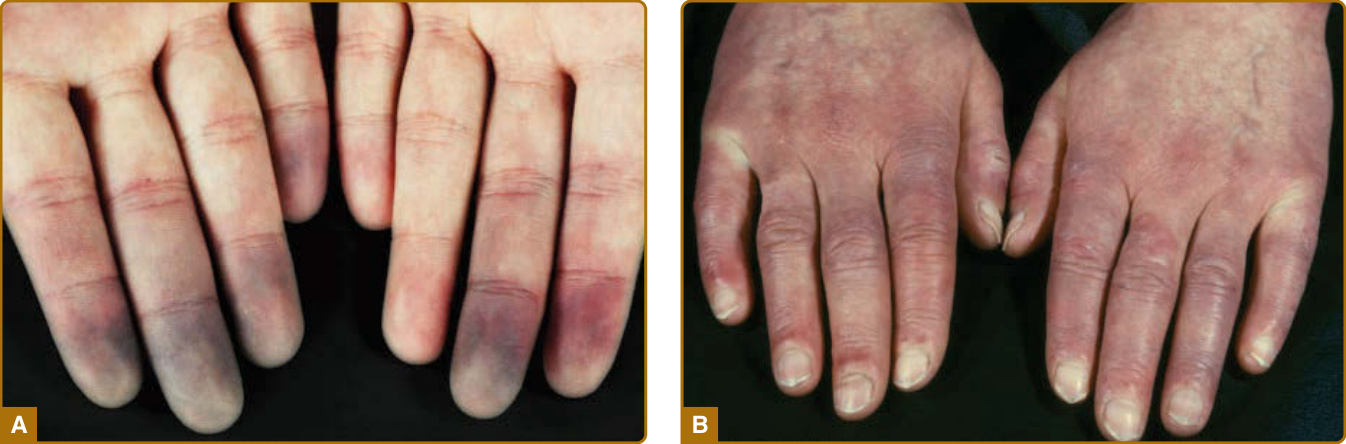
Figure 63-2 Clinical feature of patients with early disease. A, Raynaud phenomenon with typical discoloration (blue-white pallor), localized mostly at fingers and/or toes as the result of vasospasm. Coldness and emotional stress are the most frequent triggers for these attacks. B, Limited disease with puffy fingers.
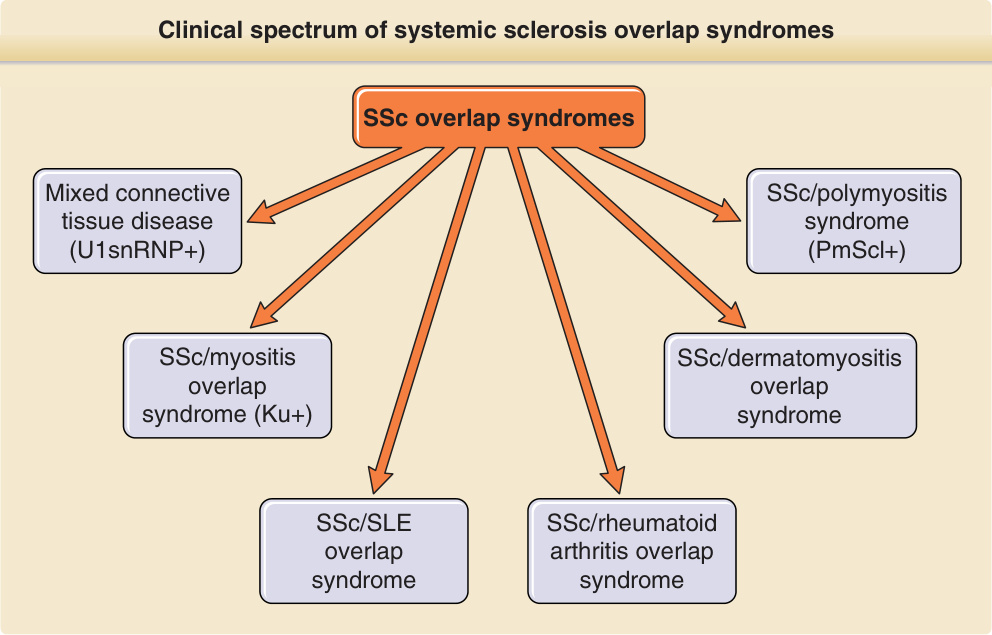
Figure 63-3 Clinical spectrum of systemic sclerosis (SSc) overlap syndromes. Patients with clinical features of scleroderma together with features of at least 1 additional autoimmune rheumatic disease are designated SSc overlap syndromes. SLE, systemic lupus erythematosus.
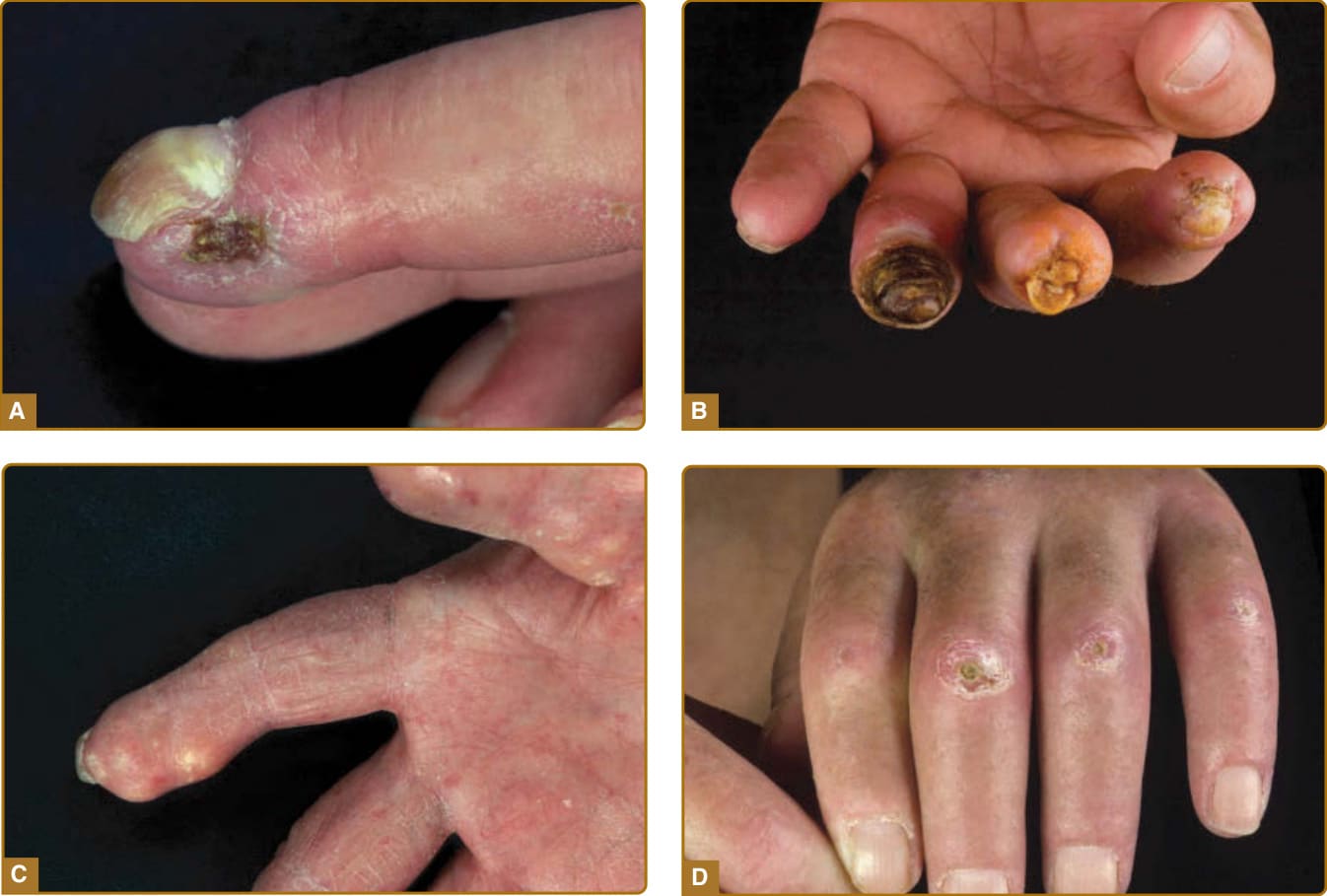
Figure 63-4 Digital alterations with complications. A, Digital ulcerations at the fingertips. B, Digital ulcerations and necrosis of the fingertips. C, Severe calcifications with deposition of subcutaneous masses. D, Multiple ulcerations at bone protuberants with inflammation in the surrounding sclerotic skin.

Figure 63-5 Pathogenesis of systemic sclerosis. The schematic shows how the development of systemic sclerosis results from a complex interplay between cells within the immune system, including adaptive and innate compartments, the vasculature, and the connective tissue. Cell–matrix interactions are important regulators of cellular functions. Early vascular events lead to later development of an autonomous population of activated fibroblasts and myofibroblasts that contract soft tissue and deposit excessive extracellular matrix proteins. These cells may develop from resident connective tissue fibroblasts; transdifferentiation from other cell types, including activated microvascular pericytes; and recruitment of circulating progenitor cells (fibrocytes). The contribution of each lineage to the fibrotic lesion is still unclear. Many growth factors and cytokines are implicated as mediators of this process, and complex reciprocal networks may lead to a profibrotic microenvironment. Potential disease-modifying therapies could target individual mediators alone or in combination (eg, tumor growth factor-β [TGF-β], endothelin [ET-1], connective tissue growth factor [CTGF], platelet-derived growth factor [PDGF]) or modulate immune cells (eg, cyclophosphamide) or the endothelial cell (eg, prostacyclin analogs). The extracellular matrix is an important repository for mediators that are later released and play a key role in pathogenesis. CCL, CC chemokine ligand; COMP, cartilage oligomeric matrix protein; EC, extracellular; ET-R, endothelin receptor; FN, fibronectin; Ig, immunoglobulin; IL, interleukin; RELM-β, resistin like molecule-β.
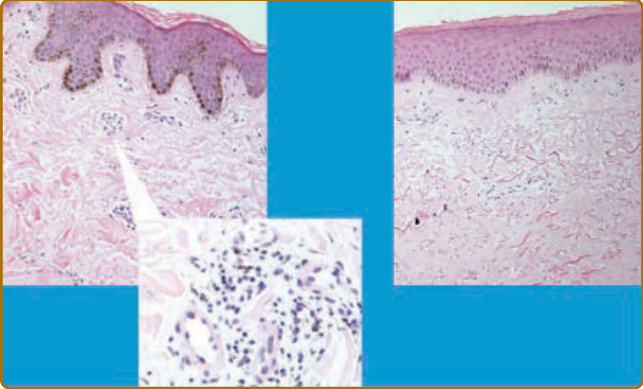
Figure 63-6 Histologic appearance of skin in early and late-stage diffuse cutaneous systemic sclerosis (SSc). In SSc, there is perivascular mononuclear cell infiltrate at the early stages of disease. This precedes the development of skin sclerosis. Perivascular changes are shown at high power in the left panel. Later stage disease is accompanied by skin sclerosis, a low density of blood vessels, and absence of inflammatory cells. At this stage, there may be associated epidermal changes with thickening and loss of secondary skin structures, including hair follicles and sweat glands. Absence of the rete ridges is also characteristic at the later stages of diffuse cutaneous SSc. Similar changes are predicted in localized cutaneous SSc, but this is rarely biopsied because of limited skin sclerosis and concerns about healing.
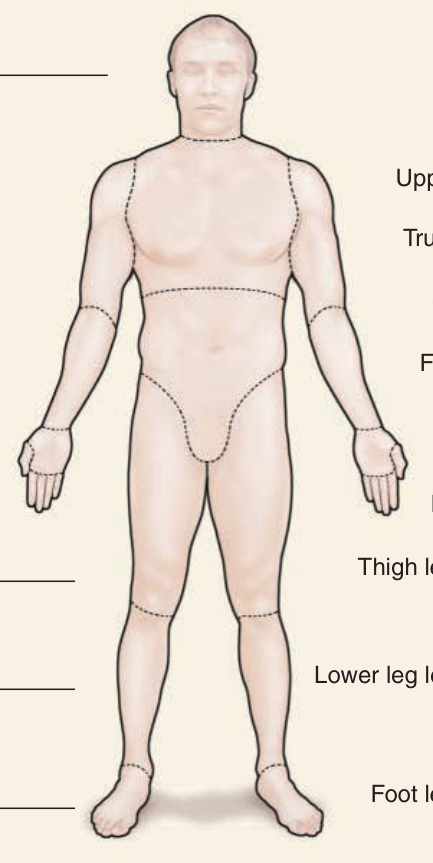
Figure 63-7 Modified Rodnan skin score (mRSS). Skin hardening evaluation using the modified mRSS is usually performed by assessing the skin thickness at 17 different areas. The skin sclerosis is categorized by palpation to grade 1, corresponding to mild, grade 2, corresponding to moderate, and grade 3, corresponding to severe. le, Left; ri, right.
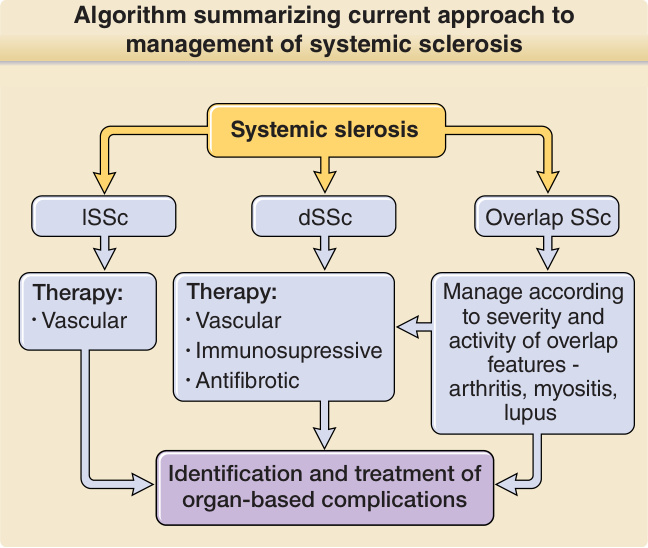
Figure 63-8 Algorithm summarizing the current approach to management of systemic sclerosis (SSc). The principles of therapy for SSc include accurate diagnosis and treatment according to the disease subset, the presence of overlap features, and the likely predominant pathologic process according to the stage of disease. In all cases, screening for and treatment of organ-based complications has a major role in successful management. Education of patients and a multidisciplinary team, including specialist nurses, physiotherapists, occupational therapists and many subspecialty physicians, and surgeons, are central to providing appropriate care for severe cases of SSc. dSSc, Diffuse cutaneous systemic sclerosis; lSSc, limited cutaneous systemic sclerosis.

TABLE 63-1 Clinical Features of Systemic Sclerosis/Myositis Overlap Syndrome

TABLE 63-2 Clinical Features of Mixed Connective Tissue Disease17,18
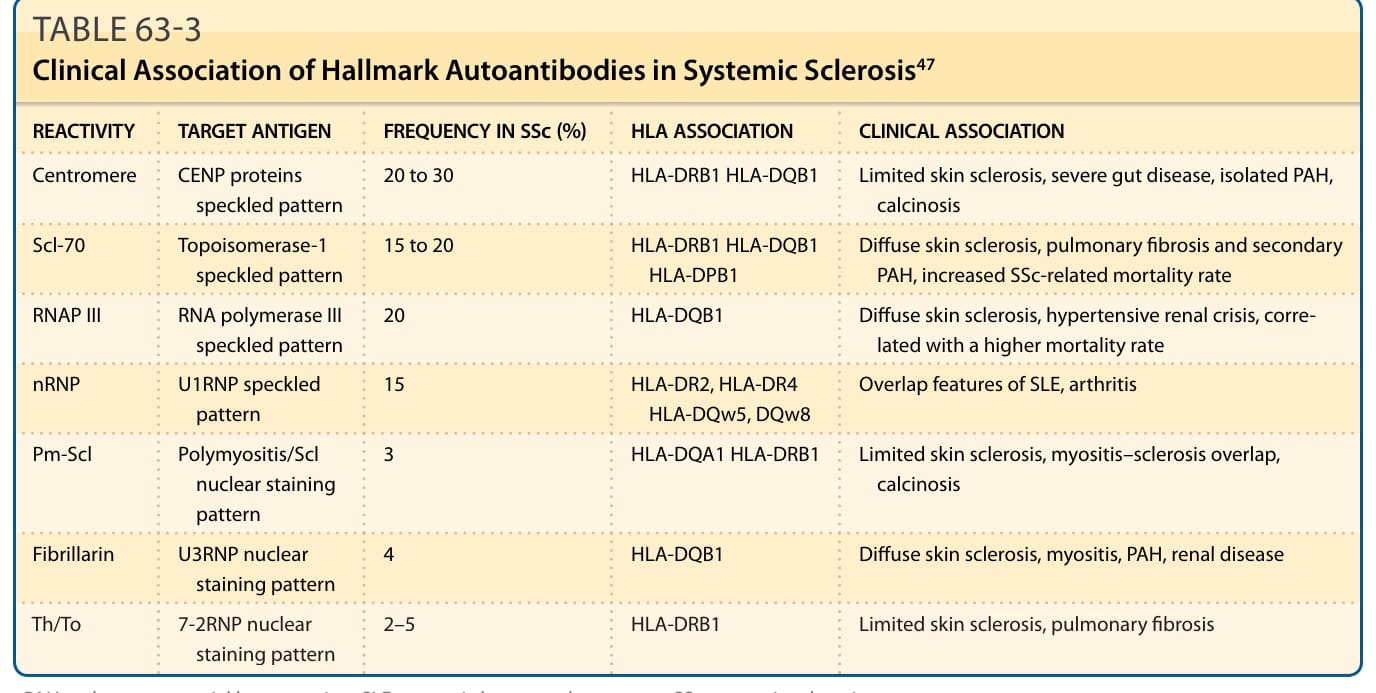
TABLE 63-3 Clinical Association of Hallmark Autoantibodies in Systemic Sclerosis47

TABLE 63-4 Functional Autoantibodies in Systemic Sclerosis and Their Association to Pathophysiology48-50
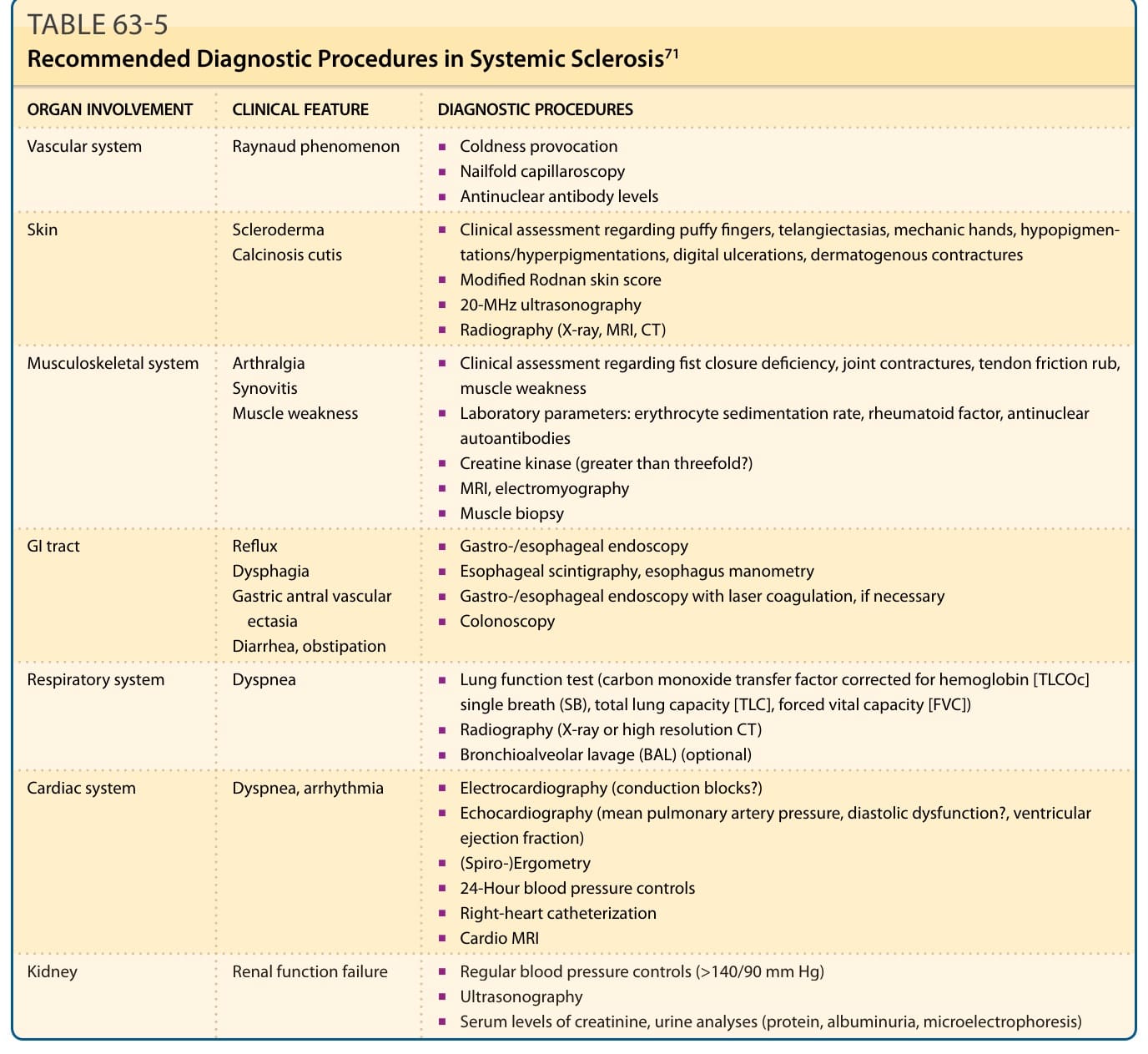
TABLE 63-5 Recommended Diagnostic Procedures in Systemic Sclerosis71

TABLE 63-6 Differential Diagnosis of Systemic Sclerosis
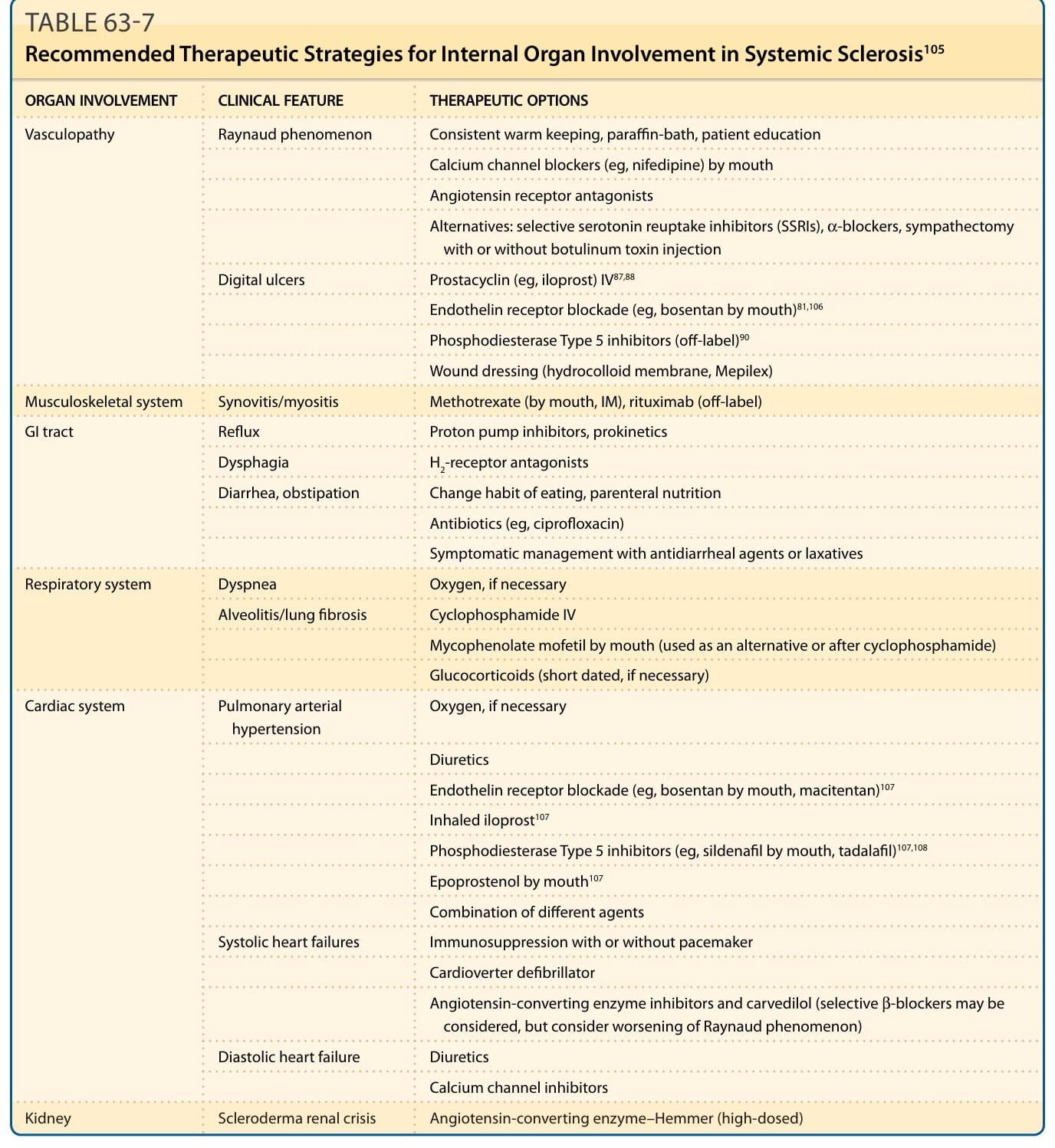
TABLE 63-7 Recommended Therapeutic Strategies for Internal Organ Involvement in Systemic Sclerosis105
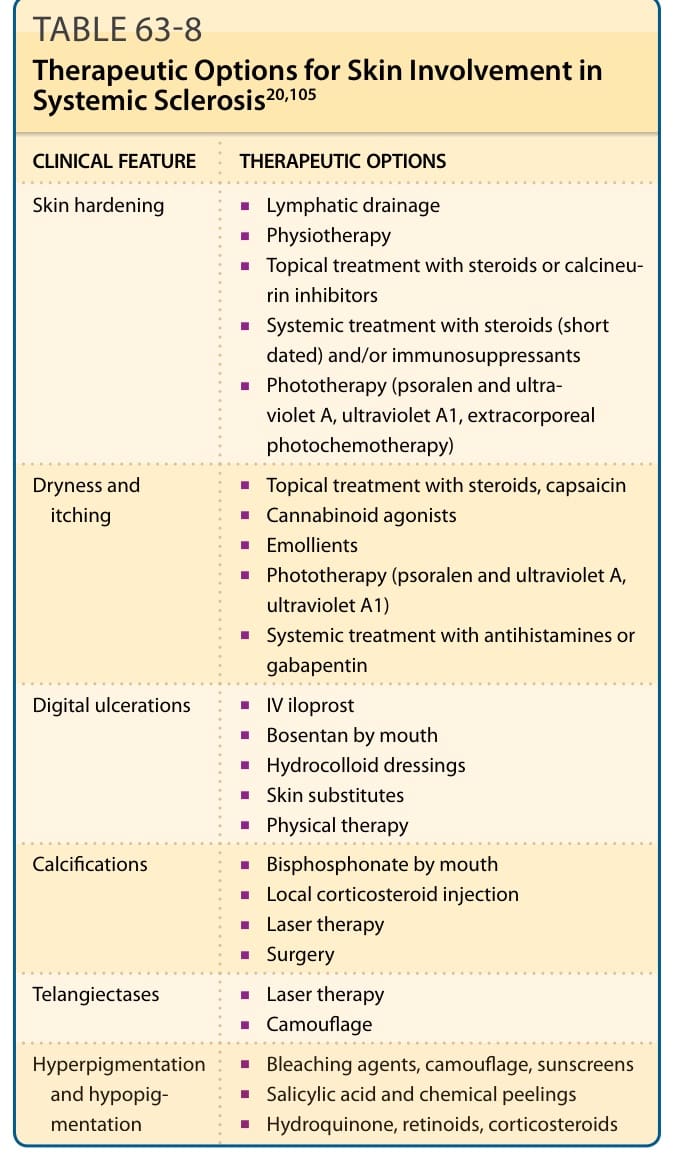
TABLE 63-8 Therapeutic Options for Skin Involvement in Systemic Sclerosis20,105

TABLE 63-9 Stem Cell Transplantation (ASCT) for Early Diffuse Cutaneous Systemic Sclerosis78-80
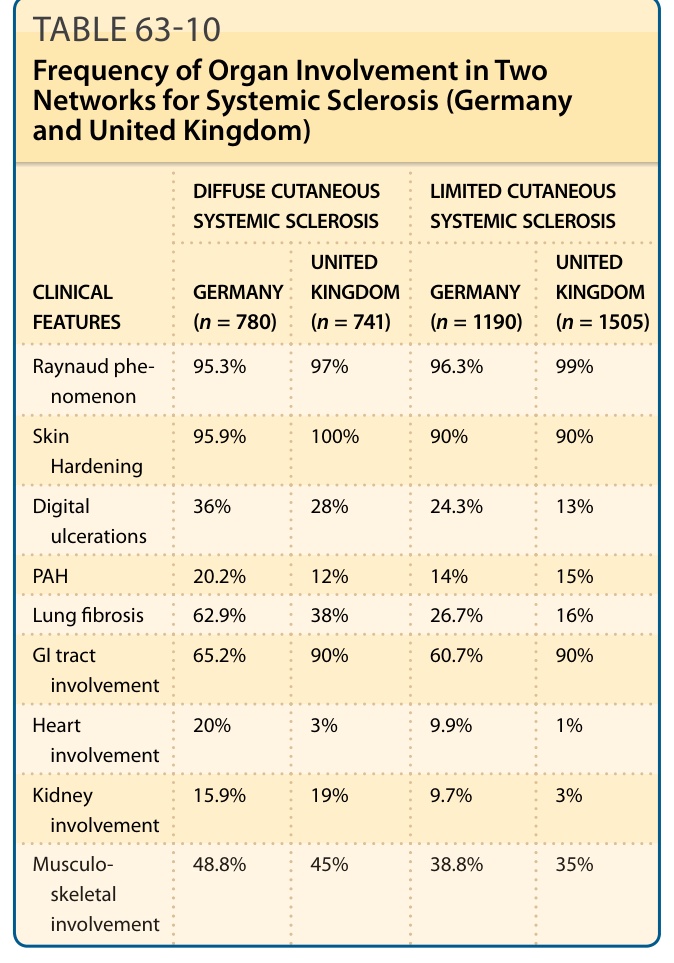
Table 63-10 illustrates the frequency of organ involvement in 2 networks for SSc (Germany and United Kingdom).