全身性硬化症 (Systemic Sclerosis)
PART 10
重點一覽 (AT-A-GLANCE)
■ 硬皮症(scleroderma,全身性硬化症 [systemic sclerosis, SSc])是一種多系統自體免疫疾病,特徵為皮膚與許多其他器官的血管病變 (vasculopathy)、發炎 (inflammation) 與纖維化 (fibrosis)。
■ SSc 的鑑別診斷包括嚴重型的局限性硬皮症 (localized scleroderma),以及許多其他類硬皮症 (scleroderma-like) 病況。
■ 雷諾現象 (Raynaud phenomenon)、循環自體抗體 (circulating autoantibodies) 與皮膚硬化 (skin sclerosis) 幾乎總是存在,對於早期診斷相當重要。
■ SSc 病人依皮膚硬化的範圍分為 2 個主要亞型(瀰漫型皮膚全身性硬化症 diffuse cutaneous systemic sclerosis 與局限型皮膚全身性硬化症 limited cutaneous systemic sclerosis)。
■ 患有重疊症候群 (overlap syndrome)(包括混合性結締組織病 mixed connective tissue disease)的病人,其特徵為具有其他風濕性疾病的額外臨床特徵。
■ 內臟器官(消化道、肺、腎與心臟)的受累可導致嚴重功能障礙,並決定預後。
■ SSc 與 SSc 重疊症候群的異質性與臨床病程,迫切需要跨學科合作 (interdisciplinary collaborations) 與定期回診追蹤。
■ 雖然本病目前仍無法治癒,但基於對病理生理學的更深入理解,在發展新療法與治療以器官為基礎的併發症方面已有顯著進展。
定義 (DEFINITIONS)
硬皮症(scleroderma,全身性硬化症 [systemic sclerosis, SSc])是一種多系統疾病,其特徵為自體免疫過程 (autoimmunologic processes)、血管內皮細胞損傷 (vascular endothelial cell injury)、發炎,以及廣泛的纖維母細胞活化 (activation of fibroblasts)。皮膚與器官受累的範圍,以及疾病進展與預後,存在巨大的個體間差異。皮膚、食道、肺、心臟與腎臟是最常受影響的器官。
流行病學 (EPIDEMIOLOGY)
女性較常受 SSc 影響,女男比介於 3:1 與 14:1 之間。¹⁻⁴ 發病年齡介於 30 至 50 歲。¹
然而,男性病人的發病較女性病人早。患有 SSc 的黑人 (Blacks) 常較白人 (whites) 年輕。SSc 是一種罕見疾病;然而,發生率 (incidence rates) 由每百萬居民 0.6 例上升至 16 例,盛行率 (prevalence rates) 則由每年每百萬居民 2 例上升至 233 例²⁻⁵,視病例定義 (case definition) 與確認方法 (ascertainment) 的方法學差異以及所研究的時間段而定。可以假定這些數字是低估的,因為輕症病人常未被診斷。SSc 在所有自體免疫風濕性疾病 (autoimmune rheumatic diseases) 中具有最高的疾病特異性死亡率 (case-specific mortality),但其因個體而異,取決於種族或族裔差異、器官受累的存在與嚴重度、SSc 亞群、診斷時的年齡,以及性別差異。雖然無法治癒,但在治療 SSc 以器官為基礎的併發症方面的治療選項已有顯著進展。
全身性硬化症的臨床特徵 (CLINICAL FEATURES OF SYSTEMIC SCLEROSIS)
SSc 通常以雷諾現象 (Raynaud phenomenon) 起始,雷諾現象可比本病早出現許多年。臨床表現在很大程度上取決於疾病的亞群與分期。已確立 SSc 的臨床特徵相當多樣,包括嚴重的皮膚纖維化以及所有其他皮膚表現。這些包括皮膚硬化、攣縮 (contractures) 的發展、指端潰瘍 (digital ulcerations) 與鈣化 (calcifications)。它們也反映了內臟器官受累的多重模式,以及血管病變、發炎與纖維化等潛在病理過程進展的後果。必須特別考量的標誌性併發症 (hallmark complications) 包括高血壓型硬皮症腎危象 (hypertensive scleroderma renal crisis, SRC)、肺動脈高壓 (pulmonary arterial hypertension, PAH)、肺纖維化 (pulmonary fibrosis, PF) 與胃腸道蠕動障礙 (GI dysmotility)。
不同 SSc 亞群的分類與定義 (CLASSIFICATION AND DEFINITION OF DIFFERENT SSc SUBSETS)
SSc 的異質性源自於病人之間在器官受累的範圍與嚴重度上各不相同的疾病表現範圍。然而,有些幾乎總是存在的臨床特徵為雷諾現象 (Raynaud phenomenon, RP) 與皮膚硬化。皮膚硬化的範圍界定了每一個主要疾病亞群,每一亞群都有其特定的臨床特性,雖然各亞群也有共同的特徵。1980 年,美國風濕病學會(American College of Rheumatology, ACR)針對已確立疾病的病人發表了 SSc 的初步分類標準⁶,該標準對 SSc 顯示 97% 的敏感度與 98% 的特異度。根據此標準,若發現 1 項主要標準或至少 2 項次要標準,則診斷成立。主要標準為掌指關節 (metacarpophalangeal) 或蹠趾關節 (metatarsophalangeal) 近端的硬皮症;次要標準包括指端硬化 (sclerodactyly)、指端潰瘍與/或點狀指端瘢痕 (pitting digital scars),以及兩肺底肺纖維化 (bibasilar PF)。雖然這些標準已使用多年,但它們無法納入早期 SSc 病人以及某些局限型皮膚全身性硬化症病人。因此,發展出了新的 ACR/歐洲抗風濕病聯盟(European League Against Rheumatism)標準,該標準以評分系統 (score system) 為基礎,並考量數項額外標準,例如異常甲褶微血管 (abnormal nailfold capillaries)、指尖病灶 (fingertip lesions) 與自體抗體。這些新標準現在能讓病人獲得早期診斷,並在廣泛纖維化發生前將這些病人納入臨床試驗。⁷
1988 年,LeRoy 引入了局限型對比瀰漫型 SSc 的描述性次分類⁸,主要與皮膚受累的範圍相關。此分類已被廣泛接受並用於臨床實務。2001 年,LeRoy 與 Medsger⁹ 發表了修訂標準,額外納入自體抗體與甲褶微血管鏡 (nailfold capillaroscopic) 的改變。此外,這些標準納入了一個獨立的早發型 SSc 病人群組,這些病人僅有極輕微的皮膚增厚。早期(局限型)SSc 病人必須具有 RP 的證據,加上硬皮症特異性自體抗體 (scleroderma-specific autoantibodies) 與/或甲褶微血管鏡表現。¹⁰,¹¹ 雖然另有數種已發表的分類,例如 Nadashkevich 與同事以及 Maricq 與 Valter 所提出者¹⁰,¹²,
最初的 LeRoy 分類在日常臨床實務中仍被廣泛使用。瀰漫型皮膚 SSc 被定義為一種進行性的 SSc 型態,RP 早期發病,通常在皮膚增厚發病後的 1 年內出現。此亞群的特徵為軀幹、顏面、上臂與大腿的快速皮膚受累,極常出現抗硬皮症 70(anti–scleroderma 70,抗拓樸異構酶-I antitopoisomerase-I)或抗 RNA 聚合酶 III(anti-RNA polymerase III)抗體。⁸ 此外,發展出肺纖維化、心臟受累與硬皮症腎危象的傾向較高(圖 63-1)。局限型皮膚 SSc 的特徵為長期既存的 RP 病史,以及膝關節與肘關節遠端肢體的皮膚變化(包括顏面皮膚)。⁸ 此 SSc 亞群變異型常(50% 至 70% 的病例)以抗著絲點抗體 (anticentromere antibodies) 表現,並常與孤立性 PAH (isolated PAH) 相關。傳統縮寫 CREST(鈣質沉著 calcinosis、RP、食道蠕動障礙 esophageal dysmotility、指端硬化 sclerodactyly 與毛細血管擴張 telangiectasias)即指 SSc 的局限型(圖 63-2)。
患有早期 SSc(亦稱為未分化 SSc undifferentiated SSc)的病人,其定義為 RP 陽性且至少有 1 項 SSc 的額外特徵(甲褶微血管改變陽性、腫脹手指 puffy fingers、肺動脈高壓),與/或可偵測到硬皮症相關自體抗體,但尚未符合 ACR 標準。¹,¹³,¹⁴
極小比例的病例(1.5%)會發展出 SSc 的血管性(RP 與/或 PAH)、免疫學(最常見為抗著絲點抗體)與以器官為基礎的纖維化特徵,但不出現皮膚硬化。¹⁵
患有此亞群的病人被歸類為無硬皮的 SSc (SSc sine scleroderma)。同時具有硬皮症特徵與另一種自體免疫風濕性疾病特徵的病人,被命名為 SSc 重疊症候群 (SSc overlap syndrome)(圖 63-3 與表 63-1)。SSc 重疊症候群的定義為一種同時出現 SSc 臨床面向(依 ACR 標準)或 SSc 主要症狀,並伴隨其他結締組織疾病或其他自體免疫疾病(如皮肌炎 dermatomyositis、修格連症候群 Sjögren syndrome、全身性紅斑性狼瘡 systemic lupus erythematosus、血管炎 vasculitis 與多發性關節炎 polyarthritis)症狀的疾病。這些病人多半呈現高效價的抗 U1RNP、抗 nRNP、抗纖維蛋白原 (antifibrillarin) 或抗 PmScl 抗體。¹⁶
此群病人包括界定明確的混合性結締組織病 (mixed connective tissue disease, MCTD) 病人,其特徵為高效價的循環抗 U1RNP 抗體(表 63-2)。關於 MCTD 究竟代表一個獨立的疾病實體,還是可能為另一種結締組織疾病的早期型態,目前仍有持續的爭論。這些 MCTD 病人具有多變的臨床特徵,伴有全身性紅斑性狼瘡或類風濕性關節炎 (rheumatoid arthritis) 的症狀加上雷諾症候群 (Raynaud syndrome),並在後期發展出硬皮樣病灶 (sclerodermatous lesions)。他們有腫脹的手指與浮腫的手 (puffy hands)。非雷諾症狀包括肢端 (acral) 區域的皮膚硬化,以及較後期出現的內臟表現。常有劇烈的發炎症狀並伴隨嚴重關節痛 (arthralgia)。MCTD 病人可發展出心包炎 (pericarditis)、胸膜炎 (pleuritis) 與肺動脈高壓。然而,他們對抗發炎/抗免疫治療反應良好,且預後明顯優於典型硬皮症病人。¹⁷,¹⁸
重疊症候群中另一個已界定明確的亞群是具有硬皮樣病灶且同時罹患劇烈肌炎 (myositis) 的病人。這些病人通常以特異性 Pm-Scl 自體抗體為特徵,具有典型的技工手 (mechanic hands),並早期發展出劇烈的皮下鈣化 (subcutaneous calcifications)。與 MCTD 病人類似,他們對早期抗發炎治療(如甲胺喋呤 methotrexate/糖皮質類固醇 glucocorticosteroids)反應良好。¹⁹
其他重疊症候群包括具有雷諾現象與紅斑性狼瘡或類風濕性關節炎症狀,並在後期發展出硬皮樣病灶的病人。這些病人中許多具有特異性循環自體抗體,可能代表不同的疾病實體。然而,仍需使用分子標記 (molecular markers) 的進一步研究來釐清它們在硬皮症相關疾病譜中的疾病身分。¹⁹
此外,SSc 不同內臟表現的頻率與時間點在主要亞群之間有所不同。然而,各亞群之間在以器官為基礎的疾病以及皮膚硬化的範圍與嚴重度方面存在一些重疊。在所有病人中,皮膚硬化的範圍與嚴重度可用改良式 Rodnan 皮膚評分 (modified Rodnan skin score) 來評估。瀰漫型皮膚 SSc 的基線皮膚評分與疾病嚴重度及結果相關。皮膚的增厚與纖維化作為 SSc 最早被辨識的現象之一,至今仍構成本疾病譜大多數分類標準與所提出亞群的基礎。也必須提到的是,已有人提出另一種完全以自體抗體為基礎的 SSc 相關疾病分類。有來自關聯性研究 (association studies) 的證據顯示這可能具有臨床意義,如表 63-3 所示。此外,使用候選基因方法 (candidate gene approach) 的遺傳關聯分析已證明,以血清學為基礎 (serologically based) 的 SSc 亞群之間存在比整體 SSc 更強的關聯性。其意義尚不確定,並應注意的是,
自體抗體反應性的遺傳基礎已被詳盡描述,導致有人提出血清學亞群可能比未經選擇的 SSc 病例或臨床定義的 SSc 亞群在遺傳上更為同質。可能需要在多國病人世代中使用分子與臨床標記進行大規模研究,以修訂目前這個異質性疾病譜的分類系統。
器官表現 (ORGAN MANIFESTATION)
雷諾現象 (RAYNAUD PHENOMENON)
本病的特徵性表現為 RP 的初始發病,其出現於超過 90% 的 SSc 病人(見圖 63-2)。¹ 它的定義為手指與腳趾的小指動脈/小動脈反覆發作的血管痙攣 (vasospasm),通常由寒冷與/或其他刺激(例如情緒壓力)所引起。RP 臨床上突然出現,明顯有界限,並伴隨單一或數個手指/腳趾的疼痛性蒼白/缺血 (pallor/ischemia),在 RP 發作結束時復溫後接著出現反應性充血 (reactive hyperemia),在某些情況下也會接著出現發紺 (cyanosis)(三相性 RP triphasic RP)(見圖 63-2)。
皮膚受累 (SKIN INVOLVEMENT)
皮膚受累是 SSc 的主要特徵 (cardinal feature),通常最先出現於手指與手部。隨著時間進展,病人發展出手指(腫脹手指 puffy fingers)、手部與四肢的非凹陷性水腫 (nonpitting edema),隨後出現逐漸增加的硬化 (induration) 與皮膚增厚(指端硬化 sclerodactyly)(見圖 63-1 與 63-2)。視皮膚增厚的部位而定,可能出現關節活動受限(皮源性攣縮 dermatogenous contractures)與/或呼吸活動度受限。典型的顏面特徵包括毛細血管擴張 (telangiectasias)、鳥喙狀鼻 (beak-shaped nose) 與口裂縮小 (microstomia)。SSc 病人典型的顏面外觀特徵為口周的放射狀皺褶 (radial furrowing)、無表情、僵硬且面具般 (mask-like) 的顏面外觀,以及繫帶 (frenulum) 的硬化。除了美觀/審美問題外,這還造成進食與口腔衛生方面的相當困難(見圖 63-1)。皮膚與/或皮下鈣質的異常沉積(皮膚鈣質沉著症 calcinosis cutis)通常發生於壓力點(肢端、關節)上方(圖 63-4)。關節旁的皮膚鈣質沉著症即為 Thibierge-Weissenbach 症候群。其他皮膚表現包括色素減退與色素沉著(鹽和胡椒狀 salt-and-pepper)皮膚(見圖 63-1),以及毛囊與汗腺的喪失(少汗症/無汗症 hypohidrosis/anhydrosis)。²⁰
約 50% 的 SSc 病人在其病程中的某個時間點受到與血管病變相關的指端潰瘍 (digital ulceration) 影響。這是結構性血管疾病 (structural vessel disease) 的主要外部特徵,可能歸因於增厚的內膜 (intima) 與管腔阻塞的血管。觸痛且疼痛的點狀瘢痕 (pitting scars) 很常見,偶爾會進展為潰瘍。這些病灶發生於手指或腳趾尖端、關節的伸側 (extensor surfaces)(因微創傷 microtrauma),或與上述皮膚鈣質沉著症相關。指端潰瘍與強烈的局部疼痛相關,並對日常功能(如穿衣、進食)方面的生活品質造成重大影響。其他併發症包括嚴重指端缺血 (critical digital ischemia)、甲溝炎 (paronychia)、感染、壞疽 (gangrene)、骨髓炎 (osteomyelitis),以及指腹喪失 (finger pulp loss) 或截肢 (amputation)。
心肺表現 (CARDIOPULMONARY MANIFESTATIONS)
心肺系統有不同的受累方式,最常表現為纖維化與 PAH。這些表現之間的區分在臨床上常很困難,因為有相似的重疊臨床特徵,例如呼吸困難 (dyspnea)、非生產性咳嗽 (nonproductive cough)、瀰散能力 (diffusion capacity) 障礙與發紺。PAH 目前是 SSc 中最常見的疾病相關死亡原因。²¹ 它發生於局限型與瀰漫型皮膚亞群中,雖然最典型的病例是局限型 SSc 合併孤立性 PAH 者。此病況與特發性 PAH (idiopathic PAH) 有實質上的相似之處。因此,SSc 中出現 2 種疾病模式。大多數病例為 PAH,但有部分病人在 SSc 後期出現廣泛的間質性肺纖維化 (interstitial lung fibrosis),並發展出真正的繼發性肺高壓 (secondary pulmonary hypertension)。²² 除了 PAH 所引起的右心惡化外,心臟也可能因瀰漫性或局灶性纖維化或發炎性心肌炎 (inflammatory myocarditis) 而受累。這可能導致舒張性 (diastolic) 或收縮性 (systolic) 功能障礙,以及心肌收縮性受限。這些病人臨床上表現出心律不整 (cardiac arrhythmia)、陣發性心搏過速 (paroxysmal tachycardia)、不完全或完全的右束支傳導阻滯 (right-heart blocks),以及心臟功能不全 (heart insufficiency)。²³
胃腸道受累 (GI INVOLVEMENT)
胃腸道受累是局限型與瀰漫型 SSc 病人中最常見的內臟器官受累(>60%)。¹ 胃腸道的許多部分都可能受損,影響蠕動 (motility)、消化 (digestion)、吸收 (absorption) 與排泄 (excretion)。²⁴
食道受累包括如吞嚥困難 (dysphagia)、因逆流 (reflux) 引起的胃灼熱 (heartburn)、噁心與/或嘔吐等症狀。下食道括約肌 (lower esophageal sphincter) 減弱與蠕動受損會增加食道炎 (esophagitis) 的風險。若未治療,這可能導致消化性食道炎 (peptic esophagitis)、胃/食道潰瘍、消化性狹窄 (peptic stricture) 形成與瘻管 (fistulae)。慢性胃食道逆流經過一段時間後可能併發較高的巴瑞特食道 (Barrett esophagus) 風險,後者可能進展為腺癌 (adenocarcinoma)。可能的胃部表現包括黏膜萎縮相關潰瘍與胃排空延遲 (delayed gastric emptying)。胃竇血管擴張 (gastric antral vascular ectasia) 也是某些 SSc 病人的重要併發症,需透過內視鏡 (endoscopy) 偵測,因為它可導致嚴重且常未被辨識的出血。²⁵
SSc 也可影響腸道,包括張力缺乏性擴張 (atonic dilation)、狹窄、吸收不良 (malabsorption)、假性阻塞 (pseudoobstruction)、腹瀉、便秘、糞便失禁 (fecal incontinence) 與嚴重營養不良 (malnutrition)。
腎臟受累 (KIDNEY INVOLVEMENT)
SRC 出現於 5% 至 10% 的 SSc 病人,可能造成顯著全身性高血壓 (>140/90 mm Hg,或收縮壓/舒張壓升高 ≥30/≥20 mm Hg) 的突然發病,伴隨血清肌酸酐 (serum creatinine) 上升、蛋白尿 (proteinuria)、血尿 (hematuria)、血小板減少 (thrombocytopenia) 或溶血 (hemolysis),隨後出現急性腎衰竭 (acute renal failure)。²⁶ 研究顯示,伴有腎絲球過濾率 (glomerular filtration rate) 降低的慢性血管病變很常見。此外,有證據顯示 SSc 中腎間質 (renal interstitium) 內纖維狀膠原蛋白 (fibrillar collagen) 沉積增加。許多病例發生於疾病的前 12 個月內,且在高達 25% 的 SRC 病人中,SSc 的診斷是在腎臟表現出現時才做出。終末器官損傷 (End-organ damage) 可導致伴有全身性癲癇發作的腦病變 (encephalopathy),或急性閃電性肺水腫 (flash pulmonary edema)。微血管病性貧血 (microangiopathic anemia) 很常見,有時會發展出瀰漫性血管內凝血 (disseminated intravascular coagulation)。SSc 病人應避免使用腎毒性藥物 (nephrotoxic drugs) 與高劑量普賴鬆龍(prednisolone, >7.5 mg/day)。²⁷
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
此複雜自體免疫疾病的致病機轉涉及多種細胞類型(內皮細胞 endothelial cells、上皮細胞 epithelial cells、纖維母細胞 fibroblasts 與淋巴細胞),它們透過多種機轉相互作用,而這些機轉取決於其微環境 (microenvironment) 與數個關鍵介質 (mediators)。本病的主要面向包括發炎、血管系統 (vasculature) 與產生結締組織細胞的活化 (activation of connective tissue-producing cells)(圖 63-5)。SSc 的臨床異質性使得不同的致病機轉很可能在特定病人或疾病亞群中佔主導地位。同樣地,關鍵途徑在 SSc 的不同階段也不一定相同。雖然致病機轉可能存在遺傳成分,且有證據支持遺傳因子決定嚴重度與易感性,但也有強力論點支持環境與化學因子作為本病的觸發因素。²⁸
遺傳因子 (GENETIC FACTORS)
支持 SSc 及相關疾病具有遺傳貢獻的最佳證據來自報告家族聚集性 (familial clustering) 的研究,以及已進行的有限雙胞胎研究。雖然家族性發生 SSc 的絕對風險相對較低,但與一般族群相比,一等親 (first-degree relatives) 的相對風險高出 13 倍。²⁹ 數個研究顯示,SSc 的陽性家族史是最強的危險因子,但族裔 (ethnicity) 也有所貢獻。²⁹ Assassi 與同事指出,受 SSc 影響家族的成員傾向呈現一致的硬皮症特異性自體抗體。³⁰ 使用候選基因方法的遺傳關聯研究提供了進一步支持。最大的成功出現在對本病個別組成成分(如自體抗體圖譜 autoantibody profiles)的遺傳分析中。這些似乎具有強烈的遺傳決定性,而這可能是 SSc 標誌性反應性 (hallmark reactivities) 明顯互斥 (mutual exclusivity) 的基礎。已證明對特定 SSc 相關抗原 (SSc-associated antigen) 產生免疫反應的能力受主要組織相容性複合體 (major histocompatibility complex) 單倍型 (haplotype) 所限制。數個研究顯示 HLA-DRB1∗1302 與 HLA-DQB1∗0604/0605 單倍型與抗纖維蛋白原陽性 (antifibrillarin-positive) 病人有關³¹,而 HLA-SRB1∗0301 出現於具有抗 Pm-Scl 抗體的病人中。³² 來自大量檢視遺傳標記研究的觀察已辨識出多個候選基因(例如貧血誘導因子 [anemia-inducing factor, AIF]-1、分化簇 [cluster of differentiation, CD] 19、CD22、CD86、細胞毒性 T 淋巴細胞抗原 [cytotoxic T-lymphocyte antigen, CTLA]-4、CCL-2、CCL-5、趨化激素配體 [chemokine ligand, CXCL]-8、趨化激素相關受體 [chemokine-related receptor, CXCR]-2、介白素 [interleukin, IL]-1α、IL-1β、IL-2、IL-10、IL-13、巨噬細胞遷移抑制因子 [macrophage migration inhibitory factor, MIF]、蛋白酪胺酸磷酸酶非受體 22 [protein tyrosine phosphatase non-receptor 22, PTPN22]、腫瘤壞死因子 [tumor necrosis factor, TNF]-α)。²⁹,³³ 大多數較近期的全基因組關聯研究 (genome-wide association studies) 已辨識出與先天免疫系統 (innate immune system) 相關的基因座;其中某些關聯已相當穩健,並可能導向新的治療方法。其他則反映了結締組織反應的改變。然而,與其他複雜疾病一樣,在很多情況下,最初看似有希望的數據並不總能被重複驗證。對遺傳上同質的族群進行的研究特別具有資訊價值,包括對美洲原住民喬克托族 (Choctaw Nation of Native Americans) 的研究。然而,有趣的是,某些關聯在分子致病機轉方面非常合理。上位效應 (epistasis) 與多個修飾基因 (modifier genes) 的效應很可能混淆了 SSc 中簡單的遺傳關聯研究,就如同其他複雜疾病一樣。³⁴ 有越來越多證據顯示,透過調節染色質結構與對免疫反應活化及/或纖維化反應至關重要之細胞激素/生長因子的基因表現的表觀遺傳機轉 (epigenetic mechanisms),是促成硬皮症發展的重要額外因子。
環境因子 (ENVIRONMENTAL FACTORS)
類硬皮症症候群 (Scleroderma-like syndromes) 已被報告與許多環境毒素及藥物相關。這些物質包括溶劑(氯乙烯 vinyl chloride、苯 benzene、甲苯 toluene、環氧樹脂 epoxy resins)、藥物(博來黴素 bleomycin、卡比多巴 carbidopa、潘他唑新 pentazocine、古柯鹼 cocaine、歐洲紫杉醇 docetaxel、間苯二胺 metaphenylenediamine),以及其他各種物質。³⁵
SSc 曾被報告發生於地下煤礦工與金礦工身上。在年齡大於 40 歲患有矽肺症 (silicosis) 的男性病人中,發展出 SSc 的可能性約為未暴露於矽 (silica) 男性的 190 倍,並為無矽肺症但暴露於矽塵 (silica dust) 男性的 50 倍。³⁶ 矽膠植入物 (silicone gel implants) 與其他矽膠產品在硬皮症發展中的角色已受到質疑。³⁷
然而,大多數流行病學研究未能顯示顯著的關聯。一種不尋常型態的硬皮症,特徵為 RP、硬斑病樣 (morphea-like) 皮膚變化、甲褶的微血管異常(類似 SSc 者)、遠端指骨 (distal phalanges) 的骨溶解 (osteolysis),以及肝臟與肺纖維化,可能發生於暴露於聚氯乙烯 (polyvinyl chloride) 的工作者。博來黴素 (Bleomycin) 也會產生肺纖維化、RP 與無法與 SSc 區分的皮膚變化。³⁷ 這些變化的發展似乎是劑量依賴性的,並在停藥後可逆。總體而言,化學物質暴露僅佔類硬皮症疾病的一小部分。大型流行病學研究尚未揭示毒素與藥物在硬皮症中的顯著角色。
組織病理學 (HISTOPATHOLOGY)
SSc 的組織病理學顯示真皮 (dermis) 下三分之二與皮下纖維小梁 (subcutaneous fibrous trabeculae) 的纖維化,這是因為細胞外基質 (extracellular matrix, ECM) 蛋白的過度沉積,最顯著的是第 I 型與第 III 型膠原蛋白 (collagen Types I and III)(圖 63-6)。²⁸
脂膜炎 (Panniculitis) 與黏液樣水腫 (mucoid edema) 在早期階段也可能是顯著特徵,此時皮下脂肪被纖維結締組織所取代。在組織學上可以區分一個早期細胞期 (cellular stage) 與一個較晚期纖維化期 (fibrotic stage)。在早期階段,真皮網狀層 (reticular dermis) 內病理性地呈現膠原束 (collagen bundles),外觀蒼白、均質、與皮膚表面平行走向、腫脹,且常有血管周圍淋巴細胞浸潤 (perivascular lymphocytic infiltrate)。這些發炎細胞浸潤位於膠原束之間,但主要在血管周圍,也可擴散至皮下脂肪組織。浸潤也可包覆汗腺 (sweat glands)。覆蓋區域的表皮 (epidermis) 常變得萎縮 (atrophic)。各種大小的血管都可能在 SSc 中受累。在早期階段,可能僅有微血管 (capillaries) 擴張,接著出現內皮增生 (endothelial proliferation) 與血管的完全阻塞。隨著硬皮症的進展,受累皮膚變得更為無血管化 (avascular),發炎減少。在較晚期階段,毛囊皮脂腺單位 (pilosebaceous units) 與外分泌腺 (eccrine glands) 消失,膠原束緊密堆積,並可能出現表皮腳 (rete ridges) 的消失。²⁰
血管病變 (VASCULOPATHY)
SSc 中的血管病變是一個早期事件,其基礎為不當的血管重塑 (vascular remodeling) 與修復過程。它涉及微循環 (microcirculation) 與小動脈,並極可能是本病致病過程中的原發事件。血管異常的特徵為血管收縮 (vasoconstriction)、外膜 (adventitial) 與內膜增生 (intimal proliferation)、發炎與血栓形成 (thrombosis)。²⁸ 血管功能障礙的最早徵象表現為血管通透性增強,伴隨血管舒張性介質(一氧化氮 nitric oxide、前列環素 prostacyclin、降鈣素基因相關肽 calcitonin gene-related peptide)與血管收縮性介質(內皮素-1 endothelin-1、血管收縮素 II angiotensin II、α2-腎上腺素受體 α2-adrenoreceptors)之間的失衡。因此,受損的血流導致組織缺氧 (tissue hypoxia),這誘導血管內皮生長因子 (vascular endothelial growth factor) 及其受體的強烈表現,並伴隨血管生成 (vasculogenesis) 的缺陷。然而,發炎細胞激素如 TNF-α 可能刺激或抑制血管新生 (angiogenesis),取決於刺激的持續時間。³⁸
除了這些功能異常外,血管內與結構性變化促成了明顯的 RP,並隨著時間進展導致血管與血流的進行性減少。這種閉塞性血管病變 (obliterative vasculopathy) 模式臨床上可能表現於幾乎所有器官的所有血管中。微循環中因結構損傷造成的早期病灶最初見於甲褶微血管,以及 RP 中的血管痙攣反應。此外,血管變化,即內皮的過度增生與瘢痕組織的沉積,產生了 SSc 的某些主要併發症,包括 PAH、SRC 與指端血管病變 (digital vasculopathy)。
免疫事件 (IMMUNE EVENTS)
SSc 病人的皮膚與肺部有早期發炎變化。病灶性皮膚中最初的發炎浸潤主要為單核球譜系 (monocyte lineage) 的細胞(T 細胞、巨噬細胞 macrophages、B 細胞與肥大細胞 mast cells)。³⁹ 有數條證據線支持先天免疫系統在 SSc 中的關鍵角色。干擾素調節因子-5(interferon regulatory factor-5)變異與硬皮症的關聯凸顯了這一點。⁴⁰,⁴¹ 數個研究也證明了巨噬細胞作為細胞激素重要貢獻者的角色,這些細胞激素影響纖維化反應。⁴²
之後,T 淋巴細胞佔主導地位,並可在循環與受累器官中偵測到。這些 T 細胞主要為 CD4+,帶有活化標記,呈現寡克隆擴增 (oligoclonal expansion)(顯示抗原驅動的增生),並顯示主要的 T 輔助細胞 2(T-helper 2)表型。⁴³,⁴⁴ 因此,在硬皮症病人中觀察到 T 輔助細胞 2 衍生細胞激素(IL-2、IL-4、IL-10、IL-13 與 IL-17)的血清濃度升高。⁴⁵,⁴⁶
除了 T 細胞外,B 細胞也存在於受累皮膚中。數個研究顯示 B 細胞能透過分泌 IL-6 與轉化生長因子-β(transforming growth factor-β, TGF-β)誘導 ECM 生成,並參與自體抗體的產生。其中數種自體抗體與本病界定明確的亞群相關,並是重要的診斷標記(見表 63-3)。⁴⁷ 自體抗體在致病機轉中的潛在角色是一個引人入勝且令人興奮的領域。大多數 SSc 病例具有循環抗體。這些包括許多標誌性反應性,以及出現於其他自體免疫風濕性疾病中的自體抗體(如抗環瓜胺酸肽 anticyclic citrullinated peptide、類風濕因子 rheumatoid factor),但也包括可能具有功能意義的抗體,因為它們針對細胞表面抗原(如抗內皮細胞抗體 antiendothelial cell antibodies、抗纖維蛋白 antifibrillin antibodies、抗血小板衍生生長因子 [platelet-derived growth factor, PDGF] 受體抗體)(表 63-4)。⁴⁸⁻⁵⁰ 然而,這些抗體的功能影響仍是一個研究領域。有越來越多證據顯示抗內皮細胞自體抗體與抗纖維母細胞反應抗體 (antifibroblast-reacting antibodies) 具有功能意義。報告也顯示有抗纖維蛋白自體抗體 (antifibrillin autoantibodies) 與和 PDGF 受體反應的刺激性自體抗體 (stimulatory autoantibodies) 的存在。⁴⁸⁻⁵⁰ 在某些病例中曾提出微嵌合 (Microchimerism) 與移植物抗宿主病 (graft-versus-host disease) 機轉,雖然微嵌合在健康個體或其他疾病狀態中的相對高頻率顯示,若它在 SSc 中有角色,可能是促成性而非因果性的。仔細的臨床研究已辨識出一個 SSc 病人亞群(以 RNA 聚合酶抗體為特徵),他們在惡性腫瘤 (malignancies) 發生的關聯下發展出本病。⁵¹⁻⁵³ 這導致了一個假說,即纖維化可能代表針對腫瘤抗原的免疫反應,並引發了關於自體免疫與惡性腫瘤一般關係的討論。仍需進一步研究來釐清此議題。
纖維化 (FIBROSIS)
SSc 是一種多系統纖維化疾病。最初的發炎與缺氧誘導纖維母細胞產生數種參與 ECM 重塑的蛋白質,例如血小板反應蛋白-1 (thrombospondin-1)、纖維連接蛋白-1 (fibronectin-1)、賴胺酸羥化酶-2 (lysylhydroxylase-2) 與 TGF-β 誘導蛋白。⁵⁴ 同時,合成與降解機轉之間的平衡被擾亂,導致特定器官中 ECM 過量,這隨後造成本病大部分的病態與死亡。纖維化發展的關鍵事件是將纖維母細胞誘導為活化的肌纖維母細胞 (activated myofibroblasts)。或者,其他細胞類型(如循環前驅細胞 circulating precursor cells、內皮細胞與上皮細胞)也可轉換為肌纖維母細胞。此過程的啟動包括許多可能代表合理治療標靶的關鍵細胞激素與生長因子。這些包括纖維化性細胞激素 (fibrogenic cytokines),如 TGF-β、結締組織生長因子 (connective tissue growth factor)、PDGF 與內皮素-1。²⁸,⁵⁵⁻⁵⁷ 尤其是 TGF-β 已被證明扮演核心角色⁵⁸,這也被使用不同疾病階段硬皮症病人皮膚切片的廣泛表現圖譜研究所強調。⁵⁹ 這已導致在早期臨床研究中使用抗 TGF-β 抗體的治療方法。⁶⁰
肌纖維母細胞的特徵為高收縮性 (contractility)、ECM 產生與細胞激素釋放。此功能連同所產生結締組織改變的生物物理特性,導致纖維母細胞的持續活化並伴隨 ECM 成分的過度沉積。然而,理解本病機轉的關鍵在於自體免疫、血管病變與纖維化之間的緊密連結。這最近在一個小鼠模型中得到證明,該模型的特徵為轉錄因子 Friend 白血病整合 1(Friend leukemia integration 1, Fli1)與 Krüppel 樣因子 5(Kruppel-like factor 5, KLF5)的下調,這些小鼠會發展出伴隨自體抗體產生的類硬皮症疾病。⁶¹
診斷 (DIAGNOSIS)
雷諾現象 (RAYNAUD PHENOMENON)
僅以 RP 表現的病人應接受微血管改變以及自體抗體狀態的檢查。所有這些都是 SSc 發展的預測因子,合在一起使得 SSc 的診斷頗為可能。
為了辨識並可視化由 SSc 造成的血管性皮膚改變,甲褶微血管鏡檢查 (nailfold capillaroscopy) 是一種非侵入性、簡單,且最有用的診斷與預後方法之一(表 63-5)。此外,它是將微血管變化分類為早期、活動期與晚期模式的有用工具。雷射都卜勒灌流影像 (Laser Doppler perfusion imaging) 也是一種非侵入性微血管影像技術,能提供皮膚血流的圖譜。⁶²
皮膚硬化 (SKIN SCLEROSIS)
皮膚受累應使用改良式 Rodnan 皮膚評分 (modified Rodnan Skin Score) 來評估。通常評估 17 個部位,並由受過訓練的檢查者依皮膚觸診將皮膚厚度分類為第 1、2 或 3 級,分別對應輕度、中度與重度(圖 63-7)。計算皮膚增厚的較新技術也已被評估。除了改良式 Rodnan 皮膚評分⁶³ 之外,20-MHz 超音波檢查 (20-MHz ultrasonography)⁶⁴、MRI⁶⁵ 與皮褶測量計 (plicometer)⁶⁶ 方法對於評估皮膚增厚也很有用(建議的診斷程序列於表 63-5)。其他監測皮膚纖維化的物理程序為硬度計 (durometer)⁶⁷、皮膚彈性測量計 (cutometer)⁶⁸ 與彈性測量計 (elastometer)⁶⁹。除了這些非侵入性方法外,伴有真皮皮膚厚度組織學評估的皮膚切片是一種適當但侵入性的方法。此方法能對發炎浸潤進行特徵描述。
心肺受累 (CARDIOPULMONARY INVOLVEMENT)
患有 SSc 且有心肺症狀的個體應至少每年使用肺功能檢查 (pulmonary function tests)、心臟超音波 (echocardiography)、6 分鐘步行測試 (6-minute walk test) 與高解析度 CT(high-resolution CT, HRCT)進行追蹤。⁷⁰ 肺功能檢查是判定可能心肺受累的最重要技術,因為肺部一氧化碳瀰散能力 (diffusion capacity of the lung for carbon monoxide, DLCO ≤75%) 受損是肺纖維化與 PAH 兩者的早期標記。⁷¹
為了判定間質性肺受累的存在,即胸膜下局部線狀陰影 (subpleural localized line opacities)、毛玻璃樣陰影 (ground-glass opacities),以及伴有蜂窩狀形成 (honeycomb formations) 的胸膜下囊腫 (subpleural cysts),應使用 HRCT 與/或胸部 X 光攝影。追蹤也應包括經胸都卜勒心臟超音波 (transthoracic Doppler echocardiography),這是一種非侵入性程序,可指出右心室肥厚(伴有或不伴有擴大)、心室間隔的矛盾運動 (paradoxical motion)、三尖瓣閉鎖不全 (tricuspid valve insufficiency) 與心包積液 (pericardial effusion)。右心導管術 (Right-heart catheterization) 確實是黃金標準,但它是判定 PAH 的侵入性診斷程序。PAH 的定義為靜息時平均肺動脈壓 (mean pulmonary artery pressure) ≥25 mm Hg,並伴有經右心導管術判定的肺微血管楔壓 (pulmonary capillary wedge pressure) ≤15 mm Hg。⁷⁰,⁷²,⁷³
心臟 MRI 也是評估 SSc 心肌受累的潛在策略。除了影像程序外,使用 N 端腦鈉肽 (N-terminal brain natriuretic peptide) 來偵測右心室損傷也有些前景(見表 63-5)。早期偵測心臟受累對於預防並能早期治療心肌病變 (cardiomyopathy) 與嚴重心律不整至關重要。⁷⁰
胃腸道受累 (GI INVOLVEMENT)
食道炎的存在可透過上消化道內視鏡 (upper GI endoscopy) 並進行組織學評估來判定。食道蠕動受損通常可透過放射性標記餐食後的閃爍掃描評估 (scintigraphic evaluation) 或 24 小時 pH 測壓 (pH manometry) 來診斷(見表 63-5)。⁷¹
腎臟受累 (KIDNEY INVOLVEMENT)
早期診斷在改善 SRC 結果方面扮演關鍵角色,做法為使用定期血壓監測、尿液分析微電泳 (urine analysis microelectrophoresis),以及肌酸酐清除率 (creatinine clearance) 的測定(見表 63-5)。²⁶
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
SSc 的診斷是臨床性的。雖然有為了促進 SSc 與其他結締組織疾病區分而發展的標準,但尚未發展出正式的診斷標準。然而,判定本病的正確亞群(包括 SSc 重疊症候群)是必要的,以便能判斷預後與特定器官的受累,並決定治療方法。有數種模擬硬皮症的鑑別診斷:局限性(局部性)硬皮症 circumscript (localized) scleroderma;嗜酸性筋膜炎 eosinophilic fasciitis;硬皮樣遺傳性皮膚病 sclerodermiform genodermatoses;慢性萎縮性肢端皮膚炎 acrodermatitis chronica atrophicans;環境因子誘發的類硬皮症症候群 sclerodermalike syndromes induced by environmental factors;成人硬腫病 scleroderma adultorum Buschke;糖尿病性硬皮病 scleroderma diabeticorum;硬化性黏液水腫 scleromyxedema;腎源性纖維化皮膚病 nephrogenic fibrosing dermopathy;遲發性皮膚紫質症 porphyria cutanea tarda;移植物抗宿主病 graft-versus-host disease;以及惡性腫瘤中的類硬皮症病灶。這些當然必須被排除。表 63-6 概述了 SSc 的鑑別診斷。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
本病的發展在很大程度上取決於特定亞群。局限型病人在其他器官表現發病前許多年即已發展出 RP。皮膚纖維化仍局限於肢端區域,主要併發症為指端潰瘍與肺高壓的發展。然而,在瀰漫型中,纖維化早期發生,並伴隨發炎、關節疼痛,並顯示出快速擴散至幾乎全部體表 (integument) 的特點。在這些病人中,肺(肺纖維化)、心臟與腎臟的表現在病程早期出現,並常決定預後。此疾病亞群仍有大量死亡發生。數個研究指出,瀰漫型皮膚 SSc 病人在最初幾年顯示出本病的快速惡化。在後期,本病的活性降低,症狀可改善。令人驚訝的是,硬化的皮膚也可變軟,攣縮可減輕。雖然 SSc 仍是一種危及生命的疾病,但對病人進行多學科管理 (multidisciplinary management) 並早期偵測與治療併發症,可在病人一生中帶來大幅改善的預後。
處置 (MANAGEMENT)
疾病修飾治療 (DISEASE-MODIFYING TREATMENT)
SSc 有三個面向可能適合進行治療調節,這提升了真正疾病修飾治療 (disease-modifying treatment) 的可能性。目前,血管療法 (vascular therapies) 與免疫調節 (immunomodulation) 擁有最廣泛的候選療法。表 63-7 與 63-8 總結了這些方法。一般性免疫抑制 (General immunosuppression) 可透過改善皮膚受累與間質性肺疾病 (interstitial lung disease) 而帶來益處。最佳證據可用於環磷醯胺 (cyclophosphamide),但近期黴酚酸酯 (mycophenolate mofetil, MMF) 已被證明與口服環磷醯胺一樣有效,並被許多中心使用。⁷⁴,⁷⁵ 也有證據顯示,若標準免疫抑制已失敗,利妥昔單抗 (rituximab) 可在一群經選擇的病人中改善疾病病程。⁷⁶ 一般免疫抑制的有效性由來自美國與歐洲、在某些經選擇病人中使用高強度免疫抑制 (high-intensity immunosuppression) 合併自體造血幹細胞移植 (autologous hemopoietic stem cell transplantation) 的試驗所證明。⁷⁷⁻⁸⁰ 然而,副作用始終必須被考量(表 63-9)。抗纖維化治療 (Antifibrotic treatment) 仍是一項挑戰,雖然在過去幾年,主要基於對潛在機轉的更深入理解,已產生了許多新方法。一項使用新的抗 TGF-β 抗體的較新研究帶來了皮膚受累嚴重度的改善,以及數個 TGF-β 依賴性基因表現的減少。⁶⁰ 特發性肺纖維化的較新臨床試驗也提供了一些鼓舞。然而,目前尚無已證實的抗纖維化藥物。圖 63-8 是一個簡化示意圖,用於整合假定的疾病修飾治療與篩檢及監測計畫,這些計畫能在 SSc 中以器官為基礎的策略下進行及時介入,而這些策略目前構成大多數 SSc 治療學的基礎。標靶疾病修飾治療的可能性取決於治療藥物的可得性以及對其在致病機轉中角色的清楚理解。在 SSc 以器官為基礎的治療學領域有更多成功,這已對許多病人的生活品質產生重大影響。需要早期偵測這些器官特異性併發症,以便能進行早期介入。
指端血管病變及其併發症 (DIGITAL VASCULOPATHY AND ITS COMPLICATIONS)
減少雷諾發作頻率的簡單但重要的建議包括:透過避免如尼古丁 (nicotine)、擬交感神經藥物 (sympathomimetics)、情緒壓力與寒冷等誘發因素來減少血管收縮,並改為有良好的家中暖氣、厚實且密不透風的衣物、熱化學或可微波的暖手器 (hand warmers)、電熱手套、鞋墊或紅外線熱療 (infrared hyperthermy)、定期石蠟浴 (paraffin wax bath) 治療,以及將手指創傷最小化。
治療需要數個醫療專科之間的密切互動,運用局部與全身性療法。目前指端潰瘍的局部處置包括非藥物照護、抗生素(感染時)、止痛 (analgesia) 與個別應用的傷口敷料(必要時)的組合。可能的藥物治療需要對 RP 的最佳化治療,包括具有血管重塑與/或擴張潛力的藥物,如鈣離子通道阻斷劑 (calcium channel blockers) 與血管收縮素 II 受體拮抗劑 (angiotensin II receptor antagonists)⁸¹,⁸²,這些應被視為一線治療。其他藥物治療選項(如地爾硫卓 diltiazem 與血管收縮素轉化酶抑制劑 angiotensin-converting enzyme inhibitors)的結果則相互矛盾。⁸³⁻⁸⁶
非腸道前列環素衍生物 (Parenteral prostacyclin derivatives),尤其是伊洛前列素 (iloprost),被廣泛使用,有助於治癒指端潰瘍並可能預防復發病灶。靜脈輸注的前列環素衍生物是嚴重指端缺血治療的主軸。⁸⁷,⁸⁸ 抗血小板藥物 (Antiplatelet agents),如阿斯匹靈 (aspirin) 與氯吡格雷 (clopidogrel),也被使用,尤其在嚴重指端缺血。對於有效治療 PAH 的療法用於指端血管病變一直有熱忱。因此,在 2 個大型對照試驗中,波生坦 (bosentan),一種口服雙重特異性內皮素受體拮抗劑 (dual-specificity endothelin receptor antagonist),被證明與安慰劑相比能顯著減少新發指端潰瘍的數量。⁸⁹ 然而,並未證明對已確立潰瘍的癒合有正面效果。其他藥物,如第 5 型磷酸二酯酶抑制劑 (phosphodiesterase Type 5 inhibitors) 西地那非 (sildenafil) 與他達拉非 (tadalafil),也被用於治療 RP 與指端潰瘍,但尚無前瞻性臨床試驗數據。⁹⁰
外科治療包括指端微動脈鬆解術 (digital microarteriolysis),可使有頑固性潰瘍的單一手指獲益。只要可能,會避免手指的外科截肢,而長期使用非腸道前列環素合併第 5 型磷酸二酯酶抑制劑與強效止痛劑可能有助於此。腰椎交感神經切除術 (Lumbar sympathectomy) 對於下肢 RP 或潰瘍可能有幫助。一般而言,會先進行一個暫時性程序以判定明確交感神經切除術可能帶來的益處。在嚴重指端缺血的病例中,常給予抗血小板療法,有軼事報告指出氯吡格雷在預防指端梗塞 (digital infarction) 方面的益處(見表 63-7)。
皮膚受累 (SKIN INVOLVEMENT)
處置 SSc 皮膚表現的關鍵要素是物理治療 (physical therapy) 與規律運動,以維持循環、關節活動度與肌力,全都旨在改善 SSc 病人的生活品質。受硬皮症影響的皮膚傾向非常乾燥、緊繃且易受創傷。皮膚硬化可透過物理治療與運動、淋巴引流 (lymphatic drainage)、局部類固醇治療、鈣調神經磷酸酶抑制劑 (calcineurin inhibitors) 與保濕乳霜 (moisturizing crèmes) 來改善。全身性療法包括免疫抑制劑、全身性類固醇(僅短期使用)與光照治療(紫外線 A1 ultraviolet A1,或補骨脂素加紫外線 A psoralen and ultraviolet A)。紫外線 A1 光照治療似乎能抑制纖維化與發炎過程,並減少硬化皮膚的量。⁹¹
乾燥與搔癢的皮膚應局部使用皮質類固醇、大麻素致效劑 (cannabinoid agonists)、辣椒素 (capsaicin)、潤膚劑 (emollients) 與光照治療(見上文)治療。局部類固醇注射以及雷射或外科治療也可嘗試用於治療皮膚鈣質沉著症。雷射療法或如遮瑕 (camouflage) 等非侵入性方法已用於毛細血管擴張。漂白劑 (Bleaching agents)、水楊酸 (salicylic acids) 與化學換膚 (chemical peels),以及遮瑕、視黃酸 (retinoids) 與皮質類固醇,可能有改善色素沉著或色素減退的潛力(見表 63-8)。兩個隨機臨床試驗顯示,甲胺喋呤能改善早期瀰漫型 SSc 的皮膚評分,而對其他器官表現的正面效果尚未被確立。⁹²,⁹³ 另一方面,在兩個隨機臨床試驗中,環磷醯胺改善了皮膚硬化。⁹⁴,⁹⁵ 為了在療效與副作用之間取得平衡,MMF 可能是影響皮膚纖維化的一個有趣選項。⁷⁴ 蛋白激酶抑制劑 (Protein kinase inhibitors)(如伊馬替尼 imatinib)已被使用,但截至撰寫本文時,對照臨床試驗結果參差不齊,並顯示耐受性不佳。⁹⁶,⁹⁷
心肺表現 (CARDIOPULMONARY MANIFESTATIONS)
人們日益認識到,一群具有某些肺纖維化的病人主要為 PAH,而此群病人可能對標準 PAH 療法有反應。過去十年間 PAH 的治療已有顯著進展。一旦 PAH 在功能上受到顯著限制(紐約心臟協會 New York Heart Association 第 III 級),大多數病例以口服藥物治療,即內皮素受體拮抗劑(如波生坦 bosentan、安立生坦 ambrisentan)或第 5 型磷酸二酯酶抑制劑(如西地那非 sildenafil、他達拉非 tadalafil)。之後,若發生進展,則使用口服治療的組合,或經靜脈或皮下途徑引入非腸道前列環素。伊洛前列素的吸入式給藥系統可供使用。雖然 PAH 在 SSc 中所造成的死亡可能多於肺纖維化,但肺纖維化仍是一個重要的併發症。SSc-PF 的治療仍具挑戰性。⁹⁸ 在大量顯示環磷醯胺對 SSc-PF 有益的非對照或回溯性數據之外,已報告了 2 個隨機、雙盲、安慰劑對照試驗的結果。兩者都顯示環磷醯胺有適度的扣除安慰劑後益處 (placebo-subtracted benefit)。⁹⁹⁻¹⁰¹ 對於用力肺活量 (forced vital capacity)(佔預測值百分比)的變化,在比較口服環磷醯胺與安慰劑的硬皮症肺研究 (Scleroderma Lung Study) 中具統計顯著性,而在靜脈注射環磷醯胺後接續口服硫唑嘌呤 (azathioprine) 的試驗中顯示強烈趨勢 (p = 0.06)。目前,大多數中心使用環磷醯胺作為嚴重或進行性 SSc-PF 的治療,並以肺功能檢查與 HRCT 來界定其範圍與嚴重度。HRCT 上的疾病範圍以及肺功能檢查上進行性限制性異常 (progressive restrictive abnormality) 的病史,是未來肺功能下降的最佳預測因子,並普遍被用於做出治療決定。較新的臨床試驗顯示 MMF 與環磷醯胺在穩定硬皮症與間質性肺疾病 (ILD) 病人肺功能的治療上具有同等療效。⁷⁴ MMF 治療與長達 36 個月的肺功能穩定相關,且其副作用特性優於接受硫唑嘌呤治療的病人。¹⁰² 其他使用中的療法包括羧甲司坦 (carbocysteine) 與低劑量皮質類固醇。其他免疫抑制策略的地位仍不確定,需要在前瞻性多中心臨床試驗中評估。值得注意的是,儘管使用內皮素受體拮抗劑波生坦作為肺纖維化治療有強烈的理論依據,但在近期一項針對 SSc-PF 病例的大型多中心研究中,波生坦並未優於安慰劑。SSc 的心臟受累也是死亡率的重要貢獻者,但仍是 SSc 內臟器官併發症中最不被充分理解且最少被辨識者之一。大量研究證實放射性核素影像 (radionuclide imaging)、電生理學 (electrophysiologic) 與功能異常在 SSc 中很常見,但這些發現的意義尚不確定。血流動力學上顯著的心臟受累發生於高達 10% 的瀰漫型皮膚 SSc 病例。心肌炎的發炎成分可能對免疫抑制治療有反應,因此有一套對心臟硬皮症處置的操作性方法。雖然這尚未基於足夠可靠的數據,但它可構成前瞻性評估 SSc 中左心室射出分率 (left ventricular ejection fraction) 受損與循環肌鈣蛋白 (troponin) 濃度升高意義的基礎。SSc 已有許多進展,包括對本病多樣性的更佳認識、對潛在病理機轉的改善理解,以及在治療以器官為基礎併發症方面的重大進步。這包括穩健臨床試驗數據的累積,這些數據證明個別療法的有效性或缺乏益處,以及疾病評估指標的驗證。¹⁰³
胃腸道受累 (GI INVOLVEMENT)
胃腸道的受累在 SSc 中頻繁發生。食道症狀對質子幫浦抑制劑 (proton pump inhibitors) 與增加下食道括約肌張力的藥物(如多潘立酮 domperidone)反應可能非常好,雖然高劑量治療可能與心律不整風險增加相關。中腸 (Midgut) 受累有多種型態。假性阻塞最初需要保守處置,但隨後可能需要非腸道營養補充 (parenteral nutritional supplementation)。小腸細菌過度生長 (Small intestinal bacterial overgrowth) 可使用廣效抗生素治療,而胰功能不全 (pancreatic insufficiency) 可能需要酵素補充。大腸受累是一項重大挑戰。肛門直腸失禁 (Anorectal incontinence) 有時對植入式薦神經刺激器 (implanted sacral nerve stimulator) 反應良好,或對較不複雜的方法(如為增加肛門內括約肌體積的生物塑料注射 bioplastic injection)反應良好。相關的直腸脫垂 (rectal prolapse) 可能需要額外的外科介入。慢性便秘(有時伴有溢出性腹瀉 overflow diarrhea)是一個常見問題。建議調整飲食並審慎使用刺激性、軟化性或膨脹性瀉劑 (aperients),但伴隨病人實質參與的個別化方法通常是最成功的方法。偶爾需要進行轉流性結腸造口術 (defunctioning colostomy),但這僅適用於非常有限的少數病例。
硬皮症腎危象 (SCLERODERMA RENAL CRISIS)
整體而言,到專科中心就診的 SRC 病例約有三分之二需要腎臟替代療法 (renal replacement therapy)。其中約有一半的病例最終恢復至足以停止透析 (dialysis)。這可發生在腎危象後超過 24 個月,因此關於腎臟移植的決定應視結果而延後。晚期恢復的可能性使 SRC 與其他末期腎衰竭原因有所區別。這些結果之所以可能,是透過使用血管收縮素轉化酶抑制劑作為 SRC 的常規療法。在它們可得之前,已確立 SRC 的 12 個月死亡率大於 90%。SRC 處置最關鍵的面向是在硬皮症的背景下迅速辨識並治療顯著高血壓,並啟動血管收縮素轉化酶抑制劑。這是一個醫療急症,任何腎功能損害或終末器官損傷的特徵都應促使住院治療。¹⁰⁴
其他支持性程序 (OTHER SUPPORTIVE PROCEDURES)
所有這些器官特異性治療方法都必須由數項一般措施支持,以幫助病人。這些一般措施包括保持家中與身體溫暖以及優化營養狀態的建議。必須提供石蠟浴與物理治療。需要教導病人應付日常生活的併發症,並早期辨識那些指出疾病進展與新器官受累的症狀。
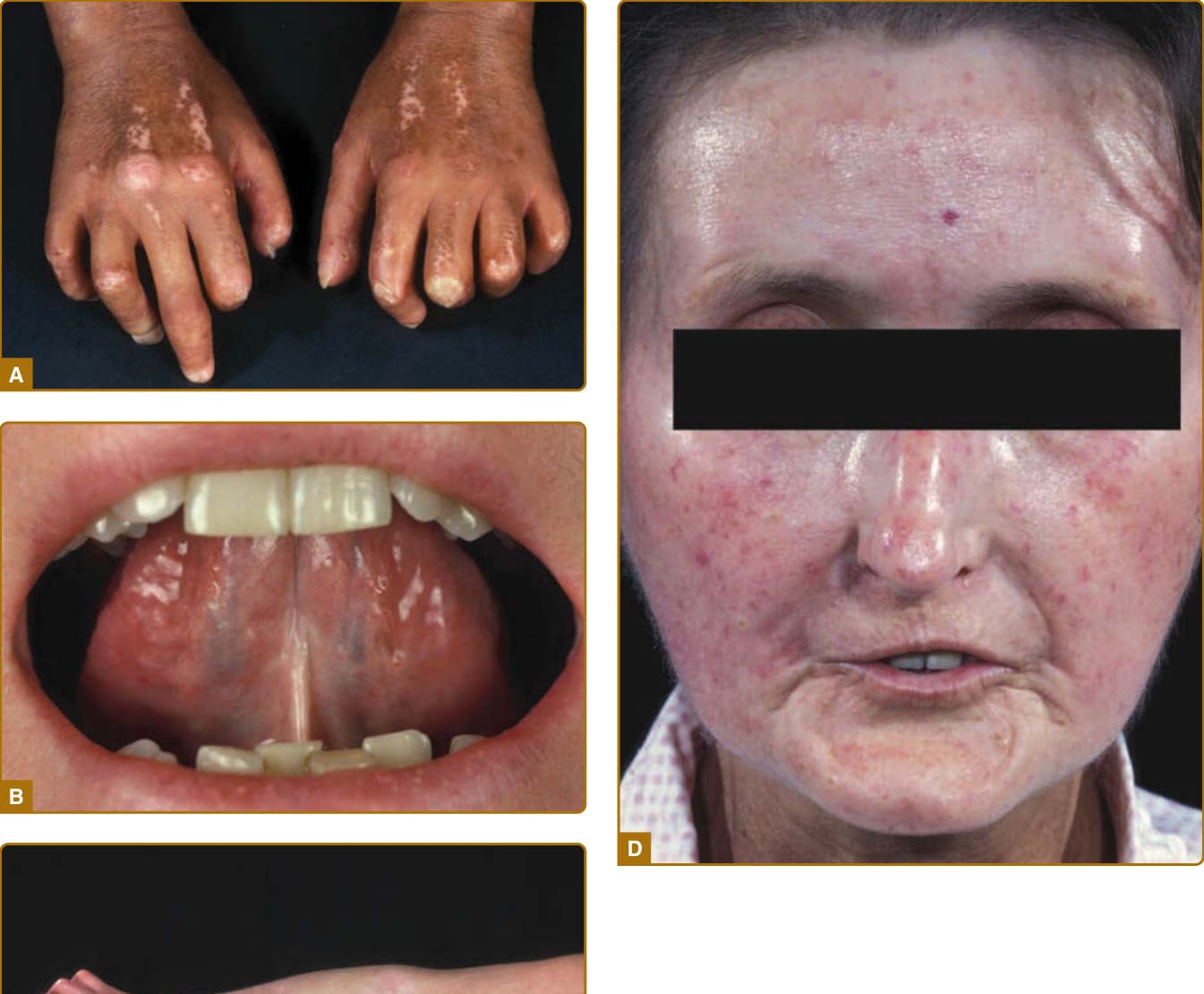
圖 63-1:瀰漫型皮膚全身性硬化症 (diffuse cutaneous systemic sclerosis) 病人的廣泛皮膚受累。A,指端硬化 (sclerodactyly) 伴皮源性攣縮 (dermatogenous contractures,指關節活動受限) 及鹽和胡椒狀 (salt-and-pepper) 色素沉著與色素減退。B,口裂縮小 (microstomia,口周放射狀皺褶) 伴繫帶硬化 (frenulum sclerosis)。C,掌指關節 (metacarpophalangeal joints) 近端的皮膚增厚。D,典型硬皮症顏面相貌 (facial physiognomy),伴有表情過度 (hypermimia)、口裂縮小、毛細血管擴張 (telangiectasias) 與鳥喙狀鼻 (beaked nose)。
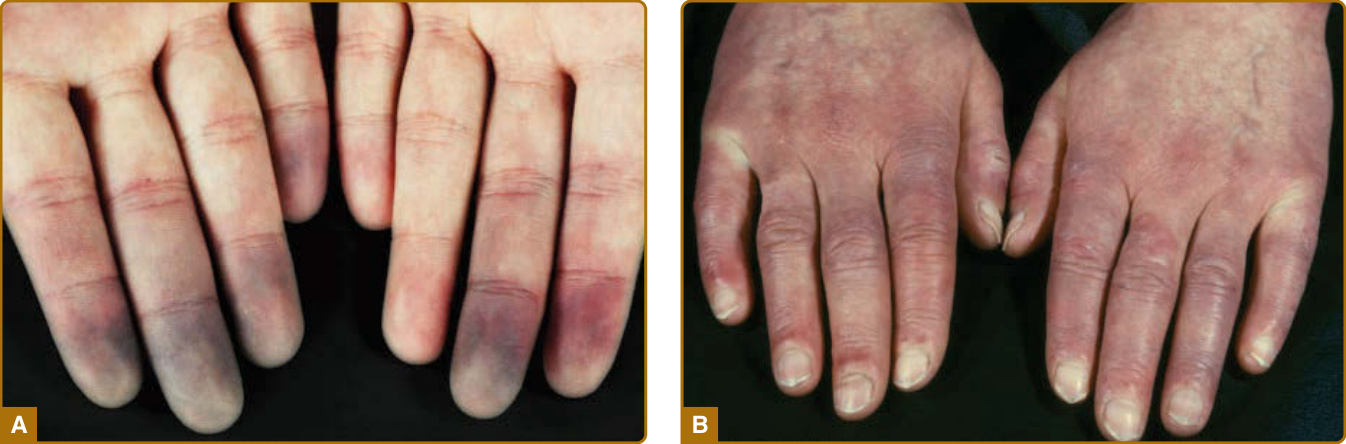
圖 63-2:早期疾病病人的臨床特徵。A,雷諾現象 (Raynaud phenomenon) 伴典型變色(藍白色蒼白 blue-white pallor),主要因血管痙攣 (vasospasm) 而局限於手指與/或腳趾。寒冷與情緒壓力是這些發作最常見的觸發因素。B,局限型疾病伴腫脹手指 (puffy fingers)。
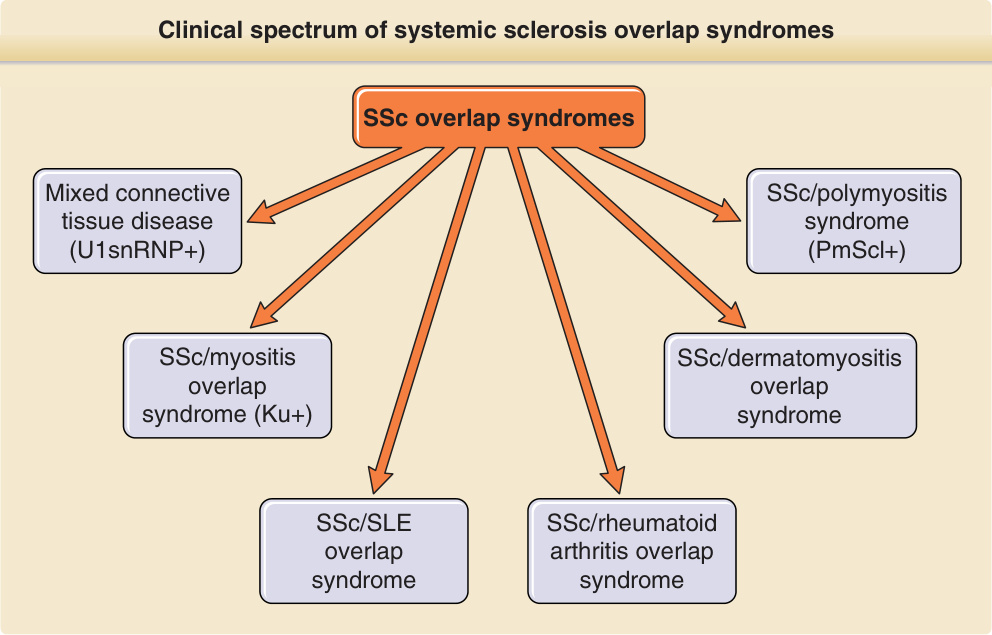
圖 63-3:全身性硬化症 (systemic sclerosis, SSc) 重疊症候群的臨床譜。同時具有硬皮症臨床特徵與至少 1 種額外自體免疫風濕性疾病特徵的病人,被命名為 SSc 重疊症候群。SLE,全身性紅斑性狼瘡 systemic lupus erythematosus。
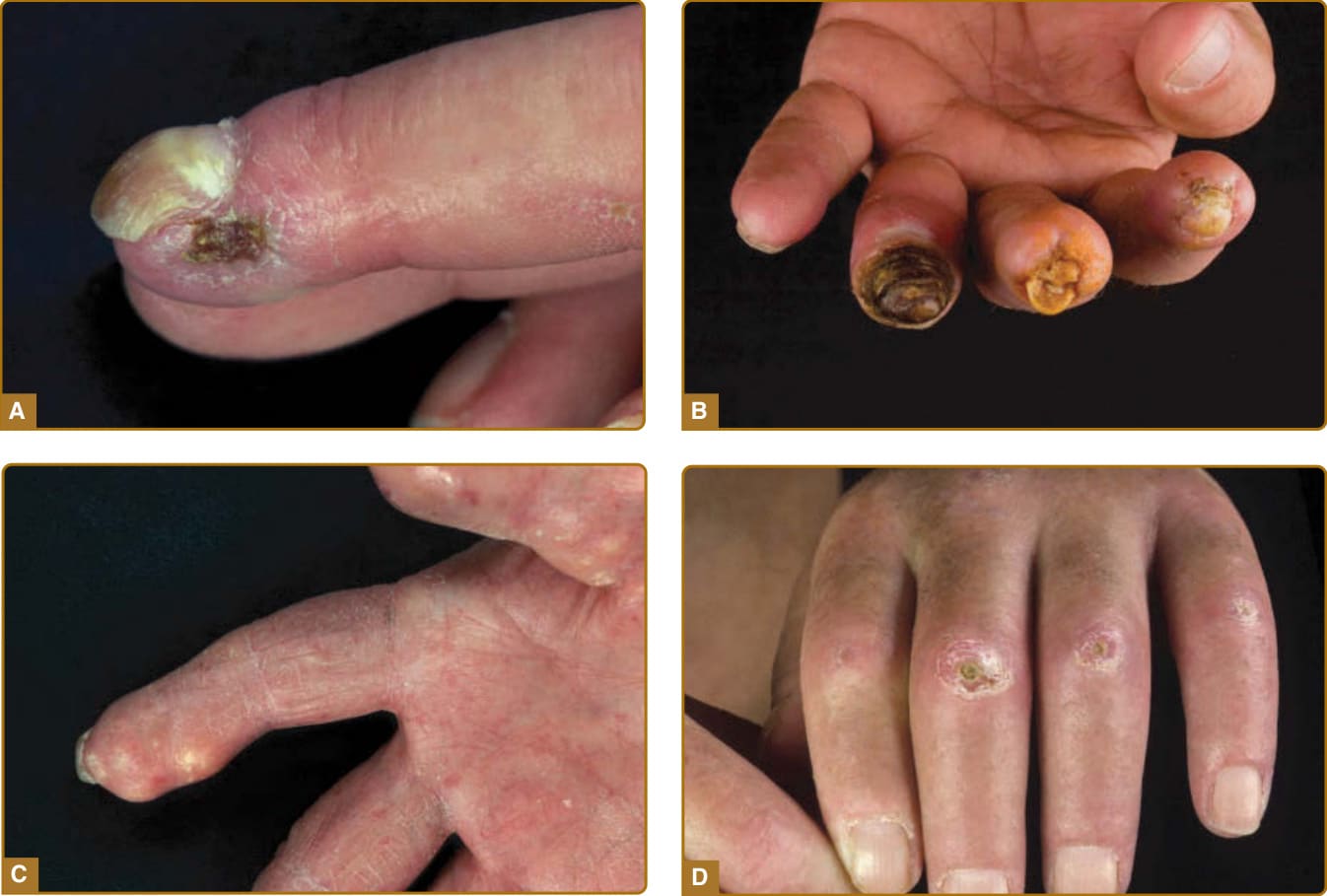
圖 63-4:伴有併發症的指端改變。A,指尖的指端潰瘍 (digital ulcerations)。B,指尖的指端潰瘍與壞死 (necrosis)。C,伴有皮下團塊沉積的嚴重鈣化 (calcifications)。D,骨突 (bone protuberants) 處多發潰瘍,伴周圍硬化皮膚的發炎。

圖 63-5:全身性硬化症的致病機轉。此示意圖顯示全身性硬化症的發展如何源自免疫系統內細胞(包括後天與先天區室)、血管系統與結締組織之間的複雜交互作用。細胞-基質交互作用 (Cell–matrix interactions) 是細胞功能的重要調節者。早期血管事件導致後期一群自主性活化纖維母細胞與肌纖維母細胞 (myofibroblasts) 的發展,這些細胞使軟組織收縮並沉積過量的細胞外基質蛋白。這些細胞可能源自常駐結締組織纖維母細胞;由其他細胞類型轉分化 (transdifferentiation)(包括活化的微血管周細胞 microvascular pericytes);以及循環前驅細胞 (circulating progenitor cells,纖維細胞 fibrocytes) 的招募。每個譜系對纖維化病灶的貢獻仍不清楚。許多生長因子與細胞激素被認為是此過程的介質,而複雜的相互網絡可能導致促纖維化 (profibrotic) 微環境。潛在的疾病修飾治療可單獨或合併針對個別介質(如腫瘤生長因子-β [tumor growth factor-β, TGF-β]、內皮素 [endothelin, ET-1]、結締組織生長因子 [connective tissue growth factor, CTGF]、血小板衍生生長因子 [platelet-derived growth factor, PDGF]),或調節免疫細胞(如環磷醯胺 cyclophosphamide)或內皮細胞(如前列環素類似物 prostacyclin analogs)。細胞外基質是介質的重要儲存庫,這些介質之後被釋放並在致病機轉中扮演關鍵角色。CCL,CC 趨化激素配體 CC chemokine ligand;COMP,軟骨寡聚基質蛋白 cartilage oligomeric matrix protein;EC,細胞外 extracellular;ET-R,內皮素受體 endothelin receptor;FN,纖維連接蛋白 fibronectin;Ig,免疫球蛋白 immunoglobulin;IL,介白素 interleukin;RELM-β,抵抗素樣分子-β resistin like molecule-β。
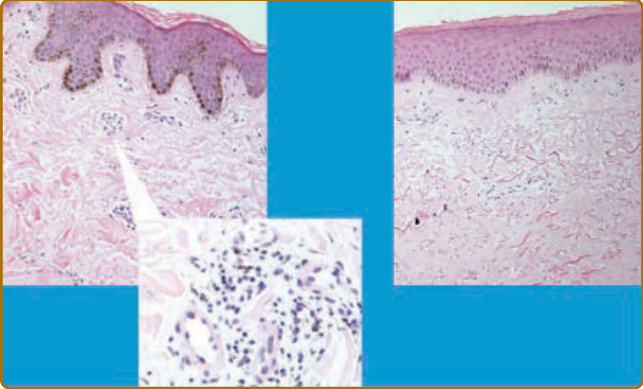
圖 63-6:早期與晚期瀰漫型皮膚全身性硬化症 (diffuse cutaneous systemic sclerosis, SSc) 皮膚的組織學外觀。在 SSc 中,疾病早期階段有血管周圍單核細胞浸潤 (perivascular mononuclear cell infiltrate)。這先於皮膚硬化的發展。血管周圍變化在左側面板以高倍率顯示。較晚期疾病伴隨皮膚硬化、低密度的血管,以及發炎細胞的缺乏。在此階段,可能有相關的表皮變化,伴增厚與次級皮膚結構(包括毛囊與汗腺)的喪失。表皮腳 (rete ridges) 的缺乏在瀰漫型皮膚 SSc 較晚期階段也是特徵性的。局限型皮膚 SSc 預期有類似變化,但因皮膚硬化有限以及對癒合的顧慮,很少進行切片。
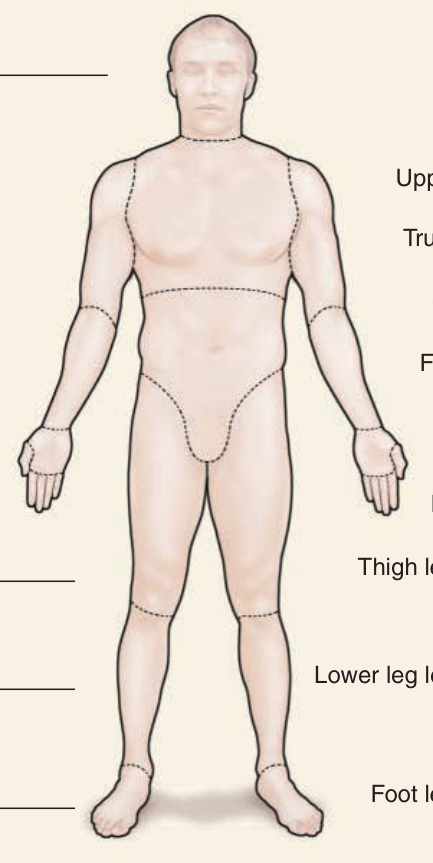
圖 63-7:改良式 Rodnan 皮膚評分 (modified Rodnan skin score, mRSS)。使用改良式 mRSS 的皮膚硬化評估通常透過評估 17 個不同區域的皮膚厚度來進行。皮膚硬化經觸診分類為第 1 級(對應輕度)、第 2 級(對應中度)與第 3 級(對應重度)。le,左 Left;ri,右 right。
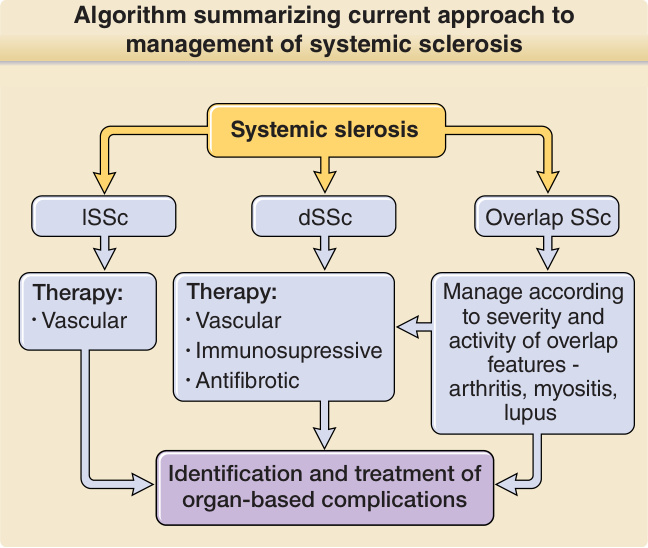
圖 63-8:總結全身性硬化症 (systemic sclerosis, SSc) 目前處置方法的演算法。SSc 的治療原則包括準確診斷,並依疾病亞群、重疊特徵的存在,以及依疾病分期判定的可能主導病理過程進行治療。在所有病例中,針對以器官為基礎併發症的篩檢與治療在成功處置中扮演重要角色。對病人的衛教,以及包含專科護理師、物理治療師、職能治療師與許多次專科醫師及外科醫師的多學科團隊,是為嚴重 SSc 病例提供適當照護的核心。dSSc,瀰漫型皮膚全身性硬化症 diffuse cutaneous systemic sclerosis;lSSc,局限型皮膚全身性硬化症 limited cutaneous systemic sclerosis。

表 63-1:全身性硬化症/肌炎重疊症候群 (Systemic Sclerosis/Myositis Overlap Syndrome) 的臨床特徵

表 63-2:混合性結締組織病 (Mixed Connective Tissue Disease) 的臨床特徵¹⁷,¹⁸
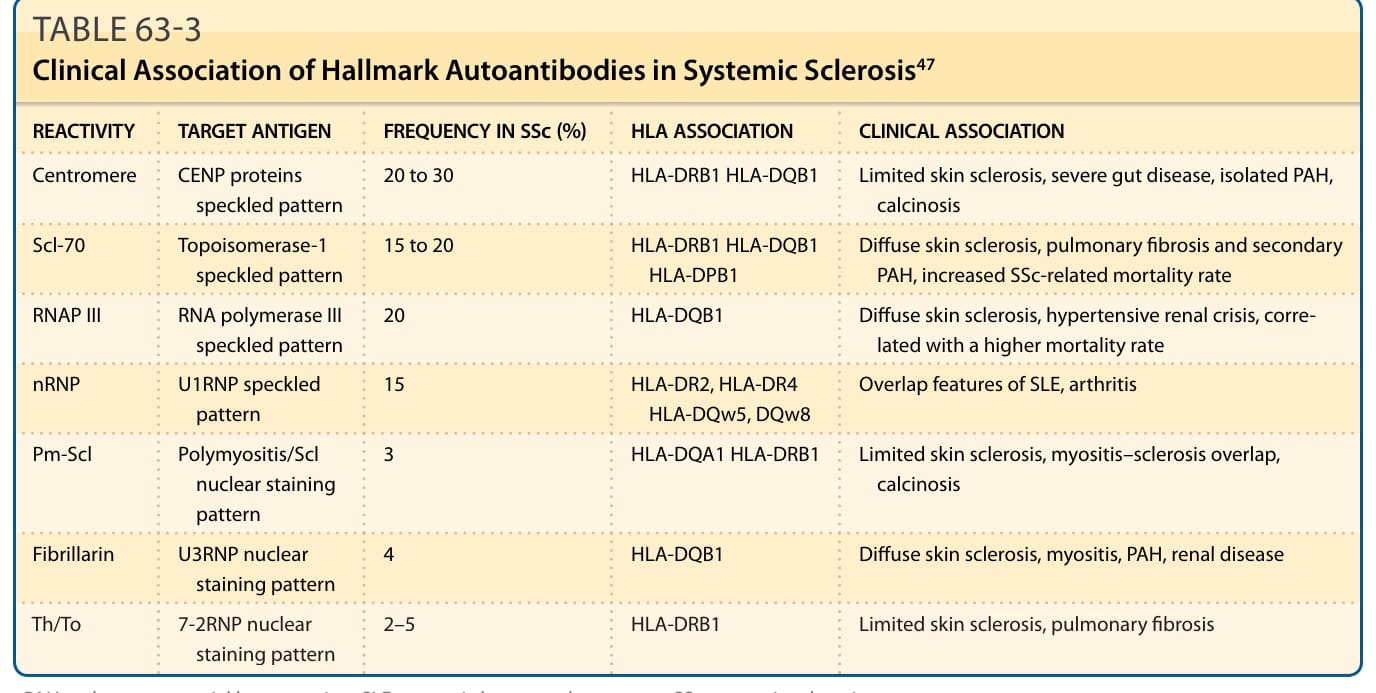
表 63-3:全身性硬化症中標誌性自體抗體 (Hallmark Autoantibodies) 的臨床關聯⁴⁷

表 63-4:全身性硬化症中的功能性自體抗體 (Functional Autoantibodies) 及其與病理生理學的關聯⁴⁸⁻⁵⁰
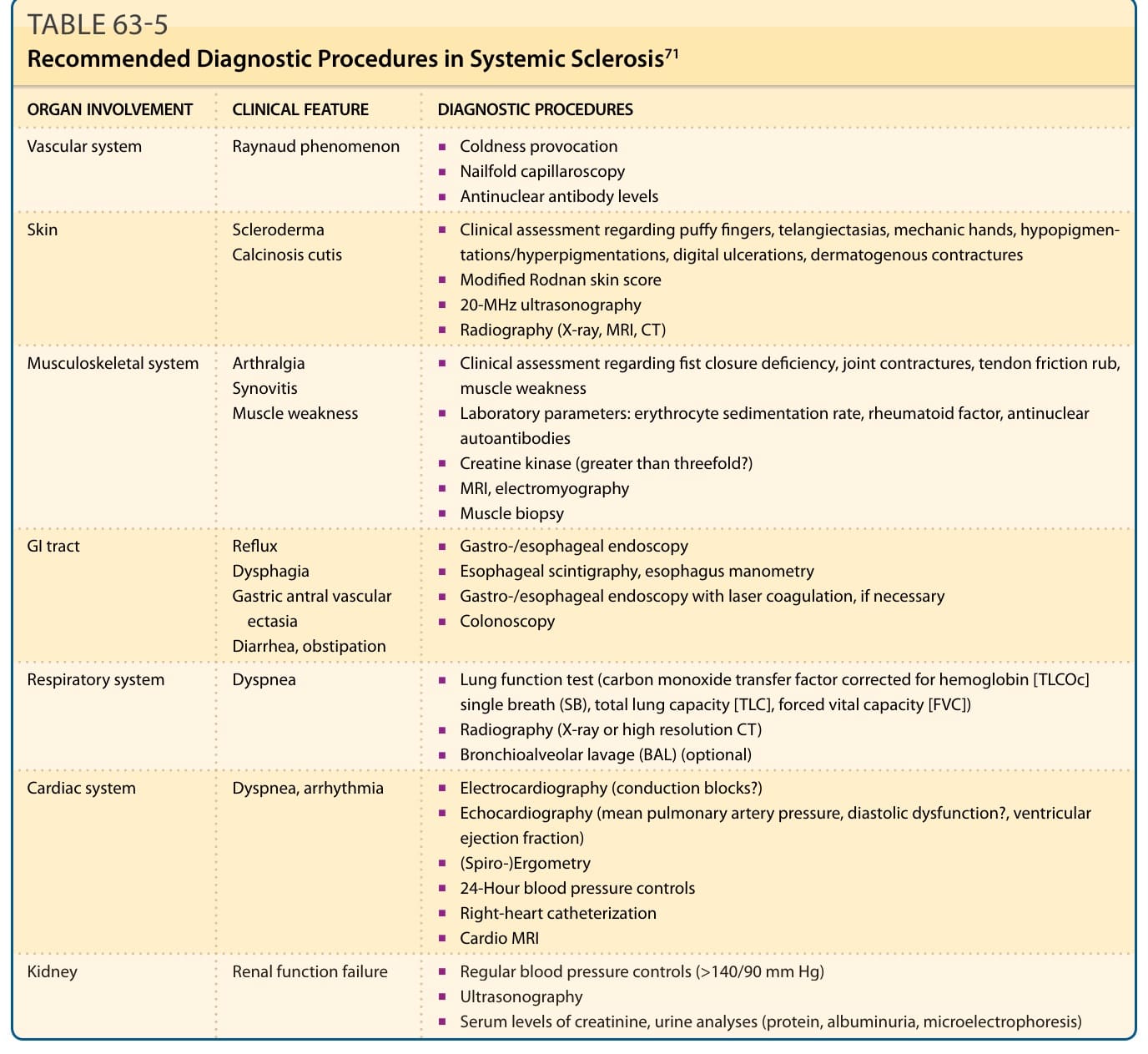
表 63-5:全身性硬化症的建議診斷程序 (Recommended Diagnostic Procedures)⁷¹

表 63-6:全身性硬化症的鑑別診斷 (Differential Diagnosis)
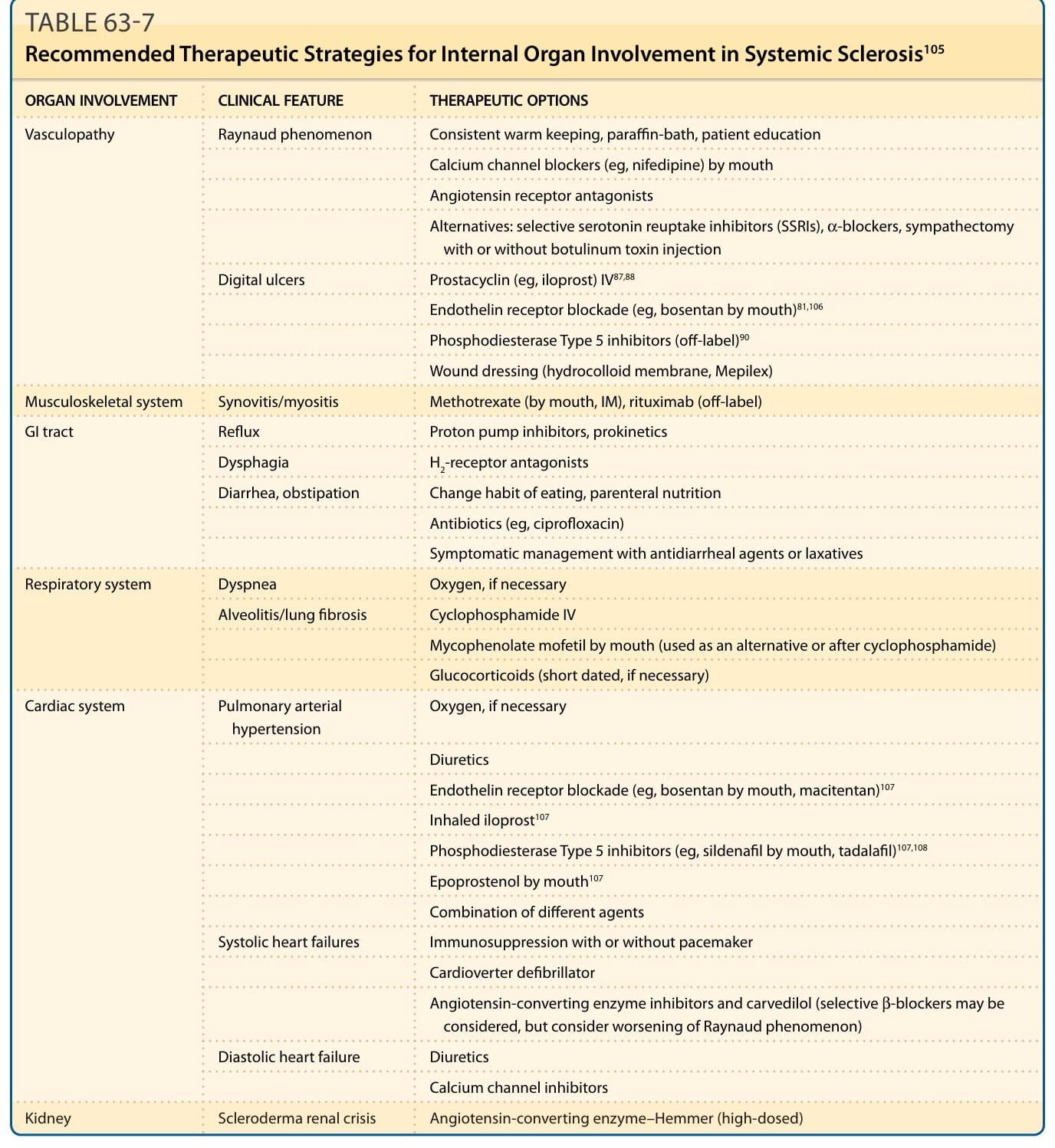
表 63-7:全身性硬化症內臟器官受累的建議治療策略 (Recommended Therapeutic Strategies for Internal Organ Involvement)¹⁰⁵
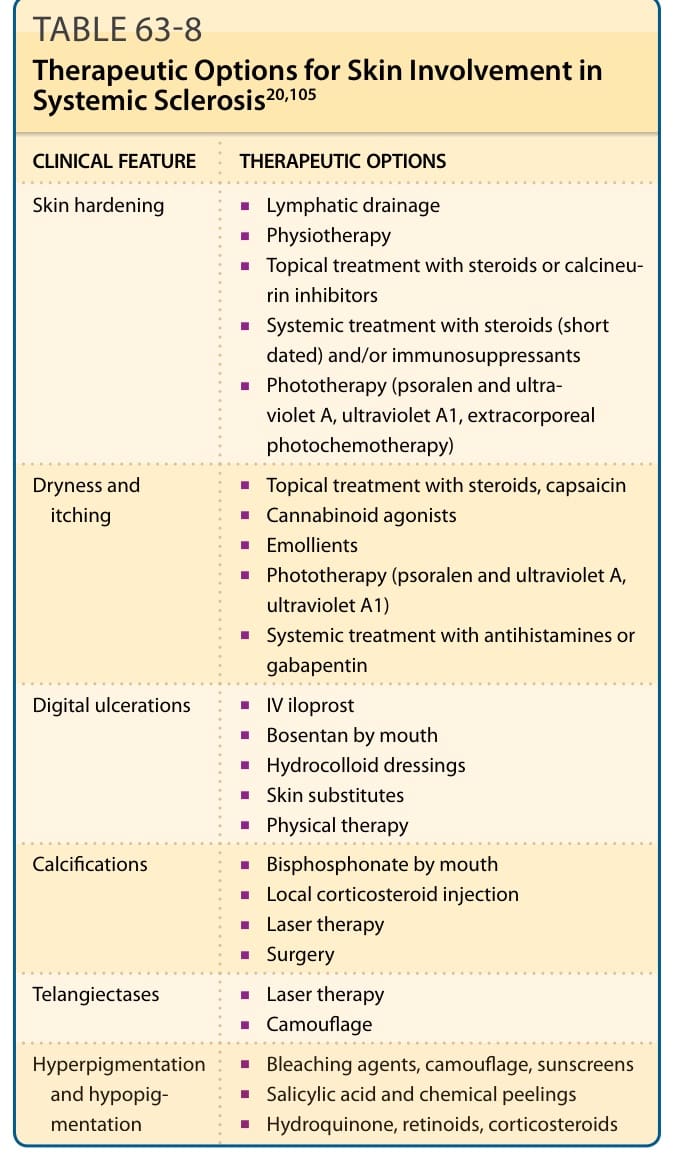
表 63-8:全身性硬化症皮膚受累的治療選項 (Therapeutic Options for Skin Involvement)²⁰,¹⁰⁵

表 63-9:早期瀰漫型皮膚全身性硬化症的幹細胞移植 (Stem Cell Transplantation, ASCT)⁷⁸⁻⁸⁰
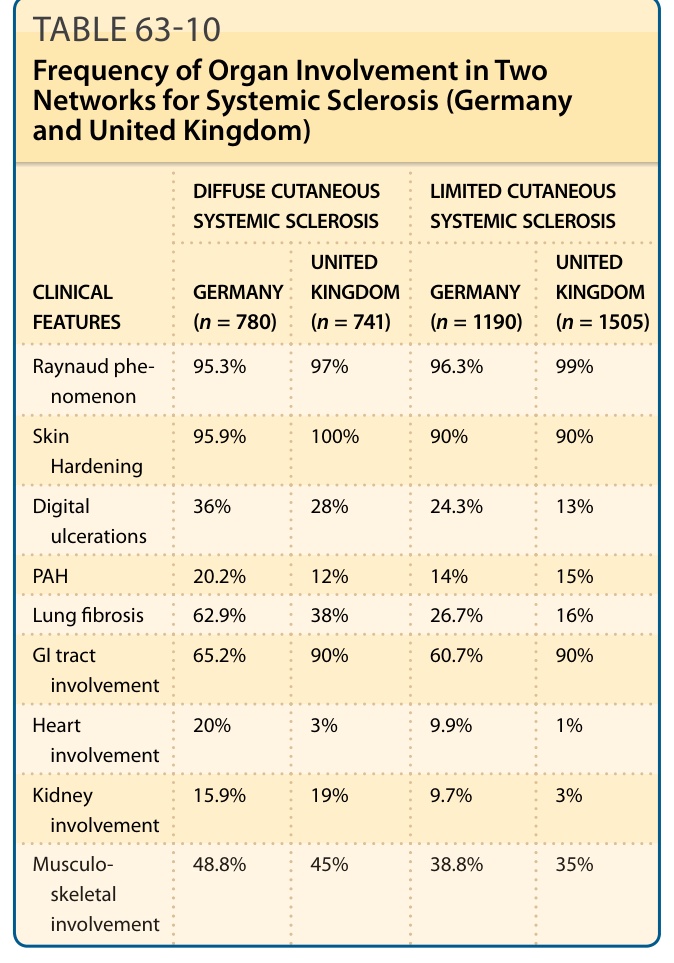
表 63-10:闡釋 2 個 SSc 網絡(德國與英國)中器官受累的頻率。