後天性表皮鬆解水皰症 (Epidermolysis Bullosa Acquisita)
PART 9
水皰大皰性疾病 (Vesiculobullous Disorders)
重點一覽 (AT-A-GLANCE)
■ 一種罕見的自體免疫性表皮下水皰病,肇因於針對第 VII 型膠原蛋白 (Type VII collagen) 的免疫球蛋白 G (immunoglobulin G) 自體抗體。
■ 病因不明。
■ 表現為皮膚脆弱、表皮下水皰、殘餘瘢痕,以及粟粒疹 (milia) 形成。好發部位為易受外傷的區域,例如手、足、肘、膝、薦部 (sacrum)、指甲與口腔。
■ 相關特徵可能包括潛在的全身性疾病,例如發炎性腸道疾病 (inflammatory bowel disease)。可能出現黏膜糜爛與食道狹窄 (esophageal stenosis)。
■ 病理顯示表皮下大皰、纖維化、粟粒疹形成,以及在表皮—真皮交界處 (epidermal–dermal junction) 對免疫球蛋白 G 沉積呈陽性的直接免疫螢光 (direct immunofluorescence)。
■ 治療選擇有限,且往往十分困難。
流行病學 (EPIDEMIOLOGY)
後天性表皮鬆解水皰症(epidermolysis bullosa acquisita, EBA)是一種病因不明的散發性自體免疫水皰病。本病無性別、種族、族裔或地理上的好發傾向。居住在美國東南部的非裔美國人對 EBA 與自體免疫可能存在某種遺傳易感性。¹ 美國東南部罹患 EBA 或大皰性全身性紅斑狼瘡 (bullous systemic lupus erythematosus, SLE) 的非裔美國病人具有高比例的 HLA-DR2 表現型 (phenotype)。在這些病人中,HLA-DR2+ 個體罹患 EBA 的計算相對風險為 13.1。這些結果亦提示 EBA 與大皰性 SLE 在免疫遺傳學上相關,且 HLA-DR2 基因要不是參與了針對錨定纖維膠原蛋白 (anchoring fibril collagen) 的自體免疫,便是某個與其處於連鎖不平衡 (linkage disequilibrium) 之其他基因的某種標記。¹
在另一項檢視 EBA 病人 HLA 基因型的研究中,發現 HLA-DRB1∗15:03 呈現過度表現。²
與人類對 EBA 的遺傳許可性 (genetic permissiveness) 相符,在一個以鼠源第 VII 型膠原蛋白免疫小鼠所建立的活動性 EBA 小鼠模型中,EBA 的發生取決於所使用的小鼠品系。³,⁴
EBA 是一種極為罕見的疾病,其發生率遠低於其他自體免疫水皰病(大皰性類天皰瘡 bullous pemphigoid、尋常性天皰瘡 pemphigus vulgaris、皰疹樣皮膚炎 dermatitis herpetiformis 等),而後者本身也相當罕見。在血液中表現抗基底膜帶 (anti–basement membrane zone) 自體抗體的未經篩選之水皰病病人中,約有 5% 被診斷為 EBA 或大皰性 SLE。⁵ 此發生率與其他研究者的報告相近。⁶ 一項使用電子顯微鏡與免疫電子顯微鏡來確認 EBA 診斷的法國前瞻性研究發現,其發生率為每百萬人口 0.17 至 0.26 例,僅佔所有表皮下自體免疫水皰病病人的 2% 至 3%。⁷ 德國某地區的 EBA 發生率為 0.17,再次確認了 EBA 之罕見性。⁸
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
EBA 是一種慢性、表皮下水皰性疾病,與針對位於真皮—表皮交界處 (dermal–epidermal junction, DEJ) 之錨定纖維 (anchoring fibril) 結構內膠原蛋白(第 VII 型膠原蛋白)的自體免疫有關。雖然 EBA 的確切病因不明,但大多數證據提示為自體免疫病因。針對第 VII 型膠原蛋白的免疫球蛋白 G (IgG) 自體抗體,與分隔表皮與真皮之基底膜帶 (basement membrane zone, BMZ) 處正常錨定纖維的稀少及表皮—真皮黏附不良有關。雖然這是一種通常始於成年期的後天性疾病,但在一百多年前即被歸入表皮鬆解水皰症 (epidermolysis bullosa, EB) 的範疇,因為醫師驚訝於 EBA 的臨床病灶與罹患遺傳性失養型 EB (hereditary dystrophic forms of EB) 的兒童所見者何其相似。EBA 病人病灶周圍皮膚切片的直接免疫螢光 (direct immunofluorescence, DIF) 顯示 DEJ 處有 IgG 沉積。⁹ EBA 抗體結合於錨定纖維內的第 VII 型膠原蛋白。¹⁰,¹¹
錨定纖維將表皮及其下方的 BMZ 錨定至乳頭層真皮 (papillary dermis)。罹患遺傳性失養型 EB(第 60 章)與 EBA 的病人其 DEJ 中錨定纖維數目減少。此錨定纖維的稀少與兩種相似的臨床表現型相關,即 EBA 與遺傳性失養型 EB,因為這兩種疾病皆以皮膚脆弱、表皮下水皰、粟粒疹形成與瘢痕為特徵。雖然 EBA 與遺傳性失養型 EB 在其潛在發病機轉上於病因學上互不相關,但兩者皆具有錨定纖維減少的共同特徵。就遺傳性失養型 EB 而言,錨定纖維減少或缺失的原因是編碼第 VII 型膠原蛋白 α 鏈之 COL7A1 基因的遺傳缺陷,最終導致錨定纖維小型化、無功能或減少。¹²,¹³ 編碼第 VII 型膠原蛋白的基因位於第 3 號染色體短臂上。¹⁴ 遺傳性失養型 EB 所涉及的基因缺陷已在不同位置被鑑定出來,但疾病的嚴重度似乎與第 VII 型膠原蛋白及錨定纖維受擾亂的程度相關。¹³ 在 EBA 中,IgG 自體抗體結合於第 VII 型膠原蛋白 α 鏈導致錨定纖維減少,但導致此減少的途徑不明。可能是新合成但被 EBA 自體抗體修飾 (decorated) 的第 VII 型膠原蛋白 α 鏈無法形成三股螺旋結構 (triple-helical structures) 與穩定的錨定纖維。以培養之角質細胞層 (cultured keratinocyte sheets) 覆蓋之痊癒燒傷傷口,在移植後第一年內錨定纖維數目亦減少,且此情形與自發性水皰形成、抽吸性水皰形成時間 (suction blistering times) 縮短及皮膚脆弱有關。¹⁵ 這些觀察結果提供了間接證據,說明錨定纖維在維持表皮與真皮之間的黏附上扮演角色。第 VII 型膠原蛋白 α 鏈的分子量為 290 kDa,該膠原蛋白由 3 條相同 α 鏈所組成的同源三聚體 (homotrimer) 構成(第 15 章)。每條 α 鏈包含一個大型球狀的非膠原性胺基末端 (noncollagenous amino terminus),稱為非膠原性 1 (noncollagenous 1, NC-1) 區域,約佔整條 α 鏈質量的一半。其次是一個具有典型甘胺酸-X-Y 重複序列 (glycine-X-Y repeats) 的螺旋區域。在羧基末端 (carboxyl terminus) 則有第二個球狀非膠原性區域 NC-2,其遠小於 NC-1。¹⁶ 大多數 EBA 自體抗體辨識 NC-1 區域內 4 個主要的抗原性表位 (antigenic epitopes),而不辨識螺旋區域或 NC-2 區域。¹⁷,¹⁸
NC-1 區域可能具有某種內在的「抗原性 (antigenic)」特質,因為現有針對第 VII 型膠原蛋白所產生的單株抗體(抗 C-VII 抗體 anti–C-VII antibodies)專一性地僅辨識 NC-1 次區域。在 EBA 病人的病灶與病灶周圍皮膚中可見錨定纖維數目減少,但導致此減少的途徑不明。數條獨立的證據線索已將自體免疫反應牽連為 EBA 發病機轉中的關鍵要素。第一,EBA 抗體的致病角色由以下觀察所提示:當 SLE 病人發展出針對第 VII 型膠原蛋白的自體抗體時,他們會出現廣泛的皮膚水皰,並落入一種稱為大皰性 SLE 的罕見 SLE 亞型。¹⁹
此「自然界的實驗 (experiment of nature)」提示 EBA 自體抗體具有致病性,並能誘發表皮與真皮之間的脫離 (disadherence)。第二,EBA 自體抗體具致病性的直接證據來自近期的被動轉移 (passive transfer) 研究。我們免疫兔子並產生針對人類第 VII 型膠原蛋白 NC-1 區域的高效價抗血清。我們將此抗體注射至無毛、免疫功能正常的小鼠體內,小鼠便發展出具有許多人類 EBA 特徵的大皰性皮膚病。²⁰ 這些小鼠發展出表皮下水皰並失去足部指甲。牠們血液中亦有循環的 NC-1 抗體,且 DEJ 處有抗 NC-1 IgG 抗體沉積。此外,這些小鼠在 DEJ 處有由自體抗體—抗原複合物所誘發的鼠源補體沉積。²⁰ Sitaru 及其同僚²¹ 的另一項研究顯示,將兔子針對小鼠第 VII 型膠原蛋白 NC-1 區域的多株抗體 (polyclonal antibodies) 注射至小鼠體內,亦誘發了類似人類 EBA 的表皮下皮膚水皰。²¹
此外,我們也以 NC-1 管柱親和純化 (affinity-purified) 人類 EBA 自體抗體並注射至小鼠體內。小鼠隨後發展出類似人類 EBA 的臨床、組織學、免疫學與超微結構 (ultrastructural) 特徵。²² 綜合而言,這些成功的被動轉移實驗與大皰性 SLE 的觀察結果強烈提示 EBA 自體抗體具「致病性」,並能在皮膚中造成表皮—真皮分離。透過臨床病人研究、體外試驗 (in vitro assays) 與 EBA 小鼠模型,已對帶有抗第 VII 型膠原蛋白自體抗體之 EBA 病人的免疫反應細節有諸多了解。EBA IgG 自體抗體無論在病人血液中或結合於病人 DEJ 的組織中,皆涵蓋全部 4 種 IgG 亞型 (subclasses),尤以 IgG1 與 IgG4 為著。²³ 循環的 EBA 自體抗體包含可結合補體與不結合補體的兩種族群。²⁴ 然而,與 BP 相較,EBA 病人的 C3b 與 C5 出現率較高(分別為 90% vs 33%、90% vs 58%)。這些研究提供了證據³,說明大多數 EBA 病人在 DEJ 處有補體活化,且 EBA 抗體較 BP 抗體為更強效的 C5 活化劑。²⁵ Gammon 及其同事發展出一種體外試驗,以觀察大皰性類天皰瘡與 EBA 免疫複合物在皮膚中的功能。製作一個玻片小室 (slide chamber),由人類皮膚(正常皮膚或來自 BP 或 EBA 病人的皮膚)的冷凍切片構成,並在有無添加白血球與補體的情況下進行。其判讀指標為白血球向人類皮膚冷凍切片之 DEJ 的遷移與趨化。²⁶ 當比較大皰性類天皰瘡皮膚與 EBA 皮膚的冷凍切片時,EBA 免疫複合物再次產生比大皰性類天皰瘡免疫複合物更多的補體活化。除了如上述「流行病學」一節所提及之以被動轉移抗第 VII 型膠原蛋白抗體所建立的 EBA 小鼠模型外,已有以鼠源第 VII 型膠原蛋白免疫某些品系小鼠所建立的活動性 EBA 小鼠模型。此活動性鼠源 EBA 模型涉及具補體固定能力 (complement fixing) 的抗第 VII 型膠原蛋白抗體,且疾病誘發需要 T 細胞。
臨床表現 (CLINICAL FINDINGS)
見圖 56-1。若病人出現皮膚大皰,而在詳盡的病史詢問與身體檢查後仍無合理解釋,則應進行 3 項檢查:取皮膚切片做常規蘇木精與伊紅 (hematoxylin and eosin) 組織學檢查、在病灶旁但外觀正常之皮膚取第二片切片做 DIF,以及抽血以間接免疫螢光 (indirect immunofluorescence, IIF) 或酵素連結免疫吸附分析 (enzyme-linked immunosorbent assay, ELISA) 檢測針對 BMZ 與/或第 VII 型膠原蛋白的抗體。EBA 的皮膚病灶可相當多變,並可模擬其他類型的後天性自體免疫水皰病。EBA 病人的共同特徵為針對第 VII 型(錨定纖維)膠原蛋白的自體免疫。至少有 5 種臨床表現:(1) 典型表現、(2) 類大皰性類天皰瘡 (bullous pemphigoid, BP) 表現、(3) 類黏膜類天皰瘡 (mucous membrane pemphigoid, MMP) 表現、(4) 類似 Brunsting–Perry 類天皰瘡、具瘢痕性病灶且以頭頸部分布為主的表現,以及 (5) 類似線狀 IgA 大皰性皮膚病 (linear IgA bullous dermatosis) 或兒童慢性大皰病 (chronic bullous disease of childhood) 的表現。典型表現與類 BP 表現是這種極罕見疾病最常被報告的表現。
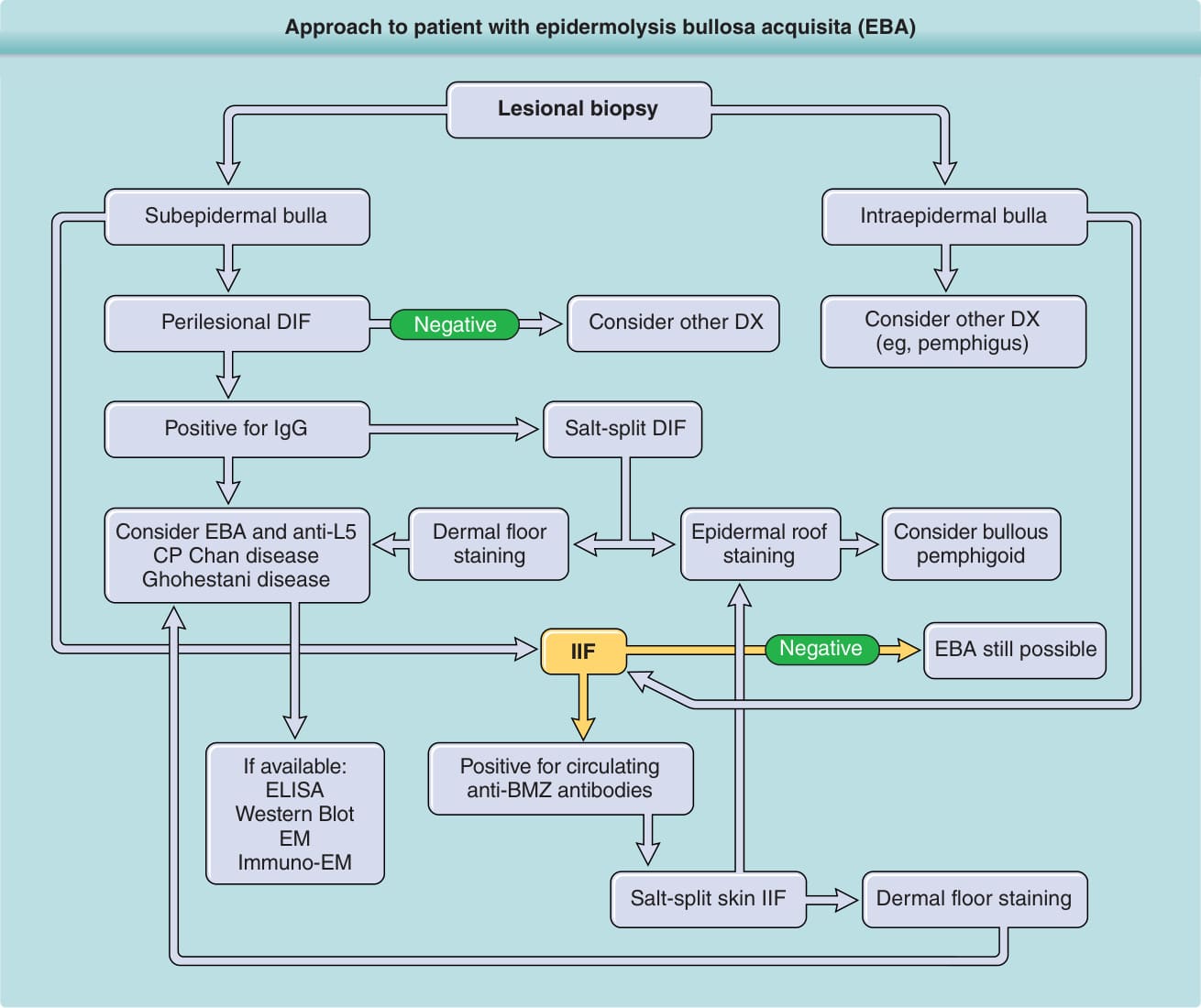
圖 56-1:後天性表皮鬆解水皰症 (epidermolysis bullosa acquisita, EBA) 病人的處理流程。以下為流程內容:病灶切片 (Lesional biopsy) → 若為表皮下大皰 (Subepidermal bulla) 則進行病灶周圍 DIF (Perilesional DIF);若為表皮內大皰 (Intraepidermal bulla) 則考慮其他診斷(如天皰瘡 pemphigus);其餘情形考慮其他診斷。病灶周圍 DIF 若為陰性 (Negative) 則考慮其他診斷;若 IgG 陽性 (Positive for IgG) 則進行鹽裂 DIF (Salt-split DIF)。鹽裂 DIF 若為真皮底染色 (Dermal floor staining),考慮 EBA 與抗 L5 (anti-L5)、CP、Chan 病、Ghohestani 病;若為表皮頂染色 (Epidermal roof staining),考慮大皰性類天皰瘡。接著進行 IIF(若可取得:ELISA、西方墨點 Western Blot、電子顯微鏡 EM、免疫電子顯微鏡 Immuno-EM)。IIF 若為陰性,EBA 仍有可能 (EBA still possible);若循環抗 BMZ 抗體陽性 (Positive for circulating anti-BMZ antibodies),則進行鹽裂皮膚 IIF (Salt-split skin IIF),呈真皮底染色 (Dermal floor staining)。BMZ,基底膜帶;DIF,直接免疫螢光;DX,診斷;ELISA,酵素連結免疫吸附分析;IgG,免疫球蛋白 G;IIF,間接免疫螢光。
典型表現 (CLASSIC PRESENTATION)
典型表現(圖 56-2 與圖 56-3A)為一種具肢端 (acral) 分布、痊癒後留下瘢痕與粟粒疹形成的非發炎性水皰病。此表現在輕度時令人聯想到遲發性皮膚卟啉症 (porphyria cutanea tarda, PCT;第 124 章),在重度時則令人聯想到遺傳性隱性失養型 EB (recessive dystrophic EB;第 60 章)。因此,典型型 EBA 是一種以皮膚脆弱為特徵的機械性水皰病 (mechanobullous disease)。這些病人在易受外傷的表面,例如手背、指節、肘、膝、薦部與腳趾,有糜爛、位於非發炎皮膚內的緊張性水皰,以及瘢痕(圖 56-2、圖 56-3A 與圖 56-4)。有些水皰可能為出血性,或發展出鱗屑、痂皮或糜爛。病灶痊癒後留下瘢痕,並常在瘢痕區域內形成珍珠樣的粟粒囊腫 (pearl-like milia cysts)(圖 56-3A)。雖然此表現可能令人聯想到 PCT,但這些病人並無 PCT 的其他標誌特徵,例如多毛症 (hirsutism)、疹子的光照分布 (photodistribution),或硬皮病樣 (scleroderma-like) 變化,且其尿中卟啉 (urinary porphyrins) 在正常範圍內。可見瘢痕性禿髮 (scarring alopecia) 與某種程度的甲營養不良 (nail dystrophy)。雖然本病通常不如遺傳性隱性失養型 EB 病人那般嚴重,但罹患典型型 EBA 的病人可能有許多相同的後遺症,例如瘢痕、頭皮毛髮脫落、指甲脫落、手部與手指的纖維化,以及食道狹窄。²⁷
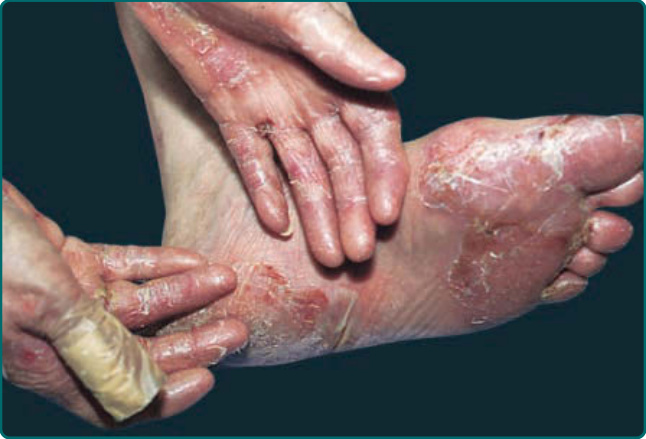
圖 56-2:一名後天性表皮鬆解水皰症病人,其皮膚易受外傷的區域有嚴重水皰、糜爛、瘢痕與粟粒疹 (milia) 形成。此為典型表現。
類大皰性類天皰瘡表現 (BULLOUS PEMPHIGOID-LIKE PRESENTATION)
EBA 的第二種臨床表現為一種廣泛、發炎性的水皰大皰性疹 (vesiculobullous eruption),除四肢外,亦侵犯軀幹、身體中央部位與皮膚皺褶處。⁶ 大皰病灶呈緊張性,並被發炎或甚至蕁麻疹樣 (urticarial) 的皮膚所環繞。可見大片發炎皮膚而無任何水皰,僅有紅斑或蕁麻疹樣斑塊。這些病人常主訴搔癢 (pruritus),且不顯現明顯的皮膚脆弱、瘢痕或粟粒疹形成。此臨床組合較令人聯想到 BP(圖 56-3B 與圖 56-4),而非機械性水皰病。與 BP 相似,病灶的分布可能在屈側 (flexural) 區域與皮膚皺褶處顯現加重。
類黏膜類天皰瘡表現 (MUCOUS MEMBRANE PEMPHIGOID-LIKE PRESENTATION)
EBA 的典型型與類 BP 型皆可能侵犯黏膜表面。然而,EBA 亦可能以如此顯著的黏膜侵犯為主要表現,致其臨床外觀令人聯想到 MMP(圖 56-3C)。²⁸ 這些病人通常在口腔、上食道、結膜 (conjunctiva)、肛門或陰道的黏膜表面有糜爛與瘢痕,無毛皮膚 (glabrous skin) 上可有或可無類似病灶。
類 Brunsting–Perry 類天皰瘡表現 (BRUNSTING–PERRY PEMPHIGOID-LIKE PRESENTATION)
Brunsting–Perry 瘢痕性 BP 是一種慢性、復發性的水皰大皰性疹,侷限於頭頸部,並以殘餘瘢痕、表皮下大皰、DEJ 處 IgG 沉積,以及極少或無黏膜侵犯為特徵。然而,IgG 自體抗體的抗原標的尚未被界定。儘管如此,一名報告有此組合表現的病人,其 IgG 自體抗體指向位於緻密板 (lamina densa) 下方的錨定纖維。²⁹ 我們另見過 3 名具有 Brunsting–Perry 類天皰瘡特徵且自體抗體指向第 VII 型膠原蛋白的病人(未發表之觀察)。因此,EBA 病人似乎可能以 Brunsting–Perry 類天皰瘡的臨床表現型呈現(圖 56-3D)。
類免疫球蛋白 A 大皰性皮膚病表現 (IMMUNOGLOBULIN A BULLOUS DERMATOSIS-LIKE PRESENTATION)
EBA 的類 IgA 大皰性皮膚病 (IgA bullous dermatosis) 表現,其特徵為表皮下水皰性疹、嗜中性球浸潤 (neutrophilic infiltrate),以及在 DIF 下於 BMZ 處所見的線狀 IgA 沉積。它可能類似線狀 IgA 大皰性皮膚病 (linear IgA bullous dermatosis, LABD)、皰疹樣皮膚炎 (dermatitis herpetiformis) 或兒童慢性大皰病,並可能呈現排列成環狀 (annular) 的緊張性水皰與黏膜侵犯。³⁰ 自體抗體通常為 IgA、IgG 或兩者皆有。這些帶有抗第 VII 型膠原蛋白 IgA 抗體、於 BMZ 處顯現線狀 IgA 沉積的表皮下水皰病例,其診斷尚有爭議。有些臨床醫師認為這些病人是純粹的 LABD,而另一些則認為他們屬於 EBA 的一個亞型。此外,大多數 EBA 病人血液中有針對第 VII 型膠原蛋白的低效價 IgA 抗體。兒童 EBA 是一種罕見疾病。它有多變的表現,包括類 LABD 疾病、類 BP 疾病,以及典型的機械性水皰型 EBA 表現。雖然黏膜侵犯在兒童 EBA 中常見且嚴重,但整體預後較成人 EBA 為佳。
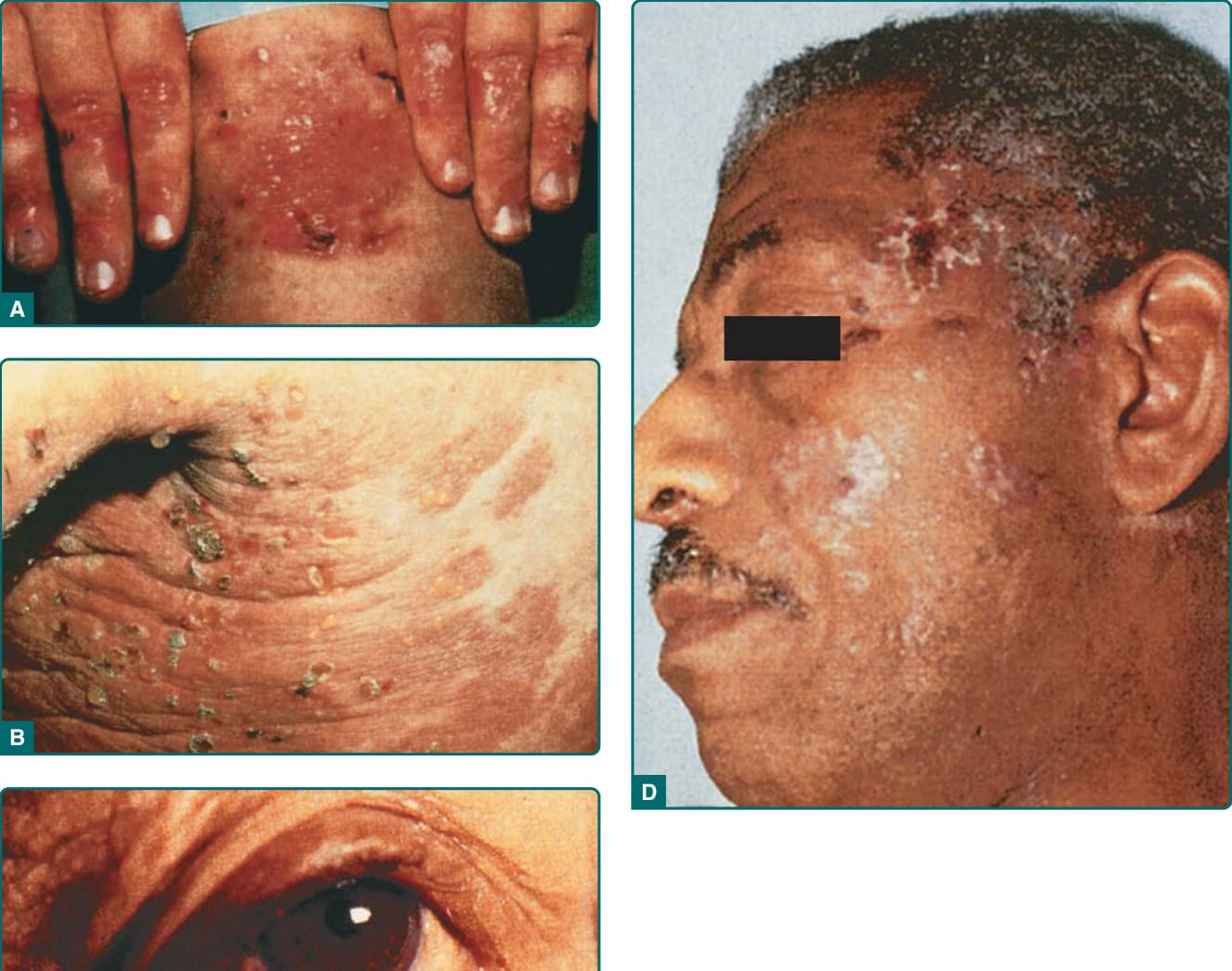
圖 56-3:A,後天性表皮鬆解水皰症的典型表現,於皮膚易受外傷的區域有瘢痕與粟粒疹 (milia)。B,後天性表皮鬆解水皰症的類大皰性類天皰瘡 (bullous pemphigoid–like) 表現,呈廣泛的發炎性水皰大皰性皮膚病。C,後天性表皮鬆解水皰症的類瘢痕性類天皰瘡 (cicatricial pemphigoid–like) 表現,呈以黏膜為中心的大皰性瘢痕性疹。D,後天性表皮鬆解水皰症的類 Brunsting–Perry 類天皰瘡 (Brunsting–Perry pemphigoid–like) 表現,水皰與瘢痕性病灶主要位於頭頸部。
後天性表皮鬆解水皰症各臨床表現之發生率 (INCIDENCE OF THE CLINICAL PRESENTATIONS OF EPIDERMOLYSIS BULLOSA ACQUISITA)
根據作者的經驗,約 25% 的 EBA 病人可能以類 BP 的臨床外觀呈現。其中部分病人的疾病最終會緩慢轉變為較非發炎性的機械性水皰型。然而,典型型與類 BP 型疾病可能在同一病人身上並存(圖 56-5)。令人聯想到純粹 CP 的 EBA 臨床表現型,發生於不到 10% 的所有 EBA 病例。
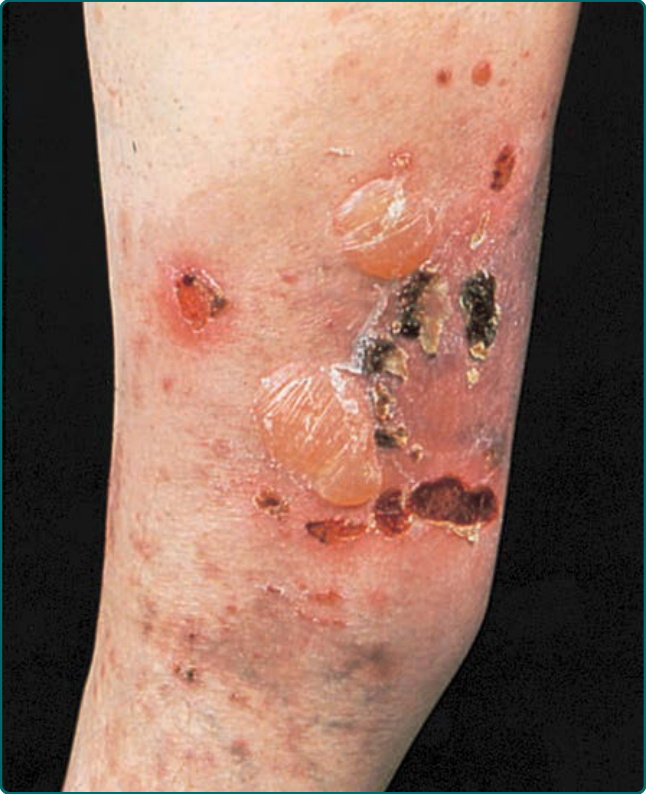
圖 56-4:一名侵犯腿部的後天性表皮鬆解水皰症病人。注意水皰、糜爛與痂皮。
相關身體檢查發現 (RELATED PHYSICAL FINDINGS)
EBA 病人可能有許多與因第 VII 型膠原蛋白基因缺陷所致之遺傳性失養型 EB 病人相似的身體檢查發現。這些包括口腔糜爛、食道狹窄 (esophageal strictures)、皮膚的色素減退與色素沉著斑駁 (hypo- and hyperpigmentation skin mottling)、指甲脫落、粟粒疹形成、瘢痕,以及某種程度的手部纖維化。多篇已發表的報告提示 EBA 可能與各種全身性疾病相關¹⁸,例如發炎性腸道疾病、SLE、澱粉樣變性 (amyloidosis)、甲狀腺炎 (thyroiditis)、多發性內分泌病變症候群 (multiple endocrinopathy syndrome)、類風濕性關節炎 (rheumatoid arthritis)、肺纖維化 (pulmonary fibrosis)、慢性淋巴球性白血病 (chronic lymphocytic leukemia)、胸腺瘤 (thymoma)、糖尿病、多發性骨髓瘤 (multiple myeloma),以及其他被認為具自體免疫發病機轉的疾病。在北卡羅來納大學 (University of North Carolina)、史丹佛 (Stanford)、西北大學 (Northwestern) 與南加州大學 (University of Southern California),合計追蹤超過 62 名 EBA 病人的經驗顯示,發炎性腸道疾病似乎是與 EBA 最常相關的全身性疾病。
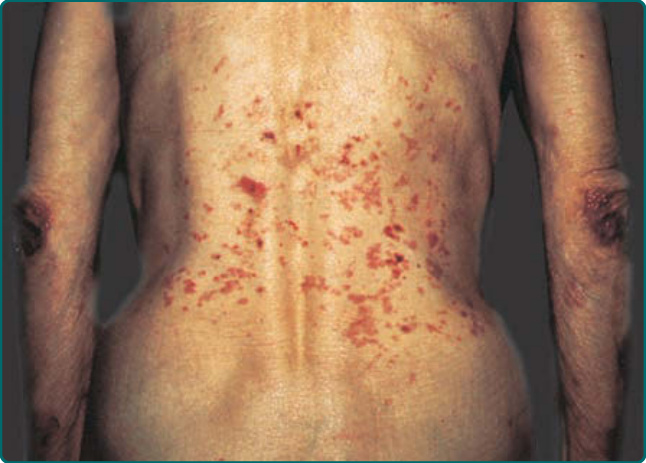
圖 56-5:一名後天性表皮鬆解水皰症病人,顯現本病的 2 種表現:肘部有糜爛、瘢痕與粟粒疹的典型機械性水皰表現,以及軀幹上較發炎的類大皰性類天皰瘡 (bullous pemphigoid–like) 病灶。
實驗室檢查 (LABORATORY TESTS)
組織病理學 (HISTOPATHOLOGY)
對取自 EBA 病人之病灶皮膚進行常規組織學檢查,顯示表皮下水皰以及表皮與真皮之間的乾淨分離 (clean separation)。真皮內發炎浸潤的程度通常反映臨床醫師所觀察到病灶的發炎程度。令人聯想到隱性失養型 EB 或 PCT 的病灶,其真皮內通常顯著缺乏發炎細胞。臨床上令人聯想到 BP 的病灶,其真皮內通常有明顯較多的發炎細胞,這些細胞可能是淋巴球、單核球、嗜中性球與嗜酸性球的混合。取自類 BP 病灶之 EBA 皮膚標本的組織學,可能難以與 BP 本身區分。
直接免疫螢光 (DIRECT IMMUNOFLUORESCENCE)
EBA 病人的皮膚 DEJ 內有 IgG 沉積。¹⁰,²⁷ 此最佳的偵測方式為對取自病灶周圍部位的切片標本進行 DIF(圖 56-6)。IgG 是主要的免疫球蛋白類別,但亦可能偵測到補體、IgA、IgM、B 因子 (factor B) 與備解素 (properdin) 的沉積。DIF 染色顯示 DEJ 處有一條強烈的線狀螢光帶。Yaoita 等人⁹ 曾提出,DIF 陽性且 IgG 沉積位於緻密板下 (sublamina densa) 區域,是診斷 EBA 的必要標準。臨床上可能模擬 EBA 的 PCT 病人,常於 DEJ 處有與 EBA 病人相似的 IgG 與補體沉積(第 124 章)。然而,可區分 PCT 與 EBA 的 DIF 特徵在於,PCT 皮膚亦於真皮血管周圍顯現免疫沉積。EBA 病人血液中可能有針對 DEJ 的自體抗體。¹⁰ 這些抗體可藉由將病人血清作用於猴或兔食道或人類皮膚之受質 (substrate) 的 IIF 來偵測,並以線狀方式染色 DEJ,其可能與 BP 血清無法區分。²²
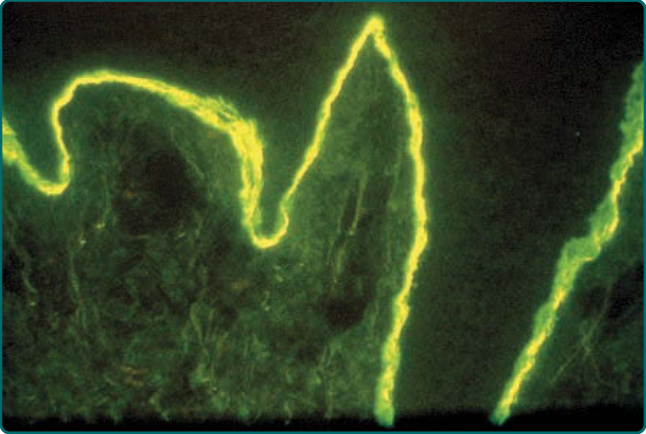
圖 56-6:一名後天性表皮鬆解水皰症病人病灶周圍皮膚對免疫球蛋白 G 沉積的直接免疫螢光 (direct immunofluorescence) 染色。注意真皮—表皮交界處 (dermal–epidermal junction) 內的緻密沉積(此切片中表皮位於上方)。
免疫電子顯微鏡 (IMMUNOELECTRON MICROSCOPY)
以免疫電子顯微鏡定位 EBA 病人皮膚 DEJ 內免疫沉積的位置,是診斷的「黃金標準 (gold standard)」。如 Nieboer 等人³¹ 與 Yaoita 等人⁹ 所示,EBA 病人在皮膚 BMZ 的緻密板下區域內有免疫沉積。此定位明顯不同於 BP 的沉積,後者位於基底膜較上方的半橋粒 (hemidesmosome) 區域或透明板 (lamina lucida) 區域。它亦不同於 CP,後者的抗原標的侷限於透明板。
間接鹽裂皮膚免疫螢光 (INDIRECT SALT-SPLIT SKIN IMMUNOFLUORESCENCE)
當人類皮膚於 1 莫耳濃度的氯化鈉中培養時,DEJ 會乾淨地沿透明板區域斷裂。此斷裂將 BP 抗原置於裂隙的表皮側,並將所有其他基底膜結構置於分離的真皮側。鹽裂皮膚受質可用於區分 EBA 與 BP 血清。¹⁰
若血清抗體為 IgG 並標記表皮頂 (epidermal roof),則該病人並非 EBA,應考慮 BP。反之,若抗體標記分離的真皮側,則該病人通常為 EBA 或大皰性 SLE。後者可藉由其他血清學與臨床標準予以排除。
直接鹽裂皮膚免疫螢光 (DIRECT SALT-SPLIT SKIN IMMUNOFLUORESCENCE)
於冷的 1 莫耳濃度氯化鈉中培養的病灶周圍皮膚,會沿 DEJ 斷裂,此舉有效地將 BP 抗原(及任何相關免疫沉積)置於表皮頂,並將 EBA 抗原(及任何相關免疫沉積)置於分離的真皮底 (dermal floor)(圖 56-7)。²⁷ 若病人罹患 EBA,則以使用螢光素標記之抗人類 IgG (fluorescein-conjugated anti-human IgG) 的常規 DIF 方法,可在分離的真皮側偵測到 IgG 免疫沉積。
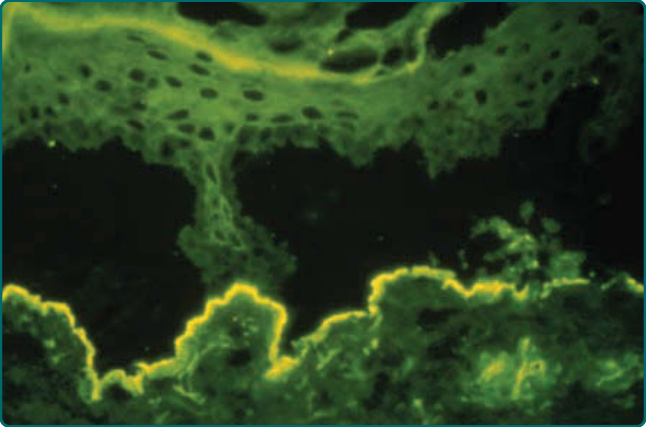
圖 56-7:一名病人病灶周圍皮膚切片在冷的 1 莫耳濃度鹽水中培養 72 小時並以抗人類 IgG 抗體探測後的直接免疫螢光 (direct immunofluorescence, DIF) 染色。冷鹽培養使皮膚標本的真皮—表皮交界處斷裂。在 EBA 病人中,IgG 沉積留存於斷裂皮膚的真皮底 (dermal floor)。相對地,在大皰性類天皰瘡病人中,沉積會留存於斷裂皮膚的表皮頂 (epidermal roof)(未顯示)。同樣地,若 EBA 病人的血清含有針對第 VII 型(錨定纖維)膠原蛋白的循環 IgG 抗體並進行間接免疫螢光 (indirect immunofluorescence, IIF),則該血清會標記鹽裂人類皮膚受質的真皮底,而表皮頂則不被標記。這些結果是因為將人類皮膚於冷的 1 莫耳濃度鹽水中培養會使真皮—表皮交界處斷裂,致使表皮的半橋粒 (hemidesmosomes) 及與半橋粒相關的大皰性類天皰瘡自體抗原留存於表皮頂,而錨定纖維與第 VII 型膠原蛋白(即 EBA 的自體抗原)則留存於真皮底。
西方免疫墨點法 (WESTERN IMMUNOBLOTTING)
EBA 血清中的抗體會結合於含第 VII 型膠原蛋白之人類皮膚基底膜蛋白西方墨點 (Western blots) 中的一條 290-kDa 條帶,而所有其他原發性水皰病的血清則不會。¹⁰
此條帶是第 VII 型膠原蛋白的 α 鏈。EBA 抗體常會標記到第二條 145 kDa 的條帶。此條帶是第 VII 型膠原蛋白 α 鏈的胺基末端球狀 NC-1 區域,富含碳水化合物,並含有 EBA 自體抗體、大皰性 SLE 自體抗體與抗第 VII 型膠原蛋白單株抗體的抗原性表位。¹⁷
酵素連結免疫吸附分析 (ENZYME-LINKED IMMUNOSORBENT ASSAY)
Chen 等人³⁰ 已在穩定轉染 (stably transfected) 的人類細胞中產生毫克量、重組、純化且經轉譯後修飾 (posttranslationally modified) 的 NC-1,並利用此 NC-1 發展出一種用於偵測 EBA 病人與大皰性 SLE 病人自體抗體的 ELISA。此 ELISA 較免疫螢光與西方墨點法更為敏感,但對第 VII 型膠原蛋白抗體仍極具專一性。目前已有市售的 ELISA 可用於偵測病人血清中的抗第 VII 型膠原蛋白。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 56-1。由於 EBA 曾在嬰兒與兒童中被描述,因此值得考慮一名被認為罹患遺傳性失養型 EB 的病人,可能正是一名罕見的兒童 EBA 病人。此可藉由「實驗室檢查」一節中所列的抗體檢測予以排除。PCT 在臨床上看起來可能與典型 EBA 十分相似,可藉由尿液或血漿的尿卟啉 (uroporphyrins) 檢測予以排除。假性 PCT (Pseudo-PCT) 通常由非類固醇抗發炎藥 (nonsteroidal anti-inflammatory agents) 等藥物所引起,看起來可能與 EBA 相似,於易受外傷區域有皮膚脆弱、糜爛與水皰、瘢痕及粟粒疹形成。儘管如此,其 DIF 表現有所不同,因假性 PCT 如同 PCT,於 DEJ 的 BMZ 處與真皮血管周圍皆顯現 IgG 沉積(而在 EBA 中真皮血管周圍不染色)。
類 BP 型 EBA 可藉由上述數種方法予以排除,但第一線檢測應為間接與直接鹽裂免疫螢光。
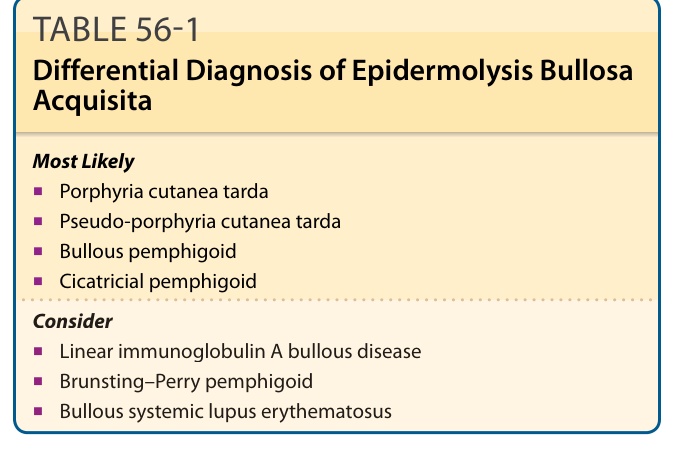
表 56-1:後天性表皮鬆解水皰症之鑑別診斷。最可能 (Most Likely):遲發性皮膚卟啉症 (porphyria cutanea tarda)、假性遲發性皮膚卟啉症 (pseudo-porphyria cutanea tarda)、大皰性類天皰瘡 (bullous pemphigoid)、瘢痕性類天皰瘡 (cicatricial pemphigoid)。考慮 (Consider):線狀免疫球蛋白 A 大皰病 (linear immunoglobulin A bullous disease)、Brunsting–Perry 類天皰瘡 (Brunsting–Perry pemphigoid)、大皰性全身性紅斑狼瘡 (bullous systemic lupus erythematosus)。
診斷 (DIAGNOSIS)
Yaoita 等人⁹ 為診斷 EBA 所制定的診斷標準至今仍然成立。這些標準經略微更新修改後,列於表 56-2。表中最後一項的替代方法為間接或直接鹽裂皮膚免疫螢光、西方墨點法與 ELISA。

表 56-2:後天性表皮鬆解水皰症之診斷標準。■ 屬於前述臨床範疇內的水皰性疾病(見「臨床表現」)。■ 無水皰性疾病的家族史。■ 組織學顯示表皮下水皰。■ 免疫球蛋白 G 沉積於真皮—表皮交界處(即病灶周圍皮膚的直接免疫螢光呈陽性)。■ 以直接免疫電子顯微鏡檢查病灶周圍皮膚時,免疫球蛋白 G 沉積侷限於真皮—表皮交界處的緻密板下層 (lower lamina densa) 與/或緻密板下 (sublamina densa) 區域。
併發症 (COMPLICATIONS)
EBA 所引起的併發症包括繼發性皮膚感染,通常由葡萄球菌 (Staphylococcus) 或鏈球菌 (Streptococcus) 所致,因水皰與糜爛損害了皮膚的屏障功能。瘢痕與粟粒疹形成是深層水皰過程自然發生的併發症或後遺症。嚴重的 EBA 病人可能發展出顯著的手部纖維化,伴隨手掌與手指活動範圍減少。由於足底與腳趾的傷口與纖維化,部分 EBA 病人行走困難。許多 EBA 病人會失去手指甲。有顯著黏膜侵犯的 EBA 病人可能發展出食道狹窄甚至喉部瘢痕 (laryngeal scarring)。
治療 (TREATMENT)
EBA 通常對治療反應不佳。所有 EBA 病人皆有支持性治療的必要。此包括開放性傷口照護的指導,以及避免外傷的策略。應警告病人不要過度清洗或過度使用熱水或刺激性肥皂,並避免以毛巾或浴巾長時間或劇烈摩擦皮膚。在某些病人中,長時間日曬似乎可能加重或促發手背與指節上的新病灶。因此,避免長時間日曬與使用防曬乳有所助益。應教育病人辨識局部皮膚感染,並在感染發生時迅速尋求醫療照護與抗生素治療。EBA 病人常對高劑量的全身性糖皮質素 (systemic glucocorticoids)、azathioprine、methotrexate 與 cyclophosphamide 反應頑強,尤其當他們罹患典型機械性水皰型疾病時。這些藥物在 EBA 表現為發炎性類 BP 疾病時,對控制病情可能稍有助益。部分 EBA 病人在使用 dapsone 後改善,尤其當其真皮浸潤中存在嗜中性球時。Cyclosporine 已被證實對 EBA 有益。³² 然而,此藥的長期毒性限制了其使用。亦有獨立報告指出 EBA 病人對高劑量 colchicine 有反應。³³ 此藥常被用作第一線藥物,因其副作用相較於其他治療選擇而言相對良性。然而,腹瀉是 colchicine 常見的副作用,這使得許多病人難以達到足以控制疾病的高劑量。此外,由於此副作用,我們對同時患有發炎性腸道疾病的 EBA 病人使用 colchicine 持保留態度。再者,也有病人對 colchicine 無反應。Colchicine 是一種眾所周知的微管抑制劑 (microtubule inhibitor),但它似乎也具有可能抑制抗原呈現給 T 細胞的特性,此舉可下調 (downregulate) 自體免疫。光分離術 (Photopheresis) 可改善 EBA 的臨床特徵,並顯著延長病人的抽吸性水皰形成時間,提示其表皮—真皮黏附的改善。³⁴
除光分離術外,血漿置換術 (plasmapheresis) 並移除 EBA 病人血漿中針對第 VII 型膠原蛋白的抗體,對於控制 EBA 病人病情有所助益,類似於天皰瘡病人。鑒於這些自體抗體具致病性,此並不令人意外,但進行血漿置換術時,必須同時讓病人接受化療藥物治療(例如 azathioprine、cyclophosphamide、mycotile mofelate、methotrexate)。靜脈注射免疫球蛋白 (Intravenous immunoglobulin, IVIG) 已被報告對 EBA 病人有效。³⁵ γ-球蛋白 (γ-globulin) 在 EBA 中可能引發正向反應的機轉不明。抗 TNF-α 生物製劑(例如 infliximab)已在 EBA 中試用,並在未對照的開放性試驗中取得部分成功。Rituximab 是一種針對 B 淋巴球表面 CD20 蛋白的單株抗體,可誘發病人 B 細胞淋巴球顯著減少,並已顯示對頑固型 EBA 病人有療效。³⁶,³⁷ 表 56-3 概述了在醫學文獻中有部分支持的 EBA 治療選擇。
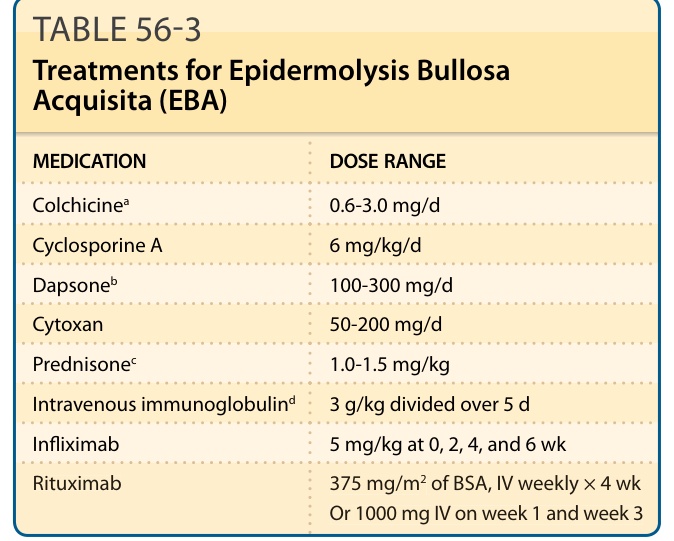
表 56-3:後天性表皮鬆解水皰症 (EBA) 之治療。藥物 (MEDICATION) 與劑量範圍 (DOSE RANGE):Colchicineᵃ 0.6-3.0 mg/d;Cyclosporine A 6 mg/kg/d;Dapsoneᵇ 100-300 mg/d;Cytoxan 50-200 mg/d;Prednisoneᶜ 1.0-1.5 mg/kg;靜脈注射免疫球蛋白 (Intravenous immunoglobulin)ᵈ 3 g/kg 分次於 5 日給予;Infliximab 5 mg/kg 於第 0、2、4、6 週給予;Rituximab 375 mg/m² 體表面積 (BSA),靜脈注射每週 1 次 × 4 週,或 1000 mg 靜脈注射於第 1 週與第 3 週。ᵃ必須從 0.4 至 0.6 mg/d 開始,每 1 至 2 週在可耐受範圍內將此劑量加倍。當病人發生腹瀉時,減少 1 顆藥錠 (0.4-0.6 mg)。ᵇ從 25 mg/d 開始,於檢查全血球計數與肝功能後每週加倍。大多數病人需要 100 至 250 mg/d 之間。緩慢增加劑量有助於病人耐受所發生的貧血(即較少姿勢性頭暈等)。在治療劑量下,預期病人血紅素會下降 1 至 2 g。ᶜ通常對發炎極少的典型機械性水皰型 EBA 沒有幫助。然而,對類大皰性類天皰瘡型 EBA 可能稍有助益。ᵈ靜脈注射免疫球蛋白於每月給予 4 至 5 日,連續 5 或 6 個月,以給予充分的試驗。