皮肌炎 (Dermatomyositis) 精華筆記
定義與分類
- 皮肌炎 (dermatomyositis, DM):全身性自體免疫疾病,特徵為皮膚與肌肉的發炎與損傷;發病年齡呈雙峰分布(兒童與成人),女性為男性的 2–3 倍。
- 傳統歸類為特發性發炎性肌病變 (idiopathic inflammatory myopathies) 之一,依 Bohan 與 Peter 標準須同時有肌炎與特徵性皮疹。
- 約 20% 病人有特徵性皮疹但無肌炎臨床徵象,稱為臨床無肌病皮肌炎 (clinically amyopathic dermatomyositis, CADM):須有典型皮疹(伴一致切片)≥6 個月、無無力證據。
- CADM 再分為:低肌病性 DM (hypomyopathic DM,至少一項肌肉檢查異常) 與無肌病性 DM (amyopathic DM,所有檢查正常)。
- CADM 與典型 DM 一樣有相同的全身性疾病(ILD)與惡性腫瘤風險,辨識同等重要。
流行病學
- 雙峰高峰:5–14 歲與 45–64 歲。
- 發生率約每年每百萬人 5–10 例;盛行率約每 10 萬人 10–20 例。
臨床表現
皮膚表現
- 特徵為多部位紫紅色 (violaceous) 斑塊,色從鮮粉紅到深紫;深膚色者紅斑常細微,需自然光與適當擺位檢查。受影響皮膚常顯著搔癢(尤其頭皮),程度常與紅斑不成比例。
- 頭皮:粉紅到紅色紅斑,常侵犯前額髮際線上下線狀帶,需與脂漏性皮膚炎、乾癬、接觸性皮膚炎鑑別。
- 向陽性徵象 (heliotrope sign):上眼瞼紫紅色紅斑,可伴眼周水腫,易誤診為過敏性接觸性皮膚炎或血管性水腫。
- 披肩徵象 (shawl sign):後頸、上背、肩部紅斑。V 領徵象 (V-neck sign):下前頸與前胸融合性紫紅色紅斑。
- Gottron 丘疹 (Gottron papules):覆蓋於 MCP 與 IP 關節之上的紫紅到粉紅色丘疹。Gottron 徵象 (Gottron sign):覆蓋於 IP 關節、鷹嘴突、髕骨、內踝的對稱斑狀紫紅色紅斑。
- 槍套徵象 (Holster sign):側髖與側大腿的紫紅色紅斑與異色症,常以毛囊為中心,易與毛囊角化症混淆。
- 技工手 (mechanic’s hands):內側拇指與外側第二、三指角化過度與龜裂;與 ILD 風險增加有關(勝算比 3.28,95% CI 1.36–7.88,P = 0.01)。
- 血管病變徵象:近端甲褶水腫/紅斑、微血管擴張與擴張的微血管環、甲皮出血與破碎甲皮(圖 62-12);亦可見齒齦微血管擴張、網狀青斑 (livedo reticularis)。
- 皮膚潰瘍:可見於約 30% 病人,常在伸側關節表面,應警覺抗 MDA5 抗體或惡性腫瘤(尤其有壞死時)。
- 卵圓形腭斑 (ovoid palatal patch):硬腭中線紫紅色斑塊,最常見於抗 TIF1-γ 抗體亞群。
- 「紅白相間 (red on white)」(圖 62-15):網狀白色斑點毗鄰紅斑或微血管擴張,具特徵性病徵,對 DM 具特異性,有助與皮膚型狼瘡鑑別。
- 異色症 (poikiloderma):晚期損傷表現(萎縮、色素增減、微血管擴張),不具診斷特異性。
- 臨床須區分活性(紅斑、搔癢、硬結、脫屑、潰瘍,可逆)與損傷(不應為損傷升階免疫抑制)。
- 鈣質沉著 (calcinosis):晚期表現,成人 DM 盛行率 20%、青少年 DM 高達 40%,與抗 NXP-2 抗體相關。
- 禿髮:多為非瘢痕性、瀰漫性;抗 MDA5 抗體者風險較高、常早發且嚴重。
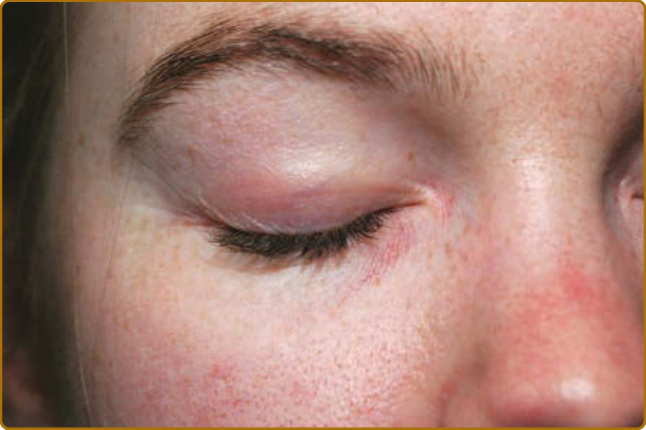
圖 62-3:向陽性徵象 (Heliotrope sign),上眼瞼紫紅到粉紅色紅斑與水腫。
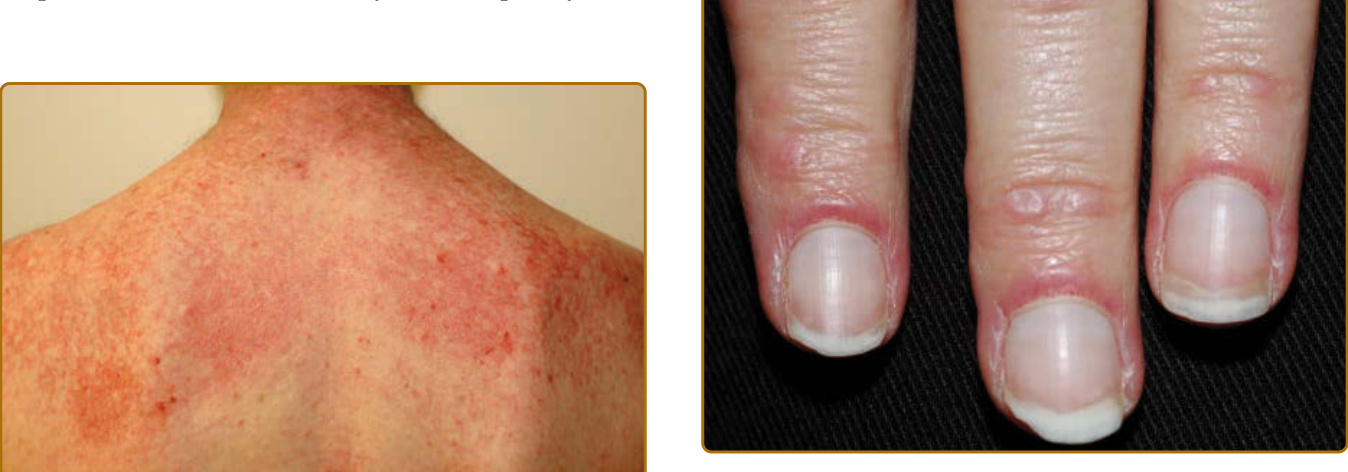
圖 62-7:Gottron 丘疹,覆蓋於指間關節之上的丘疹,伴近端甲褶紅斑、水腫與擴張甲褶微血管。
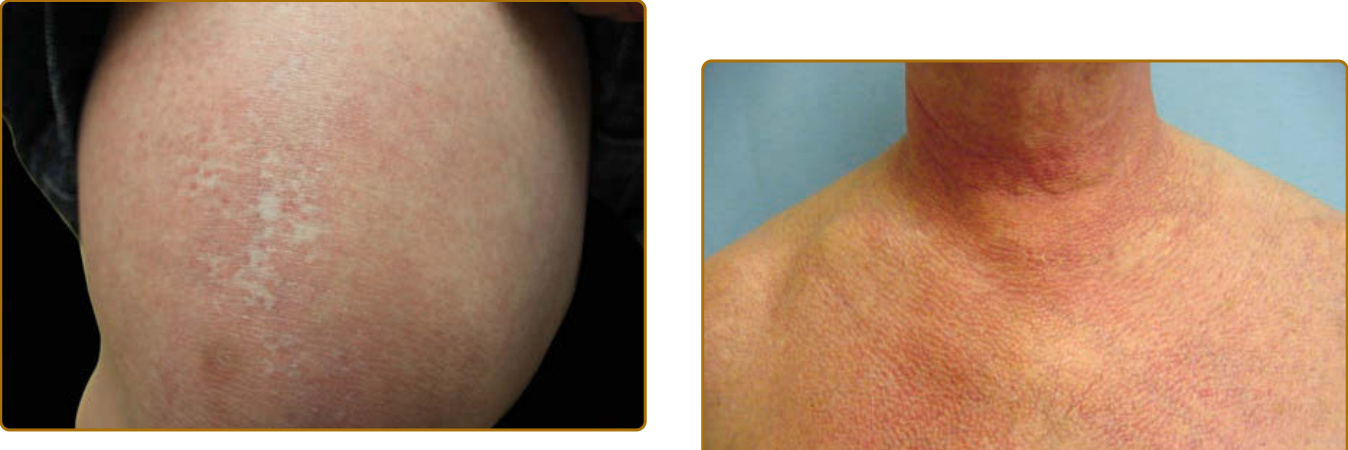
圖 62-15:「紅白相間 (Red on white)」,對皮肌炎具特異性,有助與皮膚型狼瘡鑑別。
皮膚外表現
- 肺部:ILD 是最常見肺部表現,也是病態與死亡主因,影響 15%–50% 病人。三型:無症狀(僅影像)、隱匿起病、急性起病(缺氧、呼吸衰竭)。抗合成酶抗體者 >75%–86% 發展 ILD;抗 MDA5 抗體者 50%–100%。快速進行性 ILD 對免疫抑制反應差,6 個月存活率約 40%,可影響 40%–60% 抗 MDA5 者。最常見影像/組織型態為非特異性間質性肺炎 (nonspecific interstitial pneumonia)。診斷必須做胸部高解析度 CT。
- 肌肉:典型為對稱性近端肌肉無力(肩帶、骨盆帶);發病者約 80% 於症狀發作第一年內出現無力。約 30% 病人有肌痛(觸診無壓痛)。發聲困難 (dysphonia) 達 40%;吞嚥困難 (dysphagia) 達 20%–50%。遠端手部無力多見於疾病晚期或抗 NXP2 抗體者。
- 關節:關節痛見於 30%–40% 病人,多侵犯手部小關節;真正關節炎可似類風濕性關節炎。抗 MDA5 與抗合成酶抗體者關節炎/關節痛更常見;抗合成酶(最常 Jo-1)可有非侵蝕性關節炎達 93%,屬抗合成酶症候群(發燒、關節炎、肌炎、ILD、技工手、雷諾現象)之一。
- 胃腸道:青少年 DM 約 4%,與更嚴重疾病相關,源於腸壁血管病變,可致潰瘍與穿孔。
- 心血管:多為亞臨床;最常見 ST-T 變化(12.5%–56.7%)與傳導異常(25%–38.5%)。抗 MDA5 亞型風險可能較高。
- 內臟惡性腫瘤:成人 DM 10%–20% 合併內臟惡性腫瘤,多在發病第 1–2 年內,標準化發生率比 4–6。常見實體腫瘤含乳房、肺、卵巢、攝護腺、結直腸、胃、胰;東南亞人鼻咽癌較多。危險因子:年齡漸增、男性、皮膚壞死、吞嚥困難、肌炎快速發作;保護因子:ILD、關節炎、雷諾現象。最相關抗體為抗 TIF1-γ(抗 NXP2 亦相關但較弱)。
致病機轉
- 免疫介導,先天與後天免疫皆參與。皮膚/肌肉切片見 CD3+ T 細胞、漿細胞樣樹突狀細胞、巨噬細胞與 B 細胞浸潤;皮膚為介面皮膚炎伴角質細胞損傷,肌肉為束周分布的肌纖維萎縮/退化/再生。
- 先天免疫活化關鍵:血液、肌肉、皮膚有高濃度干擾素 (IFN) 誘導基因/蛋白,與疾病活性相關;型態辨識受體被異常核酸活化。
- 後天免疫:HLA 區域多型性與 DM 高度相關(HLA-B8、HLA 8.1 祖先單倍型);BLK 與 TYK2 基因亦賦予風險。
- 血管病變可能為原發事件:內皮退化與微血管退失為最早期表現;C5b-9 膜攻擊複合體沉積於皮膚 DEJ 微血管與肌肉束周血管。
- 模型:遺傳易感個體遭環境誘發因子(UV、感染、惡性腫瘤)→ 抗原修飾/上調、細胞死亡、核酸反應與 IFN 生成、內質網壓力、先天免疫活化。
診斷
- 目前無經驗證的 DM 診斷標準;仰賴臨床醫師依病史與理學檢查的印象。
- 敏感的皮膚表現:顯微鏡下甲周微血管擴張、外側指角化過度、頭皮紅斑與感覺異常。特異性較高:「紅白相間」斑塊、卵圓形腭斑、肉眼可見甲周微血管擴張、Gottron 丘疹。
- 症候群性表現:禿髮+黏膜潰瘍+掌部紅斑性丘疹+嚴重關節痛/關節炎+喘 → 考慮抗 MDA5;技工手+關節炎+雷諾現象+肺症狀 → 抗合成酶;極度肌痛+周邊水腫+遠端無力 → 抗 NXP2。
- 自體抗體 (MSAs):TIF1-γ、NXP2、MDA5、SAE、Mi-2、Jo-1 及其他抗合成酶抗體,美國世代診斷敏感度 80%–85%,並助辨識亞群與風險分層。ANA 在 DM 約 50% 可陰性(用直接免疫螢光法)。
- 肌肉酵素:肌酸激酶 (creatine kinase)、醛縮酶 (aldolase)、LDH、AST、ALT;早期敏感,中晚期下降。應將肌酸激酶、醛縮酶、LDH 作為一組評估。劇烈活動後可升高,可於 10–14 天後重測。γ-麩胺醯轉移酶在肌炎不升高,可協助排除肝源。
- 血清鐵蛋白:抗 MDA5 病人常 >500 mg/dL,可助診斷並追蹤 ILD。
- 肌電圖:早期 70%–90% 活動性肌肉疾病可偵測;三聯表現為小振幅短時程多相運動單位電位、纖維顫動與正向銳波、複雜重複放電。
- MRI:可辨水腫/發炎/纖維化/萎縮,導引切片並評估治療反應。
病理學
- 皮膚:細胞稀少的介面皮膚炎、真皮黏蛋白增加、血管周圍淋巴球浸潤、血管擴張(圖 62-17)。介面皮膚炎缺如時(20% 病例)真皮黏蛋白增加總是存在。膜攻擊複合體(DEJ 與血管周圍)沉積加狼瘡帶陰性,對診斷 DM 相對皮膚型狼瘡 78% 敏感、93% 特異。
- 肌肉:皮膚科少需肌肉切片;典型表現為束周萎縮、退化/再生肌纖維、肌內膜微血管壁膜攻擊複合體沉積、內皮腫脹、微血管壞死。
臨床病程與預後
- 成人長期存活率約 65%–75%;緩解率報告 25%–70%,多數研究 5 年時約 20%–40%。
- 主要死因:惡性腫瘤、肺/心臟疾病、感染;抗 MDA5 抗體為死亡危險因子。
- 兒童約 60% 為慢性疾病,慢性化危險因子為治療延遲與早期持續皮膚疾病。
處置
- 原則:先評估受影響器官(皮膚、肌肉、肺);ILD 與相關癌症為主要死因,治療選擇時優先考量。癌症篩檢至關重要。皮膚疾病常與肌肉疾病反應不一致,須權衡長期毒性。多科協作(風濕、皮膚、神經、胸腔)。
- 癌症篩檢:診斷時做年齡別篩檢(大腸鏡、乳房攝影、攝護腺檢查)與血液檢查;較高風險期為診斷後 2–3 年。
局部治療
- 光防護為第一步(但達 60% 病人僅極輕度光敏感)。
- 局部皮質類固醇有緩解效果;第 I/II 級乳膏或軟膏用於肘、膝厚皮與手部角化過度,夜間可加封包增效。
- 局部鈣調神經磷酸酶抑制劑:他克莫司 0.1% 軟膏 (tacrolimus 0.1% ointment) 或匹美莫司 1% 乳膏 (pimecrolimus 1% cream),療效約同第 IV–VI 級局部類固醇,可安全用於臉部。
全身性治療
- 全身性皮質類固醇:肌炎第一線;普賴鬆 (prednisone) 單一療法 >0.5 mg/kg/day 可達肌肉發炎完全臨床反應(27%–87%)。皮膚單一療法不理想(僅部分反應)。
- 抗瘧藥:皮膚疾病第一線,30%–50% 改善;高達 30% 病人開始羥氯奎寧 (hydroxychloroquine) 時出現藥物疹;加奎納克林 (quinacrine) 可更有效。
- 甲氨蝶呤 (methotrexate):減少皮膚疾病嚴重度(50%–100%);與普賴鬆併用為肌炎第一線,常需 20 至 25 mg/wk。疑似/診斷 ILD 者應避用(恐誘發肺炎與肺纖維化)。
- 黴酚酸酯 (mycophenolate mofetil):2 至 3 g/day 有效降低皮膚疾病與肌炎;有 ILD 時為第一線口服藥;約 20% 病人在 2 g/day 出現噁心/腹瀉,可改用腸溶衣黴酚酸鈉。
- 靜脈注射免疫球蛋白 (IVIG):可能為皮膚型 DM 單一最有效藥物,70%–80% 達近完全/完全反應;標準方案 2 g/kg/mo,分 3 至 5 天投予;作用快,適用迅速惡化或因吞嚥困難/呼吸肌侵犯急性病危者。副作用:頭痛(達 56%)、無菌性腦膜炎、IgA 缺乏者過敏性休克(輸注前查血清 IgA)、靜脈血栓與腎損傷。
- 硫唑嘌呤 (azathioprine):DM/多發性肌炎合併研究中改善肌炎達 75%;隨機試驗用 2.5 mg/kg/day;常作為 ILD 維持治療(環磷醯胺誘導後)。
- 利妥昔單抗 (rituximab):皮膚型 DM 結果不一;對抗 Jo-1、抗 Mi-2 者肌炎可能有效;給藥依類風濕性關節炎方案,第 0 天與第 14 天靜脈注射 1000 mg;感染為最常見嚴重不良反應。
- 鈣調神經磷酸酶抑制劑(環孢素 cyclosporine、他克莫司):少用於肌肉/皮膚發炎;隨機試驗環孢素 3–3.5 mg/kg/day 與甲氨蝶呤 7.5–15 mg/wk 療效相當;主用於 ILD(抗 MDA5 或抗合成酶);腎毒性在 >3 mg/kg/day 最高。
- 氨苯碸 (dapsone) 與沙利竇邁 (thalidomide):證據稀少。
物理復健與特殊情況
- 肌力訓練改善肌力與功能、有氧運動改善耐力;建議診斷後及早物理治療。
- 鈣質沉著:手術切除局部病灶為最有效且確切治療;無單一藥物可靠有效,IVIG 與雙磷酸鹽 (bisphosphonates) 結果不一。
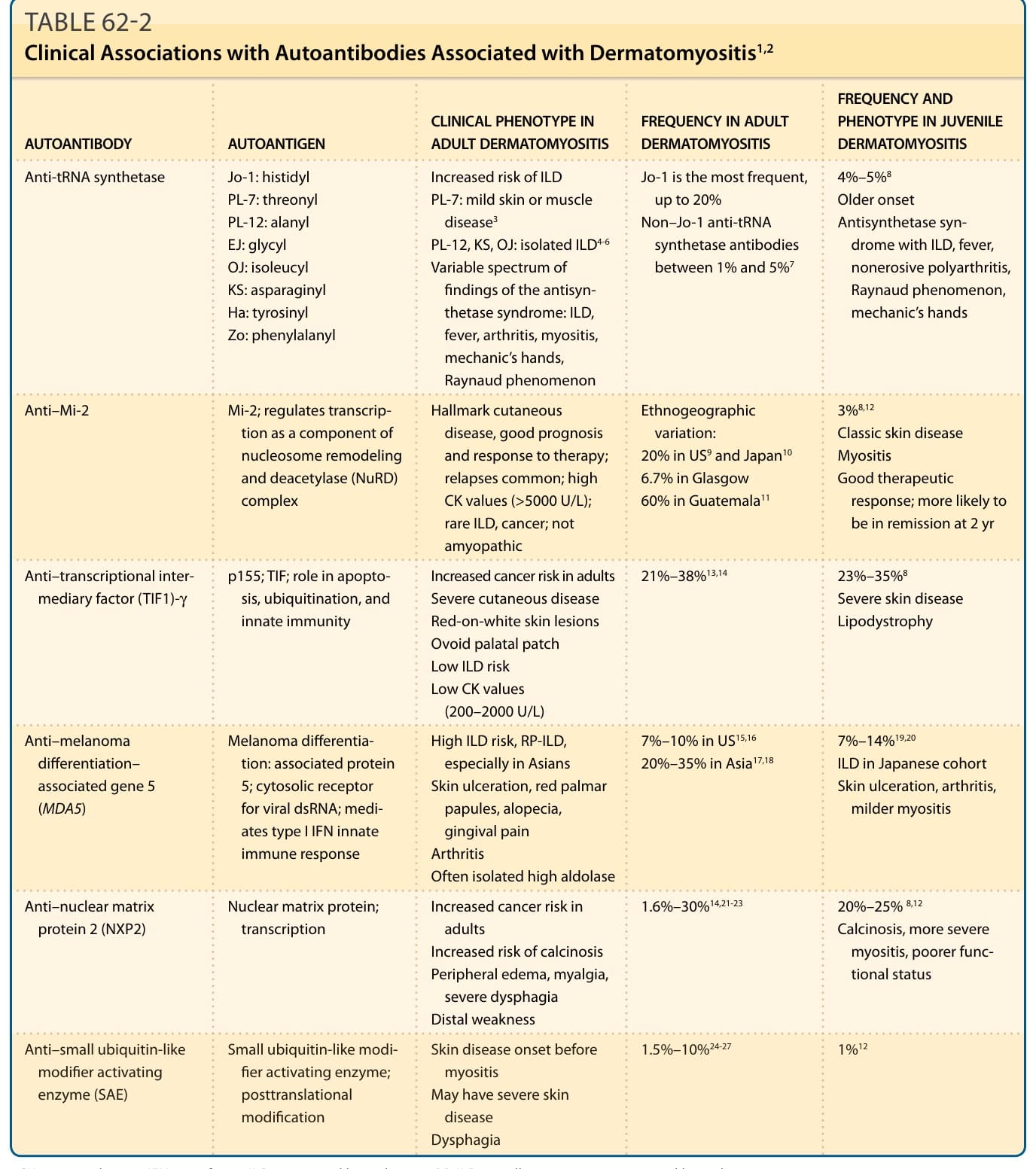
表 62-2:與皮肌炎相關之自體抗體的臨床關聯(抗合成酶、抗 Mi-2、抗 TIF1-γ、抗 MDA5、抗 NXP2、抗 SAE 的抗原、臨床表型與頻率)。
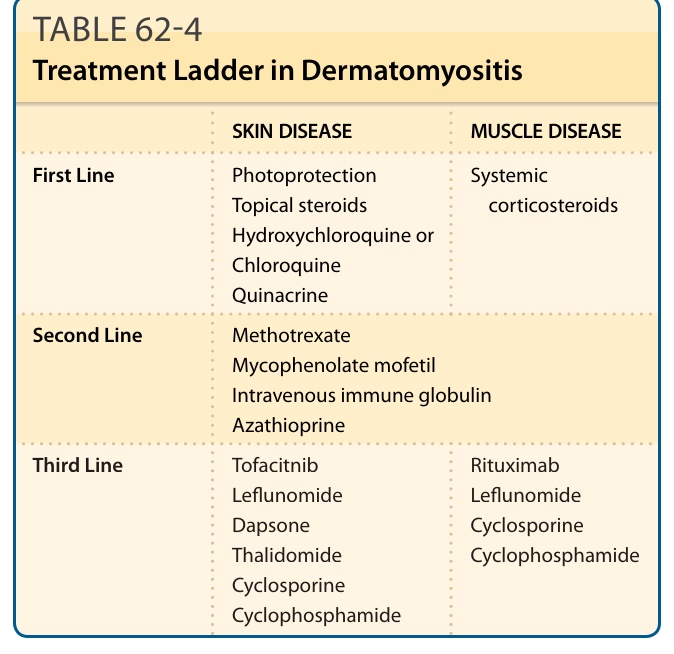
表 62-4:皮肌炎的治療階梯(皮膚疾病與肌肉疾病的第一至三線藥物)。