皮肌炎 (Dermatomyositis)
PART 10
自體免疫結締組織與風濕性疾病 (Autoimmune Connective Tissue and Rheumatologic Disorders)
重點一覽 (AT-A-GLANCE)
■ 皮肌炎(dermatomyositis, DM)是一種全身性自體免疫疾病,其發病年齡呈雙峰分布,侵犯兒童與成人,特徵為皮膚與肌肉的發炎與損傷。
■ 間質性肺病(interstitial lung disease, ILD)影響 20% 的病人,是這些病人病態與死亡的主要來源。
■ 在成人,DM 預示著 10% 至 20% 的病例合併內臟惡性腫瘤 (internal malignancy)。徹底的系統回顧 (review of systems) 與惡性腫瘤檢查(包括胸部、腹部與骨盆腔電腦斷層掃描 computed tomography scans)對於偵測在例行年齡別篩檢中被遺漏的隱匿性癌症 (occult cancers) 可能是審慎之舉。
■ 診斷的確立需結合具標誌性的皮膚表現與支持性的皮膚組織病理學,伴隨或不伴隨近端肌病變 (proximal myopathy) 的證據。診斷可能具有挑戰性,因為有 20% 的病人從未表現出具臨床意義的肌肉無力。
■ 皮膚表現以多個部位的紫紅色紅斑 (violaceous erythema) 為典型,最顯著者為眼瞼、上胸部、背部、肘部、膝部與側髖部,此外還有近端甲褶微血管擴張 (proximal nailfold capillary dilation) 與微血管周圍出血 (pericapillary hemorrhage)。
■ 特徵性自體抗體(抗合成酶 antisynthetase、抗 Mi-2、抗轉錄中介因子 [transcriptional intermediary factor, TIF1]-γ、抗黑色素瘤分化相關基因 5 [melanoma differentiation–associated gene 5, MDA5]、抗核基質蛋白 2 [nuclear matrix protein 2, NXP2],以及抗小泛素樣修飾物活化酶 [small ubiquitin-like modifier activating enzyme, SAE] 自體抗體)對於辨識臨床亞群,以及在缺乏典型臨床或組織病理學特徵的病例中建立診斷,可能有所助益。
■ 某些自體抗體的存在與否有助於器官侵犯及相關惡性腫瘤的風險分層。值得注意的是,抗 MDA5 與抗合成酶自體抗體的存在與 ILD 風險增加有關,而抗 TIF1-γ 與抗 NXP2 自體抗體的存在則與合併癌症的風險增加有關。
■ 皮膚型 DM 的治療可能具有挑戰性,多數有顯著皮膚疾病的病人需要全身性治療。要達到完全緩解可能需要多種藥物;考量到可能的長期治療療程,應仔細權衡每種藥物的風險與效益。
前言 (INTRODUCTION)
定義 (DEFINITIONS)
皮肌炎(dermatomyositis, DM)持續被歸類為肌炎 (myositis) 的一種形式,事實上,傳統上被視為特發性發炎性肌病變 (idiopathic inflammatory myopathies) 之一。此分類隱含的意義是,診斷本病必須有肌炎,以及特徵性皮疹——常被使用的 Bohan 與 Peter 標準即是此一概念的典型。¹,² 這是有問題的,因為目前廣為接受的是,有一部分病人(也許 20%)³ 具有特徵性皮疹,但沒有肌炎的臨床徵象或症狀。這些病人構成了現在被定義為臨床無肌病皮肌炎(clinically amyopathic dermatomyositis, CADM)的族群,此名詞由 Sontheimer 於 1991 年提出。⁴ CADM 病人具有特徵性 DM 皮疹(伴隨一致的皮膚切片)至少持續 6 個月,但在病史或臨床檢查上沒有無力的證據;早期的概念還要求證明肌肉酵素濃度正常⁵⁻⁷,但此定義後來被修訂為,肌力正常但肌肉酵素濃度異常的病人仍可被納入 CADM 的定義之下。⁸ 若非 CADM,則其餘的 DM 病人具有常被稱為「典型皮肌炎 (classic dermatomyositis)」者,意指傳統上認定、伴隨症狀性肌炎的疾病。CADM 對醫師而言是一個有用的名詞,因為它是以臨床方式定義,並不試圖否認亞臨床肌肉發炎 (subclinical muscle inflammation) 的存在。CADM 這把大傘還可依進一步影像學(如磁振造影 magnetic resonance imaging [MRI])、肌電圖 (electromyographic)、肌肉切片,或(現在的)肌肉酵素實驗室檢查的結果,進一步細分為剛好兩組:低肌病性 DM (hypomyopathic DM),其中至少有一項檢查結果異常;以及無肌病性 DM (amyopathic DM),其中所有檢查結果皆正常。應注意的是,數個其他團隊已提出聚焦於肌肉組織學⁹、臨床表現¹⁰,¹¹,或這些組合¹² 的 DM 分類標準。如今已清楚的是,無法定義出對 DM 具特異性的肌肉組織學與臨床表現,儘管仍有可能新型血清型 (serotypes) 具有足夠的敏感度與特異度,能在定義 DM 上扮演重要角色。「DM 皮疹」究竟由何構成的定義一直難以捉摸。Sontheimer 於 2002 年提出了診斷 CADM 的正式皮膚標準。⁵ 三項主要標準包括具特徵性病徵 (pathognomonic) 的向陽性徵象(heliotrope sign,上眼瞼的紫紅色紅斑)、Gottron 丘疹(覆蓋於掌指 [metacarpophalangeal, MCP] 與指間 [interphalangeal, IP] 關節之上的丘疹),以及 Gottron 徵象(覆蓋於膝部、肘部或 IP 關節之上的紅斑),加上 14 項額外的次要標準
各型——典型、臨床無肌病、低肌病與無肌病皮肌炎之間的關係 (Relationships among classic, clinically amyopathic, hypomyopathic, and amyopathic dermatomyositis)
皮肌炎 (Dermatomyositis)
典型 (Classic) — 臨床無肌病皮肌炎 (Clinically amyopathic dermatomyositis)
低肌病性皮肌炎 (Hypomyopathic dermatomyositis)
- 無無力 (No weakness)
- 在實驗室檢查、肌電圖或 MRI 上有肌炎證據 (Evidence of myositis on laboratory studies, EMG, or MRI)
無肌病性皮肌炎 (Amyopathic dermatomyositis)
- 無無力 (No weakness)
- 在實驗室檢查、肌電圖或 MRI 上無肌炎證據 (No evidence of myositis on laboratory studies, EMG, or MRI)
(圖 62-1)。Sontheimer 建議,若存在兩項主要標準,或一項主要標準加兩項次要標準,再加上至少一個部位的切片顯示與 DM 一致的組織病理學變化,即可做出 CADM 的診斷。⁵ 雖然這些標準看似全面且合理,但尚未以任何方式經過驗證。即使沒有證據顯示這一群病人在致病機轉、組織學或血清學上與典型組有所不同(除了沒有臨床無力之外),辨識出 CADM 病人仍將持續至關重要。即使這些病人在積極免疫治療 (aggressive immunotherapy) 方面的處置與肌炎病人相同(這未必是事實),他們仍是一群需要被辨識的重要族群,因為這些病人似乎與其典型對應者懷有相同的全身性疾病(如間質性肺病 [interstitial lung disease, ILD])與惡性腫瘤風險。⁸ 因此,目前對皮膚科醫師而言,辨識 CADM 的原型以及非典型或細微的皮膚表現尤其重要,因為這些線索可能是診斷所依據的唯一資料。
流行病學 (EPIDEMIOLOGY)
DM 在發病年齡上呈雙峰分布,發生於兩個高峰:生命中的 5 至 14 歲與 45 至 64 歲。本病侵犯女性的頻率為男性的兩到三倍。如前所述,DM 通常被歸類為特發性發炎性肌病變,此一群疾病包括多發性肌炎 (polymyositis)、包涵體肌炎 (inclusion body myositis)、非特異性肌炎 (nonspecific myositis) 與免疫介導壞死性肌病變 (immune-mediated necrotizing myopathy)。由於診斷標準缺乏精確性與標準化(許多研究甚至將 DM 與多發性肌炎歸為單一疾病)、病例確認方法不同、研究設計不同,以及可能的地理影響,估計純 DM 的發生率與盛行率具有挑戰性。¹³,¹⁴ 儘管如此,利用少數以族群為基礎的研究或現有最大型資料庫的資料,發生率與盛行率的估計值出奇地相似,發生率介於每年每 100 萬人 5 至 10 例,盛行率估計為每 10 萬人 10 至 20 例。來自挪威東南部最大型族群研究(>260 萬人)的資料估計,多發性肌炎或 DM 總計的時點盛行率 (point prevalence) 為每 10 萬人 8.7(95% 信賴區間 [confidence interval, CI],4.5–11.2),其中約半數病例為 DM。此研究估計發生率為每年每 100 萬人 6 至 10 例。¹⁵ Bendewald 與同事提供了來自明尼蘇達州 Olmstead 郡成人發病 DM 的族群估計值。年齡與性別校正後的發生率為每十年每 100 萬人 9.63(95% CI,6.09–13.17),盛行率為每 10 萬人 21.42 人(95% CI,13.07–29.77)。³ 在他們的小型世代中,20%(29 例中的 6 例)為臨床無肌病。對日本與台灣大型保險理賠資料庫資料的分析估計,年發生率分別為每年每百萬人 10 至 13 例與 6 至 10 例。¹⁶,¹⁷ 根據登錄資料,美國 2 至 17 歲兒童中青少年發病 DM 的年發生率估計介於每百萬人 2.5 至 4.1 例。¹⁸
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
病人病史的某些要素可能有助於區分 DM 與類似的皮疹。可能存在誘發因子,例如大量紫外線暴露、劇烈活動(對合併肌炎的病人而言),或近期惡性腫瘤。受影響的皮膚常合併顯著搔癢,尤其在頭皮,也可能被描述為「緊繃感」或灼熱,或具有其他感覺異常 (dysesthetic) 的特質,如爬行感或刺痛感。這種頭皮的嚴重搔癢可能由表皮小纖維神經 (epidermal small-fiber nerves) 的結構性損傷所致。¹⁹ 皮疹的自然病程為慢性且常為進行性,最終導致以色素異常 (dyspigmentation)、萎縮 (atrophy) 與微血管擴張 (telangiectasias) 為背景的表現。一過性 (evanescent) 或完全間歇性的皮疹不太可能代表 DM。然而,透過治療,許多病人最終可進入緩解,此時不再有進一步的活性,但殘留的損傷特徵(如異色症 poikiloderma)可能仍然存在。DM 受影響皮膚的特徵性皮膚表現是紫紅色斑塊 (violaceous patches) 與斑塊 (plaques),顏色從鮮粉紅色到深紫色不等。在較深膚色類型中,紅斑常很細微,仔細檢查對於偵測皮膚侵犯很重要。為了提高檢查的敏感度並察覺這些有時細微的顏色變化,病人的擺位與檢查室適當的照明至關重要。許多類型的頭頂直接照明往往會遮蔽 DM 皮膚所見的細微顏色變化,因此建議在臨床檢查時使用自然光。以斜躺或仰臥姿勢評估病人有助於減少陰影(尤其是臉部),並避免直接照明的過度曝光。頭皮是最常受侵犯的部位之一,常僅有粉紅到紅色的紅斑(圖 62-2),但可能伴隨細白色脫屑。其搔癢與感覺異常常很嚴重,且與檢查所見的紅斑程度不成比例。頭皮可在任何位置受影響,但常侵犯前額髮際線上下的線狀帶。單憑檢查表現並不總是具有足夠特異性,難以將其與脂漏性皮膚炎 (seborrheic dermatitis)、乾癬 (psoriasis) 與接觸性皮膚炎 (contact dermatitis) 區分。即使在其餘皮膚疾病處於靜止期時,頭頂頭皮或沿著髮分線或髮際線邊緣仍常可察覺到細微的紅斑。向陽性徵象(heliotrope sign,圖 62-3)可體現皮疹的粉紅到紫紅色調,類似此徵象命名所依據的花瓣顏色。眼瞼皮疹可伴隨眼周水腫 (periorbital edema),病人初期常被誤診為過敏性接觸性皮膚炎 (allergic contact dermatitis) 或血管性水腫 (angioedema)。此外,可見外眥 (lateral canthi)、內眥 (medial canthi) 與相鄰鼻側壁的紅斑(圖 62-4)。前額、臉頰、耳朵與下巴的表現可從未受侵犯到局部斑塊或斑塊,乃至瀰漫性紅斑不等。DM 的皮膚變化常分布於身體上的原型區域(表 62-1)。軀幹侵犯常見於後頸、上背與肩部,稱為披肩徵象(shawl sign,圖 62-5),可能延伸至上臂後側。病人可表現出對稱性紫紅色紅斑,常具有網狀青斑 (livedo) 的特質,對稱地出現於下背側部。融合性紫紅色紅斑出現於下前頸與前胸的陽光暴露區,稱為 V 領徵象(V-neck sign,圖 62-6)。覆蓋於 IP 與 MCP 關節之上的紫紅到粉紅色丘疹稱為 Gottron 丘疹(圖 62-7)。它們可能呈現與其他部位相同範圍的特徵,例如異色症、萎縮、色素減退 (hypopigmentation)、角化過度 (hyperkeratosis) 或潰瘍(圖 62-8)。它們可能呈現為較大、界線不清、圓形環狀的斑塊(常伴脫屑),或呈 2 至 4 mm、平滑、界線分明、常具臍狀凹陷 (umbilicated) 的丘疹。Gottron 徵象是對稱性斑狀紫紅色紅斑,覆蓋於 IP 關節、鷹嘴突(olecranon processes,圖 62-9)、髕骨 (patellae) 與內踝 (medial malleoli)。一般而言,青少年 DM 在 Gottron 徵象的典型區域更具特徵性地呈現萎縮與異色症。值得注意的是,許多皮膚型 DM 的皮膚侵犯區域未必位於紫外線(ultraviolet, UV)暴露區(即所謂「光分布 photodistributed」),包括頭皮、下背與側大腿。某些病人可表現出典型的光分布皮疹,但更常見的是即使在高 UV 暴露風險的部位,紅斑仍呈斑片狀。側髖部與側大腿的紫紅色紅斑與異色症稱為槍套徵象(Holster sign,圖 62-10)。這常呈現為紫紅色、以毛囊為中心 (folliculocentric) 的斑點或細微丘疹,可能與毛囊角化症 (keratosis pilaris) 混淆。皮疹呈紫紅色且典型為斑狀的性質,通常在缺乏毛囊角化症其他典型區域(如上臂外側)侵犯的情況下,有助於辨識 DM 的此一表現。此外,可發現鞭笞狀紅斑 (flagellate erythema),呈廣泛的線狀斑塊或蕁麻疹樣斑塊 (urticarial plaques),典型位於中下背與側腹,可能由抓痕或衣物或床單的壓印所致。其他特徵性的手部表現包括所謂的「技工手 (mechanic’s hands)」。此表現最初於 1979 年被描述為沿著內側拇指與外側第二、三指的角化過度與龜裂。²⁰ 此表現可能很細微,常需觸摸手指才能察覺其粗糙的質地(圖 62-11)。這種外側第二指角化過度似乎在質與量上不同於具有抗合成酶抗體 (antisynthetase antibodies) 病人的表現,後者合併的龜裂與脫屑往往也侵犯遠端指尖。2012 年,Sato 與同事發現,在他們的病人中,有技工手的 DM 病人(78%,9 例中的 7 例)與無技工手者(40%,30 例中的 12 例)相比,ILD 盛行率增加²¹,支持技工手是可能存在 ILD 的皮膚線索。Werth 與同事近期一項研究發現,在 101 名 DM 病人中,技工手與罹患 ILD 的勝算比 (odds ratio) 為 3.28(95% CI,1.36–7.88;P = 0.01)有關。血管病變 (vasculopathy) 的證據在皮膚上以多種方式表現。近端甲褶 (proximal nailfold) 侵犯可表現為細微的水腫或紅斑,以及顯微鏡下或臨床上明顯的微血管擴張。當這些甲褶微血管變化顯著且肉眼即可清楚看見時,相較於其他結締組織疾病,它們可高度提示 DM。典型表現包括發紅、水腫、常有觸痛的近端甲褶,伴隨分枝狀 (ramified) 與擴張的微血管環,其間夾雜蒼白到白色的無血管區與微血管退失 (capillary drop-out)、甲皮出血 (cuticular hemorrhages),以及拉長、破碎的甲皮 (cuticles)(圖 62-12)。雖然這些變化是持續性皮膚疾病活性的徵象²²,但在長期的 DM 中,損傷可能表現為持續擴張與樹枝狀的微血管。²³
敏感的甲褶可以是 DM 有用的診斷症狀。此外,DM 中亦曾描述齒齦微血管擴張 (gingival telangiectasias)。²⁴ 此外,少數病人可見網狀青斑 (livedo reticularis)。皮膚潰瘍 (cutaneous ulceration) 可以是血管病變的典型徵象,但它也可能是強烈的液化性介面皮膚炎 (liquefactive interface dermatitis) 或抓痕的結果。潰瘍可能存在於 30% 的病人中,常侵犯覆蓋於伸側關節表面之上的皮膚,但可發生於任何部位。²⁵ DM 中的皮膚潰瘍應警覺抗 MDA5 抗體或惡性腫瘤的存在(尤其在見到壞死時)。在有抗 MDA5 抗體的情況下,皮膚潰瘍常發生於 Gottron 丘疹之上(見圖 62-8)或 Gottron 徵象的區域(如伸側表面)。²⁵,²⁶ 潰瘍與 ILD 的存在相關,但這可能是透過其與抗 MDA5 抗體的關聯。²⁵,²⁷ 因此,有抗 MDA5 抗體的病人皮膚潰瘍惡化,可能是 ILD 惡化的皮膚徵象。觀察口腔黏膜(尤其是硬腭)可能提供有價值的徵象以協助 DM 的診斷。可觀察到橫跨硬腭中線的對稱性紫紅色斑塊,稱為卵圓形腭斑(ovoid palatal patch,圖 62-13),最常見於具有抗轉錄中介因子 1γ(transcriptional intermediary factor 1γ, TIF1-γ)抗體的 DM 病人亞群。²⁸ 這些病灶的切片似乎顯示介面黏膜炎 (interface mucositis),與 DM 的典型表現一致。如同稍後描述的甲褶微血管變化,這些硬腭變化似乎會隨疾病活性受控而消退。這些病灶可能與盤狀紅斑性狼瘡 (discoid lupus) 或扁平苔癬 (lichen planus) 的口腔表現混淆,但它們一致地局限於硬腭中央,可在其他皮膚特徵無診斷意義時協助 DM 的診斷。此外,其他口腔黏膜表現包括(前述的)齒齦微血管擴張,以及齒齦或頰黏膜周圍更典型的扁平苔癬樣 (lichen planus–like) 病灶。後者究竟代表共存的扁平苔癬,或是 DM 疾病譜的一部分,目前並不清楚。一般而言,臨床醫師應始終區分皮膚活性的徵象(可能透過治療逆轉)與損傷。DM 皮膚中活性與損傷的區分對臨床決策至關重要,以免因皮膚損傷的徵象而升階免疫抑制治療。皮膚疾病活性以紅斑、搔癢、硬結(丘疹或斑塊)、脫屑或潰瘍為特徵。雖然紅斑常是活性的重要徵象,但它也常可以是損傷的徵象(如瀰漫性微血管擴張性紅斑),因此必須小心不要因為沒有其他疾病活性特質的紅斑而不慎升階治療。此現象在臉部與胸部尤其可能成立,在缺乏症狀或其他皮膚變化的情況下,淡淡的紅斑常是輕度損傷,可能對傳統免疫抑制治療無反應。此外,手掌與掌側表面呈粉紅到紅色紅斑的網狀型態(更常見於抗 MDA5 組,圖 62-14),加上更典型的網狀青斑(包括出現於側腹者),代表與疾病活性關聯不明的血管變化。當曾存在大量發炎時,可見一種獨特且具特徵性病徵的型態,由網狀、有時萎縮的白色斑點,毗鄰紅斑或微血管擴張所組成,作者稱之為「紅白相間 (red on white)」(圖 62-15)。²⁹
沿著雙顳側髮際線的薄皮膚是觀察「紅白相間」型態的常見部位,但值得注意的是,此型態未必遵循陽光暴露的型態,也可能出現於有毛髮的頭皮上。日益清楚的是,這些區域中許多並不代表永久性損傷本身,因為這些病灶可能隨時間緩慢消退,即使在萎縮的區域亦然。然而,此型態是非常有用的診斷工具,因為它似乎與其他結締組織疾病(如皮膚型狼瘡 cutaneous lupus)無關。DM 造成的皮膚損傷表現為棕色、常呈網狀的發炎後色素過度沉著 (postinflammatory hyperpigmented) 斑塊,位於先前疾病活性的區域。長期的疾病活性(典型位於陽光暴露區)導致更顯著的損傷,特徵為萎縮、色素減退、色素過度沉著與微血管擴張,稱為異色症(poikiloderma,圖 62-16)。異色症(相對於前述的「紅白相間」表現)是晚期表現,且不具診斷特異性,因為許多其他後天與先天疾病都會導致異色症,例如皮膚型狼瘡、慢性光化性損傷(Civatte 異色症 poikiloderma of Civatte)、異色性蕈狀肉芽腫(血管萎縮性異色症 poikiloderma vasculare atrophicans)、伯氏疏螺旋體感染 (Borrelia infection)(慢性萎縮性肢端皮膚炎 acrodermatitis chronica atrophicans)、慢性放射性皮膚炎、移植物對抗宿主疾病 (graft-versus-host disease)、羥基脲 (hydroxyurea),以及先天性角化不良 (dyskeratosis congenita)。因此,需要輔助性皮膚表現來辨明異色症的成因。鈣質沉著 (Calcinosis) 通常是皮膚、皮下組織、筋膜或肌肉的晚期表現,典型侵犯軀幹、近端肢體,或先前疾病活性的區域。鈣質沉著的盛行率在成人 DM 為 20%³⁰,在青少年 DM 高達 40%。³¹ 鈣質沉著在青少年 DM 中也較成人 DM 在發病後更快發生(分別為 2.9 年對 7.9 年)。³² 在青少年 DM,鈣質沉著發展的危險因子包括較長的疾病病程、較年輕的發病年齡、持續的疾病活性,以及內臟器官侵犯。³³,³⁴ 鈣質沉著在 DM 中最常發生於四肢,這與系統性硬化症 (systemic sclerosis) 相反,後者最常見指端鈣質沉著。³² 在青少年與成人 DM 中,抗核基質蛋白 2(nuclear matrix protein 2, NXP-2)抗體的存在皆與鈣質沉著風險增加有關。³⁵,³⁶ 在成人,指尖潰瘍與鈣質沉著有關³⁶,顯示血管功能不足或損傷可能參與鈣質沉著的致病機轉。鈣質沉著在抗 MDA5 亞群中也常見(尤其在長期患病的病人)³⁶,此亞群與已知的血管病變有關。
脂膜炎 (Panniculitis) 反映 DM 的活性疾病,典型侵犯臀部、軀幹與近端肢體;它可能進展為鈣質沉著或脂肪萎縮 (lipoatrophy)。³⁷
組織病理學顯示小葉性脂膜炎 (lobular panniculitis),但可能具有狼瘡性脂膜炎 (lupus panniculitis) 伴脂膜樣變化 (lipomembranous changes) 的特徵,或如深部硬斑病 (deep morphea) 般的隔膜增厚。DM 中的禿髮 (Alopecia) 最常為非瘢痕性 (nonscarring) 且瀰漫性,雖然也可見斑片狀侵犯(罕見伴瘢痕)。禿髮可由 DM 疾病、共存疾病、藥物或休止期落髮 (telogen effluvium) 所致。具有抗 MDA5 抗體的病人禿髮風險較高,常為嚴重且早期發生於疾病過程中。
皮膚型皮肌炎的罕見表現 (RARE PRESENTATIONS OF CUTANEOUS DERMATOMYOSITIS)
無論是全身性或局限於四肢的皮下水腫 (Subcutaneous edema),已被認定為 DM 的罕見表現。³⁸ 有趣的是,遠端肢體的水腫近期被發現與抗 NXP2 抗體有關,並可能預示更嚴重的肌肉疾病。³⁸,³⁹ 抗 NXP2 抗體與文獻中所描述更嚴重、全身性表現之間的關係尚不清楚。罕見情況下,DM 可表現為紅皮症 (erythroderma),其中 90% 或更多的體表面積呈現融合性紅斑。全身性魚鱗癬 (Generalized ichthyosis) 也可以是 DM 的表現徵象。有一群 DM 病人的臨床表現兼具乾癬與 DM 的重疊特徵。⁴⁰,⁴¹ 他們的皮膚疾病可顯示覆蓋於 MCP 與近端指間(proximal interphalangeal, PIP)關節、肘部與膝部之上的乾癬樣 (psoriasiform)、界線分明的厚斑塊,此外還有擴張且異常的甲褶微血管。皮膚切片往往同時呈現表皮增生 (epidermal hyperplasia) 與介面皮膚炎。這些病人中有些有乾癬病史,這些病灶究竟代表並存的乾癬或 DM 的乾癬樣表現,目前並不清楚。
皮膚外表現 (EXTRACUTANEOUS FINDINGS)
肺部侵犯 (PULMONARY INVOLVEMENT)
ILD 是 DM 最常見的肺部表現,也是這些病人病態與死亡的主要原因。⁴² DM 的其他肺部表現包括吸入性肺炎 (aspiration pneumonia);藥物引起的肺炎 (drug-induced pneumonitis);以及罕見的肺高壓 (pulmonary hypertension)。ILD 影響 15% 至 50% 的 DM 病人,取決於族群與自體抗體分布。⁴³⁻⁴⁶
肺部侵犯有三種臨床描述的型態,可能先於或後於肌肉侵犯⁴⁷,⁴⁸:它可能無症狀,僅有 ILD 的放射影像證據;它可能呈隱匿起病,伴隨運動能力下降、用力時呼吸困難或乾咳的逐漸發展;最後,它可能呈急性起病,伴隨缺氧 (hypoxia) 及可能的呼吸衰竭,需要住院治療。在近期一項來自美國 438 名多發性肌炎或 DM(n = 393)大型世代的研究中,DM 病人 ILD 的存在與死亡風險增加有關,風險比 (hazard ratio) 為 2.13(95% CI,1.06–4.25;P = 0.03)。⁴⁹ 大型病例系列顯示,超過 75% 至 86% 具有抗合成酶抗體的病人會發展出 ILD。⁴⁷,⁵⁰
同樣地,具有抗 MDA5 抗體的 DM 病人發展出 ILD 的風險明顯增加,估計介於 50% 至 100% 之間²⁵,⁵¹(表 62-2)。快速進行性 ILD (Rapidly progressive ILD) 是一種對免疫抑制治療反應不佳的侵襲性型態,6 個月存活率約為 40%。⁵² 快速進行性 ILD 可能影響 40% 至 60% 具有抗 MDA5 抗體的病人。⁵³⁻⁵⁶
肺功能檢查(pulmonary function tests, PFTs)顯示限制型 (restrictive) 疾病型態,伴隨用力肺活量(forced vital capacity, FVC)下降⁵⁷ 或一氧化碳擴散量 (diffusion capacity of carbon monoxide) 下降。6 分鐘步行測試 (6-minute walk test) 的用力性血氧去飽和 (exertional oxygenation desaturation) 提供心肺功能與運動表現的整體評估。然而,諸如體能失調 (deconditioning)、肌病變、關節炎、呼吸肌無力與肺高壓等干擾因子,也可能造成 FVC 與一氧化碳擴散量降低。⁵⁸ 因此,胸部高解析度電腦斷層(computed tomography, CT)是建立 ILD 診斷的必要步驟。胸部高解析度 CT 掃描是有價值的診斷檢查,可在症狀出現前顯示亞臨床纖維化 (subclinical fibrosis)。高達 65% 的多發性肌炎與 DM 病人在 CT 上會有 ILD 的亞臨床證據。⁵⁹
DM 中最常見的放射影像與組織學型態是非特異性間質性肺炎 (nonspecific interstitial pneumonia),報告於 81.8%(22 例中的 18 例)。⁶⁰ 在放射影像上,基底部與周邊的毛玻璃樣陰影 (ground-glass opacities) 與胸膜下豁免 (subpleural sparing) 是非特異性間質性肺炎的特徵。其他侵犯型態包括尋常型間質性肺炎 (usual interstitial pneumonia)、隱原性機化性肺炎 (cryptogenic organizing pneumonia),以及瀰漫性肺泡損傷 (diffuse alveolar damage),後者預後不佳。肺動脈高壓 (Pulmonary arterial hypertension) 是 DM 的罕見表現。症狀可能包括疲勞增加、喘不過氣、用力時呼吸困難、心悸、胸痛、水腫、頭暈,以及罕見的暈厥前 (presyncopal) 或暈厥 (syncopal) 發作。PFT 顯示一氧化碳擴散量不成比例地偏低,而 FVC 相對正常,應促使進一步篩檢肺動脈高壓。若因呼吸惡化與 PFT 結果異常而檢查高解析度 CT 掃描,可能顯示肺動脈擴大但無 ILD 證據。提示肺高壓的心臟超音波 (Echocardiographic) 表現包括右心室收縮壓大於 40 mm Hg、右心室擴大、三尖瓣逆流速度峰值大於 3.0 m/s,以及心包膜積液 (pericardial effusion) 的存在。⁶¹ 經胸心臟超音波 (Transthoracic echocardiography) 偵測肺高壓的敏感度僅 82%、特異度僅 69%⁶¹,且無法區分肺動脈高壓病人與左心疾病或 ILD 相關肺高壓病人。診斷肺動脈高壓的黃金標準是右心導管檢查 (right heart catheterization),顯示靜息時平均肺動脈壓 25 mm Hg 或更高,以及吐氣末肺動脈楔壓 (end-expiratory pulmonary artery wedge pressure) 15 mm Hg 或更低。⁶²
肌肉侵犯 (MUSCLE INVOLVEMENT)
DM 的肌炎典型表現為對稱性近端肌肉無力 (symmetrical proximal muscle weakness)。約 20% 的 DM 病人(即 CADM 者)沒有無力的臨床證據或症狀,雖然這些病人中究竟有多少比例實際上可能有亞臨床肌炎尚不清楚。若 DM 病人確實發展出無力,則約 80% 的病人在症狀發作後第一年內發展出無力。⁶³ 肌炎的時間病程可能為急性、亞急性,或慢性且進行性。
病人常報告肩帶與骨盆帶周圍肌肉及近端肢體的伸肌無力。股四頭肌 (Quadriceps) 與臀肌 (gluteal muscle) 無力可表現為難以從椅子或馬桶起身、爬樓梯或踏上路緣。病人可能將肩部與上肢無力報告為難以洗頭或伸手拿頭頂櫥櫃中的物品。頸屈肌 (Neck flexor) 侵犯也很常見,表現為仰臥時難以將頭抬離床面。病人也可能抱怨肌痛 (myalgias),即使在沒有明顯臨床無力的情況下也可能發生。約 30% 的病人抱怨有或無肌肉無力的肌肉疼痛。⁹ 肌痛被描述為痠痛、肌肉緊繃或灼熱,但肌肉觸診不會有壓痛。應小心將此疼痛與其他疼痛原因(如關節痛或纖維肌痛症 fibromyalgia)區分⁶⁴,且必須小心不要為了可能是疼痛疾患的症狀而升階免疫抑制。胸壁或橫膈呼吸肌的侵犯也可能導致呼吸功能不全,偶爾導致呼吸衰竭。病人可能注意到因環杓肌 (cricoarytenoid muscle) 侵犯而聲音沙啞或刺耳(發聲困難 dysphonia),此情況發生於高達 40% 的 DM 病人。⁶⁵ 此外,吞嚥困難 (dysphagia) 可能發生於 20% 至 50% 的病例,因咽部肌肉無力,因而無法在吞嚥的咽部期推送食物。⁶⁶ 有趣的是,吞嚥困難與前頸肌(如胸鎖乳突肌 sternocleidomastoids)無力之間有高度相關。⁶⁷ 手部遠端肌肉無力(表現為難以打開罐子或握住物品)較典型地發生於疾病晚期或具有抗 NXP2 抗體的病人。
關節侵犯 (JOINT INVOLVEMENT)
關節痛 (Arthralgias) 在 DM 中很常見,報告於 30% 至 40% 的病人。⁶⁸⁻⁷⁰ 一般而言,DM 的關節痛為輕度至中度嚴重,侵犯手部小關節(包括腕關節、MCP 與 PIP 關節),以及肩部、肘部與踝部。⁷¹ 更罕見的情況下,病人可有真正的關節炎,常表現為對稱性多關節病變 (symmetric polyarthropathy),侵犯遠端關節,且臨床上常與類風濕性關節炎 (rheumatoid arthritis) 難以區分。因此,以關節炎為表現症狀的 DM 病人,可能在皮膚、肌肉或肺部表現演進之前,被診斷為患有類風濕性關節炎或類風濕性關節炎重疊疾病。⁷²,⁷³ 引出發炎性關節炎症狀的示例性病史問題包括關節腫脹、晨間僵硬持續 30 分鐘或更久,或隨活動改善的關節疼痛。對滑膜炎 (synovitis)(指示活動性關節發炎)的評估,可透過觸診溫熱感;活動範圍;腫脹或可觸及的積液;以及手部每個小關節、肘部、肩部、膝部與其他有症狀關節的壓痛來判定。
具有抗 MDA5 與抗合成酶抗體的 DM 病人,關節炎與關節痛更為常見。然而,亦曾報告侵蝕性 (erosive) 變化,較常見於抗合成酶抗體亞群。⁷⁴⁻⁷⁷ Hall 與同事發現,11 名抗 MDA5 病人中有 9 名(81.8%)相對於 149 名非 MDA5 DM 病人中有 40 名(26.7%)表現出發炎性關節炎(P <0.001)。⁷⁸
具有抗合成酶抗體(最常見者為 Jo-1)的 DM 病人,也可能表現出非侵蝕性關節炎 (nonerosive arthritis),高達 93% 的病例。⁷¹
在這些具有抗合成酶抗體的病人中,非侵蝕性關節炎可能發生於「抗合成酶症候群 (antisynthetase syndrome)」的情境下,此症候群由發燒、關節炎、肌炎、ILD、技工手或雷諾現象 (Raynaud phenomenon) 組成。關節炎可能在 50% 的病人疾病復發期間惡化。⁷¹
胃腸道侵犯 (GASTROINTESTINAL INVOLVEMENT)
胃腸道(gastrointestinal, GI)侵犯在青少年 DM 中是不常見的表現,報告於 4% 的青少年 DM 病人。⁷⁹ 它與更嚴重的疾病有關⁸⁰,且被認為是由侵犯腸壁的血管病變所致。⁸¹ 其後遺症包括潰瘍與穿孔,可能危及生命。表現症狀包括持續且惡化的腹痛。他們也可能有非特異性的伴隨症狀,例如腹瀉、嘔吐、便秘,以及更罕見的明顯吐血 (hematemesis) 或便血 (hematochezia);青少年病人初期可能沒有出血的證據,例如黑便 (melena)、糞便潛血,或穿孔的放射影像證據。⁸² 因此,對有腹痛的青少年 DM 病人進行審慎監測是有必要的,以便辨識並及早介入有 GI 侵犯的病例。
心血管侵犯 (CARDIOVASCULAR INVOLVEMENT)
DM 的心臟侵犯日益被認定為重要的臨床特徵。心臟侵犯通常為亞臨床。最常見的心電圖異常是 12.5% 至 56.7% 病人的 ST-T 節段變化,以及 25% 至 38.5% DM 病人的傳導異常。⁸³ 心臟超音波表現顯示 8% 至 15% 病人有左心室肥厚,42% 病人有左心室舒張功能障礙。⁸⁴ 心肌炎 (Myocarditis) 可能導致心肌纖維化與心室功能障礙,進而導致心肌病變 (cardiomyopathy)。Rosenbohm 與同事以心臟 MRI 篩檢 11 名 DM 病人,發現 54%(11 例中的 6 例)顯示晚期釓增強 (late gadolinium enhancement) 的證據,與心肌發炎一致。⁸⁵ 心肌旋轉蛋白 I(cardiac troponin I)可能是偵測亞臨床心肌侵犯的有用生物標記。⁸⁶ 具有抗 MDA5 抗體亞型的病人,心臟侵犯的風險可能較高。⁸⁷,⁸⁸
內臟惡性腫瘤 (INTERNAL MALIGNANCY)
DM 在 10% 至 20% 的病例中與內臟惡性腫瘤有關。⁸⁹ 惡性腫瘤往往發生於疾病發作的第一至二年內,可為多種類型。多項以族群為基礎的研究估計,相較於正常族群,惡性腫瘤的標準化發生率比 (standardized incidence ratio) 為 4 至 6。⁹⁰ 過度呈現的癌症類型可能依研究族群而異,但似乎同時涉及實體腫瘤與造血系統惡性腫瘤。常見的實體腫瘤類型包括乳房、肺、卵巢、攝護腺、結直腸、胃與胰臟癌⁹¹⁻⁹³,而鼻咽癌 (nasopharyngeal cancer) 在東南亞人中較常見。⁹⁴ DM 與惡性腫瘤之間關係的機轉尚不清楚,但可能包括免疫抑制治療情境下癌症風險增加、加強監測情境下偵測率增加,以及 DM 發生於對內臟惡性腫瘤免疫反應的情境下。有數個與惡性腫瘤風險相關的臨床危險因子,包括年齡漸增、男性、皮膚壞死、吞嚥困難,以及肌炎的快速發作。⁹⁵ 對惡性腫瘤具保護作用的因子包括 ILD、關節炎與雷諾現象。近期,惡性腫瘤也被發現與特定的 DM 特異性自體抗體更為相關。與惡性腫瘤相關的主要抗體是抗 TIF1-γ,雖然風險增加的確切程度似乎在不同研究與不同研究族群之間有所差異。⁹⁶,⁹⁷
較近期的資料顯示,抗 NXP2 抗體也可能與癌症有關,雖然此關聯不如前者強烈。⁹⁷,⁹⁸
癌症篩檢是爭議的來源,因為目前尚無指引指導新診斷 DM 病人的篩檢。多數權威至少會同意進行年齡別的例行癌症篩檢。然而,在一項小型研究中,Sparsa 與同事報告,初始的例行癌症篩檢未能發現其世代中 13 例惡性腫瘤中的 4 例。⁹⁹ 近期一項 400 名病人的研究顯示,相當數量的隱匿性癌症(29 例中的 17 例)僅透過超出「年齡別」範圍的檢查(通常是 CT 掃描)才被發現。辨識應該安排哪些檢查以及在哪些病人族群中安排,是高度優先的課題。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
DM 主要是一種免疫介導的疾病,分子與組織學證據支持先天性 (innate) 與後天性 (adaptive) 免疫皆扮演角色。肌肉與皮膚切片顯示 CD3+ T 細胞、漿細胞樣樹突狀細胞 (plasmacytoid dendritic cells) 與巨噬細胞,以及 B 細胞(尤其在肌肉中)的浸潤。¹⁰⁰,¹⁰¹ 實質細胞損傷在皮膚表現為伴隨角質細胞 (keratinocyte) 損傷的介面皮膚炎,在肌肉則表現為肌纖維的萎縮、退化與再生,典型呈束周 (perifascicular) 分布。有強力證據支持先天免疫系統的活化是疾病致病機轉與傳播的關鍵步驟。在血液、肌肉與皮膚中發現高濃度的干擾素(interferon, IFN)誘導基因與蛋白質,並已證明與疾病活性相關。¹⁰² 有一些證據顯示此反應可能由 IFN-β 所驅動,雖然這尚不清楚。有多條途徑可能導致 IFN 的活化,多數源自細胞質中所謂的型態辨識受體 (pattern recognition receptors)。許多這些受體被核酸 (nucleic acids) 的異常數量或結構所活化,這些核酸可能來自病毒或宿主,後者以受損 DNA 或不當處理的 RNA 的形式存在。高濃度的 IFN 可誘導 DM 自體抗原(如 MDA5)、活化未成熟樹突狀細胞使其成為有效的抗原呈現細胞、上調主要組織相容性(major histocompatibility, MHC)第一類表現,並活化淋巴細胞。在皮膚與肌肉中,表現這些 IFN 誘導基因產物的細胞似乎最集中於組織損傷的區域,並可能在招募與活化細胞毒性效應細胞中扮演角色。也有大量證據支持 DM 中後天免疫系統的活化。基因研究顯示,人類白血球抗原(human leukocyte antigen, HLA)區域的多型性與 DM 風險高度相關,牽涉到 T 細胞的活化。此外,DM 與數種高度特異性的循環自體抗體的存在有關,這是 T 細胞與 B 細胞活化的證據。如前所述,組織切片中可見 CD3+ T 細胞。這些抗原特異性反應的確切角色目前尚不清楚,但臨床表型與特異性自體抗體之間的相關性,顯示這些免疫反應位於疾病致病機轉的核心。此外,可透過在小鼠中誘導抗 Jo1 免疫反應來建立肌炎的動物模型。雖然其中一些抗原現已被發現,但這種免疫反應的標靶是何種細胞類型或結構仍不清楚。值得注意的是,數種 DM 自體抗原在受損與再生的肌肉細胞中表現增加,顯示肌纖維本身可能是抗原特異性 CD3+ 細胞的直接標靶。免疫反應可能不是故事的全貌,因為組織學資料支持血管病變是 DM 的原發事件。皮膚與肌肉的切片皆顯示內皮退化與微血管退失,是最早期的表現之一。補體的膜攻擊複合體 (membrane attack complex)(由 C5b-9 組成)的沉積,可見於皮膚真皮表皮交界(dermal–epidermal junction, DEJ)的微血管上,以及肌肉的束周血管上。¹⁰³ 然而,沒有其他證據顯示膜攻擊複合體沉積是免疫攻擊血管的結果。Greenberg 與同事假設,漿細胞樣樹突狀細胞與肌細胞 (myocytes) 增加 IFN-α/β 誘導蛋白的生成,也可能導致內皮損傷。¹⁰⁴
遺傳學可能在發展 DM 的風險中扮演重要角色。某些 HLA 等位基因 (alleles) 顯然是發展 DM 最大的危險因子之一。HLA-B8 等位基因是第一個被發現在 17 名青少年 DM 病人中有 12 名(75%)盛行率增加(相較於 21% 的對照參與者)的等位基因。¹⁰⁵ 隨後,多種其他 HLA 等位基因(主要由 HLA 8.1 祖先單倍型 [ancestral haplotype](HLA-A∗0101、-C∗0701、-B∗0801、-DRB1∗0301、-DQA1∗0501 與 -DQB1∗0201)組成)在 DM 病人中富集。¹⁰⁶,¹⁰⁷ 全基因組研究 (Genome-wide studies) 已證實 MHC 第一類與第二類基因的角色,但也發現了其他賦予風險的基因座 (loci);這些包括參與 B 細胞活化的 BLK 基因,以及在 IFN 受體訊號傳遞中扮演角色的 TYK2 基因。此外,其他非免疫事件可能在疾病特徵中扮演角色。目前,一個研究領域圍繞著內質網壓力反應 (endoplasmic reticulum stress response),它可能部分地造成 DM 中所見的某些無力症狀。內質網壓力可能是 MHC 第一類或其他蛋白質上調的結果,並誘導未折疊蛋白反應 (unfolded protein response),導致發炎性核因子 kappa B(nuclear factor kappa B, NF-κB)訊號傳遞途徑的活化、粒線體功能障礙與活性氧物質 (reactive oxygen species) 升高,所有這些都可能促成無力。目前的模型顯示,DM 在具有遺傳易感性的個體中啟動,此個體隨後遭受環境誘發因子。該誘發因子的本質可能各異,可能包括 UV 暴露、感染與惡性腫瘤。在細胞層面,這些誘發因子可能導致抗原修飾或上調、細胞死亡,以及核酸反應的活化,伴隨 IFN 生成增加、內質網壓力,以及先天免疫系統的活化。先天與後天免疫系統之間可能存在冗餘的交互作用,不僅允許疾病的啟動,也允許疾病的傳播。
診斷 (DIAGNOSIS)
不幸的是,目前沒有足夠、經驗證的 DM 診斷標準。如同多數風濕性疾病,DM 以不同程度的嚴重度可變地侵犯不同的器官系統。因此,難以僅以臨床或組織學標準來定義本病。我們已討論過,使用臨床、組織學、肌電圖,或肌肉侵犯的組織學標準來診斷,將無法診斷出無肌病疾病的病人族群。然而,同樣的情況也可能適用於皮膚疾病;雖然有人會主張特徵性皮疹是診斷的關鍵,但皮膚是否需要參與 DM 疾病過程,並不比肌肉更明確。一種實用的做法不是要求特定的肌肉或皮膚侵犯,而是擁有器官特異性標準,即若某器官確實受侵犯,則必須符合這些標準。不幸的是,沒有經驗證的臨床皮膚標準能用以診斷 DM 皮膚疾病。曾有人建議,血管膜攻擊複合體沉積可用以區分本病與皮膚型狼瘡,但不幸的是,沒有關於其他皮膚疾病中膜攻擊複合體沉積的資料。一般認為肌肉切片上的某些特徵能標誌 DM,即束周萎縮 (perifascicular atrophy),但近期研究挑戰了此一概念。¹⁰⁸ 近期數種 DM 特異性自體抗體的鑑定,帶來了以血液檢查做出此診斷的希望,但這些檢測在跨越多種類似皮膚與肌肉疾病的敏感度與特異度資料仍然缺乏。未來診斷標準的最佳化很可能需要結合臨床、組織學與血清學資料。因此,目前 DM 診斷的建立仍持續仰賴臨床醫師根據病史與理學檢查表現所形成的印象。
有用的皮膚表現 (HELPFUL SKIN FINDINGS)
我們發現特別具有敏感度的皮膚檢查特徵包括顯微鏡下甲周微血管擴張 (periungual telangiectasias)、外側指角化過度 (lateral digit hyperkeratosis),以及頭皮紅斑與感覺異常。一些較具特異性的要素包括「紅白相間」斑塊、卵圓形腭斑、肉眼可見的甲周微血管擴張,以及 Gottron 丘疹。
肌肉疾病 (MUSCLE DISEASE)
如前所述,肌炎並非診斷的必要條件。然而,當病人表現出可疑皮疹時,支持發炎性肌病變存在的檢查是有幫助的。如此,DM 的臨床懷疑是診斷 DM 的必要但不總是充分條件。病史上的肌肉侵犯(包括吞嚥困難、發聲困難與肌痛)與檢查上的無力,將有助於提高對肌炎的懷疑,但並非確診性的。然而,肌肉酵素的升高應是確定診斷的下一步(見後文)。在臨床上仍高度懷疑肌病變但肌肉酵素正常的情況下,可使用肌電圖或 MRI。最後,在肌肉症狀的成因仍不清楚的病例中,可進行肌肉切片。
症候群性表現 (SYNDROMIC PRESENTATIONS)
某些症狀組合雖然未必常見,但具有相當高的特異度,無論是否存在較「典型」的 DM 皮膚特徵,都有助於做出 DM 的診斷。一名表現出禿髮、皮膚或黏膜潰瘍、掌部紅斑性丘疹、嚴重關節痛或關節炎,以及喘不過氣的病人,即使只有其中幾項症狀,也應促使臨床醫師考慮抗 MDA5 DM。具有技工手、關節炎、雷諾現象與肺部症狀的病人,罹患抗合成酶疾病的風險很高,有些人認為若皮膚上有特定的 DM 樣徵象,此疾病可歸於 DM 的範疇之下。具有極度肌痛、周邊水腫與遠端無力的病人,即使皮疹細微,也應特別排除具有抗 NXP2 抗體的 DM。了解這些表型將提高臨床醫師做出 DM 診斷的敏感度。
診斷性實驗室評估 (DIAGNOSTIC LABORATORY EVALUATION)
自體抗體 (AUTOANTIBODIES)
與 DM 相關的肌炎特異性自體抗體(myositis-specific autoantibodies, MSAs)正演變為協助建立診斷的關鍵工具。這些 MSAs 包括 TIF1-γ、NXP2、MDA5、小泛素樣修飾物活化酶(small ubiquitin-like modifier activating enzyme, SAE)、Mi-2、Jo-1,以及其他抗合成酶抗體(PL-7、PL-12、EJ、OJ、SRP)。在我們的美國世代中,這些抗體檢查診斷 DM 的敏感度為 80% 至 85%。因此,這些 MSAs 可能提供關鍵資訊,以在細微或複雜的病例中建立 DM 的診斷。重要的是,MSAs 在辨識臨床亞群與疾病關聯方面日益相關(見表 62-2)。隨著臨床表型分析在疾病特徵與臨床病程方面的改善,以及 MSAs 檢測可得性的增加,此分類方法很可能透過改善預後判斷、標靶篩檢與潛在的量身治療,為目前的定義增添價值。抗核抗體(ANA)檢測若要進行,應使用直接免疫螢光法 (direct immunofluorescence)。ANA 檢查結果在 DM 中有 50% 的時間可為陰性,但在全身性紅斑性狼瘡 (systemic lupus erythematosus) 中通常為陽性,因此陰性的檢查結果若全身性狼瘡在鑑別診斷之列,可有助於排除全身性狼瘡。
肌肉酵素 (MUSCLE ENZYMES)
在肌炎病程的早期,血清肌肉酵素(即肌酸激酶 creatine kinase、醛縮酶 aldolase、乳酸脫氫酶 [lactate dehydrogenase, LDH]、天門冬胺酸轉胺酶 [aspartate aminotransferase, AST]、丙胺酸轉胺酶 [alanine aminotransferase, ALT])是肌肉發炎相當敏感的生物標記。然而,在肌炎病程的中期至晚期,其敏感度會下降。肌肉酵素敏感度隨肌炎持續而下降的原因尚不清楚,但可能是束周肌肉萎縮與纖維化的結果,導致即使發炎持續,這些檢查的變化也較不顯著。此外,單一病人常只有肌酸激酶或只有醛縮酶會升高。為了提高在實驗室檢查上偵測肌炎的敏感度,臨床醫師在檢測肌肉酵素時,應將肌酸激酶、醛縮酶與 LDH 作為一組來評估。評估肌肉酵素時,有幾個需注意的實務問題。首先,所有肌肉酵素都可能在劇烈活動後升高;在有疑問的病例中,可在活動後 10–14 天重新檢測酵素濃度。其次,醛縮酶(以及 AST 與 ALT)可能因肝臟疾病或血液檢體溶血而升高。極高的數值應重新檢測。γ-麩胺醯轉移酶(γ-Glutamyl transferase)在肝損傷時會升高,但在肌炎時不會升高,若懷疑肝臟來源,它是肌炎實驗室檢查的有用補充。
肌電圖檢查 (ELECTROMYOGRAPHIC STUDIES)
與肌肉酵素異常類似,在疾病病程早期,肌炎可在 70% 至 90% 具有活動性肌肉疾病的 DM 病人的肌電圖檢查上被偵測到,而偵測肌炎的敏感度會隨時間下降。肌炎的典型肌電圖三聯表現是:小振幅、短時程、多相運動單位電位 (polyphasic motor unit potentials);纖維顫動 (fibrillations) 與正向銳波 (positive sharp waves);以及複雜重複放電 (complex repetitive discharges)。¹⁰⁹ 這些表現反映活動性肌肉發炎,也可見於其他發炎性肌病變,如多發性肌炎。¹¹⁰ 在疾病病程晚期,敏感度會下降。
其他診斷性實驗室檢查 (OTHER DIAGNOSTIC LABORATORY TESTS)
在疾病發作時進行的多數其他實驗室檢查,是為了評估標靶器官侵犯,未必用於做出診斷。然而,血清鐵蛋白 (serum ferritin) 在抗 MDA5 DM 病人中常高度升高(>500 mg/dL)¹¹¹,並可能在血清學檢測不可得或敏感度不足時,有助於診斷抗 MDA5 疾病。此外,它可能是評估嚴重度並追蹤抗 MDA5 DM 病人 ILD 臨床反應的有用生物標記。¹¹²,¹¹³
影像學 (IMAGING)
MRI 提供肌肉解剖的詳細視野,能定位並辨別病理過程的類型(如水腫、發炎、纖維化、鈣化或萎縮)。當肌肉酵素與肌電圖檢查結果不明確時,MRI 可用以區分由損傷或類固醇肌病變 (steroid-myopathy) 引起的無力與活動性肌炎。MRI 也可用於導引診斷性切片的部位(若需要)¹¹⁴,或可用於評估對治療的臨床反應。¹¹⁵ 肌肉水腫是肌炎的敏感指標,並與肌酸激酶濃度相關。¹¹⁶ 在短 tau 反轉回復 (short tau inversion recovery)(正常肌肉為暗、發炎肌肉為亮)影像上,肌肉組織內訊號強度增加提示肌肉發炎、壞死或退化。¹¹⁷ Yoshida 與同事也在 14 名 DM 病人中的 12 名身上於 MRI 上發現筋膜炎 (fasciitis),顯示筋膜微血管系統可能是原發侵犯部位。¹¹⁸ 肌肉損傷可在 T1 加權影像上被辨識,其中有骨骼肌的脂肪取代。⁷³
病理學 (PATHOLOGY)
皮膚組織病理學 (SKIN HISTOPATHOLOGY)
DM 受影響皮膚的切片典型顯示細胞稀少的介面皮膚炎(cell-poor interface dermatitis)、真皮黏蛋白 (dermal mucin) 增加、血管周圍淋巴球浸潤,以及血管擴張 (vascular ectasia)(圖 62-17)。其他不恆定的表現包括表皮萎縮與基底膜增厚。然而,這些表現中有許多常很細微或缺如。Smith 與同事回顧了 40 個 DM 皮膚切片,並指出當介面皮膚炎缺如時(20% 的病例),真皮黏蛋白增加總是存在。¹¹⁹ 曾有人建議,DM 的特徵是漿細胞樣樹突狀細胞增加且主要位於表皮位置,而在皮膚型狼瘡中,它們主要位於真皮。¹²⁰
直接免疫螢光檢測在 DM 診斷中的角色仍不清楚。曾有報告指出,DM 切片不會在 DEJ 沿線顯示免疫反應物,即所謂的「狼瘡帶 (lupus band)」,但此表現在強度與抗體組成方面並未獲得一致定義。Magro 與同事發現,若將狼瘡帶的定義設為嚴格,則 24 名 DM 病人中只有 1 名狼瘡帶檢查結果為陽性。然而,除了多數 DM 病人外,超過 35% 的皮膚型狼瘡切片狼瘡帶檢查也呈陰性。因此,若切片上存在嚴格定義的狼瘡帶(如免疫球蛋白 [immunoglobulin, Ig] G 的間斷性沉積,或 IgM 的連續性沉積),這些資料顯示 DM 的診斷不太可能。此外,Mascaró 與同事發現,膜攻擊複合體或 C5b-9 在 DM 的 DEJ 或血管周圍的沉積發生於 77% 至 86% 的病人。¹⁰³ Magro 與同事在 24 名 DM 病人中重複此檢測,發現膜攻擊複合體在 DEJ 與血管周圍兩處的沉積,加上狼瘡帶檢查結果陰性,對於診斷 DM 相對於皮膚型狼瘡,具有 78% 的敏感度與 93% 的特異度。¹²¹
肌肉組織病理學 (MUSCLE HISTOPATHOLOGY)
在因皮疹與無力被轉診至皮膚科診所的病人中,肌肉切片很少是建立 DM 診斷所必需的。在支持 DM 的皮膚皮疹情境下,實驗室檢查、肌電圖或 MRI 上的肌炎證據常已足夠。肌肉切片可能產生偽陰性結果,這既因為免疫抑制藥物,也因為發炎的斑片狀本質。MRI 可用作選擇切片部位的指引。若需要肌肉切片來確認 DM 的診斷,則在開始任何免疫調節治療的 2 週內進行其產出率最高。DM 的典型肌肉組織病理學表現包括束周萎縮、退化與再生的肌纖維、肌內膜微血管壁中的膜攻擊複合體沉積¹²²、
內皮細胞腫脹,以及微血管壞死。發炎細胞浸潤由 CD4+ T 細胞¹²³、
分泌 IFN-α 的漿細胞樣樹突狀細胞¹⁰¹、B 淋巴球、巨噬細胞與漿細胞組成。然而,即使在明確的 DM 病例中,組織學表現也存在範圍變異,發炎性肌病變的組織學表現常依其他標準聚集,未必遵循已定義的特發性發炎性肌病變類別的臨床界線。
皮肌炎的鑑別診斷 (DIFFERENTIAL DIAGNOSIS OF DERMATOMYOSITIS)
見表 62-3。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
成人 DM 的長期存活率約為 65% 至 75%,但關於特定器官結果(如肌肉或皮膚疾病)的資料相對較少。¹²⁴ 已報告 25% 至 70% 的緩解率,但多數研究顯示,5 年時的數字可能更接近 20% 至 40%。主要死因包括惡性腫瘤、肺部或心臟疾病,以及感染,但其他預測因子的報告各異,包括較年長、男性、非白人種族,以及症狀持續時間較長。結果也可能與自體抗體狀態有關;日本與美國的研究皆顯示,抗 MDA5 抗體的存在是死亡的危險因子。¹²⁵,¹²⁶
對兒童而言,長期登錄資料提供了關於緩解率(包括皮膚疾病)較清楚的資料。多數研究顯示,約 60% 的病人有慢性疾病(如過去 5 年),慢性化的主要危險因子是治療延遲與早期持續(如 3 個月)的皮膚疾病或甲褶微血管密度下降。多數報告顯示,持續疾病活性最常見的來源是覆蓋於肌肉之上的皮膚或其他器官。¹²⁷⁻¹²⁹
處置 (MANAGEMENT)
處置原則 (PRINCIPLES OF MANAGEMENT)
DM 的治療首先需要評估可能受影響的器官,即皮膚、肌肉與肺部。應強調的是,ILD 與相關癌症是疾病相關死亡的主要原因,在治療選擇時應優先考量。篩檢惡性腫瘤至關重要,因為治療與 DM 相關的癌症可能導致疾病嚴重度降低,有時甚至疾病緩解。皮膚型 DM 常與肌肉疾病有不一致的治療反應¹³⁰,頑固性皮膚疾病在肌肉疾病緩解多年後仍持續。考量到皮膚疾病的慢性本質,值得權衡所開立治療的長期毒性與已達成或潛在的皮膚效益。考量到這一點,應首先選擇適當的藥物以針對受影響的器官,考量病人的共病與偏好,並在決定治療方案之前與病人討論每種藥物的風險與效益。與共同管理的醫療提供者(風濕科、皮膚科、神經科與胸腔科醫師)建立合作關係,對於治療多器官受侵犯的病人至關重要。
癌症篩檢 (CANCER SCREENING)
鑑於 10% 至 20% 的 DM 病人有相關惡性腫瘤,在初次診斷時進行徹底調查是合理的。除了完整的病史與理學檢查外,例行的年齡別癌症篩檢檢查(大腸鏡 colonoscopy、乳房攝影 mammogram、攝護腺檢查)與相關的篩檢血液檢查(全血球計數、腎與肝功能檢查)以及尿液分析皆有指徵。其他血液檢查(如紅血球沉降速率 erythrocyte sedimentation rate、C 反應蛋白 C-reactive protein、癌症標記、血清與尿液蛋白免疫固定電泳 protein immunofixation electrophoresis)在 DM 病人中辨識癌症的角色目前尚未確立。檢測抗 TIF1-γ 與抗 NXP-2 抗體在風險分層中的角色尚不清楚,因為事實上,至少在美國族群中,多數具有這些抗體的病人仍未懷有惡性腫瘤。更積極篩檢(如 CT 掃描、正子斷層造影 positron emission tomography)的角色尚不清楚,雖然如前所述,這種影像可能在較高風險期間(診斷後 2–3 年)偵測到在例行年齡別篩檢中未發現的隱匿性惡性腫瘤。重新篩檢的角色甚至更不清楚。對於疾病難以控制,或在持續靜止期後經歷不明原因疾病惡化的病人(尤其是有惡性腫瘤病史的病人),考慮進行盲目重新篩檢可能是審慎的做法。
皮膚外疾病的監測 (MONITORING OF EXTRACUTANEOUS DISEASE)
肌肉疾病 (MUSCLE DISEASE)
徒手肌力測試 (Manual muscle testing) 有助於評估就診間肌力的變化。即使是忙碌的皮膚科醫師,也可在每次就診時檢查幾個肌群,如頸屈肌、三角肌與股四頭肌,以評估肌力的粗略變化。若有更多時間,則可進行八個肌群子集的徒手肌力測試(MMT8),這是一項經驗證的肌肉測試,其中八個主要肌群(頸屈肌、三角肌、二頭肌、腕伸肌、臀大肌、臀中肌、股四頭肌、踝背屈肌)在特發性發炎性肌病變中產出率最高。¹³¹ 諸如疼痛、關節攣縮與疲勞等其他因子可能影響此測量。血清肌肉酵素(肌酸激酶、醛縮酶與 LDH)在每次就診時檢測,作為肌炎的生物標記。然而,隨著肌炎持續時間增加,它們成為肌炎不敏感的標記。在無力漸增而肌肉酵素正常的情況下,肌電圖與 MRI 仍是偵測活動性肌炎較敏感的檢查。臨床醫師必須考量無力的其他成因,例如先前肌炎造成的肌肉損傷相對於體能失調、類固醇肌病變、羥氯奎寧引起的肌病變(hydroxychloroquine-induced myopathy),或甲狀腺肌病變。如此,重要的是不要在辨識出活動性肌炎為成因之前,反射性地以增加免疫抑制來治療無力。
肺部疾病 (PULMONARY DISEASE)
雖然沒有正式的 ILD 篩檢指引,但取得伴隨擴散量的基準 PFTs 是有價值的,以防未來發展出肺部症狀,然後只要 DM 的其他症狀持續,即進行每年篩檢。若發展出新的肺部症狀,伴隨一氧化碳擴散量的篩檢 PFTs 可每 3 至 6 個月進行一次。PFT 結果依賴用力程度,因此受測試期間遵循指示、肌力與精力程度的影響。若 PFTs 顯示令人擔憂的新發或惡化 ILD 變化,則胸部高解析度 CT 掃描是評估肺實質的下一步。
心血管侵犯 (CARDIOVASCULAR INVOLVEMENT)
應考慮評估心肌疾病。肌酸激酶 MB 同功酶(creatine kinase-MB isoform)見於心臟以及再生的骨骼肌,因此它不是特異性檢查,雖然它可用作心臟侵犯的篩檢檢查。我們偏好使用心肌旋轉蛋白 I 檢測,因為它對心肌損傷具特異性。沒有篩檢心臟侵犯的標準建議,但有些權威建議使用心肌旋轉蛋白 I 作為亞臨床心肌侵犯的篩檢檢查,若結果為陽性,則接續以心臟超音波或心電圖檢查。具有抗 MDA5 抗體的 DM 病人,發生臨床上明顯且有時致命的心臟侵犯的風險可能較高。⁸⁷,⁸⁸
介入措施 (INTERVENTIONS)
DM 醫療治療與治療階梯 (treatment ladders) 的證據,主要來自單一中心、回溯性病例報告、病例系列與專家意見¹³²⁻¹³⁶(表 62-4)。
局部治療 (TOPICAL THERAPY)
雖然光防護 (photoprotection) 是處置的關鍵第一步,但高達 60% 的 DM 病人實際上僅有極輕度的光敏感性,而僅有少至 20% 可能報告 UV 暴露後疾病惡化。¹³⁰,¹³⁷
儘管有此發現,仔細回顧 UV 防護的基本概念仍有助益。局部皮質類固醇 (Topical corticosteroids) 可能透過減少紅斑、脫屑與搔癢,對 DM 皮膚疾病有緩解效果,並對全身性藥物扮演輔助角色。然而,除了最輕微的病例外,它們不太可能完全控制皮膚症狀。第 I 或第 II 級局部類固醇乳膏或軟膏用於肘部、膝部的厚皮膚以及手部的角化過度,並可嘗試用以減輕常見的甲褶敏感主訴,夜間可加上塑膠膜封包 (occlusion) 以進一步增加效力。
皮膚疾病 — 肌肉疾病 (SKIN DISEASE — MUSCLE DISEASE)
第一線 (First Line):光防護、局部類固醇、羥氯奎寧 (Hydroxychloroquine) 或氯奎寧 (Chloroquine)、奎納克林 (Quinacrine) | 全身性皮質類固醇 (Systemic corticosteroids)
第二線 (Second Line):甲氨蝶呤 (Methotrexate)、黴酚酸酯 (Mycophenolate mofetil)、靜脈注射免疫球蛋白 (Intravenous immune globulin)、硫唑嘌呤 (Azathioprine)
第三線 (Third Line):托法替尼 (Tofacitinib)、來氟米特 (Leflunomide)、氨苯碸 (Dapsone)、沙利竇邁 (Thalidomide)、環孢素 (Cyclosporine)、環磷醯胺 (Cyclophosphamide)|利妥昔單抗 (Rituximab)、來氟米特、環孢素、環磷醯胺
頭皮搔癢可能透過局部類固醇溶液或油劑改善,雖然嚴重病例常需全身性藥物。局部鈣調神經磷酸酶抑制劑(topical calcineurin inhibitors),如他克莫司 0.1% 軟膏(tacrolimus 0.1% ointment)或匹美莫司 1% 乳膏(pimecrolimus 1% cream),在某些皮膚型 DM 病例中可能有中等療效。¹³⁸⁻¹⁴²
在實務上,這些藥物的療效與低至中效局部皮質類固醇(第 IV 至第 VI 級)相當。它們確實提供了可安全用於臉部而不必擔心萎縮或色素減退的益處。
全身性治療 (SYSTEMIC THERAPY)
全身性皮質類固醇 (Systemic corticosteroids) 作為皮膚型 DM 的單一療法是不理想的藥物,因為它們通常只引起部分反應,且與長期副作用有關。然而,全身性皮質類固醇是治療肌炎的第一線治療。¹⁴³ 以普賴鬆 (prednisone) 單一療法在大於 0.5 mg/kg/day 的劑量下,已在 27%¹⁴⁴ 至 87%¹¹ 的 DM 病人中達成肌肉發炎的完全臨床反應。¹⁴⁵ 它們也可用於關節疾病與 ILD,雖然常需要類固醇節省劑 (corticosteroid-sparing agent)。加入類固醇節省劑可能改善肌炎與皮膚外表現的控制,但其關鍵角色是將口服皮質類固醇的毒性降至最低,包括皮質類固醇引起的肌病變 (corticosteroid-induced myopathy) 的風險。¹⁴⁶
抗瘧藥 (Antimalarials) 通常被視為皮膚疾病的第一線藥物。這些藥物對 DM 皮膚疾病有中等效益,回溯性研究顯示約 30% 至 50% 的病人可見改善。¹⁴⁷ 此外,高達 30% 的 DM 病人在開始使用羥氯奎寧時可能經歷藥物疹。¹⁴⁸ 在羥氯奎寧或氯奎寧中加入奎納克林,可能比單一藥物更有效。¹⁴⁹ 羥氯奎寧也可能對發炎性關節炎的輕度症狀有用,而一篇病例報告報告氯奎寧能改善 DM 的關節炎。¹⁵⁰
甲氨蝶呤 (Methotrexate) 在顯著降低 50% 至 100% DM 病人的皮膚疾病嚴重度上有效。¹⁵¹⁻¹⁵⁴ 同樣地,甲氨蝶呤與普賴鬆併用是肌炎的第一線治療,且常需要 20 至 25 mg/wk 的劑量來控制肌肉發炎。⁶⁸,¹⁵⁵ 當有並存的關節炎時,甲氨蝶呤也是有效的治療選擇。¹⁵⁶ 在懷疑或診斷有 ILD 的 DM 病人中,審慎的做法是選擇甲氨蝶呤以外的藥物,因為它有誘導急性肺炎與肺纖維化的潛能¹⁵⁷,¹⁵⁸,從而使 ILD 的評估與管理複雜化。黴酚酸酯 (Mycophenolate mofetil) 已證明在 2 至 3 g/day 的劑量下,能有效降低皮膚疾病嚴重度¹⁵⁹,¹⁶⁰ 與肌炎。¹⁶¹⁻¹⁶³ 當有 ILD 時,它被視為第一線口服藥物。¹⁶⁴⁻¹⁶⁸ 約 20% 的病人在 2 g/day 時經歷噁心或腹瀉。¹⁶⁹ 若 GI 副作用為劑量限制因素,則改用腸溶衣黴酚酸鈉 (enteric-coated mycophenolate sodium) 是維持病人持續治療的一個選項。¹⁷⁰
靜脈注射免疫球蛋白(intravenous immunoglobulin, IVIG)很可能是皮膚型 DM 單一最有效的藥物,一致有 70% 至 80% 的病人達成幾乎完全或完全反應。¹⁷¹,¹⁷² IVIG 對肌炎也有效。¹⁷³,¹⁷⁴ Dakalas 與同事於 1993 年對 15 名 DM 病人進行的一項隨機安慰劑對照交叉試驗¹⁷⁵ 顯示,接受 IVIG 的病人中,12 名中有 9 名(75%)肌力顯著改善,根據臨床照片,12 名中有 8 名(67%)皮膚疾病有戲劇性改善。標準給藥方案是 2 g/kg/mo,分 3 至 5 天投予。治療效果可能早至 1 週即可感知,但可能要到第二或第三個月才會顯現。由於 IVIG 作用相對快速,它可用於迅速惡化或因吞嚥困難或呼吸肌侵犯而急性病危的病人。頭痛可能發生於高達 56% 的病人,且可能嚴重而使人虛弱。輸注速率、總劑量¹⁷⁶、IVIG 的劑型¹⁷⁷,以及病人的容積狀態,可能影響頭痛的發生。無菌性腦膜炎 (Aseptic meningitis) 是一種罕見的不良事件,表現為發燒、頭痛、畏光、腦膜刺激徵 (meningismus)、腦脊髓液中的嗜中性球增多 (neutrophilic pleocytosis) 或嗜酸性球增多。¹⁷⁸,¹⁷⁹ 過敏性休克 (Anaphylaxis) 也罕見,但可能發生於原發性 IgA 缺乏症 (primary IgA deficiency);因此,建議在輸注前檢測血清 IgA 濃度。然而,沒有證據顯示具有偏低但可偵測的免疫球蛋白濃度會賦予任何過敏性休克風險的增加。¹⁷⁶ 此外,靜脈血栓 (venous thrombosis) 與腎損傷¹⁷⁹ 是 IVIG 的風險,分別尤其在具有既存血栓形成傾向 (thrombophilias) 或慢性腎臟病的病人中。硫唑嘌呤 (Azathioprine) 尚未針對皮膚型 DM 進行專門評估。在 DM 與多發性肌炎的合併研究中,硫唑嘌呤已顯示在高達 75% 的病例中改善肌炎¹⁸⁰⁻¹⁸² 並改善存活率。¹⁸³ 第一項針對 28 名多發性肌炎或 DM 病人的隨機對照試驗,比較普賴鬆龍 (prednisolone) 加硫唑嘌呤 2.5 mg/kg/day 與普賴鬆龍加甲氨蝶呤 15 mg/wk,發現兩者對肌炎的療效沒有差異。¹⁸⁷ 第二項針對 30 名多發性肌炎或 DM 病人的隨機對照交叉試驗顯示,接受口服甲氨蝶呤與硫唑嘌呤合併治療組(15 名中的 8 名;53%)的反應,較單獨使用甲氨蝶呤組(15 名中的 3 名;20%)有所改善。¹⁸⁵ 當單獨使用甲氨蝶呤而肌炎持續時,作者偶爾會將低劑量硫唑嘌呤與甲氨蝶呤併用。硫唑嘌呤常用作與特發性發炎性肌病變相關 ILD 治療的維持治療⁴⁷,⁶⁰,典型在以環磷醯胺 (cyclophosphamide) 誘導後使用。¹⁸⁶,¹⁸⁷
利妥昔單抗 (Rituximab) 在皮膚型 DM 中的結果不一,一項臨床試驗報告無效益¹⁸⁸,一篇報告則指出皮膚型 DM 有中等改善。¹⁸⁹ 利妥昔單抗可對肌炎有效益,尤其是那些具有抗 Jo-1 與抗 Mi-2 自體抗體的病人。¹⁹⁰ 一項大型隨機臨床試驗並未顯示利妥昔單抗的效益,雖然這可能圍繞著試驗設計的困難問題。¹⁹¹ 利妥昔單抗在回溯性研究中已被報告成功用於治療 ILD。¹⁹² DM 的給藥典型遵循類風濕性關節炎的方案,即在第 0 天與第 14 天靜脈注射 1000 mg。¹⁸⁸ 感染性併發症是 DM 病人最常見的¹⁹³ 嚴重不良反應,罕見有進行性多發性腦白質病變 (progressive multifocal leukoencephalopathy) 的報告。¹⁹³⁻¹⁹⁵
鈣調神經磷酸酶抑制劑(calcineurin inhibitors,如環孢素與他克莫司)不常用於治療肌肉或皮膚發炎,雖然一項針對 36 名 DM(n = 20)或多發性肌炎(n = 16)病人的隨機臨床試驗,比較環孢素(3–3.5 mg/kg/day)與甲氨蝶呤(7.5–15 mg/wk)併用口服皮質類固醇,發現在 6 個月時兩組間肌酸激酶的下降與肌力的改善相當。¹⁹⁶ 其主要用途在於 ILD 的情境,尤其是抗 MDA5 或抗合成酶抗體情境下的嚴重 ILD。¹⁹⁷,¹⁹⁸
腎毒性 (Nephrotoxicity) 風險在高於 3 mg/kg/day 時最高,需要仔細監測腎功能與血壓。¹⁹⁹,²⁰⁰
其他被報告用於 DM 皮膚疾病的治療包括氨苯碸 (dapsone) 與沙利竇邁 (thalidomide),但其療效的證據稀少。
物理醫學與復健 (PHYSICAL MEDICINE AND REHABILITATION)
肌力訓練已證明能改善肌力與功能²⁰¹⁻²⁰³,有氧運動已證明能改善慢性 DM 病人的耐力。²⁰⁴,²⁰⁵ 在活動性 DM 與多發性肌炎中,居家肌力訓練計畫已證明安全²⁰⁶,但在一項隨機臨床試驗中,相較於進行活動範圍運動的對照組,並未顯示在肌力或疾病控制上的效益。²⁰⁷ 我們建議所有病人在診斷後及早參加物理治療計畫,以預防傷害並盡可能維持活動能力與肌力。
特殊情況 (SPECIAL CIRCUMSTANCES)
鈣質沉著 (Calcinosis) 仍是 DM 中最棘手的治療挑戰之一。對局部病灶進行手術切除仍是最有效且確切的治療。²⁰⁸ 已提出多種醫療治療,包括抗發炎藥物與鈣磷調節劑,但沒有單一藥物可靠有效。³¹ IVIG 在某些病例中被報告效果良好²⁰⁹⁻²¹¹,但在其他病例中則否。²¹² 雙磷酸鹽 (Bisphosphonates) 被引述對青少年 DM 的鈣質沉著有效,雖然仍需要對照研究。²¹³⁻²¹⁵
表格 (TABLES)
表 62-1:Sontheimer 提出的皮膚型皮肌炎診斷標準
表 62-1:Sontheimer 提出的皮膚型皮肌炎診斷標準 (Sontheimer’s Proposed Diagnostic Criteria for Cutaneous Dermatomyositis)¹
皮膚型皮肌炎的診斷需要:
- 存在兩項主要標準,或一項主要標準加兩項次要標準,且
- 與皮膚型皮肌炎一致的皮膚切片變化
| 主要標準 (Major Criteria) | 次要標準 (Minor Criteria):斑狀紫紅色紅斑侵犯(每個區域計為一項次要標準) |
|---|---|
| 向陽性徵象 (Heliotrope sign) | ■ 頭皮或前髮際線 |
| Gottron 丘疹 (Gottron papules) | ■ 臉部顴隆突 (Malar eminences)、前額或下巴 |
| Gottron 徵象 (Gottron sign) | ■ 頸部或上胸部 V 區(V 領徵象 V-neck sign) |
| ■ 後頸或後肩(披肩徵象 shawl sign) | |
| ■ 上臂或前臂的伸側表面 | |
| ■ 覆蓋於手背伸肌腱之上的線狀條紋 | |
| ■ 甲周 (Periungual) 皮膚 | |
| ■ 側大腿或髖部(槍套徵象 holster sign) | |
| ■ 內踝 (Medial malleoli) | |
| 甲褶微血管擴張、出血-梗塞 (Nailfold capillary telangiectasia, hemorrhage-infarct) | |
| 異色症 (Poikiloderma) | |
| 技工手 (Mechanic’s hands) | |
| 皮膚鈣質沉著 (Cutaneous calcinosis) | |
| 皮膚潰瘍 (Cutaneous ulcers) | |
| 搔癢 (Pruritus) |
圖表 (FIGURES AND TABLES)
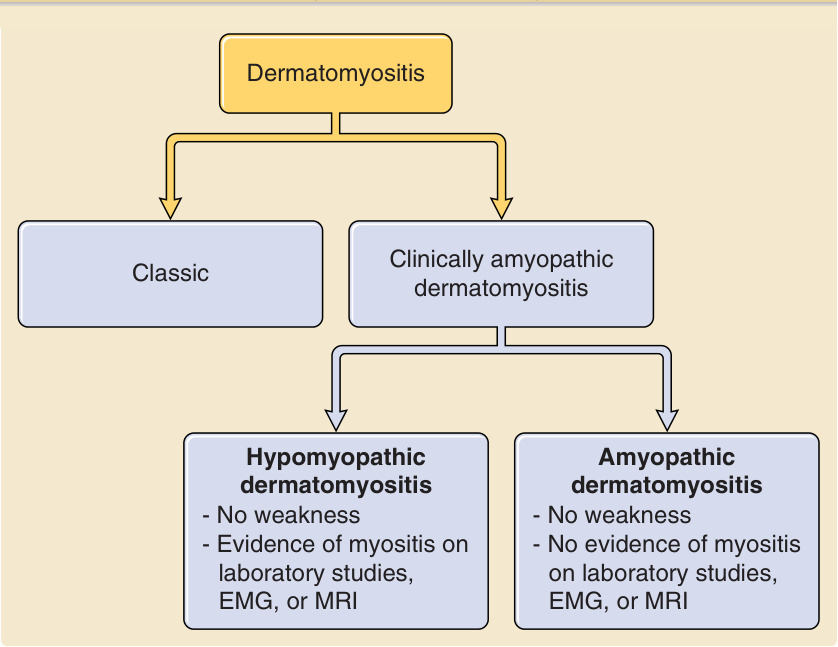
圖 62-1:展示典型、臨床無肌病、低肌病與無肌病皮肌炎之間的關係。EMG,肌電圖 (electromyography);MRI,磁振造影 (magnetic resonance imaging)。
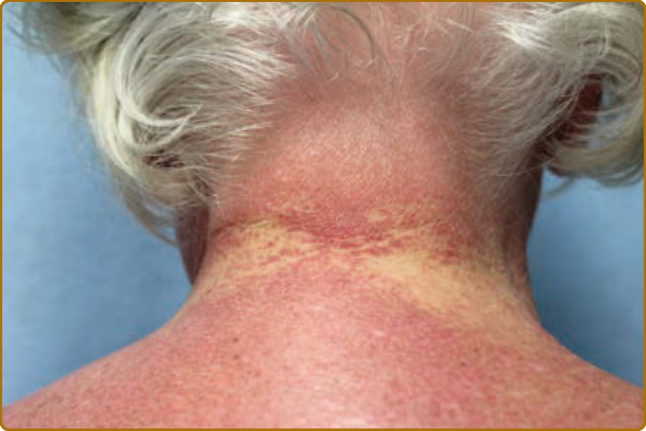
圖 62-2:後頸與項部頭皮上的瀰漫性粉紅色乾癬樣斑塊 (psoriasiform plaques)。
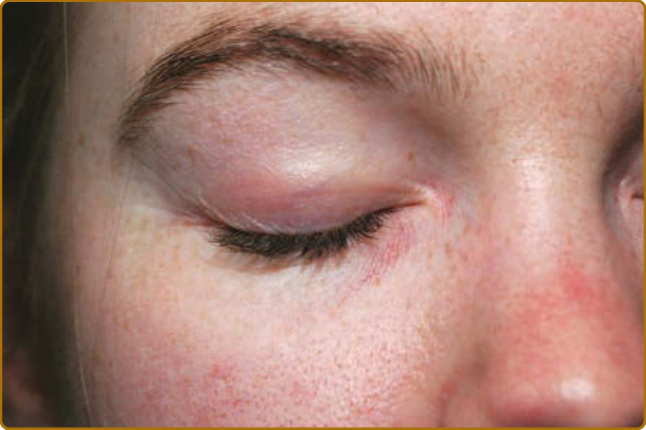
圖 62-3:向陽性徵象 (Heliotrope sign)。上眼瞼的紫紅到粉紅色紅斑與水腫。
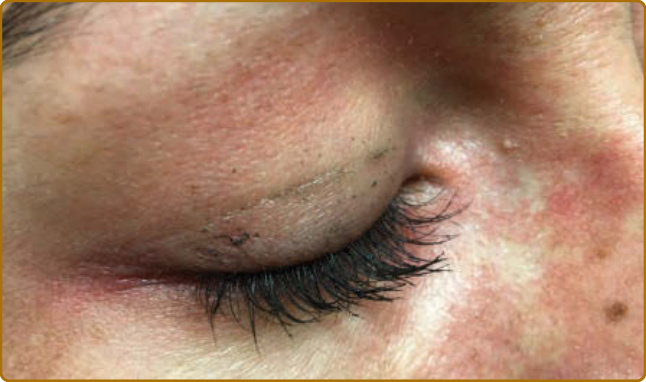
圖 62-4:內眥與外眥的紅色界線不清斑點,常與向陽性徵象一同出現。
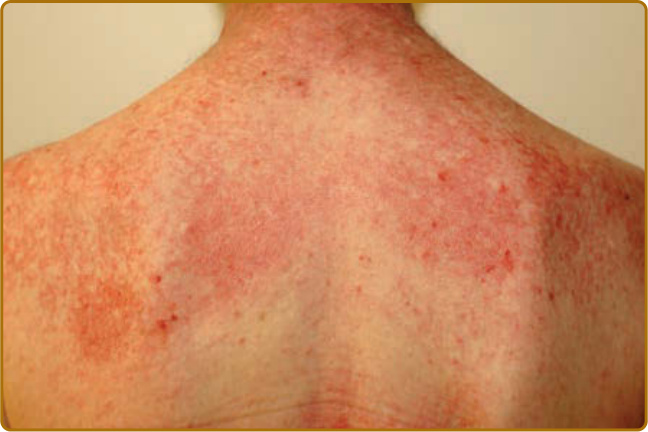
圖 62-5:披肩徵象 (Shawl sign)。上後背具特徵性的粉紅到紅色薄斑塊與斑塊,常伴薄白色脫屑。
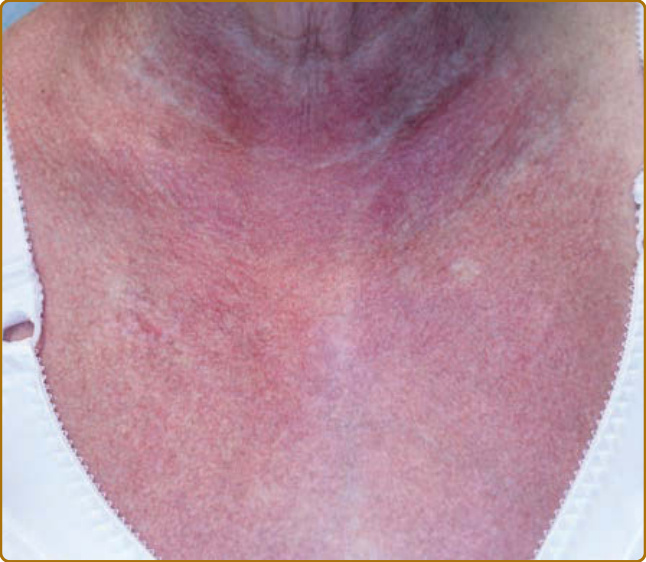
圖 62-6:V 領徵象 (V-neck sign)。上胸部紅色、界線不清的微血管擴張性斑塊。
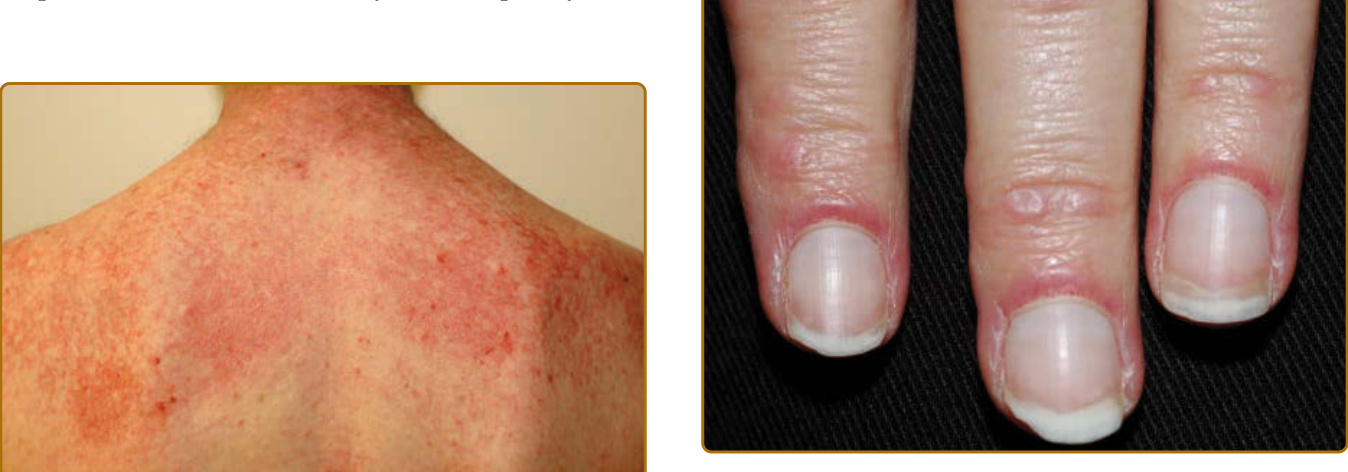
圖 62-7:Gottron 丘疹 (Gottron papules)。覆蓋於近端與遠端指間關節之上的淡粉紅色、界線不清丘疹,少數有中央臍狀凹陷。可見近端甲褶的深紅色紅斑、水腫,以及擴張的甲褶微血管。
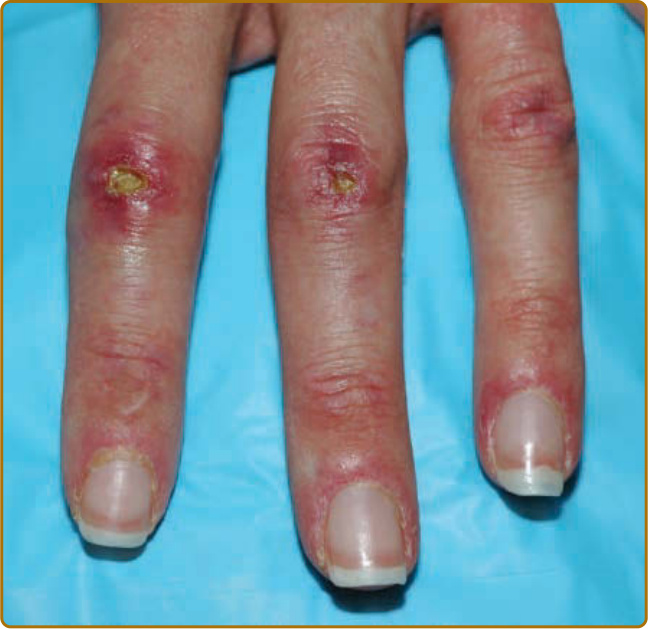
圖 62-8:潰瘍性 Gottron 丘疹 (Ulcerated Gottron papules)。第二與第三近端指間關節上界線分明的 3 至 4 mm 潰瘍,伴周圍紅斑與水腫,體現皮肌炎所見的血管病變。
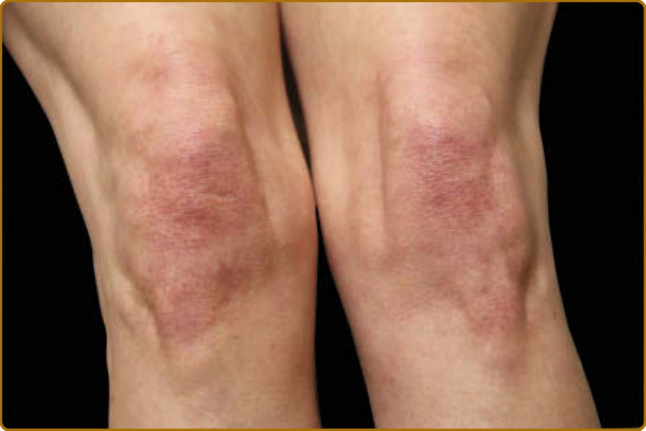
圖 62-9:Gottron 徵象 (Gottron sign)。可見膝部界線不清的紫紅色紅斑,此病人有毛囊強化 (follicular accentuation)。
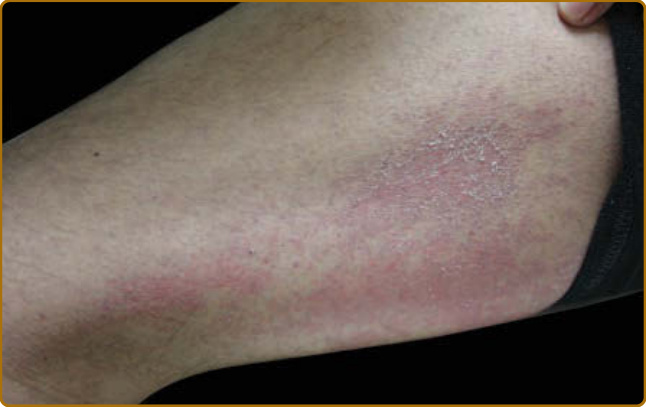
圖 62-10:槍套徵象 (Holster sign)。側大腿上可見界線不清的粉紅色丘疹融合成斑塊,伴脫屑與毛囊強化。
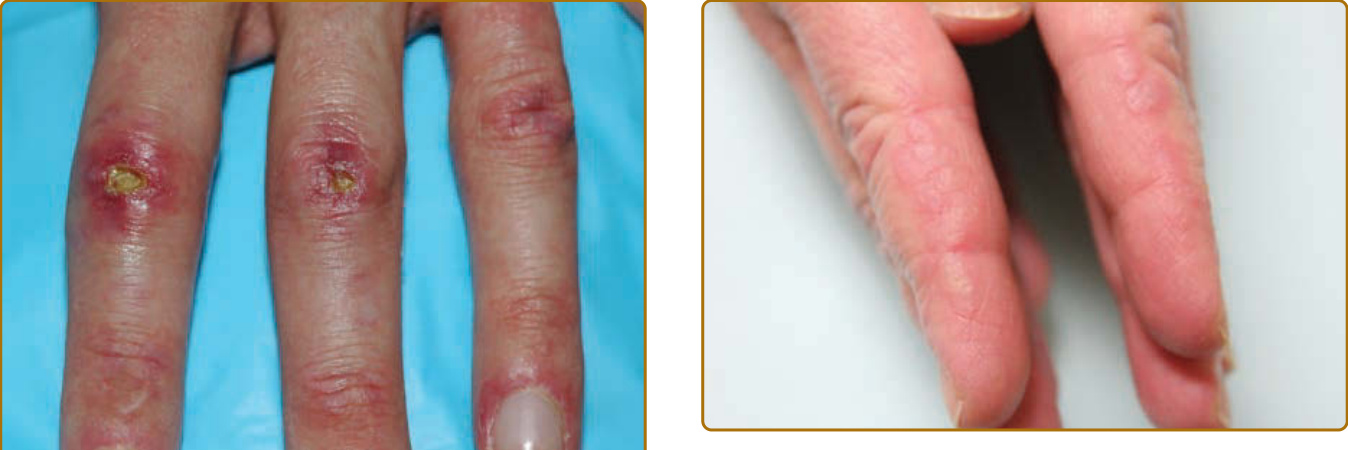
圖 62-11:技工手 (Mechanic’s hands)。外側第二指上有角化過度的粉紅色界線不清丘疹,伴脫屑。
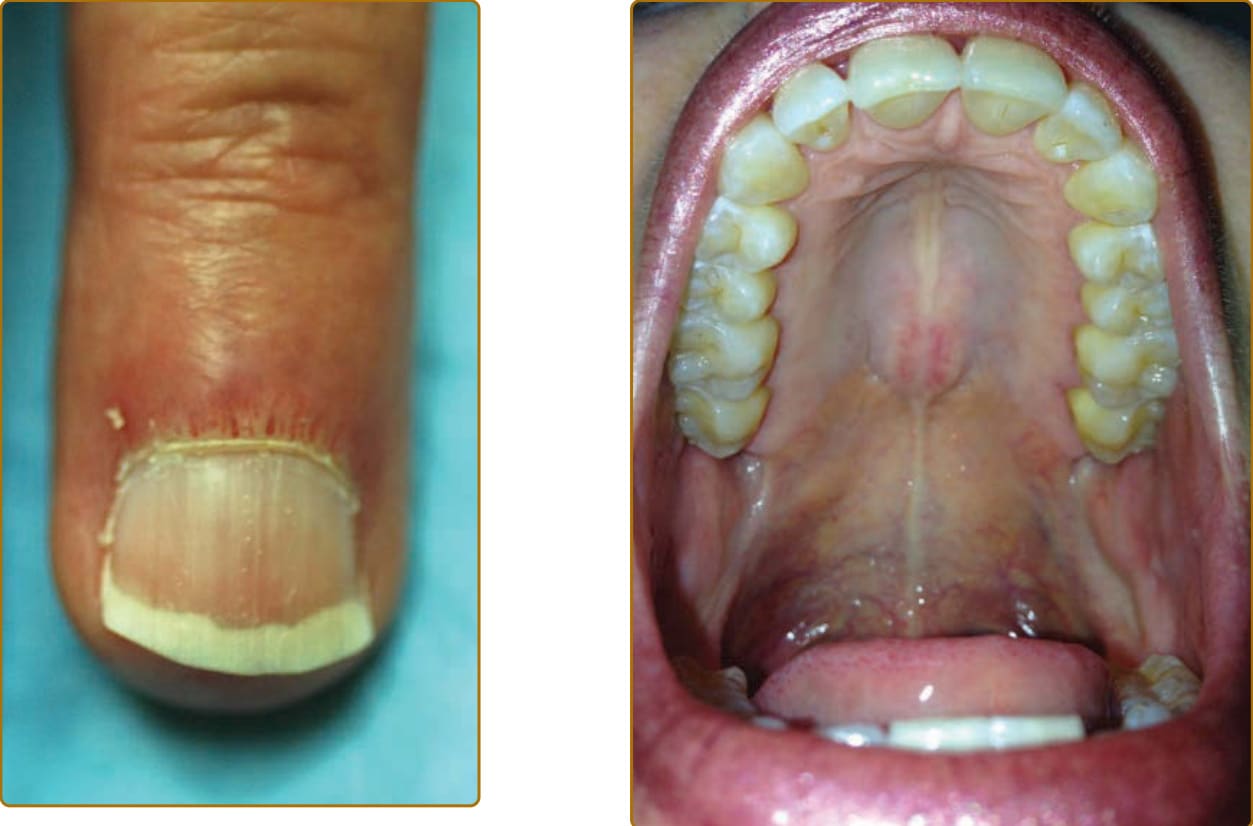
圖 62-12:擴張的近端甲褶微血管環,其間夾雜無血管區。
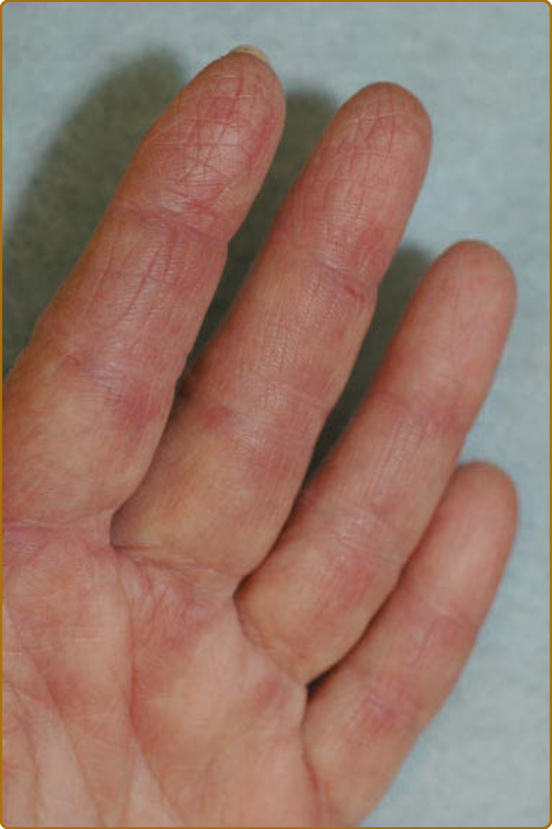
圖 62-14:掌側手指的深紅色紅斑網狀型態,更常見於抗黑色素瘤分化相關基因 5(melanoma differentiation–associated gene 5, MDA5)皮肌炎組。
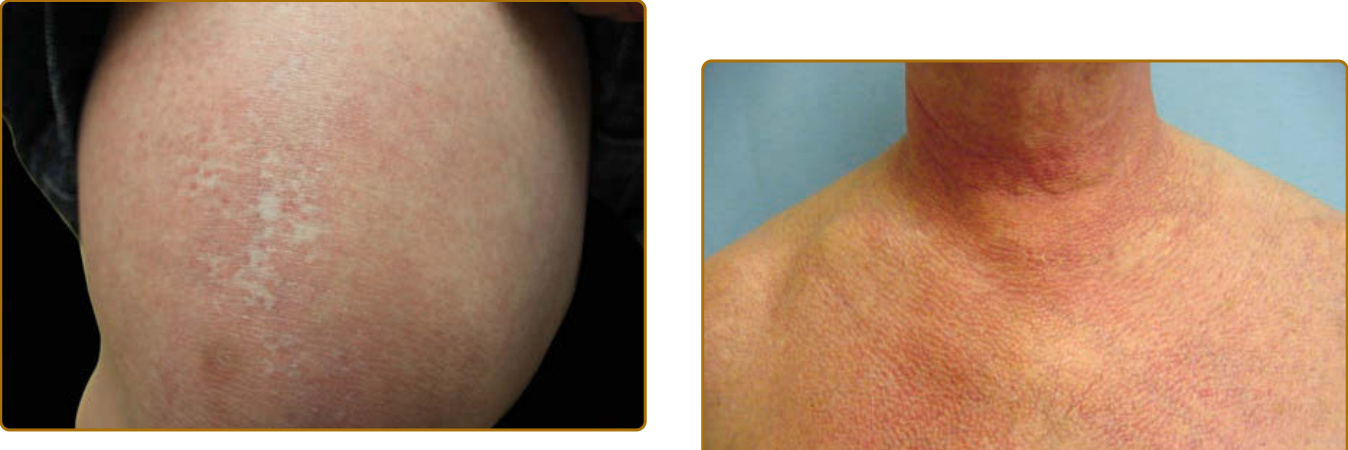
圖 62-15:「紅白相間 (Red on white)」。遠端大腿與膝部可見網狀白色斑點,周圍環繞微血管擴張性紅色斑點。此皮膚表現對皮肌炎具特異性,在區分皮肌炎與皮膚型狼瘡時很有價值。
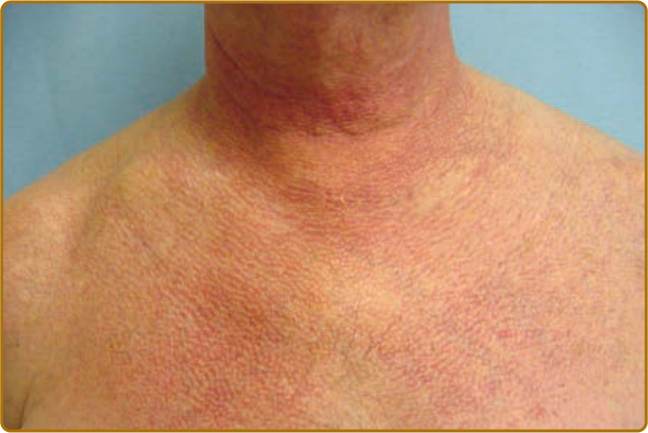
圖 62-16:異色症 (Poikiloderma)。如胸部所示,此色素過度沉著、色素減退與微血管擴張的三聯表現是損傷的徵象。
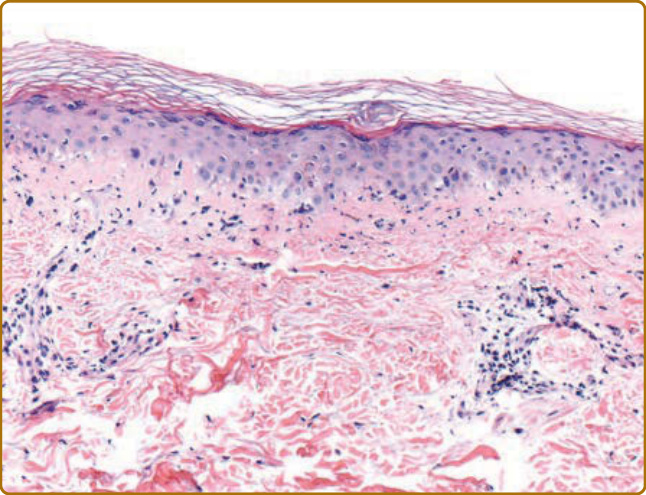
圖 62-17:皮膚型皮肌炎的組織病理學。皮膚切片顯示空泡性介面皮膚炎 (vacuolar interface dermatitis)、基底膜增厚、血管周圍淋巴球浸潤,以及真皮黏蛋白增加。
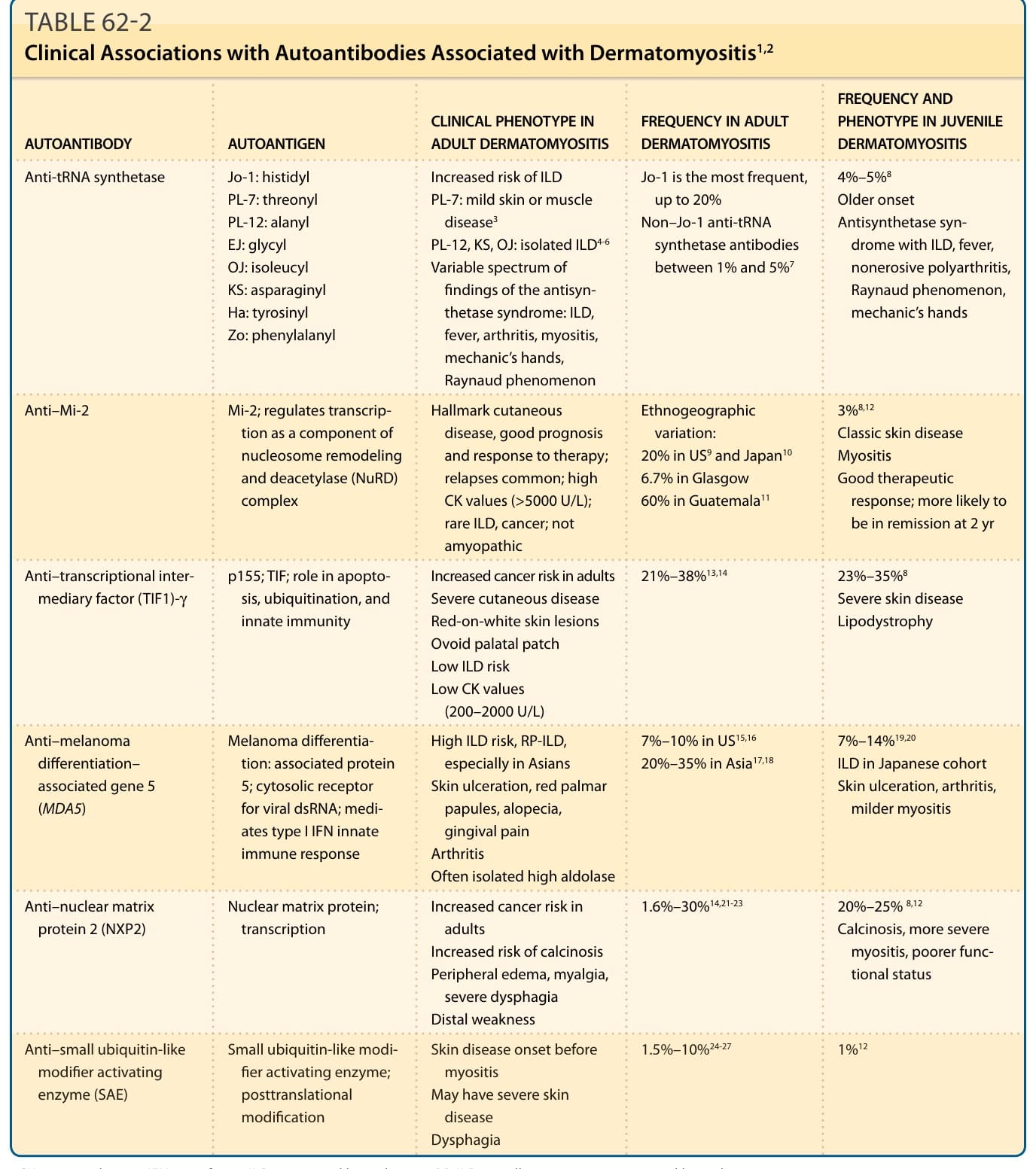
表 62-2:與皮肌炎相關之自體抗體的臨床關聯 (Clinical Associations with Autoantibodies Associated with Dermatomyositis)¹,²
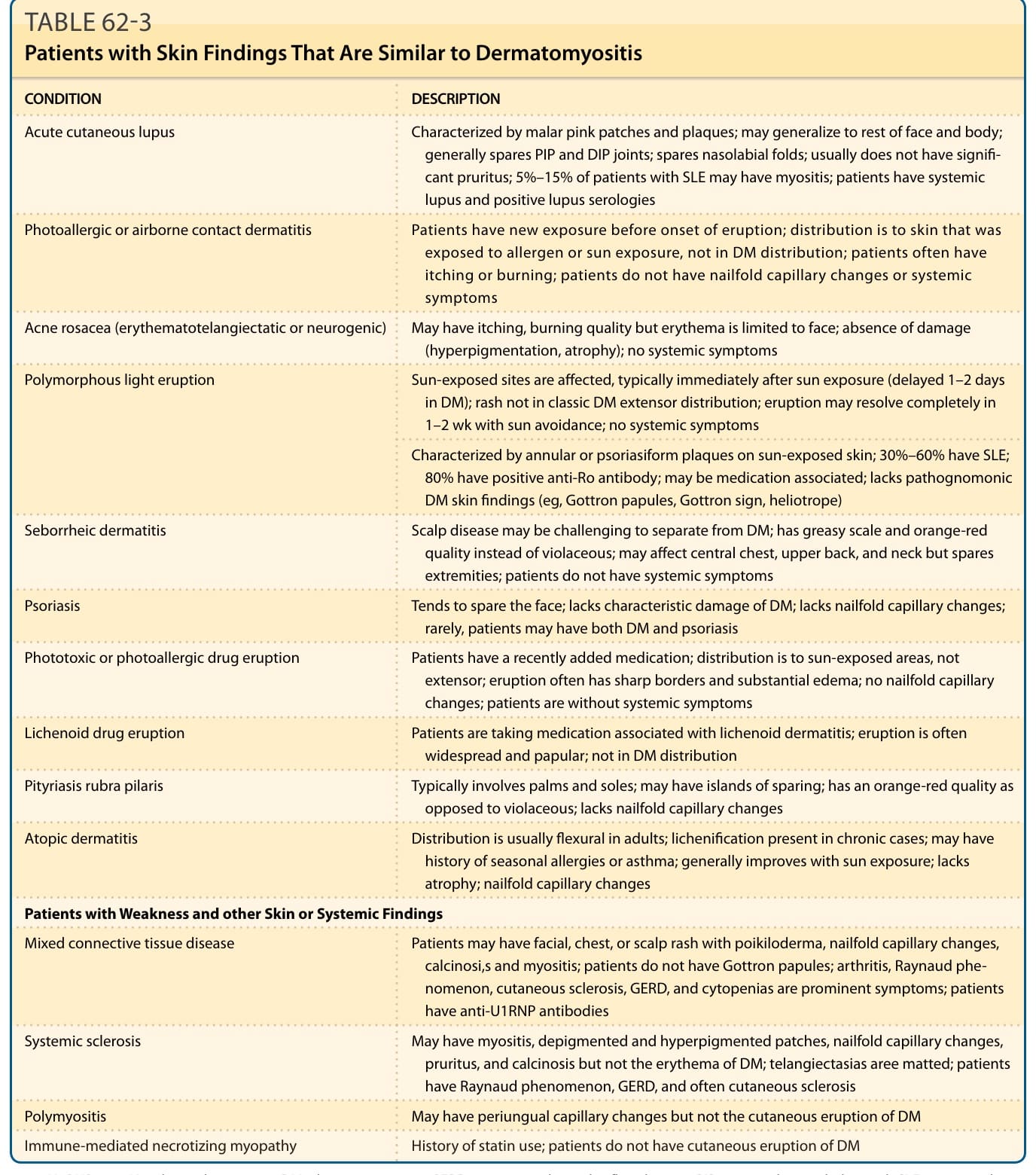
表 62-3:具有與皮肌炎相似皮膚表現的病人 (Patients with Skin Findings That Are Similar to Dermatomyositis)
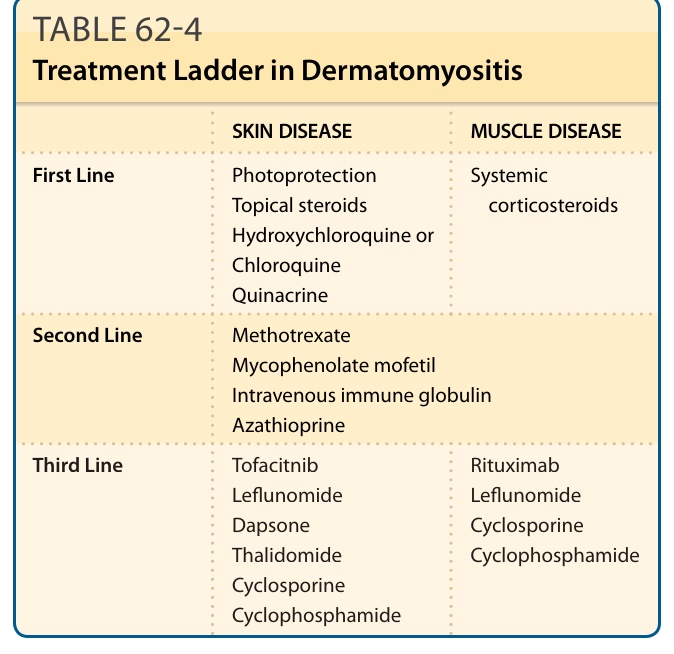
表 62-4:皮肌炎的治療階梯 (Treatment Ladder in Dermatomyositis)