Dermatomyositis
10
AT-A-GLANCE
■ Dermatomyositis (DM) is a systemic autoimmune disease with a bimodal distribution affecting children and adults, characterized by inflammation and damage to the skin and muscle.
■ Interstitial lung disease (ILD) affects 20% of patients and is a major source of morbidity and mortality in these patients.
■ In adults, DM heralds the diagnosis of a coexisting internal malignancy in 10% to 20% of cases. A thorough review of systems and malignancy workup, including computed tomography scans of the chest, abdomen, and pelvis, may be prudent to detect occult cancers that are missed on routine age-appropriate screening.
■ The diagnosis is established with a combination of the hallmark cutaneous findings and supportive skin histopathology, with or without evidence of a proximal myopathy. The diagnosis may be challenging because 20% of patients never manifest clinically significant muscle weakness.
■ The cutaneous manifestations are typified by violaceous erythema in many sites, most notably the eyelids, upper chest, back, elbows, knees, and lateral hips in addition to proximal nailfold capillary dilation and pericapillary hemorrhage.
■ Characteristic autoantibodies (antisynthetase, anti–Mi-2, anti–transcriptional intermediary factor [TIF1]-γ, anti–melanoma differentiation– associated gene 5 [MDA5], anti–nuclear matrix protein 2 [NXP2], and anti–small ubiquitin-like modifier activating enzyme [SAE] autoantibodies) may be useful in identifying clinical subsets and establishing the diagnosis in cases without the classic clinical or histopathologic features.
■ The presence or absence of certain autoantibodies assists with risk stratification for organ involvement as well as for associated malignancy. Notably, the presence of anti-MDA5 and antisynthetase autoantibodies is associated with an increased risk of ILD, and the presence of anti– TIF1-γ and anti-NXP2 autoantibodies is associated with an increased risk of having an associated cancer.
■ Treatment of cutaneous DM can be challenging, and most patients with significant skin disease require systemic therapy. Multiple agents may be necessary to achieve complete remission; the risks and benefits each agent should be considered carefully given the potentially prolonged treatment course.
INTRODUCTION
DEFINITIONS
DEFINITIONS
Dermatomyositis (DM) continues to be classified as a form of myositis and, in fact, is traditionally considered one of the idiopathic inflammatory myopathies. Implicit in this classification is that myositis is required to make this diagnosis, as well as the characteristic rash—the often used criteria of Bohan and Peter exemplify this.1,2 This is problematic because it is widely accepted now that a proportion of patients, perhaps 20%,3 have the characteristic rash but no clinical signs or symptoms of myositis. These patients make up a population now defined as clinically amyopathic dermatomyositis (CADM), a term coined by Sontheimer in 1991.4 Patients with CADM have the characteristic DM rash (with a consistent skin biopsy) for at least 6 months’ duration but no evidence of weakness by history or clinical examination; earlier concepts also require demonstration of normal muscle enzyme levels,5-7 but this definition was later refined that a patient with normal strength and abnormal muscle enzyme levels could still be included under the CADM definition.8 If not CADM, then the rest of patients with DM have what is often termed “classic dermatomyositis,” referring to the traditionally recognized disease with symptomatic myositis. CADM is a useful term for physicians because it is defined clinically and makes no attempt to deny the presence of subclinical muscle inflammation. The CADM umbrella can further be subclassified into exactly two groups depending on the results of further imaging (eg, magnetic resonance imaging [MRI]), electromyographic, muscle biopsy, or (now) laboratory studies of muscle enzymes: hypomyopathic DM, in which at least one of these test results is abnormal, and amyopathic DM, in which all the test results are normal. It should be noted that several other groups have proposed classification criteria for DM that focus on muscle histology,9 clinical findings,10,11 or combinations of these.12 It has become clear that muscle histology and clinical findings cannot be defined that are specific to DM, although it remains possible that novel serotypes may have sufficient sensitivity and specificity to play a major role in defining DM. The definition of exactly what constitutes a “DM rash” has been elusive. Sontheimer has proposed formal cutaneous criteria for making a diagnosis of CADM in 2002.5 Three major criteria include the pathognomonic heliotrope sign (violaceous erythema on the upper eyelids), Gottron papules (papules overlying the metacarpophalangeal [MCP] and interphalangeal [IP] joints), and Gottron sign (erythema overlying the knees, elbows, or IP joints) and 14 additional minor criteria
10
Relationships among classic, clinically amyopathic, hypomyopathic, and amyopathic dermatomyositis
Dermatomyositis
Classic Clinically amyopathic dermatomyositis
Hypomyopathic dermatomyositis
- No weakness
- Evidence of myositis on laboratory studies, EMG, or MRI
Amyopathic dermatomyositis
- No weakness
- No evidence of myositis on laboratory studies, EMG, or MRI
(Fig. 62-1). Sontheimer suggested that a diagnosis of CADM could be made if two major criteria were present or one major criterion and two minor criteria were present in addition to biopsy of at least one region showing histopathologic changes consistent with DM.5 Although these criteria seem comprehensive and reasonable, they have not yet been validated in any way. It will continue to be important to recognize patients with CADM even though there is no evidence that this group of patients is pathogenically, histologically, or serologically different from the classic group except with regards to absence of clinical weakness. Even if these patients were managed the same as patients with myositis regarding aggressive immunotherapy (which is not necessarily the case), they are an important group to recognize because these patients appear to harbor the same risks for systemic disease (eg, interstitial lung disease [ILD]) and malignancy as their classic counterparts.8 Thus, for now, recognizing the prototypical and also atypical or subtle cutaneous presentations of CADM is especially important for dermatologists because these clues may provide the only data upon which the diagnosis is based.
EPIDEMIOLOGY
DM has a bimodal distribution in the age of disease onset, occurring at two peaks, at 5 to 14 years and 45 to 64 years of life. The disease affects women two to three times more than often than men. As discussed earlier, DM is typically classified as an idiopathic inflammatory myopathy, a group of disorders that includes polymyositis, inclusion body myositis, nonspecific myositis, and immune-mediated necrotizing myopathy. Estimating the incidence and prevalence of pure DM is challenging because of a lack of precision and standardization
1062
regarding diagnostic criteria (with many studies even grouping DM and polymyositis into one disease), differing methods of case ascertainment, differing study design, and possible geographic influences.13,14 Despite this, using data from the few population-based studies or largest databases available, estimates of incidence and prevalence are remarkably similar, with incidence rates ranging from 5 to 10 per 1 million per year and prevalence estimates of 10 to 20 per 100,000. Data from the largest population-based study in southeast Norway (>2.6 million people) estimated a point prevalence of total polymyositis or DM of 8.7 (95% confidence interval [CI], 4.5–11.2) per 100,000, of which about half of cases were DM. This study estimated an incidence of 6 to 10 cases per 1 million each year.15 Bendewald and coworkers provided population estimates for adultonset DM from Olmstead County, Minnesota. The age- and sex-adjusted incidence was 9.63 (95% CI, 6.09–13.17) per 1 million per decade, and the prevalence was 21.42 per 100,000 persons (95% CI, 13.07–29.77).3 In their small cohort, 20% (6 of 29 cases) were clinically amyopathic. Analysis of data from large insurance claims databases in Japan and Taiwan estimated an annual incidence of 10 to 13 and 6 to 10 per million annually, respectively.16,17 Based on registry data, the annual incidence of juvenileonset DM in the United States among children ages 2 to 17 was estimated between 2.5 and 4.1 per million.18
CLINICAL FEATURES
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
Elements of the patient history may be useful in distinguishing DM from mimicking eruptions. Triggers may be present such as substantial ultraviolet exposure,
strenuous activity (for the patient with concurrent myositis), or recent malignancy. There is often significant pruritus associated with affected skin, particularly on the scalp, which may also be described as a “tightness” or burning or with other dysesthetic qualities such as crawling or tingling. This severe pruritus on the scalp may be caused by structural damage to epidermal small-fiber nerves.19 The natural history of the eruption is chronic and often progressive, ultimately resulting in a background of dyspigmentation, atrophy, and telangiectasias. An evanescent or wholly intermittent eruption is unlikely to represent DM. With treatment, however, many patients can ultimately enter remission in which no further activity is present but residual damage features (eg, poikiloderma) may remain. The characteristic cutaneous feature of affected skin in DM is violaceous patches and plaques, varying from a bright pink to a deep violet color. In darker skin types, the erythema is often subtle, and careful examination is important to detect skin involvement. To heighten the sensitivity of the exam and appreciate these, at times, subtle color changes, patient positioning, and proper lighting in the examination room are critical. Many types of overhead direct lighting tend to obscure the subtle color changes seen in DM skin, and thus natural lighting is recommended for the clinical examination. Evaluating the patient in a reclined or supine position helps minimize shadows, particularly on the face, and overexposure with direct lighting. The scalp is one of the most commonly involved sites, often only with pink to red erythema (Fig. 62-2), but there may be associated fine white scale. The pruritus and dysesthesia are often severe and out of proportion to the erythema on examination. The scalp can be affected in any location but often involves a linear band just above and below the frontal hairline. Examination findings alone are not always specific enough to differentiate it from seborrheic dermatitis, psoriasis, and contact dermatitis. Subtle erythema is often perceptible on the vertex scalp or along the hair part or borders of the hairline even when the remainder of the cutaneous disease is quiescent. The heliotrope sign (Fig. 62-3) may exemplify the pink to purple violet hue of the eruption, resembling the
10
color of the flower petals after which the sign is named. The eyelid eruption can be associated with periorbital edema, and patients are often initially misdiagnosed with allergic contact dermatitis or angioedema. In addition, erythema of the lateral canthi, medial canthi, and adjacent nasal sidewalls can be seen (Fig. 62-4). The forehead, cheeks, ears, and chin may vary from uninvolved to focal patches or plaques to diffuse erythema. The skin changes in DM are often distributed to prototypical regions on the body Table 62-1). Trunk involvement is often seen on the posterior neck, upper back, and shoulders, known as the shawl sign (Fig. 62-5), which may extend to the posterior upper arms. Patients can display symmetric violaceous erythema, often with a livedo character, symmetrically on the lateral lower back. Confluent violaceous erythema on the sun-exposed areas of the lower anterior neck and anterior chest is termed the V-neck sign (Fig. 62-6). The violaceous to pink papules over the IP and MCP joints are termed Gottron papules (Fig. 62-7). They may display the same range of features seen elsewhere such as poikiloderma, atrophy, hypopigmentation, hyperkeratosis, or ulceration (Fig. 62-8). They may take the form of larger, ill-defined, round circular plaques (often with scale) or of 2- to 4-mm, smooth, well-demarcated, often umbilicated papules. Gottron sign is symmetric macular violaceous erythema over
1063
10
TABLE-62-1 Sontheimer’s Proposed Diagnostic Criteria for Cutaneous Dermatomyositis1
Diagnosis of cutaneous dermatomyositis requires:
- Presence of two major criteria or one major criterion and two minor criteria and
- Skin biopsy changes consistent with cutaneous dermatomyositis
Major Criteria Heliotrope sign Gottron papules Gottron sign
Minor Criteria Macular violaceous erythema involving (each area counts as one minor criterion)
■Scalp or anterior hairline
■Malar eminences of face, forehead, or chin
■V-area of neck or upper chest (V-neck sign)
■Posterior neck or posterior shoulders (shawl sign)
■Extensor surfaces of arms or forearms
■Linear streaking overlying extensor tendons of dorsal hands
■Periungal skin
■Lateral thighs or hips (holster sign)
■Medial malleoli
Nailfold capillary telangiectasia, hemorrhage-infarct
Poikiloderma
Mechanic’s hands
Cutaneous calcinosis
Cutaneous ulcers
Pruritus
Pruritus
the IP joints, olecranon processes (Fig. 62-9), patellae, and medial malleoli. Generally, juvenile DM more characteristically displays atrophy and poikiloderma in classic areas of Gottron sign. Of note, many of sites of cutaneous DM skin involvement areas are not necessarily in areas of ultraviolet (UV) exposure (so-called “photodistributed”), including the scalp, lower back, and lateral thighs. Some patients can present with a classic photodistributed eruption, but more often the erythema is patchy even
1064
in sites with high risk of UV exposure. The violaceous erythema and poikiloderma on the lateral hips and lateral thighs is termed the Holster sign (Fig. 62-10). Often this is patterned as violaceous, folliculocentric macules or subtle papules and can be confused with keratosis pilaris. The violaceous and typically macular nature of the eruption, usually in the absence of involvement of other areas typical for keratosis pilaris (eg, upper outer arms) helps to characterize this finding of DM. In addition, flagellate erythema may be found as widespread linear patches or urticarial plaques, typically on the middle and lower back, and flanks,
10
possibly caused by excoriation or imprinting from clothing or bed sheets. Other characteristic hand findings include the, socalled “mechanic’s” hands. This finding was initially described as hyperkeratosis and fissuring along the medial thumb and lateral second and third digits in 1979.20 This finding may be subtle and often requires palpating the fingers to appreciate the rough texture (Fig. 62-11). This lateral second digit hyperkeratosis seems to differ in quality and severity with the findings in patients with antisynthetase antibodies, the latter associated with fissuring and scaling that tends to also involve the distal fingertips. In 2012, Sato and coworkers found an increased prevalence of ILD among their patients with DM with mechanic’s hands (78%, 7 of 9) compared with without mechanic’s hands (40%, 12 of 30),21 supporting that mechanic’s hands is a cutaneous clue to the possible presence of ILD. A recent study by Werth and coworkers found that in 101 patients with DM, mechanic’s hands was associated with an odds ratio of 3.28 (95% CI, 1.36–7.88; P = 0.01) of having ILD. Evidence of vasculopathy is manifested in many ways on the skin. Proximal nailfold involvement can present as subtle edema or erythema as well as microscopic or clinically obvious capillary dilation. When pronounced and easily visualized with the naked eye, these nailfold capillary changes can be highly suggestive of DM over other connective tissue disorders. The classic findings include red, edematous, often tender proximal nailfolds with ramified and dilated capillary loops with intervening pale to white avascular areas with capillary drop-out, cuticular hemorrhages, and elongated, ragged cuticles (Fig. 62-12). Although these changes are a sign of ongoing cutaneous disease activity,22 in longstanding DM, damage may be evident as persistently dilated and arborizing capillaries.23
Sensitive nailfolds can be a helpful diagnostic symptom for DM. In addition, gingival telangiectasias have been described in DM.24 In addition, livedo reticularis can be seen in a minority of patients. Cutaneous ulceration can be an exemplary sign of vasculopathy, although it can also be the result of robust, liquefactive interface dermatitis, or excoriation.
1065
10
Ulceration may be present in 30% of patients and often affects the skin over the extensor joint surfaces, although it can be found anywhere.25 Cutaneous ulceration in DM warrants concern for the presence of anti- MDA5 antibodies or malignancy (especially if necrosis is seen). In the setting of anti-MDA5 antibodies, cutaneous ulceration commonly occurs over Gottron papules (see Fig. 62-8) or in areas of Gottron sign (eg, extensor surfaces).25,26 Ulcers have been correlated with the presence of ILD, but this is likely through their association with anti-MDA5 antibodies.25,27 As such, worsening cutaneous ulceration in a patient with anti-MDA5 antibodies may be a cutaneous sign of worsening ILD. Visualization of the oral mucosa, particularly the hard palate, may provide a valuable sign to aid in the diagnosis of DM. One can observe a symmetric violaceous patch across the midline of the hard palate, termed the ovoid palatal patch (Fig. 62-13), most frequently in the subset of patients with DM with anti–transcriptional intermediary factor 1γ (TIF1-γ) antibodies.28 Biopsies from these lesions appear to demonstrate interface mucositis, consistent with typical findings of DM. Like nailfold capillary changes described later, these hard palate changes appear to fade with control of disease activity. These lesions could be confused with oral findings of discoid lupus or lichen planus, but their consistent localization to the center of the hard palate may aid in the diagnosis of DM when other cutaneous features are nondiagnostic. In addition, other oral mucosal findings include gingival telangiectasias (noted earlier) as well as more classic lichen planus–like lesions around the gingiva or buccal mucosa. Whether or not these latter lesions
1066
represent coexisting lichen planus or are part of the DM disease spectrum is not clear. In general, the clinician should be always differentiating signs of skin activity, which may be reversible with therapy, versus damage. The distinction between activity and damage in DM skin is critical for clinical decision making so immunosuppressive treatments are not escalated for signs of skin damage. Cutaneous disease activity is characterized by erythema, itch, induration (papules or plaques), scale, or ulceration. Although erythema is often an important sign of activity, it can often be a sign of damage as well (eg, diffuse telangiectatic erythema), and thus care must be taken not to inadvertently escalate therapy solely caused by erythema that has no other qualities of disease activity. This phenomenon can be especially true on the face and chest, and faint erythema in the absence of symptoms or other skin changes is often mild damage and may not respond to traditional immunosuppressive therapies. Also, a reticular pattern of pink to red erythema on the palms and volar surfaces (more commonly seen in the anti-MDA5 group, Fig. 62-14), in addition to more classic livedo reticularis (including that found on the lateral flanks) represents vascular changes that have unclear association with disease activity. When substantial inflammation has been present, a distinctive and pathognomonic pattern may be seen composed of reticulated, sometimes atrophic white macules adjacent to erythema or telangiectasias, which
the authors have termed “red on white” (Fig. 62-15).29
The thin skin along the bitemporal hairline is a frequent place to visualize the “red-on-white” pattern, but notably, this pattern does not necessarily follow patterns of sun exposure and may be found on the hair-bearing scalp. It is becoming increasingly clear that many of these areas do not represent permanent damage per se because these lesions may slowly resolve with time, even in areas with atrophy. However, this morphology is a very useful diagnostic tool because it does not
10
seem to be associated with other connective tissue diseases, such as cutaneous lupus. Skin damage from DM is reflected by brown, often reticulated, postinflammatory hyperpigmented patches in areas of prior disease activity. Longstanding disease activity, typically in sun-exposed areas, results in more significant damage, characterized by atrophy, hypopigmentation, hyperpigmentation, and telangiectasias, termed poikiloderma (Fig. 62-16). Poikiloderma (as opposed to the “red-on-white” manifestation discussed earlier) is a late manifestation and is not diagnostically specific because many other acquired and congenital diseases result in poikiloderma such as cutaneous lupus, chronic actinic damage (poikiloderma of Civatte), poikilodermatous mycosis fungoides (poikiloderma vasculare atrophicans), Borrelia infection (acrodermatitis chronica atrophicans), chronic radiation dermatitis, graft-versus-host disease, hydroxyurea, and dyskeratosis congenita. Therefore, ancillary cutaneous findings are necessary to discern the cause of poikiloderma. Calcinosis is typically a late manifestation in the skin, subcutaneous tissue, fascia, or muscle and typically affects the trunk, proximal extremities, or areas of previous disease activity. The prevalences of calcinosis are 20% in adult DM30 and up to 40% in juvenile DM.31 Calcinosis also occurs more rapidly after disease onset in juvenile versus adult DM (2.9 years vs 7.9 years, respectively).32 In juvenile DM, risk factors for development of calcinosis include longer disease duration, younger age of disease onset, sustained disease activity, and internal organ involvement.33,34. Calcinosis is most frequent on the extremities in DM, in contrast to systemic sclerosis in which digital calcinosis is most frequent.32 In both juvenile and adult DM, the presence of anti–nuclear matrix protein 2 (NXP-2) antibodies is associated with an increased risk of calcinosis.35,36 In adults, fingertip ulceration has been associated with calcinosis,36 suggesting that vascular insufficiency or damage may be involved in the pathogenesis of calcinosis. Calcinosis is also commonly seen in the anti-MDA5 subset (especially in patients with longstanding disease),36 which is associated with known vasculopathy.
1067
10
Panniculitis reflects active disease in DM, typically affecting the buttocks, trunk, and proximal extremities; it may progress to calcinosis or lipoatrophy.37
Histopathology shows a lobular panniculitis but may have features of lupus panniculitis with lipomembranous changes or with septal thickening as in deep morphea. Alopecia in DM is most commonly nonscarring and diffuse, although patchy involvement, rarely with scarring, can also be seen. Alopecia can be caused by DM disease, coexisting disorders, medications, or telogen effluvium. Patients with anti-MDA5 antibodies have a higher risk of alopecia, which commonly is severe and occurs early in the disease.
RARE PRESENTATIONS OF CUTANEOUS DERMATOMYOSITIS
RARE PRESENTATIONS
OF CUTANEOUS
DERMATOMYOSITIS
Subcutaneous edema that is either generalized or located in the limbs has been recognized as a rare manifestation of DM.38 Interestingly, edema in the distal extremities has recently been found to be associated with anti-NXP2 antibodies and may predict more severe muscle disease.38,39 It is unclear what the relationship is between anti-NXP2 antibodies and the more severe, generalized presentations described in the literature. Rarely, DM may present erythroderma in which 90% or more of the body surface shows confluent erythema. Generalized ichthyosis can also be a presenting sign of DM. There is a subset of patients with DM with clinical manifestations with overlapping features of both psoriasis and DM.40,41 Their skin disease may show psoriasiform, well-demarcated thick plaques over the MCPs and proximal interphalangeal (PIP) joints, elbows, and knees in addition to dilated and abnormal nailfold capillaries. Skin biopsies tend to reveal both epidermal hyperplasia and interface dermatitis. Some of these patients have a history of psoriasis, and it is unclear if these lesions represent concomitant psoriasis or a psoriasiform manifestation of DM.
EXTRACUTANEOUS FINDINGS
PULMONARY INVOLVEMENT
PULMONARY INVOLVEMENT
ILD is the most common pulmonary manifestation in DM and is a leading cause of morbidity and mortality in these patients.42 Other pulmonary manifestations in DM include aspiration pneumonia; drug-induced pneumonitis; and, rarely, pulmonary hypertension. ILD affects between 15% and 50% of patients with DM, depending on the population and autoantibody distribution.43-46
1068
There are three clinically described patterns of lung involvement, which may precede or proceed muscle involvement47,48: it may be asymptomatic with only radiologic evidence of ILD; it may have an insidious onset with gradual development of decreased exercise capacity, dyspnea on exertion, or a dry cough; finally, it may be acute onset with hypoxia and possibly respiratory failure, necessitating hospitalization. In a recent study from a large US cohort of 438 patients with polymyositis or DM (n = 393), the presence of ILD in patients with DM was associated with an increased risk of death with a hazard ratio of 2.13 (95% CI, 1.06–4.25; P = 0.03).49 Large case series have suggested that more than 75% to 86% of patients who have an antisynthetase antibody will develop ILD.47,50
Similarly, patients with DM with anti-MDA5 antibodies have a marked increased risk of developing ILD with estimates between 50% and 100%25,51 Table 62-2). Rapidly progressive ILD is an aggressive form that responds poorly to immunosuppressive therapies, having a 6-month survival rate of approximately 40%.52 Rapidly progressive ILD may affect 40% to 60% of patients with anti-MDA5 antibodies.53-56
Pulmonary function tests (PFTs) show a restrictive disease pattern with a decreased forced vital capacity (FVC)57 or a decreased diffusion capacity of carbon monoxide. Exertional oxygenation desaturation on the 6-minute walk test provides a global assessment of cardiopulmonary function and exercise performance. However, confounding factors such as deconditioning, myopathy, arthritis, respiratory muscle weakness, and pulmonary hypertension may also produce reduced FVC and diffusion capacity of carbon monoxide.58 Therefore, high-resolution computed tomography (CT) of the chest is a necessary step in establishing the diagnosis of ILD. High-resolution CT scan of the chest is a valuable diagnostic test and may show subclinical fibrosis before symptom onset. Up to 65% of patients with polymyositis and DM will have subclinical CT evidence of ILD.59
The most common radiographic and histologic pattern in DM is nonspecific interstitial pneumonia, reported in 81.8% (18 of 22 cases).60 Radiographically, basilar and peripheral ground-glass opacities and subpleural sparing characterize nonspecific interstitial pneumonia. Other patterns of involvement include usual interstitial pneumonia, cryptogenic organizing pneumonia, and diffuse alveolar damage, the latter with a poor prognosis. Pulmonary arterial hypertension is a rare manifestation in DM. Symptoms may include increased fatigue, shortness of breath, dyspnea on exertion, palpitations, chest pain, edema, lightheadedness, and rarely presyncopal or syncopal episodes. PFT revealing a disproportionately low diffusion capacity of carbon monoxide compared with a relatively normal FVC should prompt further screening for pulmonary artery hypertension. If a high-resolution CT scan is checked for worsening shortness of breath and abnormal PFT results, it may show an enlarged pulmonary artery but no evidence of ILD. Echocardiographic findings suggestive of pulmonary hypertension include a right ventricular systolic pressure greater than 40 mm Hg, right ventricular
10
FREQUENCY AND PHENOTYPE IN JUVENILE DERMATOMYOSITIS
AUTOANTIBODY AUTOANTIGEN CLINICAL PHENOTYPE IN ADULT DERMATOMYOSITIS FREQUENCY IN ADULT DERMATOMYOSITIS
Anti-tRNA synthetase Jo-1: histidyl PL-7: threonyl PL-12: alanyl EJ: glycyl OJ: isoleucyl KS: asparaginyl Ha: tyrosinyl Zo: phenylalanyl
4%–5%8
Jo-1 is the most frequent, up to 20% Non–Jo-1 anti-tRNA synthetase antibodies between 1% and 5%7
Increased risk of ILD PL-7: mild skin or muscle disease3
Older onset Antisynthetase syndrome with ILD, fever, nonerosive polyarthritis, Raynaud phenomenon, mechanic’s hands
PL-12, KS, OJ: isolated ILD4-6
Variable spectrum of findings of the antisynthetase syndrome: ILD, fever, arthritis, myositis, mechanic’s hands, Raynaud phenomenon
Anti–Mi-2 Mi-2; regulates transcription as a component of nucleosome remodeling and deacetylase (NuRD) complex
3%8,12
Hallmark cutaneous disease, good prognosis and response to therapy; relapses common; high CK values (>5000 U/L); rare ILD, cancer; not amyopathic
Anti–transcriptional intermediary factor (TIF1)-γ p155; TIF; role in apoptosis, ubiquitination, and innate immunity
Ethnogeographic variation: 20% in US9 and Japan10
Classic skin disease Myositis Good therapeutic response; more likely to be in remission at 2 yr
6.7% in Glasgow 60% in Guatemala11
21%–38%13,14 23%–35%8
Increased cancer risk in adults Severe cutaneous disease Red-on-white skin lesions Ovoid palatal patch Low ILD risk Low CK values (200–2000 U/L)
Anti–melanoma differentiation– associated gene 5 (MDA5)
Melanoma differentiation: associated protein 5; cytosolic receptor for viral dsRNA; mediates type I IFN innate immune response
Severe skin disease Lipodystrophy
20%–35% in Asia17,18 7%–14%19,20
High ILD risk, RP-ILD, especially in Asians Skin ulceration, red palmar papules, alopecia, gingival pain Arthritis Often isolated high aldolase
7%–10% in US15,16
ILD in Japanese cohort Skin ulceration, arthritis, milder myositis
1.6%–30%14,21-23 20%–25% 8,12
Anti–nuclear matrix protein 2 (NXP2) Nuclear matrix protein; transcription Increased cancer risk in adults Increased risk of calcinosis Peripheral edema, myalgia, severe dysphagia Distal weakness
Anti–small ubiquitin-like
Small ubiquitin-like modi-
Calcinosis, more severe myositis, poorer functional status
1.5%–10%24-27 1%12
Skin disease onset before
Anti–small ubiquitin-like modifier activating enzyme (SAE)
Small ubiquitin-like modi- fier activating enzyme; posttranslational modification
1.5%–10%24-27 1%12
Skin disease onset before myositis May have severe skin disease Dysphagia
modifier activating enzyme (SAE)
fier activating enzyme; posttranslational modification
myositis May have severe skin
disease Dysphagia
CK, creatine kinase; IFN, interferon; ILD, interstitial lung disease; RP-ILD, rapidly progressive interstitial lung disease.
enlargement, maximum tricuspid regurgitant velocity greater than 3.0 m/s, and presence of a pericardial effusion.61 Transthoracic echocardiography only has a sensitivity of 82% and specificity of 69% to detect pulmonary hypertension61 and cannot differentiate patients with pulmonary arterial hypertension from those with left heart disease or ILD associated pulmonary hypertension. The gold standard for the diagnosis of pulmonary arterial hypertension is right heart catheterization showing a mean pulmonary artery pressure of 25 mm Hg or greater at rest and an end-expiratory pulmonary artery wedge pressure of 15 mm Hg or less.62
MUSCLE INVOLVEMENT
MUSCLE INVOLVEMENT
Myositis in DM typically presents as symmetrical proximal muscle weakness. About 20% of patients with DM, those with CADM, do not have clinical evidence or symptoms of weakness, although it is unclear what proportion of those patients might actually have subclinical myositis. If patients with DM do develop weakness, then about 80% of patients develop weakness within the first year of symptom onset.63 The temporal course of myositis may be acute, subacute, or chronic and progressive.
1069
10
Patients often report weakness in the extensor muscles surrounding the shoulder and pelvic girdles and proximal limbs. Quadriceps and gluteal muscle weakness may manifest as difficulty rising from a chair or toilet, climbing stairs, or stepping onto curbs. Patients may report shoulder and upper extremity weakness as difficulty washing their hair or reaching for items in overhead cupboards. Neck flexor muscle involvement is also common with difficulty raising the head off the bed while laying supine. Patients may also complain of myalgias, which can occur even in the absence of frank clinical weakness. Approximately 30% of patients complain of muscle pain with or without muscle weakness.9 Myalgias are described as soreness or muscle tightness or burning, but muscles are not tender to palpation. Care should be taken to distinguish this pain from other causes of pain, such as joint pain or fibromyalgia,64 and care must be taken to not escalate immunosuppression for what may be symptoms of a pain disorder. Involvement of respiratory muscles of the chest wall or diaphragm may also lead to respiratory insufficiency and occasionally respiratory failure. Patients may note a hoarse or raspy voice (dysphonia) from cricoarytenoid muscle involvement, which occurs in up to 40% of patients with DM.65 Also, dysphagia may occur in 20% to 50% of cases because of weak pharyngeal musculature and thus an inability to propel food in the pharyngeal phase of swallowing.66 Interestingly, there is a high correlation between dysphagia and weakness of the anterior neck muscles (eg, sternocleidomastoids).67 Distal muscle weakness in the hands, manifesting as difficulty opening jars or holding onto objects, more typically occurs late in disease or in patients with anti-NXP2 antibodies.
JOINT INVOLVEMENT
JOINT INVOLVEMENT
Arthralgias are common in DM, reported in 30% to 40% of patients.68-70 In general, the arthralgias in DM are mild to moderate in severity and involve the small joints of the hands, including the wrists, MCP and PIP joints, and the shoulders, elbows, and ankles.71 More rarely, patients can have a true arthritis, often presenting as a symmetric polyarthropathy affecting the distal joints and often clinically indistinguishable from rheumatoid arthritis. Thus, patients with DM with arthritis as the presenting symptom may be diagnosed as having rheumatoid arthritis or a rheumatoid arthritis overlap disease before evolution of the skin, muscle, or pulmonary manifestations.72,73 Illustrative historical questions eliciting symptoms of an inflammatory arthritis include joint swelling, morning stiffness for 30 minutes or longer, or joint pain that improves with activity. Evaluation for synovitis, indicative of active joint inflammation, can be determined by palpation for warmth; range of motion; swelling or palpable fluid; and tenderness of each small joint of the hand, elbows, shoulders, knees, and other symptomatic joints.
1070
Arthritis and arthralgias are more common among patients with DM with anti-MDA5 and antisynthetase antibodies. However, erosive changes have been reported, more commonly in the anti– synthetase antibody subset.74-77 Hall and coworkers found that 9 of 11 (81.8%) anti-MDA5 patients versus 40 of 149 (26.7%) non-MDA5 patients with DM exhibited an inflammatory arthritis (P <0.001).78
Patients with DM with antisynthetase antibodies, the most common of which is Jo-1, may also manifest a nonerosive arthritis in up to 93% of cases.71
In these patients with antisynthetase antibodies, the nonerosive arthritis may occur in the setting of “antisynthetase syndrome” consisting of fever, arthritis, myositis, ILD, mechanic’s hands, or Raynaud phenomenon. The arthritis may flare in 50% of patients during disease relapse.71
GASTROINTESTINAL INVOLVEMENT
GASTROINTESTINAL
INVOLVEMENT
Gastrointestinal (GI) involvement is an uncommon manifestation in juvenile DM, reported in 4% of juvenile patients with DM.79 It is associated with more severe disease80 and is thought to result from a vasculopathy affecting the bowel wall.81 The sequelae include ulceration and perforation, which may be life threatening. The presenting symptoms include persistent and worsening abdominal pain. They also may have nonspecific accompanying symptoms such as diarrhea, vomiting, constipation, and more rarely frank hematemesis or hematochezia; juvenile patients may not initially have evidence of hemorrhage such as melena, occult blood in the stool, or radiographic evidence of perforation.82 Therefore, judicious monitoring of juvenile patients with DM with abdominal pain is warranted to recognize and intervene early in cases with GI involvement.
CARDIOVASCULAR INVOLVEMENT
CARDIOVASCULAR
INVOLVEMENT
Cardiac involvement in DM is increasingly being recognized as an important clinical feature. Cardiac involvement is usually subclinical. The most common electrocardiographic abnormalities are ST-T segment changes in 12.5% to 56.7% and conduction abnormalities in 25% to 38.5% of patients with DM.83 Echocardiographic findings have shown left ventricular hypertrophy in 8% to 15% and left ventricular diastolic dysfunction in 42% of patients.84 Myocarditis may lead to myocardial fibrosis and ventricular dysfunction and thus cardiomyopathy. Rosenbohm and coworkers screened 11 patients with DM with cardiac MRI and found that 54% (6 of 11) displayed evidence of late gadolinium enhancement, consistent with myocardial inflammation.85 Cardiac troponin I may
be a useful biomarker in detecting subclinical cardiac muscle involvement.86 Patients with the anti-MDA5 antibody subtype may be at higher risk for cardiac involvement.87,88
INTERNAL MALIGNANCY
INTERNAL MALIGNANCY
DM is associated with an internal malignancy in 10% to 20% of cases.89 Malignancies tend to occur within the first 1 to 2 years of disease onset and can be many types. Multiple population-based studies have estimated a standardized incidence ratio of 4 to 6 for malignancy compared with the normal population.90 The cancer types that are overrepresented may vary depending on the population studied but appear to involve both solid tumors as well as hematopoietic malignancies. Common types of solid tumors include breast, lung, ovarian, prostate, colorectal, gastric, and pancreatic,91-93 with nasopharyngeal cancer being more common in Southeast Asians.94 The mechanism of the relationship between DM and malignancy is unknown but may include increased risk of cancer in the setting of immunosuppressive therapy, increased detection in the setting of heightened surveillance, and DM occurring in the setting of an immunologic response to internal malignancy. There are several clinical risk factors associated with risk of malignancy, which include increasing age, male gender, cutaneous necrosis, dysphagia, and rapid onset of myositis.95 Factors protective of malignancy include ILD, arthritis, and Raynaud phenomenon. Recently, malignancies have also been found to be more associated with particular DM-specific autoantibodies. The major antibody associated with malignancy is anti– TIF1-γ, although the precise elevation in risk appears to vary among studies and populations studied.96,97
More recent data suggest that anti-NXP2 antibodies may also be associated with cancer, although this association is not as strong.97,98
Cancer screening is a source of controversy because no guidelines currently exist to direct screening in newly diagnosed patients with DM. Most authorities would agree with age-appropriate, routine cancer screening at a very minimum. However, in a small study, Sparsa and coworkers reported that initial routine cancer screening failed to discover 4 of 13 malignancies in their cohort.99 A recent study of 400 patients showed that a substantial number of occult cancers (17 of 29) were only discovered using tests, usually CT scans, beyond what would be considered “age appropriate.” Identifying which tests to order and in what patient population is of high priority.
ETIOLOGY AND PATHOGENESIS
DM is primarily an immune-mediated disorder with molecular and histologic evidence supporting a role for both innate and adaptive immunity. Muscle and
10
skin biopsies show infiltration of CD3+ T cells, plasmacytoid dendritic cells, and macrophages as well as B cells (especially in muscle).100,101 Parenchymal cell injury is manifested in the skin by interface dermatitis with keratinocyte injury and in muscle by atrophy, degeneration, and regeneration of muscle fibers, typically in a perifascicular distribution. There is strong evidence for activation of the innate immune system as a critical step in disease pathogenesis and propagation. High levels of interferon (IFN)- induced genes and proteins found in blood, muscle, and skin and have been shown to correlate with disease activity.102 There is some evidence that this response may be driven by IFN-β, although this is not yet clear. There are multiple pathways that could lead to activation of IFNs, most arising from so-called pattern recognition receptors in the cytoplasm. Many of these receptors are activated by aberrant quantity or structure of nucleic acids, which could come from viruses or the host, the latter in the form of damaged DNA or improperly processed RNA. High levels of IFN can induce DM autoantigens (eg, MDA5), activate immature dendritic cells to become effective antigen-presenting cells, and upregulate major histocompatibility (MHC) class I expression, and activate lymphocytes. In both skin and muscle, the cells expressing these IFN-induced gene products appear most concentrated in the areas of tissue damage and may play a role in recruiting and activating cytotoxic effector cells. There is also abundant evidence for activation of the adaptive immune system in DM. Genetic studies have shown that polymorphisms in the human leukocyte antigen (HLA) region are highly associated with risk of DM, implicating activation of T cells. In addition, DM is associated with the presence of several, highly specific, circulating autoantibodies that are evidence of T- and B-cell activation. As mentioned, CD3+ T cells are found in tissue biopsies. The exact role of these antigen-specific responses is at present unclear, but the correlation between clinical phenotypes and specific autoantibodies suggests that these immune responses lie at the heart of disease pathogenesis. In addition, an animal model of myositis can be created by inducing anti–Jo1 immune responses in mice. Although some of these antigens have now been discovered, it remains unclear what cell type(s) or structures are the targets of such an immune response. Of interest, expression of several DM autoantigens is increased in damaged and regenerating muscle cells, suggesting that muscle fibers themselves might be direct targets of antigenspecific CD3+ cells. The immune response may not be the entire story because histologic data support that a vasculopathy is a primary event in DM. Biopsies of both skin and muscle show endothelial degeneration and capillary dropout as some of the earliest findings. Deposition of the membrane attack complex of complement, consisting of C5b-9, is seen on the capillaries at the dermal–epidermal junction (DEJ) in skin and in the perifascicular blood vessels of the muscle.103 However, there is no other evidence that membrane attack complex deposition is the result of an immune attack on
1071
10
the vessels. Greenberg and coworkers have hypothesized that increased production of IFN-α/β–inducible proteins by plasmacytoid dendritic cells and myocytes can also result in endothelial damage.104
Genetics likely play a large role in the risk for developing DM. Certain HLA alleles are clearly some of the greatest risk factors for the development of DM. The HLA-B8 allele was the first allele found in increased prevalence in 12 of 17 (75%) juvenile patients with DM compared to 21% of control participants.105 Subsequently, multiple other HLA alleles, largely consisting of the HLA 8.1 ancestral haplotype (HLA-A∗0101, -C∗0701, -B∗0801, -DRB1∗0301, -DQA1∗0501, and -DQB1∗0201) have been enriched in patients with DM.106,107 Genome-wide studies have confirmed a role for MHC class I and II genes but have also uncovered other loci conferring risk; these include the BLK gene involved in B-cell activation and the TYK2 gene, which plays a role in signaling from IFN receptors. In addition, other nonimmune events may play a role in features of the disease. Currently, one area of investigation centers around the endoplasmic reticulum stress response that may be partially responsible for some of the weakness symptoms seen in DM. Endoplasmic stress could be the result of upregulation of MHC class I or other proteins and induces an unfolded protein response that results in activation of inflammatory nuclear factor kappa B (NF-κB) signaling pathways, mitochondrial dysfunction, and elevated reactive oxygen species, all of which can contribute to weakness. Current models suggest that DM is initiated in a genetically predisposed individual who is then subjected to an environmental trigger. The nature of that trigger could vary and could include UV exposure, infection, and malignancy. On the cellular level, these triggers could result in antigen modification or upregulation, cellular death, and activation of nucleic acid response with increased production of IFN, endoplasmic stress, and activation of the innate immune system. There is likely redundant interplay between the innate and adaptive immune system that allows not only initiation but also disease propagation.
DIAGNOSIS
Unfortunately, there are no adequate, validated diagnostic criteria for DM. Like most rheumatic diseases, DM variably affects different organ systems and with varying degrees of severity. Because of this, it is difficult to define this disease on either clinical or histologic criteria. We have already discussed that using clinical, histologic, electromyographic, or histologic criteria of muscle involvement for diagnosis will not diagnose the population of patients with amyopathic disease. However, the same may hold true for skin disease; although some would argue that the characteristic rash is the key to diagnosis, it is not clear that the skin needs to be involved in the DM disease process any more than does the muscle. A practical approach is not to require specific muscle or skin involvement but to have organ-specific criteria that must be met if
1072
indeed an organ is involved. Unfortunately, there are no validated clinical skin criteria that can serve to diagnose DM skin disease. It has been suggested that vascular membrane attack complex deposition may serve to distinguish this disease from cutaneous lupus, but unfortunately, there are no data regarding membrane attack complex deposition in other skin diseases. It is thought that certain features on muscle biopsy characterize DM, namely perifascicular atrophy, but recent studies challenge this notion.108 The recent characterization of several DM-specific autoantibodies gives the hope of making this diagnosis with a blood test, but sensitivity and specificity data of these assays across multiple mimicking skin and muscle diseases are still lacking. It is likely that optimization of future diagnostic criteria will require a combination of clinical, histologic, and serologic data. Thus, at present, establishing a diagnosis of DM continues to rely on the clinician’s impression based on history and physical examination findings.
HELPFUL SKIN FINDINGS
HELPFUL SKIN FINDINGS
Features of the skin examination that we find particularly sensitive are microscopic periungual telangiectasias, lateral digit hyperkeratosis, and scalp erythema and dysesthesia. Some elements that are more specific are the “red-on-white” patches, the ovoid palatal patch, grossly visible periungual telangiectasias, and Gottron papules.
MUSCLE DISEASE
MUSCLE DISEASE
As discussed, myositis is not a requirement for the diagnosis. However, tests supporting the presence of an inflammatory myopathy are helpful when a patient presents with a suspicious rash. In this way, a clinical suspicion of DM is a necessary but not always sufficient for diagnosing DM. Muscle involvement by history (including dysphagia, dysphonia, and myalgia) and weakness on examination will serve to increase suspicion for myositis but are not confirmatory. Elevation of muscle enzymes, however, should be the next step in nailing down the diagnosis (see later). Electromyography or MRI can be used in situations when the clinical suspicion is still high for myopathy but muscle enzymes are normal. Finally, a muscle biopsy can be performed in cases in which a cause for muscle symptoms is still not clear.
SYNDROMIC PRESENTATIONS
SYNDROMIC
PRESENTATIONS
Certain combinations of symptoms, although not necessarily common, have reasonably high specificity to
be helpful in making a diagnosis of DM, regardless of the presence of the more “typical” cutaneous features of DM. A patient presenting with alopecia, cutaneous or mucosal ulceration, palmar erythematous papules, severe arthralgia or arthritis, and shortness of breath, even with a few of these symptoms, should trigger the clinician to consider anti-MDA5 DM. Patients with mechanic hands, arthritis, Raynaud phenomenon, and lung symptoms are at high risk of having antisynthetase disease, which some consider to fall under the umbrella of DM if there are particular DM-like signs present on the skin. Patients with extreme myalgia, peripheral edema, and distal weakness should be ruled out for DM specifically with anti-NXP2 antibodies even if the rash is subtle. Knowledge of these phenotypes will increase the clinician’s sensitivity for making the DM diagnosis.
DIAGNOSTIC LABORATORY EVALUATION
DIAGNOSTIC LABORATORY
EVALUATION
AUTOANTIBODIES
Myositis-specific autoantibodies associated with DM are evolving as key tools in assisting in establishing the diagnosis. These MSAs include TIF1-γ, NXP2, MDA5, small ubiquitin-like modifier activating enzyme (SAE), Mi-2, Jo-1, and the other antisynthetase antibodies (PL- 7, PL-12, EJ, OJ, SRP). In our US cohort, these antibody tests have an 80% to 85% sensitivity for diagnosing DM. Thus, these MSAs may provide pivotal information to establish the diagnosis of DM in subtle or complex cases. Importantly, MSAs are becoming increasingly relevant to identify clinical subsets and disease associations (see Table 62-2). As clinical phenotyping improves with respect to disease features and clinical course and increasing availability of testing for MSAs, this classification method will likely add value to current definitions by allowing improved prognostication, targeted screening, and potentially tailored therapy. ANA testing, if performed, should be done using method of direct immunofluorescence. ANA test results can be negative in DM 50% of the time but are usually positive in systemic lupus erythematosus, and thus a negative test result can help point away from systemic lupus if that is in the differential diagnosis.
MUSCLE ENZYMES
Early in the course of myositis, serum muscle enzymes (ie, creatine kinase, aldolase, lactate dehydrogenase [LDH], aspartate aminotransferase [AST]), alanine aminotransferase [ALT]) are reasonably sensitive biomarkers of muscle inflammation. However, mid to late in the course of myositis, their sensitivity decreases. It is unclear why the sensitivity of muscle enzyme drops as myositis continues, but it may be a result of perifascicular muscle atrophy and fibrosis, resulting in
10
less dramatic changes in these tests even while inflammation persists. Additionally, often only the creatine kinase or only the aldolase will be elevated in a single patient. To increase the sensitivity of detecting myositis on laboratory testing, the clinician should evaluate creatine kinase, aldolase, and LDH as a group when testing muscle enzymes. There are a few practical issues to be aware of when evaluating muscle enzymes. First, all muscle enzymes can be elevated after strenuous activity; in questionable cases, the enzyme levels can be rechecked after 10-14 days following the activity. Second, aldolase (and AST and ALT) can be elevated with liver disease or hemolysis of the blood sample. Very high levels should be rechecked. γ-Glutamyl transferase will be elevated with hepatic injury but will not be elevated in myositis and is a helpful addition to laboratory testing for myositis if hepatic sources are suspected.
ELECTROMYOGRAPHIC STUDIES
Similar to muscle enzyme abnormalities, early in the disease course, myositis may be detectable on electromyographic studies in 70% to 90% of patients with DM with active muscle disease and the sensitivity to detect myositis decreases over time. The classic triad electromyographic findings of myositis is small amplitude, short duration, polyphasic motor unit potentials; fibrillations and positive sharp waves; and complex repetitive discharges.109 These findings reflect active muscle inflammation and may be seen in other inflammatory myopathies such as polymyositis.110 Late in the course of the disease, the sensitivity decreases.
OTHER DIAGNOSTIC LABORATORY TESTS
Most other laboratory tests performed at disease onset would be to evaluate for target organ involvement and not necessarily be used in making the diagnosis. However, serum ferritin is often highly elevated (>500 mg/dL) in anti-MDA5 patients with DM111 and may be helpful in diagnosis anti-MDA5 disease when serologic testing is unavailable or of inadequate sensitivity. In addition, it may be a useful biomarker to assess the severity and follow the clinical response of ILD in anti-MDA5 patients with DM.112,113
IMAGING
MRI provides a detailed view of the muscle anatomy, allowing for localization and discrimination of the type of pathologic process (eg, edema, inflammation, fibrosis, calcifications, or atrophy). MRI can be useful to differentiate weakness caused by damage or steroid-myopathy versus active myositis when muscle enzymes and electromyographic studies are inconclusive. MRI can also be used in directing the site of a diagnostic biopsy, if needed,114 or may be used to assess clinical response to treatment.115 Muscle edema
1073
10
is a sensitive indicator of myositis and correlates with creatine kinase levels.116 On short tau inversion recovery, in which normal muscle is dark and inflamed muscle is bright, an increased signal intensity within muscle tissue suggests muscle inflammation, necrosis, or degeneration.117 Yoshida and coworkers also identified fasciitis on MRI in 12 of 14 patients with DM, suggesting that the fascial microvasculature may be a primary site of involvement.118 Muscle damage may be identified on T1-weighted images, in which there is fatty replacement of skeletal muscle.73
PATHOLOGY
SKIN HISTOPATHOLOGY
SKIN HISTOPATHOLOGY
Skin biopsies from affected skin in DM classically show a cell-poor interface dermatitis, increased dermal mucin, a perivascular lymphocytic infiltrate, and vascular ectasia (Fig. 62-17). Other inconstant findings include epidermal atrophy and basement membrane thickening. However, frequently many of these findings are subtle or absent. Smith and coworkers reviewed 40 DM skin biopsies and noted that when interface dermatitis was absent (20% of cases), increased dermal mucin was always present.119 It has been suggested that whereas DM is characterized by increased presence of plasmacytoid dendritic cells in mostly an epidermal location, in cutaneous lupus, they are situated primarily in the dermis.120
The role of direct immunofluorescence testing in DM diagnosis is still unclear. It has been reported that DM biopsies do not demonstrate immunoreactants along the DEJ, the so-called “lupus band,” but this finding is not uniformly defined in terms of its intensity and antibody composition. Magro and coworkers found that if the definition of a lupus band is made stringent, only 1 of 24 patients with DM has a positive lupus band test result. However, in addition to most
1074
patients with DM, more than 35% of cutaneous lupus biopsies were also negative for the lupus band test. So, if a stringently defined lupus band (eg, interrupted deposition of immunoglobulin [Ig] G or continuous deposition of IgM) is present on biopsy, these data suggest that a diagnosis of DM is unlikely. In addition, Mascaró and coworkers found that deposition of the membrane attack complex or C5b-9, at either the DEJ or around vessels of DM occurs in 77% to 86% of patients.103 Magro and coworkers repeated this testing in 24 patients with DM and found that membrane attack complex deposition at both the DEJ and around vessels along with a negative lupus band test result was 78% sensitive and 93% specific for making a diagnosis of DM over cutaneous lupus.121
MUSCLE HISTOPATHOLOGY
MUSCLE HISTOPATHOLOGY
In patients referred to a dermatology clinic with a rash and weakness, muscle biopsy is seldom necessary to establish the diagnosis of DM. Evidence of myositis on laboratory testing, electromyography, or MRI is often sufficient in the setting of a cutaneous eruption supporting DM. Muscle biopsy can yield false-negative results, both because of immunosuppressive medications and the patchy nature of the inflammation. MRI can be used to as a guide to selecting a biopsy site. If a muscle biopsy is necessary to confirm the diagnosis of DM, it has the highest yield if performed within 2 weeks of beginning any immunomodulatory therapy. Typical muscle histopathology findings in DM include perifascicular atrophy, degenerating and regenerating myofibers, membrane attack complex deposition in the endomysial capillary walls,122
endothelial cell swelling, and capillary necrosis. The inflammatory cell infiltrate consists of CD4+ T cells,123
plasmacytoid dendritic cells secreting IFN-α,101 B lymphocytes, macrophages, and plasma cells. However, there is a range of histologic findings even in clear DM cases, and histologic findings of inflammatory myopathies often cluster according to other criteria and do not necessarily respect the clinical boundaries of idiopathic inflammatory myopathy categories that have been defined.
DIFFERNTIAL DIAGNOSIS OF DERMATOMYOSITIS
See Table 62-3.
CLINICAL COURSE AND PROGNOSIS
The long-term survival rate for adults with DM is approximately 65% to 75%, but there are relatively little data regarding specific organ outcomes such as muscle
10
CONDITION DESCRIPTION
Acute cutaneous lupus Characterized by malar pink patches and plaques; may generalize to rest of face and body; generally spares PIP and DIP joints; spares nasolabial folds; usually does not have signifi- cant pruritus; 5%–15% of patients with SLE may have myositis; patients have systemic lupus and positive lupus serologies
Photoallergic or airborne contact dermatitis Patients have new exposure before onset of eruption; distribution is to skin that was exposed to allergen or sun exposure, not in DM distribution; patients often have itching or burning; patients do not have nailfold capillary changes or systemic symptoms
Acne rosacea (erythematotelangiectatic or neurogenic) May have itching, burning quality but erythema is limited to face; absence of damage (hyperpigmentation, atrophy); no systemic symptoms
Polymorphous light eruption
Sun-exposed sites are affected, typically immediately after sun exposure (delayed 1–2 days in DM); rash not in classic DM extensor distribution; eruption may resolve completely in 1–2 wk with sun avoidance; no systemic symptoms
Characterized by annular or psoriasiform plaques on sun-exposed skin; 30%–60% have SLE; 80% have positive anti-Ro antibody; may be medication associated; lacks pathognomonic DM skin findings (eg, Gottron papules, Gottron sign, heliotrope)
Seborrheic dermatitis Scalp disease may be challenging to separate from DM; has greasy scale and orange-red quality instead of violaceous; may affect central chest, upper back, and neck but spares extremities; patients do not have systemic symptoms
Psoriasis Tends to spare the face; lacks characteristic damage of DM; lacks nailfold capillary changes; rarely, patients may have both DM and psoriasis
Phototoxic or photoallergic drug eruption Patients have a recently added medication; distribution is to sun-exposed areas, not extensor; eruption often has sharp borders and substantial edema; no nailfold capillary changes; patients are without systemic symptoms
Lichenoid drug eruption Patients are taking medication associated with lichenoid dermatitis; eruption is often widespread and papular; not in DM distribution
Pityriasis rubra pilaris Typically involves palms and soles; may have islands of sparing; has an orange-red quality as opposed to violaceous; lacks nailfold capillary changes
Atopic dermatitis Distribution is usually flexural in adults; lichenification present in chronic cases; may have history of seasonal allergies or asthma; generally improves with sun exposure; lacks atrophy; nailfold capillary changes
Patients with Weakness and other Skin or Systemic Findings
Mixed connective tissue disease Patients may have facial, chest, or scalp rash with poikiloderma, nailfold capillary changes, calcinosi,s and myositis; patients do not have Gottron papules; arthritis, Raynaud phenomenon, cutaneous sclerosis, GERD, and cytopenias are prominent symptoms; patients have anti-U1RNP antibodies
Systemic sclerosis May have myositis, depigmented and hyperpigmented patches, nailfold capillary changes, pruritus, and calcinosis but not the erythema of DM; telangiectasias aree matted; patients have Raynaud phenomenon, GERD, and often cutaneous sclerosis
Polymyositis May have periungual capillary changes but not the cutaneous eruption of DM
Immune-mediated necrotizing myopathy History of statin use; patients do not have cutaneous eruption of DM
Immune-mediated necrotizing myopathy History of statin use; patients do not have cutaneous eruption of DM
anti-U1RNP, anti-U1 ribonucleoprotein; DM, dermatomyositis; GERD, gastroesophageal reflux disease; PIP, proximal interphalangeal; SLE, systemic lupus erythematosus.
or skin disease.124 Remission rates of 25% to 70% have been reported, but most studies suggest that number is probably closer to 20% to 40% at 5 years. Major causes of death include malignancy, pulmonary or cardiac disease, and infection, but other predictors have variably been reported, including older age, male gender, nonwhite race, and longer duration of symptoms. Outcomes may also relate to autoantibody status; studies in both Japan and the United States have shown that presence of anti- MDA5 antibodies is a risk factor for death.125,126
For children, long-term registries have provided clearer data regarding remission rates, including that for skin disease. Most studies suggest that approximately 60% of patients have chronic disease (eg, past 5 years), with major risk factors for chronicity being a delay in therapy and early persistent (eg, 3 months) skin disease or decline in nailfold capillary density. Most reports suggest that the most common source of persistent disease activity is in the skin over muscles or other organs.127-129
1075
10
MANAGEMENT
PRINCIPLES OF MANAGEMENT
PRINCIPLES OF
MANAGEMENT
Treatment of DM first requires assessing the potentially affected organs, namely the skin, muscle, and lungs. It should be emphasized that ILD and associated cancers are leading causes of disease-related death and are prioritized during treatment selection. Screening for malignancy is critical because the treatment of a cancer associated with DM may result in a reduced disease severity or, at times, disease remission. Cutaneous DM often has discordant treatment response with the muscle disease,130 with recalcitrant skin disease continuing years after the muscle disease is in remission. Given the chronicity of the skin disease, it is worthwhile to weigh the long-term toxicity of the prescribed therapy against the achieved or potential cutaneous benefit. With that in mind, one should first select the appropriate agents to target the affected organs, consider patient comorbidities and preferences, and discuss the risks and benefits of each with the patient before deciding upon a regimen. Establishing collaborative relationships with the comanaging providers (rheumatologist, dermatologist, neurologist, and pulmonologist) is critical for the treatment of patients in whom multiple organs are involved.
CANCER SCREENING
CANCER SCREENING
Given that 10% to 20% of patients with DM have an associated malignancy, a thorough investigation is justified upon initial diagnosis. In addition to a complete history and physical exam, routine ageappropriate cancer screening studies (colonoscopy, mammogram, prostate examination) and relevant screening blood work (complete blood count, renal and liver function tests) as well as a urinalysis are indicated. The role of other blood work (eg, erythrocyte sedimentation rate, C-reactive protein, cancer markers, serum and urine protein immunofixation electrophoresis) in identifying cancer in a patient with DM is currently not established. The role of testing for anti–TIF1-γ and anti–NXP-2 antibodies in risk stratification is not yet known given the fact that, at least in the US population, most patients with these antibodies still do not harbor a malignancy. The role for more aggressive screening (eg, CT scans, positron emission tomography) is unclear, although as discussed earlier, this imaging may detect occult malignancies not found on routine age-appropriate screening in the higher risk period (2–3 years from diagnosis). The role for rescreening is even less clear. It would be prudent to consider blind rescreening be considered in patients with disease that is difficult to control or who have experienced an unexplained disease flare after
1076
a sustained quiescent period, especially in patients with a history of malignancy.
MONITORING OF EXTRACUTANEOUS DISEASE
MONITORING OF
EXTRACUTANEOUS
DISEASE
MUSCLE DISEASE
Manual muscle testing is helpful to gauge changes in strength between visits. Even a busy dermatologist can check a few muscle groups such as the neck flexors, deltoids, and quadriceps at each visit to assess gross changes in muscle strength. If more time is available, then the manual muscle testing for a subset of eight muscle groups (MMT8) may be performed, which is a validated muscle test in which eight major muscle group (neck flexors, deltoids, biceps, wrist extensors, gluteus maximus, gluteus medius, quadriceps, ankles dorsiflexors) that are highest yield in idiopathic inflammatory myopathies.131 Other factors such as pain, joint contracture, and fatigue may impact this measurement. Serum muscle enzymes (creatine kinase, aldolase, and LDH) are checked at each visit as biomarkers of myositis. However, as the duration of myositis increases, they become insensitive markers of myositis. Electromyography and MRI remain as more sensitive tests for active myositis in the setting of increasing weakness and normal muscle enzymes. The clinician must consider other causes of weakness, including muscle damage from prior myositis versus deconditioning, steroid myopathy, hydroxychloroquine-induced myopathy, or thyroid myopathy, for example. In this way, it is important not to reflexively treat weakness with increased immunosuppression until active myositis is identified as the cause.
PULMONARY DISEASE
Although there are no formalized guidelines for screening for ILD, it is valuable to obtain baseline PFTs with diffusion capacity in case there is future development of pulmonary symptoms and then annual screening as long as other symptoms of DM continue. Screening PFTs with diffusion capacity of carbon monoxide can be performed up to every 3 to 6 months if new pulmonary symptoms develop. PFT results are effort dependent and therefore influenced by following instructions during the test, muscle strength, and energy level. If PFTs show concerning changes for new or worsening ILD, then a high-resolution CT scan of the chest is the next step to evaluate the lung parenchyma.
CARDIOVASCULAR INVOLVEMENT
Consideration should be given to evaluating for cardiac muscle disease. The creatine kinase-MB isoform is found in both cardiac as well as regenerating skeletal
muscle, so it is not a specific test, although it could be used as a screening test for cardiac involvement. We prefer to use cardiac troponin I testing because it is specific for myocardial damage. There are no standard recommendations for screening for cardiac involvement, but some authorities suggest using cardiac troponin I as a screening test for subclinical myocardial involvement followed by echocardiography or electrocardiography if the result is positive.86 Patients with DM with anti-MDA5 antibodies may be at higher risk for clinically evident and, at times, fatal cardiac involvement.87,88
INTERVENTIONS
Evidence for medical therapies and treatment ladders in DM arise largely from single-center, retrospective case reports, case series, and expert opinion132-136
(Table 62-4).
TOPICAL THERAPY
TOPICAL THERAPY
Although photoprotection is a key first step in management, up to 60% of patients with DM are actually minimally photosensitive, and as few as 20% may report disease exacerbation after UV exposure.130,137
Despite this finding, it is helpful to carefully review the basic concepts of UV protection. Topical corticosteroids may have a palliative effect on DM skin disease by reducing erythema, scale, and pruritus and play an adjunctive role to systemic agents. However, they are unlikely to fully control skin symptoms except in the mildest cases. Class I or II topical steroid creams or ointments are used on the thick skin of the elbows, knees, and hyperkeratosis on the hands and can be tried to mitigate the common complaint of nailfold sensitivity, and occlusion with plastic wrap can be added at night to increase the potency further.
SKIN DISEASE MUSCLE DISEASE
First Line Photoprotection Topical steroids Hydroxychloroquine or Chloroquine Quinacrine
Systemic corticosteroids
Second Line Methotrexate Mycophenolate mofetil Intravenous immune globulin Azathioprine
Third Line Tofacitnib Leflunomide Dapsone Thalidomide Cyclosporine Cyclophosphamide
Third Line Tofacitnib Leflunomide Dapsone Thalidomide Cyclosporine Cyclophosphamide
Rituximab Leflunomide Cyclosporine Cyclophosphamide
Rituximab Leflunomide Cyclosporine Cyclophosphamide
10
Scalp pruritus may improve with topical steroid solutions or oils, although systemic agents are often needed in severe cases. Topical calcineurin inhibitors such as tacrolimus 0.1% ointment or pimecrolimus 1% cream may have modest efficacy in some cases of cutaneous DM.138-142
In practice, these agents have comparable efficacy to low- to midpotency topical corticosteroids (class IV to class VI). They do offer the benefit of being used safely on the face without concern for atrophy or hypopigmentation.
SYSTEMIC THERAPY
SYSTEMIC THERAPY
Systemic corticosteroids are undesirable agents as monotherapy for cutaneous DM because they usually elicit only partial responses and are associated with long-term side effects. However, systemic corticosteroids are first-line therapy in the treatment of myositis.143 Complete clinical responses in muscle inflammation with prednisone monotherapy at doses greater than 0.5 mg/kg/day have been achieved in 27%144 to 87%11 of patients with DM.145 They can also be used for joint disease and ILD, although often a corticosteroid-sparing agent is required. The addition of corticosteroid-sparing agents may improve control of myositis and extracutaneous manifestations, but their critical role is to minimize toxicities of oral corticosteroids, including the risk for corticosteroid-induced myopathy.146
Antimalarials are typically regarded as first-line agents for skin disease. These agents have modest benefit for skin disease in DM with retrospective studies suggesting that improvement is seen in approximately 30% to 50% of patients.147 In addition, up to 30% of patients with DM may experience a drug eruption on initiation of hydroxychloroquine.148 The addition of quinacrine to hydroxychloroquine or chloroquine may be more effective than a single agent.149 Hydroxychloroquine may also be useful for mild symptoms of inflammatory arthritis, and chloroquine has been reported to ameliorate arthritis in DM in one case report.150
Methotrexate is effective in significantly reducing cutaneous disease severity in 50% to 100% of patients with DM.151-154 Similarly, methotrexate is a first-line treatment for myositis in combination with prednisone, and often dosages of 20 to 25 mg/wk are typically necessary to control muscle inflammation.68,155 Methotrexate is also an effective treatment choice when concomitant arthritis is present.156 In patients with DM with suspected or diagnosed ILD, it is prudent to select a different agent than methotrexate because of its potential to induce acute pneumonitis and pulmonary fibrosis,157,158 thereby complicating the evaluation and management of ILD. Mycophenolate mofetil has been shown to be effective at dosages of 2 to 3 g/day in reducing cutaneous disease severity159,160 and myositis.161-163 It is considered a first-line oral agent when ILD is present.164-168 About 20% of patients experience nausea or diarrhea
1077
10
at 2 g/day.169 If GI side effects are dose limiting, then switching to enteric-coated mycophenolate sodium is an option for maintaining a patient on therapy.170
Intravenous immunoglobulin (IVIG) is most likely the single most effective agent for cutaneous DM, with consistently 70% to 80% of patients achieving an almost complete or complete response.171,172 IVIG is also effective for myositis.173,174 A randomized placebo-controlled crossover trial of 15 patients with DM by Dakalas and coworkers in 1993175 showed a significant improvement in muscle strength in 9 of 12 patients (75%) and a dramatic improvement in skin disease based on clinical photographs in 8 of 12 patients (67%) who received IVIG. The standard dosing regimen is 2 g/kg/mo divided over 3 to 5 days. The therapeutic effect may be perceived as early as 1 week, but it may not be apparent until the second or third month. Because IVIG works relatively quickly, it can be used in patients who are rapidly declining or are acutely ill with dysphagia or respiratory muscle involvement. Headaches may occur in up to 56% of patients and may be severe and debilitating. The rate of infusion, the total dose,176 the formulation of IVIG,177 and the volume status of the patient may influence the occurrence of headache. Aseptic meningitis is a rare adverse event manifesting as fever, headache, photophobia, meningismus, neutrophilic pleocytosis, or eosinophilia in the cerebrospinal fluid.178,179 Anaphylaxis is also rare but may occur in primary IgA deficiency; therefore, checking serum IgA levels before infusion is recommended. However, there is no evidence that having low but detectable immunoglobulin levels confers any increased risk for anaphylaxis.176 Also, venous thrombosis and renal injury179 are risks of IVIG, particularly among patients with preexisting thrombophilias or chronic kidney disease, respectively. Azathioprine has not been assessed specifically for cutaneous DM. In combined studies of DM and polymyositis, azathioprine has shown efficacy in improving myositis in up to 75% of cases180-182 and improving survival.183 The first randomized controlled trial of 28 patients with polymyositis or DM compared prednisolone with azathioprine 2.5 mg/kg/day versus prednisolone with methotrexate 15 mg/wk and found no difference in efficacy for myositis.187 The second randomized controlled crossover trial of 30 patients with polymyositis or DM showed improved response in the group receiving combination oral methotrexate and azathioprine (8 of 15; 53%) compared with methotrexate alone (3 of 15; 20%).185 The authors occasionally combine low dose azathioprine with methotrexate when myositis is persistent with methotrexate alone. Azathioprine is commonly used as maintenance therapy in the treatment of ILD associated with idiopathic inflammatory myopathies,47,60 typically after induction with cyclophosphamide.186,187
Rituximab has mixed results in cutaneous DM with a clinical trial reporting no benefit188 one report of moderate improvement in cutaneous DM.189 Rituximab can be of benefit for myositis, especially those with anti–Jo-1 and anti–Mi-2 autoantibodies.190 A
1078
large randomized clinical trial did not show benefit with rituximab, although this may center around difficult issues with trial design.191 Rituximab has been reported to be successful in retrospective studies for the treatment of ILD.192 Dosing in DM typically follows the rheumatoid arthritis protocol of 1000 mg intravenously on days 0 and 14.188 Infectious complications are the most frequent193 serious adverse effects in patients with DM, with rare reports of progressive multifocal leukoencephalopathy.193-195
Calcineurin inhibitors (eg, cyclosporine and tacrolimus) are not commonly used to treat muscle or skin inflammation, although a randomized clinical trial of 36 patients with DM (n = 20) or polymyositis (n = 16) comparing cyclosporine (3–3.5 mg/kg/day) with methotrexate (7.5–15 mg/wk) in addition to oral corticosteroids found decreases in creatine kinase, and improvements in strength were comparable between the groups at 6 months.196 Its primary use is in the setting of ILD, especially severe ILD in the setting of anti-MDA5 or antisynthetase antibodies.197,198
Nephrotoxicity risk is highest above 3 mg/kg/day and requires careful monitoring of renal function and blood pressure.199,200
Other treatments reported for use in DM skin disease include dapsone and thalidomide, but evidence of their efficacy is scant.
PHYSICAL MEDICINE AND REHABILITATION
PHYSICAL MEDICINE AND
REHABILITATION
Strength training has been shown to improve muscle strength and function,201-203 and aerobic exercise has been shown to improve endurance204,205 in patients with chronic DM. In active DM and polymyositis, a home strength training program has been shown to be safe206 but did not show benefit in strength or disease control over the control group that performed range of motion exercises in a randomized clinical trial.207 We advise all patients to enroll in a physical therapy program early after diagnosis so as to prevent injury and to maintain as much mobility and muscle strength as possible.
SPECIAL CIRCUMSTANCES
SPECIAL CIRCUMSTANCES
Calcinosis remains one of the most formidable therapeutic challenging in DM. Surgical excision for localized lesions remains the most effective and definitive therapy.208 Multiple medical therapies have been proposed, including anti-inflammatory medications and calcium and phosphate modulators, but no single agent is reliably effective.31 IVIG has been reported to work well in some cases209-211 but not in others.212 Bisphosphonates have been cited as effective for calcinosis in juvenile DM, although controlled studies are needed.213-215
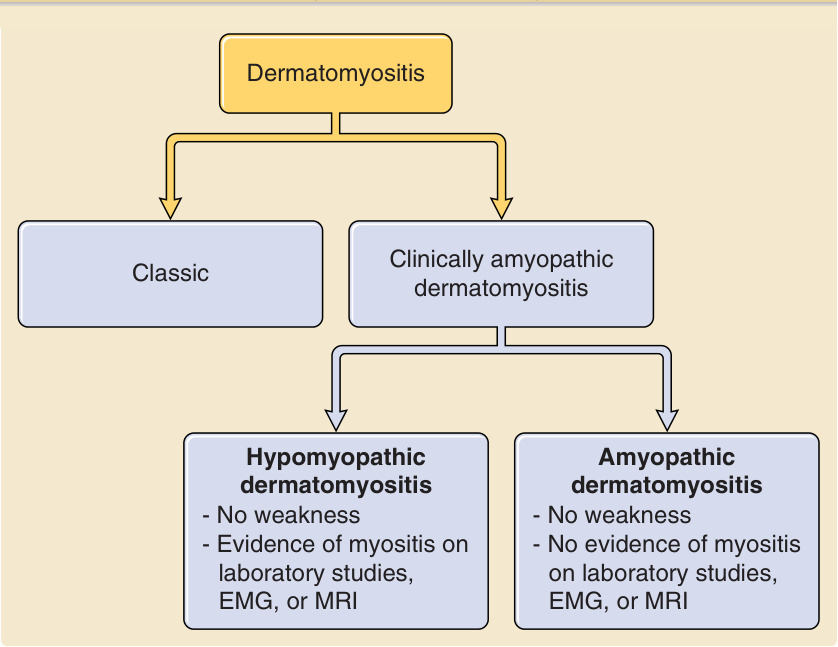
Figure 62-1 Display of the relationships among classic, clinically amyopathic, hypomyopathic, and amyopathic dermatomyositis. EMG, electromyography; MRI, magnetic resonance imaging.
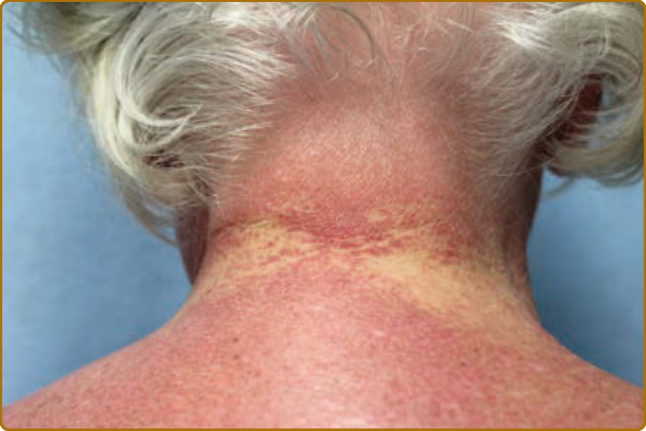
Figure 62-2 Diffuse pink psoriasiform plaques on the posterior neck and nuchal scalp.
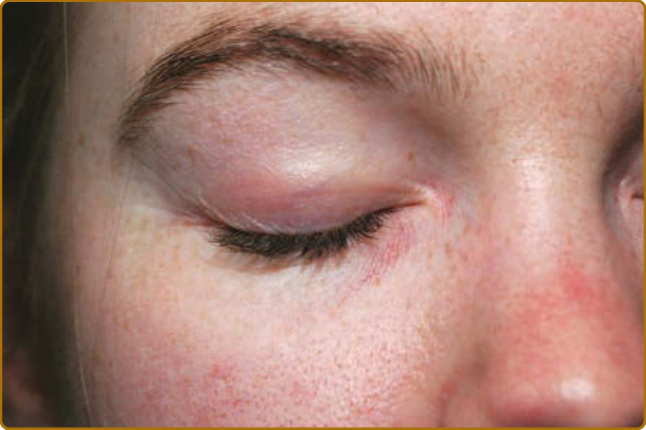
Figure 62-3 Heliotrope sign. Violaceous to pink erythema and edema on the upper eyelid.
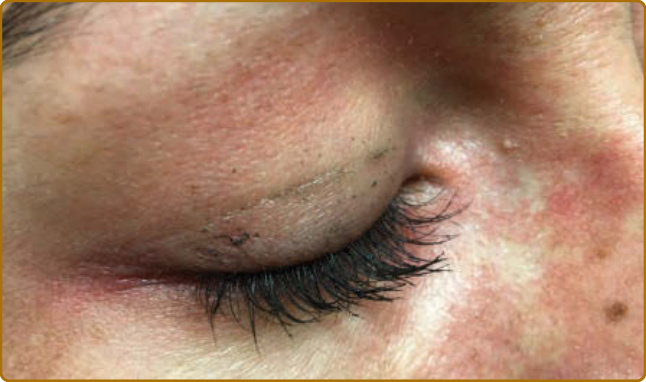
Figure 62-4 Red ill-defined macules on the medial and lateral canthi, often seen in conjunction with the heliotrope sign.
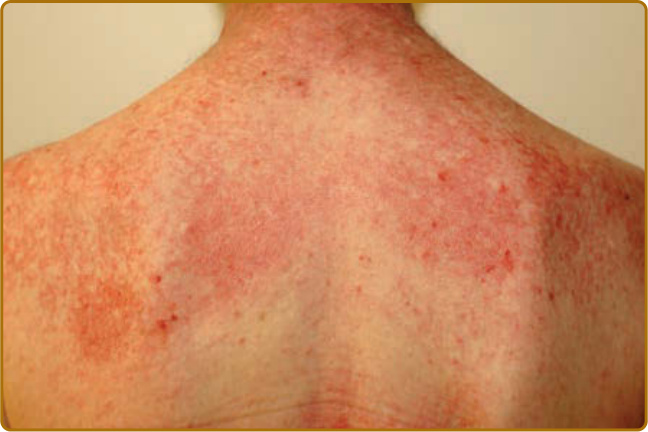
Figure 62-5 Shawl sign. Pink to red thin patches and plaques, often with thin white scale, on the upper posterior back characteristic of the shawl sign.
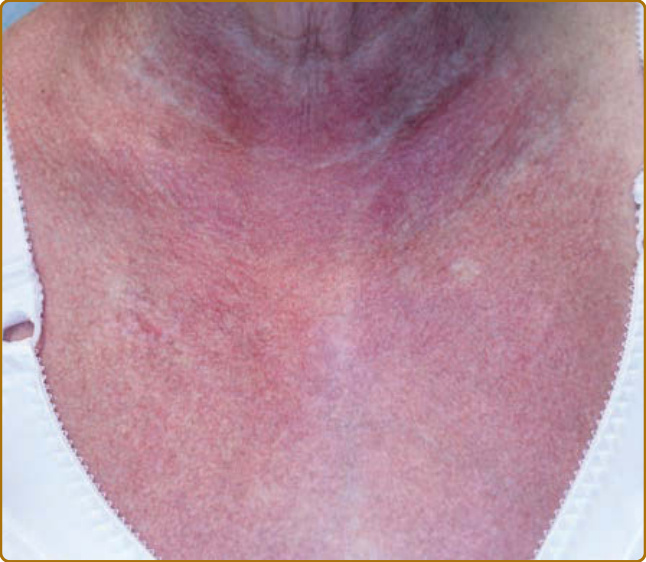
Figure 62-6 V-neck sign. Red, ill-defined telangiectatic patches on the upper chest.
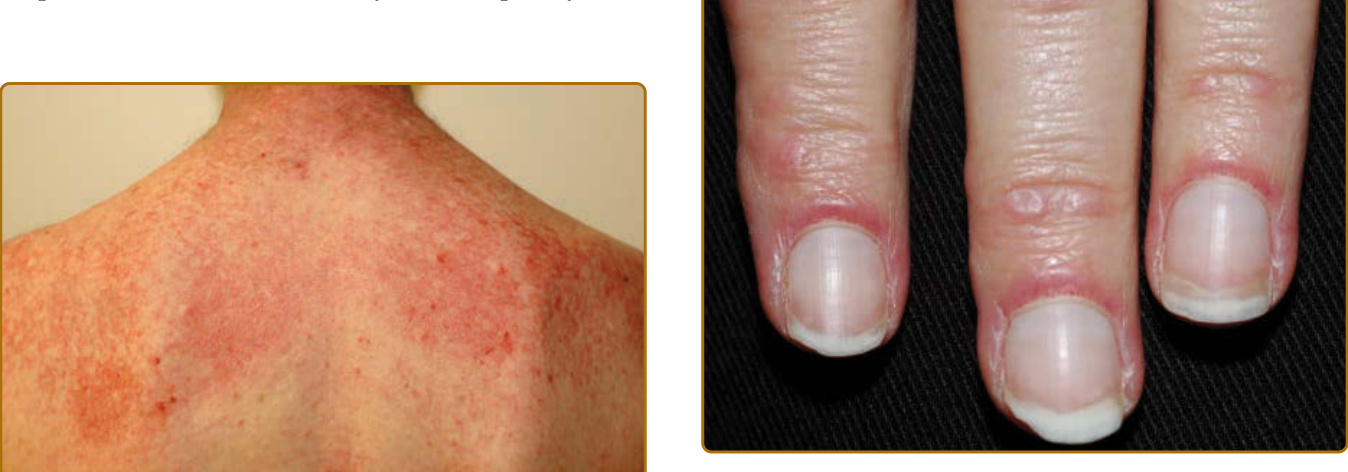
Figure 62-7 Gottron papules. Light pink, ill-defined papules over the proximal and distal interphalangeal joints, few with central umbilication. Deep red erythema, edema of the proximal nailfolds, and dilated nailfold capillaries are evident.
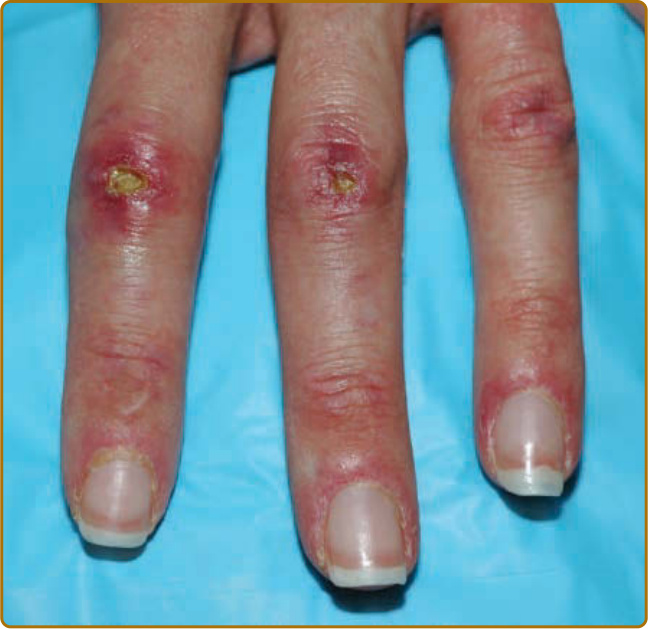
Figure 62-8 Ulcerated Gottron papules. Well-demarcated 3- to 4-mm ulcers with surrounding erythema and edema on the second and third proximal interphalangeal joints exemplify the vasculopathy seen in dermatomyositis.
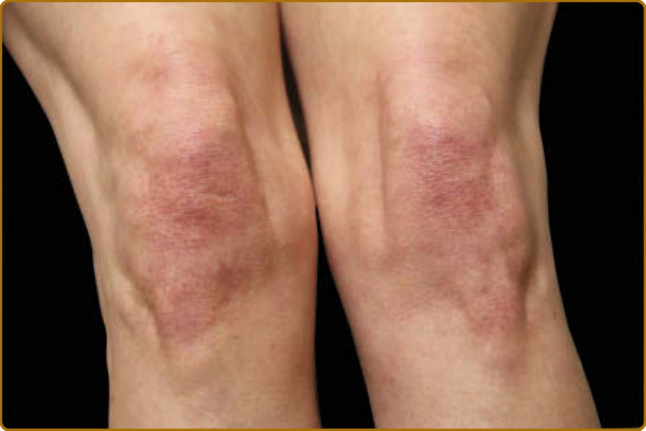
Figure 62-9 Gottron sign. Ill-defined violaceous erythema on the knees is seen, which has follicular accentuation in this patient.
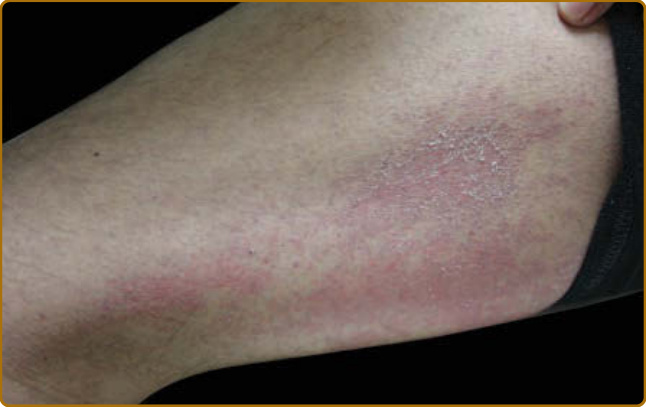
Figure 62-10 Holster sign. On the lateral thigh are illdefined pink papules coalescing into plaques are seen with scale and follicular accentuation.
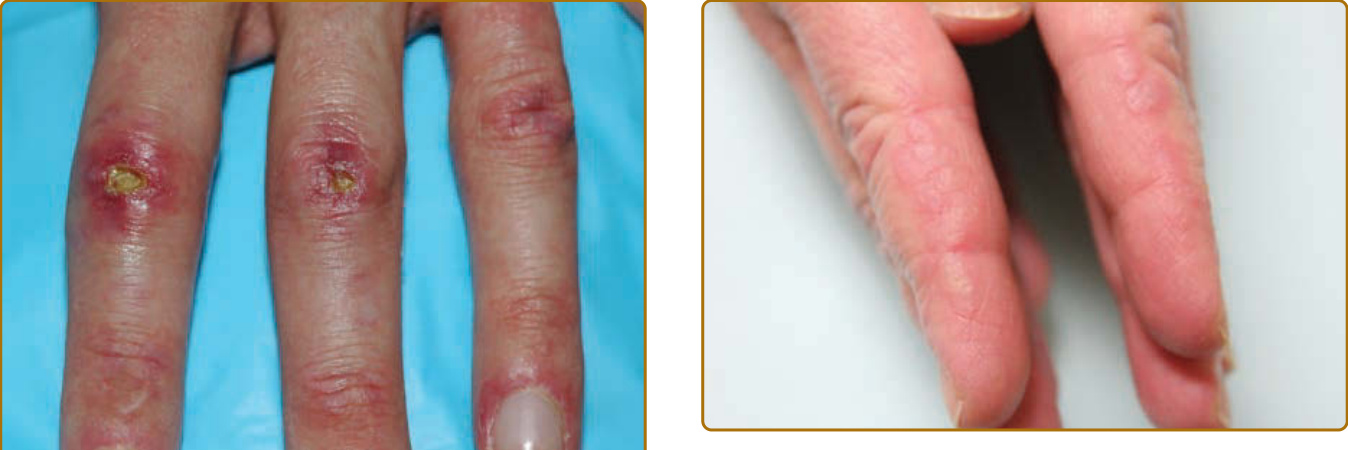
Figure 62-11 Mechanic’s hands. On the lateral second digits are hyperkeratotic pink ill-defined papules with scale.
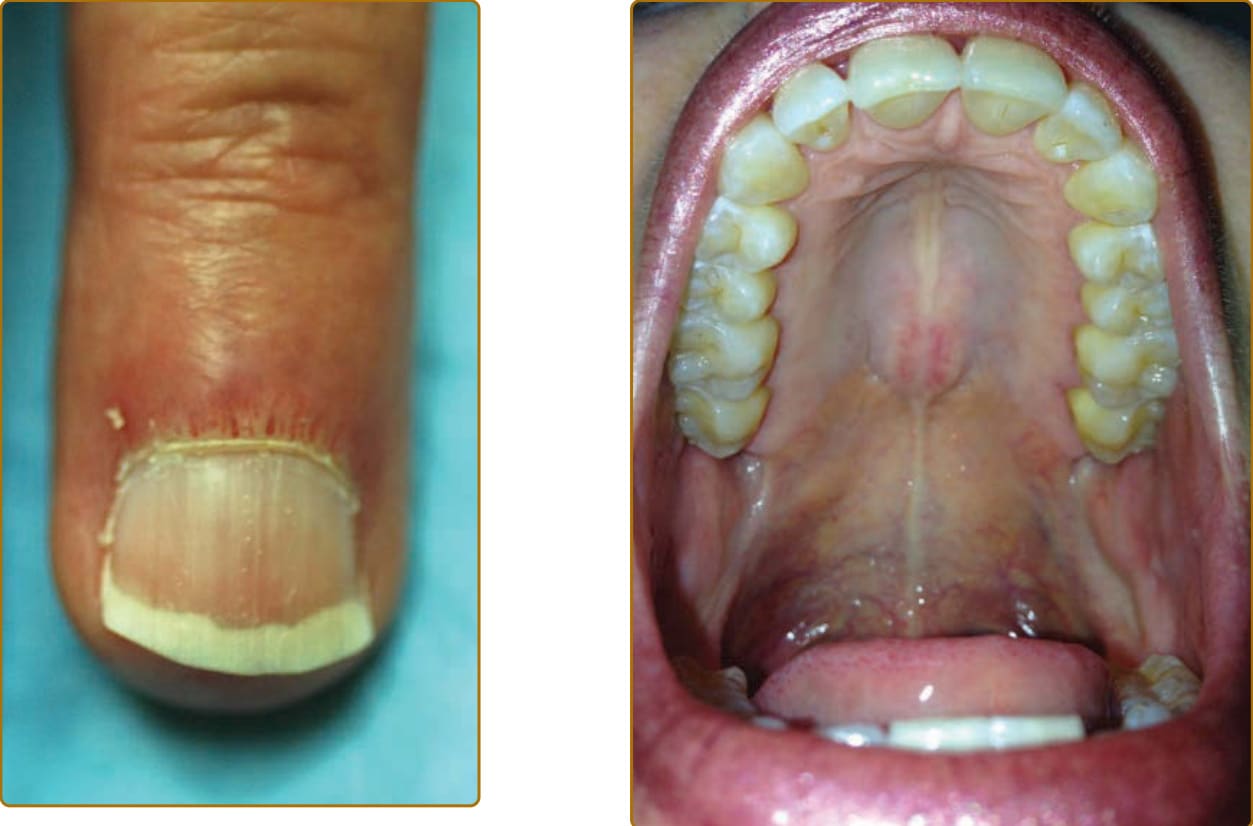
Figure 62-12 Dilated proximal nailfold capillary loops with intervening avascular areas.
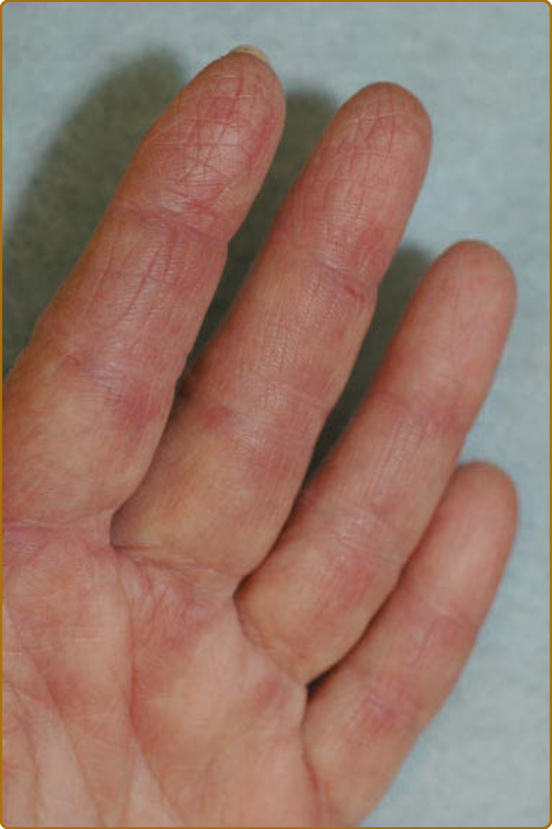
Figure 62-14 A reticular pattern of deep red erythema on the palmar fingers is more commonly seen in the anti– melanoma differentiation–associated gene 5 (MDA5) dermatomyositis group.
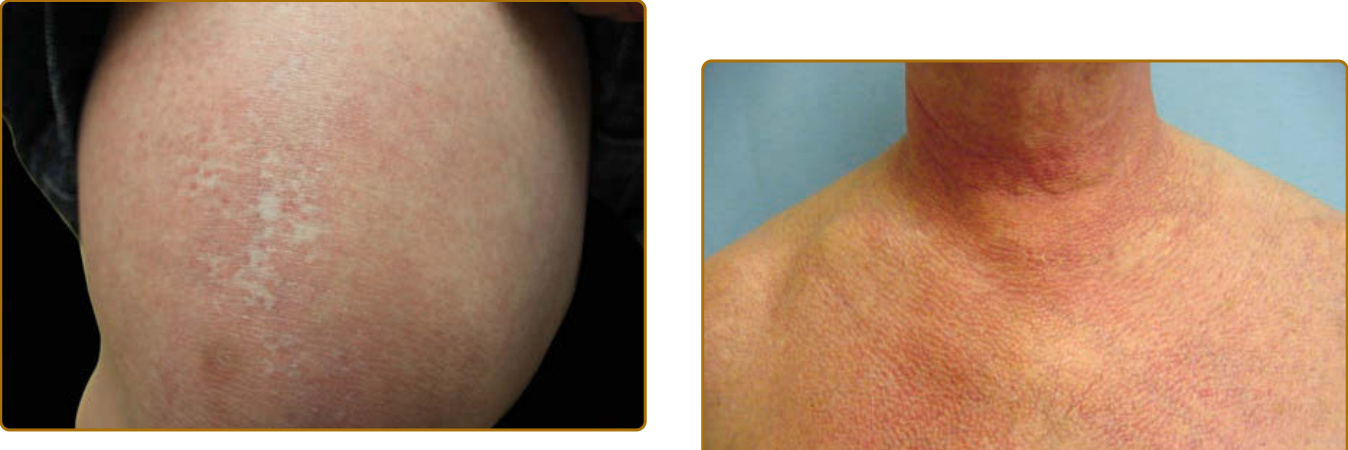
Figure 62-15 “Red on white.” Reticulated white macules surrounded by telangiectatic red macules are seen on the distal thigh and knee. This cutaneous finding is specific to dermatomyositis and is valuable in differentiating dermatomyositis from cutaneous lupus.
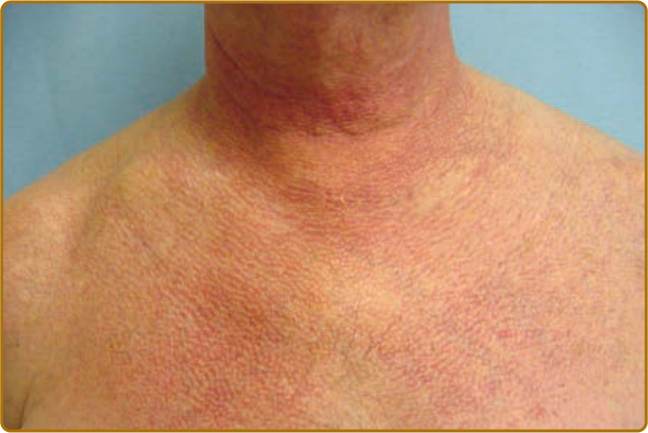
Figure 62-16 Poikiloderma. This triad of hyperpigmentation, hypopigmentation, and telangiectasias is a sign of damage, as shown on the chest.
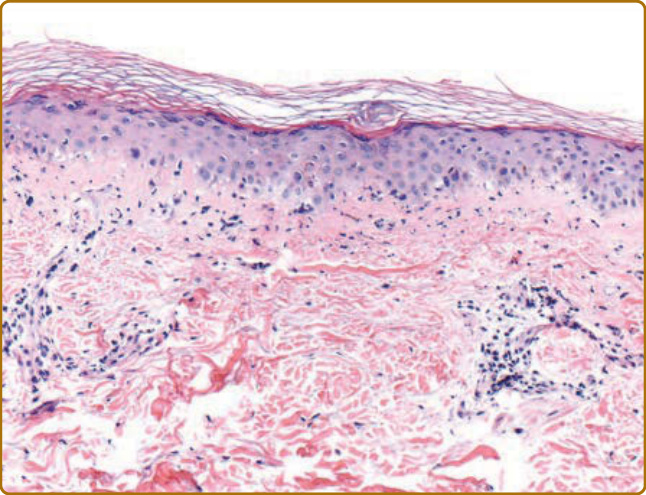
Figure 62-17 Histopathology of cutaneous dermatomyositis. Skin biopsy shows vacuolar interface dermatitis, basement membrane thickening, perivascular lymphocytic infiltrate, and increased dermal mucin.
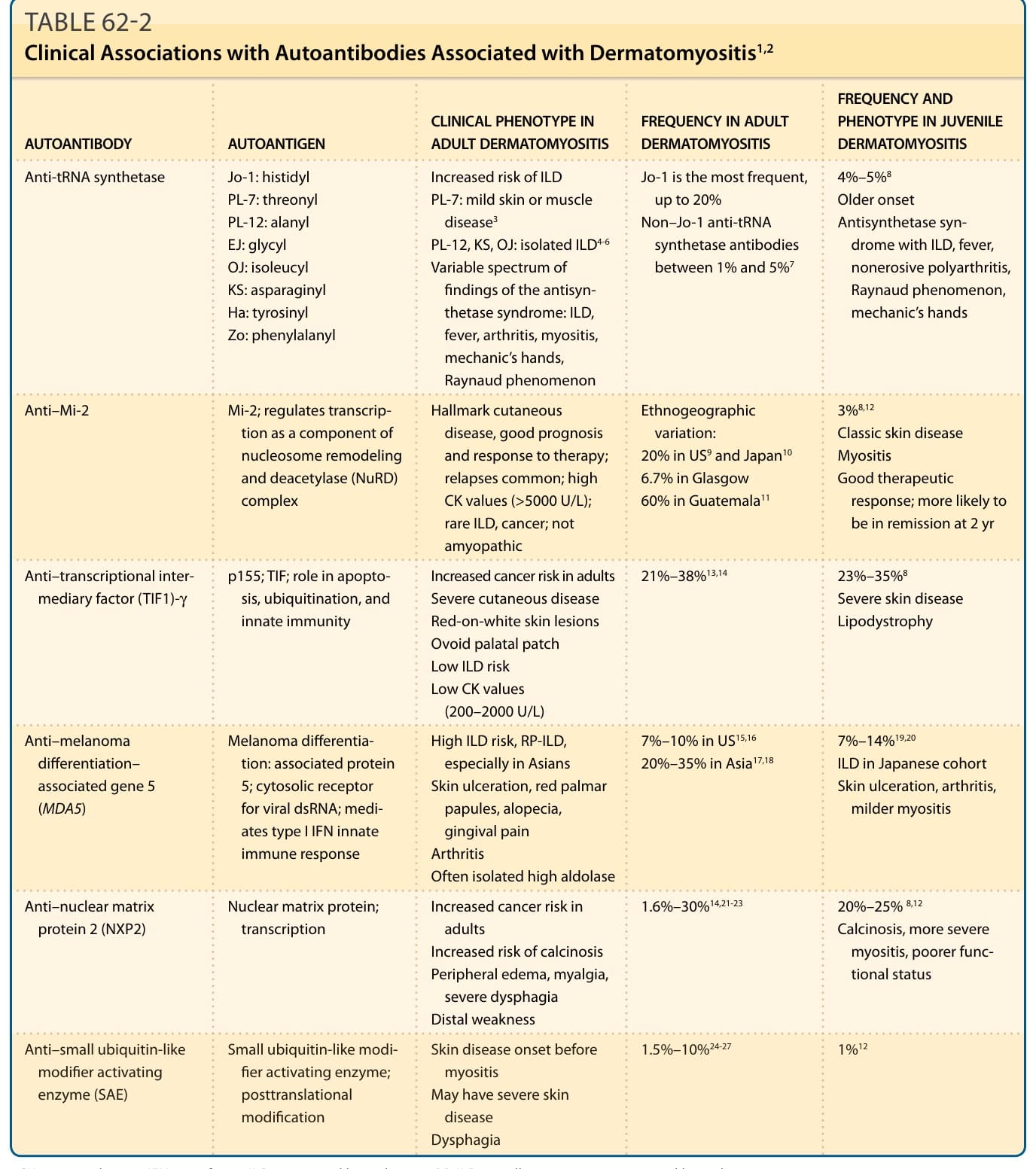
TABLE 62-2 Clinical Associations with Autoantibodies Associated with Dermatomyositis1,2
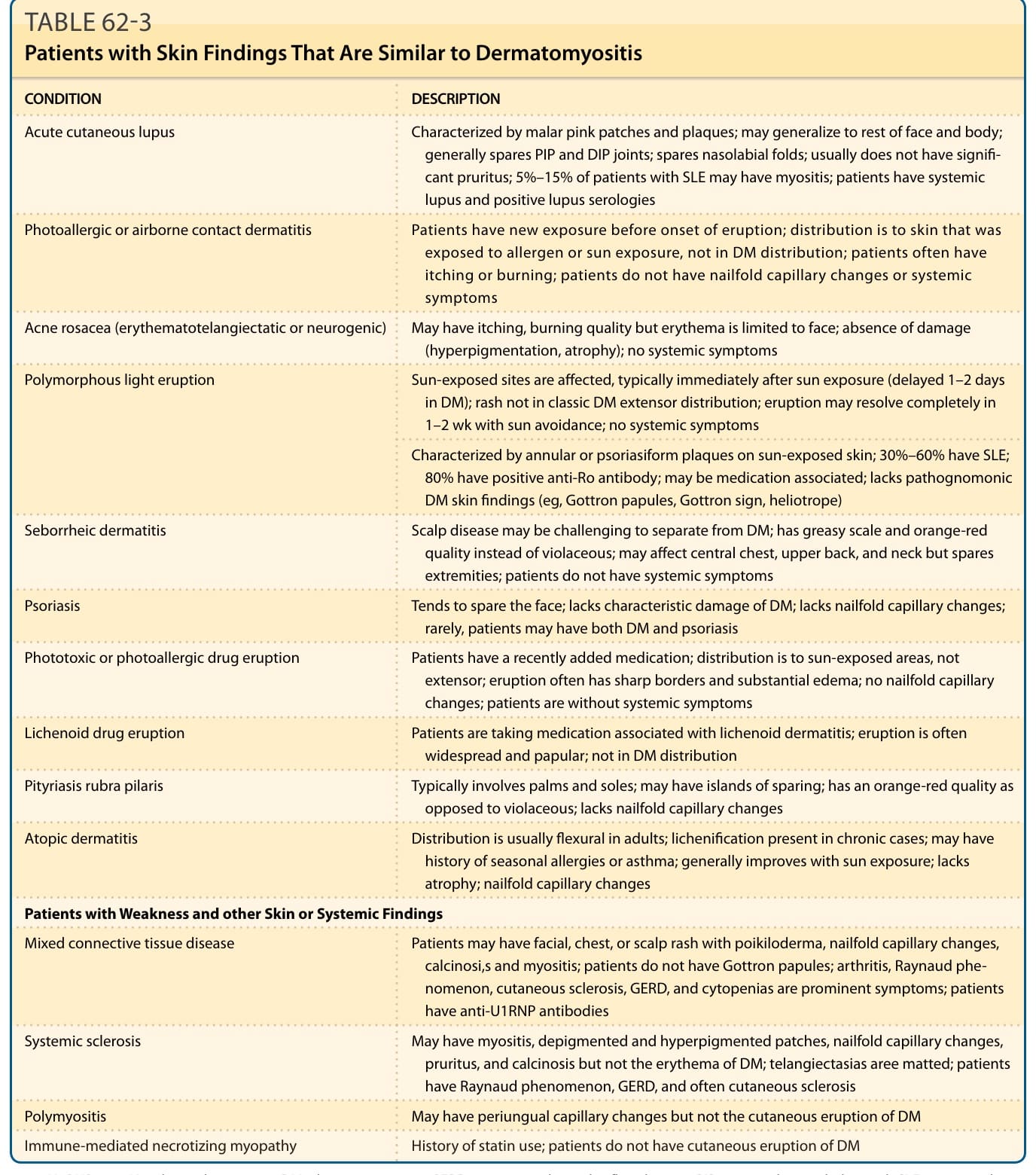
TABLE 62-3 Patients with Skin Findings That Are Similar to Dermatomyositis
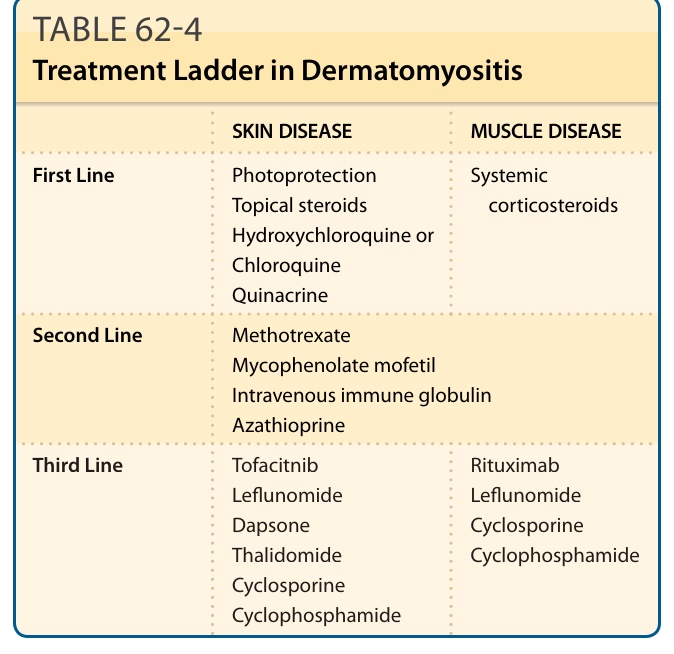
TABLE 62-4 Treatment Ladder in Dermatomyositis