Inherited Epidermolysis Bullosa
9
AT-A-GLANCE
■ Epidermolysis bullosa is a family of inherited genodermatoses characterized by blistering in response to minor trauma.
■ Blistering level categories are the simplex, hemidesmosomal, junctional, and dystrophic subtypes.
■ Cutaneous involvement varies from localized to widespread blistering depending on subtype.
■ Extracutaneous involvement varies from none to severely debilitating or lethal.
■ The oropharynx, trachea, esophagus, eyes, teeth, nails, hair can be involved depending on subtype.
■ Diagnosis is made by immunofluorescent or electron microscopy followed by DNA analysis.
INTRODUCTION
Inherited epidermolysis bullosa (EB, is a family of diseases with the common feature of blistering in response to mild trauma. Patients with EB can show blistering in the form of small vesicles or larger bullae, occurring on cutaneous surfaces and, in the severe forms, on mucosal tissues as well. Although skin and mucosa fragility and trauma-induced painful blisters are hallmarks across the EB spectrum, the distribution of the involvement, the depth of blister formation, any associated extracutaneous involvement, and the severity of the blistering process vary with the different EB subtypes and depend on the underlying heritable molecular defect. EB diseases vary also in the way in which blistered areas heal. The wound repair responses are often abnormal and can eventuate into chronic erosions, hypertrophic granulation tissue, scarring, or even invasive carcinoma. Although the milder EB subtypes are associated with a normal lifespan and little or no internal involvement, the most severe recessively inherited forms are mutilating, multiorgan disorders that threaten both the quality and length of life. A number of early studies identified the major subtypes of EB. Studies of von Hebra.1-3 were the first to distinguish pemphigus from inherited blistering, and the term epidermolysis bullosa hereditaria was first suggested by Koebner.4 Hallopeau was the first to distinguish between simplex (nonscarring) and dystrophic (scarring) forms of the disease,5 and Weber,6
and Cockayne,7 Dowling and Meara,8 and Koebner4 each described unique forms of epidermolysis bullosa simplex. Hoffman,9 Cockayne,10 Touraine,11, Pasini,12
and Bart13 provided much of the information about
subtypes of DEB. Herlitz described epidermolysis bullosas letalis,14 which was later found to be a part of the third major category of EB: the junctional form. The use of electron microscopy in EB diagnosis led to the studies of Pearson and collaborators,15 who classified the patients not only on the basis of clinical findings but also on the existence of ultrastructural changes. A comprehensive classification of EB based on a combination of ultrastructural and clinical findings was completed in an early landmark treatise by Gedde-Dahl.16 Recent major advances have led to the identification of protein and genetic abnormalities in most types of EB patients. These studies have led to an improved understanding of the biological basis of EB and culminating in the current EB classification based on genetic and protein defects,17 which provides a rational approach to specific molecular therapy.
ETIOLOGY AND PATHOGENESIS
OVERVIEW
OVERVIEW
EB arises from defects of attachment of basal keratinocytes to the underlying dermis. These defects can arise from inside the keratinocyte plasma membrane or extracellularly in the dermal-epidermal basement membrane zone (BMZ). Many tissues such as the skin and cornea, which are subjected to external disruptive forces, contain a complex BMZ composed of a group of specialized components that assemble into anchoring complexes (Fig. 60-1). At the most superior aspect of the BMZ, keratin-containing intermediate filaments of the basal cell cytoskeleton insert upon electrondense condensations of the basal cell plasma membrane termed hemidesmosomes. Anchoring filaments span the lamina lucida, connecting hemidesmosomes with the lamina densa and anchoring filaments. At the most inferior aspect of the BMZ, collagen VII– containing anchoring fibrils extend from the lamina densa into the papillary dermis and combine with the lamina densa and anchoring plaques, trapping interstitial collagen fibrils. Thus, the cutaneous BMZ connects the extensive basal cell cytoskeletal network with the abundant network of interstitial collagen fibrils in the dermis.18,19
KERATIN FILAMENTS
KERATIN FILAMENTS
Keratins are obligate heteropolymers that are composed of pairs of acidic and basic monomers. The
9
Components of the dermal-epidermal basement membrane
Hemidesmosome
Keratin 5/14
Plectin
BP230
α6β4 Integrin
Cell membrane
Collagen XVII
Laminin-332/ laminin-311
Lamina densa
Collagen VII
Interstitial collagen fibrils
Intermediate filaments
Hemidesmosome
Anchoring filaments
Lamina densa
Anchoring fibrils
keratin pair 5 and 14 assemble to form the extensive intermediate filament network of the basal cell cytoskeleton.20 Keratins contain a central α-helical rod with several nonhelical interruptions as well as nonhelical carboxy and amino terminal regions. The regions of highest conservation between the keratins are located on the ends of the keratin rod in the helix boundary motifs. Whereas extensive mutagenesis studies suggest that helical regions near the ends of the central rod are important in keratin filament elongation, the nonhelical domains may be important in forming lateral associations.21 Keratin intermediate filaments insert upon electron-dense structures known as hemidesmosomes.
HEMIDESMOSOMES
HEMIDESMOSOMES
Hemidesmosomes contain intracellular proteins including plectin and BP230. Plectin is a 500-kD protein that acts as an intermediate filament binding protein. It is possible that plectin also interacts with microfilaments because plectin contains a domain with similarity to the actin binding domain of spectrin.22,23 BP230, also known as BPAG1, is a 230-kD protein that has homology both to desmoplakin24 and to plectin. Several splicing variants of BP230 are of vital importance in the nervous system.25-27 BP230 localizes to a region referred to as the inner plate on the cytoplasmic surface of the hemidesmosome
1012
and like plectin functions in the connection between hemidesmosomes and intermediate filaments. BP230 negative transgenic mice lack a hemidesmosomal inner plate and the connection between hemidesmosomes and intermediate filaments is severed, creating a cytoplasmic zone of mechanical fragility just above the hemidesmosomes.
ANCHORING FILAMENTS
ANCHORING FILAMENTS
Hemidesmosomes also contain the transmembrane proteins collagen XVII (also termed BPAG2 and BP180)28 and α6β4 integrin.29 The cytoplasmic portions of these molecules make up part of the hemidesmosome-dense plaque, and the extracellular portions of these molecules make up portions of the anchoring filament and probably contribute to the structure known as the subbasal dense plate that underlies hemidesmosomes in the lamina lucida region. β4 Integrin only pairs with the α6 subunit, but the α6 subunit can combine either with the β4 integrin or with the β1 integrin. Both the α6β1 or α6β4 integrin combinations have been shown to act as receptors for laminins, and α6β4 integrin acts as a specific receptor for laminin-332. α6β4 Integrin plays a central role in organization of the hemidesmosome. The β4 integrin contains an especially large cytoplasmic domain, which functions in the interaction with other proteins of the hemidesmosomal plaque including collagen XVII and
plectin.30 Skin from transgenic mice lacking β4 integrin is devoid of hemidesmosomes and shows severe deficits in cell adhesion.31 Interactions between plectin and α6β4 integrin appear to be critical both in the assembly as well as the disassembly of hemidesmosomes.32
Collagen XVII (BPAG2, BP180) is a collagenous protein with a type II transmembrane orientation. Based on electron microscopy and crosslinking studies, collagen XVII assembles into a triple-helical homotrimer and contains three main regions: an intracellular amino-terminal globular head, a central rod, and an extracellular flexible tail.33 Collagen XVII associates with laminin-332 and α6β4 integrin in adhesion structures termed stable anchoring contacts formed by keratinocytes in vivo, which are thought to represent prehemidesmosomes.34 The autoantigen in linear immunoglobulin (Ig) A bullous dermatosis, LAD-1,35,36
is a 120-kD protein that has been shown by peptide sequencing to be the cleaved exodomain of collagen XVII.37 Collagen XVII undergoes processing in keratinocyte cultures and in skin through the action of sheddases, membrane-associated proteases that solubilize cell surface receptors.38,39
In addition to α6β4 integrin and collagen XVII, anchoring filaments contain the molecules laminin-332 and laminin-311. Like all members of the family of laminin proteins,40-42 laminin-332 is a large heterotrimeric molecule and contains α3, β3, and γ2 chains.43,44
The first laminins to be described contained 3 short arms and 1 long arm, forming a cross shape as shown by rotary shadowing analysis. In contrast, laminin-332 contains truncations of each short arm.45-47 Because of these short arm truncations, laminin-332 cannot self-polymerize with other laminins or bind to nidogen. Instead, laminin-332 forms a disulfide bonded attachment to laminin-311,48 the other known anchoring filament laminin43 that contains α3, β1, and γ1 chains. Laminin-332 also undergoes processing of its γ2 and α3 chains.49 Although rat laminin γ2 chain has been previously been shown to be processed by metalloproteinase-250 and membrane-type metalloproteinase type 1,51 the predominant site of cleavage by these enzymes is not conserved in human laminin-332.52
Other studies have shown that processing of laminin γ2 chain takes place through a special class of proteins termed C-proteinases, which also process C-terminal domains of procollagen molecules.53 Although one member of this class of proteins, bone morphogenic protein 1,54 is capable of performing this action, a splice variant of this protein termed mammalian tolloid is the enzyme that is predominantly expressed in keratinocytes and fibroblasts, and mammalian tolloid is the thus the enzyme that likely performs this function in the skin.52 Mammalian tolloid also processes the laminin α3 chain,52 although other enzymes such as plasmin,55 matrix metalloproteinase (MMP)-250 and membrane type MMP-151 are also capable of this function. The γ2 chain short arm appears important in the assembly of laminin-332 into basement membrane.56 The antigen recognized by monoclonal antibody (mAb) 19-DEJ-157 also localizes to anchoring filaments, but its molecular identity remains unknown.
9
ANCHORING FIBRILS
ANCHORING FIBRILS
Collagen VII is the major constituent of anchoring fibrils. Analysis of the deduced amino acid sequence of collagen VII58 reveals the presence of a long central collagenous region characterized by repeating Gly-X-Y sequences that contains a number of noncollagenous interruptions, including a 39–amino acid noncollagenous segment in the center of the helix that corresponds to the “hinge region” predicted by biochemical studies.59,60 These interruptions account for the flexibility of the collagen VII molecule and explain its ability to loop around and entrap dermal matrix molecules to provide its function of stabilizing the basement membrane to the underlying papillary dermis.61 A 50-kD component of anchoring fibrils has also been identified that appears to localize to the insertion sites of anchoring fibrils to the lamina densa.62
The 145-kD N-terminal end of collagen VII contains the largest noncollagenous domain that inserts onto the lamina densa and anchoring plaques. Collagen IV, the most abundant component of these structures, binds to the collagen VII NC1 domain. A direct interaction between anchoring filaments and anchoring fibrils exists from a specific interaction between the anchoring filament component laminin-332 and collagen VII NC1 domain.63,64 Collagen VII binds the β3 chain on laminin-332.63,65-67 This appears to be a critical factor in the maintenance of dermal–epidermal cohesion. Like all collagens, collagen VII assembles into a triple helix. Only one chain of collagen VII, the α1 chain, has been identified; one gene codes for an entire molecule, and thus collagen VII is a homotrimer. Collagen VII triple helices are joined together at their processed NC-2 globular domains to form antiparallel dimers.61,68 Processing of the NC-2 domains takes place via the same family of C-proteinases (bone morphogenic protein 1 and/or mammalian tolloid) that are known to process laminin-332, a closely associated molecule. Anchoring fibrils may derive from lateral associations of collagen VII antiparallel dimers.
CLINICAL FINDINGS
A classification of EB is made based on the ultrastructural level within which the cleavage plane of the blister occurs. EB has been traditionally classified according to the level of BMZ separation on transmission electron microscopy into simplex, junctional, and dystrophic subtypes69 (Fig. 60-2). Although traditionally the diagnostic “gold standard” for grouping EB has been electron microscopy, immunomapping of basement membrane antigens as viewed by indirect immunofluorescence can also be quite useful in distinguishing subtypes of EB. Within each of these groups, there are several distinct types of EB based on clinical, genetic, histologic, and biochemical evaluation.17 When possible, molecular characterization at the protein or DNA levels should be appended to the clinical subtype (termed the “onion skin approach”) to better delineate the diagnosis and to aid in delivery of emerging molecular therapies. These clinical subtypes are summarized in Table 60-1.
1013
9
Comparison levels of skin separation in EB with clinical findings
A B C
EBS
Keratins 5/14
EBS JEB
Plectin BP230
D
Integrin `6a4
Laminin-332/ Laminin-311
Collagen VII
DEB
E
Collagen XVII
F
JEB
Collagen I/ Collagen III
G H
I J K
1014
DISEASE AFFECTED GENE AFFECTED PROTEIN
Simplex
Generalized severe KRT5/KRT14 Keratin 5/keratin 14
Generalized intermediate RT5/KRT14 Keratin 5/keratin 14
Localized RT5/KRT14 Keratin 5/keratin 14
Mottled pigmentation KRT5 Keratin 5
EB with muscular dystrophy PLEC1 Plectin
Superficialis Unknown Unknown
Acantholytic DSP, JUP Desmoplakin, plakoglobin
Plakophillin deficiency PKP1 Plakophillin 1
Plakoglobin deficiency JUP Plakoglobin
Ogna PLEC1 Plectin
BP230 deficiency DST-e BP230/BPAG1
Exophillin-5 deficiency EXPH5 Exophillin 5
Acral peeling skin syndrome TGM5 Transglutaminase 5
Junctional
EB with pyloric atresiaa ITGΒ4/ITGΑ6/PLEC1 Integrin α6, integrin β4, plectin
Generalized severe LAMΒ3/LAMΑ3/ LAMC2 Laminin-332
Generalized intermediate LAMΒ3/LAMΑ3/ LAMC2/COL17A1 Laminin-332, collagen XVII
Localized LAMΒ3/LAMΑ3/ LAMC2/COL17A1 Laminin-332, collagen XVII
Inversa LAMΒ3/LAMΑ3/ LAMC2 Laminin-332
Laryngo-onychocutaneous LAMA3A Laminin α3
Respiratory and Renal ITGA3 Integrin α3
Dystrophic
Generalized dominant COL7A1 Collagen VII
Localized dominant COL7A1 Collagen VII
Generalized severe recessive COL7A1 Collagen VII
Generalized intermediate recessive COL7A1 Collagen VII
Localized recessive COL7A1 Collagen VII
Inversa recessive COL7A1 Collagen VII
Bullous dermolysis of the newborn COL7A1 Collagen VII
Kindler syndrome KIND1 Kindlin
Kindler syndrome KIND1 Kindlin
aAlternatively classified as simplex. EB, epidermolysis bullosa.
9
EPIDERMOLYSIS BULLOSA SIMPLEX
EPIDERMOLYSIS BULLOSA
SIMPLEX
Epidermolysis bullosa simplex (EBS) is a disease group characterized by intraepidermal blistering and most often is associated with keratin gene mutations. The range of disease phenotypes range from mild to severe among different subgroups.70 The common EBS types are dominantly inherited and include generalized severe (Dowling-Meara), generalized intermediate (Koebner), and localized (Weber-Cockayne). There are several uncommon varieties, which include EBS Ogna, EBS with muscular dystrophy, EBS with mottled pigmentation, and a group of suprabasal EBS subtypes.
GENERALIZED SEVERE EPIDERMOLYSIS BULLOSA SIMPLEX
This subtype is also known as the Dowling Meara, herpetiformis variant of EBS. It presents at birth, has a generalized distribution, and is regarded as the most severe of the EBS subtypes (Fig. 60-3). It differs from the generalized intermediate variant in that the oral mucosa is more often involved, occasionally showing extensive erosions. Milium formation may sometimes occur in infancy in patients with this subtype; however, this postwound phenomenon usually resolves later in childhood. The disease can often be associated with spontaneous appearance of grouped or “herpetiform” blisters. These occur on the trunk and proximal extremities and heal without scarring. It should be noted that often when patients show a generalized blistering, this herpetiform pattern is not seen; therefore, its absence should not be used as a basis to exclude this EB subtype. Generalized severe EBS often shows nail involvement. In this disease, nails may become shed and may regrow with dystrophy. Interestingly, although heat exacerbates the blistering in
1015
9
other EBS subtypes, it does not appear to have a major impact in generalized severe EBS. Hyperkeratosis of the palms and soles often develops beginning in early childhood and can progress to confluent keratoderma of the palms and soles. These can be quite painful, and occasionally interference with ambulation has led to flexural contractures. Occasionally, involvement of the esophagus in generalized severe EBS ranging from erosions to pyloric atresia71 has been reported. The upper respiratory tract can also be affected, including the laryngeal mucosa.72
GENERALIZED INTERMEDIATE EPIDERMOLYSIS BULLOSA SIMPLEX
The other common form of generalized EBS is the intermediate form, also known as the Koebner EBS. This subtype shows an onset of generalized blistering at birth or at latest during early infancy. The hands, feet, and extremities usually show the most involvement. Lesions often heal with postinflammatory hyper or hypopigmentation, and although occasional atrophy and milia can occur, they are rare and much less frequent than in generalized severe EBS. Palmar-plantar hyperkeratosis and erosions may be present (Fig. 60-4). Thickening of the soles is common but often does not present until later childhood. The oral mucosa sometimes shows mild erosive activity, but these usually improve with increasing age. Usually there is not a severe growth retardation in this EB subtype.
LOCALIZED EPIDERMOLYSIS BULLOSA SIMPLEX
This is a mild form of EB, previously referred to as the Weber-Cockayne subtype of EBS (Fig. 60-5). This disease
1016
is the most common form of EB and often presents during infancy or childhood. Occasionally, it presents in early adulthood, such as when blisters are noted after marching during military service. It is speculated that there are a number of undiagnosed cases of this form of EB because it can be mild enough to escape reporting or detection during clinical visits. Hyperhidrosis of the palms and soles is a common association. Blisters can occasionally become secondarily infected. Postinflammatory pigmentary abnormalities occur with this variant, but milia and scarring as a rule are absent. Blistering activity usually follows areas of trauma, with the hands and feet being the most common and the scalp being the least common. Mild oral erosions are present only rarely and usually resolve with increasing age. Nail involvement is rare with this EB subtype.
ADDITIONAL VARIANTS OF EPIDERMOLYSIS BULLOSA SIMPLEX Epidermolysis Bullosa Simplex of Ogna: Onset in infancy is common with seasonal blistering (summer) on the acral areas. Small hemorrhagic and serous blisters occur primarily on the extremities. Healing occurs without scarring. This disease was originally reported in patients from Norway. These patients also show a characteristic onychogryphosis of the great toenails.
Epidermolysis Bullosa Simplex with Muscular Dystrophy: This rare clinical entity is an autosomal recessive disorder that consists of generalized blistering of the skin at birth or shortly thereafter. This is accompanied by a progressive muscular dystrophy.73 It presents with generalized blistering
similar to generalized intermediate EBS. These patients have been shown to harbor mutations in the gene coding for HD1/plectin.74-76
Epidermolysis Bullosa Simplex with Mottled Pigmentation: This form of EB, as its name implies, is characterized by mottled hyperpigmentation of the trunk and proximal extremities. There is blistering in a generalized distribution beginning at birth or early infancy. Pigmentary alterations and blistering may improve with increasing age. Mild oral mucosal involvement may be present in infancy. This condition is distinct from the large melanocytic nevi, which can be seen in all three EB types.77
Epidermolysis Bullosa Simplex Superficialis: This is an uncommon form of EBS is named after the subcorneal separation that produces the blisters in this disease.78 Erosions and crusts, rather than intact bullae, are usually seen in these patients, and heal with postinflammatory pigmentary changes. Despite the superficial cleavage plane, nail involvement, atrophic scarring, and milia have been observed in this disease.
Acantholytic Epidermolysis Bullosa Simplex: This subtype79 is a rare recessively inherited and lethal disorder characterized by generalized erosions at birth. As in the superficialis subtype, blisters are not normally seen because of the very superficial level of epidermal separation, which has been described as sheetlike. Nails are dystrophic. Alopecia, neonatal teeth, oral erosions, and respiratory involvement distinguish this disorder from other superficial EBS subtypes.
Plakophilin Deficiency: This subtype, also known as ectodermal dysplasia skin fragility syndrome, is another inherited disorder of suprabasilar epidermal separation characterized by generalized erosions and sometimes superficial blisters at birth.80
Alopecia is also very common, as are palmoplantar keratoderma, painful fissures, and nail dystrophy. Patients may sometimes demonstrate failure to thrive, cheilitis, hypohidrosis, and pruitis. Like the other superficial EBS subtypes, this disorder is associated with dystrophic nails.
BP230 Deficiency: This is an extremely rare variant of EB caused by mutations of the DST-e gene, which has been described in a small collection of Kuwaiti and Iranian families. Blistering onset is usually at birth, and the phenotype is localized, often affecting the feet, without significant mucosal or other extracutaneous involvement. Like other forms of EBS, blisters may heal with postinflammatory pigment changes but no scarring. Inheritance appears autosomal recessive, but some heterozygous carriers have also reported selflimiting mild blistering in the first and second decades of life.
Exophillin-5 Deficiency: This is a very rare autosomal recessive EBS variant caused by mutations of the EXPH5 gene. Blistering is high in the epidermis, resulting in trauma-induced crusting or fragile
9
blisters and vesicles. The disease is overall mild and has no extracutaneous involvement, and remission or improvement during childhood is characteristic of this disease.81
Acral Peeling Skin Syndrome: This EB variant82
is characterized by superficial and painless skin peeling occurring most commonly on the hands and feet. Humidity, heat, and water exposure can exacerbate this condition. The level of separation histologically is between the stratum granulosum and stratum corneum.
MOLECULAR PATHOLOGY OF EPIDERMOLYSIS BULLOSA SIMPLEX
Most of the patients with EB simplex analyzed at the genetic level have been found to be associated with mutations of the genes coding for keratins 5 and 14.21,70 The level of separation of the skin in these patients is at the midbasal cell, shown in Fig. 60-4, associated with variable intermediate filament clumping. Hemidesmosomes and other BMZ structures are normal by electron microscopy. The majority of keratin gene mutations associated with EBS are dominantly inherited because of abnormalities in the multimeric assembly of keratin filaments. There is a smaller subset of patients with recessively inherited disease of varying severity.83,84
Mutations coding for the most conserved regions of keratins 5 and 14, the helix boundary domains.85 correlate with the most severe forms of EBS, such as the Dowling-Meara subtype, which exhibits intermediate filament clumping seen by transmission electron microscopy. On the other hand, milder types of disease, such as the Weber-Cockayne subtype, are associated with mutations coding for regions of keratins 5 and 14, which are less conserved. Mutations that code for a specific region of the amino terminus of keratin 5 are present in patients having EBS with mottled pigmentation.86 Although significance of this type of mutation and its association with pigmentary abnormalities remains unclear,87,88 it has been suggested that the keratin 5 globular head domain is responsible for keratin filament insertion onto melanosomes. Recent studies have shown that some mutations of the keratin 5 gene may produce protein that is unstable under increased temperatures.89 This could help to explain the well observed exacerbation of some subtypes of EBS to warm temperatures. The mutations associated with EBS with muscular dystrophy produce premature termination codons, splice-site, or other mutations that result in lack of expression or defective expression of plectin.90 Although the form of EB associated with plectin abnormalities is classified as simplex, it has an identical level of skin separation to that seen in junctional epidermolysis bullosa (JEB) with pyloric atresia.69 Specifically, the separation is present just above the level of the hemidesmosome in the intracellular part of the BMZ. This separation of EBS with muscular dystrophy and JEB with pyloric atresia, diseases with identical levels
1017
9
of separation, into two distinct EB categories illustrates the limitations of the current EB classification system. Plectin defects, like α6β4 integrin defects, can also be associated with pyloric atresia.90
Plectin is normally expressed in a wide range of tissues, including muscle.22 Although the mechanism of muscular dystrophy in plectin-deficient patients is unknown, it has been observed that disorganization of muscle sarcomeres occurs in the absence of plectin. It is possible that absence of plectin’s spectrin-like domain, which may normally interact with actin filaments in muscle, may be a key factor in the muscle pathology.91
The underlying molecular defect of ectodermal dysplasia skin fragility syndrome has been shown to be loss of function of the desmosomal protein plakophilin 1.80
Plakophilin is expressed mainly in suprabasilar keratinocytes and outer root sheath cells. Microscopic findings in this disease usually show intraepidermal acantholysis located in the areas where plakophilin 1 is normally expressed. The molecular defect involves loss-of-function mutations in the PKP1 gene coding for plakophilin 1.92-97
Deficiency of BP230 (an alternatively spliced dystrophic variant sometimes termed DST-e) is characterized by decreased or absent expression of BP230 by immunofluorescence microscopy and loss of the inner hemidesmosomal plate as seen by transmission electron microscopy. Homozygous premature termination codon mutations as well as point mutations of the DST-e gene in the area coding for the coiledcoil domain of BP230 have been associated with this disorder.81 Exophillin-5 deficiency is associated with loss-of-function mutations of the EXPH5 gene. This disease can show a complete absence of expression of this protein in the skin by immunofluorescence microscopy, and electron microscopy may show abnormal collections of basal keratinocyte perinuclear vesicles.98,99
Mutations of TGM5, which encodes a member of the multigene transglutaminase family, produces alterations of transglutaminase expression associated with the underlying cause of acral peeling skin syndrome.82
JUNCTIONAL EPIDERMOLYSIS BULLOSA
JUNCTIONAL
EPIDERMOLYSIS BULLOSA
All patients with JEB share the common histopathologic feature of blister formation within the lamina lucida of the BMZ caused by either a defect of anchoring filaments located in the lamina lucida and superior lamina densa. This group of diseases is inherited in an autosomal recessive manner, and there is considerable variation of the individual clinical phenotypes depending on the molecular defect. Three principal forms of JEB are most common. Patients with severe generalized JEB (previously termed Herlitz disease, JEB gravis, or lethal JEB) present with the most severe disease phenotype.14,100 It is unfortunate that this most severe subtype of JEB is also the most common.
1018
GENERALIZED SEVERE JUNCTIONAL EPIDERMOLYSIS BULLOSA
Generalized severe JEB is one of the most severe EB subtypes, resulting in lethality during infancy or early childhood.101 This disease is characterized by generalized and often extensive blistering at birth (Fig. 60-6). Later during infancy, a distinctive periorificial granulation tissue manifests. This can occur around the mouth, eyes, and nares most commonly but also can be present in the scalp; around the ears; and less frequently, in other locations besides the head and neck. This hypertrophic granulation tissue can also occur in nonlethal subtypes. Nails are usually severely affected and often are lost during infancy. When nails are still present, they are usually dystrophic and often associated with hypertrophic granulation tissue. Dental defects are present in this disease characterized by pitting of the tooth enamel. Oropharyngeal mucosal erosions are usually present and may be widespread. Erosions of all stratified squamous epithelial tissues, including nasal, conjunctival, esophageal, tracheal, laryngeal, rectal, and urethral mucosa can be affected. Associated systemic findings in severe cases are important factors in the lethality of this disease. Involvement of the large airways including tracheolaryngeal stenosis or obstruction are commonly associated with Herlitz JEB disease and hoarseness in early infancy is an ominous sign. There is a characteristic failure to thrive and growth retardation in this disease, often with a mixed anemia. Sepsis is a common and often lethal complication.
GENERALIZED INTERMEDIATE JUNCTIONAL EPIDERMOLYSIS BULLOSA
Sometimes patients initially presenting with a generalized severe phenotype survive infancy. These
patients eventually prove to have a severity of blistering and oral erosions less than the lethal form. In particular, a lack of significant hoarseness is regarded as a favorable prognostic sign, indicative of less severe internal disease manifestations. Scalp and nail lesions and periorificial nonhealing erosions are among the most common findings in these patients during childhood. Despite the lack of lethality in infancy, these patients nonetheless can have severe epithelial adhesion abnormalities, and tracheostomies or gastrostomy tubes may help in patient survival (Fig. 60-7). The nonlethal status of these patients distinguishes them from the Herlitz group and the terms nonlethal JEB or JEB mitis have been used in the past to describe these patients. They are much less common than severe generalized JEB patients. There are other rare variants of nonlethal JEB that present with localized junctional blistering of the extremities or with blistering in an inversa distribution on the trunk and intertriginous areas. A distinct subset of generalized intermediate JEB (previously termed generalized atrophic benign EB) presents at birth with generalized cutaneous involvement.102 However, despite the widespread cutaneous blistering, there is a relative paucity of oral erosions or other mucosal disease. Although enamel pitting is present, resulting in extensive dental caries, and nail dystrophy can often be severe, there is little other extracutaneous involvement noted in these patients. The blisters in these patients, which heal with a characteristic atrophic scarring, are debilitating and can be widespread, but nonetheless these patients generally have a normal lifespan. Blistering improves with age, growth is normal, and anemia is rarely seen. Some patients with this disease subtype have undergone normal uncomplicated pregnancies and deliveries. One peculiar characteristic of these patients is a progressive alopecia of the scalp and terminal hairs elsewhere in the body. The hair loss starts to become severe after the onset of puberty, and although patchy hair loss associated
9
with atrophic scarring has been described, often the alopecia is quite diffuse, and scarring is subtle or nonexistent.
LOCALIZED JUNCTIONAL EPIDERMOLYSIS BULLOSA
A rare subtype of JEB is the localized variant, previously known as minimus JEB. These patients generally show mild disease, which can be accentuated in localized areas, most often the hands, feet, and pretibial regions. Nails can sometimes be shed or become dystrophic, and enamel pitting can occur. Oral or nasal erosions can also occur, but there is an absence of any internal involvement. These patients generally have a favorable prognosis and a normal lifespan.
JUNCTIONAL EPIDERMOLYSIS BULLOSA WITH PYLORIC ATRESIA
These patients exhibit extreme mucosal and cutaneous fragility and may also have various urologic abnormalities, including hydronephrosis and nephritis. Mutations of the genes coding for the β4 and α6 integrin have been associated with EB,103 and in this group of diseases, pyloric atresia is present. Although pyloric stenosis is relatively more common, the distinct association of pyloric atresia, a rare condition, with EB makes this disease particularly unique. In these patients, hemidesmosomes are usually absent or rudimentary, and the level of separation is a low intrabasal epidermal at the level of the hemidesmosome as seen in EBS with muscular dystrophy described earlier. Most cases of this disease are quite severe and lethal in infancy caused by extensive extracutaneous epithelial sloughing in addition to widespread blistering of the skin and mucosa. Rare nonlethal cases of this disease have been characterized that appear to result from a partial loss of function of β4 integrin.104 Interestingly, nonlethal JEB can sometimes ameliorate itself through alterations in mRNA splicing.105,106
JUNCTIONAL EPIDERMOLYSIS BULLOSA WITH RESPIRATORY AND RENAL INVOLVEMENT
Blistering of the skin starts early in life, is generally mild or even absent, and may be associated with a diffuse alopecia and nail dystrophy. Extracutaneous involvement, however, presents at birth and is quite severe, consisting of interstitial lung disease, resulting in respiratory distress and lung infections. Kidney manifestations of this disease include renal failure, nephrotic syndrome, renal hypoplasia, and hydronephrosis. Despite aggressive neonatal supportive care, patients die in infancy because of these severe respiratory or renal complications.
1019
9
LARYNGO-ONYCHO- CUTANEOUS SYNDROME
This rare autosomal recessive condition107 is characterized by nail dystrophy, cutaneous erosions, and extensive granulation tissue, especially localizing to the conjunctiva and larynx. Sites of cutaneous involvement include areas of repeated trauma or pressure such as the elbows, knees, fingers, and toes.
MOLECULAR PATHOLOGY OF JUNCTIONAL EPIDERMOLYSIS BULLOSA
JEB can be associated with mutations of the genes coding for the α3, β3, or γ2 subunits of laminin-332.108,109 Absence of any of the three chains results in a lack of trimeric laminin-332 assembly and secretion, which results in a similar blistering phenotype. Patients with mutations of genes coding for the α3 or γ2 laminin subunits still show normal expression of laminin-311, which contains α3, β1, and γ1 chains.44 Therefore, positive linear BMZ staining of JEB skin for α3 chain on indirect immunofluorescence microscopy (IDIF) with absent expression of the other chains is an indication of either a α3 or γ2 chain defect. Conversely, absence of α3 staining on IDIF can be indicative of a mutation of the α3 gene. About 80% of laminin-332 mutations can be traced to one of two recurrent nonsense mutations in the LAMB3 gene, making prenatal testing for laminin-332 lesions easier than other EB candidate genes.110 In patients with Herlitz disease, all of the mutations so far detected have been those producing premature termination codons, resulting in absence of expression of laminin-332. Although patients with Herlitz disease generally show a complete lack of expression of laminin-332, patients without Herlitz disease who have abnormal granulation tissue and significant mucosal involvement often show reduced expression of laminin-332. This is due to expression of a molecule with deletions or missense mutations resulting in partial loss of laminin-332 function. This can lead to cases clinically classified as non-Herlitz JEB, especially cases with significant mucosal involvement.76,111,112 In some cases, spontaneous amelioration of blistering in severe JEB cases has taken place, which has been associated with genetic mechanisms that result in the reexpression of laminin-332.106
In the non-Herlitz JEB variant, generalized atrophic benign junctional EB (GABEB), blistering occurs in the lamina lucida region and abnormalities of hemidesmosomes or anchoring filaments are usually present (Fig. 60-8). Although laminin-332 mutations underlie a subset of GABEB patients, the majority of these patients have abnormalities of the hemidesmosomal protein collagen XVII (also known as BP180 or BPAG2). A number of mutations of the gene coding for collagen XVII have been described in patients with GABEB, including premature termination codon mutations, missense mutations, splice-site mutations, truncations, and a glycine substitution mutation.113,114
1020
Although intralamina lucida or junctional skin separation has been shown in all patients with this disease, one patient was described with a cytoplasmic deletion of collagen XVII who showed intrabasal epidermal skin separation.115 Localized JEB has been shown to be associated with COL17A1 mutations.116
Laryngo-onycho-cutaneous syndrome is characterized by N-terminal mutations of the LAMA3 gene leading to deletion of the laminin α3 LE domain.107
Of interest, mosaic GABEB patients have been identified who demonstrate well defined areas of blistering associated with absence of collagen XVII expression as well as areas of nonblistering skin associated with normal collagen XVII expression. Careful analysis of these patients’ keratinocytes revealed reversion of one of the two alleles of the mutation, most likely caused by a mitotic gene conversion involving nonreciprocal exchange of parental allele DNA.117,118 Understanding how EB can undergo spontaneous molecular correction in these cases could help in the design of future molecular therapeutic strategies. JEB with respiratory and renal involvement is caused by mutations of the ITGA3 gene.119,120 These patients can show an absence of expression of integrin α3 in skin biopsies by immunofluorescence microscopy. Electron microscopy shows intralamina lucida separation, like other JEB subtypes.119
DYSTROPHIC EPIDERMOLYSIS BULLOSA
DYSTROPHIC
EPIDERMOLYSIS BULLOSA
Dystrophic EB (DEB) is characterized by blisters that heal with scarring and milium formation. DEB can be inherited either in an autosomal recessive (RDEB) or dominant fashion (DDEB). One of the most important
reasons to distinguish between these two subtypes is the increased prevalence of invasive squamous cell carcinoma (SCC) associated with the recessive but not the dominant form. Regardless of the mode of inheritance, DEB is derived from defects of the ultrastructural entity known as the anchoring fibril, which results in sublamina densa separation.
LOCALIZED AUTOSOMAL DOMINANT DYSTROPHIC EPIDERMOLYSIS BULLOSA
The localized subtype of dominant DDEB (sometimes called the Cockayne-Touraine of DDEB) can present at birth, but occasionally it is not appreciated until childhood. Although generalized blistering can sometimes take place, especially early in life, the blistering usually becomes localized to repetitively traumatized areas such as the knees, sacrum, and acral surfaces (Figs. 60-9 and 60-10). These areas show a characteristic scarred, dystrophic appearance. Often the scarring is hypertrophic. Milia are common accompanying features of the healing process in these patients (Fig. 60-11). Nail
9
dystrophy and nail loss with atrophic scarring of the distal digits are common. Occasionally, nail abnormalities can be the only presenting abnormality in DDEB. Oral lesions are not common, and teeth are usually unaffected. These patients have a good prognosis and a normal lifespan.
GENERALIZED AUTOSOMAL DOMINANT DYSTROPHIC EPIDERMOLYSIS BULLOSA
The generalized form of DDEB (sometimes referred to as the Pasini subtype of DDEB) presents at birth with a generally more severe and widespread blistering phenotype compared with the localized subtype. Blisters in generalized DDEB heal with scarring plaques and milia in a fashion similar to other DEB subtypes. In addition, this disease is sometimes distinguished by the spontaneous appearance of distinctive scarlike, flesh-colored papules on the truck. These albopapuloid lesions are not pathognomonic because these lesions can also be seen in other EB subtypes. As patients get older, the generalized blistering may eventually localize to the extremities. Patients often show dystrophic or absent nails. Oral erosions can often be present but usually are not extensive, and enamel defects can be seen in some patients. A rare variant of self-remitting generalized DDEB, termed bullous dermolysis of the newborn, consists of generalized blistering that gradually recedes after infancy.121 Skin biopsies from these patients often show basal epidermal intracytoplasmic accumulations of collagen VII when examined by immunofluorescence microscopy.122
RECESSIVE DYSTROPHIC EPIDERMOLYSIS BULLOSA
Recessive DEB (RDEB) can be quite variable in its severity. Although the severe subtype is the most common, a localized form can occasionally be seen that has been previously termed RDEB mitis. Similar to localized DDEB, localized RDEB is usually confined to repetitively traumatized skin surfaces, most often in an acral distribution. Scarring and milium formation
1021
9
accompany the healing of blisters. Mucosal involvement in localized RDEB, if present, is mild. Severe RDEB, previously known by the eponym the Hallopeau-Siemens, is a devastating disease (Fig. 60-12). This disease presents with generalized blistering at birth. Occasionally, there is extensive
denudation of an entire region of skin at birth, often involving one of the limbs. This congenital absence of skin is sometimes termed Bart syndrome. Healing and blistering cycles occurring during infancy can lead to a progressive scarring, which can become quite extensive. Pseudosyndactyly resulting from a closure of the digits in a “mitten” of skin is an extremely common feature of this disease (Fig. 60-13). Scarring can lead to flexion contracture of the hands (Fig. 60-14), as well as the limbs. In contrast to the patients with severe JEB, these patients do not show significant periorificial involvement. Instead, the scalp is the most commonly affected area on the head and neck of these patients. The oropharynx can be extensively involved in both dominant and recessive DEB (Fig. 60-15) with generalized erosions evolving into a scarring that limits the movement of the tongue and narrows the opening of the oral cavity. The teeth can show significant enamel pitting, and caries can be extensive, leading to loss of
Approach to patients with epidermolysis bullosa
Review of systems: gastrointestinal, respiratory, ocular, or growth abnormalities?
Personal history: onset of trauma-induced blisters at birth or childhood?
Biopsy (light microscopy): Cannot be used to Dx EB but can rule out other disorders Biopsy suspicious or nonhealing lesions in postpubertal RDEB patients to rule out SCC
Family history: siblings or parents with disease?
Laboratory: examine CBC, albumin, and other laboratory values as needed to evaluate anemia and malnutrition
Physical exam: Scarring, atrophy, or milia? Nails, eyes, mucosa, hair, teeth involved?
Biopsy (electron microscopy) can show cleavage plane, clumped tonofilaments, abnormal hemidesmosomes, and anchoring filaments
Biopsy (immunofluorescent microscopy) can show cleavage plane and defective expression of EB-associated proteins
DNA analysis and genetic counseling: Patient and family members DNA analyzed can show specific DNA defect Useful for family planning prenatal diagnosis and future molecular therapy
Therapy: Support wound care and nutrition; instruct family Treat infection, anemia, and extracutaneous disease through multidisciplinary approach Implementation of molecular therapies in the future
1022
teeth. Involvement of the trachea or larynx can lead to a narrowing of the airway, which could require intervention with a tracheostomy. Mucosal erosions of the esophagus can lead to stricture formation and webbing. The combination of oral lesions, dental caries, esophageal strictures, and increased caloric needs from extensive wound healing can lead these patients toward malnutrition and growth retardation. These patients usually have problems with anemia and may show a deficiency of iron absorption. In the more distant past, most severe RDEB patients died in infancy of sepsis and other complications of extensive blistering. More recently, with improved nutritional, infection, and wound support, these patients usually can survive into their teens or into adulthood. However, after puberty, another devastating complication, SCC, can and often does appear (Fig. 60-16).
9
It is estimated that 50% to 80% of patients with severe- RDEB eventually develop these carcinomas, and many of these die of metastatic disease. RDEB-associated carcinomas are distinct from most other cutaneous SCCs in that they are extremely aggressive with strong tendencies for invasion and metastasis.
MOLECULAR PATHOLOGY OF DYSTROPHIC EPIDERMOLYSIS BULLOSA
Abnormalities of anchoring fibrils are present in DEB patients, which range from subtle changes in some patients with dominant disease to absence of anchoring fibrils in patients with the severe recessive form of this disease, are present, and a sublamina densa plane of blister cleavage is present (see Fig. 60-4). These observations correlate with indirect immunofluorescent microscopic analysis of patients with DEB, which demonstrates varying degrees of linear collagen VII staining at the dermal–epidermal junction in dominant patients and totally absent staining in severe recessive patients. In some patients, there is a cytoplasmic retention of collagen VII, which can be demonstrated in patient biopsy sections and analysis of patient keratinocytes.123
DEB has been shown to be associated in all cases thus far with mutations of the gene coding for collagen VII (COL7A1). In the recessive forms, mutations usually cause premature termination codons, which result in lack of collagen VII in tissue. It is known that mRNAs bearing premature stop codons show accelerated turnover.124 In addition, truncated proteins that are not secreted or not assembled into anchoring fibrils may also show accelerated turnover. Either or both of these mechanisms can explain the lack of detectable collagen VII in the tissue of individuals with severe RDEB associated with mutations that produce premature termination codons.61,125,126 However, even in individuals who show absent collagen VII tissue staining by immunofluorescence microscopy, analysis of patient keratinocytes can in many cases still demonstrate low levels of mutant collagen VII expression.127,128 Recently, in addition to cases reported in JEB patients, a revertant mosaicism phenotype has also been reported in RDEB.129
1023
9
Generally, COL7A1 mutations that do not cause premature termination codons produce less severe disease.108,114 For example, mutations that produce glycine substitutions of the triple helical region interfere with triple helical assembly of the collagen VII molecule. These types of mutations are present in many patients with milder dominant forms of this disease. In these patients, collagen VII molecules may not be able to assume the proper conformation needed to polymerize into anchoring fibrils. One subtype of DEB associated with increased pruritis, EB pruriginosa, has also been described to be associated with glycine mutations.130 Other COL7A1 mutations have been shown to be associated with impaired secretion of collagen VII, resulting in intracellular accumulation of this molecule. In one study, DEB patient mutations that involve the area of the gene coding for the collagen VII NC2 domain were shown to interfere with NC2 processing and the assembly of anchoring fibrils.131
KINDLER SYNDROME
KINDLER SYNDROME
New advances in our understanding of the molecular pathology of the skin have brought to light the underlying pathophysiology of a disease related to EB, Kindler syndrome. Kindler syndrome was first described by Theresa Kindler in 1954.132 It is characterized by EB-like trauma-induced blistering at birth and during infancy, with atrophic changes during healing reminiscent or JEB or DEB.133-139 However, in late childhood, the blistering usually subsides and gives way to a progressive poikiloderma, which distributes to sun-exposed areas. The poikiloderma may show areas of atrophy and hyperkeratosis, as well as hypopigmentation, hyperpigmentation, and telangiectasias. These patients often show photosensitivity. Nail changes and webbing of the toes and fingers are also sometimes present. Internal complications include oral inflammation, esophageal or ureteral strictures, and ectropion. Ultrastructurally, these patients show reduplication of the basement membrane, which is the most consistent feature seen. Although there is often a sublamina densa split with anchoring fibril abnormalities, sometimes lamina lucida or intraepidermal separation can be seen associated with the blistering phenotype. Molecular investigation of this disease led to the discovery of a new epidermal protein, kindlin-1, which shows decreased expression by immunofluorescent microscopy in this disease. Kindlin-1 appears to have some homology to signaling proteins such as talin, which suggests a signaling function, but its role in Kindler syndrome remains unclear.140 A number of mutations of the gene coding for kindlin-1, KIND1, have been described.141-143 These include nonsense, frameshift, and splice-site mutations that underlie the observed decreased kindlin-1 expression in affected skin. Why the disease evolves from a blistering to a poikilodermatous phenotype and the exact function
1024
of kindlin-1 in epidermal homeostasis remains to be fully elucidated.
DIAGNOSIS
The first step toward making the diagnosis of EB begins with a thorough history and physical examination (see Fig. 60-13). Useful historical information includes the age of onset of blistering and the presence of blistering in other family members. A review of gastrointestinal (GI), respiratory, ocular, dental, and genitourinary systems is important as is evaluation of general growth and development. Physical examination requires not only a complete skin examination but also a thorough evaluation of mucosal tissues, hair, nails, and teeth. Laboratory measurements of importance in the initial visit include evaluation for anemia and for measures of nutrition, such as serum albumin. Skin biopsy is another important diagnostic step. Routine histologic analysis cannot be used to diagnose EB but can be useful for excluding other causes of blistering. The dermal–epidermal BMZ is simply too small to be visualized by light microscopy. To differentiate levels of BMZ separation in skin biopsies, transmission electron microscopy (TEM) or indirect immunofluorescent microscopy (IDDF) must be used. The interior of blisters rapidly reepithelialize, which can obscure correct determination of blister levels. For this reason, it is critical to biopsy a blister that is as fresh as possible. One way to ensure a fresh blister is for the clinician to induce it in the office. This can be accomplished by gently rotating a pencil eraser over an intact area of patient’s skin until separation of the epidermis from the dermis can be observed. This is more easily performed with patients who have more severe disease variants compared with patients with milder disease. When actually doing the biopsy, its best to place the circular biopsy punch so that only 10% of the punch covers the visible blister with 90% covering intact skin. This is because it is helpful to have both intact and blistered skin on the same biopsy specimen, and extension of the blister is likely to occur either during the biopsy process or during shipping. TEM has been regarded for many years as the gold standard for determining the level of blistering in EB subtypes. In addition to determining the level of blistering, ultrastructural entities can also be analyzed by TEM for characteristic alterations. For example, clumping of keratin intermediate filaments in basal keratinocyte cytoplasm is a pathognomonic finding for severe generalized (Dowling-Meara) EBS. Rudimentary hemidesmosomes can be an important clue to the diagnosis of JEB. Absent or altered anchoring fibrils often occur in DEB subtypes, especially the recessive forms. IDDF microscopy can provide additional information on the level of blistering, as well as important clues to the underlying molecular defects. In this technique, an antibody panel against known BMZ antigens is applied to frozen sections of blistered patient
skin. The localization of the antigens to the epidermal or dermal portion of the blister indicates the level of skin separation in the BMZ. In EBS samples, for example, intracellular hemidesmosomal components such as BP230 and a lamina densa protein such as type IV collagen would each localize to the floor of the blister. In JEB cases, BP230 would localize to the roof of the blister, and type IV collagen would localize to the floor. In DEB, collagen VII and BP230 would localize to the roof of the blister. The specific absence or presence of staining with a particular antibody in frozen sections of intact portions of patient skin give important clues to the specific molecular defect. Whereas samples that lack staining with antibodies specific to laminin-332 would further support a JEB diagnosis, a lack of staining for collagen VII would support a DEB diagnosis. Absence of staining for collagen XVII would support a generalized intermediate JEB diagnosis. A complete panel of antibodies to support IDDFbased diagnosis of EB would include antibodies against laminin-332 (Herlitz and non-Herlitz JEB), as well as antibodies against BP180/collagen XVII (non- Herlitz JEB or generalized atrophic benign EB), collagen VII (RDEB), α6 and β4 integrins (JEB with pyloric atresia), plectin (EBS with muscular dystrophy), and keratins 5 and 14 (recessive EBS). Antibodies against the individual chains α3, γ2, and β3 chains of laminin-332 are especially helpful. Its known that laminin-311 (which shares the same laminin α3 chain as laminin-332) is expressed in Herlitz JEB associated with null mutations of genes coding for β3 chain (LAMB3) and γ2 chain (LAMC2) but not in Herlitz JEB associated with null mutations of the α3 chain gene (LAMA3).44 Therefore, if laminin β3 and γ2 antibodies are negative and α3 antibody is positive, this could point the genetic analysis to examine the LAMB3 and LAMC2. Conversely, if all three laminin-332 chains are absent by IDIF, this could point the genetic investigation toward LAMΑ3, saving time and effort in arriving at the final molecular diagnosis. Gene mutation analysis has revolutionized our understanding of the EB family of diseases and is considered the ultimate final step in arriving at the molecular diagnosis in EB. Concurrent advances in our knowledge of the biochemical structure and supramolecular assembly of BMZ proteins have both facilitated and complemented the molecular biology studies. Thus, EB patient diagnosis requires both clinical and molecular information. Blood samples or buccal swabs are taken from the patient as well as the parents and siblings for genetic analysis. Whole-exome sequencing will likely make EB mutation detection easier, faster and less expensive as this practice becomes more widespread.144
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
See Table 60-2.
9
Most Likely
■Pompholyx
■Insect bites
■Friction blisters
■Thermal burns
■Bullous impetigo
Consider
■Chronic bullous dermatosis of childhood (linear immunoglobulin A disease)
■Bullous pemphigoid
■Epidermolysis bullosa acquisita
■Bullous systemic lupus erythematosus
■Cicatritial pemphigoic
■Pemphigus vulgaris
Always Rule Out
Always Rule Out
■Stevens-Johnson syndrome
■Stevens-Johnson syndrome
■Toxic epidermal necrolysis
■Toxic epidermal necrolysis
GENETIC COUNSELING
GENETIC COUNSELING
DNA mutation analyses can be extremely helpful to EB patients. The prognostic benefit to the patient can often be highly significant. For example, recessive DEB cases may have equivalent blistering activity compared with the more severe dominant cases, but their risk of invasive SCC is much greater. Through DNA diagnosis, these two groups can be distinguished, thereby identifying patients potentially at risk for invasive SCC. Prenatal diagnosis of EB in affected families can be an extremely accurate technique, especially if the original proband has previously had mutational analysis or identification of the defective gene. Fetal skin biopsies and fetoscopy with their increased risk of pregnancy loss can now be avoided by analysis of either chorionic villus sampling as early as 8 to 10 weeks145 or gestation or amniocentesis in the second trimester.146 The development of highly informative intragenic and flanking polymorphic DNA markers in EB candidate genes together with rapid screening of genetic “hotspots” makes genetic screening of at-risk pregnancies a viable option.110,147 Coupling of the technique of in vitro fertilization with EB prenatal diagnosis, preimplantation diagnosis has now been successfully performed for EB cases.148 Another promising area of prenatal diagnosis with potential future applications to EB is the detection and analysis of fetal cells in the maternal circulation.149
TREATMENT
Most therapy for EB is supportive in nature. The regimen is tailored to the severity and extent of skin and systemic involvement and usually entails a combination of wound management, infection control, surgical management as needed, and nutritional support. Skin care and supportive care for other organ systems
1025
9
in certain EB subtypes are most optimally coordinated through a multidisciplinary approach. Comprehensive topical therapy is a mainstay of treatment in EB, with avoidance of trauma as a primary goal. Wound healing is impaired by endogenous factors, including foreign bodies, bacteria, nutritional deficiencies, anemia, and repeated trauma. Therefore, optimizing wound healing in patients with EB involves control of all of these factors.150
SUPPORTIVE SKIN CARE
SUPPORTIVE SKIN CARE
Extensive areas of denuded skin can result in the loss of the barrier provided by the stratum corneum. Subsequent microbial penetration can result in the accumulation of serum and moisture that further enhances bacterial propagation. The above factors combined with immunosuppressive therapy facilitate development of infections. Prevention of infection is obviously the preferred strategy. A modified Dakin solution (0.025% w/v sodium hypochlorite) can be helpful in reducing the bacterial load in patient skin. Soaking wounds in this solution for 20 minutes before dressing changes also helps to free adherent bandages that have dried onto the wound bed. After soaking, wounds can be dressed with mupirocin or other topical antibiotics and covered with semiocclusive nonadhesive dressings. Tape causes further blistering and peeling of the skin, so it is essential to use self-adhering (clinging) gauze or self-adherent paper to hold nonadherent dressings in place. For patients with generalized or localized subtypes of EBS, controlling exposure to heat may prove helpful in controlling blister formation. Advising patients to use soft, well-ventilated shoes is also recommended. Patients with Herlitz JEB, lacking functional laminin-332, an extracellular matrix protein shown to be involved in keratinocyte adhesion and migration, may have especially difficult problems with wound healing. For patients with DEB, use of finger splinting or diligent hand wrapping and appropriate hand protection against trauma are helpful, especially after hand surgery (see later).
INFECTION
INFECTION
Management of skin infections is a critical part of EB patient care. Large areas of denuded skin, a common finding in patients with EB, provides an inadequate barrier to microbial penetration and can lead to both skin infections as well as the more devastating complication of sepsis. Staphylococcus aureus and Streptococcus pyogenes are common infectious agents. Gram-negative infections with Pseudomonas aeruginosa can also occur. Skin cultures and the use of the appropriate systemic antibiotics are indicated for wound infection. Gentle whirlpool therapy, frequent
1026
(daily) dressing changes, dilute chlorine baths, rotation of topical antibiotics, and use of topical disinfectants such as iodine–povidone are all helpful ways to reduce resistant bacteria.
SURGICAL TREATMENT
SURGICAL TREATMENT
Among the EB patient population, those with the severe recessive DEB (Hallopeau-Siemens) variant are generally the most in need of surgical intervention.151
Mitten pseudosyndactyly in these patients can be surgically released; however, this procedure may have to be repeated periodically because of the strong tendency of this condition to recur.151-154 Splinting after surgery is essential to reducing recurrence of hand deformities. Surgery may also be used to correct limb, perioral, and perineal contracture deformities, but unfortunately, a high rate of recurrence is common. Extra care must be taken to minimize trauma to oral mucosa in EB patients during intubation.
TUMORS
TUMORS
SCC often arises after puberty in patients with recessive DEB. SCC may arise in multiple primary sites, especially in nonhealing areas. Careful surveillance of nonhealing areas is of utmost importance because patients often die from metastatic disease.155
Surgical excision using either Mohs or non-Mohs approaches is an important first-line modality with radiation therapy as a useful adjunct in some cases.156
Isotretinoin has been used for patients with RDEB for chemoprevention of SCC. Although it appears well tolerated, it is not clear whether in can increase the overall survival rate of these patients.157 Cetuximab therapy has been demonstrated to offer some benefit,155,158 but the cutaneous side effects can sometimes be problematic.
CARE FOR EXTRACUTANEOUS INVOLVEMENT
CARE FOR
EXTRACUTANEOUS
INVOLVEMENT
GASTROINTESTINAL MANAGEMENT
Esophageal lesions are often the most disabling complication found in recessive DEB and JEB of both the severe and intermediate generalized variants. Esophageal strictures usually respond to dilation; however, recurrence of strictures after dilation is common.159 Colonic interposition has proven effective in advanced cases but is rarely used. Gastrostomy tube insertion has been effective in providing nutrition to individuals with esophageal strictures.160
Increased fluid and fiber intake and stool softeners may also be of value in EB patients with constipation and colitis.161
EYE LESIONS
Patients with EBS, particularly those with the Dowling-Meara subtype, can experience recurrent inflammation of the eyelid, with bullous lesions in the conjunctivae. Patients with JEB and patients with recessive DEB can experience corneal ulcerations with scarring, obliteration of tear ducts, and eyelid lesions.162 Cicatricial conjunctivitis can also occur in patients with recessive DEB. Corneal erosions are treated supportively with application of antibiotic ointments and use of cycloplegic agents to reduce ciliary spasms and provide comfort. Moisture chambers and ocular lubricants are also commonly used. Severely affected upper eyelids may be surgically managed with full-thickness skin grafting. Complete correction of any eye disorder in EB patients is difficult to achieve. Proper management of eye lesions in EB patients must include the assistance of an ophthalmologist to prevent serious visual compromise.163
Surgical and ocular surface reconstruction can help reduce granulation tissue in laryngo-onychocutaneous syndrome.164
OROPHARYNGEAL LESIONS
Good dental hygiene is essential for EB patients, and regular visits to the dentist are especially important. Enamel defects in JEB patients and pain on brushing and flossing in patients with severe JEB and DEB often lead to dental carries.165,166 The softest brush available should be used for regular cleansing. Oral mucosal blistering may also accompany forms of JEB and DEB, especially in severe DEB subtypes such as the severe generalized recessive form, narrowing of the mouth opening (microstomia), and scarring induced limitation of movement of the tongue (ankyloglossia) and be particularly debilitating. Normal saline rinses are effective for gentle cleaning of the mucosal surfaces. Mouthwashes containing alcohol or other harsh agents should be avoided. Erosions and scarring involving the trachea and larynx with resultant narrowing of the airway. In patients with airway involvement, there is danger of pulmonary aspiration.167 Surgical intubation should be performed gently with small bore airways by anesthesiologists experienced in the care of EB patients.168
NUTRITION, ANEMIA, AND CARDIOVASCULAR DISEASE
Nutritional assessment and support can be critical in patients with EB169 for several reasons. Extensive cutaneous injury is associated with marked alterations in hemodynamic and metabolic responses, with increased caloric and protein requirements. Oropharyngeal
9
and GI lesions provide the greatest overall threat to nutritional well-being. These include oral blistering, abnormal esophageal motility, strictures, dysphagia, diarrhea, malabsorption, and dental problems. Nutritional assessment must take into account these factors to develop a supplemental regimen to replenish nutritional deficiencies.170 Patients are often unable to increase their food intake to balance this increased caloric need. For example, hypoplastic enamel formation in JEB may lead to tooth decay, mucosal blistering, and oral candidiasis. All of these potential complications may compromise patients’ ability to eat. Extensive internal mucosal disadhesion in the GI tract may cause abnormal GI motility, strictures, and diarrhea are complications that may lead to malabsorption of iron and other nutrients. Anemia of chronic disease can certainly affect all severe EB subtypes. Recessive DEB patients often show a particularly severe iron deficiency that may not responsive to oral iron supplementation. For these patients, parenteral iron can be helpful. Furthermore, if a lack of a reticulocyte response to iron supplementation is seen in iron-deficient patients, it is helpful to assess the erythropoietin level and to treat with recombinant erythropoietin if necessary.171 Transfusion is also useful in the treatment of anemia in EB, especially when symptoms require a rapid correction. Dilated cardiomyopathy is a devastating and potentially fatal complication of severe DEB and JEB patients that is highly associated with chronic anemia.172
PSYCHOLOGICAL ASPECTS OF EPIDERMOLYSIS BULLOSA
Patients with EB, especially the severe subtypes, can be plagued with chronic pain.173-175 Although many, despite extremely adverse conditions, seem to find a way to maintain a surprisingly positive outlook on life, others lapse into depression.176,177 Patients with severe EB can also create stress for their families and loved ones.178,179 Thus, it is important to identify the warning signs of depression when they arise and to work in a multidisciplinary approach with psychiatrists and clinical psychologists as needed. Supportive psychotherapy and patient support group meetings can help patients and their families in this regard. An additional source of support for patients and families include several important patient-based organizations that assist with education and support, including the Dystrophic Epidermolysis Bullosa Research Association and Epidermolysis Bullosa Medical Research Foundation.
ANTIINFLAMMATORY THERAPIES
ANTIINFLAMMATORY
THERAPIES
A number of new therapies are emerging for EB, which are listed in Fig. 60-17 and described later.
1027
9
Current studied or proposed therapies for epidermolysis bullosa
EBS Inflammation
Dominant EBS Gene editing KRT14/KRT5 Keratin 5/14
Alantoin antiinflammatory therapy Diacerin antiinflammatory therapy
Recessive EBS viral KRT14/KRT5 epidermal grafts
Plectin BP230
Integrin α6β4
Laminin-332/ Laminin-311
Recessive JEB
Retroviral LAMB3 epidermal grafts Bone marrow replacement
Recessive DEB
Collagen Vll
Gene replacement strategies Retroviral COL7A1 epidermal grafts Lentiviral COL7A1 fibroblast injections Bone marrow replacement Collagen VII protein topical, intradermal, or intravenous Gentamycin or other read-through therapeutics Allogenic cell therapies
Collagen XVll
Recessive JEB Viral COLXVII epidermal grafts Extension of revertant mosaicism
Dominant DEB
Gene editing COL7A1 Exon Skipping COL7A1
Collagen I/ Collagen Ill
DEB scarring Losartan antifibrotic therapy
Repetitive scratching–induced trauma and an exuberant inflammatory wound each contribute to wound chronicity, especially in patients with inflammatory subtypes of EBS or EB pruriginosa. Although topical corticosteroids can produce shortterm relief from itching and reduce inflammation, in the long term, these agents induce skin atrophy, which exacerbates the intrinsic fragility of EB skin and worsens blistering/healing. Tetracycline and phenytoin have been used in the past for EB but are not currently indicated treatment.180 The naturally occurring component of rhubarb root, and interleukin-β blocker diacerein181 was found to be well tolerated and to reduce blister formation in patients with severe EBS with inflammatory skin phenotypes.182 Losartan, a small-molecule angiotensin II type I receptor antagonist used to treat hypertension, has shown promise in preclinical studies in reducing the fibrosis accompanying DEB wounding. Losartan achieves this effect by reducing transforming growth factor-β expression in the skin.183
1028
ALLOGENEIC CELL THERAPIES
ALLOGENEIC CELL
THERAPIES
Allogeneic cell therapies including, allogeneic keratinocyte skin equivalents,184 allogeneic fibroblast injections,185,186 and mesenchymal stem cell infusions,187
have shown positive short-term effects on EB wound healing. However, they fail to show any long-term benefits. Allogeneic bone marrow transplantation performed on a group of seven children with recessive DEB. Some patients demonstrated noticeable clinical benefit, as well as collagen VII staining at the dermal–epidermal junction for 1 year or longer; however, restoration of anchoring fibrils was incomplete.188 Of note, this procedure demonstrated a mortality rate of approximately 30%. Contributing to the mortality rate observed may have been the combination of widespread cutaneous erosions and the immunomyeloablative-induced immunosuppression required for successful bone marrow transplant take.
9
AGE OF ONSET DISTRIBUTION CLINICAL MANIFESTATIONS SEQUELAE AND COMPLICATIONS
Epidermolysis Bullosa Simplex (EBS)
Generalized severe (Dowling-Meara) Birth Generalized; oral mucosal involvement; nail involvement
Generalized intermediate (Koebner)
Birth Generalized but most prominent on hands, feet, and extremities; mild oral mucosal involvement
Localized (Weber-Cockayne) Infancy, childhood, occasionally adulthood
Areas of friction or trauma, most commonly the hands and feet; mild oral involvement
Junctional Epidermolysis Bullosa (JEB)
Generalized severe JEB (Herlitz) Birth Generalized; mucosal involvement (nasal, conjunctival, esophageal, tracheal, laryngeal, rectal, urethral); nail involvement; dental involvement
Generalized intermediate JEB Birth Generalized; minimal oral or other mucosal involvement; dental involvement
Localized JEB
Localized (most often hands, feet, pretibial regions); oral mucosal involvement (minimal); nail involvement; dental involvement
JEB with pyloric atresia Birth Generalized; mucosal involvement; systemic involvement
Dystrophic Epidermolysis Bullosa (DEB)
Localized DDEB (Cockayne- Touraine)
Birth or childhood Generalized early in life but localizes to sites of trauma (knees, sacrum, acral areas); nail involvement
Generalized DDEB (Pasini) Birth Generalized early in life but may localize to extremities; nail involvement
Sudden onset of grouped or herpetiform blisters, extensive erosions; nail shedding and dystrophy palmoplantar hyperkeratosis in older children
Milia formation, contractures, esophageal erosions, pyloric atresia
Erosions, palmoplantar hyperkeratosis Postinflammatory hyperpigmentation or hypopigmentation, milia formation
Blistering after trauma; palmoplantar hyperhidrosis No milia formation; no scarring sequelae
Extensive blistering; periorificial granulation tissue (mouth, eyes, nares); loss of nails or dystrophic nails with granulation tissue; pitted tooth enamel
Tracheolaryngeal stenosis or obstruction, failure to thrive, sepsis
Widespread blistering; extensive dental caries; nail dystrophy; may have progressive alopecia of scalp
Atrophic scars, normal growth
Localized erosions; nail shedding and dystrophy; enamel pitting; oral and nasal erosions
Extensive cutaneous sloughing, with widespread blistering Pyloric stenosis, hydronephrosis, nephritis
Blisters; nail dystrophy or nail loss may be presenting symptoms
Milia, hypertrophic scarring
Widespread severe blistering; albopauloid lesions; oral erosions; dystrophic or absent nails; enamel defects
Milia, scarring plaques
Localized RDEB Birth Acral distribution Blisters; mild oral mucosal erosions Milia, scarring
Severe RDEB
Birth Generalized; mucosal
Severe RDEB (Hallopeau- Siemens)
Birth Generalized; mucosal involvement; dental involvement
(Hallopeau- Siemens)
involvement; dental involvement
Extensively denuded skin;
Pseudosyndactyly, flexion con-
Extensively denuded skin; enamel pitting with caries and loss of teeth
Pseudosyndactyly, flexion contractures of hands and limbs, tracheal and laryngeal involvement, esophageal structures, malnutrition, growth retardation, anemia, iron deficiency, SCC
enamel pitting with caries and loss of teeth
tractures of hands and limbs, tracheal and laryngeal involvement, esophageal structures, malnutrition, growth retardation, anemia, iron deficiency, SCC
DDEB, dominant dystrophic epidermolysis bullosa; RDEB, recessive dystrophic epidermolysis bullosa; SSC, squamous cell carcinoma.
1029
9
AUTOLOGOUS GENETIC THERAPIES
AUTOLOGOUS GENETIC
THERAPIES
Retroviral ex vivo gene therapy using autologous keratinocytes was performed for one patient with JEB.189 In this study, a patient with a missense mutation of laminin-332 was grafted with genetically corrected keratinocyte monolayers expressing wildtype laminin-332. After 6.5 years of postgrafting follow-up, grafts still showed positive expression of laminin-332 and resistance to blistering.190 More recently, this therapy was extended in a widespread and remarkable manner to another JEB patient with genetically corrected epidermal grafts covering more than 80% of skin surfaces.191 A similar approach was more recently performed on a clinical trial of four patients with recessive DEB.127 In this study, fulllength COL7A1 gene, coding for collagen VII was transferred ex vivo to primary recessive DEB patient keratinocytes. Collagen VII engineered patient cells were expanded then grafted as monolayers to patient wounds (six grafts per patient). Results showed clinical improvement as well as restoration of collagen VII expression and anchoring fibril formation as long as 1 year. Gene editing via CRISPER/Cas9 combined with the use of induced pluripotent stem cells192-194 is another promising future therapy for Exon skipping has shown promise in as a potential future therapy in both JEB195 as well as DEB skin.196,197 The potential to extend skin in patients with COL17A1 and COL7A1 self-correcting mutations by autologous grafting has also been proposed as a novel therapy.198
SUMMARY
A summary of the types of EB and their clinical manifestations is shown in Table 60-3.
Figure 60-1 Schematic of the components of the dermal–epidermal basement membrane on the left compared with ultrastructural appearance of basement membrane morphological entities on the right.
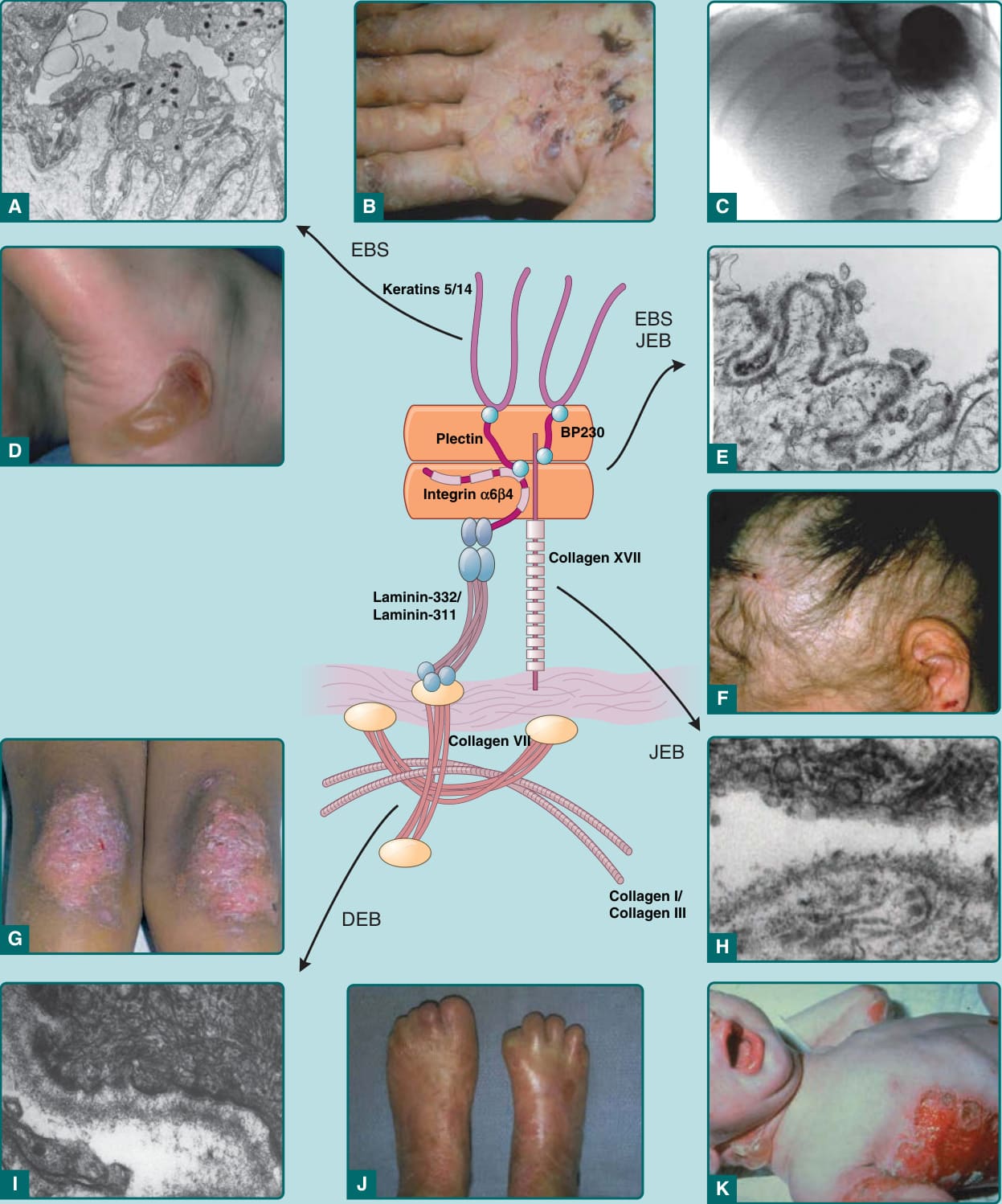
Figure 60-2 Comparison of levels of skin separation in epidermolysis bullosa (EB) with clinical findings. A, Transmission electron micrograph showing typical intraepidermal separation in epidermolysis bullosa simplex (EBS). B, Palmar hyperkeratosis and erosions in EB herpetiformis. C, Radiograph showing pyloric atresia associated with EB. D, Localized blister on heel in EBS Weber-Cockayne. E, Transmission electron micrograph showing typical separation at level of hemidesmosome in EB with pyloric atresia, alternatively classified as EBS or junctional epidermolysis bullosa (JEB). F, Nonscarring diffuse alopecia and scalp erosions in generalized atrophic benign epidermolysis bullosa. G, Localized dystrophic changes with milia in dominant dystrophic epidermolysis bullosa (DEB). H, Transmission electron microscopy showing typical intralamina lucida separation in JEB. I, Transmission electron microscopy showing typical sublamina densa separation in DEB. J, Pseudosyndactly in recessive DEB. K, Generalized blistering in Herlitz JEB.
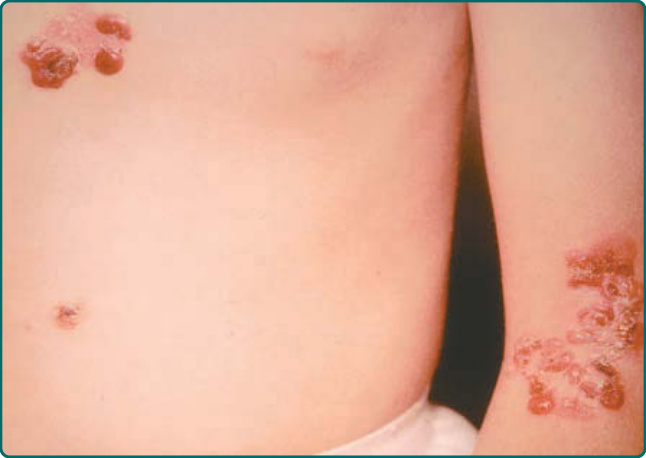
Figure 60-3 Characteristic blister formation on trunk and arm in patient with Dowling-Meara epidermolysis bullosa simplex.
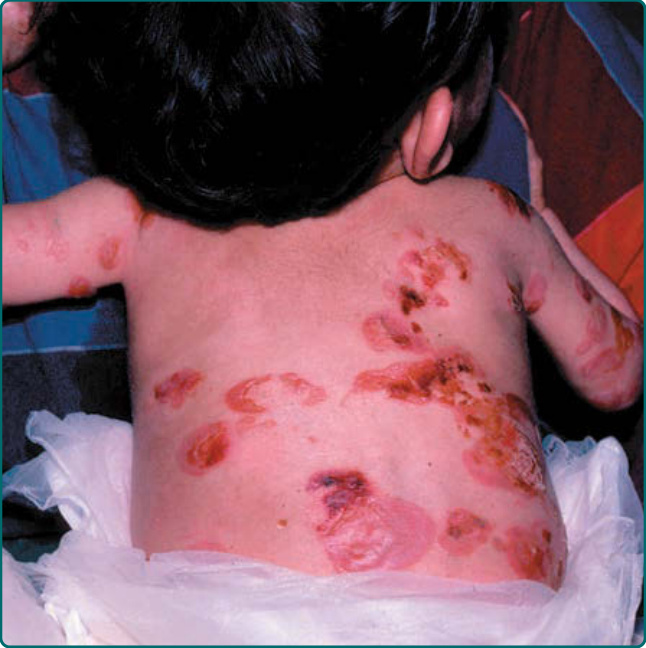
Figure 60-4 Widespread blistering in an infant with generalized epidermolysis bullosa simplex.
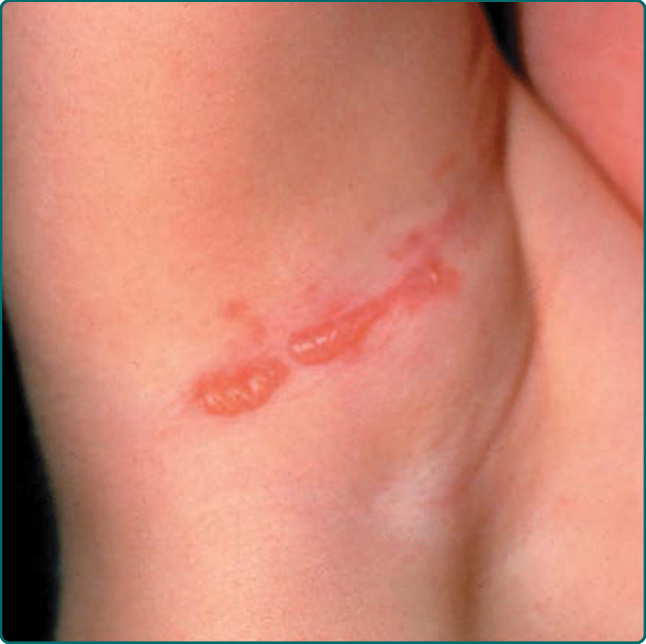
Figure 60-5 Trauma-induced blistering from clothing in patient with localized epidermolysis bullosa simplex.
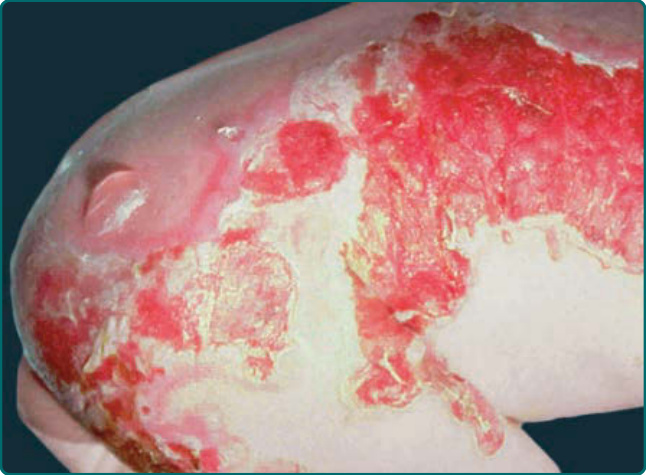
Figure 60-6 Widespread blistering at birth in an infant with Herlitz junctional epidermolysis bullosa.
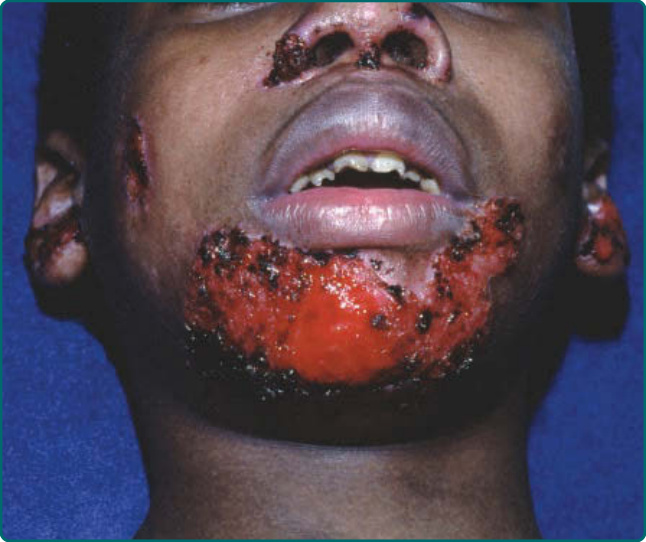
Figure 60-7 Periorificial erosions and hypertrohic granulation tissue in a patient with non-Herlitz junctional epidermolysis bullosa.
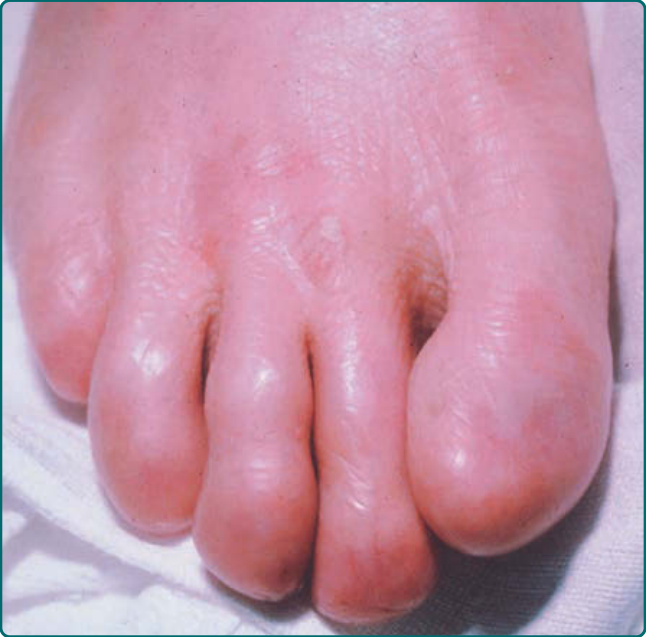
Figure 60-8 Loss of nails and skin atrophy a patient with non-Herlitz junctional epidermolysis bullosa or generalized atrophic benign epidermolysis bullosa.
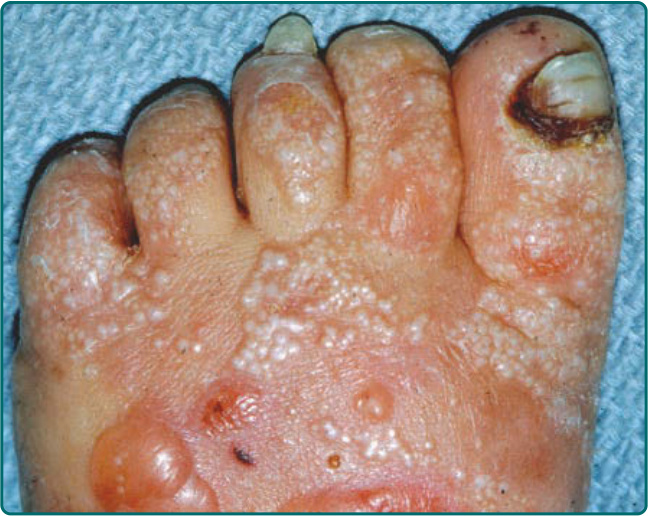
Figure 60-9 Localized trauma-induced blistering with secondary milia in a patient with dominant dystrophic epidermolysis bullosa.
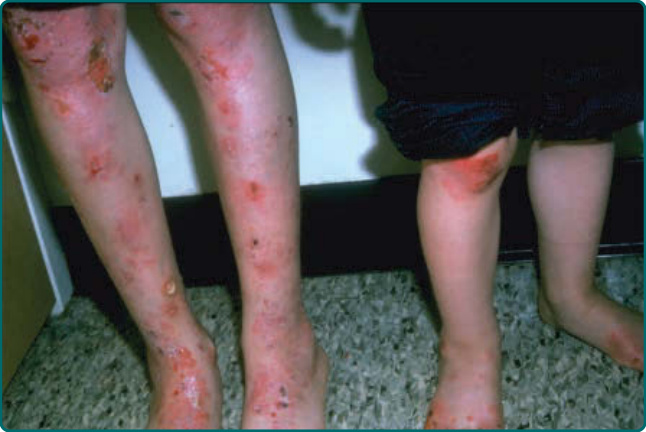
Figure 60-10 Cutaneous lesions in siblings with dominant dystrophic epidermolysis bullosa.
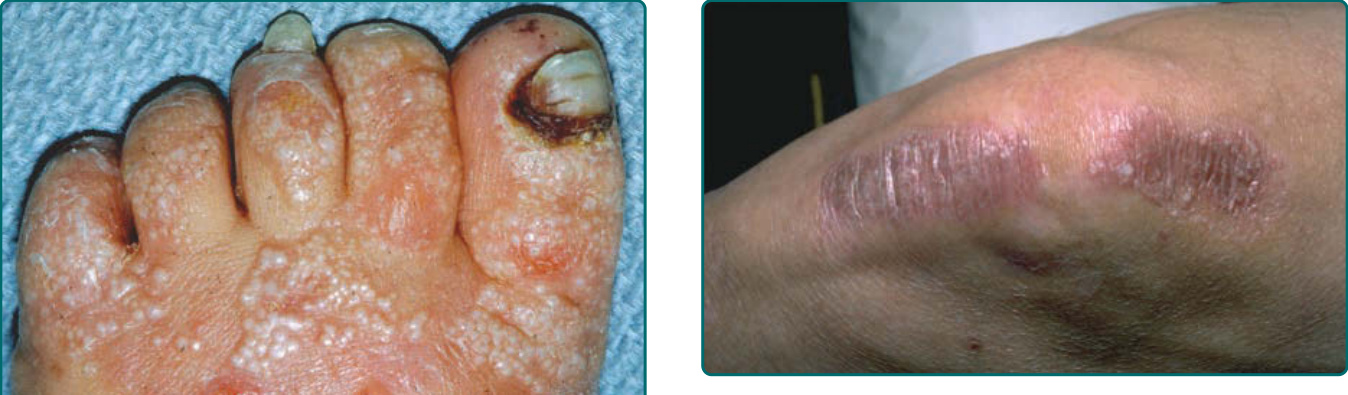
Figure 60-11 Milia in a patient with dominant dystrophic epidermolysis bullosa.
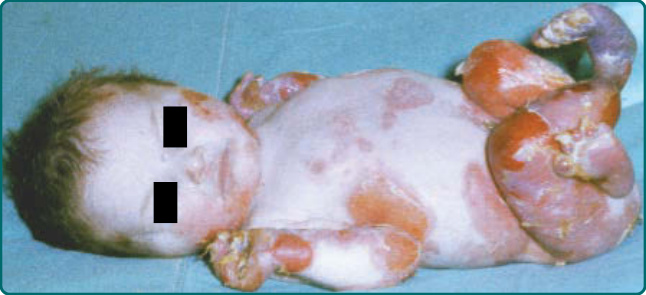
Figure 60-12 Widespread blistering with localized absence of skin at birth in a patient with recessive dystrophic epidermolysis bullosa.
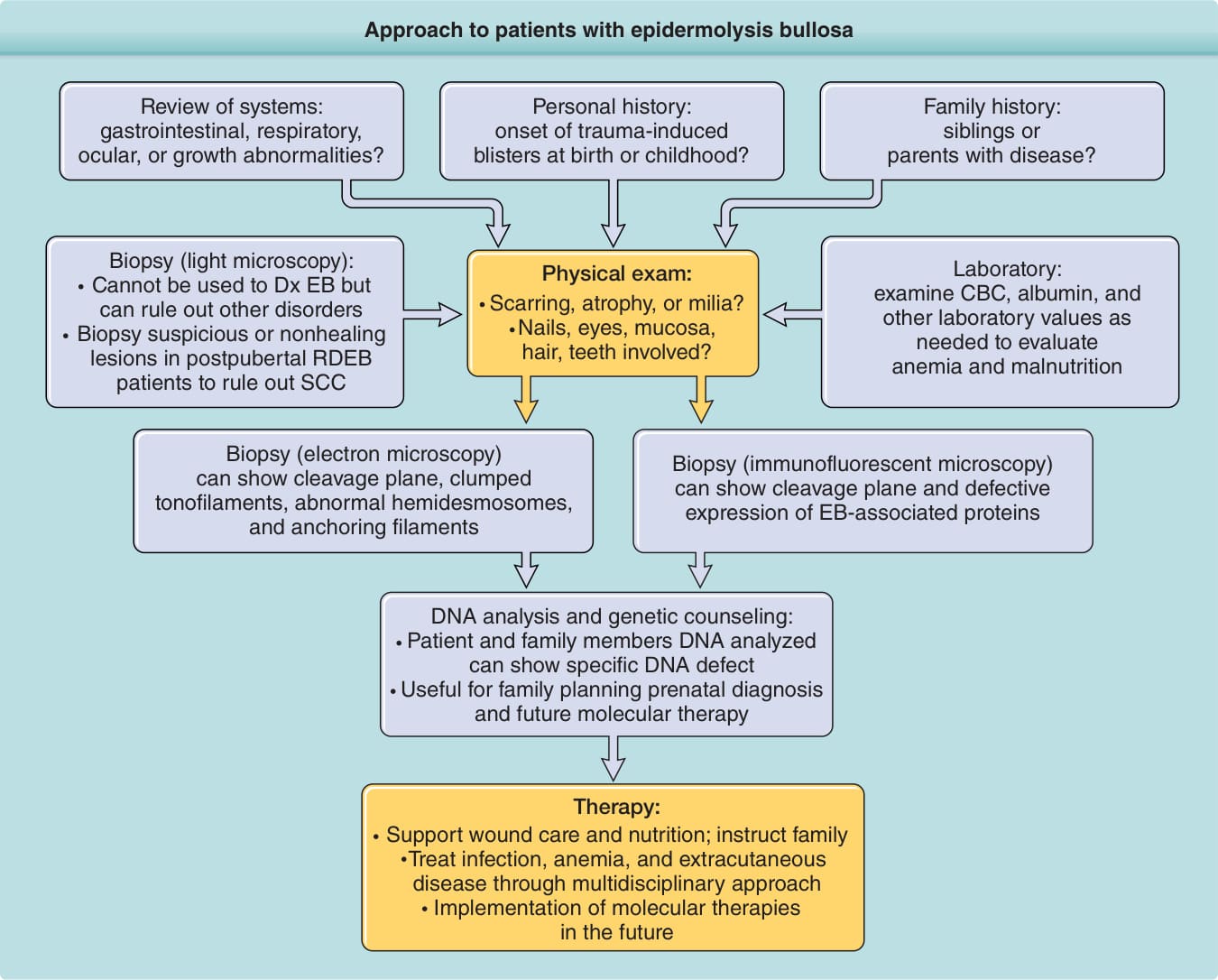
Figure 60-13 Approach to patients with epidermolysis bullosa (EB). A complete history, including family history and review of systems, is essential. Physical examination can provide important clues, and laboratory values can help identify associated anemia and malnutrition. Electron microscopy or indirect immunofluorescent microscopy is required for diagnosis. DNA analysis is helpful for determining prognosis and family planning. Therapy is mainly supportive. Teaching and working with nursing staff and especially families is critical, as are interdisciplinary interactions with other specialists in the treatment of extracutaneous complications. CBC, complete blood count; Dx, diagnosis; RDEB, autosomal recessive epidermolysis bullosa; SCC, squamous cell carcinoma.
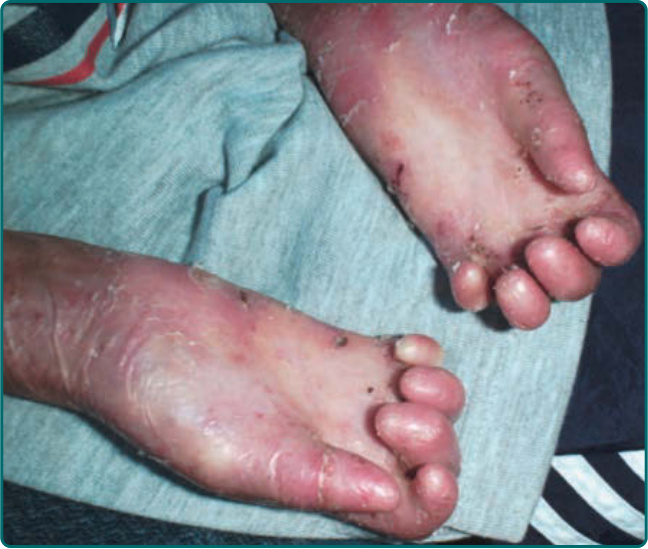
Figure 60-14 Scarring of the hands in patient with recessive dystrophic epidermolysis bullosa.
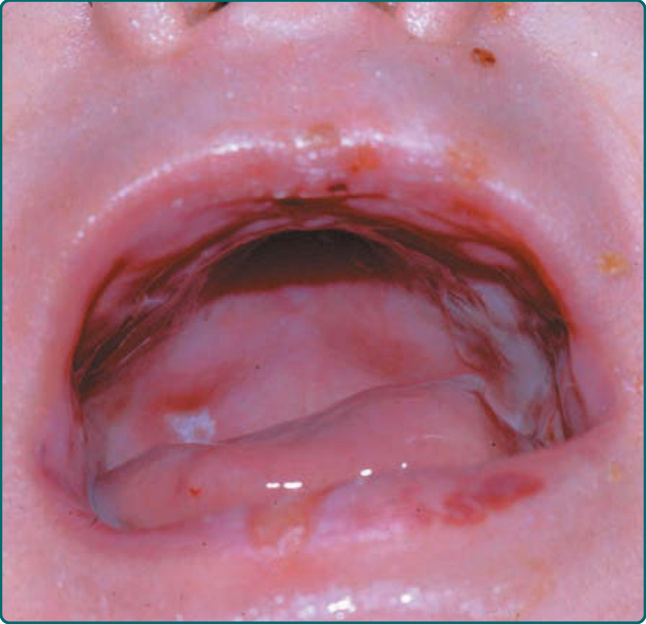
Figure 60-15 Oral erosions in a patient with dominant dystrophic epidermolysis bullosa.
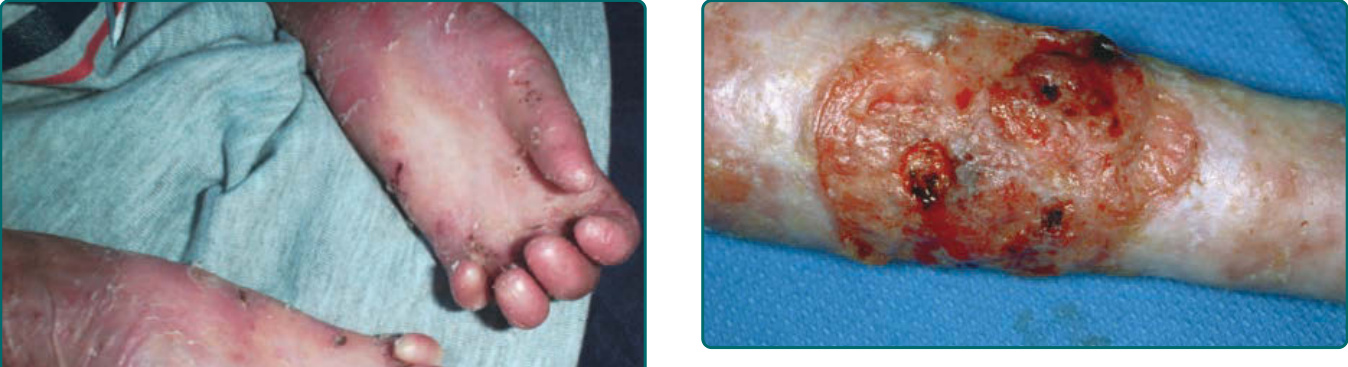
Figure 60-16 Squamous cell carcinoma in patient with recessive dystrophic epidermolysis bullosa.
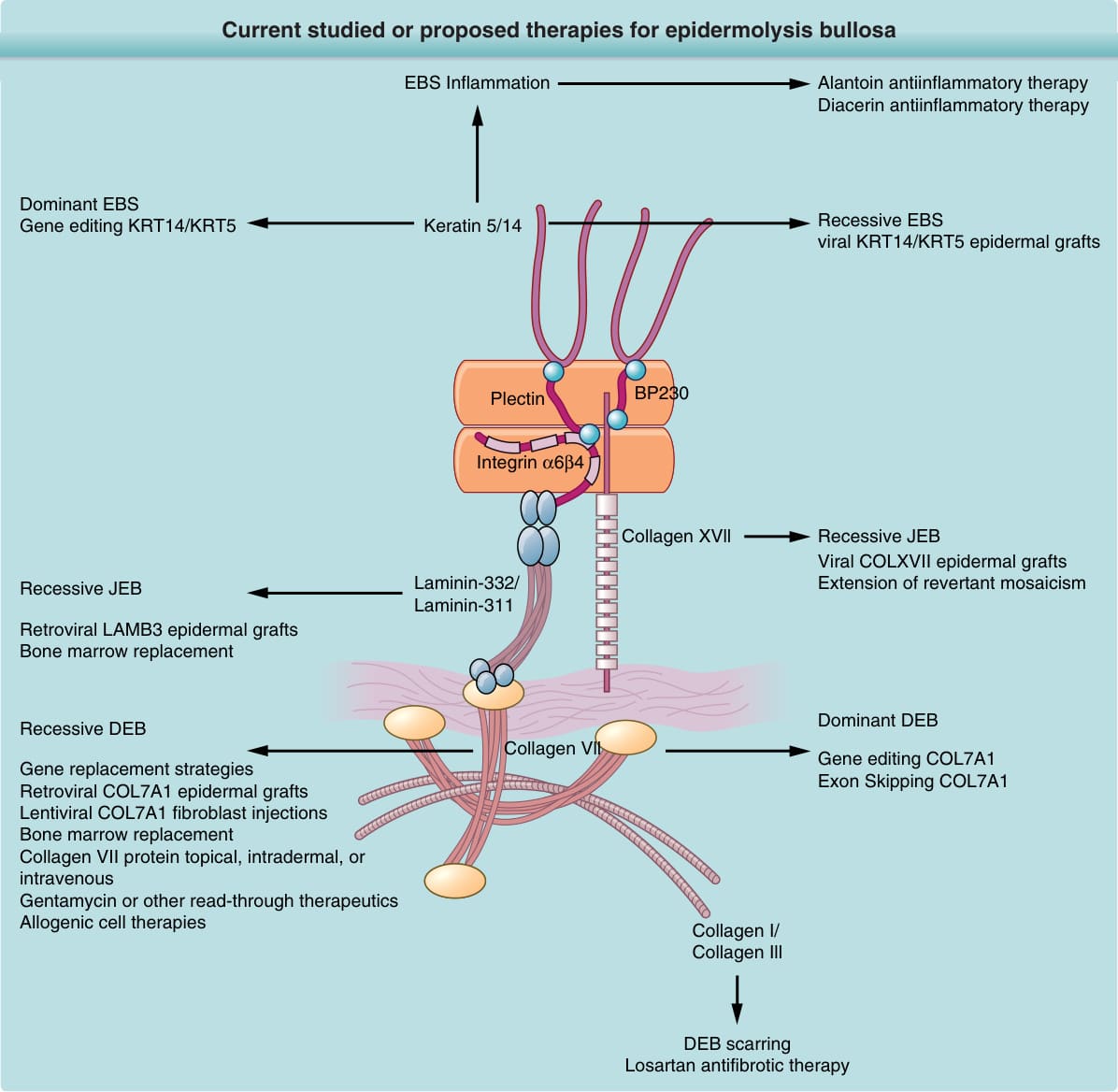
Figure 60-17 Current studied or proposed therapies for epidermolysis bullosa. DEB, dystrophic epidermolysis bullosa; EBS, epidermolysis bullosa simplex; JBS, junctional epidermolysis bullosa.
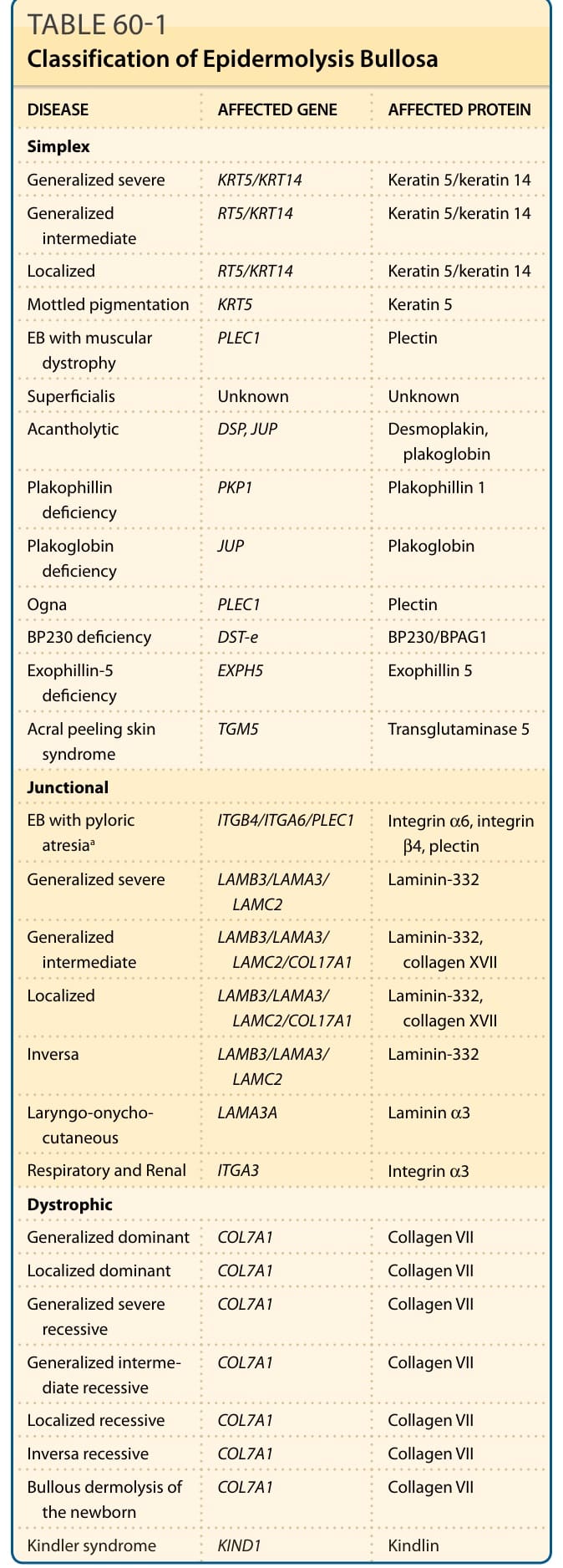
TABLE 60-1 Classification of Epidermolysis Bullosa

TABLE 60-2 Differential Diagnosis of Epidermolysis Bullosa
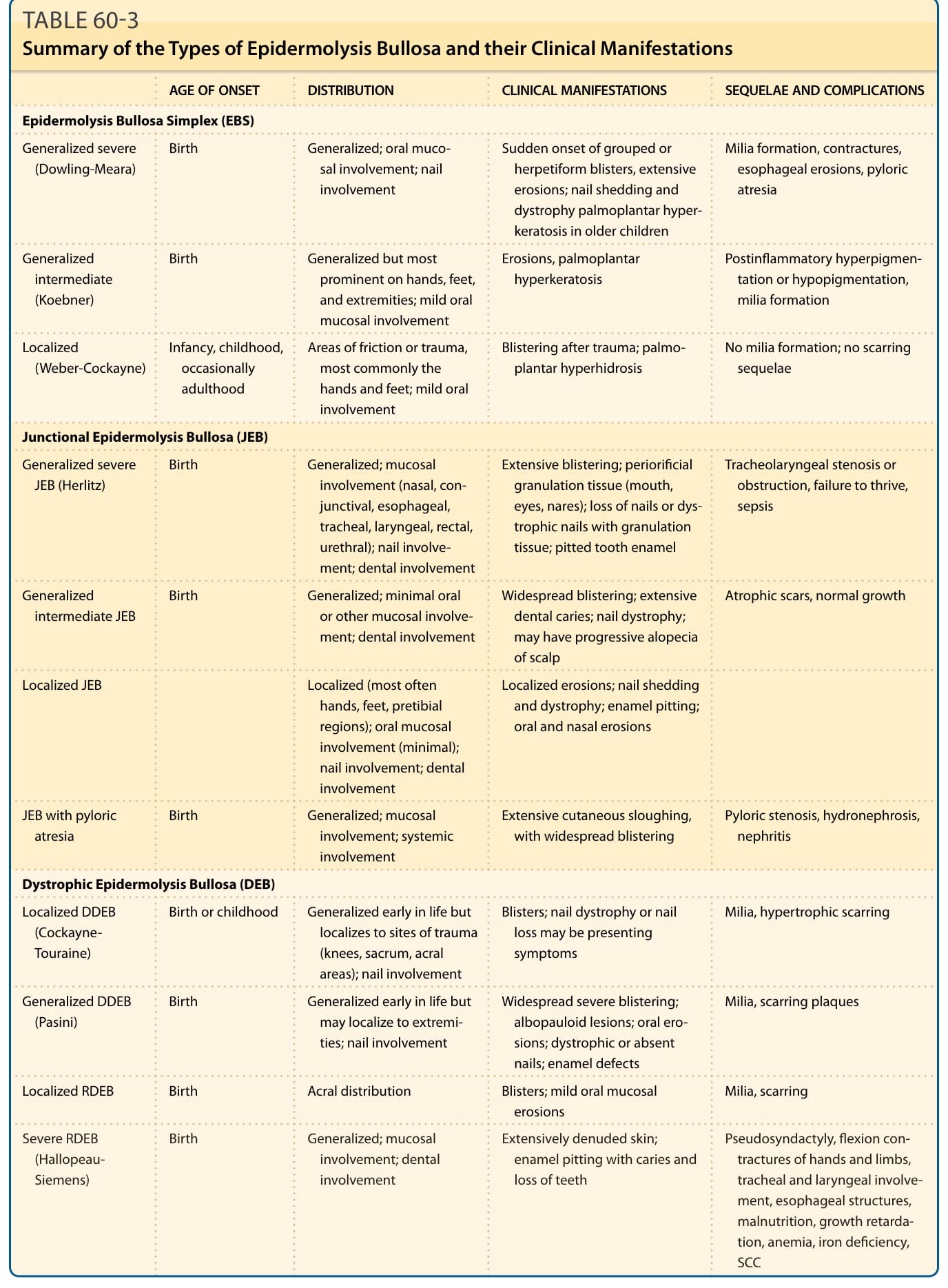
TABLE 60-3 Summary of the Types of Epidermolysis Bullosa and their Clinical Manifestations