Epidermolysis Bullosa Acquisita
9
AT-A-GLANCE
■ Rare, autoimmune subepidermal bullous disease due to immunoglobulin G autoantibodies to Type VII collagen.
■ Etiology is unknown.
■ Skin fragility, subepidermal blisters, residual scarring, and milia formation. Common sites are trauma-prone areas such as hands, feet, elbows, knees, sacrum, nails, and mouth.
■ Related features may include an underlying systemic disease such as inflammatory bowel disease. May have erosions of the mucosa and esophageal stenosis.
■ Pathology shows subepidermal bulla, fibrosis, milia formation, and positive direct immunofluorescence for immunoglobulin G deposits at the epidermal–dermal junction.
■ Treatment options are limited and often difficult.
EPIDEMIOLOGY
Epidermolysis bullosa acquisita (EBA) is a sporadic autoimmune bullous disease of unknown etiology. The disease has no gender, racial, ethnic, or geographical predisposition. There may be some genetic predisposition to EBA and autoimmunity in African Americans who live in the southeastern part of the United States.1 African American patients in the southeastern part of the United States who have either EBA or bullous systemic lupus erythematosus (SLE) have a high incidence of the HLA-DR2 phenotype. The calculated relative risk for EBA in HLA-DR2+ individuals is 13.1 in these patients. These results also suggest that EBA and bullous SLE are immunogenetically related and that the HLADR2 gene either is involved with autoimmunity to anchoring fibril collagen or is some sort of a marker for some other gene that exists in linkage disequilibrium with it.1
In another study examining HLA genotypes in EBA patients, it was found that HLA-DRB1∗15:03 was overrepresented.2
In concordance with the genetic permissiveness towards EBA in humans, an active murine model of EBA in which mice are immunized with murine Type VII collagen, the development of EBA depends on the strain of mouse used.3,4
EBA is an exceedingly rare disease and considerably less frequent than other autoimmune bullous diseases (bullous pemphigoid, pemphigus vulgaris, dermatitis herpetiformis, etc.), which themselves are quite rare. In unselected bullous disease patients who exhibited an
anti–basement membrane zone autoantibody in their blood, approximately 5% had the diagnosis of EBA or bullous SLE.5 This incidence is similar to those reported by other investigators.6 A prospective French study using electron and immune-electron microscopy to confirm the diagnosis of EBA, found an incidence rate as 0.17 to 0.26 per million people and accounted for only 2% to 3% of all patients with a subepidermal autoimmune bullous disease.7 The incidence of EBA in a region of Germany was 0.17, again confirming the rarity of EBA.8
ETIOLOGY AND PATHOGENESIS
EBA is a chronic, subepidermal blistering disease associated with autoimmunity to the collagen (Type VII collagen) within anchoring fibril structures that are located at the dermal–epidermal junction (DEJ). Although the precise etiology of EBA is unknown, most of the evidence suggests an autoimmune etiology. The immunoglobulin G (IgG) autoantibodies to Type VII collagen are associated with a paucity of normal anchoring fibrils at the basement membrane zone (BMZ) separating the epidermis from the dermis and poor epidermal–dermal adherence. Although it is an acquired disease that usually begins in adulthood, it was placed in the category epidermolysis bullosa (EB) more than 100 years ago because physicians were struck by how similar the clinical lesions of EBA were to those seen in children with hereditary dystrophic forms of EB. Direct immunofluorescence (DIF) of perilesional skin biopsies from EBA patients reveals IgG deposits at the DEJ.9 EBA antibodies bind to Type VII collagen within anchoring fibrils.10,11
Anchoring fibrils anchor the epidermis and its underlying BMZ to the papillary dermis. Patients with hereditary forms of dystrophic EB (Chap. 60) and EBA have decreased numbers of anchoring fibrils in their DEJ. This paucity of anchoring fibrils is associated with 2 similar clinical phenotypes, EBA and dystrophic forms of hereditary EB, because both diseases are characterized by skin fragility, subepidermal blisters, milia formation, and scarring. Although both EBA and hereditary forms of dystrophic EB are etiologically unrelated in terms of their underlying pathogenesis, they share the common feature of decreased anchoring fibrils. In the case of dystrophic forms of hereditary EB, the cause of decreased or absent anchoring fibrils is a genetic defect in the COL7A1 gene, which encodes for Type VII collagen α chains, that ultimately results in small, nonfunctional, or decreased anchoring fibrils.12,13 The gene coding for Type VII collagen is located on the short arm of chromosome 3.14 The gene defects involved in hereditary forms of dystrophic EB have been identified
9
at variable locations, but the severity of the disease appears to correlate with the degree of Type VII collagen and anchoring fibril perturbations.13 In EBA, the IgG autoantibodies binding to the Type VII collagen α chains result in decreased anchoring fibrils, but the pathway leading to this reduction is unknown. It may be that Type VII collagen α chains that are newly synthesized but decorated with EBA autoantibodies cannot form triple-helical structures and stable anchoring fibrils. Healed burn wounds that have been covered with cultured keratinocyte sheets also have decreased numbers of anchoring fibrils within the first year after transplantation, and this is associated with spontaneous blister formation, shortened suction blistering times, and skin fragility.15 These observations provide indirect evidence that anchoring fibrils play a role in maintaining adherence between the epidermis and dermis. The Type VII collagen α chain has a molecular mass of 290 kDa and the collagen consists of a homotrimer of 3 identical α chains (Chap. 15). Each α chain consists of a large globular noncollagenous amino terminus called the noncollagenous 1 (NC-1) domain that is approximately one-half the entire mass of the α chain. Next, there is a helical domain with typical glycine-X-Y repeats. At the carboxyl terminus is a second globular noncollagenous domain, NC-2, that is much smaller than NC-1.16 Most EBA autoantibodies recognize 4 predominant antigenic epitopes within the NC-1 domain and do not recognize the helical or NC-2 domains.17,18
There may be something intrinsically “antigenic” about the NC-1 domain because the available monoclonal antibodies that have been generated against Type VII collagen (anti–C-VII antibodies) specifically recognize only NC-1 subdomains. A reduction in the number of anchoring fibrils is seen in the lesional and perilesional skin of EBA patients, but the pathway leading to this reduction is unknown. Several independent lines of evidence have implicated autoimmune responses as a key element in the pathogenesis of EBA. First, the pathogenic role of EBA antibodies is suggested by the observation that when patients with SLE develop autoantibodies to the Type VII collagen, they develop widespread skin blisters and fall into a rare subset of SLE called bullous SLE.19
This “experiment of nature” suggests that EBA autoantibodies are pathogenic and capable of inducing disadherence between the epidermis and dermis. Second, direct proof that EBA autoantibodies are pathogenic comes from recent passive transfer studies. We immunized rabbits and raised a high-titer antiserum to the NC-1 domain of human Type VII collagen. We injected this antibody into hairless immune-competent mice, and the mice developed bullous skin disease with many of the features of EBA in humans.20 The mice developed subepidermal blisters and lost nails on their feet. They also had circulating NC-1 antibodies in their blood and anti–NC-1 IgG antibody deposits at their DEJ. In addition, the mice had murine complement deposits at the DEJ induced by the autoantibody–antigen complex.20 Another study by Sitaru and colleagues21 showed that the injection of rabbit
972
polyclonal antibodies to the NC-1 domain of mouse Type VII collagen into mice also induced subepidermal skin blisters that were reminiscent of human EBA.21
Further, we have also affinity-purified human EBA autoantibodies against an NC-1 column and injected them into mice. The mice then developed clinical, histologic, immunologic, and ultrastructural features akin to human EBA.22 Taken together, these successful passive transfer experiments and the observations with bullous SLE strongly suggest that EBA autoantibodies are “pathogenic” and capable of causing epidermal– dermal separation in skin. Using clinical patient studies, in vitro assays and murine models of EBA, much has been learned about the details of the immune responses of EBA patients harboring anti–Type VII collagen autoantibodies. EBA IgG autoantibodies in both the patients’ blood and tissue bound in the patients’ DEJ represent all 4 subclasses of IgG, particulary IgG1 and IgG4.23 Circulating EBA autoantibodies consist of both complement binding and non– complement binding populations.24 Nevertheless, when compared with BP, EBA patients had a higher presence of C3b and C5 (90% vs 33%, 90% vs 58%, respectively). These studies provided evidence3 for complement activation at the DEJ in the majority of EBA patients and that EBA antibodies were more potent activators of C5 than BP antibodies.25 Gammon and co-workers developed an in vitro assay to look at the functions of bullous pemphigoid and EBA immune complexes in skin. A slide chamber was made consisting of a cryostat section of human skin (normal skin or skin from patients with BP or EBA) with and without the additions of leukocytes and the additions of complement. The readout was the migration and attraction of the leukocytes to the DEJ of the human skin cryosections.26 When cryostat sections of bullous pemphigoid skin and EBA skin were compared, the EBA immune complexes again generated more complement activation than bullous pemphigoid immune complexes. In addition to murine models of EBA created by the passive transfer of anti–Type VII collagen antibodies, as noted in the sections “Epidemiology” above, an active murine model of EBA has been established by immunizing certain strains of mice with murine Type VII collagen. This active murine EBA model invokes complement fixing anti-Type VII collagen antibodies and requires T cells for disease induction.
CLINICAL FINDINGS
See Fig. 56-1. If a patient presents with bullae on the skin with no reasonable explanation despite a thorough history and physical examination, 3 tests should be done: a skin biopsy for routine hematoxylin and eosin histology, a second biopsy juxtaposed to a lesion but on normal-appearing skin for DIF and a blood draw to test for antibodies against the BMZ and/or Type VII collagen by indirect immunofluorescence (IIF) or enzyme-linked immunosorbent assay (ELISA). The cutaneous lesions of EBA can be quite varied and can mimic other types of acquired autoimmune
9
Approach to patient with epidermolysis bullosa acquisita (EBA)
Lesional biopsy
Subepidermal bulla
Intraepidermal bulla
Perilesional DIF Consider other DX (eg, pemphigus) Consider other DX
Negative
Positive for IgG
Salt-split DIF
Dermal floor staining Epidermal roof staining Consider bullous pemphigoid
Consider EBA and anti-L5 CP Chan disease Ghohestani disease
IIF
If available: ELISA Western Blot EM Immuno-EM
Negative
EBA still possible
Positive for circulating anti-BMZ antibodies
Salt-split skin IIF Dermal floor staining
bullous diseases. The common denominator for patients with EBA is autoimmunity to Type VII (anchoring fibril) collagen. There are at least 5 clinical presentations: (1) a classic presentation, (2) a bullous pemphigoid (BP)-like presentation, (3) a mucous membrane pemphigoid (MMP)-like presentation, (4) a presentation reminiscent of Brunsting–Perry pemphigoid with scarring lesions and a predominant head and neck distribution, and (5) a presentation reminiscent of linear IgA bullous dermatosis or chronic bullous disease of childhood. The classical presentation and the BP-like presentation are the most commonly reported presentations of this very rare disease.
CLASSIC PRESENTATION
CLASSIC PRESENTATION
The classic presentation (Figs. 56-2 and 56-3A) is of a noninflammatory bullous disease with an acral distribution that heals with scarring and milia formation. This presentation is reminiscent of porphyria cutanea tarda (PCT; Chap. 124) when it is mild and of the hereditary form of recessive dystrophic EB when it is severe
(Chap. 60). The classic form of EBA is thus a mechanobullous disease marked by skin fragility. These patients have erosions, tense blisters within noninflamed skin, and scars over trauma-prone surfaces such as the backs
973
9
A
B
C
of the hands, knuckles, elbows, knees, sacral area, and toes (Figs. 56-2, 56-3A, and 56-4). Some blisters may be hemorrhagic or develop scales, crusts, or erosions. The lesions heal with scarring and frequently with the formation of pearl-like milia cysts within the scarred areas (Fig. 56-3A). Although this presentation may be reminiscent of PCT, these patients do not have other hallmarks of PCT, such as hirsutism, a photodistribution of the eruption, or scleroderma-like changes, and their urinary porphyrins are within normal limits. A scarring alopecia and some degree of nail dystrophy may be seen. Although the disease is usually not as severe as that of patients with hereditary forms of recessive dystrophic EB, EBA patients with the classic form of the disease may have many of the same sequelae, such as scarring, loss of scalp hair, loss of nails, fibrosis of the hands and fingers, and esophageal stenosis.27
974
D
BULLOUS PEMPHIGOID– LIKE PRESENTATION
BULLOUS PEMPHIGOID–
LIKE PRESENTATION
A second clinical presentation of EBA is of a widespread, inflammatory vesiculobullous eruption involving the trunk, central body, and skin folds in addition to the extremities.6 The bullous lesions are tense and surrounded by inflamed or even urticarial skin. Large areas of inflamed skin may be seen without any blisters and only erythema or urticarial plaques. These patients often complain of pruritus and do not demonstrate prominent skin fragility, scarring, or milia formation. This clinical constellation is more reminiscent of BP (Figs. 56-3B and 56-4) than a mechanobullous disorder. Similar to BP, the distribution of the lesions may show an accentuation within flexural areas and skin folds.
MUCOUS MEMBRANE PEMPHIGOID-LIKE PRESENTATION
MUCOUS MEMBRANE
PEMPHIGOID-LIKE
PRESENTATION
Both the classic and BP-like forms of EBA may have involvement of mucosal surfaces. However, EBA also may present with such predominant mucosal involvement that the clinical appearance is reminiscent of MMP (Fig. 56-3C).28 These patients usually have erosions and scars on the mucosal surfaces of the mouth, upper esophagus, conjunctiva, anus, or vagina with or without similar lesions on the glabrous skin.
BRUNSTING–PERRY PEMPHIGOID–LIKE PRESENTATION
BRUNSTING–PERRY
PEMPHIGOID–LIKE
PRESENTATION
Brunsting–Perry cicatricial BP is a chronic, recurrent vesiculobullous eruption localized to the head and neck and characterized by residual scars, subepidermal bullae, IgG deposits at the DEJ, and minimal or no mucosal involvement. The antigenic target for the IgG autoantibodies, however, has not been defined. Nevertheless, a patient reported with this constellation of findings had IgG autoantibodies directed to anchoring fibrils below the lamina densa.29 We have seen 3 additional patients with the features of Brunsting– Perry pemphigoid and autoantibodies directed to
9
Type VII collagen (unpublished observations). Therefore, it appears that EBA patients may present with a clinical phenotype of Brunsting–Perry pemphigoid (Fig. 56-3D).
IMMUNOGLOBULIN A BULLOUS DERMATOSIS– LIKE PRESENTATION
IMMUNOGLOBULIN A
BULLOUS DERMATOSIS–
LIKE PRESENTATION
IgA bullous dermatosis–like presentation of EBA is manifested by a subepidermal bullous eruption, a neutrophilic infiltrate, and linear IgA deposits at the BMZ when viewed by DIF. It may resemble linear IgA bullous dermatosis (LABD), dermatitis herpetiformis, or chronic bullous disease of childhood and may feature tense vesicles arranged in an annular fashion and involvement of mucous membranes.30 The autoantibodies are usually IgA, IgG, or both. The diagnosis of these subepidermal blistering cases with IgA anti–Type VII collagen antibodies showing linear IgA deposition at the BMZ is disputable. Some clinicians regard the patients as having purely LABD, whereas others regard them as having a subset of EBA. Further, the majority of EBA patients have low-titer IgA antibodies in their blood directed against Type VII collagen. Childhood EBA is a rare disease. It has a variable presentation, including an LABD-like disease, a BPlike disease, and the classic mechanobullous EBA presentation. Although mucosal involvement is frequent and severe in childhood EBA, the overall prognosis is more favorable than in adult EBA.
INCIDENCE OF THE CLINICAL PRESENTATIONS OF EPIDERMOLYSIS BULLOSA ACQUISITA
INCIDENCE OF THE
CLINICAL PRESENTATIONS
OF EPIDERMOLYSIS
BULLOSA ACQUISITA
According to the authors’ experience, approximately 25% of patients with EBA may present with a BPlike clinical appearance. The disease of some of these patients eventually smolders into a more non-inflammatory mechanobullous form. However, both the classic and BP-like forms of the disease may coexist in the same patient (Fig. 56-5). The clinical phenotype of EBA that is reminiscent of pure CP occurs in fewer than 10% of all EBA cases.
RELATED PHYSICAL FINDINGS
RELATED PHYSICAL
FINDINGS
EBA patients may have many physical findings similar to patients with hereditary dystrophic EB due to
975
9
gene defects in the Type VII collagen gene. These include oral erosions, esophageal strictures, hypo- and hyperpigmentation skin mottling, nail loss, milia formation, scarring, and a degree of fibrosis of the hands. A number of published reports suggest that EBA may be associated with various systemic diseases18
such as inflammatory bowel disease, SLE, amyloidosis, thyroiditis, multiple endocrinopathy syndrome, rheumatoid arthritis, pulmonary fibrosis, chronic lymphocytic leukemia, thymoma, diabetes, multiple myeloma and other diseases in which an autoimmune pathogenesis has been implicated. At the University of North Carolina, Stanford, Northwestern, and University of Southern California, with a combined experience of following more than 62 EBA patients, it appears that inflammatory bowel disease is the systemic disease most frequently associated with EBA.
LABORATORY TESTS
HISTOPATHOLOGY
HISTOPATHOLOGY
Routine histologic examination of lesional skin obtained from EBA patients shows a subepidermal blister and a clean separation between the epidermis and dermis. The degree of inflammatory infiltrate within the dermis usually reflects the degree of inflammation of the lesion observed by the clinician. Lesions that are reminiscent of recessive dystrophic EB or PCT usually have a notable scarcity of inflammatory cells within the dermis. Lesions that are clinically reminiscent of BP usually have significantly more inflammatory cells within the dermis, and these cells may be a mixture of lymphocytes, monocytes, neutrophils, and eosinophils. The histology of EBA skin specimens obtained from BP-like lesions may be difficult to distinguish from BP itself.
976
DIRECT IMMUNOFLUORESCENCE
DIRECT
IMMUNOFLUORESCENCE
Patients with EBA have IgG deposits within the DEJ of their skin.10,27 This is best detected by DIF of a biopsy specimen obtained from a perilesional site (Fig. 56-6). IgG is the predominant immunoglobulin class, but deposits of complement, IgA, IgM, factor B, and properdin also may be detected. The DIF staining demonstrates an intense linear fluorescent band at the DEJ. Yaoita et al.9 have suggested that a positive DIF and IgG deposits within the sublamina densa zone are necessary criteria for the diagnosis of EBA. Patients with PCT, which may mimic EBA clinically, frequently have IgG and complement deposits at the DEJ similar to those of EBA patients (Chap. 124). However, the DIF feature that distinguishes PCT from EBA is that PCT skin also demonstrates immune deposits around the dermal blood vessels. Patients with EBA may have autoantibodies in their blood directed against the DEJ.10 These antibodies can be detected by IIF of the patient’s serum on a substrate of monkey or rabbit esophagus or human skin and stain the DEJ in a linear fashion that may be indistinguishable from BP sera.22
IMMUNOELECTRON MICROSCOPY
IMMUNOELECTRON
MICROSCOPY
The localization of the immune deposits within the DEJ of the skin of EBA patients by immunoelectron microscopy is the “gold standard” for the diagnosis. As demonstrated by Nieboer et al.31 and Yaoita et al.,9
patients with EBA have immune deposits within the sublamina densa zone of the cutaneous BMZ. This localization is clearly distinct from the deposits in BP, which are higher up in the hemidesmosome area or lamina lucida area of the basement membrane. It is
also distinct from CP, which has antigenic targets confined to the lamina lucida.
INDIRECT SALT-SPLIT SKIN IMMUNOFLUORESCENCE
INDIRECT SALT-SPLIT SKIN
IMMUNOFLUORESCENCE
When human skin is incubated in 1 molar sodium chloride, the DEJ fractures cleanly through the lamina lucida zone. This fracture places the BP antigen on the epidermal side of the split and all other basement membrane structures on the dermal side of the separation. Salt-split skin substrate can be used to distinguish EBA and BP sera.10
If the serum antibody is IgG and labels the epidermal roof, the patient does not have EBA, and BP should be considered. If, on the other hand, the antibody labels the dermal side of the separation, the patient usually has either EBA or bullous SLE. The latter can be ruled out by other serology and by clinical criteria.
DIRECT SALT-SPLIT SKIN IMMUNOFLUORESCENCE
DIRECT SALT-SPLIT SKIN
IMMUNOFLUORESCENCE
Perilesional skin incubated in cold 1 molar sodium chloride is fractured through the DEJ, which effectively places the BP antigen (and any associated immune deposits) on the epidermal roof and the EBA antigen (and any associated immune deposits) on the dermal floor of the separation (Fig. 56-7).27 If the patient has EBA, IgG immune deposits are detected on the dermal side of the separation by a routine DIF method using fluorescein-conjugated anti-human IgG.
WESTERN IMMUNOBLOTTING
Antibodies in EBA sera bind to a 290-kDa band in Western blots of human skin basement membrane proteins containing Type VII collagen, whereas sera from all other primary blistering diseases do not.10
This band is the α chain of Type VII collagen. Often, a second band of 145 kDa is labeled with EBA antibodies. This band is the amino-terminal globular NC-1 domain of the Type VII collagen α chain, which is rich in carbohydrate and contains the antigenic epitopes of EBA autoantibodies, bullous SLE autoantibodies, and monoclonal antibodies against Type VII collagen.17
ENZYME-LINKED IMMUNOSORBENT ASSAY
ENZYME-LINKED
IMMUNOSORBENT ASSAY
Chen et al.30 have produced milligram quantities of recombinant, purified, posttranslationally modified NC-1 in stably transfected human cells and have
9
used this NC-1 to develop an ELISA for autoantibody detection in EBA patients and in patients with bullous SLE. This ELISA is more sensitive than immunofluorescence and Western blotting, and yet it is very specific for antibodies to Type VII collagen. There is now a commercially available ELISA for the detection of anti–Type VII collagen in the sera of patients.
DIFFERENTIAL DIAGNOSIS
See Table 56-1. Because EBA has been described in infants and children, it is worth considering that a patient thought to have genetic dystrophic EB just might be a rare childhood patient with EBA. This can be ruled out by the antibody tests outlined in the section “Laboratory Tests.” PCT can look clinically very much like classic EBA and can be ruled out by a urine or plasma test for uroporphyrins. Pseudo- PCT, usually caused by drugs such as nonsteroidal anti-inflammatory agents, can look similar to EBA with skin fragility, erosions, and blisters over traumaprone areas, scarring, and milia formation. Nevertheless, the DIF appears different in that pseudo-PCT, like PCT, shows IgG deposits at both the BMZ at the DEJ and around dermal blood vessels (which are not stained in EBA).
977
9
Most Likely
■Porphyria cutanea tarda
■Pseudo-porphyria cutanea tarda
■Bullous pemphigoid
■Cicatricial pemphigoid
Consider
Consider
■Linear immunoglobulin A bullous disease
■Linear immunoglobulin A bullous disease
■Brunsting–Perry pemphigoid
■Brunsting–Perry pemphigoid
■Bullous systemic lupus erythematosus
■Bullous systemic lupus erythematosus
The BP-like EBA can be eliminated by several methods listed above, but the first-line test would be indirect and direct salt-split immunofluorescence.
DIAGNOSIS
The diagnostic criteria developed by Yaoita et al.9
for the diagnosis of EBA still stand. These criteria, with slightly updated modifications, are shown in Table 56-2. Alternatives for the last item in the table are indirect or direct salt-split skin immunofluorescence, Western blotting, and ELISA.
COMPLICATIONS
The complications caused by EBA include secondary skin infections, usually due to Staphylococcus or Streptococcus, because the blisters and erosions compromise the skin’s barrier. Scarring and milia formation are naturally occurring complications or sequelae of the deep blistering process. Severe EBA patients may develop significant fibrosis of the hands with decreased range of motion of the palm and digits. Because of wounds and fibrosis of the soles of the feet and toes, some EBA patients have difficulty walking. Many patients with EBA lose their fingernails. EBA patients with significant mucosal involvement may develop esophageal strictures and even laryngeal scarring.
■A bullous disorder within the clinical spectrum outlined earlier (see “Clinical Findings”).
■A bullous disorder within the clinical spectrum outlined earlier
(see “Clinical Findings”).
■No family history of a bullous disorder.
■No family history of a bullous disorder.
■Histology showing a subepidermal blister.
■Histology showing a subepidermal blister.
■Deposition of immunoglobulin G deposits within the dermal– epidermal junction (ie, a positive direct immunofluorescence of perilesional skin).
■Deposition of immunoglobulin G deposits within the dermal–
epidermal junction (ie, a positive direct immunofluorescence of perilesional skin).
■Immunoglobulin G deposits localized to the lower lamina densa and/or sublamina densa zone of the dermal–epidermal junction when perilesional skin is examined by direct immunoelectron microscopy.
■Immunoglobulin G deposits localized to the lower lamina densa
and/or sublamina densa zone of the dermal–epidermal junction when perilesional skin is examined by direct immunoelectron microscopy.
978
TREATMENT
EBA usually responds poorly to treatment. Supportive therapy is warranted in all patients with EBA. This includes instruction in open wound care and strategies for avoiding trauma. Patients should be warned not to over wash or overuse hot water or harsh soaps and to avoid prolonged or vigorous rubbing of their skin with a washcloth or towel. In some patients, it appears that prolonged sun exposure may aggravate or promote new lesions on the dorsal hands and knuckles. Thus, avoidance of prolonged sun exposure and the use of sunscreens are helpful. The patient should be educated to recognize localized skin infections and to seek medical care and antibiotic therapy promptly when they occur. EBA patients are often refractory to high doses of systemic glucocorticoids, azathioprine, methotrexate, and cyclophosphamide, especially when they have the classic mechanobullous form of the disease. These agents may be somewhat helpful in controlling EBA when it appears as an inflammatory BP-like disease. Some EBA patients improve on dapsone, especially when neutrophils are present in their dermal infiltrate. Cyclosporine has been shown to be beneficial in EBA.32 However, the long-term toxicity of this drug limits its use. There are also independent reports of EBA patients responding to high doses of colchicine.33 This is often used as a first-line drug because its side effects are relatively benign compared with other therapeutic choices. Diarrhea is a common side effect of colchicine, however, which makes it difficult for many patients to achieve a high enough dose to control the disease. Moreover, because of this side effect, we are hesitant to use colchicine in EBA patients who also have inflammatory bowel disease. In addition, there are patients who do not respond to colchicine. Colchicine is a wellknown microtubule inhibitor, but it also appears to have properties that have the potential to inhibit antigen presentation to T cells, which could downregulate autoimmunity. Photopheresis improves the clinical features of EBA and remarkably lengthens the suction blistering times of the patients, suggesting an improvement in their dermal–epidermal adherence.34
In addition to photophoresis, plasmapheresis and removal of the antibodies to Type VII collagen in an EBA patient’s plasma is useful for gaining control of EBA patients similar to pemphigus patients. Given that the autoantibodies are pathogenic, this is not surprising, but when plasmapheresis is performed it is necessary to have the patient also treated with a chemotherapy agent (such as azathioprine, cyclophosphamide, mycotile mofelate, methotrexate). Intravenous immunoglobulin (IVIG) has been reported to be effective in patients with EBA.35 The mechanism by which γ-globulin may invoke a positive response in EBA is unknown. The anti–TNF-α biologics (such as infliximab) have been tried in EBA with some success in uncontrolled open trials. Rituximab, a monoclonal antibody against
MEDICATION DOSE RANGE
Colchicinea 0.6-3.0 mg/d
Cyclosporine A 6 mg/kg/d
Dapsoneb 100-300 mg/d
Cytoxan 50-200 mg/d
Prednisonec 1.0-1.5 mg/kg
Intravenous immunoglobulind 3 g/kg divided over 5 d
Infliximab 5 mg/kg at 0, 2, 4, and 6 wk
Rituximab 375 mg/m2 of BSA, IV weekly × 4 wk Or 1000 mg IV on week 1 and week 3
Rituximab 375 mg/m2 of BSA, IV weekly × 4 wk Or 1000 mg IV on week 1 and week 3
aMust start with 0.4 to 0.6 mg/d and each 1 to 2 weeks double this dose as tolerated. When patient develops diarrhea, back off 1 tablet (0.4-0.6 mg).
bBegin at 25 mg/d and double each week after the complete blood count and liver function tests. Most patients need between 100 and 250 mg/d. Increasing the dose slowly helps the patient tolerate the anemia that develops (ie, less orthostatic light-headedness, etc.). Expect a 1- to 2-g drop in the patient’s hemoglobin on therapeutic doses.
cUsually does not help the classic, mechanobullous type of EBA with minimal inflammation. However, it may be somewhat helpful in the bullous pemphigoid–like type of EBA.
dIntravenous immunoglobulin is given over 4 to 5 days every month for 5 or 6 months to give it an adequate trial.
the CD20 protein on the surface of B lymphocytes induces markedly decreased B-cell lymphocytes in the patients and has shown efficacy in recalcitrant EBA patients.36,37 Table 56-3 outlines treatment options in EBA that have some support in the medical literature.
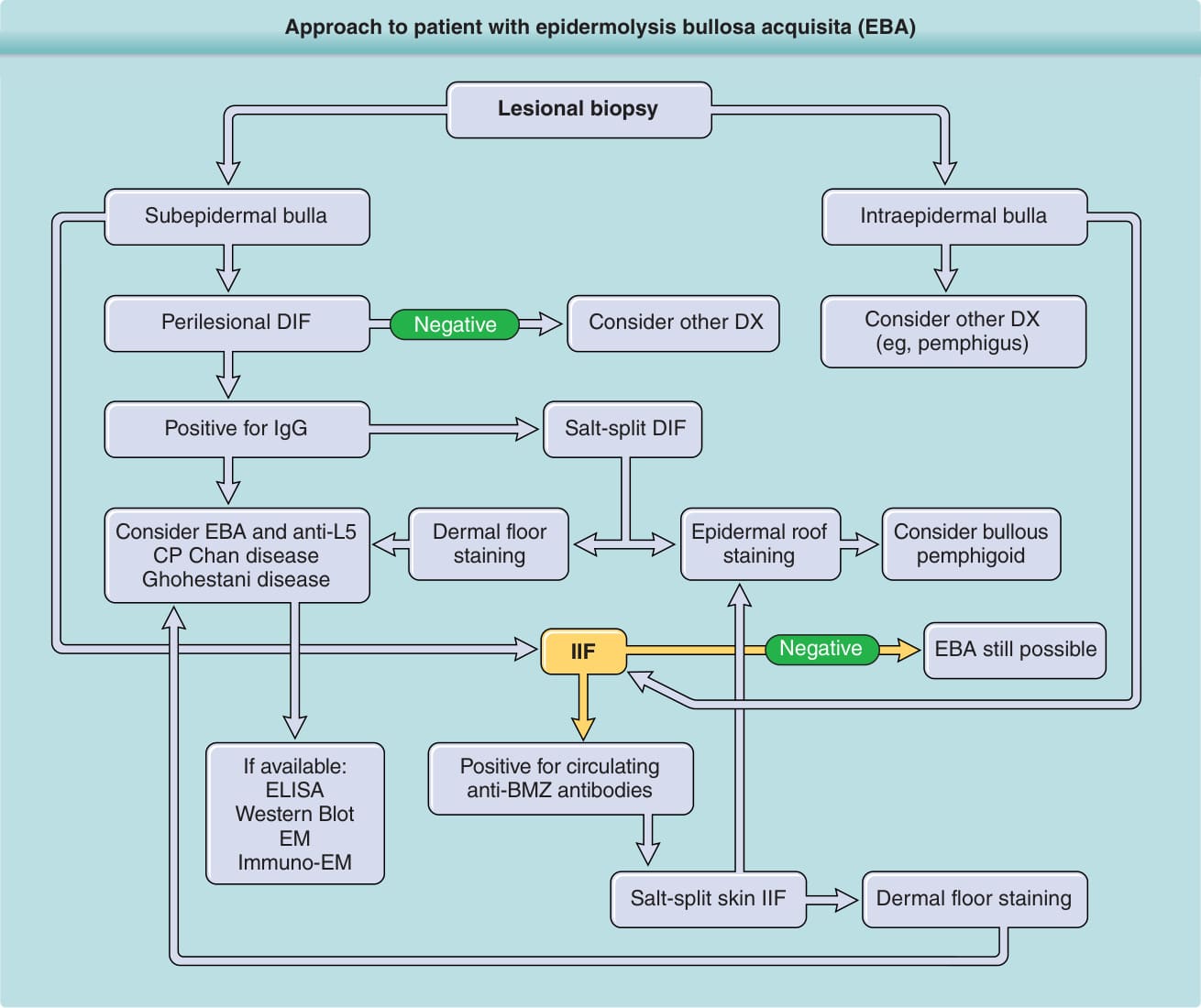
Figure 56-1 Approach to the patient with epidermolysis bullosa acquisita (EBA). BMZ, basement membrane zone; DIF, direct immunofluorescence; DX, diagnosis; ELISA, enzyme-linked immunosorbent assay; IgG, immunoglobulin G; IIF, indirect immunofluorescence.
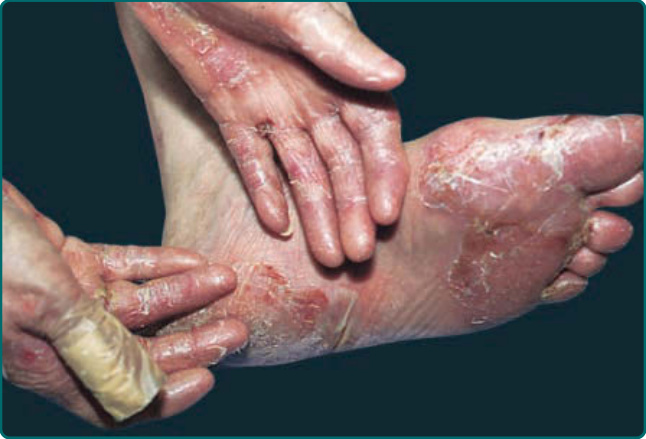
Figure 56-2 Patient with epidermolysis bullosa acquisita who has severe blistering, erosions, scarring, and milia formation on trauma-prone areas of her skin. This is the classic presentation.
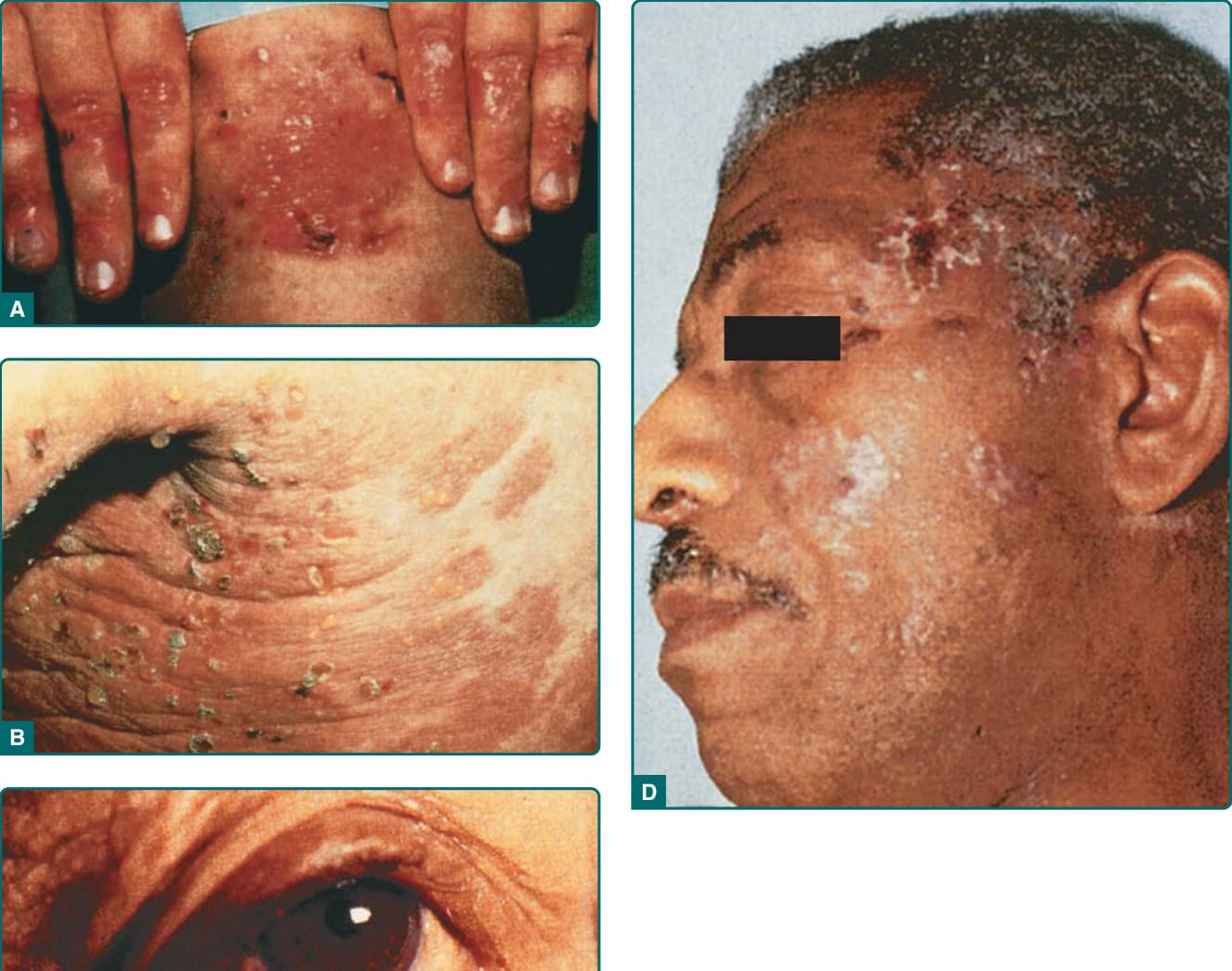
Figure 56-3 A, Classic presentation of epidermolysis bullosa acquisita with scarring and milia over trauma-prone areas of skin. B, Bullous pemphigoid–like presentation of epidermolysis bullosa acquisita with a widespread inflammatory vesiculobullous dermatosis. C, Cicatricial pemphigoid–like presentation of epidermolysis bullosa acquisita with a mucosal-centered bullous scarring eruption. D, Brunsting–Perry pemphigoid–like presentation of epidermolysis bullosa acquisita with bullous and scarring lesions predominantly on the head and neck.
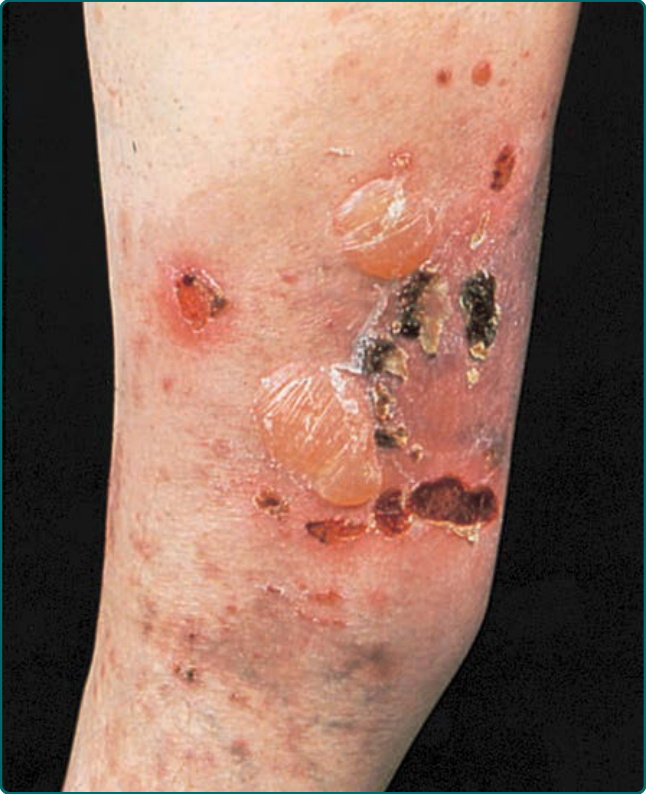
Figure 56-4 An epidermolysis bullosa acquisita patient with involvement of the leg. Note bullae, erosions, and crusts.
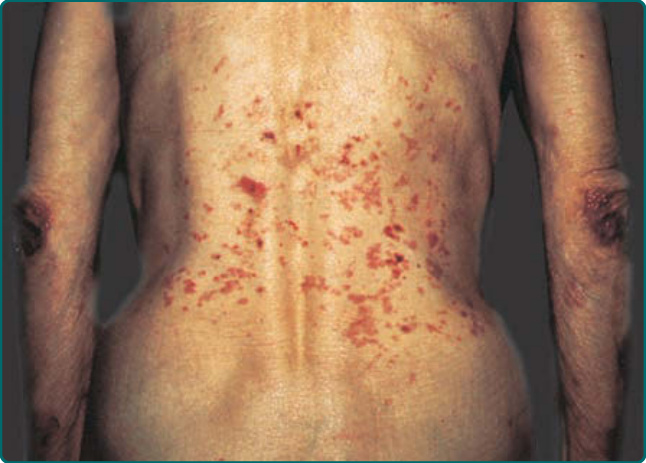
Figure 56-5 An epidermolysis bullosa acquisita patient demonstrating 2 presentations of the disease: The classic mechanobullous presentation with erosions, scarring, and milia over the elbows and the more inflammatory bullous pemphigoid–like lesions on her trunk.
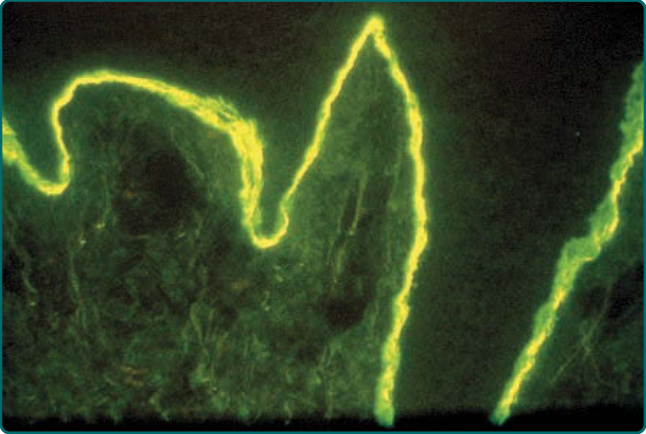
Figure 56-6 Direct immunofluorescence staining for immunoglobulin G deposits in perilesional skin of an epidermolysis bullosa acquisita patient. Note the dense deposits within the dermal–epidermal junction (the epidermis is on top in this section).
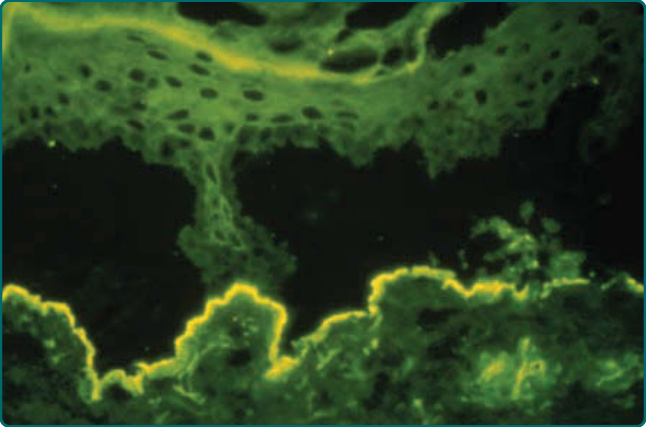
Figure 56-7 Direct immunofluorescence (DIF) staining of a patient’s perilesional skin biopsy after incubation in 1 molar cold saline for 72 hours and probed with antihuman IgG antibodies. The cold salt incubation fractures the dermal–epidermal junction of the skin specimen. In EBA patients, the IgG deposits remain with the dermal floor of the fractured skin. In contrast, in patients with bullous pemphigoid, the deposits would remain with the epidermal roof of the fractured skin (not shown). Likewise, if the serum of an EBA patient has a circulating IgG antibody to Type VII (anchoring fibril) collagen and indirect immunofluorescence (IIF) is performed, the serum will label the dermal floor of salt-split human skin substrate and leave the epidermal roof unlabeled. These results are because incubating human skin in cold 1 molar salt fractures the dermal–epidermal junction such that the hemidesmosomes of the epidermis and the bullous pemphigoid autoantigens associated with hemidesmosomes remain with the epidermal roof, whereas anchoring fibrils and Type VII collagen, the autoantigen in EBA, remain with the dermal floor.
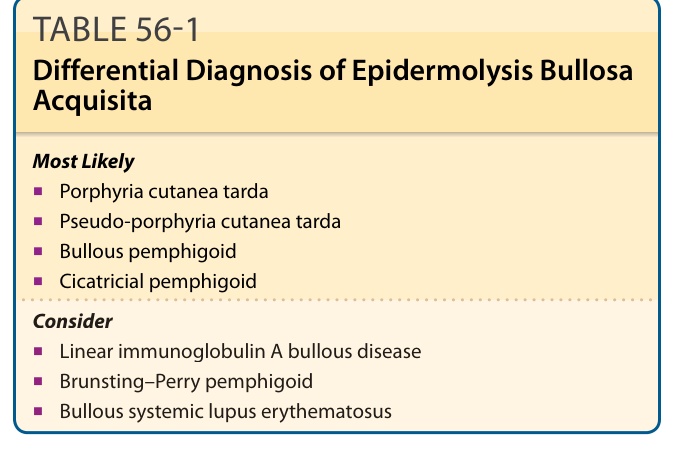
TABLE 56-1 Differential Diagnosis of Epidermolysis Bullosa Acquisita

TABLE 56-2 Diagnostic Criteria for Epidermolysis Bullosa Acquisita
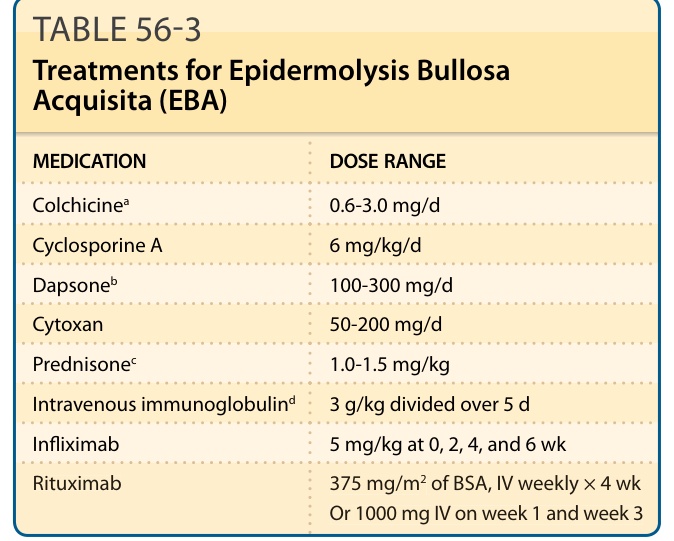
TABLE 56-3 Treatments for Epidermolysis Bullosa Acquisita (EBA)