遺傳性表皮鬆解症 (Inherited Epidermolysis Bullosa)
PART 9
水疱大疱性疾病 (Vesiculobullous Disorders)
重點一覽 (AT-A-GLANCE)
■ 表皮鬆解症 (Epidermolysis bullosa) 是一群遺傳性遺傳性皮膚病 (genodermatoses),特徵為對輕微創傷產生水疱。
■ 依水疱形成的層次可分為單純型 (simplex)、半橋粒型 (hemidesmosomal)、接合部型 (junctional) 與營養不良型 (dystrophic) 亞型。
■ 皮膚侵犯依亞型不同,從局部到廣泛的水疱皆有。
■ 皮膚外侵犯依亞型不同,從無侵犯到嚴重失能或致命皆有。
■ 依亞型不同,口咽 (oropharynx)、氣管 (trachea)、食道 (esophagus)、眼睛、牙齒、指甲、毛髮皆可能受侵犯。
■ 診斷藉由免疫螢光顯微鏡 (immunofluorescent microscopy) 或電子顯微鏡 (electron microscopy),繼而進行 DNA 分析來確立。
前言 (INTRODUCTION)
遺傳性表皮鬆解症(inherited epidermolysis bullosa, EB)是一群疾病,其共同特徵為對輕微創傷產生水疱。EB 病人的水疱可表現為小水疱 (small vesicles) 或較大的大疱 (larger bullae),發生於皮膚表面,而在嚴重型中亦發生於黏膜組織上。雖然皮膚與黏膜的脆弱性 (fragility) 以及創傷誘發的疼痛性水疱是整個 EB 疾病譜的標誌,但受侵犯的分布、水疱形成的深度、任何相關的皮膚外侵犯,以及水疱過程的嚴重度,皆隨不同 EB 亞型而異,並取決於潛在的可遺傳分子缺陷。EB 疾病在水疱區域癒合的方式上也有所不同。其傷口修復反應 (wound repair responses) 常為異常,並可能演變為慢性糜爛 (chronic erosions)、增生性肉芽組織 (hypertrophic granulation tissue)、瘢痕形成 (scarring),甚至侵襲性癌 (invasive carcinoma)。雖然較輕微的 EB 亞型伴隨正常壽命且極少或無內部侵犯,但最嚴重的隱性遺傳型則是會造成毀損性、多器官的疾病,威脅生命的品質與長度。
數個早期研究辨識出 EB 的主要亞型。von Hebra 的研究¹⁻³ 最先區分了天疱瘡 (pemphigus) 與遺傳性水疱,而「epidermolysis bullosa hereditaria」一詞最先由 Koebner 提出。⁴ Hallopeau 最先區分了本病的單純型(非瘢痕性 nonscarring)與營養不良型(瘢痕性 scarring)形式⁵,而 Weber⁶、Cockayne⁷、Dowling 與 Meara⁸,以及 Koebner⁴ 各自描述了表皮鬆解症單純型 (epidermolysis bullosa simplex) 的獨特形式。Hoffman⁹、Cockayne¹⁰、Touraine¹¹、Pasini¹²,以及 Bart¹³ 提供了關於營養不良型 EB (DEB) 亞型的大部分資訊。Herlitz 描述了致死性表皮鬆解症 (epidermolysis bullosa letalis)¹⁴,後來發現這是 EB 第三大類——接合部型 (junctional form) 的一部分。電子顯微鏡在 EB 診斷上的應用催生了 Pearson 與其合作者的研究¹⁵,他們不僅依臨床發現,也依超微結構變化 (ultrastructural changes) 的存在來對病人進行分類。一個基於超微結構與臨床發現結合的全面性 EB 分類,由 Gedde-Dahl 在一篇早期的里程碑論著中完成。¹⁶ 近期的重大進展已使大多數類型 EB 病人的蛋白質與遺傳異常得以辨識。這些研究促成了對 EB 生物學基礎的更佳理解,並最終形成了目前以遺傳與蛋白質缺陷為基礎的 EB 分類¹⁷,此分類為特定分子治療提供了合理的途徑。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
概述 (OVERVIEW)
EB 源自基底角質細胞 (basal keratinocytes) 與其下真皮附著的缺陷。這些缺陷可能源自角質細胞質膜內部,或細胞外的真皮-表皮基底膜帶(dermal-epidermal basement membrane zone, BMZ)。許多承受外部破壞性力量的組織,例如皮膚與角膜,含有一個複雜的 BMZ,由一群組裝成錨定複合體 (anchoring complexes) 的特化組分所構成(圖 60-1)。在 BMZ 最上方,基底細胞細胞骨架中含角蛋白的中間絲 (intermediate filaments) 嵌入基底細胞質膜上稱為半橋粒 (hemidesmosomes) 的電子緻密凝聚物上。錨定絲 (Anchoring filaments) 橫跨透明板 (lamina lucida),連接半橋粒與緻密板 (lamina densa) 及錨定絲。在 BMZ 最下方,含第 VII 型膠原蛋白 (collagen VII) 的錨定纖維 (anchoring fibrils) 從緻密板延伸至乳頭真皮 (papillary dermis),並與緻密板及錨定斑 (anchoring plaques) 結合,捕捉間質膠原纖維 (interstitial collagen fibrils)。因此,皮膚 BMZ 連接了廣泛的基底細胞細胞骨架網絡與真皮中豐富的間質膠原纖維網絡。¹⁸,¹⁹
圖 60-1:左側為真皮-表皮基底膜各組分的示意圖,與右側基底膜形態學實體的超微結構外觀比較。
角蛋白絲 (KERATIN FILAMENTS)
角蛋白 (Keratins) 是專性異聚物 (obligate heteropolymers),由酸性與鹼性單體 (monomers) 對所構成。角蛋白 5 與 14 的配對組裝形成基底細胞細胞骨架廣泛的中間絲網絡。²⁰ 角蛋白含有一個中央 α 螺旋桿 (α-helical rod),帶有數個非螺旋中斷,以及非螺旋的羧基 (carboxy) 與胺基 (amino) 末端區域。角蛋白之間保守性最高的區域位於角蛋白桿末端的螺旋邊界基序 (helix boundary motifs) 上。雖然廣泛的突變誘發研究顯示中央桿末端附近的螺旋區域在角蛋白絲延長中很重要,但非螺旋結構域可能在形成側向結合中很重要。²¹ 角蛋白中間絲嵌入稱為半橋粒的電子緻密結構上。
半橋粒 (HEMIDESMOSOMES)
半橋粒含有細胞內蛋白,包括網蛋白 (plectin) 與 BP230。網蛋白是一個 500-kD 的蛋白質,作為中間絲結合蛋白。網蛋白也可能與微絲 (microfilaments) 交互作用,因為網蛋白含有一個與血影蛋白 (spectrin) 肌動蛋白結合結構域相似的結構域。²²,²³ BP230,又稱 BPAG1,是一個 230-kD 的蛋白質,與橋粒斑蛋白 (desmoplakin)²⁴ 及網蛋白都有同源性。BP230 的數個剪接變異體 (splicing variants) 在神經系統中至關重要。²⁵⁻²⁷ BP230 定位於半橋粒細胞質表面稱為內板 (inner plate) 的區域,並如同網蛋白般在半橋粒與中間絲之間的連接中發揮功能。BP230 陰性的基因轉殖小鼠缺乏半橋粒內板,且半橋粒與中間絲之間的連接被切斷,在半橋粒上方形成一個機械脆弱的細胞質區。
錨定絲 (ANCHORING FILAMENTS)
半橋粒也含有跨膜蛋白第 XVII 型膠原蛋白(collagen XVII,又稱 BPAG2 與 BP180)²⁸ 與 α6β4 整合素 (integrin)。²⁹ 這些分子的細胞質部分構成半橋粒緻密斑 (hemidesmosome-dense plaque) 的一部分,而這些分子的細胞外部分構成錨定絲的部分,並可能促成透明板區域中位於半橋粒下方、稱為次基底緻密板 (subbasal dense plate) 的結構。β4 整合素只與 α6 次單元配對,但 α6 次單元可與 β4 整合素或 β1 整合素結合。α6β1 或 α6β4 整合素的組合皆已被證明作為層黏連蛋白 (laminins) 的受體,而 α6β4 整合素作為層黏連蛋白-332 (laminin-332) 的特異性受體。α6β4 整合素在半橋粒的組織中扮演核心角色。β4 整合素含有一個特別大的細胞質結構域,其功能在於與半橋粒斑的其他蛋白質交互作用,包括第 XVII 型膠原蛋白與網蛋白。³⁰ 缺乏 β4 整合素的基因轉殖小鼠皮膚沒有半橋粒,並顯示嚴重的細胞黏附缺陷。³¹ 網蛋白與 α6β4 整合素之間的交互作用,在半橋粒的組裝與拆解中似乎都至關重要。³²
第 XVII 型膠原蛋白(BPAG2、BP180)是一種具有第 II 型跨膜方向 (type II transmembrane orientation) 的膠原性蛋白。根據電子顯微鏡與交聯研究 (crosslinking studies),第 XVII 型膠原蛋白組裝成三股螺旋同源三聚體 (triple-helical homotrimer),並含有三個主要區域:細胞內胺基末端球狀頭部、中央桿,以及細胞外可撓性尾部。³³ 第 XVII 型膠原蛋白與層黏連蛋白-332 及 α6β4 整合素結合於由角質細胞在體內形成、稱為穩定錨定接觸 (stable anchoring contacts) 的黏附結構中,這些被認為代表前半橋粒 (prehemidesmosomes)。³⁴ 線狀免疫球蛋白(immunoglobulin, Ig)A 大疱性皮膚病 (linear IgA bullous dermatosis) 中的自體抗原 LAD-1³⁵,³⁶,是一個 120-kD 的蛋白質,已透過胜肽定序 (peptide sequencing) 證明為第 XVII 型膠原蛋白被切割的外結構域 (exodomain)。³⁷ 第 XVII 型膠原蛋白在角質細胞培養與皮膚中,透過脫落酶 (sheddases)——一種使細胞表面受體溶解的膜結合蛋白酶——的作用而進行加工。³⁸,³⁹
除了 α6β4 整合素與第 XVII 型膠原蛋白外,錨定絲還含有層黏連蛋白-332 與層黏連蛋白-311 分子。如同層黏連蛋白家族的所有成員⁴⁰⁻⁴²,層黏連蛋白-332 是一個大型異源三聚體分子,含有 α3、β3 與 γ2 鏈。⁴³,⁴⁴ 最先被描述的層黏連蛋白含有 3 條短臂與 1 條長臂,如旋轉投影分析 (rotary shadowing analysis) 所示,形成十字形。相對地,層黏連蛋白-332 在每條短臂上都有截短。⁴⁵⁻⁴⁷ 由於這些短臂截短,層黏連蛋白-332 無法與其他層黏連蛋白自我聚合,也無法與巢蛋白 (nidogen) 結合。取而代之的是,層黏連蛋白-332 與層黏連蛋白-311 形成二硫鍵結 (disulfide bonded) 附著⁴⁸,後者是另一種已知的錨定絲層黏連蛋白⁴³,含有 α3、β1 與 γ1 鏈。層黏連蛋白-332 也會進行其 γ2 與 α3 鏈的加工。⁴⁹ 雖然大鼠層黏連蛋白 γ2 鏈先前已被證明由金屬蛋白酶-2 (metalloproteinase-2)⁵⁰ 與第 1 型膜型金屬蛋白酶 (membrane-type metalloproteinase type 1)⁵¹ 加工,但這些酵素的主要切割位點在人類層黏連蛋白-332 中並不保守。⁵²
其他研究顯示,層黏連蛋白 γ2 鏈的加工是透過一類稱為 C-蛋白酶 (C-proteinases) 的特殊蛋白質進行,後者也加工前膠原蛋白 (procollagen) 分子的 C 末端結構域。⁵³ 雖然此類蛋白質中的一員——骨形態發生蛋白 1 (bone morphogenic protein 1)⁵⁴——能夠執行此作用,但此蛋白質的一個剪接變異體稱為哺乳動物 tolloid (mammalian tolloid) 才是主要在角質細胞與纖維母細胞中表現的酵素,因此哺乳動物 tolloid 是皮膚中可能執行此功能的酵素。⁵² 哺乳動物 tolloid 也加工層黏連蛋白 α3 鏈⁵²,雖然其他酵素如血漿素 (plasmin)⁵⁵、基質金屬蛋白酶 (matrix metalloproteinase, MMP)-2⁵⁰ 與膜型 MMP-1⁵¹ 也能執行此功能。γ2 鏈短臂在層黏連蛋白-332 組裝進入基底膜中似乎很重要。⁵⁶ 由單株抗體(monoclonal antibody, mAb)19-DEJ-1⁵⁷ 所辨識的抗原也定位於錨定絲,但其分子身分仍屬未知。
錨定纖維 (ANCHORING FIBRILS)
第 VII 型膠原蛋白是錨定纖維的主要組分。對第 VII 型膠原蛋白推導出的胺基酸序列分析⁵⁸ 顯示存在一個長的中央膠原性區域,其特徵為重複的 Gly-X-Y 序列,並含有若干非膠原性中斷,包括螺旋中心一個 39 個胺基酸的非膠原性片段,對應於生化研究預測的「鉸鏈區 (hinge region)」。⁵⁹,⁶⁰ 這些中斷說明了第 VII 型膠原蛋白分子的可撓性,並解釋其能環繞並捕捉真皮基質分子,以發揮其穩定基底膜至下方乳頭真皮之功能的能力。⁶¹ 也已辨識出錨定纖維的一個 50-kD 組分,其似乎定位於錨定纖維與緻密板的嵌入位點。⁶²
第 VII 型膠原蛋白的 145-kD N 末端含有最大的非膠原性結構域,嵌入緻密板與錨定斑上。第 IV 型膠原蛋白 (collagen IV) 是這些結構中最豐富的組分,與第 VII 型膠原蛋白 NC1 結構域結合。錨定絲與錨定纖維之間存在直接交互作用,源自錨定絲組分層黏連蛋白-332 與第 VII 型膠原蛋白 NC1 結構域之間的特異性交互作用。⁶³,⁶⁴ 第 VII 型膠原蛋白與層黏連蛋白-332 上的 β3 鏈結合。⁶³,⁶⁵⁻⁶⁷ 這似乎是維持真皮-表皮黏合 (dermal-epidermal cohesion) 的關鍵因子。如同所有膠原蛋白,第 VII 型膠原蛋白組裝成三股螺旋。第 VII 型膠原蛋白只辨識出一條鏈,即 α1 鏈;一個基因編碼整個分子,因此第 VII 型膠原蛋白是同源三聚體。第 VII 型膠原蛋白三股螺旋在其加工後的 NC-2 球狀結構域處連接在一起,形成反平行二聚體 (antiparallel dimers)。⁶¹,⁶⁸ NC-2 結構域的加工是透過與加工層黏連蛋白-332(一個密切相關的分子)相同的 C-蛋白酶家族(骨形態發生蛋白 1 及/或哺乳動物 tolloid)進行。錨定纖維可能源自第 VII 型膠原蛋白反平行二聚體的側向結合。
臨床發現 (CLINICAL FINDINGS)
EB 的分類是根據水疱裂隙平面 (cleavage plane) 發生所在的超微結構層次來進行。EB 傳統上依穿透式電子顯微鏡 (transmission electron microscopy) 下 BMZ 分離的層次,被分類為單純型、接合部型與營養不良型亞型⁶⁹(圖 60-2)。雖然傳統上分組 EB 的診斷「黃金標準」是電子顯微鏡,但以間接免疫螢光 (indirect immunofluorescence) 觀察基底膜抗原的免疫定位 (immunomapping) 也相當有助於區分 EB 亞型。在每一組內,根據臨床、遺傳、組織學與生化評估,有數種不同類型的 EB。¹⁷ 在可能的情況下,應將蛋白質或 DNA 層次的分子特徵附加於臨床亞型上(稱為「洋蔥皮取徑 onion skin approach」),以更佳地描繪診斷並協助提供新興的分子治療。這些臨床亞型摘要於表 60-1。
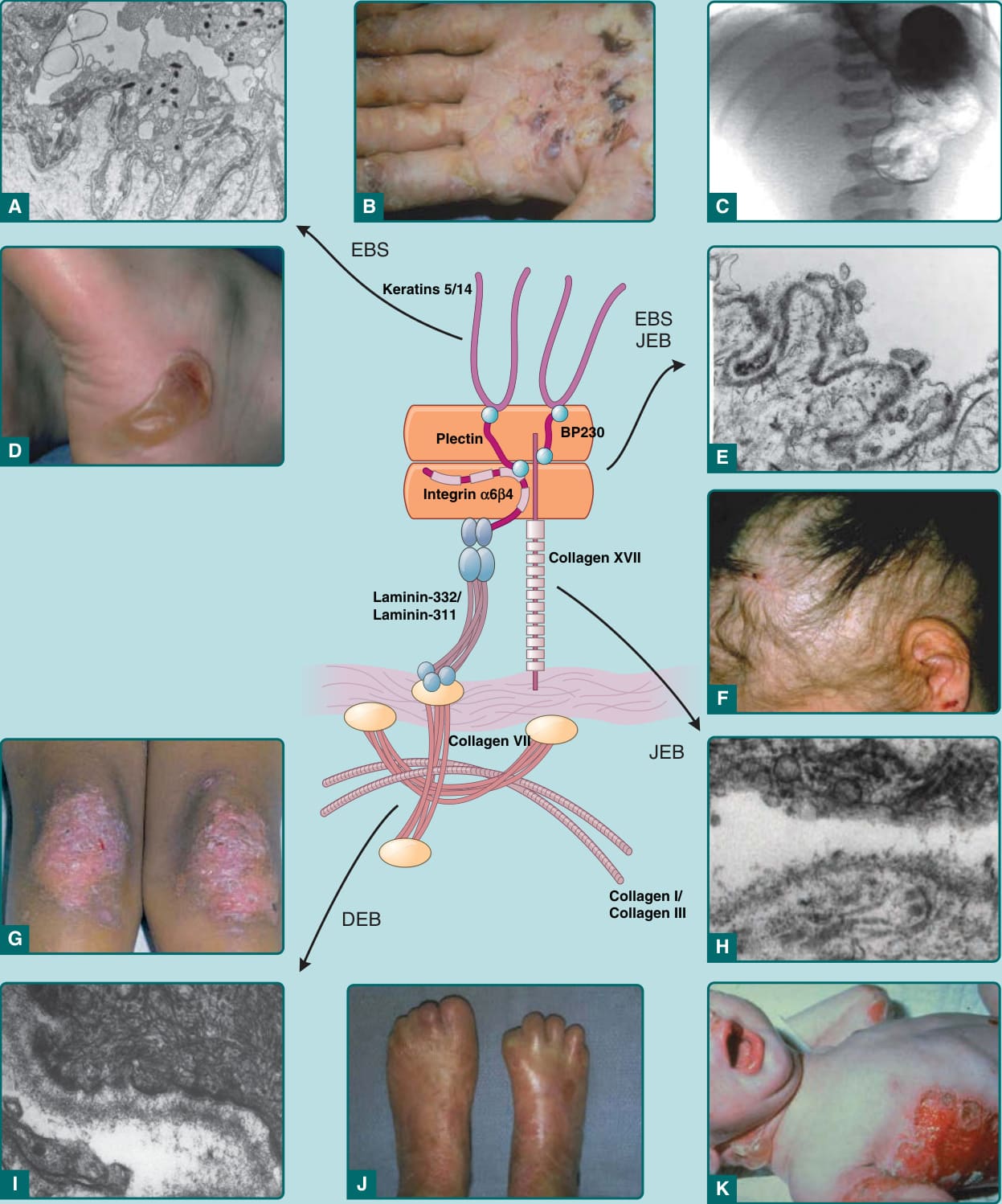
圖 60-2:表皮鬆解症 (EB) 皮膚分離層次與臨床發現的比較。A,穿透式電子顯微照片顯示表皮鬆解症單純型 (EBS) 中典型的表皮內分離。B,EB 疱疹樣型 (EB herpetiformis) 的掌部角化過度與糜爛。C,X 光片顯示與 EB 相關的幽門閉鎖 (pyloric atresia)。D,EBS Weber-Cockayne 型足跟上的局部水疱。E,穿透式電子顯微照片顯示 EB 伴幽門閉鎖(可分類為 EBS 或接合部型表皮鬆解症 JEB)中半橋粒層次的典型分離。F,廣泛性萎縮性良性表皮鬆解症 (generalized atrophic benign EB) 的非瘢痕性瀰漫性禿髮與頭皮糜爛。G,顯性營養不良型表皮鬆解症 (DEB) 中伴有粟粒疹 (milia) 的局部營養不良性變化。H,穿透式電子顯微鏡顯示 JEB 中典型的透明板內 (intralamina lucida) 分離。I,穿透式電子顯微鏡顯示 DEB 中典型的緻密板下 (sublamina densa) 分離。J,隱性 DEB 中的假性併指 (pseudosyndactyly)。K,Herlitz JEB 的廣泛性水疱。
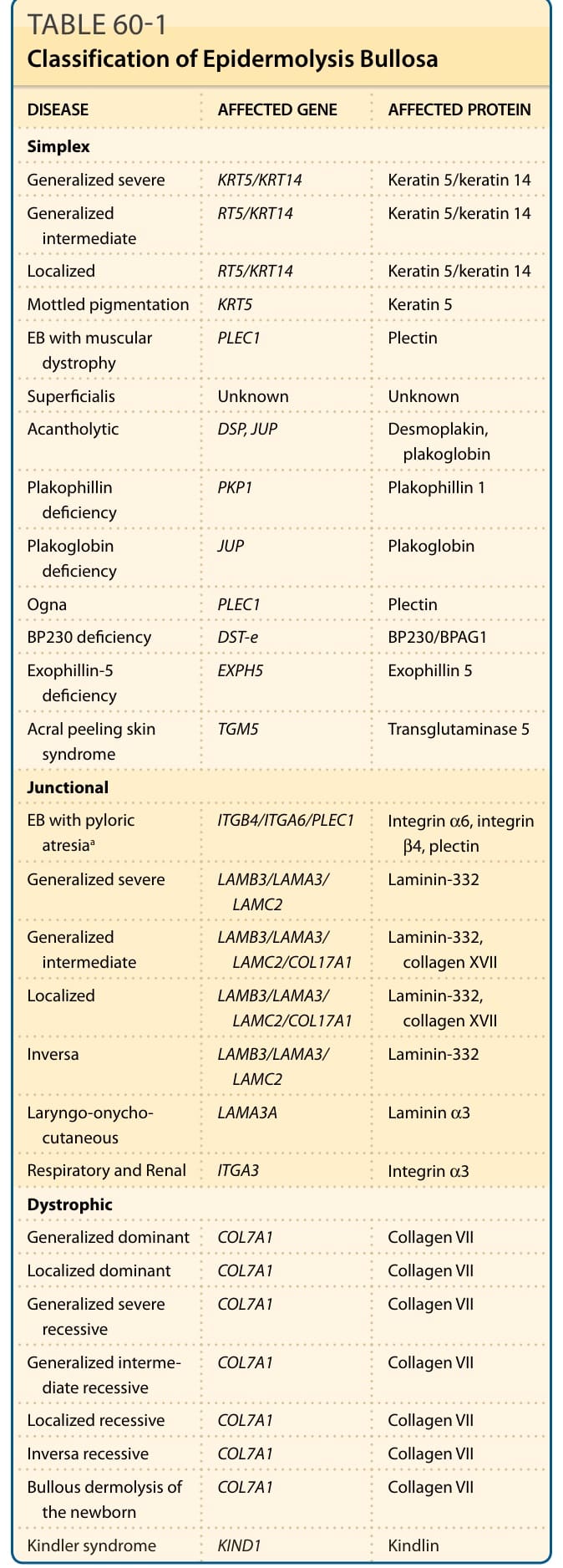
表 60-1:表皮鬆解症的分類 (Classification of Epidermolysis Bullosa)
下表為各 EB 疾病亞型及其受影響基因與受影響蛋白質的整理(依原表 60-1 譯出):
| 疾病 (DISEASE) | 受影響基因 (AFFECTED GENE) | 受影響蛋白質 (AFFECTED PROTEIN) |
|---|---|---|
| 單純型 (Simplex) | ||
| 廣泛性重度 (Generalized severe) | KRT5/KRT14 | 角蛋白 5/角蛋白 14 (Keratin 5/keratin 14) |
| 廣泛性中度 (Generalized intermediate) | KRT5/KRT14 | 角蛋白 5/角蛋白 14 |
| 局部型 (Localized) | KRT5/KRT14 | 角蛋白 5/角蛋白 14 |
| 斑駁性色素沉著 (Mottled pigmentation) | KRT5 | 角蛋白 5 (Keratin 5) |
| EB 伴肌肉失養症 (EB with muscular dystrophy) | PLEC1 | 網蛋白 (Plectin) |
| 淺表型 (Superficialis) | 未知 (Unknown) | 未知 (Unknown) |
| 棘層鬆解型 (Acantholytic) | DSP, JUP | 橋粒斑蛋白、血漿珠蛋白 (Desmoplakin, plakoglobin) |
| 斑菲素蛋白缺乏 (Plakophilin deficiency) | PKP1 | 斑菲素蛋白 1 (Plakophilin 1) |
| 血漿珠蛋白缺乏 (Plakoglobin deficiency) | JUP | 血漿珠蛋白 (Plakoglobin) |
| Ogna 型 | PLEC1 | 網蛋白 (Plectin) |
| BP230 缺乏 (BP230 deficiency) | DST-e | BP230/BPAG1 |
| 外簇蛋白-5 缺乏 (Exophillin-5 deficiency) | EXPH5 | 外簇蛋白 5 (Exophillin 5) |
| 肢端脫皮症候群 (Acral peeling skin syndrome) | TGM5 | 轉麩醯胺酸酶 5 (Transglutaminase 5) |
| 接合部型 (Junctional) | ||
| EB 伴幽門閉鎖ᵃ (EB with pyloric atresia) | ITGB4/ITGA6/PLEC1 | 整合素 α6、整合素 β4、網蛋白 (Integrin α6, integrin β4, plectin) |
| 廣泛性重度 (Generalized severe) | LAMB3/LAMA3/LAMC2 | 層黏連蛋白-332 (Laminin-332) |
| 廣泛性中度 (Generalized intermediate) | LAMB3/LAMA3/LAMC2/COL17A1 | 層黏連蛋白-332、第 XVII 型膠原蛋白 (Laminin-332, collagen XVII) |
| 局部型 (Localized) | LAMB3/LAMA3/LAMC2/COL17A1 | 層黏連蛋白-332、第 XVII 型膠原蛋白 |
| 反向型 (Inversa) | LAMB3/LAMA3/LAMC2 | 層黏連蛋白-332 |
| 喉-甲-皮型 (Laryngo-onychocutaneous) | LAMA3A | 層黏連蛋白 α3 (Laminin α3) |
| 呼吸與腎臟型 (Respiratory and Renal) | ITGA3 | 整合素 α3 (Integrin α3) |
| 營養不良型 (Dystrophic) | ||
| 廣泛性顯性 (Generalized dominant) | COL7A1 | 第 VII 型膠原蛋白 (Collagen VII) |
| 局部性顯性 (Localized dominant) | COL7A1 | 第 VII 型膠原蛋白 |
| 廣泛性重度隱性 (Generalized severe recessive) | COL7A1 | 第 VII 型膠原蛋白 |
| 廣泛性中度隱性 (Generalized intermediate recessive) | COL7A1 | 第 VII 型膠原蛋白 |
| 局部性隱性 (Localized recessive) | COL7A1 | 第 VII 型膠原蛋白 |
| 反向型隱性 (Inversa recessive) | COL7A1 | 第 VII 型膠原蛋白 |
| 新生兒大疱性真皮鬆解 (Bullous dermolysis of the newborn) | COL7A1 | 第 VII 型膠原蛋白 |
| Kindler 症候群 (Kindler syndrome) | KIND1 | Kindlin |
ᵃ 亦可分類為單純型。EB,表皮鬆解症。
表皮鬆解症單純型 (EPIDERMOLYSIS BULLOSA SIMPLEX)
表皮鬆解症單純型(epidermolysis bullosa simplex, EBS)是一個疾病群,特徵為表皮內水疱 (intraepidermal blistering),最常與角蛋白基因突變相關。在不同亞群之間,疾病表型 (phenotypes) 的範圍從輕微到嚴重不等。⁷⁰ 常見的 EBS 類型為顯性遺傳,包括廣泛性重度(Dowling-Meara)、廣泛性中度(Koebner)與局部型(Weber-Cockayne)。有數種罕見的變異型,包括 EBS Ogna 型、EBS 伴肌肉失養症、EBS 伴斑駁性色素沉著,以及一群基底上型 (suprabasal) EBS 亞型。
廣泛性重度表皮鬆解症單純型 (GENERALIZED SEVERE EPIDERMOLYSIS BULLOSA SIMPLEX)
此亞型又稱為 Dowling-Meara 型、EBS 的疱疹樣 (herpetiformis) 變異型。它在出生時即出現,呈廣泛性分布,並被視為 EBS 亞型中最嚴重者(圖 60-3)。它與廣泛性中度變異型的不同處在於,口腔黏膜更常受侵犯,偶爾顯示廣泛的糜爛。具有此亞型的病人在嬰兒期有時可能出現粟粒疹 (milium) 形成;然而,這種傷後現象通常在兒童期後期消退。本病常可伴隨群聚或「疱疹樣 (herpetiform)」水疱的自發性出現。這些水疱發生於軀幹與近端肢體,並癒合而不留瘢痕。應注意,當病人顯示廣泛性水疱時,此疱疹樣型態往往並不可見;因此,其缺乏不應作為排除此 EB 亞型的依據。廣泛性重度 EBS 常顯示指甲侵犯。在本病中,指甲可能脫落,並可能伴隨營養不良地重新生長。有趣的是,雖然熱會加重其他 EBS 亞型的水疱,但它在廣泛性重度 EBS 中似乎沒有重大影響。掌蹠的角化過度 (hyperkeratosis of the palms and soles) 常從兒童早期開始發展,並可進展為掌蹠的融合性角皮症 (confluent keratoderma)。這些可能相當疼痛,偶爾因干擾行走而導致屈曲攣縮 (flexural contractures)。偶爾,已有報告廣泛性重度 EBS 中食道侵犯的情形,範圍從糜爛到幽門閉鎖 (pyloric atresia)。⁷¹ 上呼吸道也可能受影響,包括喉黏膜。⁷²
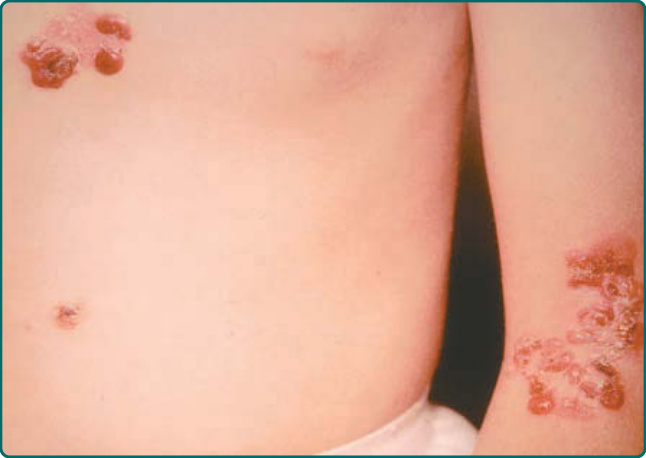
圖 60-3:Dowling-Meara 型表皮鬆解症單純型 (epidermolysis bullosa simplex) 病人軀幹與手臂上特徵性的水疱形成。
廣泛性中度表皮鬆解症單純型 (GENERALIZED INTERMEDIATE EPIDERMOLYSIS BULLOSA SIMPLEX)
另一種常見的廣泛性 EBS 形式是中度型,又稱 Koebner EBS。此亞型於出生時或最晚於嬰兒早期出現廣泛性水疱。手、足與四肢通常顯示最嚴重的侵犯。病灶常以發炎後色素過度沉著或色素減退 (postinflammatory hyper or hypopigmentation) 癒合,雖然偶爾可發生萎縮 (atrophy) 與粟粒疹,但很罕見,且遠少於廣泛性重度 EBS。可能出現掌蹠角化過度 (palmar-plantar hyperkeratosis) 與糜爛(圖 60-4)。足底增厚很常見,但往往要到兒童期後期才出現。口腔黏膜有時顯示輕微的糜爛活性,但這些通常隨年齡增長而改善。此 EB 亞型通常沒有嚴重的生長遲滯。
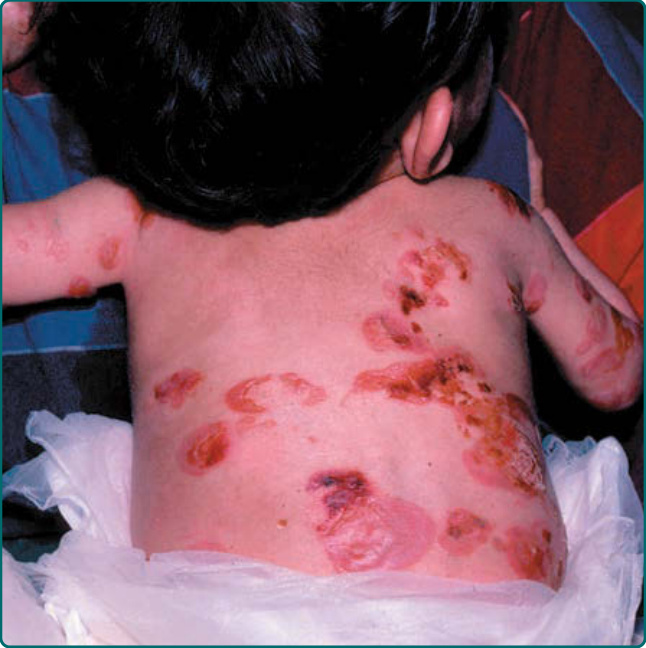
圖 60-4:一名罹患廣泛性表皮鬆解症單純型 (generalized epidermolysis bullosa simplex) 的嬰兒的廣泛性水疱。
局部型表皮鬆解症單純型 (LOCALIZED EPIDERMOLYSIS BULLOSA SIMPLEX)
這是 EB 的一種輕微形式,先前稱為 EBS 的 Weber-Cockayne 亞型(圖 60-5)。本病是最常見的 EB 形式,常於嬰兒期或兒童期出現。偶爾它在成年早期出現,例如在軍隊服役行軍後注意到水疱。據推測,本型 EB 有許多未被診斷的病例,因為它可能輕微到在臨床就診時逃過通報或偵測。掌蹠多汗症 (hyperhidrosis) 是常見的相關情形。水疱偶爾可能繼發性感染。發炎後色素異常出現於此變異型,但粟粒疹與瘢痕通常缺如。水疱活性通常隨創傷區域而出現,以手與足最常見,頭皮最少見。輕微的口腔糜爛僅罕見出現,並通常隨年齡增長而消退。此 EB 亞型的指甲侵犯很罕見。
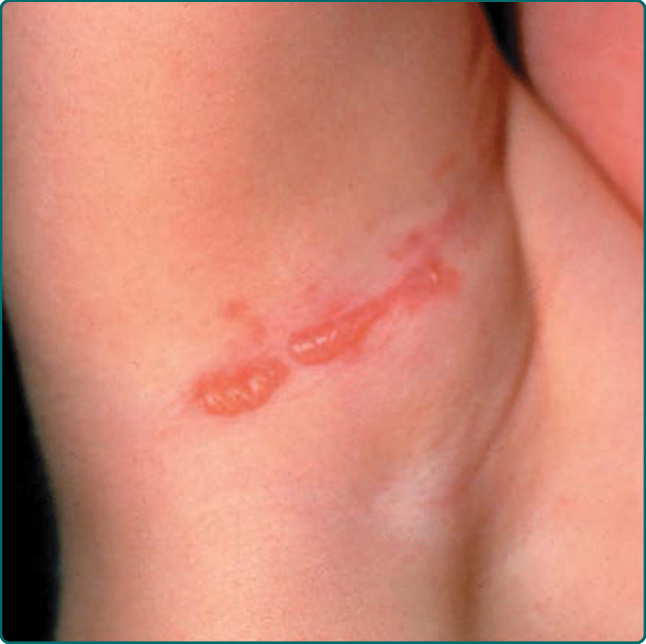
圖 60-5:局部型表皮鬆解症單純型 (localized epidermolysis bullosa simplex) 病人因衣物造成的創傷誘發水疱。
表皮鬆解症單純型的其他變異型 (ADDITIONAL VARIANTS OF EPIDERMOLYSIS BULLOSA SIMPLEX)
Ogna 型表皮鬆解症單純型 (Epidermolysis Bullosa Simplex of Ogna):常於嬰兒期發病,伴隨肢端區域的季節性水疱(夏季)。小型出血性與漿液性水疱主要發生於四肢。癒合不留瘢痕。本病最初報告於來自挪威 (Norway) 的病人。這些病人也顯示出大腳趾甲特徵性的甲彎曲 (onychogryphosis)。
表皮鬆解症單純型伴肌肉失養症 (Epidermolysis Bullosa Simplex with Muscular Dystrophy):這種罕見的臨床實體是一種自體隱性遺傳疾病,包含出生時或不久後皮膚的廣泛性水疱。這伴隨進行性肌肉失養症 (progressive muscular dystrophy)。⁷³ 它表現出類似廣泛性中度 EBS 的廣泛性水疱。這些病人已被證明帶有編碼 HD1/網蛋白 (HD1/plectin) 之基因的突變。⁷⁴⁻⁷⁶
表皮鬆解症單純型伴斑駁性色素沉著 (Epidermolysis Bullosa Simplex with Mottled Pigmentation):本型 EB,如其名所示,特徵為軀幹與近端肢體的斑駁性色素過度沉著 (mottled hyperpigmentation)。出生時或嬰兒早期開始有廣泛性分布的水疱。色素改變與水疱可能隨年齡增長而改善。嬰兒期可能出現輕微的口腔黏膜侵犯。本病況與大型黑色素細胞痣 (large melanocytic nevi) 不同,後者可見於所有三型 EB。⁷⁷
表皮鬆解症單純型淺表型 (Epidermolysis Bullosa Simplex Superficialis):這是一種不常見的 EBS 形式,得名於本病中產生水疱的角質層下 (subcorneal) 分離。⁷⁸ 在這些病人中,通常見到糜爛與結痂而非完整的大疱,並以發炎後色素變化癒合。儘管裂隙平面淺表,但本病已觀察到指甲侵犯、萎縮性瘢痕 (atrophic scarring) 與粟粒疹。
棘層鬆解型表皮鬆解症單純型 (Acantholytic Epidermolysis Bullosa Simplex):此亞型⁷⁹ 是一種罕見的隱性遺傳致死性疾病,特徵為出生時的廣泛性糜爛。如同淺表型亞型,由於表皮分離的層次非常淺表(被描述為片狀 sheetlike),通常見不到水疱。指甲為營養不良性。禿髮 (Alopecia)、新生兒牙 (neonatal teeth)、口腔糜爛與呼吸道侵犯使本病有別於其他淺表型 EBS 亞型。
斑菲素蛋白缺乏 (Plakophilin Deficiency):此亞型,又稱外胚層發育不良-皮膚脆弱症候群 (ectodermal dysplasia skin fragility syndrome),是另一種基底上表皮分離 (suprabasilar epidermal separation) 的遺傳疾病,特徵為出生時的廣泛性糜爛,有時伴有淺表水疱。⁸⁰ 禿髮也很常見,掌蹠角皮症 (palmoplantar keratoderma)、疼痛性皸裂 (painful fissures) 與指甲營養不良亦然。病人有時可能表現出生長遲滯 (failure to thrive)、唇炎 (cheilitis)、少汗症 (hypohidrosis) 與搔癢。如同其他淺表型 EBS 亞型,本病伴隨營養不良性指甲。
BP230 缺乏 (BP230 Deficiency):這是一種極為罕見的 EB 變異型,由 DST-e 基因突變所引起,已在一小群科威特 (Kuwaiti) 與伊朗 (Iranian) 家族中被描述。水疱發病通常在出生時,且表型為局部型,常侵犯足部,無顯著的黏膜或其他皮膚外侵犯。如同其他形式的 EBS,水疱可能以發炎後色素變化癒合,但無瘢痕。遺傳似乎為自體隱性,但某些異型合子帶因者 (heterozygous carriers) 也報告在第一與第二個十年中有自限性的輕微水疱。
外簇蛋白-5 缺乏 (Exophillin-5 Deficiency):這是一種非常罕見的自體隱性 EBS 變異型,由 EXPH5 基因突變所引起。水疱位於表皮高處,導致創傷誘發的結痂或脆弱的水疱與小水疱。本病整體輕微,無皮膚外侵犯,兒童期的緩解或改善是本病的特徵。⁸¹
肢端脫皮症候群 (Acral Peeling Skin Syndrome):此 EB 變異型⁸² 特徵為淺表且無痛的皮膚脫皮,最常發生於手與足。濕度、熱與水的暴露可加重此病況。組織學上分離的層次介於顆粒層 (stratum granulosum) 與角質層 (stratum corneum) 之間。
表皮鬆解症單純型的分子病理學 (MOLECULAR PATHOLOGY OF EPIDERMOLYSIS BULLOSA SIMPLEX)
在遺傳層次進行分析的 EBS 病人中,大多數已被發現與編碼角蛋白 5 與 14 之基因的突變相關。²¹,⁷⁰ 這些病人皮膚分離的層次在基底細胞中部 (midbasal cell),如圖 60-4 所示,伴隨可變的中間絲叢集 (intermediate filament clumping)。半橋粒與其他 BMZ 結構在電子顯微鏡下正常。與 EBS 相關的大多數角蛋白基因突變為顯性遺傳,因為角蛋白絲的多聚體組裝異常。有一較小的隱性遺傳病人亞群,其嚴重度不一。⁸³,⁸⁴
編碼角蛋白 5 與 14 最保守區域(即螺旋邊界結構域 helix boundary domains)的突變⁸⁵,與最嚴重的 EBS 形式相關,例如 Dowling-Meara 亞型,後者在穿透式電子顯微鏡下顯示中間絲叢集。另一方面,較輕微的疾病類型,例如 Weber-Cockayne 亞型,則與編碼角蛋白 5 與 14 較不保守區域的突變相關。編碼角蛋白 5 胺基末端特定區域的突變,存在於 EBS 伴斑駁性色素沉著的病人中。⁸⁶ 雖然這類突變的意義及其與色素異常的關聯仍不清楚⁸⁷,⁸⁸,但有人認為角蛋白 5 球狀頭部結構域負責角蛋白絲嵌入黑色素體 (melanosomes) 上。近期研究顯示,某些角蛋白 5 基因突變可能產生在溫度升高下不穩定的蛋白質。⁸⁹ 這可能有助於解釋某些 EBS 亞型在溫暖溫度下被充分觀察到的惡化。與 EBS 伴肌肉失養症相關的突變產生提早終止密碼子 (premature termination codons)、剪接位點 (splice-site) 或其他突變,導致網蛋白缺乏表現或表現缺陷。⁹⁰ 雖然與網蛋白異常相關的 EB 形式被分類為單純型,但它的皮膚分離層次與接合部型表皮鬆解症 (junctional epidermolysis bullosa, JEB) 伴幽門閉鎖所見者相同。⁶⁹ 具體而言,分離存在於 BMZ 細胞內部分中半橋粒層次的正上方。將 EBS 伴肌肉失養症與 JEB 伴幽門閉鎖——這兩種分離層次相同的疾病——分為兩個不同的 EB 類別,說明了現行 EB 分類系統的限制。網蛋白缺陷如同 α6β4 整合素缺陷,也可與幽門閉鎖相關。⁹⁰
網蛋白通常在廣泛的組織中表現,包括肌肉。²² 雖然網蛋白缺乏病人發生肌肉失養症的機轉尚屬未知,但已觀察到在網蛋白缺乏的情況下會發生肌節 (muscle sarcomeres) 的紊亂。網蛋白的血影蛋白樣結構域(其在肌肉中通常可能與肌動蛋白絲交互作用)的缺乏,可能是肌肉病理的關鍵因素。⁹¹
外胚層發育不良-皮膚脆弱症候群的潛在分子缺陷已被證明為橋粒蛋白斑菲素蛋白 1 (plakophilin 1) 的功能喪失。⁸⁰ 斑菲素蛋白主要表現於基底上角質細胞與外根鞘 (outer root sheath) 細胞。本病的顯微發現通常顯示位於斑菲素蛋白 1 正常表現區域的表皮內棘層鬆解 (intraepidermal acantholysis)。分子缺陷涉及編碼斑菲素蛋白 1 之 PKP1 基因的功能喪失突變。⁹²⁻⁹⁷
BP230 缺乏(一種有時稱為 DST-e 的、選擇性剪接的營養不良性變異)特徵為免疫螢光顯微鏡下 BP230 表現減少或缺如,以及穿透式電子顯微鏡下半橋粒內板的喪失。DST-e 基因中編碼 BP230 捲曲螺旋結構域 (coiled-coil domain) 區域的同型合子提早終止密碼子突變以及點突變,已與本病相關。⁸¹ 外簇蛋白-5 缺乏與 EXPH5 基因的功能喪失突變相關。本病在免疫螢光顯微鏡下可顯示皮膚中此蛋白質的完全缺乏表現,且電子顯微鏡可顯示基底角質細胞核周圍囊泡 (perinuclear vesicles) 的異常聚集。⁹⁸,⁹⁹
TGM5(編碼轉麩醯胺酸酶多基因家族成員)的突變,產生與肢端脫皮症候群潛在病因相關的轉麩醯胺酸酶表現改變。⁸²
接合部型表皮鬆解症 (JUNCTIONAL EPIDERMOLYSIS BULLOSA)
所有 JEB 病人共享一個共同的組織病理特徵,即水疱形成於 BMZ 的透明板內,由位於透明板與緻密板上部的錨定絲缺陷所引起。這群疾病以自體隱性方式遺傳,而依分子缺陷不同,個別臨床表型有相當大的變異。三種主要的 JEB 形式最為常見。罹患重度廣泛性 JEB(先前稱為 Herlitz 病、JEB gravis 或致死性 JEB)的病人,表現出最嚴重的疾病表型。¹⁴,¹⁰⁰ 不幸的是,這種最嚴重的 JEB 亞型也是最常見的。
廣泛性重度接合部型表皮鬆解症 (GENERALIZED SEVERE JUNCTIONAL EPIDERMOLYSIS BULLOSA)
廣泛性重度 JEB 是最嚴重的 EB 亞型之一,導致嬰兒期或兒童早期死亡。¹⁰¹ 本病特徵為出生時的廣泛性且常為廣大範圍的水疱(圖 60-6)。在嬰兒期後期,會出現一種獨特的孔口周圍肉芽組織 (periorificial granulation tissue)。這最常發生於嘴巴、眼睛與鼻孔周圍,但也可能出現於頭皮、耳朵周圍,較少出現於頭頸部以外的其他部位。這種增生性肉芽組織也可發生於非致死性亞型中。指甲通常嚴重受影響,並常於嬰兒期喪失。當指甲仍存在時,通常為營養不良性,且常伴隨增生性肉芽組織。本病存在牙齒缺陷,特徵為齒釉質 (tooth enamel) 的點蝕 (pitting)。口咽黏膜糜爛通常存在,並可能廣泛。所有複層鱗狀上皮組織 (stratified squamous epithelial tissues) 的糜爛,包括鼻腔、結膜、食道、氣管、喉、直腸與尿道黏膜皆可能受影響。嚴重病例中相關的全身性發現是本病致死性的重要因素。大氣道侵犯,包括氣管喉狹窄 (tracheolaryngeal stenosis) 或阻塞,常與 Herlitz JEB 疾病相關,而嬰兒早期的聲音嘶啞 (hoarseness) 是不祥之兆。本病有特徵性的生長遲滯與生長遲緩,常伴有混合性貧血 (mixed anemia)。敗血症 (Sepsis) 是常見且常為致死性的併發症。
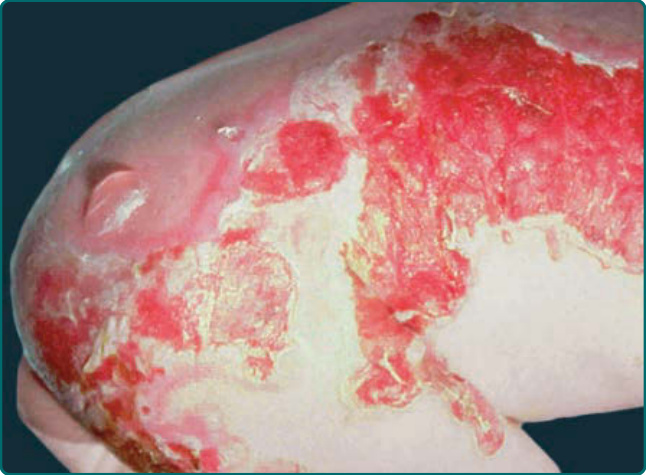
圖 60-6:一名罹患 Herlitz 接合部型表皮鬆解症 (Herlitz junctional epidermolysis bullosa) 的嬰兒出生時的廣泛性水疱。
廣泛性中度接合部型表皮鬆解症 (GENERALIZED INTERMEDIATE JUNCTIONAL EPIDERMOLYSIS BULLOSA)
有時最初以廣泛性重度表型表現的病人會存活過嬰兒期。這些病人最終證明其水疱與口腔糜爛的嚴重度低於致死型。特別是,缺乏顯著的聲音嘶啞被視為一個有利的預後徵象,指示內部疾病表現較不嚴重。頭皮與指甲病灶以及孔口周圍不癒合糜爛,是這些病人兒童期最常見的發現之一。儘管嬰兒期沒有致死性,這些病人仍可能有嚴重的上皮黏附異常,而氣切 (tracheostomies) 或胃造口管 (gastrostomy tubes) 可能有助於病人存活(圖 60-7)。這些病人的非致死狀態使他們有別於 Herlitz 組,過去曾用「非致死性 JEB (nonlethal JEB)」或「JEB mitis」等名詞來描述這些病人。他們遠少於重度廣泛性 JEB 病人。還有其他罕見的非致死性 JEB 變異型,表現為四肢局部的接合部型水疱,或軀幹與間擦部位 (intertriginous areas) 反向分布 (inversa distribution) 的水疱。一個獨特的廣泛性中度 JEB 亞群(先前稱為廣泛性萎縮性良性 EB generalized atrophic benign EB)於出生時出現廣泛性皮膚侵犯。¹⁰² 然而,儘管有廣泛的皮膚水疱,口腔糜爛或其他黏膜疾病卻相對稀少。雖然存在齒釉質點蝕,導致廣泛的齲齒 (dental caries),且指甲營養不良往往可能很嚴重,但這些病人很少注意到其他皮膚外侵犯。這些病人的水疱以特徵性的萎縮性瘢痕癒合,雖造成失能且可能廣泛,但這些病人通常壽命正常。水疱隨年齡改善,生長正常,且很少見到貧血。具有此疾病亞型的某些病人曾經歷正常、無併發症的懷孕與分娩。這些病人的一個特殊特徵是頭皮的進行性禿髮,以及身體其他部位的終毛 (terminal hairs) 脫落。毛髮脫落在青春期開始後變得嚴重,雖然已有描述伴隨萎縮性瘢痕的斑塊狀脫髮,但禿髮往往相當瀰漫,且瘢痕細微或不存在。
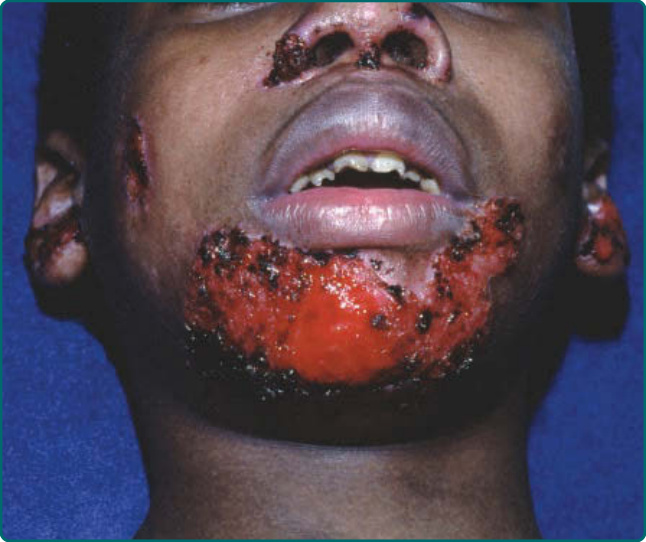
圖 60-7:一名罹患非 Herlitz 接合部型表皮鬆解症 (non-Herlitz junctional epidermolysis bullosa) 病人的孔口周圍糜爛與增生性肉芽組織。
局部型接合部型表皮鬆解症 (LOCALIZED JUNCTIONAL EPIDERMOLYSIS BULLOSA)
JEB 的一種罕見亞型是局部型變異,先前稱為 minimus JEB。這些病人通常顯示輕微疾病,可能在局部區域加重,最常見於手、足與脛前 (pretibial) 區域。指甲有時可能脫落或變得營養不良,並可能發生齒釉質點蝕。也可發生口腔或鼻腔糜爛,但無任何內部侵犯。這些病人通常預後良好且壽命正常。
接合部型表皮鬆解症伴幽門閉鎖 (JUNCTIONAL EPIDERMOLYSIS BULLOSA WITH PYLORIC ATRESIA)
這些病人表現出極度的黏膜與皮膚脆弱,並可能也有各種泌尿系統異常,包括腎水腫 (hydronephrosis) 與腎炎 (nephritis)。編碼 β4 與 α6 整合素之基因的突變已與 EB 相關¹⁰³,而在這群疾病中,幽門閉鎖存在。雖然幽門狹窄 (pyloric stenosis) 相對較常見,但幽門閉鎖(一種罕見病況)與 EB 的獨特關聯使本病格外獨特。在這些病人中,半橋粒通常缺如或發育不全,而分離的層次在半橋粒層次的低位基底內表皮 (low intrabasal epidermal),如前述 EBS 伴肌肉失養症所見。本病大多數病例相當嚴重,因廣泛的皮膚外上皮脫落 (extracutaneous epithelial sloughing) 以及皮膚與黏膜的廣泛水疱,於嬰兒期致死。已有特徵描述的罕見非致死性病例,似乎源自 β4 整合素的部分功能喪失。¹⁰⁴ 有趣的是,非致死性 JEB 有時可透過 mRNA 剪接的改變而自我改善。¹⁰⁵,¹⁰⁶
接合部型表皮鬆解症伴呼吸與腎臟侵犯 (JUNCTIONAL EPIDERMOLYSIS BULLOSA WITH RESPIRATORY AND RENAL INVOLVEMENT)
皮膚水疱在生命早期開始,通常輕微甚至缺如,並可能伴隨瀰漫性禿髮與指甲營養不良。然而,皮膚外侵犯於出生時出現且相當嚴重,包含間質性肺病 (interstitial lung disease),導致呼吸窘迫與肺部感染。本病的腎臟表現包括腎衰竭、腎病症候群 (nephrotic syndrome)、腎發育不全 (renal hypoplasia) 與腎水腫。儘管採取積極的新生兒支持照護,病人仍因這些嚴重的呼吸或腎臟併發症於嬰兒期死亡。
喉-甲-皮症候群 (LARYNGO-ONYCHO-CUTANEOUS SYNDROME)
這種罕見的自體隱性病況¹⁰⁷ 特徵為指甲營養不良、皮膚糜爛與廣泛的肉芽組織,尤其定位於結膜與喉。皮膚侵犯部位包括反覆創傷或受壓的區域,例如手肘、膝蓋、手指與腳趾。
接合部型表皮鬆解症的分子病理學 (MOLECULAR PATHOLOGY OF JUNCTIONAL EPIDERMOLYSIS BULLOSA)
JEB 可與編碼層黏連蛋白-332 α3、β3 或 γ2 次單元之基因的突變相關。¹⁰⁸,¹⁰⁹ 三條鏈中任何一條的缺如,皆導致三聚體層黏連蛋白-332 組裝與分泌的缺乏,進而產生相似的水疱表型。具有編碼 α3 或 γ2 層黏連蛋白次單元之基因突變的病人仍顯示層黏連蛋白-311 的正常表現,後者含有 α3、β1 與 γ1 鏈。⁴⁴ 因此,在間接免疫螢光顯微鏡(indirect immunofluorescence microscopy, IDIF)下,JEB 皮膚對 α3 鏈呈陽性線狀 BMZ 染色,而其他鏈的表現缺如,即為 α3 或 γ2 鏈缺陷的指標。相反地,IDIF 下 α3 染色的缺如可指示 α3 基因的突變。約 80% 的層黏連蛋白-332 突變可追溯至 LAMB3 基因中兩個復發性無義突變 (nonsense mutations) 之一,使得層黏連蛋白-332 病變的產前檢測比其他 EB 候選基因更為容易。¹¹⁰ 在 Herlitz 病的病人中,迄今偵測到的所有突變皆為產生提早終止密碼子者,導致層黏連蛋白-332 表現缺如。雖然 Herlitz 病病人通常顯示層黏連蛋白-332 完全缺乏表現,但具有異常肉芽組織與顯著黏膜侵犯而無 Herlitz 病的病人,往往顯示層黏連蛋白-332 表現減少。這是由於表現了帶有缺失或誤義突變 (missense mutations) 的分子,導致層黏連蛋白-332 部分功能喪失。這可導致臨床上分類為非 Herlitz JEB 的病例,尤其是有顯著黏膜侵犯的病例。⁷⁶,¹¹¹,¹¹² 在某些病例中,重度 JEB 病例已發生水疱的自發性改善,這與導致層黏連蛋白-332 重新表現的遺傳機轉相關。¹⁰⁶
在非 Herlitz JEB 變異型,即廣泛性萎縮性良性接合部型 EB(generalized atrophic benign junctional EB, GABEB)中,水疱發生於透明板區域,且通常存在半橋粒或錨定絲的異常(圖 60-8)。雖然層黏連蛋白-332 突變是一部分 GABEB 病人的根本原因,但大多數這類病人有半橋粒蛋白第 XVII 型膠原蛋白(又稱 BP180 或 BPAG2)的異常。已在 GABEB 病人中描述了若干編碼第 XVII 型膠原蛋白之基因的突變,包括提早終止密碼子突變、誤義突變、剪接位點突變、截短,以及甘胺酸取代突變 (glycine substitution mutation)。¹¹³,¹¹⁴ 雖然在本病所有病人中皆已顯示透明板內或接合部皮膚分離,但已描述一名病人具有第 XVII 型膠原蛋白的細胞質缺失,並顯示基底內表皮 (intrabasal epidermal) 皮膚分離。¹¹⁵ 局部型 JEB 已被證明與 COL17A1 突變相關。¹¹⁶
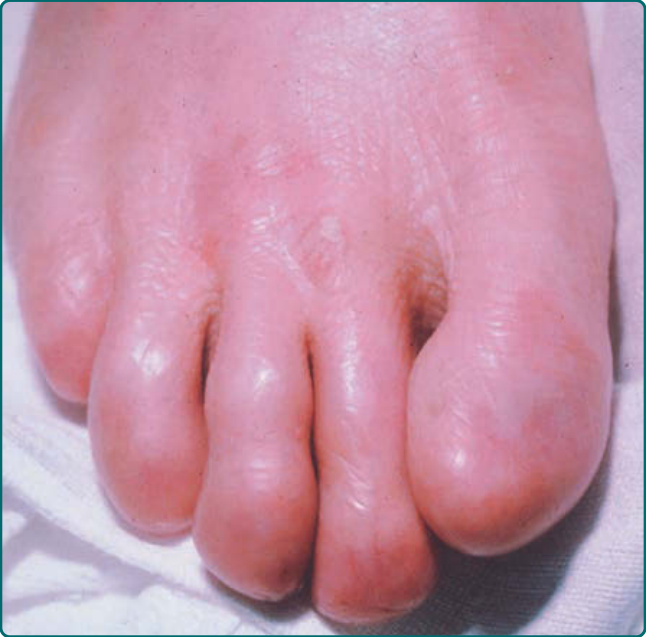
圖 60-8:一名罹患非 Herlitz 接合部型表皮鬆解症或廣泛性萎縮性良性表皮鬆解症 (non-Herlitz junctional epidermolysis bullosa or generalized atrophic benign epidermolysis bullosa) 病人的指甲喪失與皮膚萎縮。
喉-甲-皮症候群的特徵為 LAMA3 基因的 N 末端突變,導致層黏連蛋白 α3 LE 結構域的缺失。¹⁰⁷ 值得注意的是,已辨識出鑲嵌型 (mosaic) GABEB 病人,他們表現出與第 XVII 型膠原蛋白表現缺如相關的明確水疱區域,以及與正常第 XVII 型膠原蛋白表現相關的非水疱皮膚區域。對這些病人角質細胞的仔細分析揭示了兩個突變等位基因之一的回復 (reversion),最可能由涉及親代等位基因 DNA 非互惠交換的有絲分裂基因轉換 (mitotic gene conversion) 所引起。¹¹⁷,¹¹⁸ 理解 EB 在這些病例中如何能進行自發性分子矯正,可能有助於設計未來的分子治療策略。JEB 伴呼吸與腎臟侵犯由 ITGA3 基因突變所引起。¹¹⁹,¹²⁰ 這些病人在皮膚切片的免疫螢光顯微鏡下可顯示整合素 α3 表現的缺如。電子顯微鏡顯示透明板內分離,如同其他 JEB 亞型。¹¹⁹
營養不良型表皮鬆解症 (DYSTROPHIC EPIDERMOLYSIS BULLOSA)
營養不良型 EB(dystrophic EB, DEB)特徵為水疱癒合時伴隨瘢痕形成與粟粒疹形成。DEB 可以自體隱性(RDEB)或顯性(DDEB)方式遺傳。區分這兩種亞型最重要的原因之一,是侵襲性鱗狀細胞癌(squamous cell carcinoma, SCC)的盛行率增加與隱性型相關,但與顯性型無關。無論遺傳模式為何,DEB 源自稱為錨定纖維的超微結構實體的缺陷,導致緻密板下 (sublamina densa) 分離。
局部型自體顯性營養不良型表皮鬆解症 (LOCALIZED AUTOSOMAL DOMINANT DYSTROPHIC EPIDERMOLYSIS BULLOSA)
顯性 DDEB 的局部型亞型(有時稱為 DDEB 的 Cockayne-Touraine 型)可於出生時出現,但偶爾要到兒童期才被察覺。雖然有時可發生廣泛性水疱,尤其在生命早期,但水疱通常局限於反覆創傷的區域,例如膝蓋、薦部 (sacrum) 與肢端表面(圖 60-9 與 60-10)。這些區域顯示特徵性的瘢痕性、營養不良性外觀。瘢痕往往為增生性。粟粒疹是這些病人癒合過程的常見伴隨特徵(圖 60-11)。指甲營養不良與指甲喪失伴隨遠端指趾的萎縮性瘢痕很常見。偶爾,指甲異常可能是 DDEB 唯一的表現異常。口腔病灶不常見,牙齒通常不受影響。這些病人預後良好且壽命正常。
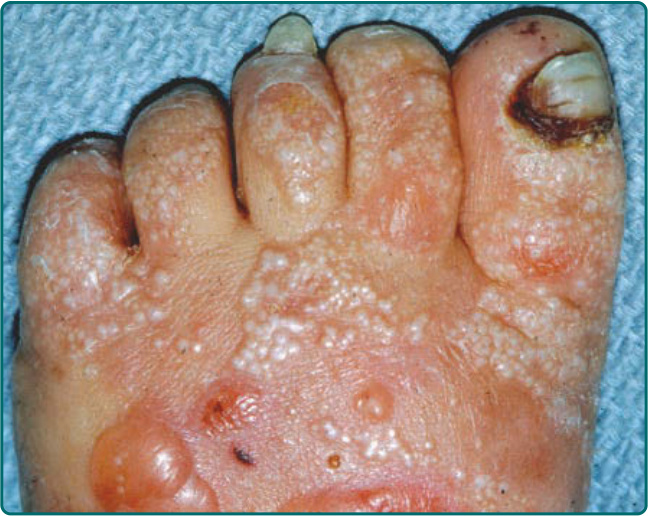
圖 60-9:一名罹患顯性營養不良型表皮鬆解症 (dominant dystrophic epidermolysis bullosa) 病人的局部創傷誘發水疱伴繼發性粟粒疹。
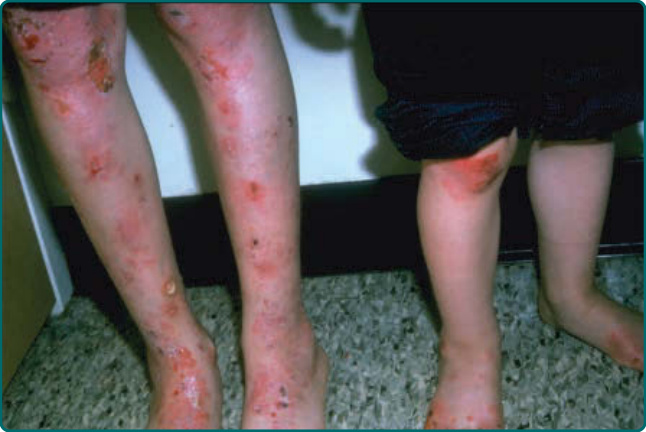
圖 60-10:罹患顯性營養不良型表皮鬆解症 (dominant dystrophic epidermolysis bullosa) 的兄弟姊妹的皮膚病灶。
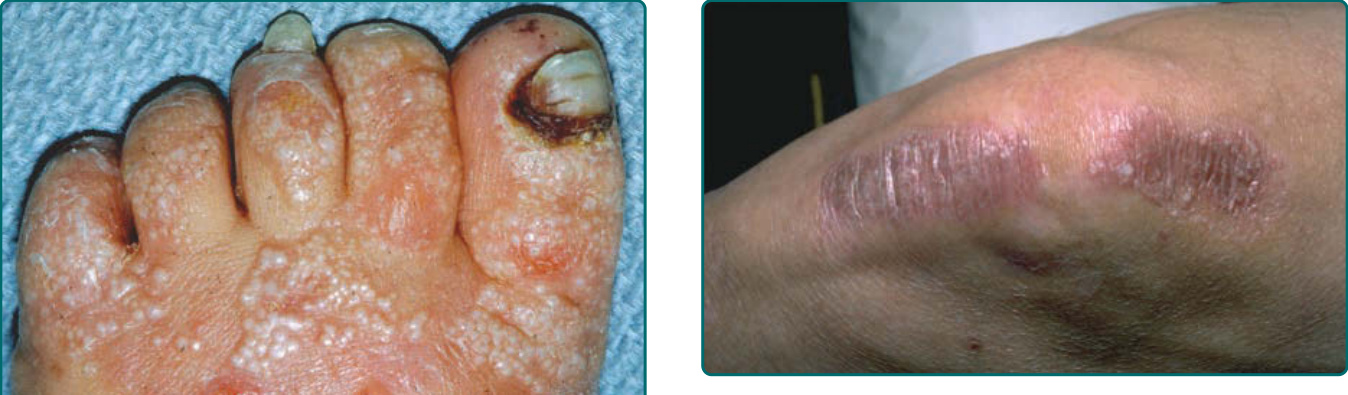
圖 60-11:一名罹患顯性營養不良型表皮鬆解症 (dominant dystrophic epidermolysis bullosa) 病人的粟粒疹 (milia)。
廣泛性自體顯性營養不良型表皮鬆解症 (GENERALIZED AUTOSOMAL DOMINANT DYSTROPHIC EPIDERMOLYSIS BULLOSA)
DDEB 的廣泛性形式(有時稱為 DDEB 的 Pasini 亞型)於出生時出現,與局部型亞型相比,水疱表型通常更嚴重且更廣泛。廣泛性 DDEB 的水疱以瘢痕斑塊與粟粒疹癒合,方式與其他 DEB 亞型相似。此外,本病有時以軀幹上獨特的瘢痕樣、膚色丘疹的自發性出現為特徵。這些白丘疹樣 (albopapuloid) 病灶並非特異性病徵,因為這些病灶也可見於其他 EB 亞型。隨著病人年齡增長,廣泛性水疱可能最終局限於四肢。病人常顯示營養不良或缺如的指甲。口腔糜爛往往可能存在,但通常不廣泛,而某些病人可見齒釉質缺陷。一種罕見的自發緩解性廣泛性 DDEB 變異型,稱為新生兒大疱性真皮鬆解 (bullous dermolysis of the newborn),包含於嬰兒期後逐漸消退的廣泛性水疱。¹²¹ 這些病人的皮膚切片在免疫螢光顯微鏡檢查時,常顯示基底表皮細胞質內第 VII 型膠原蛋白的堆積。¹²²
隱性營養不良型表皮鬆解症 (RECESSIVE DYSTROPHIC EPIDERMOLYSIS BULLOSA)
隱性 DEB(recessive DEB, RDEB)的嚴重度可能變化很大。雖然重度亞型最常見,但偶爾可見局部型,先前稱為 RDEB mitis。如同局部型 DDEB,局部型 RDEB 通常局限於反覆創傷的皮膚表面,最常為肢端分布。瘢痕與粟粒疹形成伴隨水疱的癒合。局部型 RDEB 的黏膜侵犯,若存在,為輕微。重度 RDEB,先前以人名命名為 Hallopeau-Siemens 型,是一種毀滅性的疾病(圖 60-12)。本病於出生時出現廣泛性水疱。偶爾,出生時即有整片皮膚區域的廣泛脫落 (denudation),常侵犯其中一個肢體。這種先天性皮膚缺如有時稱為 Bart 症候群 (Bart syndrome)。嬰兒期發生的癒合與水疱循環可導致進行性瘢痕,可能變得相當廣泛。手指在皮膚「連指手套 (mitten)」中閉合所造成的假性併指 (pseudosyndactyly),是本病極為常見的特徵(圖 60-13)。瘢痕可導致手部(圖 60-14)以及肢體的屈曲攣縮。與重度 JEB 病人不同,這些病人不顯示顯著的孔口周圍侵犯。取而代之的是,頭皮是這些病人頭頸部最常受影響的區域。口咽可在顯性與隱性 DEB 中廣泛受侵犯(圖 60-15),廣泛性糜爛演變為限制舌頭活動並使口腔開口變窄的瘢痕。牙齒可顯示顯著的齒釉質點蝕,齲齒可能廣泛,導致牙齒喪失。氣管或喉的侵犯可導致氣道狹窄,可能需要以氣切介入。食道的黏膜糜爛可導致狹窄 (stricture) 形成與蹼狀 (webbing)。口腔病灶、齲齒、食道狹窄,以及廣泛傷口癒合所需的熱量需求增加的組合,可使這些病人趨向營養不良與生長遲滯。這些病人通常有貧血問題,並可能顯示鐵吸收缺陷。在較早的過去,大多數重度 RDEB 病人於嬰兒期死於敗血症與廣泛水疱的其他併發症。較近期,隨著改善的營養、感染與傷口支持,這些病人通常可存活至青少年期或成年期。然而,青春期後,另一種毀滅性併發症 SCC 可能且往往會出現(圖 60-16)。
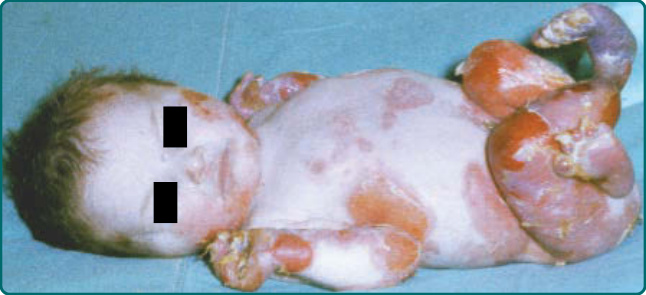
圖 60-12:一名罹患隱性營養不良型表皮鬆解症 (recessive dystrophic epidermolysis bullosa) 病人出生時的廣泛性水疱伴局部皮膚缺如。
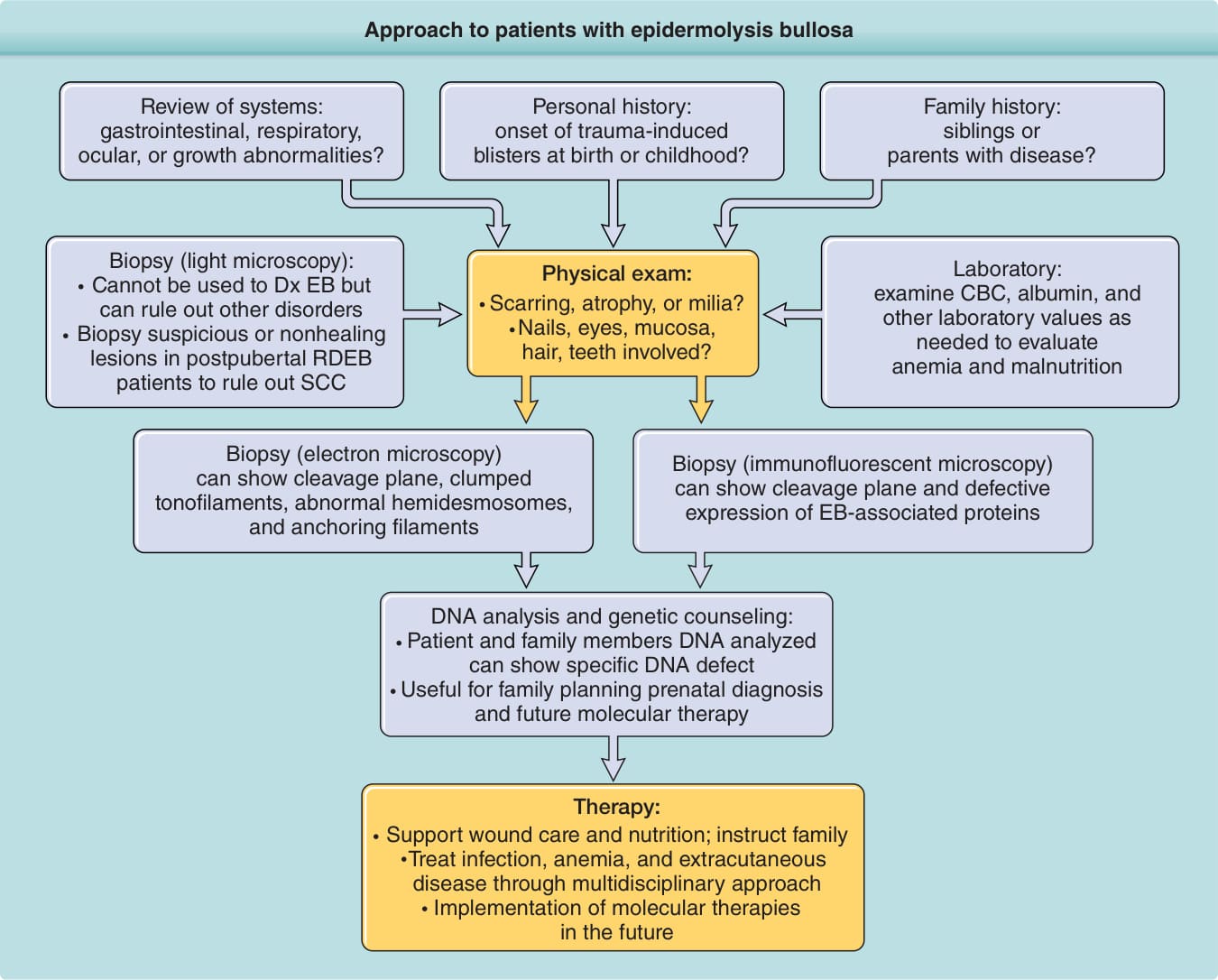
圖 60-13:表皮鬆解症 (EB) 病人的處理取徑。完整的病史,包括家族史與系統回顧,至關重要。理學檢查可提供重要線索,而實驗室數值可協助辨識相關的貧血與營養不良。電子顯微鏡或間接免疫螢光顯微鏡為診斷所必需。DNA 分析有助於決定預後與家庭計畫。治療主要為支持性。對護理人員,尤其是家屬的教導與合作至關重要,與其他專科在治療皮膚外併發症方面的跨領域互動亦然。CBC,全血球計數;Dx,診斷;RDEB,自體隱性表皮鬆解症;SCC,鱗狀細胞癌。
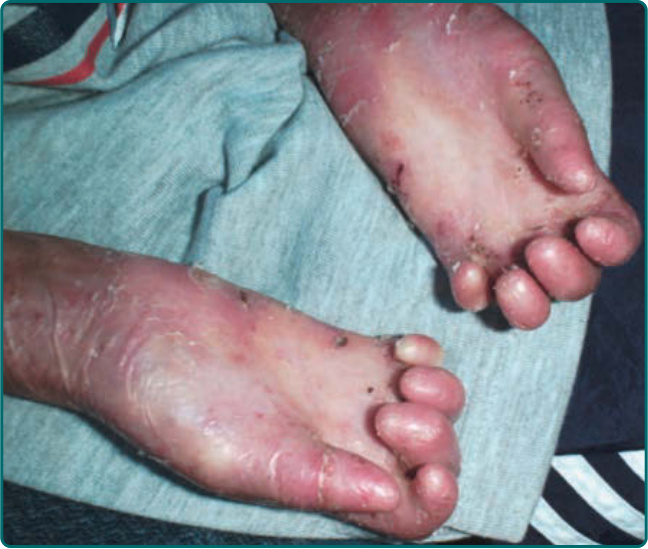
圖 60-14:一名罹患隱性營養不良型表皮鬆解症 (recessive dystrophic epidermolysis bullosa) 病人手部的瘢痕。
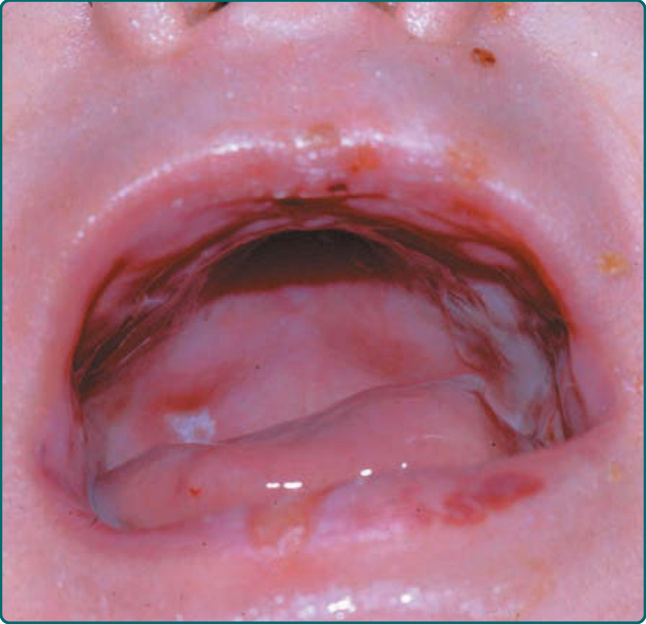
圖 60-15:一名罹患顯性營養不良型表皮鬆解症 (dominant dystrophic epidermolysis bullosa) 病人的口腔糜爛。
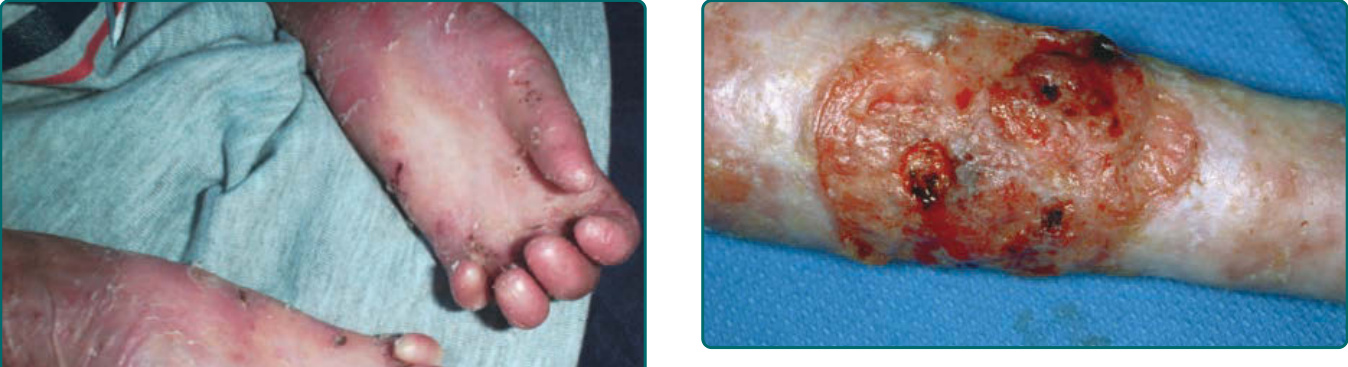
圖 60-16:一名罹患隱性營養不良型表皮鬆解症 (recessive dystrophic epidermolysis bullosa) 病人的鱗狀細胞癌 (squamous cell carcinoma)。
據估計,50% 至 80% 的重度 RDEB 病人最終會發展出這些癌,而其中許多人死於轉移性疾病。RDEB 相關的癌與大多數其他皮膚 SCC 不同,因為它們極具侵襲性,有強烈的侵犯與轉移傾向。
營養不良型表皮鬆解症的分子病理學 (MOLECULAR PATHOLOGY OF DYSTROPHIC EPIDERMOLYSIS BULLOSA)
DEB 病人存在錨定纖維的異常,範圍從某些顯性疾病病人的細微變化,到本病嚴重隱性型病人錨定纖維的缺如,且存在緻密板下水疱裂隙平面(見圖 60-4)。這些觀察與 DEB 病人的間接免疫螢光顯微分析相符,後者顯示顯性病人在真皮-表皮接合處有不同程度的線狀第 VII 型膠原蛋白染色,而重度隱性病人則完全缺乏染色。在某些病人中,存在第 VII 型膠原蛋白的細胞質滯留,可在病人切片與病人角質細胞分析中顯示出來。¹²³
迄今所有 DEB 病例皆已被證明與編碼第 VII 型膠原蛋白之基因(COL7A1)的突變相關。在隱性形式中,突變通常造成提早終止密碼子,導致組織中缺乏第 VII 型膠原蛋白。已知帶有提早終止密碼子的 mRNA 顯示加速的代謝週轉 (turnover)。¹²⁴ 此外,未被分泌或未組裝進錨定纖維的截短蛋白質也可能顯示加速的代謝週轉。這兩種機轉之一或兩者皆可解釋帶有產生提早終止密碼子之突變的重度 RDEB 個體組織中缺乏可偵測之第 VII 型膠原蛋白。⁶¹,¹²⁵,¹²⁶ 然而,即使在免疫螢光顯微鏡下顯示第 VII 型膠原蛋白組織染色缺如的個體中,病人角質細胞的分析在許多情況下仍可顯示低量的突變型第 VII 型膠原蛋白表現。¹²⁷,¹²⁸ 近期,除了 JEB 病人中報告的病例外,RDEB 中也報告了回復性鑲嵌 (revertant mosaicism) 表型。¹²⁹
一般而言,不造成提早終止密碼子的 COL7A1 突變產生較不嚴重的疾病。¹⁰⁸,¹¹⁴ 例如,產生三股螺旋區域甘胺酸取代的突變會干擾第 VII 型膠原蛋白分子的三股螺旋組裝。這類突變存在於許多罹患本病較輕微顯性形式的病人中。在這些病人中,第 VII 型膠原蛋白分子可能無法呈現聚合成錨定纖維所需的正確構形。一種與搔癢增加相關的 DEB 亞型,即癢疹性 EB (EB pruriginosa),也已被描述與甘胺酸突變相關。¹³⁰ 其他 COL7A1 突變已被證明與第 VII 型膠原蛋白分泌受損相關,導致此分子的細胞內堆積。在一項研究中,涉及編碼第 VII 型膠原蛋白 NC2 結構域之基因區域的 DEB 病人突變,被證明會干擾 NC2 加工與錨定纖維的組裝。¹³¹
Kindler 症候群 (KINDLER SYNDROME)
我們對皮膚分子病理學理解的新進展,揭示了一種與 EB 相關疾病——Kindler 症候群——的潛在病理生理學。Kindler 症候群最早由 Theresa Kindler 於 1954 年描述。¹³² 其特徵為出生時與嬰兒期創傷誘發的類 EB 水疱,癒合過程中有令人聯想到 JEB 或 DEB 的萎縮性變化。¹³³⁻¹³⁹ 然而,在兒童期後期,水疱通常消退,讓位給一種進行性異色症 (poikiloderma),分布於日曬部位。異色症可顯示萎縮與角化過度的區域,以及色素減退、色素過度沉著與微血管擴張 (telangiectasias)。這些病人常顯示光敏感性 (photosensitivity)。有時也存在指甲變化以及腳趾與手指的蹼狀。內部併發症包括口腔發炎、食道或輸尿管狹窄,以及瞼外翻 (ectropion)。在超微結構上,這些病人顯示基底膜的重複 (reduplication),這是所見最一致的特徵。雖然往往有伴隨錨定纖維異常的緻密板下裂隙,但有時可見到與水疱表型相關的透明板或表皮內分離。本病的分子研究促成了一種新表皮蛋白——kindlin-1 的發現,其在本病中以免疫螢光顯微鏡顯示表現減少。kindlin-1 似乎與訊號傳遞蛋白(如 talin)有些同源性,這暗示其有訊號傳遞功能,但其在 Kindler 症候群中的角色仍不清楚。¹⁴⁰ 已描述若干編碼 kindlin-1 之基因 KIND1 的突變。¹⁴¹⁻¹⁴³ 這些包括無義、移碼 (frameshift) 與剪接位點突變,這些是受影響皮膚中所觀察到的 kindlin-1 表現減少的根本原因。為何本病從水疱表型演變為異色症表型,以及 kindlin-1 在表皮恆定中的確切功能,仍有待充分闡明。
診斷 (DIAGNOSIS)
診斷 EB 的第一步從詳盡的病史與理學檢查開始(見圖 60-13)。有用的病史資訊包括水疱發病的年齡,以及其他家族成員是否存在水疱。胃腸道(GI)、呼吸、眼部、牙齒與泌尿生殖系統的回顧很重要,整體生長與發育的評估亦然。理學檢查不僅需要完整的皮膚檢查,也需要對黏膜組織、毛髮、指甲與牙齒進行徹底評估。初次就診時重要的實驗室測量包括貧血評估,以及營養指標(例如血清白蛋白 serum albumin)的評估。皮膚切片是另一個重要的診斷步驟。常規組織學分析不能用以診斷 EB,但可用於排除其他水疱原因。真皮-表皮 BMZ 實在太小,無法以光學顯微鏡可視化。要區分皮膚切片中 BMZ 分離的層次,必須使用穿透式電子顯微鏡(transmission electron microscopy, TEM)或間接免疫螢光顯微鏡(indirect immunofluorescent microscopy, IDDF)。水疱內部會迅速再上皮化 (reepithelialize),這可能模糊水疱層次的正確判定。因此,切片一個盡可能新鮮的水疱至關重要。確保水疱新鮮的一種方法,是由臨床醫師在診間誘發它。這可藉由在病人完整皮膚區域上輕輕轉動鉛筆橡皮擦 (pencil eraser),直到可觀察到表皮與真皮的分離來達成。與較輕微疾病的病人相比,這在較嚴重疾病變異型的病人身上較易執行。實際進行切片時,最好放置圓形切片打孔器 (circular biopsy punch),使其僅 10% 覆蓋可見水疱、90% 覆蓋完整皮膚。這是因為在同一切片標本上同時有完整與水疱皮膚是有幫助的,且水疱很可能在切片過程中或運送過程中延伸。TEM 多年來被視為判定 EB 亞型水疱層次的黃金標準。除了判定水疱層次外,超微結構實體也可由 TEM 分析其特徵性改變。例如,基底角質細胞細胞質中角蛋白中間絲的叢集,是重度廣泛性(Dowling-Meara)EBS 的特異性病徵 (pathognomonic finding)。發育不全的半橋粒可能是診斷 JEB 的重要線索。錨定纖維缺如或改變常發生於 DEB 亞型,尤其是隱性型。IDDF 顯微鏡可提供關於水疱層次的額外資訊,以及對潛在分子缺陷的重要線索。在此技術中,將針對已知 BMZ 抗原的抗體組套施用於病人水疱皮膚的冷凍切片上。抗原定位於水疱的表皮或真皮部分,指示 BMZ 中皮膚分離的層次。例如,在 EBS 樣本中,細胞內半橋粒組分(如 BP230)與緻密板蛋白(如第 IV 型膠原蛋白)會各自定位於水疱底部。在 JEB 病例中,BP230 會定位於水疱頂部,而第 IV 型膠原蛋白會定位於水疱底部。在 DEB 中,第 VII 型膠原蛋白與 BP230 會定位於水疱頂部。病人完整皮膚冷凍切片中以特定抗體染色的特異性缺如或存在,提供了關於特定分子缺陷的重要線索。缺乏與層黏連蛋白-332 特異性抗體染色的樣本會進一步支持 JEB 診斷,而缺乏第 VII 型膠原蛋白染色會支持 DEB 診斷。缺乏第 XVII 型膠原蛋白染色會支持廣泛性中度 JEB 診斷。支持 IDDF 為基礎之 EB 診斷的完整抗體組套,會包括針對層黏連蛋白-332(Herlitz 與非 Herlitz JEB)的抗體,以及針對 BP180/第 XVII 型膠原蛋白(非 Herlitz JEB 或廣泛性萎縮性良性 EB)、第 VII 型膠原蛋白(RDEB)、α6 與 β4 整合素(JEB 伴幽門閉鎖)、網蛋白(EBS 伴肌肉失養症),以及角蛋白 5 與 14(隱性 EBS)的抗體。針對層黏連蛋白-332 個別 α3、γ2 與 β3 鏈的抗體尤其有幫助。已知層黏連蛋白-311(與層黏連蛋白-332 共享相同的層黏連蛋白 α3 鏈)在與編碼 β3 鏈(LAMB3)及 γ2 鏈(LAMC2)之基因無效突變相關的 Herlitz JEB 中表現,但在與 α3 鏈基因(LAMA3)無效突變相關的 Herlitz JEB 中則不表現。⁴⁴ 因此,若層黏連蛋白 β3 與 γ2 抗體為陰性而 α3 抗體為陽性,這可將基因分析導向檢查 LAMB3 與 LAMC2。相反地,若 IDIF 下三條層黏連蛋白-332 鏈皆缺如,這可將基因研究導向 LAMA3,節省抵達最終分子診斷的時間與精力。基因突變分析徹底改變了我們對 EB 疾病家族的理解,並被視為抵達 EB 分子診斷的最終步驟。我們對 BMZ 蛋白生化結構與超分子組裝知識的並行進展,既促進也補充了分子生物學研究。因此,EB 病人診斷需要臨床與分子兩方面的資訊。從病人以及父母與兄弟姊妹採集血液樣本或頰拭子 (buccal swabs) 進行基因分析。隨著此作法日益普及,全外顯子定序 (whole-exome sequencing) 可能會使 EB 突變偵測更容易、更快速且更便宜。¹⁴⁴
EB 病人的處理取徑包括下列要素(依圖 60-13 所示流程譯出):
- 系統回顧:是否有胃腸道、呼吸、眼部或生長異常?
- 個人史:創傷誘發的水疱是於出生時或兒童期發病?
- 家族史:兄弟姊妹或父母是否罹病?
- 切片(光學顯微鏡):不能用以診斷 EB,但可排除其他疾病。對青春期後 RDEB 病人的可疑或不癒合病灶進行切片,以排除 SCC。
- 理學檢查:有瘢痕、萎縮或粟粒疹嗎?指甲、眼睛、黏膜、毛髮、牙齒是否受侵犯?
- 實驗室:檢查 CBC、白蛋白與其他必要的實驗室數值,以評估貧血與營養不良。
- 切片(電子顯微鏡):可顯示裂隙平面、叢集的張力絲 (tonofilaments)、異常的半橋粒與錨定絲。
- 切片(免疫螢光顯微鏡):可顯示裂隙平面與 EB 相關蛋白的表現缺陷。
- DNA 分析與遺傳諮詢:對病人與家族成員的 DNA 進行分析,可顯示特定的 DNA 缺陷。對家庭計畫、產前診斷與未來分子治療有用。
- 治療:支持性傷口照護與營養;教導家屬。透過多領域取徑治療感染、貧血與皮膚外疾病。未來實施分子治療。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 60-2。

表 60-2:表皮鬆解症的鑑別診斷 (Differential Diagnosis of Epidermolysis Bullosa)
依原表 60-2 譯出:
最可能 (Most Likely)
- 汗皰疹 (Pompholyx)
- 蟲咬 (Insect bites)
- 摩擦性水疱 (Friction blisters)
- 熱燒傷 (Thermal burns)
- 大疱性膿痂疹 (Bullous impetigo)
應考慮 (Consider)
- 兒童慢性大疱性皮膚病(線狀免疫球蛋白 A 疾病)(Chronic bullous dermatosis of childhood [linear IgA disease])
- 大疱性類天疱瘡 (Bullous pemphigoid)
- 後天性表皮鬆解症 (Epidermolysis bullosa acquisita)
- 大疱性全身性紅斑性狼瘡 (Bullous systemic lupus erythematosus)
- 瘢痕性類天疱瘡 (Cicatricial pemphigoid)
- 尋常性天疱瘡 (Pemphigus vulgaris)
務必排除 (Always Rule Out)
- 史蒂芬斯-強生症候群 (Stevens-Johnson syndrome)
- 毒性表皮壞死溶解症 (Toxic epidermal necrolysis)
遺傳諮詢 (GENETIC COUNSELING)
DNA 突變分析對 EB 病人極為有幫助。對病人的預後益處往往可能相當顯著。例如,隱性 DEB 病例的水疱活性可能與較嚴重的顯性病例相當,但其侵襲性 SCC 的風險大得多。透過 DNA 診斷,這兩組可被區分,從而辨識出潛在有侵襲性 SCC 風險的病人。在受影響家族中對 EB 進行產前診斷可能是一種極為準確的技術,尤其是若原始先證者 (proband) 先前已進行突變分析或缺陷基因的鑑定。具有增加流產風險的胎兒皮膚切片與胎兒鏡 (fetoscopy),現在可藉由早至妊娠 8 至 10 週的絨毛膜絨毛採樣 (chorionic villus sampling)¹⁴⁵ 或第二孕期的羊膜穿刺 (amniocentesis)¹⁴⁶ 分析而避免。EB 候選基因中高度資訊性的基因內與側翼多型性 DNA 標記的發展,連同遺傳「熱點 (hotspots)」的快速篩檢,使高風險懷孕的基因篩檢成為一個可行的選項。¹¹⁰,¹⁴⁷ 將體外受精 (in vitro fertilization) 技術與 EB 產前診斷結合,胚胎著床前診斷 (preimplantation diagnosis) 現已成功施行於 EB 病例。¹⁴⁸ 另一個產前診斷的有前景領域,具有對 EB 的潛在未來應用,是偵測與分析母體循環中的胎兒細胞。¹⁴⁹
治療 (TREATMENT)
EB 的大多數治療在本質上為支持性。治療方案依皮膚與全身侵犯的嚴重度及範圍量身訂做,通常包含傷口管理、感染控制、必要時的外科處理,以及營養支持的組合。某些 EB 亞型的皮膚照護與其他器官系統的支持照護,最理想是透過多領域取徑來協調。全面的局部治療是 EB 治療的支柱,以避免創傷為首要目標。傷口癒合會因內源性因子而受損,包括異物、細菌、營養缺乏、貧血與反覆創傷。因此,優化 EB 病人的傷口癒合涉及對所有這些因子的控制。¹⁵⁰
支持性皮膚照護 (SUPPORTIVE SKIN CARE)
廣大的皮膚脫落區域可導致角質層所提供之屏障的喪失。隨後的微生物滲透可導致血清與濕氣的累積,進一步增強細菌繁殖。上述因子結合免疫抑制治療,促進了感染的發展。預防感染顯然是較佳的策略。改良型 Dakin 溶液(0.025% w/v 次氯酸鈉 sodium hypochlorite)可有助於減少病人皮膚的細菌負荷。在更換敷料前將傷口浸泡於此溶液中 20 分鐘,也有助於鬆開已乾燥黏附於傷口床上的繃帶。浸泡後,傷口可敷以莫匹羅星 (mupirocin) 或其他局部抗生素,並以半閉合性非黏性敷料覆蓋。膠帶會造成皮膚進一步水疱與脫皮,因此使用自黏(緊貼)紗布或自黏紙來固定非黏性敷料至關重要。對於罹患廣泛性或局部型 EBS 亞型的病人,控制熱暴露可能有助於控制水疱形成。也建議建議病人使用柔軟、通風良好的鞋子。罹患 Herlitz JEB、缺乏功能性層黏連蛋白-332(一種已被證明參與角質細胞黏附與遷移的細胞外基質蛋白)的病人,傷口癒合可能有特別困難的問題。對於罹患 DEB 的病人,使用手指夾板 (finger splinting)、勤於包裹手部,以及適當的手部創傷防護有所助益,尤其在手部手術後(見後文)。
感染 (INFECTION)
皮膚感染的管理是 EB 病人照護的關鍵部分。廣大的皮膚脫落區域(EB 病人的常見發現)對微生物滲透提供了不充分的屏障,並可導致皮膚感染以及更具毀滅性的併發症——敗血症。金黃色葡萄球菌 (Staphylococcus aureus) 與化膿性鏈球菌 (Streptococcus pyogenes) 是常見的感染原。綠膿桿菌 (Pseudomonas aeruginosa) 的格蘭氏陰性感染也可能發生。傷口感染時有指徵進行皮膚培養與使用適當的全身性抗生素。溫和的漩渦浴療法 (whirlpool therapy)、頻繁(每日)更換敷料、稀釋氯浴 (dilute chlorine baths)、輪替局部抗生素,以及使用如碘-優碘 (iodine-povidone) 等局部消毒劑,都是減少抗藥性細菌的有用方法。
外科治療 (SURGICAL TREATMENT)
在 EB 病人族群中,罹患重度隱性 DEB(Hallopeau-Siemens)變異型者,通常最需要外科介入。¹⁵¹ 這些病人的連指手套樣假性併指可手術鬆解;然而,由於此病況強烈的復發傾向,此手術可能必須週期性地重複。¹⁵¹⁻¹⁵⁴ 手術後的夾板固定對減少手部畸形的復發至關重要。手術也可用於矯正肢體、口周與會陰攣縮畸形,但不幸的是,高復發率很常見。EB 病人在插管期間必須格外小心,以盡量減少對口腔黏膜的創傷。
腫瘤 (TUMORS)
SCC 常於青春期後出現於隱性 DEB 病人中。SCC 可能在多個原發部位出現,尤其在不癒合區域。對不癒合區域的仔細監測至關重要,因為病人往往死於轉移性疾病。¹⁵⁵ 使用 Mohs 或非 Mohs 取徑的外科切除是重要的第一線方式,在某些病例中放射治療作為有用的輔助。¹⁵⁶ 異維 A 酸 (Isotretinoin) 已用於 RDEB 病人以化學預防 (chemoprevention) SCC。雖然它似乎耐受性良好,但尚不清楚它是否能增加這些病人的整體存活率。¹⁵⁷ 西妥昔單抗 (Cetuximab) 治療已被證明能提供一些益處¹⁵⁵,¹⁵⁸,但其皮膚副作用有時可能造成問題。
皮膚外侵犯的照護 (CARE FOR EXTRACUTANEOUS INVOLVEMENT)
胃腸道處理 (GASTROINTESTINAL MANAGEMENT)
食道病灶往往是隱性 DEB 以及重度與中度廣泛性變異型 JEB 中最致殘的併發症。食道狹窄通常對擴張術 (dilation) 有反應;然而,擴張後狹窄復發很常見。¹⁵⁹ 結腸間置術 (Colonic interposition) 已被證明在晚期病例有效,但很少使用。胃造口管置入已能有效為食道狹窄的個體提供營養。¹⁶⁰ 增加液體與纖維攝取以及糞便軟化劑,對於有便秘與結腸炎的 EB 病人也可能有價值。¹⁶¹
眼部病灶 (EYE LESIONS)
罹患 EBS 的病人,特別是 Dowling-Meara 亞型者,可能經歷眼瞼的反覆發炎,伴隨結膜的大疱性病灶。罹患 JEB 的病人與罹患隱性 DEB 的病人,可能經歷伴隨瘢痕的角膜潰瘍、淚管 (tear ducts) 閉塞與眼瞼病灶。¹⁶² 瘢痕性結膜炎 (Cicatricial conjunctivitis) 也可發生於隱性 DEB 病人。角膜糜爛以支持性方式治療,施用抗生素藥膏,並使用睫狀肌麻痺劑 (cycloplegic agents) 以減少睫狀肌痙攣並提供舒適。保濕腔室 (Moisture chambers) 與眼部潤滑劑也常用。嚴重受影響的上眼瞼可以全層皮膚移植 (full-thickness skin grafting) 進行外科處理。EB 病人任何眼部疾患的完全矯正都難以達成。EB 病人眼部病灶的妥善處理必須包括眼科醫師的協助,以預防嚴重的視力損害。¹⁶³ 外科與眼表面重建可有助於減少喉-甲-皮症候群中的肉芽組織。¹⁶⁴
口咽病灶 (OROPHARYNGEAL LESIONS)
良好的牙齒衛生對 EB 病人至關重要,而定期看牙醫尤其重要。JEB 病人的齒釉質缺陷,以及重度 JEB 與 DEB 病人刷牙與使用牙線時的疼痛,往往導致齲齒。¹⁶⁵,¹⁶⁶ 應使用市面上最柔軟的牙刷進行常規清潔。口腔黏膜水疱也可能伴隨 JEB 與 DEB 的形式,尤其在重度 DEB 亞型(如重度廣泛性隱性型)中,口腔開口變窄(小口症 microstomia)以及瘢痕誘發的舌頭活動受限(舌繫帶過短 ankyloglossia)可能特別致殘。生理食鹽水沖洗對黏膜表面的溫和清潔有效。應避免含酒精或其他刺激性製劑的漱口水。涉及氣管與喉的糜爛與瘢痕,伴隨隨之而來的氣道狹窄。在有氣道侵犯的病人中,有肺部吸入 (pulmonary aspiration) 的危險。¹⁶⁷ 外科插管應由具有照護 EB 病人經驗的麻醉科醫師,以小口徑氣道輕柔地進行。¹⁶⁸
營養、貧血與心血管疾病 (NUTRITION, ANEMIA, AND CARDIOVASCULAR DISEASE)
營養評估與支持在 EB 病人中可能至關重要¹⁶⁹,原因有數個。廣泛的皮膚損傷伴隨血流動力學與代謝反應的顯著改變,熱量與蛋白質需求增加。口咽與 GI 病灶對營養福祉構成最大的整體威脅。這些包括口腔水疱、異常的食道蠕動、狹窄、吞嚥困難 (dysphagia)、腹瀉、吸收不良 (malabsorption) 與牙齒問題。營養評估必須將這些因子納入考量,以發展補充方案來補足營養缺乏。¹⁷⁰ 病人往往無法增加食物攝取以平衡此增加的熱量需求。例如,JEB 中的齒釉質發育不全可能導致蛀牙、黏膜水疱與口腔念珠菌病 (oral candidiasis)。所有這些潛在併發症都可能損害病人進食的能力。GI 道中廣泛的內部黏膜脫黏可能造成異常的 GI 蠕動,狹窄與腹瀉是可能導致鐵與其他營養素吸收不良的併發症。慢性病貧血 (Anemia of chronic disease) 確實可影響所有重度 EB 亞型。隱性 DEB 病人往往顯示特別嚴重的缺鐵,可能對口服鐵補充無反應。對於這些病人,非經腸鐵劑 (parenteral iron) 可有所幫助。此外,若在缺鐵病人中見到對鐵補充缺乏網狀紅血球反應 (reticulocyte response),評估紅血球生成素 (erythropoietin) 濃度並在必要時以重組紅血球生成素治療會有所幫助。¹⁷¹ 輸血在 EB 貧血的治療中也有用,尤其當症狀需要快速矯正時。擴張型心肌病 (Dilated cardiomyopathy) 是重度 DEB 與 JEB 病人一種毀滅性且潛在致命的併發症,與慢性貧血高度相關。¹⁷²
表皮鬆解症的心理層面 (PSYCHOLOGICAL ASPECTS OF EPIDERMOLYSIS BULLOSA)
罹患 EB 的病人,尤其是重度亞型,可能受慢性疼痛所苦。¹⁷³⁻¹⁷⁵ 雖然許多人儘管處於極為不利的條件下,似乎仍找到方法對生活維持出奇正面的展望,但其他人則陷入憂鬱。¹⁷⁶,¹⁷⁷ 罹患重度 EB 的病人也可能為其家庭與摯愛之人製造壓力。¹⁷⁸,¹⁷⁹ 因此,重要的是在憂鬱的警訊出現時加以辨識,並依需要與精神科醫師及臨床心理師以多領域取徑合作。支持性心理治療與病友支持團體聚會可在此方面幫助病人及其家庭。病人與家庭額外的支持來源,包括數個重要的病友組織,協助提供教育與支持,包括營養不良型表皮鬆解症研究協會 (Dystrophic Epidermolysis Bullosa Research Association) 與表皮鬆解症醫學研究基金會 (Epidermolysis Bullosa Medical Research Foundation)。
抗發炎治療 (ANTIINFLAMMATORY THERAPIES)
許多新興治療正為 EB 出現,列於圖 60-17 並於後文描述。
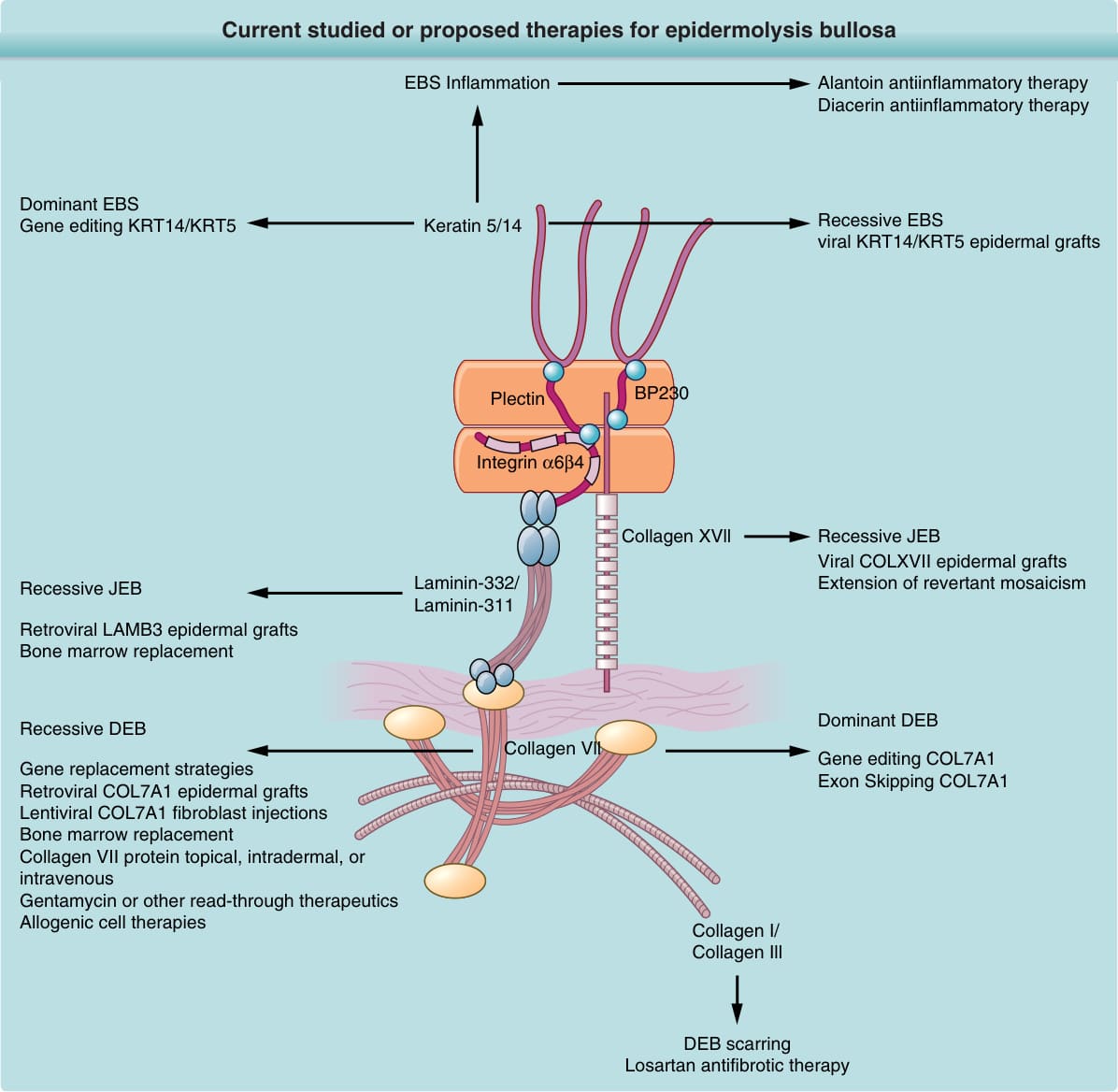
圖 60-17:目前研究中或建議用於表皮鬆解症的治療。DEB,營養不良型表皮鬆解症;EBS,表皮鬆解症單純型;JEB,接合部型表皮鬆解症。
反覆抓搔誘發的創傷以及旺盛的發炎性傷口,各自促成傷口的慢性化,尤其在罹患 EBS 發炎性亞型或癢疹性 EB 的病人中。雖然局部皮質類固醇 (topical corticosteroids) 可對搔癢產生短期緩解並減少發炎,但長期而言,這些製劑會誘導皮膚萎縮,加重 EB 皮膚的內在脆弱性,並惡化水疱/癒合。四環黴素 (Tetracycline) 與苯妥英 (phenytoin) 過去曾用於 EB,但目前並非適應的治療。¹⁸⁰ 大黃根 (rhubarb root) 的天然成分、介白素-β 阻斷劑雙醋瑞因 (diacerein)¹⁸¹ 被發現耐受性良好,並能減少罹患具發炎性皮膚表型之重度 EBS 病人的水疱形成。¹⁸² 洛沙坦 (Losartan),一種用於治療高血壓的小分子第 I 型血管收縮素 II 受體拮抗劑,在臨床前研究中已顯示出減少伴隨 DEB 傷口之纖維化 (fibrosis) 的前景。洛沙坦藉由減少皮膚中轉化生長因子-β (transforming growth factor-β) 的表現來達成此效果。¹⁸³
同種異體細胞治療 (ALLOGENEIC CELL THERAPIES)
同種異體細胞治療,包括同種異體角質細胞皮膚替代物 (allogeneic keratinocyte skin equivalents)¹⁸⁴、同種異體纖維母細胞注射 (allogeneic fibroblast injections)¹⁸⁵,¹⁸⁶,以及間質幹細胞輸注 (mesenchymal stem cell infusions)¹⁸⁷,已對 EB 傷口癒合顯示出正面的短期效果。然而,它們未能顯示任何長期益處。對一組七名隱性 DEB 兒童施行同種異體骨髓移植 (allogeneic bone marrow transplantation)。某些病人展現出顯著的臨床益處,以及真皮-表皮接合處第 VII 型膠原蛋白染色持續 1 年或更久;然而,錨定纖維的恢復並不完全。¹⁸⁸ 值得注意的是,此手術展現約 30% 的死亡率。促成所觀察到死亡率的因素,可能是廣泛皮膚糜爛與成功骨髓移植所需之免疫骨髓清除誘導的免疫抑制 (immunomyeloablative-induced immunosuppression) 兩者的組合。
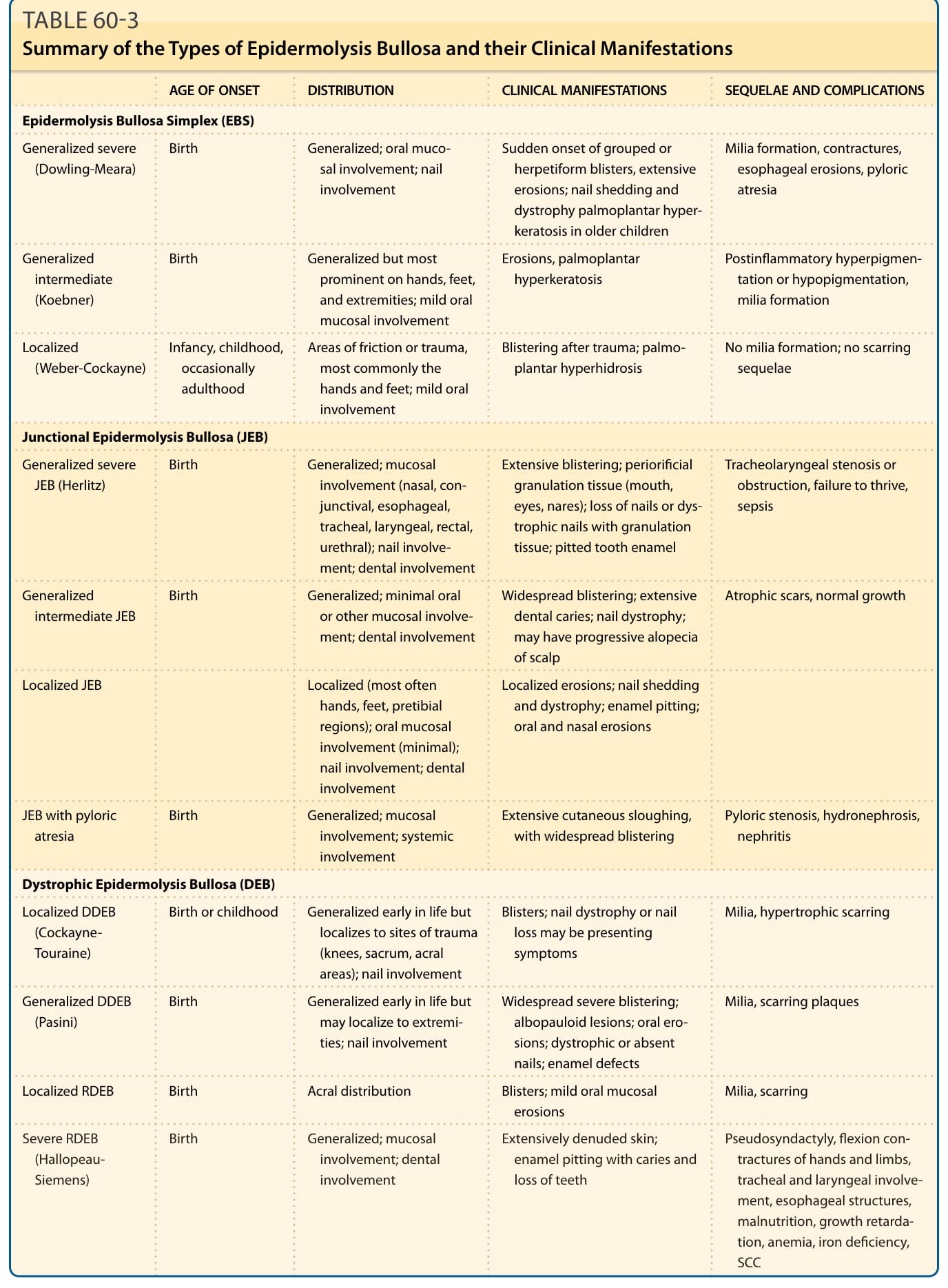
表 60-3:表皮鬆解症的類型及其臨床表現摘要 (Summary of the Types of Epidermolysis Bullosa and their Clinical Manifestations)
下表依原表 60-3 譯出(發病年齡、分布、臨床表現、後遺症與併發症):
| 類型 | 發病年齡 (AGE OF ONSET) | 分布 (DISTRIBUTION) 與臨床表現 (CLINICAL MANIFESTATIONS) | 後遺症與併發症 (SEQUELAE AND COMPLICATIONS) |
|---|---|---|---|
| 表皮鬆解症單純型 (EBS) | |||
| 廣泛性重度(Dowling-Meara) | 出生時 | 廣泛性;口腔黏膜侵犯;指甲侵犯 | 群聚或疱疹樣水疱的突然發作、廣泛糜爛;指甲脫落與營養不良;較大兒童的掌蹠角化過度 |
| 廣泛性中度(Koebner) | 出生時 | 廣泛性,但最顯著於手、足與四肢;輕微口腔黏膜侵犯 | 糜爛、掌蹠角化過度;發炎後色素過度沉著或色素減退、粟粒疹形成 |
| 局部型(Weber-Cockayne) | 嬰兒期、兒童期,偶爾成年期 | 摩擦或創傷區域,最常見於手與足;輕微口腔侵犯 | 創傷後水疱;掌蹠多汗症;無粟粒疹形成;無瘢痕後遺症 |
| 接合部型表皮鬆解症 (JEB) | |||
| 廣泛性重度 JEB(Herlitz) | 出生時 | 廣泛性;黏膜侵犯(鼻腔、結膜、食道、氣管、喉、直腸、尿道);指甲侵犯;牙齒侵犯 | 廣泛水疱;孔口周圍肉芽組織(嘴巴、眼睛、鼻孔);指甲喪失或伴肉芽組織的營養不良性指甲;點蝕齒釉質;氣管喉狹窄或阻塞、生長遲滯、敗血症 |
| 廣泛性中度 JEB | 出生時 | 廣泛性;極少口腔或其他黏膜侵犯;牙齒侵犯 | 廣泛水疱;廣泛齲齒;指甲營養不良;可能有頭皮進行性禿髮;萎縮性瘢痕、正常生長 |
| 局部型 JEB | — | 局部型(最常為手、足、脛前區域);口腔黏膜侵犯(極少);指甲侵犯;牙齒侵犯 | 局部糜爛;指甲脫落與營養不良;齒釉質點蝕;口腔與鼻腔糜爛 |
| JEB 伴幽門閉鎖 | 出生時 | 廣泛性;黏膜侵犯;全身性侵犯 | 廣泛皮膚脫落,伴廣泛水疱;幽門狹窄、腎水腫、腎炎 |
| 營養不良型表皮鬆解症 (DEB) | |||
| 局部型 DDEB(Cockayne-Touraine) | 出生時或兒童期 | 生命早期為廣泛性,但局限於創傷部位(膝蓋、薦部、肢端區域);指甲侵犯 | 水疱;指甲營養不良或指甲喪失可能為表現症狀;粟粒疹、增生性瘢痕 |
| 廣泛性 DDEB(Pasini) | 出生時 | 生命早期為廣泛性,但可能局限於四肢;指甲侵犯 | 廣泛重度水疱;白丘疹樣病灶;口腔糜爛;營養不良或缺如的指甲;齒釉質缺陷;粟粒疹、瘢痕斑塊 |
| 局部型 RDEB | 出生時 | 肢端分布 | 水疱;輕微口腔黏膜糜爛;粟粒疹、瘢痕 |
| 重度 RDEB(Hallopeau-Siemens) | 出生時 | 廣泛性;黏膜侵犯;牙齒侵犯 | 廣泛皮膚脫落;齒釉質點蝕伴齲齒與牙齒喪失;假性併指、手部與肢體屈曲攣縮、氣管與喉侵犯、食道狹窄、營養不良、生長遲滯、貧血、缺鐵、SCC |
DDEB,顯性營養不良型表皮鬆解症;RDEB,隱性營養不良型表皮鬆解症;SCC,鱗狀細胞癌。
自體基因治療 (AUTOLOGOUS GENETIC THERAPIES)
使用自體角質細胞的反轉錄病毒體外 (retroviral ex vivo) 基因治療曾施行於一名 JEB 病人。¹⁸⁹ 在此研究中,一名具有層黏連蛋白-332 誤義突變的病人,被移植了表現野生型層黏連蛋白-332、經基因矯正的角質細胞單層。經過 6.5 年的移植後追蹤,移植物仍顯示層黏連蛋白-332 的陽性表現以及對水疱的抗性。¹⁹⁰ 較近期,此治療以一種廣泛而卓越的方式擴展至另一名 JEB 病人,以經基因矯正的表皮移植物覆蓋超過 80% 的皮膚表面。¹⁹¹ 較近期對四名隱性 DEB 病人的臨床試驗施行了類似的取徑。¹²⁷ 在此研究中,編碼第 VII 型膠原蛋白的全長 COL7A1 基因被體外轉移至原代隱性 DEB 病人角質細胞。第 VII 型膠原蛋白工程化的病人細胞被擴增,然後以單層移植至病人傷口(每名病人六個移植物)。結果顯示臨床改善以及第 VII 型膠原蛋白表現的恢復與錨定纖維的形成,長達 1 年。透過 CRISPER/Cas9 的基因編輯結合誘導性多能幹細胞 (induced pluripotent stem cells) 的使用¹⁹²⁻¹⁹⁴,是另一種有前景的未來治療。外顯子跳躍 (Exon skipping) 已在 JEB¹⁹⁵ 以及 DEB 皮膚¹⁹⁶,¹⁹⁷ 中顯示出作為潛在未來治療的前景。藉由自體移植來擴展具有 COL17A1 與 COL7A1 自我矯正突變病人皮膚的潛力,也已被提議作為一種新型治療。¹⁹⁸
總結 (SUMMARY)
EB 類型及其臨床表現的摘要顯示於表 60-3。