後天性表皮鬆解水皰症 (Epidermolysis Bullosa Acquisita) 精華筆記
定義與分類
- 後天性表皮鬆解水皰症 (epidermolysis bullosa acquisita, EBA) 是一種罕見、慢性、表皮下水皰的自體免疫疾病,肇因於針對第 VII 型膠原蛋白 (Type VII collagen) 的免疫球蛋白 G (IgG) 自體抗體;病因不明,通常始於成年期。
- 至少有 5 種臨床表現:(1) 典型 (classic) 表現、(2) 類大皰性類天皰瘡 (bullous pemphigoid, BP) 表現、(3) 類黏膜類天皰瘡 (mucous membrane pemphigoid, MMP) 表現、(4) 類 Brunsting–Perry 類天皰瘡 (頭頸部為主、有瘢痕)、(5) 類線狀 IgA 大皰性皮膚病 (linear IgA bullous dermatosis) 表現。典型型與類 BP 型最常見。
流行病學
- 散發性、極罕見;無性別、種族、族裔或地理好發傾向。發生率約每百萬人口 0.17–0.26 例,僅佔表皮下自體免疫水皰病的百分之二至三。
- 美國東南部非裔美國人 EBA 與大皰性全身性紅斑狼瘡 (bullous SLE) 病人 HLA-DR2 表現型比例高,HLA-DR2+ 個體 EBA 相對風險為 13.1;另研究發現 HLA-DRB1∗15:03 過度表現。
致病機轉 (pathogenesis)
- 第 VII 型膠原蛋白構成真皮—表皮交界處 (dermal–epidermal junction, DEJ) 的錨定纖維 (anchoring fibrils),將表皮與基底膜帶 (basement membrane zone, BMZ) 錨定至乳頭層真皮;EBA 與遺傳性失養型 EB 皆有錨定纖維減少 (但前者為自體免疫、後者為 COL7A1 基因缺陷)。
- IgG 自體抗體結合於第 VII 型膠原蛋白 α 鏈 (分子量 290 kDa,由 3 條相同 α 鏈組成同源三聚體) 的胺基末端球狀 NC-1 區域;大多數自體抗體辨識 NC-1 內 4 個主要抗原性表位,不辨識螺旋區或 NC-2。
- 致病性證據:大皰性 SLE「自然界的實驗」、兔抗血清與親和純化人類自體抗體的被動轉移實驗皆能在小鼠誘發類 EBA 表皮下水皰;自體抗體涵蓋全部 4 種 IgG 亞型 (尤以 IgG1、IgG4 為主)。EBA 較 BP 有更高比例的 C3b 與 C5 (90% vs 33%、90% vs 58%),是更強效的 C5 活化劑。
臨床表現
- 典型表現:非發炎性、肢端 (acral) 分布的機械性水皰病 (mechanobullous disease),皮膚脆弱;好發手背、指節、肘、膝、薦部、腳趾等易受外傷處,見糜爛、緊張性水皰、瘢痕及痊癒後珍珠樣粟粒囊腫 (milia)。輕度像遲發性皮膚卟啉症 (porphyria cutanea tarda, PCT),重度像隱性失養型 EB,但無多毛症、光照分布或硬皮病樣變化,尿中卟啉正常。可見瘢痕性禿髮與甲營養不良。
- 類 BP 表現:廣泛、發炎性水皰大皰疹,侵犯軀幹、身體中央與皮膚皺褶;緊張性大皰被發炎或蕁麻疹樣皮膚環繞,常搔癢,無明顯皮膚脆弱、瘢痕或粟粒疹。
- 類 MMP 表現:以黏膜為主,口腔、上食道、結膜、肛門或陰道有糜爛與瘢痕。
- 類 Brunsting–Perry 表現:慢性復發、侷限頭頸部,有殘餘瘢痕、表皮下大皰,極少或無黏膜侵犯。
- 類 IgA 大皰性皮膚病表現:表皮下水皰疹、嗜中性球浸潤、BMZ 線狀 IgA 沉積,可呈環狀緊張性水皰。
- 發生率:約百分之二十五病人以類 BP 表現呈現,部分日後轉為機械性水皰型,兩型可並存;類 CP 表現不到百分之十。兒童 EBA 罕見,黏膜侵犯常見且嚴重,但整體預後較成人佳。
- 相關全身性疾病:發炎性腸道疾病 (inflammatory bowel disease) 最常相關;亦可見 SLE、澱粉樣變性、甲狀腺炎、類風濕性關節炎、肺纖維化、慢性淋巴球性白血病、胸腺瘤、糖尿病、多發性骨髓瘤等。
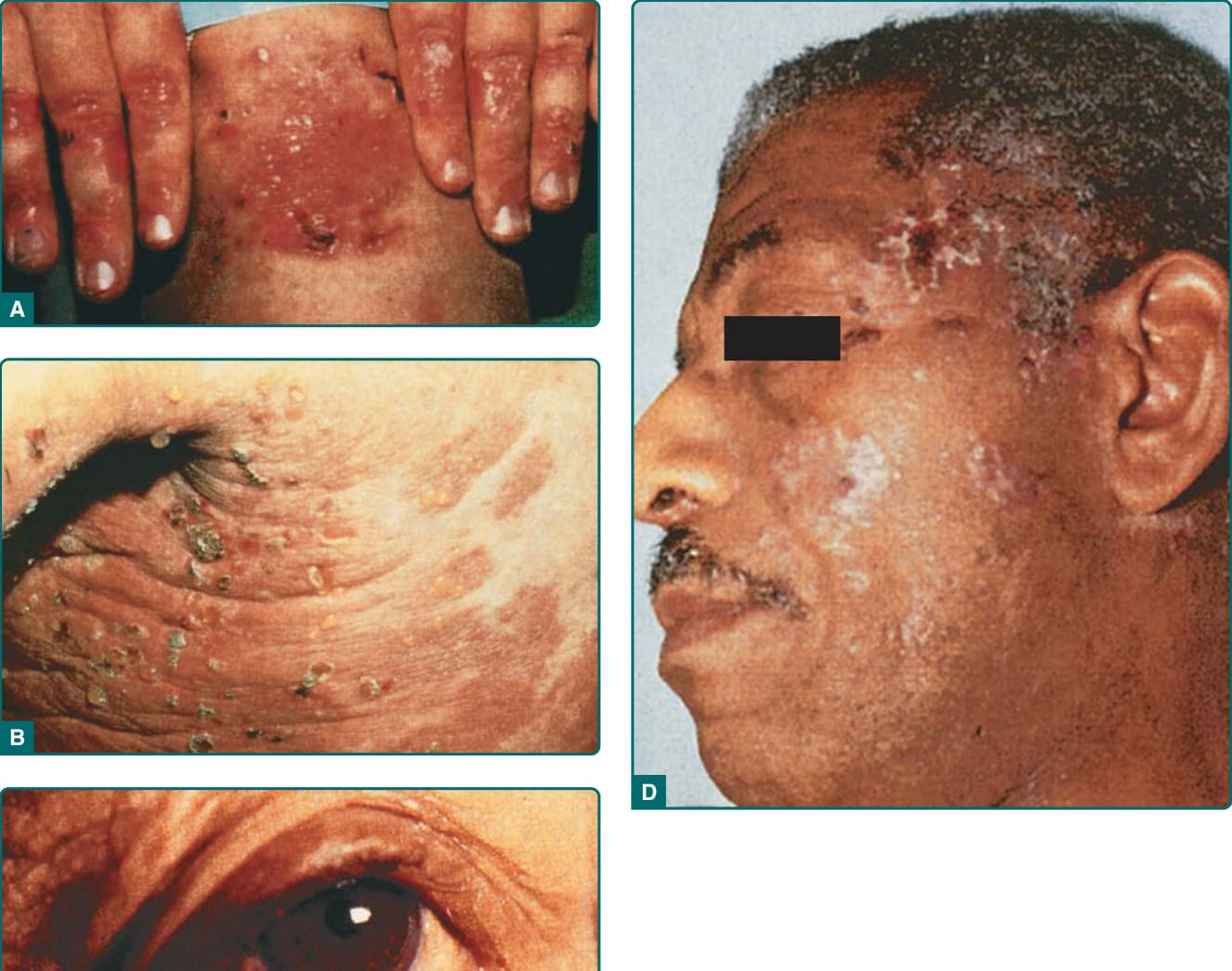
圖 56-3:EBA 四種臨床表現——A 典型 (瘢痕與粟粒疹)、B 類 BP、C 類瘢痕性類天皰瘡、D 類 Brunsting–Perry。
診斷與鑑別診斷
- 不明原因皮膚大皰應做 3 項檢查:常規 H&E 組織學、病灶旁正常皮膚 DIF、抽血測抗 BMZ/第 VII 型膠原蛋白抗體 (IIF 或 ELISA)。
- 組織病理:表皮下水皰、表皮真皮乾淨分離;發炎細胞多寡反映臨床發炎程度 (類 BP 病灶可能難與 BP 區分)。
- 直接免疫螢光 (DIF):病灶周圍皮膚 DEJ 處強烈線狀 IgG 沉積 (主要免疫球蛋白),亦可見補體、IgA、IgM、B 因子、備解素。
- 免疫電子顯微鏡:診斷黃金標準;免疫沉積位於緻密板下 (sublamina densa) 區域,異於 BP (半橋粒/透明板) 與 CP (透明板)。
- 鹽裂皮膚免疫螢光:人類皮膚於 1 莫耳氯化鈉中沿透明板斷裂;EBA 抗體標記分離後的真皮底 (dermal floor),BP 抗體標記表皮頂 (epidermal roof),為區分 EBA 與 BP 的第一線檢測。
- 西方墨點法:EBA 血清結合 290-kDa 條帶 (α 鏈),常另標記 145-kDa 條帶 (NC-1 區域)。ELISA (重組 NC-1) 較免疫螢光與墨點法敏感且具專一性,已有市售。
- 鑑別診斷 (表 56-1):最可能為 PCT、假性 PCT、BP、瘢痕性類天皰瘡;其次考慮線狀 IgA 大皰病、Brunsting–Perry 類天皰瘡、大皰性 SLE。PCT 與假性 PCT 的 DIF 在真皮血管周圍亦有 IgG 沉積 (EBA 不染色),可資區別;PCT 以尿/血漿尿卟啉檢測排除。
- 診斷標準 (表 56-2):屬前述臨床範疇的水皰病、無水皰病家族史、組織學示表皮下水皰、DEJ 處 IgG 沉積 (病灶周圍 DIF 陽性)、免疫電顯示 IgG 侷限於緻密板下層/緻密板下區。
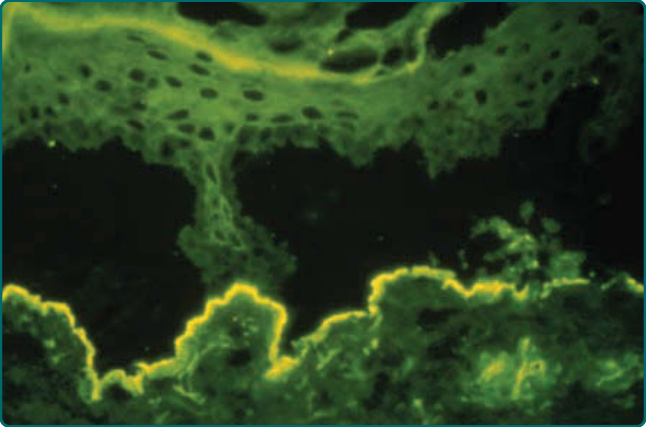
圖 56-7:冷鹽裂 DIF——EBA 的 IgG 沉積留存於真皮底,BP 則留存於表皮頂。
併發症
- 繼發性皮膚感染 (多為葡萄球菌或鏈球菌)、瘢痕與粟粒疹形成;嚴重者手部纖維化致活動範圍減少、足底與腳趾傷口纖維化致行走困難、指甲脫落;黏膜侵犯者可發展食道狹窄甚至喉部瘢痕。
治療
- EBA 通常治療反應不佳。支持性治療為所有病人所必須:開放性傷口照護、避免外傷、勿過度清洗或用熱水/刺激性肥皂、避免摩擦皮膚、避免長時間日曬並用防曬乳、辨識並及早處理皮膚感染。
- 典型機械性水皰型常對高劑量全身性糖皮質素、azathioprine、methotrexate、cyclophosphamide 頑強;這些藥對發炎性類 BP 型較有幫助。
- Dapsone 在真皮有嗜中性球時較有效。Cyclosporine 有益但長期毒性限制使用。Colchicine 常作第一線 (副作用相對良性),但腹瀉常見、發炎性腸道疾病病人應保留使用。光分離術 (photopheresis) 可改善臨床並延長抽吸性水皰形成時間。
- 血漿置換術 (plasmapheresis) 移除抗第 VII 型膠原蛋白抗體有助控制,但須併用化療藥物 (azathioprine、cyclophosphamide、mycophenolate mofetil、methotrexate)。IVIG、抗 TNF-α 生物製劑 (infliximab)、rituximab (抗 CD20,對頑固型有效) 亦有報告。
- 治療劑量 (表 56-3):Colchicine 0.6–3.0 mg/d;Cyclosporine A 6 mg/kg/d;Dapsone 100–300 mg/d;Cytoxan 50–200 mg/d;Prednisone 1.0–1.5 mg/kg;靜脈注射免疫球蛋白 3 g/kg 分次於 5 日給予;Infliximab 5 mg/kg 於第 0、2、4、6 週;Rituximab 375 mg/m² 體表面積靜脈注射每週 1 次 × 4 週,或 1000 mg 靜脈注射於第 1 與第 3 週。
預後
- 慢性難治;典型機械性水皰型可遺留瘢痕、頭皮毛髮脫落、指甲脫落、手部與手指纖維化及食道狹窄等後遺症。兒童 EBA 整體預後較成人佳。