副腫瘤性天疱瘡 (Paraneoplastic Pemphigus) 精華筆記
一覽
- 副腫瘤性天疱瘡 (paraneoplastic pemphigus, PNP) 是幾乎總與潛在淋巴增生性疾病 (lymphoproliferative disorder) 相關的自體免疫疾病。
- 核心三聯特徵:疼痛性口腔炎 (stomatitis) + 多形性皮膚疹(可為水疱性、苔癬樣或類似多形性紅斑 erythema multiforme)+ 血清抗斑蛋白 (plakin) 自體抗體。
- 死亡率高,常死於敗血症 (sepsis)、治療併發症或閉塞性細支氣管炎 (bronchiolitis obliterans)。
定義性特徵
- (a) 疼痛性口腔炎與多形性皮膚疹(水疱性、苔癬樣或似多形性紅斑)。
- (b) 組織學呈棘層鬆解 (acantholysis)、苔癬樣或界面 (interface) 變化。
- (c) 直接免疫螢光 (direct immunofluorescence):IgG 與補體沉積於表皮細胞間隙,常伴基底膜帶顆粒狀/線狀補體沉積。
- (d) 血清自體抗體以天疱瘡 (pemphigus) 模式結合皮膚黏膜細胞表面,並結合單層、柱狀與移行上皮 (simple, columnar and transitional epithelia)。
- (e) 自體抗體辨識橋粒芯糖蛋白 1 與 3 (desmogleins 1 and 3),及斑蛋白家族成員(橋粒斑蛋白 desmoplakins、包斑蛋白 envoplakins、周斑蛋白 periplakins)。
流行病學
- 發生率未知,較尋常型天疱瘡 (pemphigus vulgaris) 或落葉型天疱瘡 (pemphigus foliaceus) 少見。
- 多數病例未被正確診斷;常見誤診為多形性紅斑、史蒂芬斯-強森症候群 (Stevens-Johnson syndrome)、毒性表皮壞死溶解症 (toxic epidermal necrolysis, TEN) 與藥物反應。
相關腫瘤(致病關聯)
- 依 140 例確診估計頻率:44% 非何杰金氏淋巴瘤 (NHL)、19% 慢性淋巴球白血病 (CLL)、16% 卡斯爾曼氏病 (Castleman disease)、8% 胸腺瘤 (thymoma)、7% 腹膜後肉瘤 (sarcomas)、4% 瓦爾登斯特倫巨球蛋白血症 (Waldenström macroglobulinemia)、2% 無法分類。
- 卡斯爾曼氏病雖整體罕見卻不成比例地代表性高;兒童 PNP 幾乎總是卡斯爾曼氏病。
- 較常見癌症(乳房/腸道/肺腺癌、皮膚基底細胞癌與鱗狀細胞癌)一般不相關。
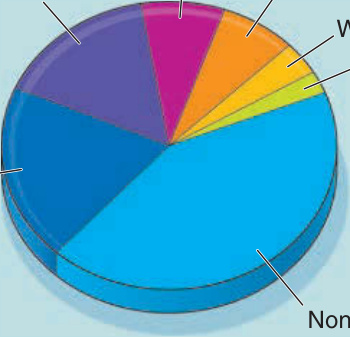
圖 53-1:與副腫瘤性天疱瘡相關聯的腫瘤。
致病機轉
- 假說:腫瘤異常表現上皮蛋白,抗腫瘤免疫與正常上皮蛋白交叉反應 (cross-reacts)(類似神經性副腫瘤症候群)。
- 遺傳易感性:HLA 第 II 類 DRB03 與第 I 類 Cw14 顯著優勢,且不同於尋常型天疱瘡,支持 PNP 為獨特實體。
- 失調的細胞激素:病人介白素 (IL)-6 顯著升高;腫瘤細胞(NHL、CLL、卡斯爾曼氏)體外分泌大量 IL-6,促進 B 細胞分化與 Ig 產生。抗 IL-6 受體單株抗體可逆轉卡斯爾曼氏病全身表現。
- 致病性自體抗體結合橋粒芯糖蛋白 3 的細胞外結構域 2 與 3(與尋常型天疱瘡結合結構域 1 不同),純化後注射新生小鼠可誘發棘層鬆解病灶。
- 細胞媒介自體免疫(界面皮膚炎)由橋粒芯糖蛋白 3 特異性 CD4+ T 細胞、經干擾素-γ (interferon-γ) 媒介,為苔癬樣變異型的主要反應;單靠注射 Ig 無法重現肺臟侵犯與淋巴球媒介上皮損傷。
- 肺臟異位性橋粒芯糖蛋白 3 表現(鱗狀化生)可能是致命閉塞性細支氣管炎的原因。
臨床表現
- 口腔炎為最恆定特徵:頑固性、最早出現、貫穿病程且極度抗治療;其缺如時不應考慮 PNP。糜爛與潰瘍可侵犯整個口咽,較尋常型天疱瘡有更多壞死與苔癬樣變化,偏好局限於舌側緣 (lateral borders of the tongue) 並延伸至嘴唇唇紅 (vermilion of the lips)。
- 皮膚病灶多變:易破裂之水疱、留下糜爛;四肢水疱可緊繃似大疱性類天疱瘡 (bullous pemphigoid),或周圍紅斑似多形性紅斑;上胸背可融合糜爛似 TEN。
- 與多形性紅斑/TEN 不同,PNP 為無情進展、歷時數月(後者自限、數週消退)。
- 苔癬樣疹常見,可為唯一皮膚徵象;存在時必伴嚴重口腔炎。慢性期苔癬樣疹可較水疱主導。
- 掌蹠與甲周組織 (paronychial tissues) 同受侵犯,有助與尋常型天疱瘡區分(後者肢端與甲周少見)。
- 少數病人無可證明的循環自體抗體(以苔癬樣病灶為主,即苔癬樣變異型),但腫瘤關聯與結局相同。
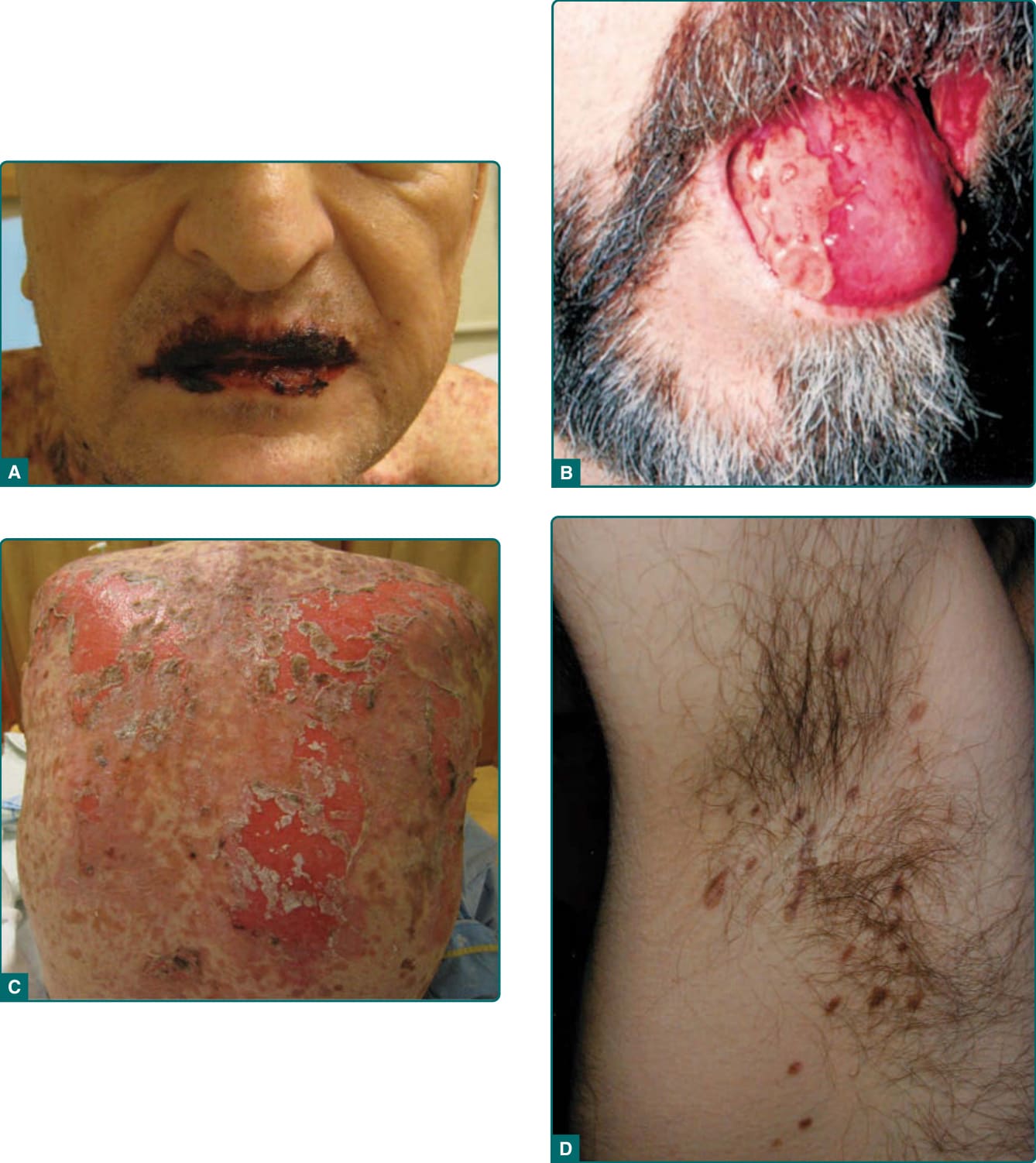
圖 53-2:A,嘴唇唇紅廣泛糜爛;B,疼痛性潰瘍局限於舌側緣;C,背部廣泛糜爛與脫落;D,腋窩病灶似扁平苔癬。
相關臨床表現(肺與肌無力)
- PNP 是唯一侵犯非複層鱗狀上皮 (nonstratified squamous epithelium) 的天疱瘡型式。
- 約 30% 至 40% 發展肺部損傷,常致命;最早為進行性呼吸困難 (progressive dyspnea),初期胸部 X 光無發現;大小氣道氣流阻塞,進展為閉塞性細支氣管炎。
- 與重症肌無力 (myasthenia gravis) 關聯:59 名病人世代中 20 名(35%)。
實驗室與診斷
- 關鍵:血清辨識針對斑蛋白的多克隆 IgG 自體抗體,多數亦針對橋粒芯糖蛋白 1 與 3。
- 最具特徵且最一致辨識的斑蛋白抗原為包斑蛋白 (envoplakin,210 kDa) 與周斑蛋白 (periplakin,190 kDa);其次為橋粒斑蛋白 I 與 II(250 與 210 kDa)。
- 抗 α2-巨球蛋白樣-1(α2-macroglobulin-like-1,170 kDa)見於 70% 病人,可能藉降低角質細胞黏附而致病。
- 篩檢:以齧齒類膀胱上皮 (rodent urinary bladder epithelium) 間接免疫螢光 (indirect immunofluorescence) 檢測 IgG,陽性意味斑蛋白抗體存在;但敏感度約 75%、特異度約 83%。
- 已有以包斑蛋白/周斑蛋白重組蛋白之 ELISA 試劑套組,敏感度與特異度高。更特異/敏感但耗時受限的檢查:免疫墨點法 (immunoblotting)、免疫沉澱法 (immunoprecipitation)。
- PNP 自體抗體圖譜較尋常/落葉型複雜,可能代表抗原表位擴散 (epitope spreading)。
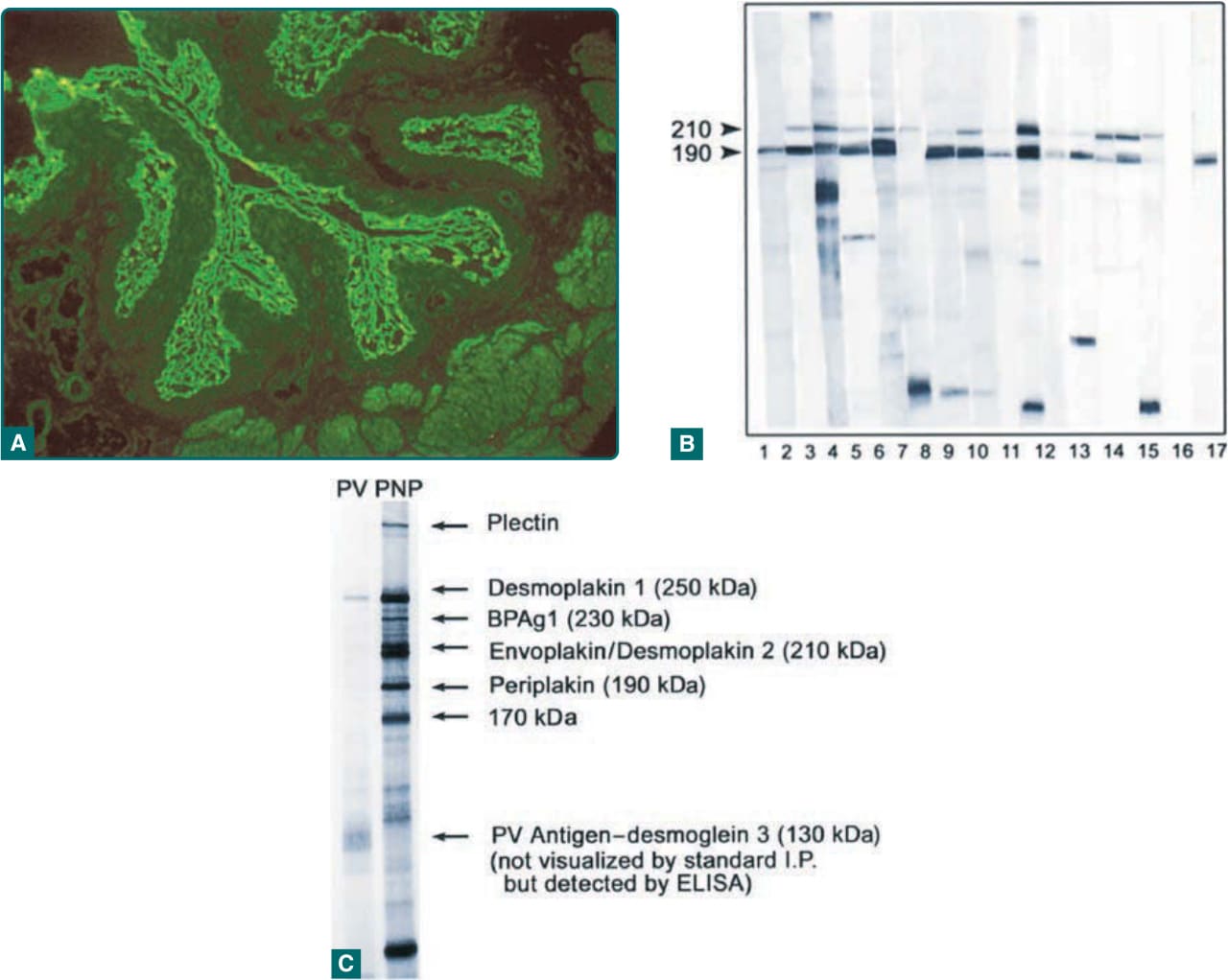
圖 53-3:抗斑蛋白抗體的證明——A 間接免疫螢光(齧齒類膀胱);B 免疫墨點法偵測包斑蛋白/周斑蛋白;C 免疫沉澱法。
組織病理學
- 因臨床多形性而組織學變異大,且常見細胞媒介細胞毒性 (cell-mediated cytotoxicity) 的發現(不見於尋常/落葉型天疱瘡)。
- 病灶周圍上皮可見苔癬樣黏膜炎 (lichenoid mucositis) 併個別細胞壞死與基底上棘層鬆解 (suprabasilar acantholysis)。
- 不同臨床型態對應不同組織學:非發炎水疱以基底上棘層鬆解為主;紅斑斑塊/丘疹以界面與苔癬樣皮膚炎為主;混合型兼具。
- 界面/苔癬樣變化光譜:(a) 角質細胞壞死併淋巴球浸潤(似多形性紅斑或移植物對抗宿主病);(b) 空泡性界面變化(似皮膚紅斑性狼瘡或皮肌炎);(c) 真皮-表皮交界厚苔癬樣帶(似扁平苔癬 lichen planus)。
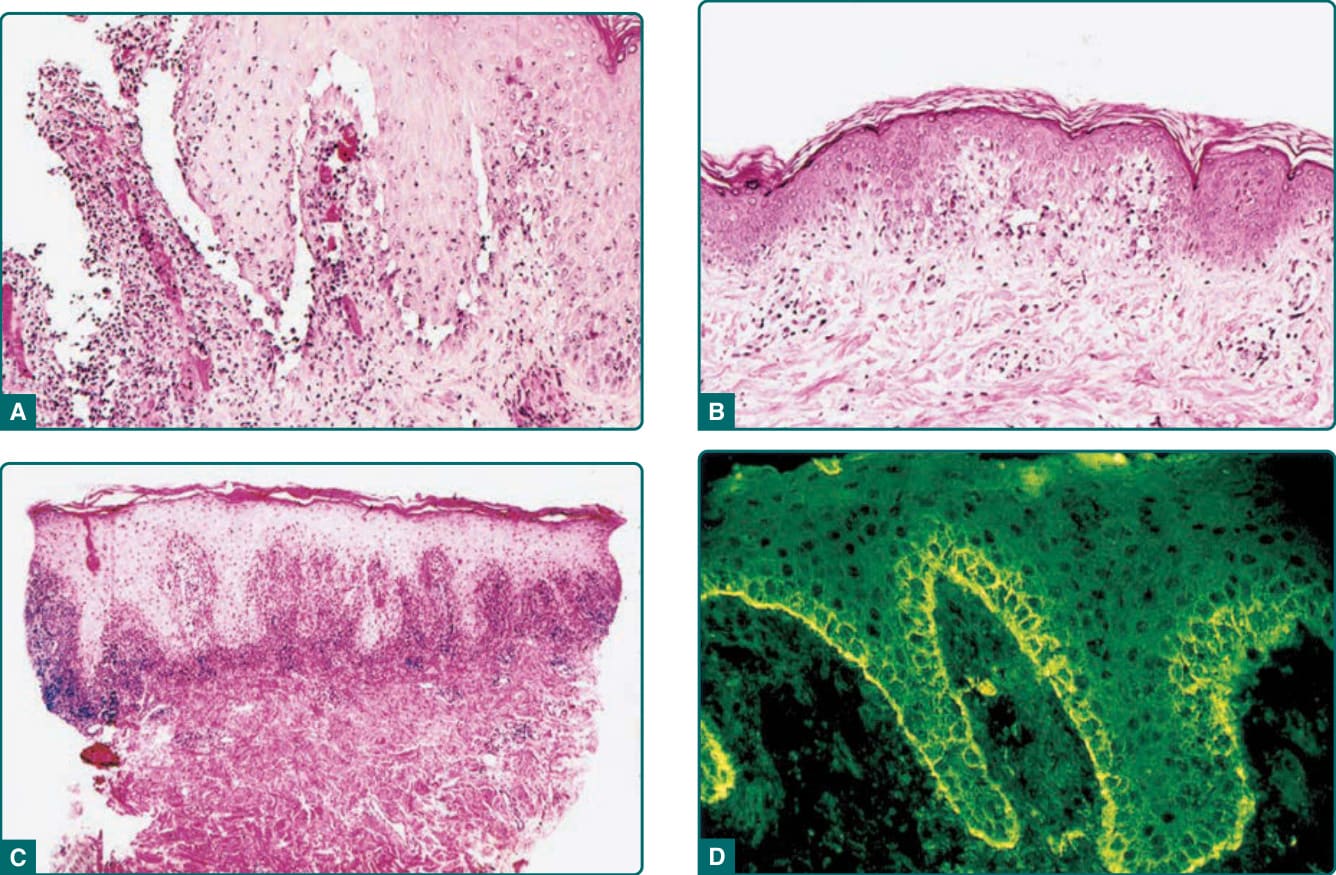
圖 53-4:A 水疱性病灶之空泡性界面變化與基底上棘層鬆解;B 斑塊/丘疹之空泡性界面變化;C 苔癬樣浸潤;D 直接免疫螢光顯示 IgG 與補體沉積。
免疫病理學
- 直接免疫螢光應見 IgG 結合於受影響上皮細胞表面;偽陰性較尋常型天疱瘡更常見,可能需重複切片並檢查附屬器結構(可能為唯一陽性部位)。
- 少數病例可見細胞表面與基底膜帶同時沉積 IgG/補體,但缺乏此合併染色不否定診斷。
特殊檢查
- 約三分之一病人發病時即有隱匿性惡性腫瘤 (occult malignancy),最可能為腹腔內淋巴瘤、胸腔/腹膜後卡斯爾曼氏腫瘤或腹膜後肉瘤。
- 篩檢首選:頸部至膀胱底部的電腦斷層或 MRI;可用時氟去氧葡萄糖 PET/CT 對隱匿性淋巴瘤更具特異性。內視鏡非必需。
診斷與鑑別診斷
- 依圖 53-5 演算法診斷。鑑別診斷見表 53-4:口腔病灶含尋常型天疱瘡、史蒂芬斯-強森症候群、黏膜類天疱瘡、口腔扁平苔癬、化療性口腔炎、重型口瘡性口腔炎;皮膚病灶含多形性紅斑/SJS/TEN、尋常型天疱瘡、藥物疹、扁平苔癬、表皮下水疱性疾病。
預後與病程
- 對通常有效的免疫抑制治療異常難治;死因含敗血症、消化道出血、多重器官衰竭與呼吸衰竭。
- 突發無法解釋的嗜中性球減少 (neutropenia) 可能導致致命敗血症。
- 呼吸衰竭為常見終末事件:阻塞性疾病進展至閉塞性細支氣管炎;呼吸短促與低血氧是不祥預後徵兆,肺損傷對藥物反應不佳。
- 法國 18 年多中心回溯研究:以類似多形性紅斑病灶表現、皮膚組織學有角質細胞壞死,為顯著負向預後因子。
- 腫瘤負荷與自體免疫活性無明確相關;治療原發腫瘤不影響自體免疫疾病活性(過程一旦啟動即獨立進展)。
治療
- 首選:若可能,外科手術完整切除腫瘤;完整移除後常大幅改善或完全緩解 (complete remission),但緩解可能需術後 1 至 2 年,期間需持續免疫抑制。常用合併 prednisone 與 rituximab。
- 兒科伴呼吸道疾病者,術後持續自體免疫可致持續肺損傷,長期存活可能需肺臟移植 (lung transplantation)。
- 惡性腫瘤病人無一致有效方案:幾乎所有 NHL 或 CLL 病人在診斷後 1 個月至 2 年內不治。
- 口服皮質類固醇 0.5 至 1.0 mg/kg 可部分改善但無法完全消退;皮膚病灶反應快,口腔炎極難治。
- 常嘗試但常失敗:cyclophosphamide、mycophenolate mofetil 或 azathioprine、金製劑 (gold)、dapsone、血漿置換術 (plasmapheresis)、光置換術 (photopheresis)。
- 部分病人對針對體液與細胞媒介自體免疫的合併治療反應良好:口服 prednisone、rituximab,加 daclizumab 或 basiliximab(皆為針對 CD25 的非耗竭性單株抗體),毒性較低,早期結果令人鼓舞。
預防
- 對已知淋巴樣惡性腫瘤病人,無已知介入可預防 PNP;較可能由腫瘤本身觸發自體免疫。