Pemphigus
PART9
Vesiculobullous Disorders
AT-A-GLANCE
■ Two major types: pemphigus vulgaris and pemphigus foliaceus.
■ Pemphigus vulgaris: erosions on mucous membranes and skin; flaccid blisters on skin.
■ Pemphigus foliaceus: crusted, scaly skin lesions.
■ Pemphigus vulgaris histology: suprabasal acantholysis.
■ Pemphigus foliaceus histology: subcorneal acantholysis.
■ Autoantigens are desmogleins, transmembrane desmosomal adhesion molecules.
■ Diagnosis depends on histology showing intraepidermal acantholysis, immunofluorescence studies documenting the presence of cell surface autoantibodies, either bound to patient skin or in the serum, and/or enzyme-linked immunosorbent assay showing anti-desmoglein antibodies
■ Therapy includes topical and systemic corticosteroids, oral immunosuppressive agents, intravenous immunoglobulin, and rituximab (anti-CD20 monoclonal antibody).
INTRODUCTION
DEFINITIONS
DEFINITIONS
The term pemphigus refers to a group of autoimmune blistering diseases of skin and mucous membranes that are characterized histologically by intraepidermal blisters due to acantholysis (ie, separation of epidermal cells from each other) and immunopathologically by in vivo bound and circulating
immunoglobulin directed against the cell surface of keratinocytes. The nosology of this group of diseases is outlined in Table 52-1. Essentially, pemphigus can be divided into 4 major types: vulgaris, foliaceus (Table 52-2), paraneoplastic (Chap. 53), and IgA pemphigus (Chap. 57). In pemphigus vulgaris (PV), the blister occurs in the deeper part of the epidermis, just above the basal layer, and in pemphigus foliaceus (PF), also called superficial pemphigus, the blister is in the granular layer.
HISTORICAL PERSPECTIVE
HISTORICAL PERSPECTIVE
The history of the discovery of pemphigus, and its various forms, is covered in Walter Lever’s classic monograph Pemphigus and Pemphigoid.1 Both PV and PF display a spectrum of disease. Various points along these spectra have been given unique names, but because the presentation of these diseases is fluid, patients’ disease usually crosses these artificial designations over time. Thus, patients with PV may present with more localized disease, one form of which is called pemphigus vegetans of Hallopeau. This may become slightly more extensive and may merge into pemphigus vegetans of Neumann. Finally, with more severe disease, full-blown PV may appear. Similarly, patients with PF may present with more localized disease, represented by pemphigus erythematosus. However, these patients often go on to more widespread PF. The discovery by Ernst Beutner and Robert Jordon in 1964 of circulating antibodies against the cell surface of keratinocytes in the sera of patients with PV pioneered our understanding that PV is a tissuespecific autoimmune disease of skin and mucosa.2 Ultimately, their work led the way to the discoveries of autoantibodies in other autoimmune bullous diseases of the skin.
9
Pemphigus Subtypes
■Pemphigus vulgaris
■Pemphigus vegetans
■Pemphigus foliaceus
■Pemphigus erythematosus
■Endemic pemphigus foliaceus (eg, fogo selvagem)
■Immunoglobulin A (IgA) pemphigus
■Subcorneal pustular dermatosis
■Intraepidermal neutrophilic dermatosis
■Paraneoplastic pemphigus
Intraepidermal Blistering Diseases Without Autoantibodies
■Familial benign pemphigus (Hailey-Hailey disease)
■Bullous impetigo, staphylococcal scalded-skin syndrome
■Blisters from herpes simplex and zoster
■Allergic contact dermatitis (eg, rhus dermatitis)
■Epidermolysis bullosa simplex
■Incontinentia pigmenti
Mouth Ulcers/Erosion Without Autoantibodies
■Aphthous ulcers
■Candidiasis
■Lichen planus
■Behçet disease
Subepidermal Blistering Diseases With Autoantibodies
■Bullous pemphigoid
■Herpes gestationis
■Cicatricial pemphigoid
■Epidermolysis bullosa acquisita
■Linear IgA disease and chronic bullous disease of childhood
■Dermatitis herpetiformis
■Bullous lupus erythematosus
Subepidermal Blistering Diseases Without Autoantibodies
Subepidermal Blistering Diseases Without Autoantibodies
■Erythema multiforme
■Erythema multiforme
■Toxic epidermal necrolysis
■Toxic epidermal necrolysis
■Porphyria
■Porphyria
■Junctional or dystrophic epidermolysis bullosa
■Junctional or dystrophic epidermolysis bullosa
EPIDEMIOLOGY
INCIDENCE, PREVALENCE, AND SEX RATIO
INCIDENCE, PREVALENCE,
AND SEX RATIO
A few prospective and several retrospective surveys of patients with pemphigus clearly indicate that the epidemiology of pemphigus is dependent on both the area in the world that is studied and the ethnic population in that area.3-11 PV is more common in Jews and probably in people of Mediterranean descent and from the Middle East. This same ethnic predominance does not exist for PF. Therefore, in areas where the Jewish, Middle Eastern, and Mediterranean population predominates, the ratio of PV to PF cases tends to be higher. For example, in New York, Los Angeles, and Croatia, the ratio of PV to PF cases is approximately 5:1; in Iran the ratio is 12:1; whereas in Singapore it is 2:1 and in Finland, it is only 0.5:1. Similarly, the incidence of pemphigus varies by region. In Jerusalem, the incidence of
910
PV has been estimated to be 1.6 per 100,000 people per year and in Iran approximately 10.0 per 100,000 people per year. In Europe, the incidences are lower, ranging from a high of 0.7 PV cases per 100,000 person years in the United Kingdom to 10-fold less, 0.5 to 1.0 per million person years, in Finland, France, Germany, and Switzerland. In Taiwan, the incidence is 4.7 per million per year. The prevalence and incidence of PF are also very dependent on its location, as best exemplified by the finding of endemic foci of PF in Brazil, Colombia, and Tunisia. The first recognition of endemic PF was in Brazil and is called fogo selvagem, which means “wild fire” in Portuguese. It is a disease that is clinically, histologically, and immunopathologically the same as sporadic PF in any individual patient, but its epidemiology is unique.12,13 Fogo selvagem is endemic in the rural areas of Brazil, especially along inland riverbeds. The prevalence in some well-studied Indian reservations in rural Brazil can be as high as 3.4%, with the incidence up to 0.8 to 4.0 new cases per 1000 people per year.13,14 On the reservation in Limao Verde, up to 55% of unaffected individuals have a low-level IgG1 antibody response against desmoglein 1, the PF autoantigen, which becomes an IgG4 response of higher titer against a more pathogenic epitope in disease.13
Fogo selvagem occurs often in children and young adults, unlike sporadic PF, which is a disease of mostly middle-aged and older patients. Also unlike PF, fogo selvagem occurs not infrequently in genetically related family members, although it is not contagious. There is no known racial or ethnic predominance, and anyone moving into an endemic area may be susceptible to disease. Furthermore, the urban development of rural endemic areas of Brazil decreased the incidence of disease (Fig. 52-1). These epidemiologic associations suggest that an environmental agent may trigger a low-level autoantibody response that in some genetically susceptible individuals becomes pathogenic against desmoglein 1 by intermolecular epitope spreading. With this theory in mind, it is interesting that 40% to 80% of patients from Brazil with the insect-borne diseases onchocerciasis, leishmaniasis, and Chagas disease have low-level anti-desmoglein 1 antibodies, but patients with other infectious diseases from Brazil rarely have such antibodies.15 Subsequent studies have shown that IgG4 and IgE antibodies from fogo selvagem patients recognize salivary gland antigens from the sand fly Lutzomyia longipalpis, the vector of leishmaniasis.16 The current theory is that fogo selvagem may initiate with an IgE response against the sand fly salivary antigen LJM11, which cross-reacts with desmoglein 1 and then generalizes to an IgG4 response.17,18 This fascinating disease holds clues to understanding how this autoimmune response is triggered. The sex ratio of pemphigus in women versus men ranges from 1.33 to 2.25.7,9,19-24 Notable outliers to this estimate are the predominance of women (4:1) in an endemic focus of PF in Tunisia,6 and a predominance of men (19:1) in an endemic focus of PF in Colombia.25
Desmogleins 1, 3 (mucosal and skin involvement) Desmoglein 3 (mucosal dominant)
Desmoglein 1†
Desmoglein 1
Guinea pig esophagus or human skin substrate ideal IgG in intercellular pattern
Monkey esophagus substrate ideal IgG in cell surface pattern
IMMUNOFLUORESCENCE
Pemphigus erythematosusb Crusted lesions in seborrheic distribution Rare Similar to pemphigus foliaceus IgG and C3 deposition at granular basement membrane zone, IgG with intercellular pattern
IgG bound to surface of keratinocytes (intercellular pattern)
Oral involvement is common∗ Suprabasal acantholysis, with papillomatosis of the dermal papillae and downward growth of epidermal strands into the dermis; presence of hyperkeratosis and scale-crust; eosinophilic or neutrophilic intraepidermal abscesses
Very rare Early lesions show eosinophilic spongiosis; histopathology demonstrates acantholysis below stratum corneum; epidermis beneath the granular layer remains intact; subcorneal pustules containing neutrophils and acantholytic epidermal cells in the blister cavity
Oral and nasal mucous membranes most commonly affected Esophageal Vulvar, cervical, vaginal Ocular
Pemphigus vulgaris Flaccid blisters, typically sparing palmoplantar surface Large erosions are common presentations (+) Nikolsky sign
Pemphigus vegetansa Erosions develop excessive papillomatosis tissue and crusting; intertriginous areas, scalp, or face
Pemphigus foliaceus Scaly, crusted lesions on erythematous base; seborrheic distribution (face, scalp, upper trunk); small flaccid blisters are transient primary lesions
9
Similar to pemphigus foliaceus; characterized by burning sensation, and exacerbation on sun exposure
Similar to pemphigus foliaceus;
sensation, and exacerbation
characterized by burning
on sun exposure
bVariant of pemphigus foliaceus.
aVariant of pemphigus vulgaris.
Endemic pemphigus foliaceus (fogo selvagem)c
(fogo selvagem)c
pemphigus
foliaceus
Endemic
9
A B
AGE OF ONSET
AGE OF ONSET
The average age of disease onset also varies by region. In Turkey, Saudi Arabia, Tunisia, and Iran, the mean age of onset is approximately 40 years.6,19,21,26 Studies in the United States, Europe, and Taiwan demonstrate an average age of onset between 50 and 70 years.5,6,9-11,20,22,27-29 Pemphigus rarely occurs in children,30 except in regions of endemic disease.
CLINICAL FEATURES
PEMPHIGUS VULGARIS
PEMPHIGUS VULGARIS
CUTANEOUS FINDINGS
The skin lesions in PV can be pruritic or painful. Exposure to ultraviolet radiation may exacerbate disease activity.31,32 The primary lesion of PV is a flaccid blister, which may occur anywhere on the skin surface, but typically not the palms and soles (Fig. 52-1A, Table 52-2). Usually, the blister arises on normalappearing skin, but it may develop on erythematous skin. Because PV blisters are fragile, the most common skin lesions observed in patients are erosions resulting from broken blisters. These erosions are often quite large, as they have a tendency to spread at their periphery (Fig. 52-2). A characteristic finding in pemphigus patients is that erosions can be extended into visibly normal skin by pulling the remnant of the blister wall or rubbing at the periphery of active lesions; additionally, erosions can be induced in normal-appearing skin distant from active lesions by pressure or mechanical shear force. This phenomenon is known as the Nikolsky sign.33 This sign helps differentiate pemphigus from other blistering diseases of the skin such as pemphigoid (Table 52-1); however, similar findings also can be elicited in staphylococcal scalded skin syndrome, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
912
In certain patients, erosions have a tendency to develop excessive papillimatosis and crusting, referred to as vegetating lesions (Fig. 52-3). This type of lesion tends to occur more frequently in intertriginous areas, in the scalp, or on the face. Generally, the prognosis for these so-called pemphigus vegetans patients is thought to be better, with milder disease and a higher chance of remission compared to typical PV patients.34
Some ordinary PV lesions heal with a vegetating morphology and can remain for long periods of time in one place. Thus, vegetating lesions seem to be one reactive pattern of the skin to the autoimmune insult of PV.
A B
9
Although hair loss is not a usual feature in pemphigus, temporary hair loss can be seen in about 5% of patients and can rarely be a presenting sign of disease.35
MUCOUS MEMBRANE LESIONS
The mucous membranes most often affected by PV are those of the oropharyngeal cavity (Fig. 52-1B) and nasal mucosa.36,37 Endoscopic evaluation revealed that 87% of PV patients had ear, nose, or throat lesions, with involvement of nasal mucosa, pharynx, and larynx being most common at 76%, 66%, and 55%, respectively.38 Laryngeal involvement was often asymptomatic. As with cutaneous lesions, intact blisters are rare. Oropharyngeal erosions can be so painful that the patient is unable to eat or drink. The inability to eat or drink adequately may require inpatient hospitalization for disease control and intravenous fluid and nutrient repletion. In the majority of patients, painful mucous membrane erosions are the presenting sign of PV and may be the only sign for an average of 5 months before skin lesions develop.3 However, the presenting symptoms may vary; in a study from Croatia, painful oral lesions were the presenting symptom in 32% of patients.23 Most of these patients progressed to a more generalized eruption in 5 months to 1 year; however, some had oral lesions for more than 5 years before generalization. On the other hand, in Tehran, 62% of patients presented with oral lesions only.7 Skin involvement without mucous membrane involvement in PV is less common, accounting in one study for 11% of PV cases.39
GI tract involvement with PV has been described in the esophagus, stomach, duodenum, and anus, although only biopsies of the esophagus have proven the lesions were due to suprabasal acantholysis.7,40,41
About 27% of PV patients demonstrate such PV histology on blind biopsies of the esophagus.41 Furthermore, esophageal involvement may be asymptomatic but may also lead to esophagitis dissecans, evidenced by sloughing of esophageal casts.41,42 Involvement of other mucous membranes can also occur, including
the vulvovaginal and ocular epithelia.43-46 Vulvar and cervicovaginal lesions may be found in up to 51% of women with active disease but these lesions may be asymptomatic. Vulvar lesions are most common and may cause severe burning with urination. Even without obvious lesions, Pap smears may be positive in women with pemphigus and the acantholytic cells may be misinterpreted as indicative of cervical dysplasia.47,48 There is ocular involvement in about 16% of PV patients, some with erosions of the conjunctiva, but findings may be nonspecific.49 There are rare case reports of corneal erosions in PV patients, but without histologic confirmation of acantholysis.50
PEMPHIGUS FOLIACEUS
PEMPHIGUS FOLIACEUS
CUTANEOUS FINDINGS
The characteristic clinical lesions of PF are scaly, crusted erosions, often on an erythematous base. In more localized and early disease, these lesions are usually well demarcated and scattered in a seborrheic distribution, including the face, scalp, and upper trunk (Fig. 52-4A, Table 52-2). The primary lesions of small flaccid blisters are typically not found. Disease may stay localized for years, or it may rapidly progress to generalized involvement, resulting in an exfoliative erythroderma (Fig. 52-4B). Like PV, PF may be exacerbated by ultraviolet radiation.32,51,52 Patients with PF often complain of pain and burning in the skin lesions. In contrast to patients with PV, those with PF very rarely, if ever, have mucous membrane involvement, even with widespread disease. The colloquial term for Brazilian endemic pemphigus, fogo selvagem (Portuguese for “wild fire”), takes into account many of the clinical aspects of this disease: the burning feeling of the skin, the exacerbation of disease by the sun, and the crusted lesions that make the patients appear as if they had been burned.
913
9
B A
PEMPHIGUS ERYTHEMATOSUS
In 1926, Francis Senear and Barney Usher described 11 patients with features of a pemphigus-lupus erythematosus overlap (Senear-Usher syndrome).53 Over the next several decades, debate over whether these patients had lupus erythematosus, pemphigus, seborrheic dermatitis, or features of all 3 disorders continued, with Senear concluding that the disease is best considered a variant of pemphigus, termed pemphigus erythematosus.54 As these observations were made prior to the development of immunofluorescence testing for both pemphigus and lupus, the diagnosis was primarily based on the clinical presentation: crusted erosions in a seborrheic distribution, at times concurrent with more lupuslike discoid lesions with “carpet-tack” scale. Walter Lever noted that many patients initially categorized as pemphigus erythematosus went on to develop systemic lupus, or more widespread pemphigus foliaceus, or even pemphigus vulgaris, in some cases because of incorrect initial diagnosis. Therefore, rather than perpetuate the use of one term for different diseases, he proposed that pemphigus erythematosus be used to describe a localized form of PF with better prognosis.1 After the development of immunofluorescence and antinuclear antibody testing for pemphigus and lupus, it was discovered that pemphigus erythematosus patients demonstrate immunologic overlap features; by definition, all demonstrate the cell surface staining pattern classic for pemphigus, approximately 30% have positive antinuclear antibody titers, and 80% have positive lupus band tests, although the latter test is only positive in 20% to 40% of biopsies on non–sun-exposed skin.55 Subsequent studies have shown that the positive “lupus band” test in pemphigus erythematosus patients is due to granular deposits of IgG and cleaved desmoglein 1 ectodomain deposited at the basement membrane zone, which is thought to occur after ultraviolet light exposure.56 Thus, because most patients with pemphigus erythematosus do not develop systemic signs or symptoms of lupus, and some may progress from localized disease to generalized PF,57 the diagnosis of the pemphigus
914
erythematosus variant of PF is largely one of historic, rather than clinical, significance.
NEONATAL PEMPHIGUS
NEONATAL PEMPHIGUS
Infants born to mothers with PV may display clinical, histologic, and immunopathologic signs of pemphigus vulgaris.58,59 The degree of involvement varies from none to severe enough to result in a stillbirth. If the infant survives, disease tends to remit as maternal antibody is catabolized. Mothers with PF may also transmit their autoantibodies to the fetus, but, as discussed in the section “Pathophysiology of Acantholysis”, neonatal PF occurs only rarely.60-62 Neonatal pemphigus should be distinguished from PV and PF that occur in childhood, which are similar to the autoimmune diseases seen in adults.63
DRUG-INDUCED PEMPHIGUS
DRUG-INDUCED
PEMPHIGUS
Although there are sporadic case reports of pemphigus associated with the use of several different drugs, the association with penicillamine, and perhaps captopril, is the most significant.64 The prevalence of pemphigus in penicillamine users is estimated to be approximately 7%. PF (including pemphigus erythematosus) is more common than PV in these penicillamine-treated patients, although either may occur. The findings of direct and indirect immunofluorescence are positive in most of these patients. Three patients with druginduced PF and one with drug-induced PV have been shown to have autoantibodies to the same molecules involved in sporadic pemphigus, namely, desmoglein 1 and desmoglein 3, respectively.65 Therefore, by immunofluorescence and immunochemical determinations, these patients with drug-induced pemphigus resemble those with sporadic disease.
Both penicillamine and captopril contain sulfhydryl groups that are postulated to interact with the sulfhydryl groups in desmoglein 1, 3, or both, thereby causing pemphigus either by directly interfering with these adhesion molecules66 or, alternatively, by modifying them so that they become more antigenic. The use of these drugs may also lead to a more generalized dysregulation of the immune response, as penicillamine has been associated with the onset of several other autoantibody-mediated diseases including myasthenia gravis,67 Goodpasture syndrome,68 and antineutrophil cytoplasmic antibody vasculitis.69 Some, but not all, patients with drug-induced pemphigus go into remission after they stop taking the offending drug. Additionally, rare anecdotal reports have suggested the association of dietary intake and pemphigus, proposing the hypothesis that thiol-containing foods such as garlic, leeks, and onions may precipitate disease.70,71
Some patients may note that certain foods aggravate oral lesions, but it is unlikely that dietary intervention alone will remit disease in most patients. Interestingly, anecdotal case reports have reported improvement of PV with cigarette smoking,72 as well as with the cholinergic agonists pyridostigmine, carbachol, and pilocarpine.73,74 Studies suggest that activation of cholinergic receptors may regulate signaling pathways modulated by PV IgG, thereby affecting cell adhesion.75 These results are intriguing given the clinical benefit of nicotine noted in other inflammatory diseases, such as ulcerative colitis.76
ASSOCIATED DISEASES
ASSOCIATED DISEASES
Myasthenia gravis, thymoma, or both have been associated with pemphigus.77-79 Approximately onehalf of thymoma-associated pemphigus cases are vulgaris; one-half, foliaceus or erythematosus. Most of these data, however, were reported before the recognition of paraneoplastic pemphigus as a distinct entity. Therefore, although thymoma may clearly be associated with PV and PF, it also may be associated with paraneoplastic pemphigus (Chap. 53). Myasthenia gravis is a tissue-specific autoantibody-mediated disease leading to skeletal muscle weakness. Early disease usually affects facial muscles, leading to symptoms of dysarthria, dysphagia, ptosis, or diplopia. Disease may then progress to affect the larger muscles of the trunk and extremities, with potential fatal complications from respiratory muscle involvement. Thymoma, in contrast, is typically asymptomatic in adults. In children, thymomas are more likely to be symptomatic with cough, chest pain, superior vena cava syndrome, dysphagia, and/or hoarseness from localized tumor encroachment. Myasthenia gravis would be best evaluated by a neurologist, who can complete a full neurologic examination and may test for the presence of serum acetylcholine receptor autoantibodies. The course of myasthenia gravis and
9
the course of pemphigus appear to be independent of each other. Likewise, thymic abnormalities may either precede or follow the onset of pemphigus. Posteroanterior and lateral chest radiographs with or without computed tomography follow-up can detect most thymomas. Irradiation of the thymus or thymectomy, although clearly beneficial for myasthenia gravis, may or may not improve the pemphigus disease activity.80
Recent epidemiologic studies have identified that pemphigus vulgaris patients have a higher prevalence of autoimmune thyroid disease, rheumatoid arthritis, and Type 1 diabetes compared with the general population.81 Higher prevalences of these autoimmune diseases are also observed in the family members of PV patients, suggesting a common genetic element that may underlie autoimmune susceptibility.
ETIOLOGY AND PATHOGENESIS
The discovery of pemphigus as an organ-specific, autoantibody-mediated disease of desmosomes highlights the synergy between clinical care and basic science research. The development of light microscopy and electron microscopy allowed dermatologists to identify the morphology and immunopathology of disease. Patient serum IgG served as a key reagent to help identify both the PF and PV antigens.82-84 The cloning and characterization of the pemphigus antigens have subsequently led to the development of enzyme-linked immunosorbent assays (ELISAs) to improve the sensitivity and specificity of disease diagnosis, and continued studies on pemphigus pathophysiology aim to develop safer and effective therapies for these potentially fatal diseases. A recent in-depth review discusses these issues.85
PEMPHIGUS AUTOANTIGENS
PEMPHIGUS
AUTOANTIGENS
Pemphigus antigens are desmogleins, transmembrane glycoproteins of desmosomes (cell-to-cell adhesion structures, reviewed in Chap. 15).86,87 Desmogleins are part of the cadherin superfamily of calcium-dependent cell-adhesion molecules. The original members of this family (eg, E-cadherin) demonstrate homophilic adhesive interactions (binding between like molecules). Desmogleins similarly demonstrate homophilic binding but likely participate in predominantly heterophilic adhesion by binding desmocollins, the other major transmembrane glycoprotein of desmosomes.88-90
The PF antigen (as well as the fogo selvagem antigen) is desmoglein 1, a 160-kDa protein.82,83,91 The PV antigen is desmoglein 3, a 130-kDa protein that is 64%
915
9
similar and 46% identical in amino acid sequence to desmoglein 1.84 All patients with PV have anti– desmoglein 3 antibodies, and some of these patients also have anti–desmoglein 1 antibodies.92,93 Patients with mucosal-dominant PV tend to have only anti– desmoglein 3 antibodies, whereas those with mucocutaneous disease usually have both anti–desmoglein 3 and anti–desmoglein 1 antibodies.94-96 PF patients typically have antibodies against only desmoglein 1. However, these rules are not true for every patient.97,98
IgG antibodies against another desmosomal cadherin, desmocollin, can be found infrequently in PV, pemphigus vegetans, and atypical pemphigus patients.99,100 Furthermore, mice with a targeted deletion of desmocollin 3 have PV-like skin lesions,101 and anti-desmocollin antibodies can cause acantholysis in vitro.99,102
Other cell surface molecules such as acetylcholine receptors and E-cadherin also have been identified as immunologic targets of pemphigus serum, although their direct involvement in the pathophysiology of pemphigus has not been well validated.103-106
ELECTRON MICROSCOPY
ELECTRON MICROSCOPY
Early ultrastructural studies of the blisters in pemphigus vulgaris and foliaceus focused on the appearance of desmosomes, because these are the most prominent cell-to-cell adhesion junctions in stratified squamous epithelia (Chap. 15). Almost all studies confirm that at various time points during acantholysis, the desmosome is affected and ultimately destroyed, consistent with the cell biologic data discussed in the section below. However, conclusions from electron microscopy studies as to the mechanism of desmosome destruction have varied. Several groups have proposed that the first pathologic event in pemphigus is intercellular widening of interdesmosomal cell membranes, with intact desmosomal junctions.107-110 Other studies have demonstrated half-split desmosomes without keratin tonofilament retraction, suggesting that pemphigus autoantibodies directly interfere with the trans-adhesive interface of desmosomes, and that keratin retraction is secondary to the loss of intercellular adhesion. Half-desmosomes without tonofilament collapse also have been observed in a mouse model of PV.111,112 Others have proposed that keratin retraction is a primary pathogenic event in pemphigus, triggered by cellular signaling after PV autoantibody binding.113 As a potential reconciliation of these findings, one study found that electron microscopic findings may differ depending on the site analyzed: in early blisters, halfdesmosomes without keratin retraction are observed; in well-developed lesions, keratin retraction from half-desmosomes occurs; and in spongiotic non-blistered skin, intercellular widening with intact desmosomal junctions can be found, similar to electron microscopy findings in other spongiotic epidermal diseases.114 Similarly, large-scale electron microscopic maps (“nanotomy”) have identified evidence of both half desmosomes and smaller desmosomes in pemphigus patients,115 and
916
super-resolution microscopy indicates that desmosomes are smaller in PV patients,116 consistent with a desmoglein nonassembly/desmosome depletion model for pathogenesis as discussed below. Currently, electron microscopy studies are not part of the clinical diagnostic workup for pemphigus.
PATHOPHYSIOLOGY OF ACANTHOLYSIS
PATHOPHYSIOLOGY OF
ACANTHOLYSIS
Autoantibodies in pemphigus are pathogenic as suggested by neonatal pemphigus, discussed above, in which mothers with even mild PV can pass IgG autoantibodies to the fetus, causing blistering oral and skin disease that resolves by approximately 6 months, concurrent with the disappearance of maternal IgG from the circulation.58
Several lines of evidence indicate that it is the antidesmoglein 1 and 3 antibodies in pemphigus patients who directly cause blisters and hence are the etiologic agents of disease. Passive transfer of PV or PF IgG to neonatal mice or human skin causes blisters that clinically and histologically mimic the corresponding type of pemphigus in patients.117-119 The anti-desmoglein antibodies are responsible for blister formation in the passive transfer model, because affinity purified antidesmoglein 1 and 3 autoantibodies cause PF and PV blisters, respectively, and adsorption of desmogleinreactive autoantibodies from PF or PV IgG abrogates disease.120-124 Finally, mice with a targeted deletion of the desmoglein 3 gene have clinical and histologic lesions similar to PV patients,125 suggesting that inactivation of desmoglein 3 results in a PV-like blister. Similarly, exfoliative toxin, the staphylococcus toxin that causes blisters in bullous impetigo and staphylococcal scalded skin syndrome, cleaves desmoglein 1 and results in a blister with a histology similar or identical to PF.126
Unlike many other autoantibody-mediated diseases, such as pemphigoid and epidermolysis bullosa acquisita, in which the constant region of the antibody is required for blister formation to activate complement or bind antibody receptors on inflammatory cells, in pemphigus the variable region of the antibody is sufficient to cause blisters in neonatal mice or human skin.119,127-129 Furthermore, IgG4, which does not fix complement, has been shown to be both pathogenic and the predominant IgG subclass in both PF and PV.130-132 For this reason, a significant amount of research on disease pathophysiology has focused on the epitopes bound by pathogenic autoantibodies, as these regions are likely critical for maintaining desmosomal cell adhesion. Epitope mapping studies have shown that pathogenic PV and PF autoantibodies bind calcium-sensitive, conformational epitopes in the amino terminal extracellular domains of desmogleins, whereas nonpathogenic antibodies tend to bind more membrane proximal extracellular domains.133-136 The amino terminal domains bound by pathogenic autoantibodies are the same domains that are predicted
to form the key molecular interactions for desmoglein intercellular adhesion, based on studies of cadherin ultrastructure.90,137,138 Furthermore, PV IgG have been shown to directly inhibit desmoglein 3–mediated trans-interactions by atomic force microscopy.139
Collectively, these data form the basis for the “steric hindrance” hypothesis, which proposes that pathogenic antibodies directly interfere with desmoglein adhesive interactions, causing acantholysis. Studies on cultured keratinocytes have indicated that loss of intercellular adhesion by pathogenic autoantibodies leads to internalization and degradation of desmogleins,140-143 thereby amplifying the loss of desmoglein function. As discussed above, loss of desmoglein 3 and 1 function results in PV- and PF-like blisters in model systems in which desmogleins are targeted genetically or by enzyme cleavage. If inactivation of desmoglein isoforms results in blistering, then why do blisters in PV and PF have specific tissue localizations that do not necessarily correlate with the sites at which the antibodies bind by immunofluorescence? In PF, for example, the anti–desmoglein 1 antibodies bind throughout the epidermis and mucous membranes,144 yet blisters occur only in the superficial epidermis. This apparent paradox can be explained by desmoglein compensation, as outlined in Fig. 52-5. The concept of desmoglein compensation originates in the assumption that autoantibodies against one desmoglein isoform inactivate only that isoform and that another isoform co-expressed in the same area can compensate in adhesion.145-147 Desmoglein compensation explains why neonatal PF is so unusual, because even though the maternal anti–desmoglein 1 antibodies cross the placenta, in neonatal skin, but not in adult skin, desmoglein 3 is co-expressed with desmoglein 1 in the superficial epidermis, thereby providing protection against the loss of desmoglein 1-based adhesion.146,148 Desmoglein compensation also offers an explanation for the differing sites of blister formation in PV and PF, both in regard to the histology (ie, suprabasal or superficial), as well as the areas of involvement (mucosa and/or skin). Further studies have suggested that perturbation of cell signaling pathways can mediate and/or modulate blister formation in pemphigus. For example, inhibition of the p38 mitogen activated protein kinase (MAPK) pathway and activation of Rho GTPases, among others, can prevent blister formation after passive transfer of pemphigus IgG in the neonatal mouse model.149-151 The p38 MAPK pathway has been most studied in this regard. It activates EGF receptor after PV antibody binding, and blocking of this receptor also blocks loss of cell adhesion.152 Although p38 activation may be secondary to loss of cell adhesion, blocking it does decrease the degree of acantholysis, suggesting that it would be a useful target in pemphigus therapy.153,154 A more specific target in this pathway, downstream of p38, is MAPK activated protein kinase 2 (MK2), which also can be blocked to modulate pemphigus blister activity, especially spontaneous blistering as opposed to blistering due to trauma (ie, Nikolsky blistering).155 Finally, in vitro studies with
9
monoclonal anti-desmoglein 3 antibodies and human pemphigus antibodies suggest that acantholysis is a net result of both direct steric hindrance of pathogenic monoclonal antibodies and polyclonal antibodies that may not directly cause steric hindrance but that cause internalization of desmogleins by crosslinking them. This latter effect is dependent on MAPK signaling.156
The depletion of desmogleins by pemphigus antibodies may lead to loss of desmogleins in desmosomes resulting in smaller desmosomes and/or their defective function in adhesion, a scenario referred to as the Dsg nonassembly depletion hypothesis.115,140,141,157,158 Consistent with this idea is the observation by direct immunofluorescence of clustering of Dsgs in both PV and PF patients’ skin and that forced increased expression of Dsg3 can prevent acantholysis by pemphigus antibodies.159
The current general consensus is that desmosomal adhesion is a dynamic process that is perturbed by pemphigus autoantibodies both directly and by signaling pathways.160 Therefore, therapies that aim to strengthen keratinocyte adhesion by modulation of signaling pathways may have a beneficial effect on pemphigus, regardless of whether cell signaling is a primary pathologic cause of disease.
GENETIC CHARACTERIZATION OF THE PEMPHIGUS IMMUNE RESPONSE
GENETIC
CHARACTERIZATION
OF THE PEMPHIGUS
IMMUNE RESPONSE
Compared to a matched population, patients with PV have a markedly increased frequency of certain class II major histocompatibility complex (MHC) antigens. Among Ashkenazi Jews with PV, the serologically defined HLA-DR4 haplotype is predominant, whereas in other ethnic groups with PV, the DQ1 allele is more common.161 However, the association with disease susceptibility becomes even more striking in an analysis of these MHC alleles at a genetic level. Patients with the DR4 serotype almost all have the unusual allele DRB1∗0402, and patients with the DQ1 serotype almost all have the rare allele DQB1∗0503. Similar, but less restricted, HLA-DR alleles are associated with PF.162 The protein chains encoded by these PV MHC II alleles vary from those found in HLA-DR4 and DQ1 controls without disease by only a few amino acids. Other studies have confirmed that the immune response in pemphigus is restricted to certain desmoglein peptides and MHC class II alleles.163-166
MHC class II alleles encode cell surface molecules that are necessary for antigen presentation to the immune system; therefore, it is hypothesized that PVassociated MHC class II molecules facilitate presentation of desmoglein 3 peptides to T cells.167 Consistent with this hypothesis, certain peptides from desmoglein 3, predicted to fit into the DRB1∗0402 peptidebinding pocket, were found to stimulate T cells from patients.168 More direct proof of the importance of the
917
9
Desmoglein (DSG) compensation
Pemphigus foliaceus
Skin
Mucous membrane
1
anti-Dsg1
3 3 1
Pemphigus vulgaris
Skin Mucous membrane
1
anti-Dsg3 only
1 3 3
Skin Mucous membrane
1
3
1
anti-Dsg3 + anti-Dsg1
3
918
DRB1∗0402 allele comes from a study of a humanized HLA-transgenic mouse for this allele in which presentation of the immunodominant Dsg3 peptides to human T cells results in production of pemphigus antibodies and the epidermal pathology of PV.169
Expression of Dsg3 in the thymus, mediated by the Aire transcription factor, promotes tolerance of CD4 T cells to stimulation by Dsg3.170 An unexpected observation was that T cells of normal people with the DRB1∗0402 or DQB1∗0503 respond just as well as those of pemphigus patients to the same desmoglein 3 peptides,167,171 indicating that T-cell reactivity to desmoglein 3 peptides is not sufficient for disease onset. Dsgspecific CD4+ T cells provide help to simulate anti-Dsg B cells to produce antibodies.172 In this context, CD4+ T follicular helper cells are thought to be particularly important. Concordantly, there are increased T follicular helper cells with their associated cytokine, IL-27, in PV patients.173 Another factor that may determine who gets pemphigus and who does not has been proposed to be the presence of regulatory T cells that can suppress the autoimmune response in those who do not.174 Likely, multiple factors prevent T-cell Dsg responsiveness and subsequent help for B-cell anti-Dsg antibody production. Cloning of anti-desmoglein B-cell repertoires from PV and PF patients has elucidated the diversity of the antibody variable region genes contributing to the pemphigus immune response.128,129,175 Shared VH1-46 gene usage has been identified in anti-Dsg3 B cells among PV patients, probably because antibodies using this gene require few or no mutations to bind Dsg3, which may favor the selection of VH1-46 B cells early in the autoimmune response.176 However, multiple other VH genes have been identified in anti-Dsg3 antibodies among PV patients that are more highly mutated and may not be shared,128,177-179 although anti-Dsg antibodies from different PV patients have been shown to bind at or near common epitopes on desmoglein 3.177 Analysis of anti-Dsg1 IgG B-cell repertoires in fogo selvagem patients has identified enrichment for VH3-23 heavy chain gene usage, as well as IGKV1D-39 light chain gene usage, with evidence of shared mutations as well as an increased number of somatic mutations in the antigen-binding regions of Dsg1-reactive antibodies, suggesting antigen-driven selective pressure.180
Cross-reactivity in the B-cell repertoire to foreign and self-antigen has been proposed as a mechanism for the onset of autoimmunity in both PV and PF. In addition to the autoantibody cross-reactivity to LJM11 sand fly antigen and Dsg1 discussed previously,16 Dsg3 autoantibodies in PV have been shown to cross-react with rotavirus VP6 coat protein, and VH1-46 mAbs that both induce suprabasal blisters and inhibit rotavirus infectivity have been identified.176 Unmutated IgM VH1-46 B cells have a propensity to bind both rotavirus VP6 coat protein,181 as well as the PV autoantigen Dsg3.176 However, cross-reactivity to VP6 and Dsg3 in the IgG compartment is rare because of the differing nature of somatic mutations that confer VP6 versus Dsg3 reactivity, thus preventing the onset of pemphigus after rotavirus exposure.
9
To produce anti-Dsg autoantibodies, B cells must lose tolerance to Dsg, which is a self-antigen. Studies of cloned anti-Dsg B cells have shown that throughout the course of PV, over the years, the same clones of anti-Dsg B cells persist and new ones generally are not produced.182 These data suggest that some event causes a time-limited loss of tolerance of B cells to Dsg, and if these B-cell clones can be completely eliminated, patients might be cured, thus explaining the rationale for B-cell depletion therapy in pemphigus.
DIAGNOSIS
Diagnosis of pemphigus relies on skin biopsy of a fresh lesion for histology to determine the site of blister formation, as well as a confirmatory immunochemical study to document the presence of skin autoantibodies, either by direct immunofluorescence of perilesional skin, or indirect immunofluorescence or ELISA of patient serum.
LABORATORY TESTING
LABORATORY TESTING
IMMUNOFLUORESCENCE
The hallmark of pemphigus is the finding of immunoglobulin G (IgG) autoantibodies against the cell surface of keratinocytes. These autoantibodies were first discovered in patients’ sera by indirect immunofluorescence techniques and soon thereafter were discovered by direct immunofluorescence of patients’ skin.183
Direct Immunofluorescence: Essentially all patients with active PV or PF have a positive finding on a direct immunofluorescence study, which tests for IgG bound to the cell surface of keratinocytes in perilesional skin (Fig. 52-6A, Table 52-2).184 This is a non-quantitative test (either negative or positive). The diagnosis of pemphigus should be seriously questioned if the test result of direct immunofluorescence is negative. It is important that the biopsy for direct immunofluorescence be performed on normal-appearing perilesional skin, as the immune reactants can be difficult to detect in blistered inflamed epidermis (leading to a false negative result). In some cases of pemphigus erythematosus, IgG and C3 are deposited at the basement membrane zone of erythematous facial skin, in addition to the epidermal cell surface IgG, representing a positive lupus band test in addition to the typical pemphigus intercellular pattern.185 In at least some cases, this band may be the result of UV-induced cleavage of Dsg1 with its accumulation at the basement membrane.56
Indirect Immunofluorescence: Indirect immunofluorescence is performed by incubating serial dilutions of patients’ sera with epithelial substrates. It is reported as a semiquantitative titer (indicating the last dilution at which the serum demonstrates a positive cell surface staining pattern). The test is offered by most major national laboratories and can remain positive for
919
9
A B
weeks to months after healing of skin lesions, making it a good diagnostic test if a patient should present with no active skin lesions, for example due to empiric treatment with prednisone by a referring physician. Depending on the substrate used for indirect immunofluorescence, more than 80% of patients with pemphigus have circulating anti–epithelial cell surface IgG (Fig. 52-6B, Table 52-2).186 The substrate used to detect pemphigus antibody binding in indirect immunofluorescence greatly influences the sensitivity of the test. In general, monkey esophagus is more sensitive for detecting PV antibodies, and guinea pig esophagus or normal human skin is a superior substrate for detecting PF antibodies. Patients with early localized disease and those in remission are most likely to have negative findings on an indirect immunofluorescence test; for these patients, the increased sensitivity of ELISA may help in diagnosis (see below). Patients with PV and PF usually display similar direct and indirect immunofluorescence findings with IgG on the cell surface of epidermal cells throughout the epidermis, despite the different autoantigen profiles in these 2 diseases. Therefore, it is usually not possible to differentiate the 2 diseases by the pattern of immunofluorescence. There is a positive, but imperfect, correlation between the titer of circulating anti–cell surface antibody and the disease activity in PV and in PF.187 Although this correlation may hold in general, and although patients in remission often show serologic remission with negative direct and indirect immunofluorescence findings,188,189 disease activity in individual patients does not necessarily correlate with indirect immunofluorescence titer. Therefore, in the day-to-day management of these patients, following disease activity is more important than following antibody titer.
ENZYME-LINKED IMMUNOSORBENT ASSAY
For diagnosis of disease, antigen-specific ELISAs have been shown to be more sensitive and specific
920
than immunofluorescence, and their titer correlates better than that of indirect immunofluorescence with disease activity.92,190,191 Additionally, ELISAs are easier to perform and less subjective than immunofluorescence, and have for many physicians replaced the latter as the preferred first diagnostic test for pemphigus (Table 52-2). These assays use desmogleins 1 and 3 bound to plates, which are then incubated with patient sera and developed with anti-human IgG reagents (Fig. 52-7). As an advantage over indirect
Enzyme-linked immunosorbent assay (ELISA) for desmoglein 3
Irrelevant antibodies
αDsg3
Dsg3
HRP-anti-human IgG
HRP-substrate
immunofluorescence, ELISAs can help differentiate between PV and PF because of the different autoantigen profiles in these 2 diseases.92,190 In most cases, ELISA is positive for desmoglein 3 (but not desmoglein 1) in mucosal PV, is positive for both desmogleins 3 and 1 in PV with both mucosal and significant skin involvement, and is positive for only desmoglein 1 in PF. PV has rarely evolved into PF, and vice versa, as determined by clinical, histologic, and immunochemical criteria.192-194 A small minority of PF patients may also demonstrate autoantibodies to desmoglein 3195; therefore, diagnosis should be made based on the clinicalserologic correlation. Additionally, some patients (eg, those with bullous pemphigoid) may demonstrate a low level of anti–desmoglein 3 autoantibodies,190
which are detectable because of the high sensitivity of the ELISA. Therefore, a result in the indeterminate range should be interpreted carefully, as this may represent a true positive or a false positive, the latter presumably because of formation of nonpathogenic bystander autoantibodies after epidermal damage. As with indirect immunofluorescence, the correlation of ELISA index value with disease activity is not perfect. For example, although ELISA titers correlate with clinical activity, patients in clinical remission (often still on corticosteroid therapy) whose titers have dropped from peak values may still have positive results.196 In making treatment decisions, a negative result on desmoglein ELISA is more helpful than a positive result, as a patient with the former is more likely to achieve remission off immunosuppressives, whereas a patient with the latter may or may not. In other words, disease activity is the mainstay for determining treatment.
PATHOLOGY
The characteristic histopathologic finding in PV is a suprabasal blister with acantholysis (Fig. 52-8, Table 52-2). Just above the basal cell layer, epidermal cells lose their normal cell-to-cell contacts and form a blister. Often, a few rounded up (acantholytic) keratinocytes are in the blister cavity. The basal cells stay attached to the basement membrane, but may lose the contact with their neighbors; as a result, they may
9
appear to be a “row of tombstones,” symbolic of the potentially fatal prognosis of this disease. Usually, the upper epidermis (from 1 or 2 cell layers above the basal cells) remains intact, as these cells maintain their cell adhesion. Pemphigus vegetans shows not only suprabasal acantholysis but also papillomatosis of the dermal papillae and downward growth of epidermal stands into the dermis, with hyperkeratosis and scalecrust formation. In addition, pemphigus vegetans lesions may show intraepidermal abscesses composed of eosinophils and/or neutrophils.197 Early PV lesions may show eosinophilic spongiosis.198
The histopathology of early blisters in PF patients demonstrates acantholysis (loss of cell-to-cell contact) just below the stratum corneum and in the granular layer (Fig. 52-9A). The stratum corneum is often lost from the surface of these lesions. The deeper epidermis, below the granular layer, remains intact. Another frequent finding is subcorneal pustules, with neutrophils and acantholytic epidermal cells in the blister cavity (Fig. 52-9B). Histologic findings in PF are often indistinguishable from those seen in bullous impetigo/staphylococcal scalded skin syndrome, because blisters in these latter diseases also result from dysfunction of desmoglein 1, in these cases due to proteolytic cleavage by Staphylococcal exfoliative toxins.87
Therefore, immunochemical studies are essential to confirm a diagnosis of PF, as these would be negative in Staphylococcal-mediated skin blisters. The site of
A
B
921
9
blister formation in pemphigus erythematosus is identical to PF. As in PV lesions, very early PF lesions may show eosinophilic spongiosis.198
DIFFERENTIAL DIAGNOSIS
See Table 52-1.
CLINICAL COURSE AND PROGNOSIS
Before the advent of glucocorticoid therapy, PV was almost invariably fatal due to severe blistering of the skin and mucous membranes, leading to malnutrition, dehydration, and sepsis. PF was fatal in approximately 60% of patients. PF was almost always fatal in elderly patients with concurrent medical problems; however, in other patients, its prognosis, without therapy, was much better than PV.199,200
The systemic administration of glucocorticoids and the use of immunosuppressive therapy have dramatically improved the prognosis for patients with pemphigus; however, pemphigus is still a disease associated with a significant morbidity and mortality.201,202 In the United States, the annual mortality rate from pemphigus (age-adjusted to the standard population) is estimated to be 0.023 deaths per 100,000.203 The inpatient mortality from pemphigus has been estimated at 1.6% to 3.2%, with increased mortality likely due to numerous comorbid health conditions.79 The risk of death in pemphigus vulgaris patients is 2.36 to 3.3 times greater than for controls in the United Kingdom and Taiwan.9,11
Infection is often the cause of death, and by causing the immunosuppression necessary in the treatment of active disease, therapy is frequently a contributing factor.11,204 With glucocorticoid and oral immunosuppressive therapy, the mortality (from disease or therapy) of PV patients followed from 4 to 10 years is approximately 10% or less, whereas that of PF is probably even less. In a study of 40 patients with PV, 2 patients (5%) died of sepsis and 17%, after an average of 18 months of therapy, went into a complete and longlasting (>4 years, average, thought to be permanent) remission requiring no further therapy.205 Another 37% of patients achieved remission but relapsed at times after therapy was stopped; most of these also eventually achieved long-lasting remissions. The remainder of patients required continual therapy. In a group of 159 patients with PV from Croatia, only approximately 12% went into long-term remission after therapy with glucocorticoids and immunosuppressives, but most relapsed.23 In a study from Tehran of 1206 pemphigus patients seen over 20 years, 6.2% of PV and 0.2% of PF patients died, mostly of septicemia; only 9.3% were in complete remission without therapy.7 Another study showed that; with the use of corticosteroids, about 50% of patients went into at least transient complete remission off therapy after a mean of 3 years.206 Immunosuppressive adjuvant therapy may reduce the risk
922
of relapse.207 With the advent of rituximab therapy, complete remission in pemphigus has become more common and is quicker to achieve. Approximately 50-90% of PV patients go into remission off all other immunosuppressive therapy, after 1 or more courses of rituximab.208 Similar remission rates are found in PF.209
MANAGEMENT
Several consensus guidelines for pemphigus disease therapy have been published.211-214 It is generally agreed that PV, even if initially limited in extent, should be treated at its onset, because it will ultimately generalize and the prognosis without therapy is very poor. In addition, it is probably easier to control early disease than widespread disease, and mortality may be higher if therapy is delayed.215 Because PF may be localized for many years, and the prognosis without systemic therapy may be good, patients with this type of pemphigus do not necessarily require treatment with systemic therapy; the use of topical corticosteroids may suffice. When the disease is active and widespread, however, the therapy for PF is, in general, similar to that for PV. A consensus statement on disease definitions and endpoints was proposed by an international committee of pemphigus experts.216 Additionally, clinical instruments have been developed for tracking disease activity that have been shown to be reliable and valid.217,218 The standardization of disease definitions and activity scoring will facilitate future clinical trials for pemphigus. There has been a tremendous advance in the armamentarium of therapies for pemphigus since the time before the development of glucocorticoids when PV was a fatal disease. Thanks to these advances, the “row of tombstones” seen in the pathology of PV no longer alludes to its prognosis for most patients.
CORTICOSTEROIDS
CORTICOSTEROIDS
The systemic administration of glucocorticoids, usually prednisone, remains the mainstay of therapy for pemphigus. Before adjuvant immunosuppressive therapy was available, very high initial doses of prednisone (>2.0 mg/kg/d) were used for treatment, although such regimens have retrospectively been associated with significant morbidity and mortality from therapy.204,219,220 The full systemic dose of glucocorticoids has been defined in the consensus guidelines as 1.5 mg/kg/d of prednisone equivalent for 2 to 3 weeks.216 However, many patients can be brought under control with a 0.5- to 1.0-mg/kg/d single daily dose, especially if used in combination with adjunctive immunosuppressive therapy, which is thought to result in fewer complications and decreased mortality as compared to higher-dose glucocorticoid regimens.221,222 For patients who do not initially respond or worsen, splitting the dose using a twice- or 3-timesdaily schedule may achieve disease control.
Once disease activity is controlled, tapering prednisone to as low a dose as possible should be the goal. Minimal therapy is defined as 5 to 10 mg daily of prednisone equivalent. Although there are no set guidelines, if disease activity can be fully controlled on minimal-dose prednisone or lower, then glucocorticoid monotherapy may be feasible depending on the patient’s other comorbidities and contraindications to alternative immunosuppressive agents. If patients have continued relapses, with daily prednisone doses approaching or exceeding 5 to 10 mg, adjunctive immunosuppressive agents are warranted. Interestingly, prednisone can control blistering within days, at a time when the autoantibody titer would be unchanged. A possible explanation is that prednisone increases the synthesis of desmogleins or other cell-adhesion molecules or change their posttranscriptional processing to prolong their half-life.223,224 If pemphigus IgG depletes desmosomes of desmogleins, as discussed above, then prednisone could counteract this effect. Topical corticosteroids may be used as monotherapy in mild forms of disease, especially PF, or as adjunctive therapy to help heal new lesions. Patients with mucosal disease may benefit from the use of glucocorticoid elixirs as a swish and spit for dental trays to help apply class I corticosteroid gels or ointments to the gingiva. Additionally, class I-IV corticosteroids can be used as topical therapy to help resolve new blisters, even in patients on systemic glucocorticoids.
RITUXIMAB
RITUXIMAB
A very effective therapy for pemphigus, even in cases refractory to standard immunosuppressive therapy, is a monoclonal anti-CD20 antibody, rituximab, which was approved by the FDA for therapy of pemphigus vulgaris in 2018. Rituximab targets B cells, the precursors of antibody-producing plasmablasts. The B cell also acts to process autoantigen and present it to T cells that provide “help” in stimulating the autoantibody response.225 Rituximab is infused intravenously at a dose of 1000 mg on day 1 and day 15. A maintenance dose of 500 mg can be infused at 12 months and every 6 months thereafter based on clinical evaluation, or a 1000 mg dose if clinical relapse occurs. A pivotal clinical trial comparing first line rituximab therapy plus short-term prednisone (0.5-1.0 mg/kg/day) to high dose (1.5 mg/kg/day) prednisone alone for moderate to severe pemphigus showed superior rates of complete remission off prednisone in the rituximab-treated group (89% versus 34%), although maintenance dosing appears to be required to prevent disease relapse. Although not the FDA-approved dose, a lymphoma dose regimen of 375 mg/m2 once weekly for four weeks has also been evaluated in a prospective clinical trial.226 A single cycle of rituximab using the lymphoma dose has been shown to be effective even in relapsed and refractory pemphigus, with 86% of patients experiencing complete healing of skin lesions on or off
9
prednisone.226 Approximately 80% of patients relapse and require additional therapy to regain disease control,209 but with repeat cycles of rituximab and/or additional prednisone therapy after clinical relapse, 48% of PV and PF patients, including those with previously refractory disease, are ultimately able to achieve complete remission off prednisone. Disease activity usually begins to remit within 3 months of rituximab infusion; with first-line rituximab therapy, the median time to complete remission off steroids is 9 months. Rituximab has also been shown to be an effective therapy for children with refractory pemphigus.227-229
Therapy with rituximab, as with other immunosuppressive treatments, has significant risks.230 37% of PV patients treated with rituximab plus prednisone experienced treatment-related infections, similar to the rate (42%) observed in patients treated with high-dose prednisone alone. Grade 3 or higher infectious adverse events requiring hospitalization were observed in 8% of rituximab and prednisone-treated patients, compared to 3% treated with prednisone alone. Fatal infections with rituximab and prednisone therapy have been observed with chronic rituximab therapy, including sepsis, Pneumocystis pneumonia, reactivation of hepatitis B, and JC virus infection reactivation causing progressive multifocal leukoencephalopathy.209,231-233 In addition, 56% of PV patients treated with rituximab develop anti-drug antibodies. Although the clinical significance of this immunogenicity is currently unclear, one PV patient was reported to develop neutralizing anti-chimeric antibodies to rituximab, associated with infusion reactions and lack of clinical efficacy.234
ORAL IMMUNOSUPPRESSIVE AGENTS
ORAL
IMMUNOSUPPRESSIVE
AGENTS
As rituximab is not accessible to all pemphigus patients, many experts still use oral immunosuppressive therapy, usually with, but also without, prednisone, from the beginning of therapy. Certainly, when continued doses of glucocorticoids greater than 5 to 10 mg are required for disease control, or if there are contraindications to or intolerable side effects from oral glucocorticoids, adjunctive immunosuppressive agents are used for pemphigus therapy. Prospective randomized studies have shown that immunosuppressive agents such as mycophenolate mofetil and azathioprine have a steroid-sparing effect; retrospective studies suggest decreased mortality with use of adjuvants plus steroids compared to steroids alone.199,235-238
Because patients may die from complications of therapy, it is important to monitor all patients closely for potential side effects, such as blood count, liver and kidney laboratory abnormalities, GI ulcer disease, high blood pressure, diabetes, glaucoma, cataracts, osteoporosis, and infection. The decision to use immunosuppressive agents, particularly in young patients, must also take into account the potential incidence of malignancies that
923
9
might be associated with the long-term use of these drugs, as well as the risks of teratogenicity (for mycophenolate mofetil, azathioprine, and methotrexate). Use of mycophenolate in women of child-bearing potential requires counseling and monitoring regarding pregnancy.
AZATHIOPRINE
Azathioprine is an effective adjunctive immunosuppressive agent for pemphigus, with clinical remission rates of approximately 50% in retrospective studies.189,229 In a prospective randomized trial of high-dose methylprednisolone (2.0 mg/kg/d) plus azathioprine (2.0 mg/kg/d), 72% of patients achieved clinical remission within a mean of 74 days, although 33% experienced significant adverse effects of therapy, including hyperglycemia, dizziness, abnormal liver enzyme tests, and infection.235
Azathioprine is a prodrug, which is converted to active mercaptopurine, thioguanine, and thioinosine metabolites, in part by thiopurine methyltransferase (TPMT), an enzyme whose levels can vary widely in the population. Overall, 89% of whites demonstrate normal to high levels of TPMT, 11% are intermediate, and 0.3% are deficient for TPMT, the latter group representing those who do not tolerate azathioprine therapy.240 Additionally, 1% to 2% of whites may have “super high” levels of TPMT, which is correlated with both treatment resistance and increased hepatotoxicity from excessive metabolite production.241 Azathioprine toxicity has also been associated with genetic polymorphisms in nucleotide triphosphate diphosphatase (NUDT15), particularly in Asian populations, and inosine triphosphate pyrophosphatase (ITPA).242 Altogether, it is estimated that approximately 5% of patients will be azathioprine intolerant.243
In patients with normal TPMT levels, the consensus dosing regimen that defines treatment failure is 2.5 mg/kg/d for 12 weeks.216 From a practical standpoint, however, not all laboratories offer TPMT testing. Additionally, because patients with normal levels of TPMT may also experience azathioprine toxicity, it is reasonable to start all patients at a lower dose (eg, 50 to 100 mg daily) and titrate upward until clinical remission, the target dose of 2.5 mg/kg/d, or unacceptable side effects result. Frequent blood and liver monitoring should continue, particularly over the first 8 to 12 weeks when delayed toxicity from the accumulation of metabolites may emerge.
MYCOPHENOLATE MOFETIL
Mycophenolate mofetil is also an effective steroidsparing agent for pemphigus. Typical doses range from 30 to 40 mg/kg/d (maximum dose 3 g/d) dosed twice daily (2.0 to 3.0 g/d), although certain patients such as the elderly may achieve disease control with doses as low as 1.0 g/d. In case series, mycophenolate mofetil has been shown to have a rapid effect in lowering pemphigus antibody titers and decreasing disease activity,
924
even in patients whose disease is unresponsive to azathioprine.244,245 A prospective randomized trial comparing methylprednisolone (2 mg/kg/d) with azathioprine (2 mg/kg/d) or mycophenolate mofetil (2.0 g/d) in pemphigus patients showed 72% in the azathioprine group and 95% in the mycophenolate mofetil group went in clinical remission in a mean of 74 and 91 days, respectively.235 Nineteen percent of patients experienced significant side effects of mycophenolate mofetil therapy, compared to 33% in the azathioprine group. None of these differences were statistically significant. Another trial showed that mycophenolate mofetil adjuvant therapy decreased the time to initial response and increased the duration of response to therapy and tended to decrease prednisone dosage, although the latter was not statistically significant.238 A prospective randomized study indicated that azathioprine was significantly more effective than mycophenolate mofetil as a steroid-sparing agent, although this study compared a full dose of azathioprine (2.5 mg/kg/d) to a partial dose of mycophenolate mofetil (2.0 g/d).224
Mycophenolate mofetil also has been reported to be an effective therapy for pediatric PV.246
Caution with use of mycophenolate mofetil is warranted, as fatal infection and sepsis occurred in 2% to 5% of transplant patients receiving mycophenolate mofetil, and increased risk of infection with or reactivation of cytomegalovirus, herpes zoster, atypical mycobacteria, tuberculosis, and JC virus (in progressive multifocal leukoencephalopathy) have been noted in postmarketing surveillance.247 Interestingly, mycophenolate mofetil may offer protection against Pneumocystis carinii infection.248
METHOTREXATE
Once-a-week use of methotrexate is another option for an immunosuppressive therapy in pemphigus, although it is not used as frequently as azathioprine or mycophenolate mofetil.249
INTRAVENOUS IMMUNOGLOBULIN
INTRAVENOUS
IMMUNOGLOBULIN
Another method of decreasing serum autoantibodies is the intravenous use of γ-globulin (IVIg) in high doses. IVIg is thought to function by saturating neonatal Fc receptor, thereby increasing catabolism of the patient’s serum antibodies, which include the pathogenic autoantibodies.250-252 It may be useful as adjuvant therapy in those pemphigus patients whose condition does not respond to more conventional therapy.253,254 A multicenter, randomized, placebo-controlled, double blind study has confirmed its efficacy in pemphigus,255
but it is expensive and probably requires continued infusions for maintenance of remission. There also can be significant side effects with this therapy, including stroke, deep venous thrombosis, and aseptic meningitis.256 Some centers will use IVIg to establish
initial control of blistering in severely affected patients because it does not increase the risk of infection as do corticosteroids and immunosuppressants. IVIg also has been used in combination with rituximab,257,258
although it is unclear whether the combination is safer or more effective compared to either alone.
OTHER THERAPIES
OTHER THERAPIES
There are additional therapies that can be used as adjuncts to standard treatments, or have historically been used for severe or refractory cases of pemphigus. Cyclophosphamide, although more toxic than azathioprine, mycophenolate mofetil, or rituximab, is thought to be very effective in controlling severe disease, with one report of 19 of 23 patients with pemphigus achieving complete remission in a median time of 8.5 months.259 A variety of small case series have evaluated different cyclophosphamide regimens for pemphigus, including daily oral therapy (1.1-2.5 mg/kg/d), daily oral therapy (50 mg) with intermittent high-dose intravenous dexamethasone and cyclophosphamide, and immunoablative intravenous cyclophosphamide.239,260-263 All methods were effective in the short term, although none were curative. Significant side effects, including hematuria, infection, and transitional cell carcinoma of the bladder, were observed with higher dose regimens, although one study using a lower daily dose of cyclophosphamide (1.0-1.5 mg/kg/d) did not report a significantly different safety profile compared with other immunosuppressive agents. Together with the risk of infertility, cyclophosphamide is not considered a firstline steroid-sparing agent in the treatment of PV. In a case series and randomized double-blind trial, dapsone demonstrated a trend toward efficacy as a steroid-sparing drug in maintenance phase PV, although these results were not statistically significant.264,265 Dapsone may be used in conjunction with other immunosuppressive agents, particularly rituximab, where it offers the additional benefit of Pneumocystis pneumonia prophylaxis. Plasmapheresis is sometimes used for severe pemphigus, or for pemphigus that is unresponsive to a combination of prednisone and immunosuppressive agents. Although one controlled study found it to be ineffective,266 other studies have found that it both reduces serum levels of pemphigus autoantibodies and controls disease activity.267 Plasmapheresis plus intravenous pulse therapy with cyclophosphamide has been reported to result in remissions of PV.268 For maximum effectiveness, it is necessary to have patients take immunosuppressive agents to prevent the antibodyrebound phenomenon that can follow the removal of IgG. Protein A immunoadsorption, which removes IgG selectively from plasma, also has been used.269
Intravenous, pulse administration of methylprednisolone 250 to 1000 mg given over approximately 3 hours daily for 4 to 5 consecutive days, can result in long-term remissions and decrease the total dose
9
of glucocorticoids necessary to control disease.270
Although the purpose of this therapy is to decrease the incidence of complications of long-term steroid use, it can result in all the usual glucocorticoid complications, as well as cardiac arrhythmias with sudden death, and its use is controversial.271 Furthermore, a controlled trial found that adjuvant oral dexamethasone pulse therapy in addition to standard therapy with prednisolone and azathioprine for PV is not beneficial.272 It may be that simply giving divided lower doses of prednisone could accomplish the same result with fewer side effects.
PERSPECTIVES FOR FUTURE THERAPEUTIC STRATEGIES
PERSPECTIVES FOR FUTURE
THERAPEUTIC STRATEGIES
Given the well-defined nature of disease in pemphigus and unmet need for safe and effective therapies, clinical trials in pemphigus are increasing. As discussed above, inhibitory anti-chimeric antibodies can cause resistance to rituximab. Fc gamma receptor polymorphisms, which can cause decreased antibody-dependent cellular cytotoxic killing of B lymphocytes after rituximab therapy, also have been proposed as a cause of rituximab resistance,273-276 although these polymorphisms may not always correlate with therapeutic outcome in patients with B-cell cancers.277,278 Newer, fully humanized or glycoengineered anti-CD20 antibodies may be useful in these situations279,280, although none are currently in clinical development for pemphigus. PRN1008, an oral covalent inhibitor of the Bruton tyrosine kinase (Btk) required for mature peripheral B cell survival, has entered Phase 2 trials for pemphigus (NCT02704429). The successful clinical development of these B cell–targeted therapies may offer novel strategies for pemphigus treatment in the future. Agents that block the neonatal Fc receptor (FcRn) have also recently entered clinical trials in pemphigus, including SYNT001 (NCT03075904) and ARGX-113 (NCT03334058). FcRn maintains the serum half-life of IgG and regulates cross-presentation of immune complexes, among other immune functions. Additionally, a phase 1 study (NCT03239470) has been initiated to evaluate the safety and preliminary efficacy of ex vivo expansion and infusion of autologous polyclonal regulatory T cells, which are hypothesized to restore immune tolerance in pemphigus patients. Finally, a novel approach to targeted therapy for pemphigus, called chimeric autoantibody receptor T cell or CAAR- T therapy, has demonstrated preclinical efficacy in experimental PV models.281 CAAR-T therapy uses the disease autoantigen, Dsg3, as part of a chimeric immunoreceptor to program a patient’s own T cells to specifically kill Dsg-specific B cells, and offers the hope of a potentially lasting remission of disease without generalized immune suppression due to the potential for long-term CAART cell engraftment. Efforts are underway to determine the clinical efficacy of CAAR-T technology in pemphigus patients.
925
9
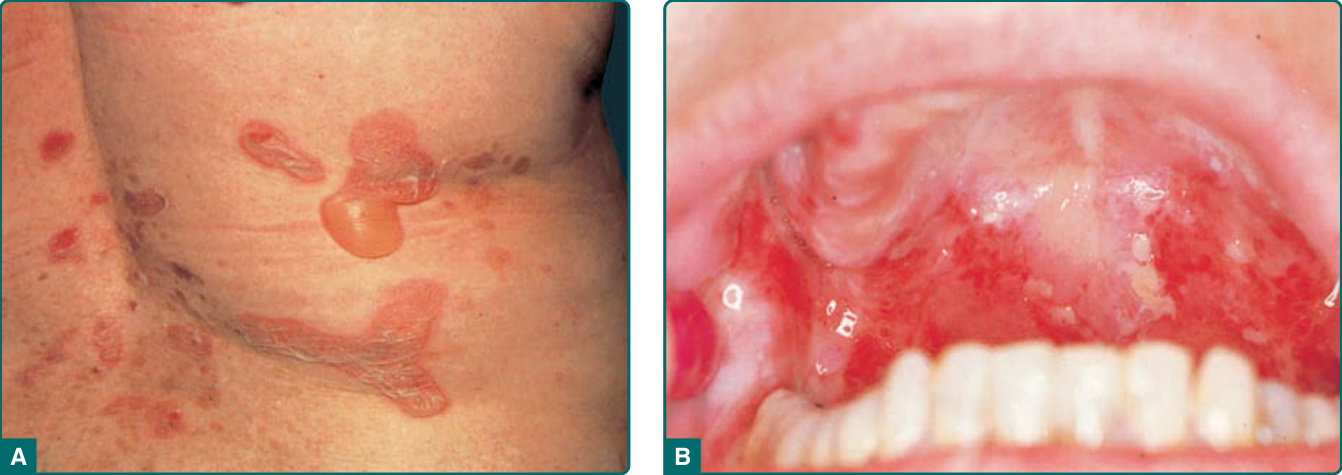
Figure 52-1 Pemphigus vulgaris. A, Flaccid blisters. (Used with permission from Lawrence Lieblich, MD.) B, Oral erosions.
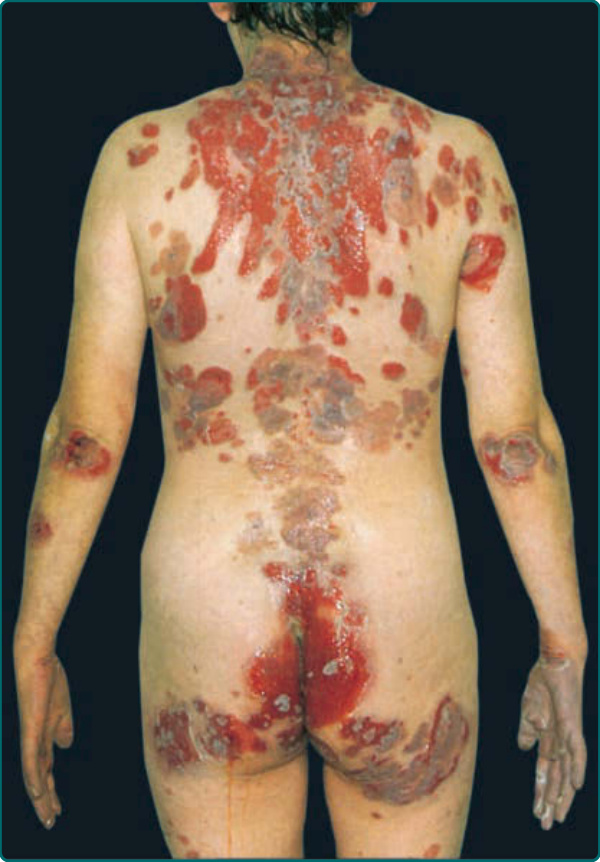
Figure 52-2 Pemphigus vulgaris. Extensive erosions due to blistering. Almost the entire back is denuded. Note intact, flaccid blisters at the lower border of eroded lesions.
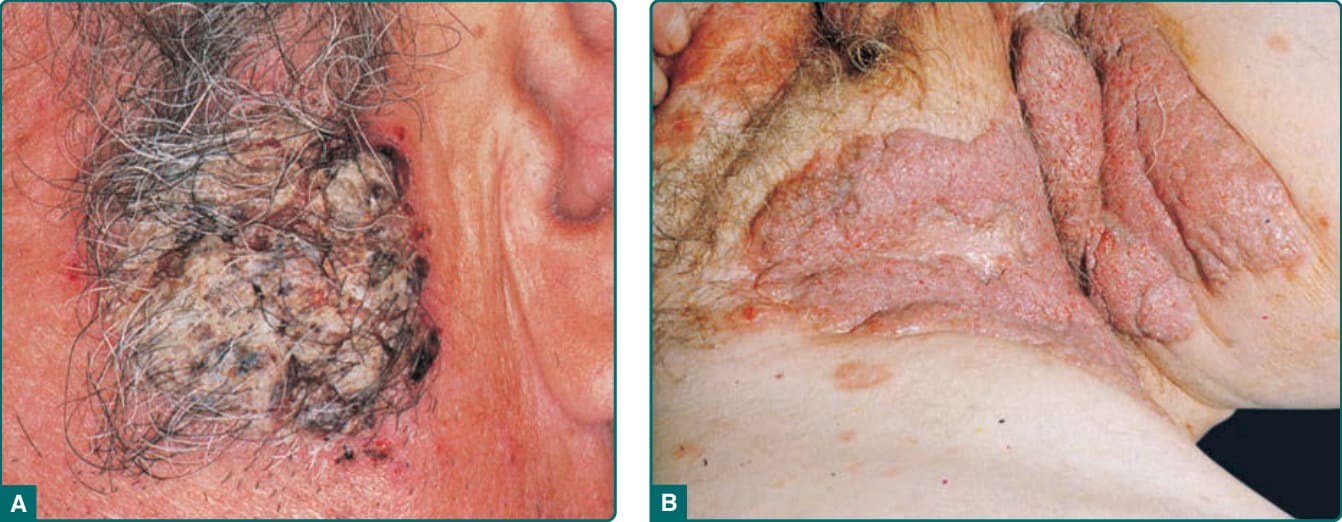
Figure 52-3 A, Crusted, vegetating lesions in pemphigus vulgaris. B, Extensive, vegetating lesions in intertriginous regions in pemphigus vegetans-type pemphigus vulgaris.
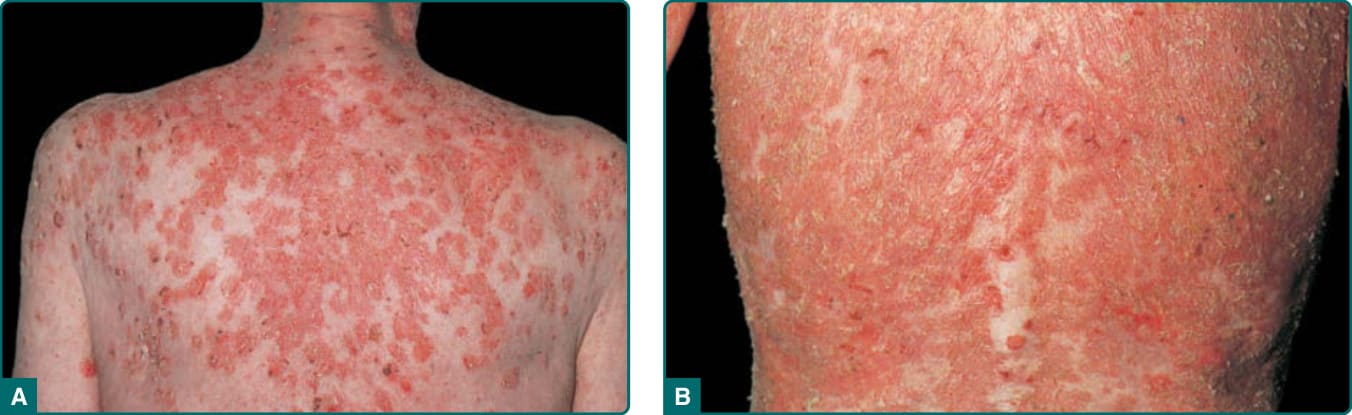
Figure 52-4 Pemphigus foliaceus. A. Scaly, crusted lesions on upper back. B. Exfoliative erythroderma due to confluent lesions.
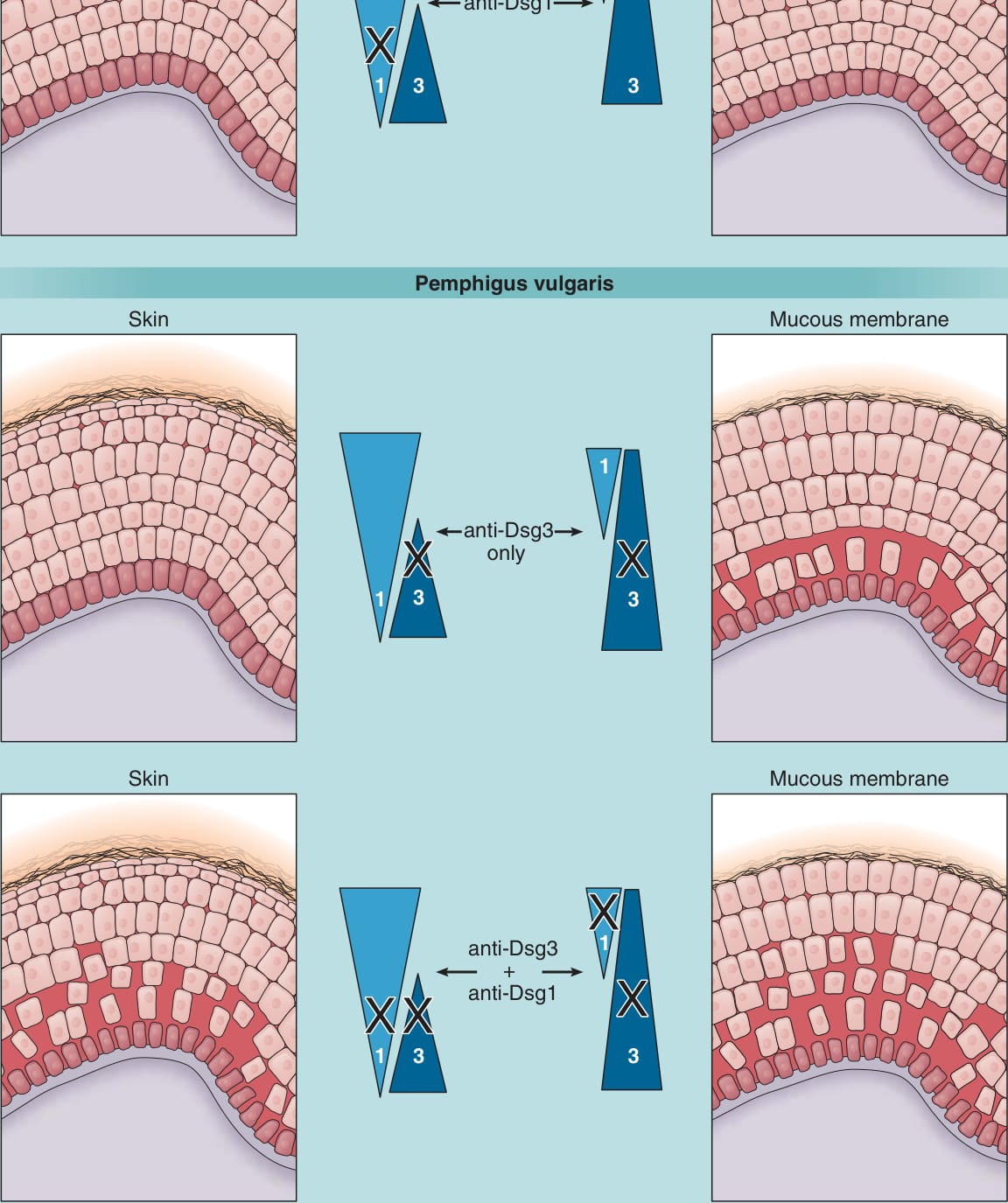
Figure 52-5 Desmoglein (Dsg) compensation. Triangles represent the distribution of Dsg1 and Dsg3 in skin and mucous membranes. Anti-Dsg1 antibodies in pemphigus foliaceus cause acantholysis only in the superficial epidermis of skin. In the deep epidermis and in mucous membranes, Dsg3 compensates for antibody-induced loss of function of Dsg1. In mucosal pemphigus vulgaris, antibodies against Dsg3 are predominant, which cause blisters only in the deep mucous membrane where Dsg3 is present without compensatory Dsg1. However, in mucocutaneous pemphigus, antibodies against both Dsg1 and Dsg3 are present, and blisters form in both mucous membrane and skin. The blister is deep probably because antibodies diffuse from the dermis and interfere first with the function of desmosomes at the base of the epidermis.
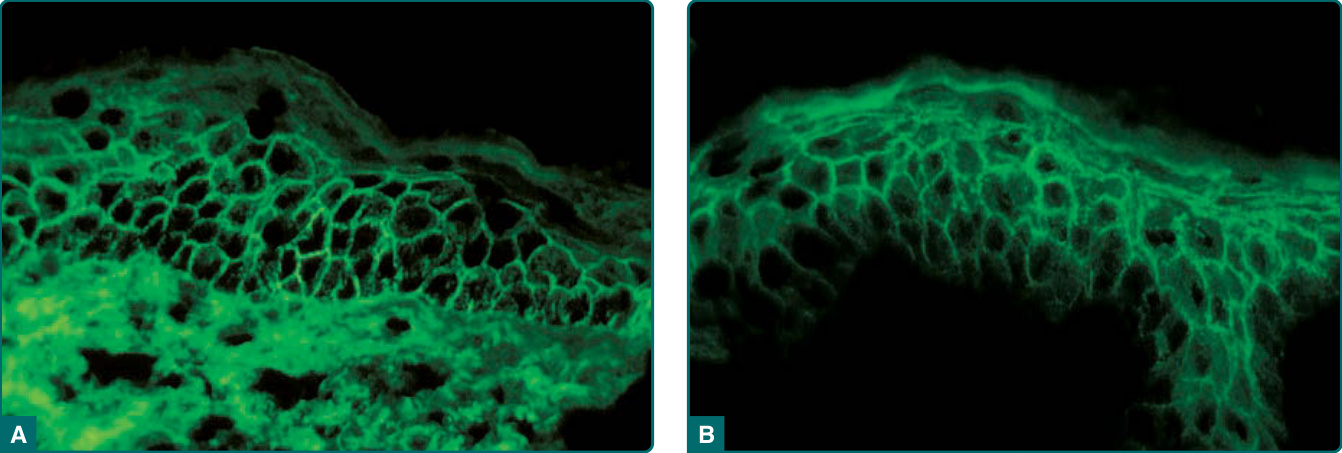
Figure 52-6 Immunofluorescence in pemphigus. A, Direct immunofluorescence for immunoglobulin G (IgG) of perilesional skin from a patient with pemphigus vulgaris. Cell surface staining is observed throughout the epidermis with a slight basal predominance. B, Indirect immunofluorescence with the serum from a patient with pemphigus foliaceus on normal human skin. IgG is observed on the cell surface throughout the epidermis with a slight superficial predominance.
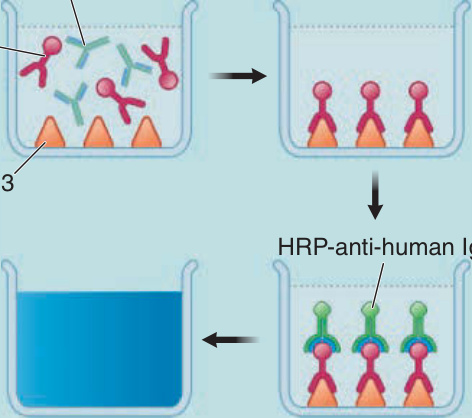
Figure 52-7 Enzyme-linked immunosorbent assay (ELISA) for desmoglein 3. Anti-Dsg3 antibodies (αDsg3) from pemphigus serum binds Dsg3 on the ELISA plate; irrelevant antibodies, that do not bind, are washed off. The plate is then incubated with horseradish peroxidase (HRP) conjugated anti-human IgG, which binds the anti-Dsg3 IgG that is on the plate. HRP is an enzyme that turns a clear substrate blue, and the amount of color, read on spectrophotometer, correlates with the amount of pemphigus (ie, anti-Dsg3) antibody in the patient’s serum.
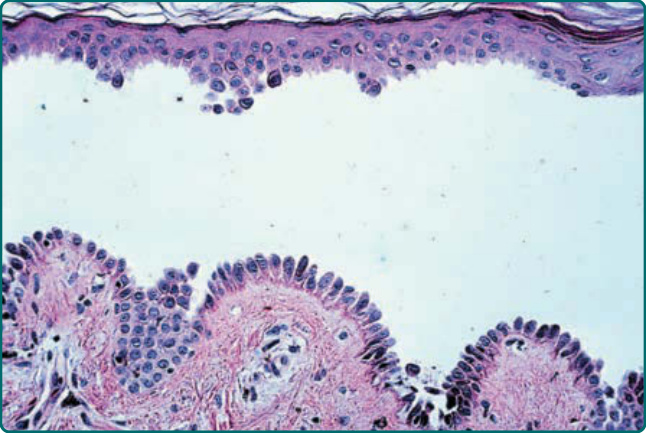
Figure 52-8 Histopathology of pemphigus vulgaris. Suprabasal acantholysis. The row of tombstones.
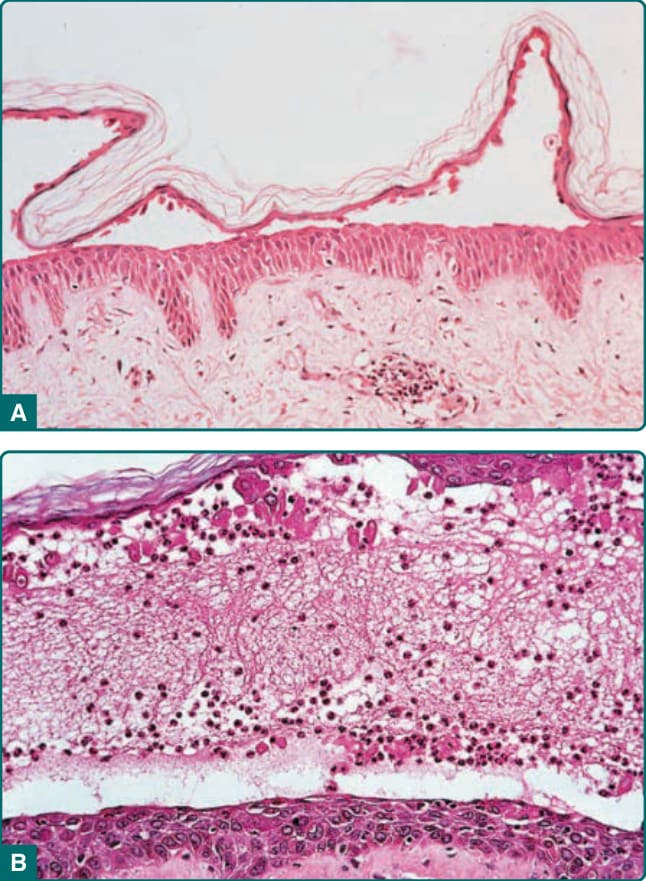
Figure 52-9 Histopathology of pemphigus foliaceus. A, Acantholysis in the granular layer. B, Subcorneal pustule with acantholysis.
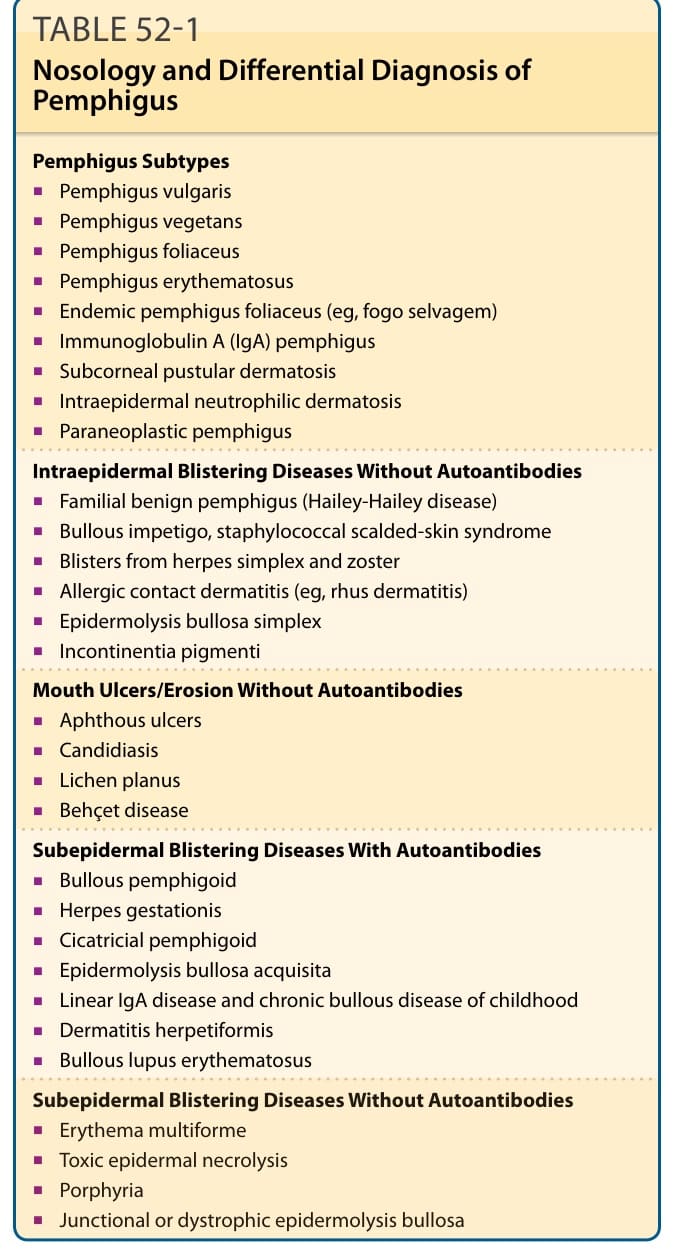
TABLE 52-1 Nosology and Differential Diagnosis of Pemphigus
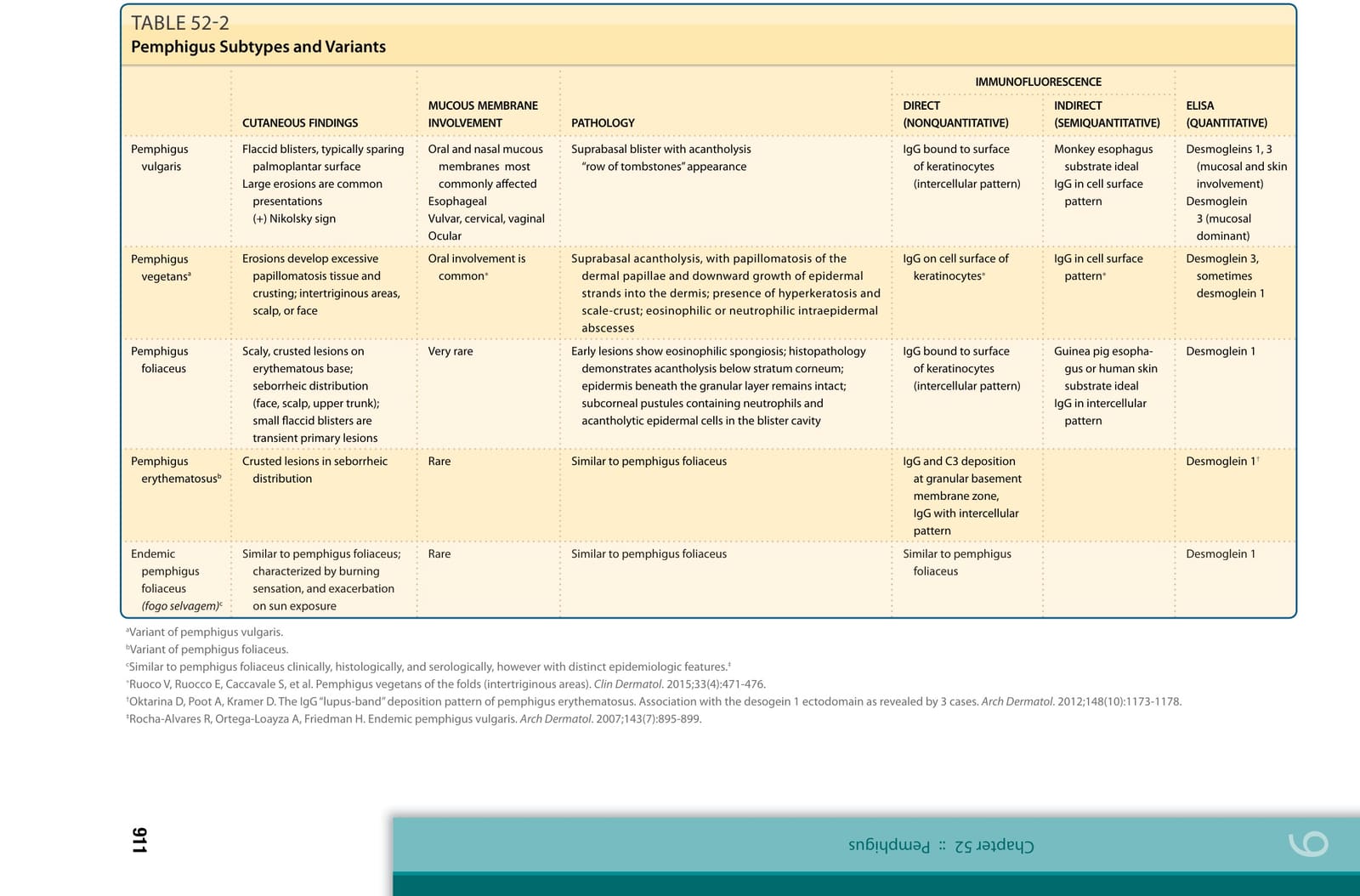
TABLE 52-2