遺傳性表皮鬆解症 (Inherited Epidermolysis Bullosa) 精華筆記
定義與概論
- 遺傳性表皮鬆解症 (inherited epidermolysis bullosa, EB) 是一群遺傳性皮膚病 (genodermatoses),共同特徵為對輕微創傷產生水疱;可表現為小水疱 (vesicles) 或大疱 (bullae),嚴重型亦侵犯黏膜。
- 受侵犯分布、水疱形成深度、皮膚外侵犯與嚴重度,皆隨亞型與潛在分子缺陷而異;異常傷口修復可演變為慢性糜爛、增生性肉芽組織、瘢痕,甚至侵襲性癌。
- 依水疱裂隙平面 (cleavage plane) 的超微結構層次分為:單純型 (simplex)、半橋粒型 (hemidesmosomal)、接合部型 (junctional)、營養不良型 (dystrophic)。
致病機轉 (Pathogenesis)
- EB 源自基底角質細胞 (basal keratinocytes) 與下方真皮附著的缺陷,可發生於角質細胞質膜內或細胞外的真皮-表皮基底膜帶 (BMZ)。
- 由上而下的錨定複合體:角蛋白中間絲 (keratin 5/14) → 半橋粒 (hemidesmosome;含網蛋白 plectin、BP230) → 跨膜的第 XVII 型膠原蛋白 (collagen XVII/BP180) 與 α6β4 整合素 → 錨定絲 (anchoring filaments;含層黏連蛋白-332 laminin-332) 橫跨透明板 (lamina lucida) → 緻密板 (lamina densa) → 含第 VII 型膠原蛋白 (collagen VII) 的錨定纖維 (anchoring fibrils) 延伸入乳頭真皮。
- 各亞型對應特定突變基因/蛋白:EBS 多為 KRT5/KRT14;JEB 多為層黏連蛋白-332 (LAMA3/LAMB3/LAMC2)、collagen XVII (COL17A1)、整合素;DEB 全部源自 COL7A1。
圖 60-1:真皮-表皮基底膜各組分示意圖與其超微結構形態的比較。
分類 (見表 60-1)
- 單純型 (Simplex):表皮內水疱,多與角蛋白基因突變相關。常見三型皆為顯性遺傳——廣泛性重度 (Dowling-Meara)、廣泛性中度 (Koebner)、局部型 (Weber-Cockayne)。
- 接合部型 (Junctional):透明板內水疱,自體隱性遺傳;三主型為廣泛性重度 (Herlitz)、廣泛性中度、局部型。
- 營養不良型 (Dystrophic):緻密板下水疱,可顯性 (DDEB) 或隱性 (RDEB);全與 COL7A1 相關。
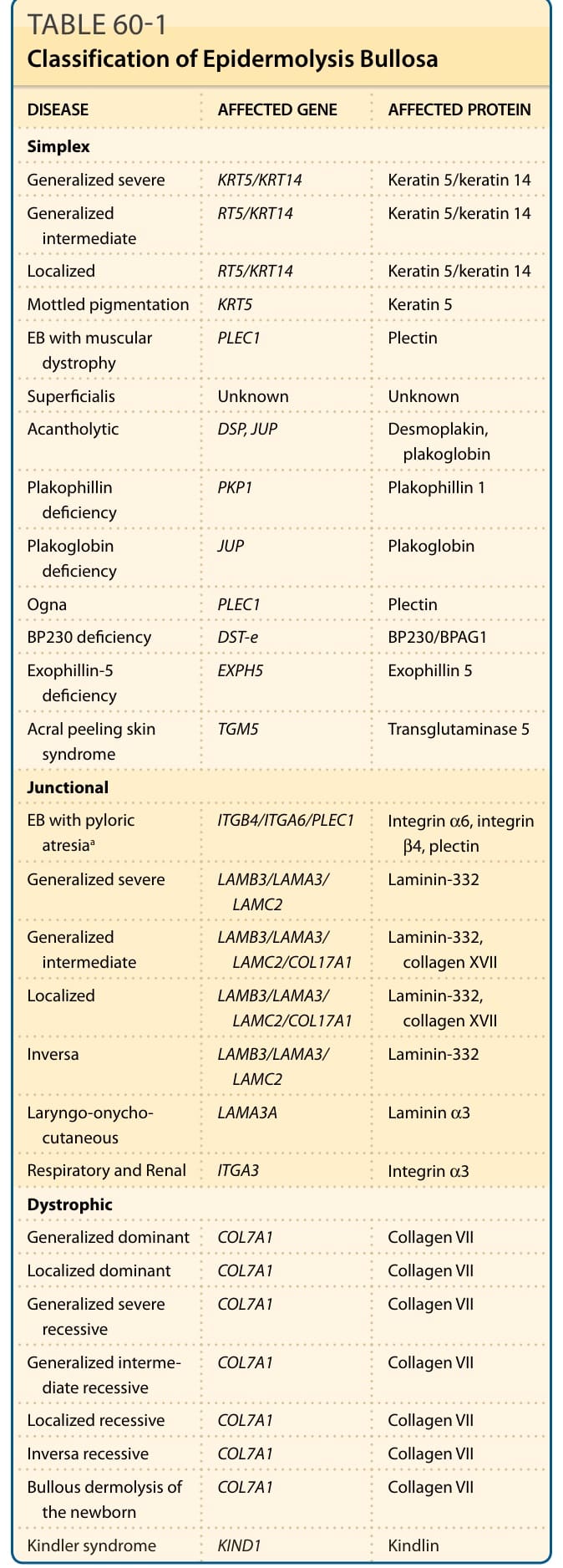
表 60-1:表皮鬆解症的分類(疾病、受影響基因與蛋白)。
臨床表現 — 單純型 EBS
- 廣泛性重度 (Dowling-Meara):出生即廣泛分布,最嚴重;口腔黏膜常受侵、可廣泛糜爛;嬰兒期可有粟粒疹 (milium);軀幹與近端肢體可出現群聚「疱疹樣 (herpetiform)」水疱、癒合不留瘢痕;常有指甲侵犯;掌蹠角化過度 (hyperkeratosis) 自兒童早期出現、可進展為融合性角皮症。與其他亞型不同,熱對本型水疱無重大影響。
- 廣泛性中度 (Koebner):出生時或嬰兒早期廣泛水疱,手、足、四肢最重;以發炎後色素過度沉著/減退癒合,萎縮與粟粒疹罕見;掌蹠角化過度與糜爛可見;通常無嚴重生長遲滯。
- 局部型 (Weber-Cockayne):最常見、最輕;嬰兒期或兒童期發病,偶於成年(如行軍後);掌蹠多汗症 (hyperhidrosis) 常見;通常無粟粒疹與瘢痕;指甲侵犯罕見。
- 其他罕見變異型:Ogna 型 (PLEC1、甲彎曲);伴肌肉失養症 (PLEC1/網蛋白、進行性肌肉失養);伴斑駁性色素沉著 (KRT5);淺表型 (subcorneal 分離);棘層鬆解型 (隱性致死);斑菲素蛋白缺乏/外胚層發育不良-皮膚脆弱症候群 (PKP1);BP230 缺乏 (DST-e);外簇蛋白-5 缺乏 (EXPH5);肢端脫皮症候群 (TGM5)。
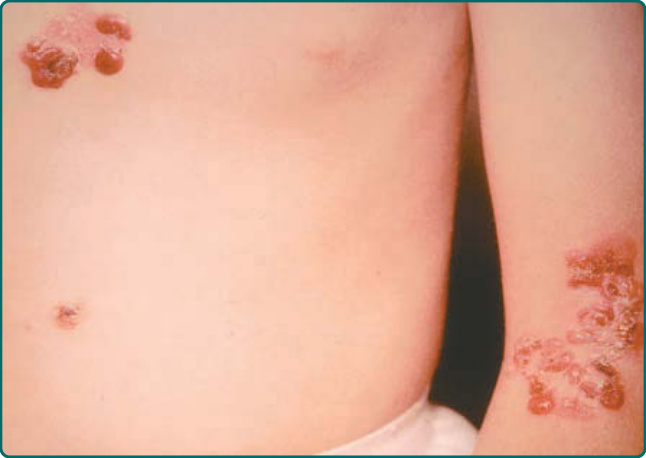
圖 60-3:Dowling-Meara 型 EBS 軀幹與手臂特徵性水疱。
臨床表現 — 接合部型 JEB
- 廣泛性重度 (Herlitz):最嚴重亦最常見,嬰兒期或兒童早期死亡;出生即廣泛大範圍水疱;特徵性孔口周圍肉芽組織 (periorificial granulation tissue,嘴、眼、鼻孔);指甲常於嬰兒期喪失;齒釉質點蝕;口咽與各複層鱗狀上皮(鼻、結膜、食道、氣管、喉、直腸、尿道)糜爛;氣管喉狹窄/阻塞、生長遲滯、混合性貧血、敗血症 (sepsis) 為致死關鍵;嬰兒早期聲音嘶啞為不祥之兆。
- 廣泛性中度:存活過嬰兒期,水疱與口腔糜爛較輕;缺乏明顯嘶啞為有利預後徵象;頭皮與指甲病灶、孔口周圍不癒合糜爛常見。先前稱廣泛性萎縮性良性 EB (GABEB) 者出生即廣泛皮膚侵犯但黏膜疾病少、以萎縮性瘢痕癒合、壽命多正常,特徵為頭皮進行性禿髮。
- 局部型 (minimus):輕微、可局部加重(手、足、脛前);預後良好、壽命正常。
- 伴幽門閉鎖 (pyloric atresia):極度黏膜與皮膚脆弱,可伴泌尿異常(腎水腫、腎炎);ITGB4/ITGA6 突變;多於嬰兒期致死。
- 伴呼吸與腎臟侵犯 (ITGA3):皮膚水疱輕,但出生即嚴重間質性肺病與腎臟表現(腎衰竭、腎病症候群),嬰兒期死亡。
- 喉-甲-皮症候群 (LAMA3):指甲營養不良、皮膚糜爛、結膜與喉廣泛肉芽組織。
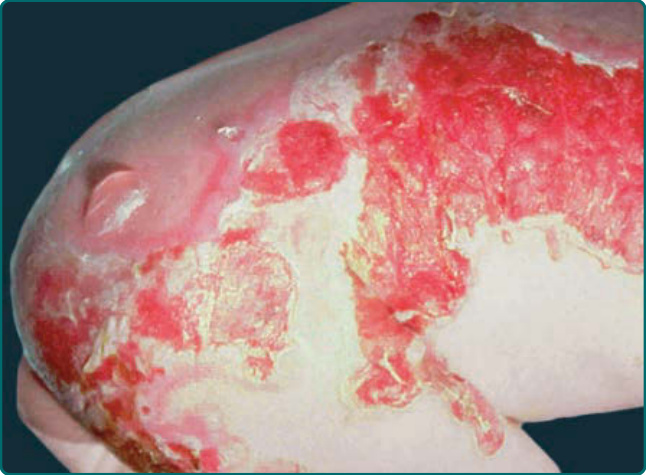
圖 60-6:Herlitz 接合部型 EB 嬰兒出生時的廣泛性水疱。
臨床表現 — 營養不良型 DEB
- 共同特徵:水疱以瘢痕與粟粒疹癒合;源自錨定纖維缺陷導致緻密板下分離。隱性 (RDEB) 與侵襲性鱗狀細胞癌 (SCC) 風險增加相關,顯性 (DDEB) 則否。
- 局部型 DDEB (Cockayne-Touraine):水疱局限於反覆創傷處(膝、薦部、肢端),瘢痕常增生性;指甲營養不良常見;預後良好、壽命正常。
- 廣泛性 DDEB (Pasini):出生即較廣泛;軀幹可出現白丘疹樣 (albopapuloid) 病灶(非特異性)。自發緩解的新生兒大疱性真皮鬆解 (bullous dermolysis of the newborn) 為罕見變異。
- 局部型 RDEB (mitis):肢端分布、黏膜侵犯輕。
- 重度 RDEB (Hallopeau-Siemens):毀滅性;出生即廣泛水疱,偶有整片皮膚先天缺如 (Bart 症候群);進行性瘢痕;「連指手套 (mitten)」狀假性併指 (pseudosyndactyly)、手與肢體屈曲攣縮;頭皮最常受侵(非孔口周圍);口咽廣泛糜爛致小口症與舌繫帶過短;食道狹窄、營養不良、生長遲滯、貧血與鐵吸收缺陷。青春期後常出現高度侵襲、易轉移的 SCC。
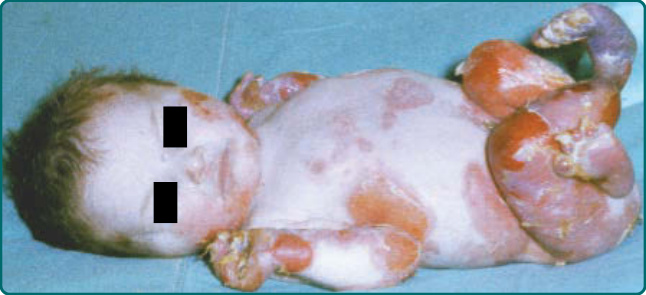
圖 60-12:隱性營養不良型 EB 病人出生時廣泛水疱伴局部皮膚缺如。
病程與預後
- 較輕亞型(局部型 EBS、局部型 JEB、局部型 DDEB)壽命正常、極少內部侵犯。
- 最嚴重隱性型(Herlitz JEB、JEB 伴幽門閉鎖、重度 RDEB)為毀損性、多器官、威脅生命。
- 重度 RDEB:估計百分之五十至百分之八十的病人最終發展出 SCC,許多人死於轉移性疾病;RDEB 相關 SCC 極具侵襲性、強烈侵犯與轉移傾向。
Kindler 症候群
- 與 EB 相關,KIND1 基因 (kindlin-1) 突變;出生與嬰兒期創傷誘發水疱、萎縮性癒合,兒童期後讓位給日曬部位的進行性異色症 (poikiloderma) 與光敏感性;超微結構特徵為基底膜重複 (reduplication)。
診斷
- 始於詳盡病史(水疱發病年齡、家族史)與理學檢查(皮膚、黏膜、毛髮、指甲、牙齒),並評估貧血與營養指標(血清白蛋白 serum albumin)。
- 常規組織學不能診斷 EB,僅能排除其他疾病;BMZ 太小無法以光學顯微鏡可視化。
- 須以穿透式電子顯微鏡 (TEM) 或間接免疫螢光顯微鏡 (IDIF) 判定分離層次。切片務必取最新鮮水疱(可用鉛筆橡皮擦在完整皮膚旋轉誘發),打孔器約 10% 覆蓋水疱、90% 覆蓋完整皮膚。
- TEM 為黃金標準:角蛋白中間絲叢集為 Dowling-Meara EBS 的特異性病徵;發育不全半橋粒提示 JEB;錨定纖維缺如/改變見於 DEB(尤隱性型)。
- IDIF 免疫定位以抗體組套(層黏連蛋白-332、collagen XVII、collagen VII、α6/β4 整合素、網蛋白、角蛋白 5/14)判讀抗原於水疱頂或底,指示分子缺陷;基因突變分析(血液或頰拭子,含父母與手足)為最終診斷步驟,全外顯子定序 (whole-exome sequencing) 漸普及。
鑑別診斷 (見表 60-2)
- 最可能:汗皰疹、蟲咬、摩擦性水疱、熱燒傷、大疱性膿痂疹。
- 應考慮:兒童慢性大疱性皮膚病(線狀 IgA 病)、大疱性類天疱瘡、後天性表皮鬆解症、大疱性紅斑性狼瘡、瘢痕性類天疱瘡、尋常性天疱瘡。
- 務必排除:史蒂芬斯-強生症候群、毒性表皮壞死溶解症。
遺傳諮詢
- DNA 突變分析可區分水疱活性相當但 SCC 風險迥異的隱性與顯性 DEB。
- 產前診斷可藉妊娠 8 至 10 週絨毛膜絨毛採樣或第二孕期羊膜穿刺,取代風險較高的胎兒皮膚切片/胎兒鏡;體外受精結合胚胎著床前診斷已成功施行。
治療
- 大多為支持性,目標為避免創傷;傷口癒合受異物、細菌、營養缺乏、貧血與反覆創傷影響。
- 支持性皮膚照護:改良型 Dakin 溶液(0.025% w/v 次氯酸鈉 sodium hypochlorite)減少細菌負荷;換藥前浸泡 20 分鐘鬆開黏附敷料;之後敷莫匹羅星 (mupirocin) 等局部抗生素並以半閉合性非黏性敷料覆蓋;以自黏紗布/紙固定(勿用膠帶);EBS 病人控制熱暴露、穿柔軟通風鞋;DEB 病人手指夾板與手部防護。
- 感染:常見金黃色葡萄球菌、化膿性鏈球菌,亦可綠膿桿菌;皮膚培養與適當全身性抗生素;漩渦浴、每日換藥、稀釋氯浴、輪替局部抗生素、碘-優碘消毒。
- 外科:重度 RDEB (Hallopeau-Siemens) 最需介入;連指手套樣假性併指可手術鬆解但易復發、術後夾板固定關鍵;亦矯正攣縮畸形;插管須極小心減少口腔黏膜創傷。
- 腫瘤:隱性 DEB 青春期後常見 SCC、可多原發、好發不癒合區,須密切監測。外科切除(Mohs 或非 Mohs)為第一線、放療為輔;異維 A 酸 (isotretinoin) 用於 RDEB 化學預防(耐受良好但對整體存活率不明);西妥昔單抗 (cetuximab) 有些益處但皮膚副作用。
- 胃腸道:食道狹窄以擴張術治療(易復發),晚期可結腸間置術(少用)、胃造口管供營養;纖維與糞便軟化劑治便秘。
- 眼部:支持性治療角膜糜爛(抗生素藥膏、睫狀肌麻痺劑、保濕腔室、潤滑劑);嚴重上眼瞼可全層皮膚移植;須眼科協助。
- 口咽:最柔軟牙刷、生理食鹽水沖洗、避免含酒精漱口水;重度 DEB 可小口症與舌繫帶過短;氣道侵犯有吸入危險,插管須由具經驗麻醉科醫師以小口徑氣道輕柔進行。
- 營養、貧血與心血管:熱量與蛋白質需求增加、進食受口咽與 GI 病灶限制;慢性病貧血影響所有重度亞型,隱性 DEB 缺鐵常嚴重、可需非經腸鐵劑、必要時重組紅血球生成素 (erythropoietin)、輸血;擴張型心肌病為與慢性貧血高度相關的致命併發症。
- 心理層面:慢性疼痛、憂鬱風險;支持性心理治療與病友團體。
- 抗發炎治療:局部皮質類固醇短期止癢但長期致皮膚萎縮、惡化水疱;四環黴素與苯妥英已非適應治療;雙醋瑞因 (diacerein,大黃根成分、介白素-β 阻斷劑) 耐受良好並減少發炎性重度 EBS 水疱;洛沙坦 (losartan) 在臨床前研究減少 DEB 傷口纖維化(降低 TGF-β 表現)。
- 同種異體細胞治療:同種異體角質細胞替代物、纖維母細胞注射、間質幹細胞輸注僅短期有效;對七名隱性 DEB 兒童施行同種異體骨髓移植,部分有臨床益處與第 VII 型膠原蛋白染色持續≥1 年,但錨定纖維恢復不完全,且手術死亡率約百分之三十。
- 自體基因治療:反轉錄病毒體外基因矯正自體角質細胞已用於 JEB(一例移植後 6.5 年仍表現層黏連蛋白-332 並抗水疱;另一例經基因矯正表皮移植物覆蓋超過百分之八十皮膚表面);四名隱性 DEB 病人以全長 COL7A1 轉移之角質細胞移植,恢復第 VII 型膠原蛋白與錨定纖維達 1 年。CRISPR/Cas9 基因編輯結合誘導性多能幹細胞、外顯子跳躍 (exon skipping)、擴展自我矯正突變皮膚等為未來方向。
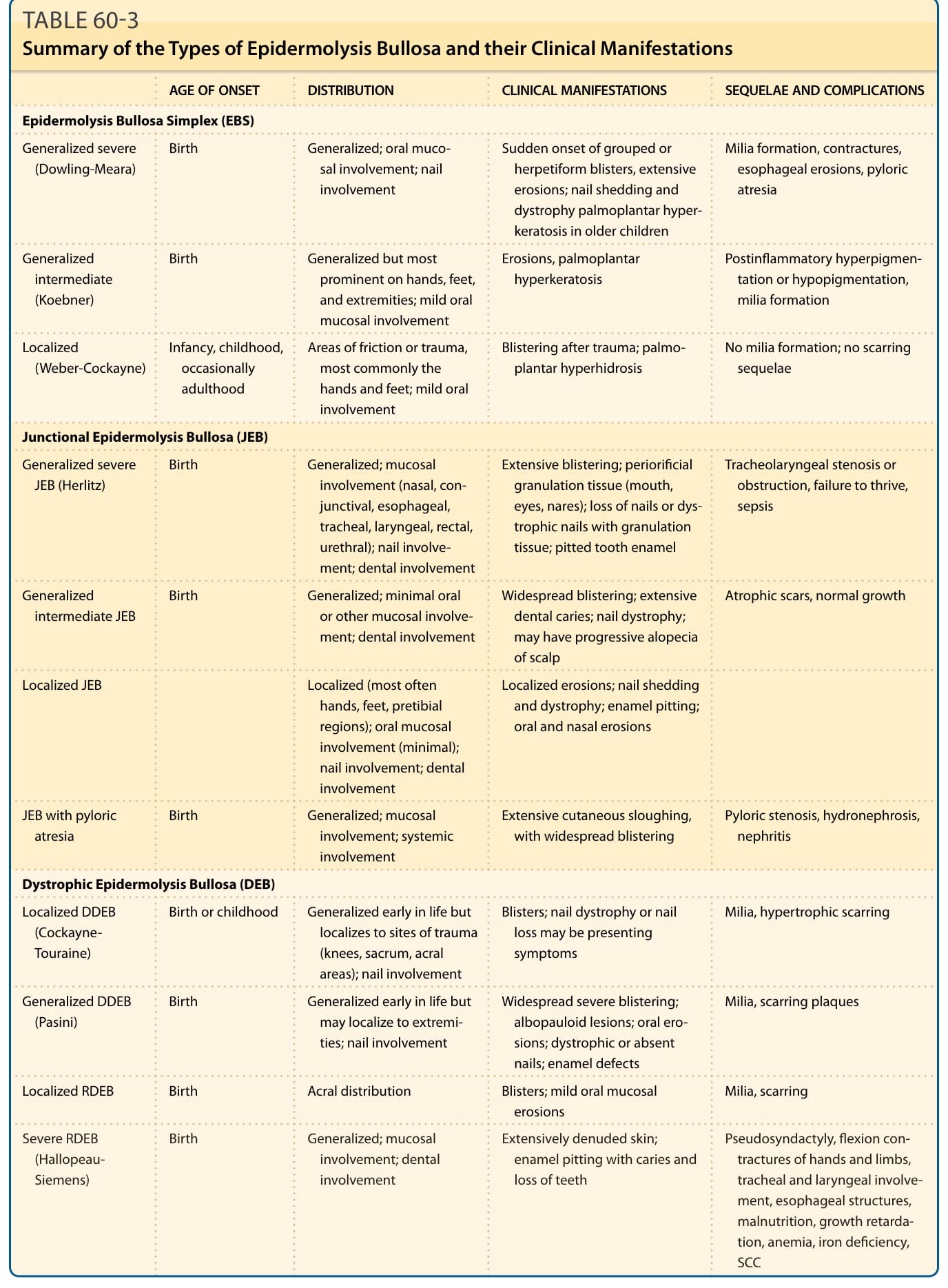
表 60-3:表皮鬆解症各類型及其臨床表現(發病年齡、分布、後遺症與併發症)摘要。