Paraneoplastic Pemphigus
9
AT-A-GLANCE
■ Rare complication of malignancy, most commonly non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease.
■ Painful, erosive stomatitis and polymorphous cutaneous lesions that may be blistering and erosive (resembling erythema multiforme), morbilliform, or lichenoid.
■ Serum autoantibodies directed against plakin proteins that are detected by indirect immunofluorescence against rodent bladder epithelium.
■ High mortality rate, with death from sepsis, complications of treatment, or bronchiolitis obliterans.
■ No consistently effective therapy, but some success with the combined use of rituximab, systemic corticosteroids, and other immunosuppressive agents.
Paraneoplastic pemphigus (PNP) is an autoimmune disorder that is almost always linked to an underlying lymphoproliferative disorder. These features define it (Table 53-1): (a) Painful stomatitis and a polymorphous cutaneous eruption with lesions that may be blistering, lichenoid, or resemble erythema multiforme. (b) Histologic findings that reflect the variability of the cutaneous lesions, showing acantholysis, lichenoid, or interface change. (c) Direct immunofluorescence findings of immunoglobulin (Ig) G and complement deposition in the epidermal intercellular spaces, and, often, granular/ linear complement deposition along the epidermal basement membrane zone. (d) Serum autoantibodies that bind to the cell surface of skin and mucosae in a pattern typical of pemphigus, which, in addition, also bind to simple, columnar and transitional epithelia. (e) The serum autoantibodies identify desmogleins 1 and 3, in addition to members of the plakin family of epithelial proteins, such as desmoplakins, envoplakins, and periplakins.1 Non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and Castleman disease are the neoplasms most commonly associated with PNP. There is no regularly effective treatment. Most patients die from complications of the disease such as pulmonary involvement with respiratory failure.
EPIDEMIOLOGY
The incidence of PNP is unknown, although it is less common than pemphigus vulgaris or foliaceus (see Chap. 52). In an adverse events reporting analysis
including 100,000 patients with known NHL and CLL, 12 were found to have PNP.2 Only 3 of the 12 patients were identified by the reporting physician, and the remainder were identified only by retrospective data analysis, suggesting that the majority of cases of PNP are not being properly diagnosed. In this series, the most common misdiagnoses were erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis (TEN), and drug reaction.
ETIOLOGY AND PATHOGENESIS
In almost all cases, PNP is associated with a limited number of lymphoproliferative neoplasms. On the basis of 140 cases of PNP confirmed by immunoprecipitation findings of the characteristic autoantibody profile, the estimated frequencies of specific neoplasms are 44% NHL, 19% CLL, 16% Castleman disease (giant follicular hyperplasia), 8% thymoma (malignant and benign), 7% sarcomas that are retroperitoneal and often poorly differentiated, 4% Waldenström macroglobulinemia, and in 2% the neoplasms were too poorly differentiated to categorize (Fig. 53-1). The disproportionate representation of Castleman disease is notable, given its overall rarity. In children with PNP, Castleman disease is almost always the underlying neoplasm.3 Before the description of PNP, many cases of Castleman disease associated with atypical forms of pemphigus had been reported, and we suspect that most were PNP. Furthermore, this association was emphasized in a retrospective study of 114 patients with Castleman disease identified 37 cases of PNP confirmed by the specific autoantibodies.4
With very rare exceptions, more common cancers, such as adenocarcinomas of breast, bowel, and lung, or basal cell and squamous cell carcinoma of skin, have not been associated with PNP. There are a few reports of PNP and squamous cell carcinoma, but in most of them, the diagnosis was made with immunofluorescent techniques only, which have significant false-positive and false-negative rates, and were not confirmed immunochemically, so the association remains unproven. The mechanisms by which these tumors induce autoimmunity against epithelial proteins remain speculative. One hypothesis states that the tumors constitutively or anomalously express epithelial proteins. These proteins are targeted by the antitumor immune response that cross-reacts with normal constitutive epithelial proteins of the host. This mechanism occurs in several neurologic paraneoplastic syndromes.5 This antitumor immune response may be initiated by reactivity against plakin proteins, and the antitumor immune response may cross-react with normal
a. Painful stomatitis and a polymorphous cutaneous eruption b. Histologic findings that reflect cutaneous lesions (acantholysis, lichenoid or interface changes) c. IgG complement deposition in the epidermal intercellular spaces and/or along basement membrane zone with granular/linear component (Direct Immunofluorescence) d. Serum IgG autoantibodies that bind of cell surface of skin and mucosae, but also to simple, columnar and transitional epithelia e. Serum IgG autoantibodies to desmogleins 1 and 3, desmoplakins, envoplakin and periplakin
a. Painful stomatitis and a polymorphous cutaneous eruption b. Histologic findings that reflect cutaneous lesions (acantholysis,
lichenoid or interface changes) c. IgG complement deposition in the epidermal intercellular spaces
and/or along basement membrane zone with granular/linear component (Direct Immunofluorescence) d. Serum IgG autoantibodies that bind of cell surface of skin and
mucosae, but also to simple, columnar and transitional epithelia e. Serum IgG autoantibodies to desmogleins 1 and 3, desmoplakins,
envoplakin and periplakin
constitutive proteins of epithelia. However, to date, there are no data to support this hypothesis. It is more likely that this autoimmune disease is a result of more complex interactions between the tumor cells, the immune system, and specific genetic background. In many autoimmune diseases, specific genetic predispositions have been found, and human leukocyte antigen (HLA) studies performed on 2 different series of PNP patients revealed a significant predominance of HLA-class II DRB∗03 and HLA-class I Cw∗14 genes. It is interesting to note that these 2 HLA molecules are not those associated with development of pemphigus vulgaris, providing another argument to consider PNP as a distinct entity.6,7
There is evidence that dysregulated cytokine production by tumor cells drives the development of autoimmunity. Patients with PNP have evidence of markedly elevated levels of interleukin (IL)-6.8 It has been observed that in a subset of cases of NHLs,9 CLL, and Castleman tumors, the tumor cells secrete massive amounts of IL-6 in vitro. IL-6 is known to promote B-cell differentiation and to drive Ig production, and dysregulated IL-6 production has been implicated in certain autoimmune diseases. Castleman tumors are known to be associated with other autoimmune phenomena such as myasthenia gravis and autoimmune
Tumors associated with paraneoplastic pemphigus
Thymoma Castleman disease
Retroperitoneal sarcoma
Other Waldenstom
Chronic lymphocytic leukemia
Non-Hodgkin lymphoma
9
cytopenias, and these patients also have high serum levels of IL-6. Symptoms attributable to Castleman tumors are routinely reversed by complete excision of the affected node(s), and, coincidentally, serum IL-6 levels revert to normal. Administration of anti– IL-6 receptor monoclonal antibodies also effectively reverses systemic manifestations of Castleman disease and is currently being studied in clinical trials.10
It had also been proposed that the autoantibody may derive from the lymphoid tumor itself. Cultures of Castleman tumors have been shown to contain B cells that produce specific autoantibody.11 However, Castleman tumors are unique in that they are not clonal neoplasms, and these studies showed the expansion of several immunologically active B-cell clones within the tumors. In Waldenström macroglobulinemia, the autoantibody is polyclonal and IgG class, not IgM, and therefore, cannot be produced by the tumor cells. Almost all patients with PNP have autoantibodies against desmogleins, demonstrable by enzyme-linked immunosorbent assay (ELISA), and when the desmoglein autoantibodies from these patients are affinity purified and injected into neonatal mice, acantholytic skin lesions are induced.12 It also has been demonstrated that pathogenic autoantibodies from PNP patients bind to the middle portion of desmoglein 3—extracellular domains 2 and 3—in contrast to pemphigus vulgaris patients where the pathogenic autoantibodies bind to extracellular domain 1.13 However, none of features of the disease that appear to be induced by cell-mediated autoimmunity are recreated by the Ig injections. No internal organs, like the lungs are involved, and there are no findings of lymphocyte-mediated lichenoid or interface epithelial injury. This is another indication that humoral immunity alone reproduces features of acantholysis, but passive transfer of autoimmune cells from these patients may be necessary to induce the complete spectrum of the disease in animals. It has been demonstrated in a mouse model that the cellular autoimmune response, which is represented by interface dermatitis, is caused by desmoglein 3–specific CD4+ T cells. This specific response is mediated by interferon-γ and is the dominant response in the lichenoid variant of PNP.14
There is evidence that ectopic desmoglein 3 expression is present in the lungs in the PNP mouse model. This squamous metaplasia of the lungs is probably the cause for the fatal bronchiolitis obliterans involvement in PNP.15
CLINICAL FINDINGS
HISTORY
HISTORY
CUTANEOUS LESIONS
The most constant clinical feature of the disease is the presence of intractable stomatitis (Fig. 53-2A,B). It is the earliest presenting sign and the one feature that persists throughout the course of the disease, even
935
9
A
C
B
D
after treatment and is extremely resistant to therapy. This finding is so consistent that in its absence, PNP should not be considered in the differential diagnosis. This stomatitis consists of erosions and ulcerations that can affect all surfaces of the oropharynx. The lesions differ from those seen in pemphigus vulgaris in that they show more necrosis and lichenoid change. They also preferentially localize to the lateral borders of the tongue, and characteristically extend onto and
936
involve the vermilion of the lips. Occasionally, oral lesions are the only manifestation of the disease. The cutaneous lesions of PNP are quite variable, and different morphologies may occur in an individual patient according to the stage of the disease (see Fig. 53-2C,D). The initial patients reported with the syndrome had episodes of waves of blistering affecting the upper trunk, head and neck, and proximal extremities. These lesions consisted of blisters that ruptured easily,
leaving erosions. The blisters on the extremities were sometimes quite tense, resembling those seen in bullous pemphigoid, or they had surrounding erythema, clinically resembling erythema multiforme (see Fig. 53-2D). On the upper chest and back, confluent erosive lesions can develop, producing a picture resembling TEN. The similarity of the mucocutaneous features to erythema multiforme and TEN explains why this is the most common differential diagnosis for PNP. However, it is important to note that erythema multiforme and TEN are self-limited events that evolve and resolve over several weeks, whereas PNP is a relentlessly progressive and evolves continuously over months. Cutaneous lichenoid eruptions are very common, and they may be the only cutaneous signs of the disease, or may develop in lesions that had previously been blistered. When cutaneous lichenoid lesions are present, severe stomatitis is also invariably present. In the chronic form of the disease and after treatment, this lichenoid eruption may predominate over blistering on the cutaneous surface. The common presence of both blisters and lichenoid lesions affecting the palms and the soles as well as the paronychial tissues helps distinguish PNP from pemphigus vulgaris, in which acral and paronychial lesions are uncommon. There are a small number of patients who appear to have PNP but who do not have demonstrable circulating autoantibodies.16 These patients tend to have predominantly lichenoid skin and mucosal lesions, but behave in every other way like antibody-positive patients. They have the same underlying neoplasms, and frequently develop bronchiolitis obliterans. Because the definition of the disease relies so heavily on demonstration of the specific autoantibody markers, further study is required to determine the exact classification of what is presently termed the lichenoid variant of paraneoplastic pemphigus. The disease also has been identified in a horse and 2 dogs. In animal species, the disease is associated with the same neoplasms and has the same clinical outcomes.17
RELATED CLINICAL FINDINGS
RELATED CLINICAL
FINDINGS
PNP is the only form of pemphigus that involves nonstratified squamous epithelium. Approximately 30% to 40% of cases develop pulmonary injury, often with a fatal outcome.18 The earliest symptoms are progressive dyspnea associated initially with an absence of findings on chest radiography. Pulmonary functions studies show airflow obstruction in large and small airways. Inflammation of the large airways evolves and is evidenced by endoscopic biopsy showing acantholysis of bronchial respiratory epithelium. Pulmonary function deteriorates in most cases despite immunosuppressive therapy, and radiologic, histologic, and functional changes characteristic of bronchiolitis obliterans develop. Another interesting clinical association of PNP is with myasthenia gravis, diagnosed in 20 (35%) of patients in a recent cohort of 59 patients with PNP.19
9
LABORATORY TESTS
The key finding is the serologic identification of polyclonal IgG autoantibodies against plakin proteins and, in most cases, desmogleins 1 and 3. The plakins are a group of sequence-related proteins that form the intracellular plaque of desmosomes and hemidesmosomes, and mediate attachment of cytoskeletal intermediate filaments to transmembrane adhesion molecules such as the desmogleins (see Chap. 48). Autoantibodies against these proteins are the most characteristic surrogate markers for the disease. The pattern of antigens recognized by individual patients shows considerable variability, but the most characteristic and consistently recognized plakin antigens are envoplakin20 and periplakin21 (210 and 190 kDa, respectively; Fig. 53-3). The next most frequently detected are antibodies against desmoplakin I and desmoplakin II (250 and 210 kDa, respectively). Autoantibodies against α2- macroglobulin-like-1 a 170-kDa protein, was identified in the sera of 70% of PNP patients. This recently reported autoantibody may be pathogenic by decreasing the normal keratinocyte adhesion.22 Less commonly, patients recognize bullous pemphigoid Ag 1 (230 kDa), plectin (400 kDa), and plakoglobin (82 kDa) (Table 53-2). PNP patients may also have clinical and serologic evidence of other autoimmune phenomena such as myasthenia gravis and autoimmune cytopenias (Fig. 53-3). To screen for PNP autoantibodies, one can test for IgG autoantibodies by indirect immunofluorescence reactive with rodent urinary bladder epithelium. A positive result implies the presence of plakin autoantibodies; however, the sensitivity and specificity of this serologic test are only approximately 75% and 83%, respectively.23 ELISA kits using recombinant proteins containing subdomains of envoplakin and periplakin have been developed and demonstrate high sensitivity and specificity for the diagnosis of PNP.24,25 More specific and sensitive tests, which are more timeconsuming, technically demanding, and of limited availability, include immunoblotting against epidermal cell extracts that can effectively detect antibodies against envoplakin, periplakin, and desmoplakin, and immunoprecipitation, using radiolabeled keratinocyte extracts, which can detect antibodies against any of the plakin proteins. The combination of rodent urinary bladder and immunoblotting has equal specificity and sensitivity to the detection of autoantibodies against the plakin proteins and the α2-macroglobulin-like-1 protein.26
The PNP autoantibody profile is more complex than that observed in pemphigus vulgaris or foliaceus, where there are autoantibodies produced only against the desmogleins. The humoral immunity in PNP may represent an example of epitope spreading in which patients develop autoantibodies against structurally related plakin proteins and structurally unrelated transmembrane cell-surface proteins (the desmogleins) that are physically linked to the plakin proteins in the desmosome and hemidesmosome.
937
9
A
C
B
AUTOANTIGEN MOLECULAR WEIGHT (KDA)
Desmoglein 1 160
Desmoglein 3 130
Desmoplakin 1 250
Desmoplakin 2 210
Bullous pemphigoid antigen 1 230
Envoplakin 210
Periplakin 190
Plectin 500
938
α2-Macroglobulin-like-1 protease inhibitor 170
α2-Macroglobulin-like-1 protease inhibitor 170
HISTOPATHOLOGY
HISTOPATHOLOGY
The histopathology of PNP is distinctive from that of pemphigus vulgaris and foliaceus for 2 reasons. First, because the lesions can be clinically very polymorphous, there is substantial variability in the histologic findings.27 Second, findings resulting from cell-mediated cytotoxicity are frequently observed. Owing to the severe mucositis, many biopsies of oral lesions yield nonspecific changes of inflammation and ulceration. If one can biopsy perilesional epithelium, a lichenoid mucositis with variable degrees of individual cell necrosis and suprabasilar acantholysis can be observed.
RESULTING FROM HUMORAL IMMUNE RESPONSE RESULTING FROM CELLULAR IMMUNE RESPONSE
Suprabasilar acantholysis
Suprabasilar acantholysis Individual keratinocyte necrosis Thick lichenoid band along the
Individual keratinocyte necrosis Thick lichenoid band along the dermal–epidermal junction Basal cell vacuolar changes
dermal–epidermal junction Basal cell vacuolar changes
When evaluating biopsies from skin lesions, one must recognize that lesions with different clinical morphologies yield differing histologic findings (Table 53-3). In noninflammatory cutaneous blisters, suprabasilar acantholysis is expected to be more prominent than the interface/lichenoid change (Fig. 53-4A). When erythematous macules and papules are sampled, interface and lichenoid dermatitis is predominant (Fig. 53-4B,C). Lesions with a mixed clinical pattern also show mixed histologic features of concomitant suprabasilar acantholysis and interface/lichenoid dermatitis. There is also observed variability of the interface and lichenoid dermatitis. The spectrum of changes
A
C
9
can include: (a) individual keratinocyte necrosis with lymphocytic infiltration into the epidermis, reminiscent of that seen in erythema multiforme or graftversus-host disease; (b) vacuolar interface change with sparse lymphocytic infiltrate of the basilar epithelium, resembling cutaneous lupus erythematosus or dermatomyositis; and (c) a thick lichenoid band along the dermal–epidermal junction similar to that seen in lichen planus. Although most of the specimens show a complex overlap of histologic patterns, there is a relatively good correlation between the clinical and the predominant histologic pattern. The histopathologic variability of this disease may be related to the fact that it is a presumed antitumor immune response. If this speculation is correct, one would expect to observe a combination of both humoral and cell-mediated immunity that is aberrantly directed against normal epithelium. In such a setting, one would expect to see changes of the sort described in the previous paragraph. This degree of cell-mediated immunity is not seen in pemphigus vulgaris or foliaceus (see Chap. 52); hence the unique histopathologic features, and, presumably, the unique clinical features as well. the presence of T-cell–mediated epithelial cytotoxicity has been demonstrated in histologic studies.2
B
D
939
9
IMMUNOPATHOLOGY
IMMUNOPATHOLOGY
Patients with PNP should have evidence of IgG autoantibodies bound to the cell surface of affected epithelium by direct immunofluorescence. However, false negatives are more common in PNP than in pemphigus vulgaris, and repeated biopsies may be necessary, as well as careful investigation of the adnexal structures, which may be the only positive site.28 In a minority of cases, one might also see a combination of both cell-surface and basement membrane zone deposition of IgG and complement components, but the absence of this combined cell-surface/ basement membrane zone staining does not negate the diagnosis (see Fig. 53-4D).
SPECIAL TESTS
SPECIAL TESTS
Approximately one-third of patients have an occult malignancy at the time they develop PNP. Neoplasms
that would not be detected by routine complete blood count, serum chemistries, and physical examination are most likely to be intraabdominal lymphoma, intrathoracic or retroperitoneal Castleman tumors, or retroperitoneal sarcomas. The most effective and efficient method for screening for these tumors is either computer-aided tomography or MRI of the body from the neck to the base of the bladder. If available, positron emission tomography/CT using fluorodeoxyglucose as a biologically active molecule, can be a more specific technique for identifying an occult lymphoma. Other studies, such as endoscopy, are not required.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The diagnosis can best be made if the algorithm shown in Figure 53-5 is followed. Table 53-4 summarizes the clinical differential diagnosis.
Approach to patient with paraneoplastic pemphigus
Absent
Painful stomatitis
Clinical evaluation
Present
Skin lesions
Biopsy for histology
Laboratory evaluation
Direct immunofluorescence
Indirect immunofluorescence
Immunochemical testing
No known tumor
Special testing
Preexisting neoplasm
Castleman tumor, thymoma, or sarcoma
Therapy
Non-Hodgkin lymphoma and chronic lymphocytic leukemia
940
Diagnosis excluded
Diffuse epithelial necrosis, tongue, and vermilion of lips
Blisters & erosions often resembling erythema multiforme or toxic epidermal necrolysis, lichenoid
Acantholytic, lichenoid, or vacuolar interface change
Immunoglobulin G and C3 on cell surfaces and along the basement membrane (false negatives common)
Cell-surface binding on monkey esophagus and murine bladder epithelium (negative result - possibly lichenoid variant)
Antibodies against desmogleins 1 & 3, desmoplakins 1 & 2, envoplakin & periplakin
Imaging of chest, abdomen, & pelvis (most common tumors - non-Hodgkin lymphoma, chronic lymphocytic leukemia, Castleman disease, retroperitoneal sarcoma, or thymoma)
Proceed to treatment
Excise completely if possible, followed by immunosuppressive therapy for up to 2 years
Reducing tumor does not stop autoimmune disease Immunosuppressive therapy with prednisone, rituximab, or other agents High mortality rate - best treatment regimen still unclear
■Oral lesions
■Oral lesions
■Pemphigus vulgaris
■Pemphigus vulgaris
■Stevens-Johnson syndrome
■Stevens-Johnson syndrome
■Mucous membrane pemphigoid
■Mucous membrane pemphigoid
■Oral lichen planus
■Oral lichen planus
■Chemotherapy-induced stomatitis
■Chemotherapy-induced stomatitis
■Major aphthous stomatitis
■Major aphthous stomatitis
■Cutaneous lesions
■Cutaneous lesions
■Erythema multiforme/Stevens-Johnson syndrome/toxic epidermal necrolysis
■Erythema multiforme/Stevens-Johnson syndrome/toxic
epidermal necrolysis
■Pemphigus vulgaris
■Pemphigus vulgaris
■Drug eruption
■Drug eruption
■Lichen planus
■Lichen planus
■Subepidermal blistering disorders
■Subepidermal blistering disorders
PROGNOSIS AND CLINICAL COURSE
It is not known why PNP is so refractory to the type of immunosuppressive treatments that are usually effective in pemphigus vulgaris and other autoimmune diseases. In those patients who do succumb, death has been attributed in individual cases to multiple factors, including sepsis, GI bleeding, “multiorgan failure,” and respiratory failure. Patients with autoimmune disease associated with B-cell neoplasms are known to have a high frequency of autoimmune cytopenias, and some fatal episodes of sepsis are suspected to have occurred because of sudden and unexplained neutropenia, possibly caused by this mechanism. Respiratory failure is a common terminal event. The development of shortness of breath with obstructive disease progressing to bronchiolitis obliterans is a terminal complication in most cases. Because these patients have autoantibodies that react with desmoplakins, and because desmoplakins are present in respiratory epithelium, respiratory failure may be a result of autoantibody-mediated
9
injury to bronchial epithelium, with plugging of terminal bronchioles, resulting in airflow obstruction and ventilation–perfusion abnormalities. Additionally, direct damage to alveolar epithelium could cause a diffusion barrier and subsequent intractable hypoxia. One autopsy study showed an absence of autoantibodies and a marked infiltration of bronchioles with cytotoxic T cells in a patient who died from PNP and bronchiolitis obliterans. This shows that there may be similar complex humoral and cell-mediated autoimmune injury to the lung, similar to what is seen in the skin. The pulmonary injury does not respond well to medical treatment, and the development of shortness of breath and hypoxia in a patient with this syndrome is an ominous prognostic sign. A recent long-term multicenter retrospective cohort study with 18 years of followup performed in France found that presentation with erythema multiforme–like lesions and keratinocyte necrosis in skin histology examination with were significant negative prognostic factors29
There is no definite correlation between tumor burden and the activity of the autoimmune syndrome in patients with malignant neoplasms. Treatment of the primary malignancy does not affect the activity of the autoimmune disease. It seems that once the process is initiated by the malignancy, the autoimmunity progresses independently. An example of the disconnect between tumor burden and autoimmunity is found in the case reported by Fullerton et al,30 in which PNP occurred after successful autologous bone marrow transplantation for NHL. This patient was free of detectable tumor burden at the time of his death, but died from pulmonary injury secondary to PNP. It is notable that the patient underwent autologous bone marrow transplantation, and therefore received his own memory T cells, or possibly individual malignant lymphoid cells that were not detectable by routine autopsy methods.
TREATMENT
DRUG USUAL DOSE OTHER DOSING
First line
Prednisone 0.5-1.0 mg/kg Methylprednisolone, 1000 mg
IV daily × 3 days
Rituximab 375 mg/m2 IV weekly × 4 wk, repeat every 6 months 1000 mg IV weekly × 2 wk
Daclizumab 2 mg/kg IV weekly × 4 wk, then every other week indefinitely
Basiliximab 20 mg IV on day 0 and day 4, repeat every 3-4 months
Second line
Cyclosporine 5 mg/kg daily
Cyclophosphamide 2.5 mg/kg daily
Mycophenolate mofetil 1000 mg by mouth twice daily
High-dose IV immunoglobulin 2 g/kg IV, repeat every 3-4 wk
941
Plasmapheresis Every other day for 6 total treatments
Plasmapheresis Every other day for 6 total treatments
9
them surgically excised. If the entire lesion is removed, the disease generally improves substantially or goes into complete remission. The remission of the autoimmune disease may take 1 to 2 years after surgery, so continued immunosuppression during this period is required. The usual treatment involves combined use of prednisone and rituximab.31 In pediatric cases with respiratory disease, the persistent autoimmunity immediately after surgery can cause ongoing pulmonary injury, and lung transplantation might be required for long-term survival.32
In patients with malignant neoplasms, there is no consensus regarding a therapeutic regimen that is consistently effective. Despite scattered individual reports of long-term survivors, almost all patients with NHL or CLL succumb in a period of 1 month to 2 years after diagnosis. Oral corticosteroids in a dose of 0.5 to 1.0 mg/kg produce partial improvement, but not complete resolution of lesions. Cutaneous lesions respond quickly to therapy, but the stomatitis is generally quite refractory to any treatment. Systemic corticosteroids and many other agents have been tried in individual cases, but none has proven to be particularly effective. Methods that have been tried and often failed include immunosuppression with cyclophosphamide, mycophenolate mofetil or azathioprine, gold, dapsone, plasmapheresis, and photopheresis. A small number of patients have shown a good response to combination treatment directed at both humoral and cell-mediated autoimmunity. These patients received oral prednisone, rituximab, and daclizumab or basiliximab (both are nondepleting monoclonal antibody against CD25, the high-affinity IL-2 receptor of T cells). This appears to be a less toxic way of downregulating both humoral and cell-mediated autoimmunity, with promising early results.
PREVENTION
There is no known intervention that may prevent the development of PNP in a patient with a known lymphoid malignancy. Although there have been individual case reports of PNP perhaps being triggered by certain drugs, radiation therapy, or cytokine administration, it is still not clear that any of these treatments triggered the autoimmune disease, and it appears more likely that the neoplasm itself triggers the autoimmunity.
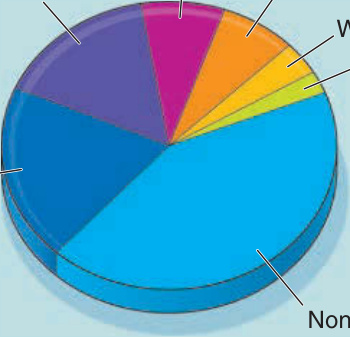
Figure 53-1 Tumors associated with paraneoplastic pemphigus.
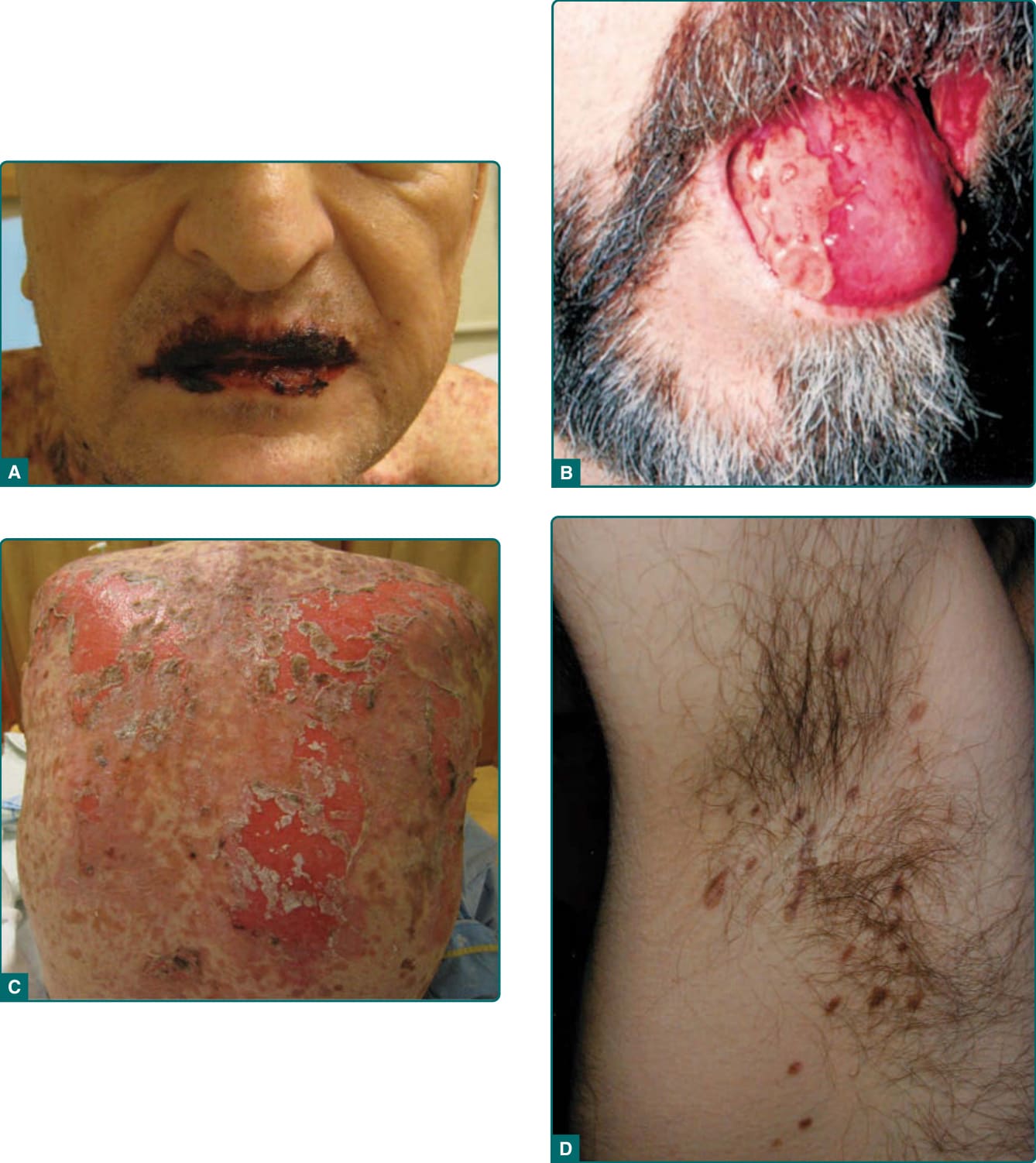
Figure 53-2 A, Extensive erosions involving the vermilion of the lips in a patient presenting with paraneoplastic pemphigus and lymphoma. The characteristic severe stomatitis, accompanied by polymorphous cutaneous lesions, is the most consistent feature of the disease. B, Painful ulcerations tend to localize to the lateral border of the tongue. C, Widespread erosions and sloughing involving the majority of the back from the same patient pictured in A. D, Lesions from the axilla of a patient with paraneoplastic pemphigus. These lesions clinically resemble lichen planus.
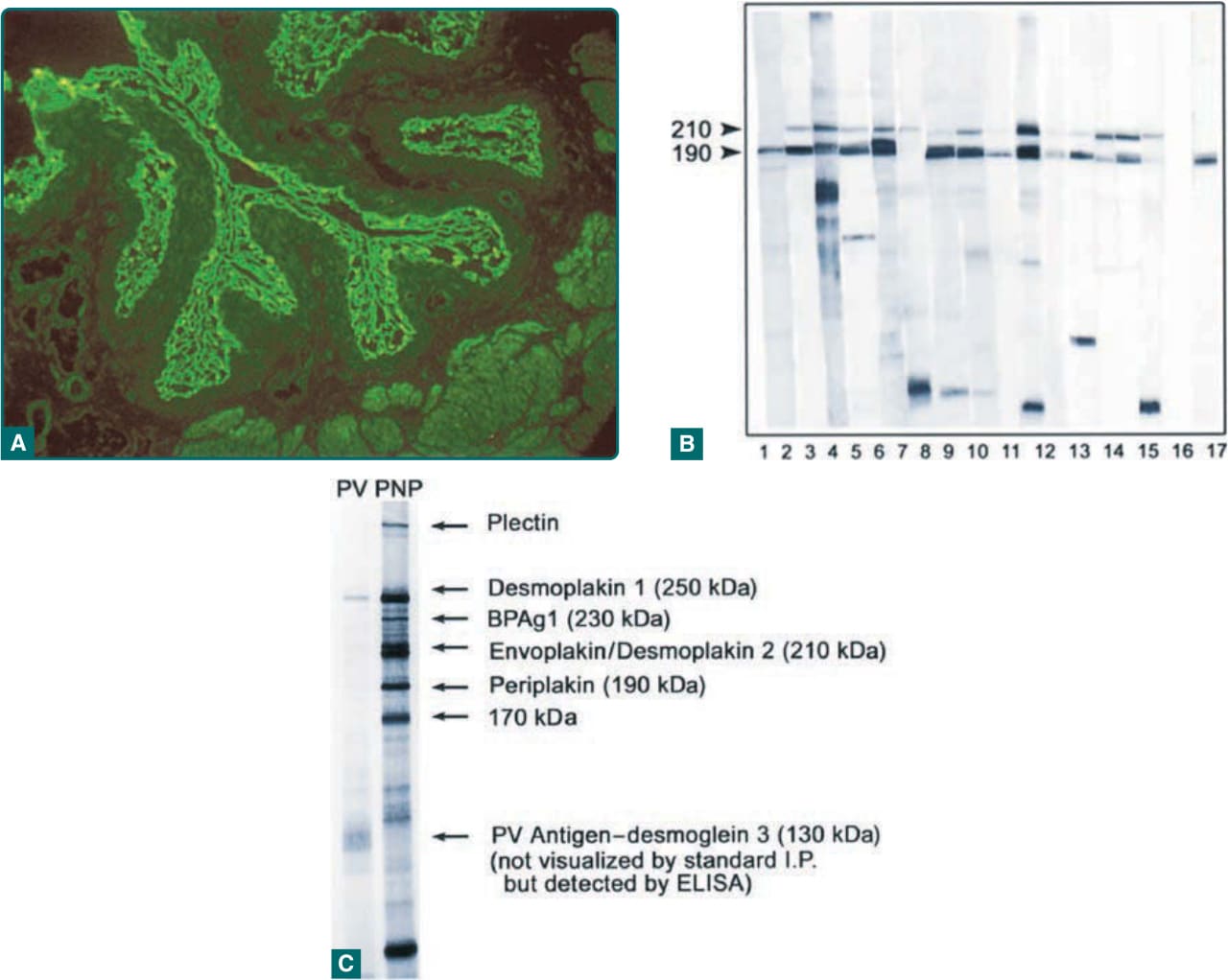
Figure 53-3 Diagnosis of paraneoplastic pemphigus (PNP) depends on the demonstration of antiplakin antibodies. A, This can be accomplished by indirect immunofluorescence of patient serum on rodent urinary bladder demonstrating binding of immunoglobulin G to the cell surface of transitional epithelial cells. A positive result implies the presence of antiplakin antibodies. This technique, although easily performed, has the lowest sensitivity and specificity. B, Immunoblotting against epidermal cell extracts is much more sensitive and specific. This shows detection of envoplakin (210 kDa) and/or periplakin (190 kDa) in 15 patients with PNP. Lane 16 is a normal control, and lane 17 shows a monoclonal antibody against periplakin. This technique uses denatured antigen extracts, so it does not reliably detect some of the PNP antigens, but antibodies against the most characteristic plakin antigens, envoplakin and periplakin, are easily detected. C, Immunoprecipitation using radiolabeled, nondenatured epidermal extracts and serum from a patient with PNP and pemphigus vulgaris (PV). In this case, the PNP patient’s serum identifies all the plakin antigens. Envoplakin and desmoplakin II migrate as a doublet at 210 kDa. This technique is the most sensitive and specific test for demonstration of antiplakin antibodies in PNP, but has limited availability. Although this technique readily detects the antiplakin antibodies, desmoglein 3 is not always efficiently identified, and this is best shown by using enzyme-linked immunosorbent assay (ELISA).
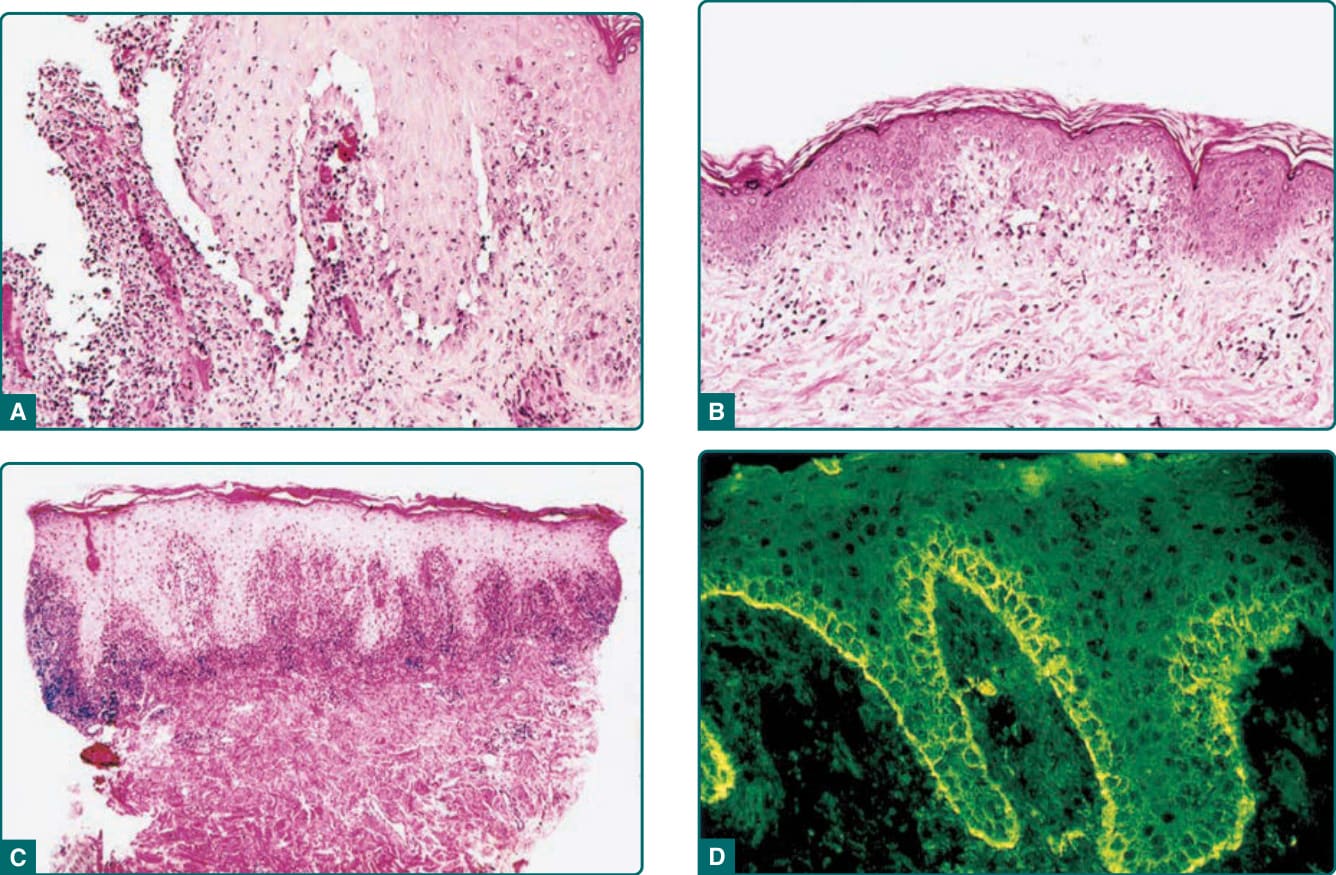
Figure 53-4 A, Histopathology of a blistering cutaneous lesion in paraneoplastic pemphigus. This demonstrates the characteristic presence of vacuolar interface change and suprabasilar acantholysis (Hematoxylin and eosin [H&E], ×200). B, Macular and papular lesions may show just vacuolar interface change (H&E, ×100). C, Lichenoid lesions demonstrate lichenoid infiltrates on histologic examination (H&E, ×40). The presence of these varied histologic findings help differentiate paraneoplastic pemphigus from pemphigus vulgaris. D, Direct immunofluorescence can be negative in a significant number of cases, but when positive, the most characteristic changes are those of deposition of immunoglobulin G and complement components on both the surface of basilar and suprabasilar keratinocytes and along the epidermal basement membrane zone (immunofluorescence with fluoresceinated anti–immunoglobulin G, ×200).
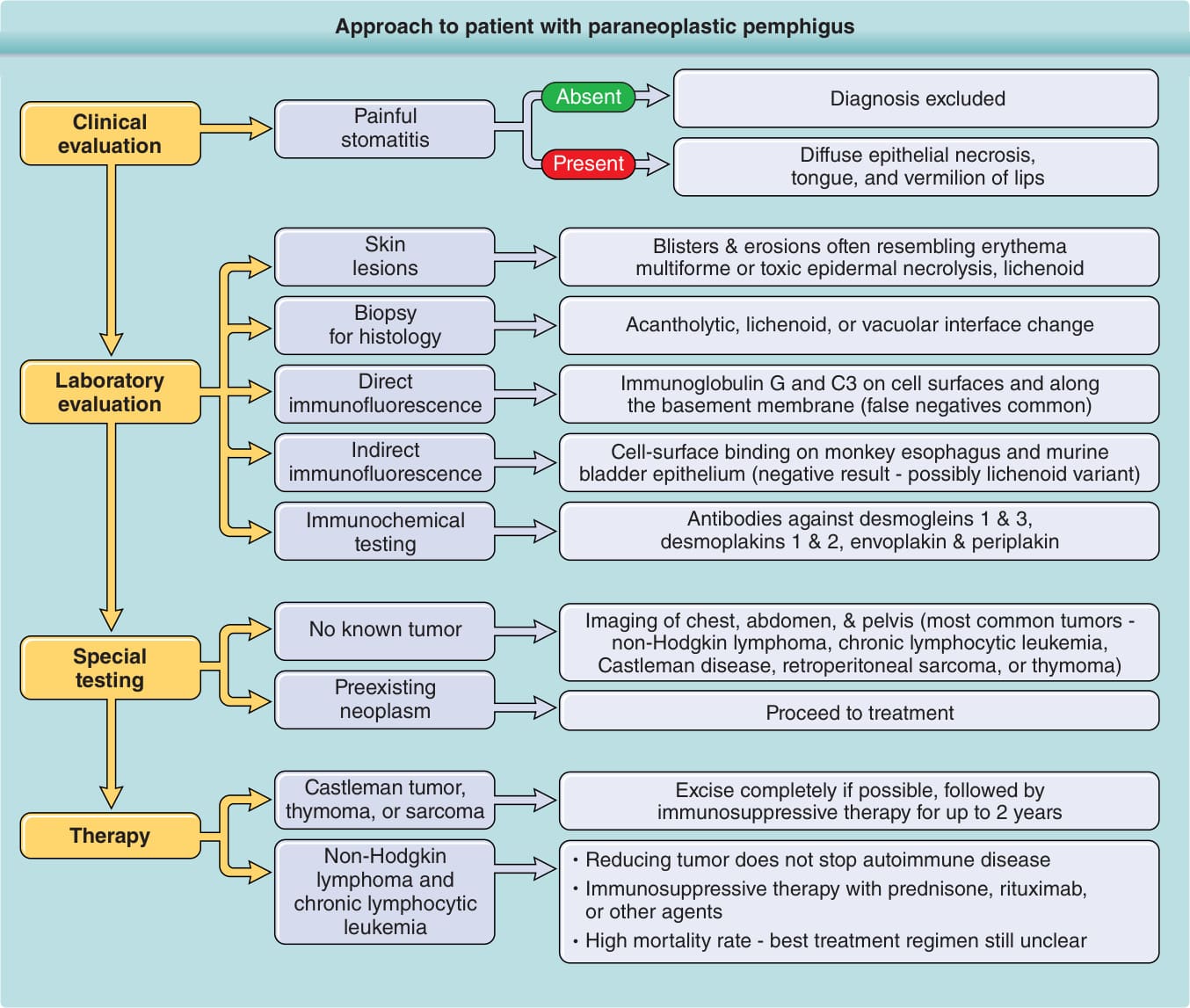
Figure 53-5 Approach to a patient with paraneoplastic pemphigus.

TABLE 53-1 Defining Features of Paraneoplastic Pemphigus
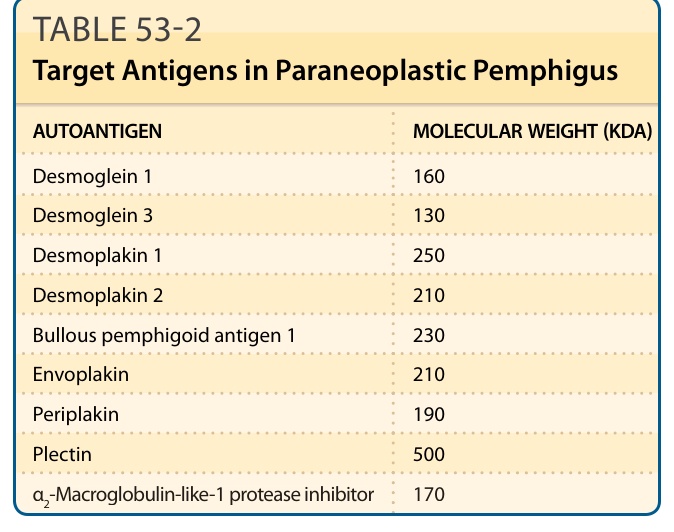
TABLE 53-2 Target Antigens in Paraneoplastic Pemphigus
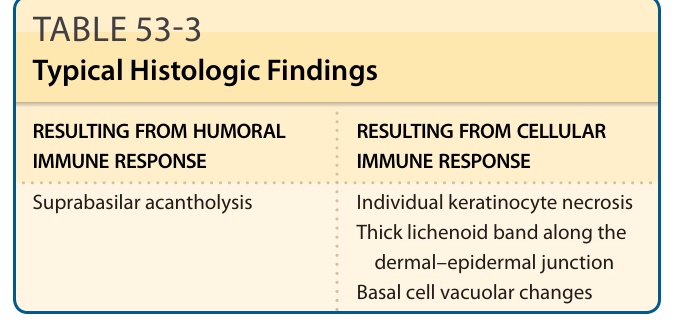
TABLE 53-3 Typical Histologic Findings

TABLE 53-4 Differential Diagnosis of Paraneoplastic Pemphigus
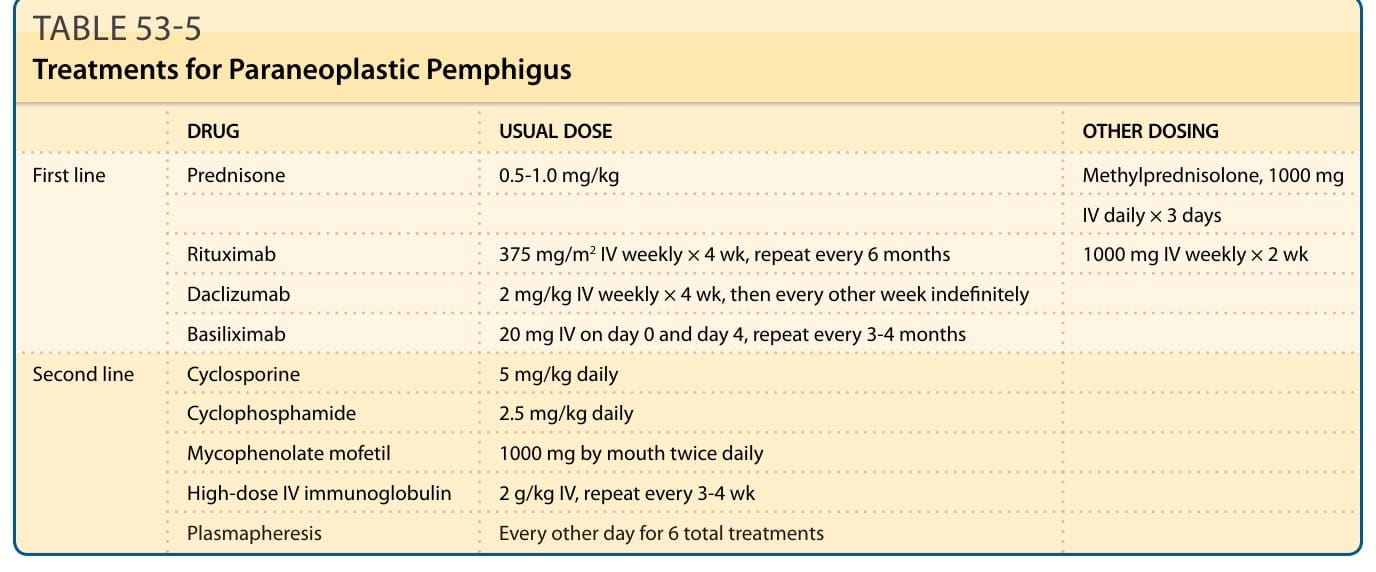
TABLE 53-5 Treatments for Paraneoplastic Pemphigus