天疱瘡 (Pemphigus) 精華筆記
定義與分類
- 天疱瘡 (pemphigus) 是一群皮膚與黏膜的自體免疫水疱性疾病,組織學特徵為棘層鬆解 (acantholysis,表皮細胞彼此分離) 所致的表皮內水疱,免疫病理特徵為針對角質細胞 (keratinocytes) 細胞表面的 IgG 自體抗體(活體內結合與循環中)。
- 四大主要型別:尋常型 (vulgaris)、落葉型 (foliaceus)、副腫瘤型 (paraneoplastic,見第 53 章)、IgA 天疱瘡(見第 57 章)。
- 尋常型天疱瘡 (pemphigus vulgaris, PV):水疱位於表皮較深處,基底層 (basal layer) 正上方。
- 落葉型天疱瘡 (pemphigus foliaceus, PF,又稱淺表型天疱瘡):水疱位於顆粒層 (granular layer)。
- 變異型:增殖型天疱瘡 (pemphigus vegetans,PV 變異型)、紅斑型天疱瘡 (pemphigus erythematosus,PF 變異型)、地方流行性 PF(如 fogo selvagem)。
流行病學
- PV 在猶太人 (Jews)、地中海後裔與中東人群較常見;PF 無此族裔優勢。PV 對 PF 比例:紐約/洛杉磯/克羅埃西亞約 5:1,伊朗 12:1,新加坡 2:1,芬蘭 0.5:1。
- 地方流行性 PF:巴西 (fogo selvagem,葡萄牙文「野火」)、哥倫比亞、突尼西亞;臨床、組織學與免疫病理與散發性 PF 相同,但流行病學獨特。常見於兒童與年輕成人,可發生於有血緣關係的家庭成員(但不具傳染性)。理論:環境媒介(如白蛉 Lutzomyia longipalpis 唾液抗原 LJM11)觸發低層級自體抗體反應,經分子間表位擴散對橋粒芯蛋白 1 (desmoglein 1) 變得具致病性。
- 發病年齡:土耳其、沙烏地阿拉伯、突尼西亞、伊朗約 40 歲;美國、歐洲、台灣 50–70 歲。兒童罕見(地方流行區除外)。
臨床表現
尋常型天疱瘡 (PV)
- 皮膚:原發病灶為鬆弛性水疱 (flaccid blister),可發生於任何部位,通常不侵犯掌蹠 (palmoplantar)。因水疱脆弱,最常見的是水疱破裂所致的糜爛 (erosions),常相當大且向周邊擴散。
- 尼可斯基徵象 (Nikolsky sign):對殘餘水疱壁或活動病灶周邊施加壓力或機械性剪切力,可使糜爛延伸進入外觀正常皮膚;有助與類天疱瘡區分(但也見於葡萄球菌燙傷樣皮膚症候群、史蒂芬斯-強森症候群、毒性表皮壞死溶解)。
- 約 5% 病人可見暫時性掉髮。
- 黏膜:最常侵犯口咽腔與鼻黏膜;疼痛的黏膜糜爛常為 PV 的表現徵象,可在皮膚病灶前平均達 5 個月為唯一徵象。亦可侵犯食道、外陰陰道與眼部;外陰與子宮頸陰道病灶可見於高達 51% 活動性疾病女性。
落葉型天疱瘡 (PF)
- 特徵性病灶為脫屑、結痂的糜爛,常在紅斑基底上,呈脂漏性分布 (seborrheic distribution,顏面、頭皮、上軀幹);小型鬆弛性水疱通常不被發現。
- 可侷限多年,或迅速進展為剝脫性紅皮症 (exfoliative erythroderma)。常抱怨疼痛與灼熱感;可因紫外線加劇。與 PV 相比,PF 極少有黏膜侵犯。
其他型別
- 紅斑型天疱瘡:脂漏性分布結痂糜爛,與狼瘡有免疫學重疊(約 30% 抗核抗體陽性、80% 狼瘡帶試驗陽性);現視為侷限型 PF,診斷主要具歷史意義。
- 新生兒天疱瘡:PV 母親經胎盤傳遞 IgG 給胎兒,隨母體抗體分解代謝(約 6 個月)而緩解;新生兒 PF 罕見(橋粒芯蛋白代償保護)。
- 藥物誘發性天疱瘡:與青黴胺 (penicillamine,使用者盛行率約 7%) 及 captopril 關聯最重要,PF 較 PV 常見;兩藥皆含硫醇基 (sulfhydryl groups)。部分病人停藥後緩解。
- 相關疾病:重症肌無力 (myasthenia gravis)、胸腺瘤 (thymoma);PV 病人自體免疫甲狀腺疾病、類風濕性關節炎、第 1 型糖尿病盛行率較高。
病因與致病機轉
- 自體抗原為橋粒芯蛋白 (desmogleins),橋粒的跨膜醣蛋白,屬鈣黏蛋白超家族。
- PF 抗原(及 fogo selvagem)= 橋粒芯蛋白 1 (160-kDa)。
- PV 抗原 = 橋粒芯蛋白 3 (130-kDa)。
- 抗體分布:黏膜為主的 PV 多只有抗橋粒芯蛋白 3 抗體;黏膜皮膚型 PV 通常同時有抗橋粒芯蛋白 3 與 1 抗體;PF 通常只有抗橋粒芯蛋白 1 抗體。
- 自體抗體具致病性:被動轉移 PV/PF IgG 至新生小鼠或人類皮膚可模擬對應疾病;天疱瘡中抗體可變區 (variable region) 即足以致病,IgG4(不固定補體)為主要且致病的亞類。
- 立體阻礙假說 (steric hindrance):致病抗體結合橋粒芯蛋白胺基末端對鈣敏感的構象性表位,直接干擾黏附,並導致橋粒芯蛋白內化與降解。
- 橋粒芯蛋白代償 (desmoglein compensation):解釋 PV/PF 不同的水疱部位與新生兒 PF 罕見——共表現的另一異構型可代償單一異構型的功能喪失(圖 52-5)。
- 訊號傳遞(p38 MAPK、Rho GTPases、MK2)參與調節水疱形成。Dsg 非組裝耗竭假說:抗體耗竭橋粒中橋粒芯蛋白使橋粒變小、功能缺損。
- 遺傳:PV 與第二類 MHC 強相關——德系猶太人 HLA-DR4 (DRB1∗0402),其他族群 DQ1 (DQB1∗0503)。抗 Dsg3 B 細胞常共享 VH1-46 基因。同一抗 Dsg B 細胞純系於病程中持續存在,為 B 細胞清除療法提供理論依據。
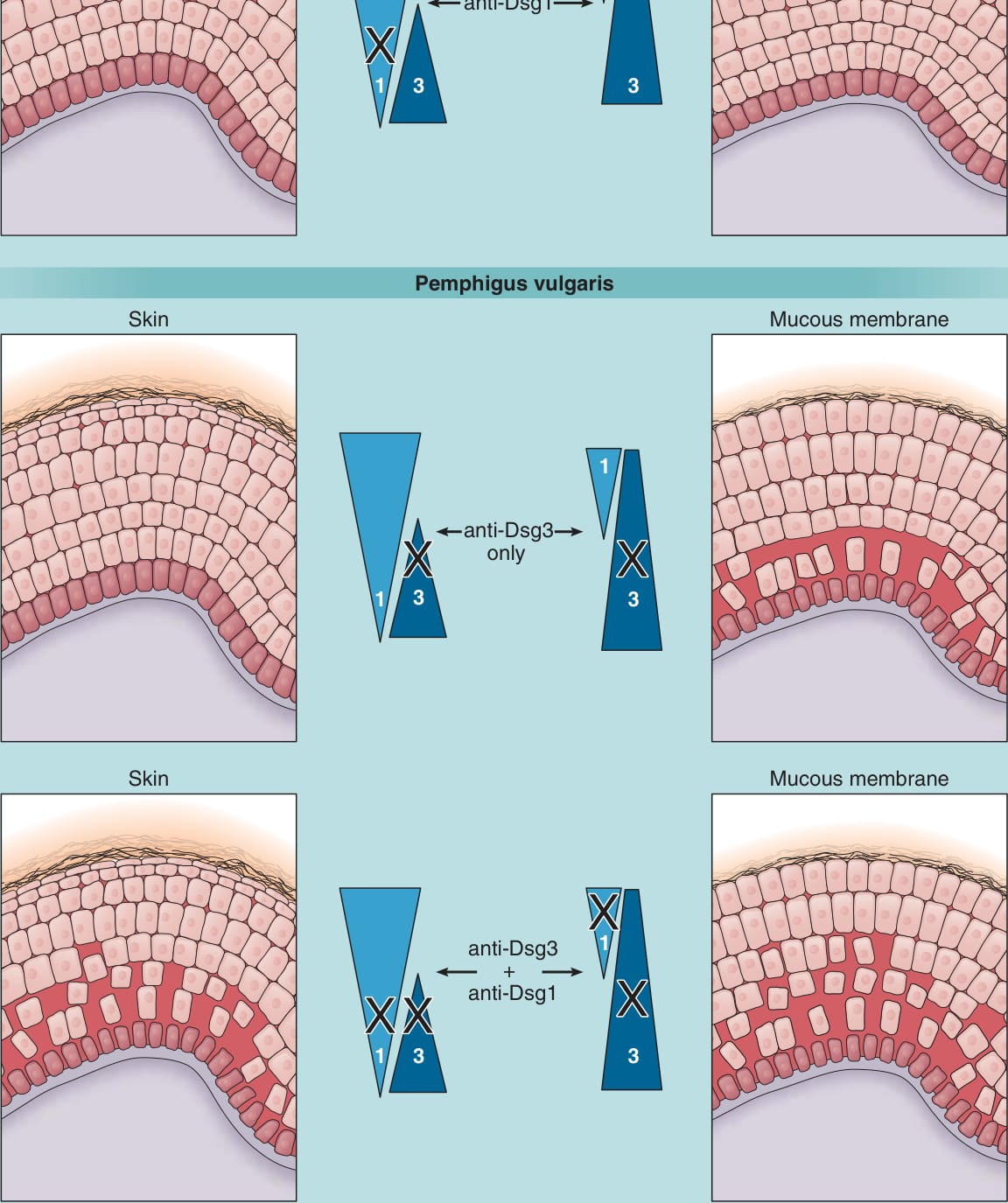
圖 52-5:橋粒芯蛋白 (Dsg) 代償——解釋 PV/PF 不同水疱部位與侵犯區域。
診斷
- 有賴新鮮病灶皮膚切片組織學,加上免疫化學研究確認皮膚自體抗體。
- 直接免疫螢光 (DIF):實質上所有活動性 PV/PF 病人呈陽性(病灶周圍皮膚角質細胞表面 IgG);非定量。切片應取自外觀正常的病灶周圍皮膚(避免偽陰性)。DIF 陰性應認真質疑診斷。
- 間接免疫螢光 (IIF):半定量效價;超過 80% 病人有循環抗上皮細胞表面 IgG。基質選擇影響敏感度——猴食道對 PV 較敏感,天竺鼠食道或正常人類皮膚對 PF 較佳。無法藉由型態區分 PV 與 PF。
- ELISA:比免疫螢光更敏感、特異,效價與疾病活性相關性較佳,且能區分 PV 與 PF。多數黏膜型 PV 僅 Dsg3 陽性;黏膜皮膚型 PV 對 Dsg3 與 Dsg1 皆陽性;PF 僅 Dsg1 陽性。治療決策上陰性結果比陽性更有幫助。疾病活性(而非抗體效價)是決定治療的主要依據。
- 病理:PV 為基底上水疱伴棘層鬆解,基底細胞呈「墓碑列 (row of tombstones)」;PF 為角質層下/顆粒層棘層鬆解,常見角質層下膿疱。PF 組織學常與大疱性膿痂疹/葡萄球菌燙傷樣皮膚症候群難以區分(後者亦因橋粒芯蛋白 1 功能障礙),故須免疫化學研究確認。
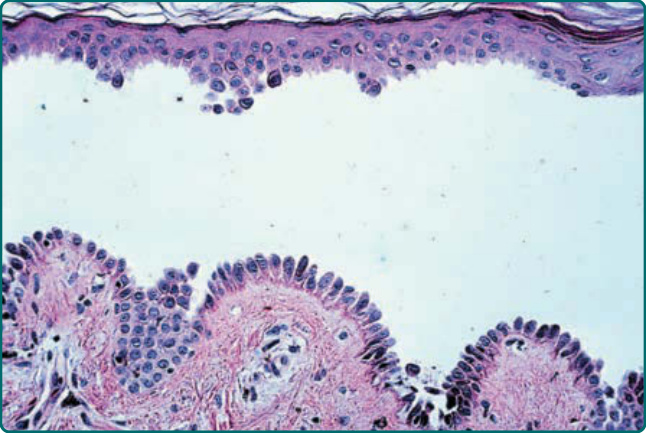
圖 52-8:尋常型天疱瘡組織病理——基底上棘層鬆解,墓碑列。
臨床病程與預後
- 糖皮質素 (glucocorticoid) 問世前,PV 幾乎無一例外致命(營養不良、脫水、敗血症);PF 約 60% 致命。
- 全身性糖皮質素與免疫抑制療法戲劇性改善預後,但仍有顯著病態與死亡率。住院死亡率估計 1.6%–3.2%;PV 病人死亡風險為對照組的 2.36–3.3 倍(英國、台灣)。感染常為死因。
- 糖皮質素加口服免疫抑制療法下,PV 4–10 年死亡率約 10% 或更低,PF 更低。
- rituximab 問世後緩解更常見、更快:約 50–90% PV 病人在一或多療程後緩解(停用其他免疫抑制療法),PF 緩解率相似。
治療管理
- PV 即使初期侷限也應在發病時治療(終將泛化);PF 可侷限多年,侷限時局部皮質類固醇 (topical corticosteroids) 可能即足夠,活動廣泛時治療同 PV。
皮質類固醇
- 全身性糖皮質素(通常 prednisone)為主軸。共識:完整劑量為 prednisone 當量 1.5 mg/kg/d,持續 2 至 3 週;但許多病人可以 0.5 至 1.0 mg/kg/d 控制(尤其併用輔助免疫抑制劑)。初期無反應者可一日兩次或三次分次給藥。
- 控制後逐漸減量。最低療法 (minimal therapy) 定義為每日 prednisone 當量 5 至 10 mg。若每日劑量接近或超過 5 至 10 mg 仍復發,宜加輔助免疫抑制劑。
Rituximab
- 抗 CD20 單株抗體,2018 年 FDA 核准用於 PV。劑量:第 1 天與第 15 天各靜脈輸注 1000 mg;維持劑量可於 12 個月時 500 mg、此後每 6 個月一次,臨床復發則 1000 mg。
- 關鍵試驗:第一線 rituximab 加短期 prednisone (0.5-1.0 mg/kg/day) 對單獨高劑量 prednisone (1.5 mg/kg/day),停用 prednisone 完全緩解率 89% 對 34%。
- 淋巴瘤劑量方案(每週一次 375 mg/m²,持續四週):單一週期使 86% 病人皮膚病灶完全癒合;約 80% 復發需額外治療,重複週期後 48% 病人最終達停用 prednisone 完全緩解。第一線療法達停類固醇完全緩解中位時間 9 個月。
- 風險:治療相關感染(37%)、致命感染(敗血症、肺囊蟲肺炎、B 型肝炎再活化、進行性多灶性白質腦病變);56% 病人發展抗藥物抗體。
口服免疫抑制劑
- 因非所有病人可取得 rituximab,許多專家仍從開始即用口服免疫抑制療法(常併 prednisone)。當糖皮質素持續劑量需大於 5 至 10 mg 或有禁忌/不耐受時加用。
- 硫唑嘌呤 (azathioprine):緩解率約 50%。前驅藥,部分經 TPMT 代謝(0.3% 白人 TPMT 缺乏、不耐受;1%–2% 「超高」濃度與抗性及肝毒性相關;亞洲人 NUDT15 多型性)。共識定義治療失敗劑量為 2.5 mg/kg/d 持續 12 週;實務可從每日 50 至 100 mg 起始向上滴定至目標 2.5 mg/kg/d。需頻繁血液與肝臟監測(尤其前 8 至 12 週)。
- 黴酚酸酯 (mycophenolate mofetil):典型劑量 30 至 40 mg/kg/d(最大 3 g/d),一日兩次(2.0 至 3.0 g/d);老年人可低至 1.0 g/d。試驗中緩解率 95%(vs 硫唑嘌呤 72%)。注意感染風險(巨細胞病毒、帶狀疱疹、結核、JC 病毒)。
- 甲氨蝶呤 (methotrexate):每週一次,較少使用。
靜脈注射免疫球蛋白 (IVIg)
- 高劑量靜脈 γ-球蛋白,藉飽和新生兒 Fc 受體增加自體抗體分解代謝。作為難治病例的輔助療法;不增加感染風險(可用於建立嚴重病人初始控制)。副作用:中風、深部靜脈血栓、無菌性腦膜炎。
其他療法
- 環磷醯胺 (cyclophosphamide):較毒但對嚴重疾病有效;因膀胱移行細胞癌與不孕風險,非第一線類固醇節省藥。
- 胺苯碸 (dapsone):可作維持期 PV 類固醇節省藥(趨勢但無統計顯著);併用 rituximab 時提供肺囊蟲肺炎預防。
- 血漿置換術 (plasmapheresis)、蛋白 A 免疫吸附法:移除 IgG,須併免疫抑制劑以防抗體反彈現象。
- 靜脈脈衝甲基培尼皮質醇 250 至 1000 mg/天、連續 4 至 5 天:可長期緩解,但有心律不整猝死風險,使用具爭議。
未來治療策略
- Btk 抑制劑 PRN1008、抗 FcRn 藥物 (SYNT001、ARGX-113)、自體調節性 T 細胞輸注、以 Dsg3 為標靶的嵌合自體抗體受體 T 細胞 (CAAR-T) 療法——後者可特異性殺死 Dsg 特異性 B 細胞,有望達持久緩解而不全身性免疫抑制。