汗孔角化症 (Porokeratosis)
定義與分類
- 汗孔角化症 (porokeratosis) 是型態上獨特的角化障礙,臨床特徵為過度角化的丘疹或斑塊,周圍環繞一條線狀隆起邊緣,並向外離心性擴展。
- 組織學上以圓錐狀板層 (cornoid lamella) 為標誌——一條薄柱狀角化不全細胞 (parakeratotic cells) 貫穿整個角質層 (stratum corneum),見於所有變異型,對應臨床上隆起的過度角化邊緣。
- 至少有六種臨床變異型;近期遺傳學進展可依潛在突變分類(表 51-1)。各型常共存或在同一家族出現,臨床次分類宜保有彈性。
- 遺傳上具異質性 (genetically heterogeneous),多數為體染色體顯性 (autosomal dominant) 遺傳。
流行病學
- 最常見型為播散性淺表性光化性汗孔角化症 (DSAP) 與 Mibelli 汗孔角化症 (PM);其餘罕見。
- 多見於膚色較淺者,深膚色者罕見。
- Mibelli 型與播散性掌蹠型 (porokeratosis palmaris et plantaris disseminata) 男性發生率為兩倍;DSAP 女性為兩倍;線狀型男女相等。
- 發病年齡:PM 多兒童期;DSAP/DSP 多第三至第四個十年;播散性掌蹠型與線狀型可見於出生至成年任何年齡。
臨床表現(各變異型)
- Mibelli 汗孔角化症 (PM):嬰兒/兒童期起,無症狀、細小、棕色至膚色的環狀丘疹,邊緣高度多 >1 mm 並帶縱向溝紋;中央可過度色素沉著、色素減退、凹陷、萎縮或無汗。直徑數毫米至數公分,偶見大至 20 cm 的巨大病灶(好發小腿與足,較大病灶惡性潛能較高)。病灶通常單側、局限,持續存在。
- 播散性淺表性光化性汗孔角化症 (DSAP):最常見型。一致細小、環狀、無症狀或輕癢的丘疹,直徑 2–5 mm,對稱分布於四肢、陽光暴露部位,常 >50 個病灶;通常不侵犯手掌、足底、黏膜。邊緣較 PM 不明顯,中央區隨進展變萎縮無汗。最早發病 7 歲,第三至第四個十年完全外顯。致病機轉仍未知。
- 播散性淺表性汗孔角化症 (DSP):型態同 DSAP,但不避開避光部位;可 >100 個病灶,好發伸側 (extensor surfaces)。DSAP 與 DSP 侵犯顏面皆罕見;兩播散型女男比約 2:1。
- 免疫抑制相關 DSP:見於腎/肝/心移植後、電子束照射、免疫抑制化療、單株抗體、合併血液惡性腫瘤之全身性皮質類固醇、HIV 感染,近期亦見於第 2 型糖尿病;亦曾於骨髓移植後無持續免疫抑制下發生。分布型態似 DSAP,但陽光暴露史較不明顯。
- 線狀汗孔角化症 (linear porokeratosis):不常見,漸被視為其他型的鑲嵌型 (mosaic) 表現。多兒童早期出現,常為沿 Blaschko 線單側分布;罕見廣泛型可波及數肢與軀幹。惡性退化潛能在所有型中最高。可由 DSAP 等位基因的雜合性缺失 (loss of heterozygosity) 解釋,為體染色體顯性疾病第 2 型節段性表現之例。
- 播散性掌蹠汗孔角化症:體染色體顯性遺傳性皮膚病,細小一致病灶先見於手掌足底,後擴及他處(含黏膜與非陽光暴露部位);掌蹠病灶較過度角化,縱向溝紋顯著。多青春期/成年早期出現,雙側對稱,男性為女性兩倍。
- 點狀汗孔角化症 (punctate porokeratosis):青春期/成年期出現,手掌足底多發微小離散的點狀過度角化病灶,邊緣薄而隆起;可呈線狀或聚成斑塊。須與點狀掌蹠角化症 (punctate keratoderma) 鑑別。
- CDAGS 症候群:罕見體染色體隱性 (autosomal recessive) 遺傳性皮膚病(顱縫早閉與鎖骨發育不全、囟門延遲閉合、肛門異常、泌尿生殖畸形、皮膚皮疹),可能連鎖 22q12–13。自 1 個月大起出現廣泛汗孔角化性丘疹,主侵犯顏面與四肢,有光照加重 (photoaggravation)。
致病機轉
- 多基因座;PMVK(膽固醇生合成途徑中之磷酸甲羥戊酸激酶)剪接突變致 PM;MVK(甲羥戊酸激酶,12q24)為 DSAP 致病基因,其產物調節鈣誘導角質細胞分化並可能抗 UVA 凋亡 (apoptosis)。
- 其他基因座:DSAP1(12q23.2–24.1)、DSAP2(15q25)、DSP(18p11.3);點狀掌蹠型對應 12q24.1–24.2,與 DSAP1 重疊,提示二者可能為等位基因 (allelic)。
- 病灶離心性擴展推測反映突變角質細胞克隆 (mutant clone) 的遷移。p53 與 pRb 在圓錐狀板層下方角質細胞過度表現,但未發現 p53 突變。
- 免疫抑制與 UV 暴露為許可性/誘發因子,凸顯受累角質細胞的異生性 (dysplastic) 潛能;除點狀型外,所有變異型均有惡性退化報告。
組織病理學
- 各型型態相似,特徵見於隆起推進邊緣:角質層過度角化,圓錐狀板層(薄柱狀染色不良角化不全細胞)貫穿其中(圖 51-6)。
- 其下角質細胞水腫、海綿狀水腫 (spongiosis)、核皺縮,可見明顯真皮淋巴細胞型態;板層下方顆粒層 (granular layer) 缺如或明顯減少。中央表皮可正常、增生或萎縮。
- 圓錐狀板層為特徵但非特異性 (not pathognomonic),亦見於病毒疣、某些魚鱗癬、痣樣過度角化。
鑑別診斷
- 經典病灶臨床獨特,多可臨床診斷;非典型病灶需鑑別(表 51-2)。
- 光化性角化症 (actinic keratoses) 可有圓錐狀板層但伴細胞學非典型性;尋常疣 (verruca vulgaris) 角化不全堆積可與板層相同但常有挖空細胞 (koilocytosis)。線狀型須與其他線狀病灶鑑別(皆無圓錐狀板層)。
治療
- 病灶慢性、緩慢進行、相對無症狀,通常不需介入,以疾病監測為標準;偶有劇烈搔癢。
- 困擾或美觀問題可選:強效局部類固醇、角質溶解劑、局部類維生素 A、局部 5-氟尿嘧啶、imiquimod 5%、calcipotriol、anthralin、冷凍治療、二氧化碳雷射、脈衝染料雷射、Nd:YAG 雷射(表 51-3)。
- Tacrolimus 0.1% 對線狀汗孔角化症有用;病灶內注射 bleomycin 的電化學療法可治線狀型相關鱗狀細胞癌 (SCC)。
- 為防殘餘/復發需達真皮中層 (middermis) 的消融措施(刮除、切除、磨皮術,成效不一)。口服類維生素 A 結果最可重現,但停藥後常復發。
- 巨大或線狀病灶及免疫抑制者宜更密切監測,對可疑病灶採低切片閾值。
病程與預後
- 一般慢性進行性,病灶隨時間增大增多;PM 歷時數十年,DSAP 在陽光暴露後可很快速。免疫受損者嚴重度可隨免疫狀態波動,切除原發惡性腫瘤後有緩解報告。
- 一般視為良性,但可惡性退化,惡性腫瘤估發生於 7% 至 11%(可能高估)。最常見為 SCC(可具侵襲性),亦有波文氏病 (Bowen disease) 與基底細胞癌報告。病灶自發消退極罕見。
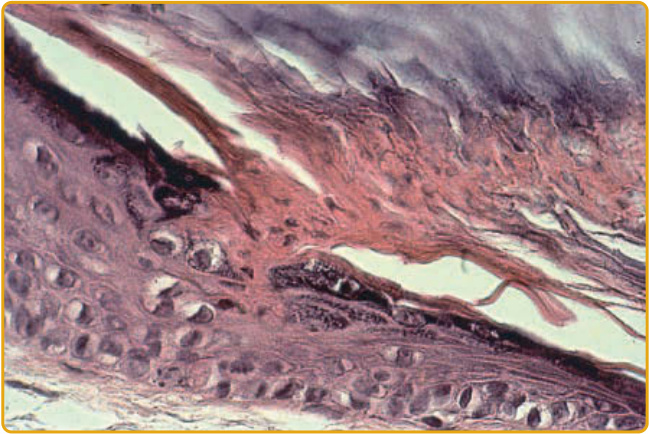
圖 51-6:圓錐狀板層起源於表皮內陷,以薄柱貫穿整個角質層,其下顆粒層缺如或減少。
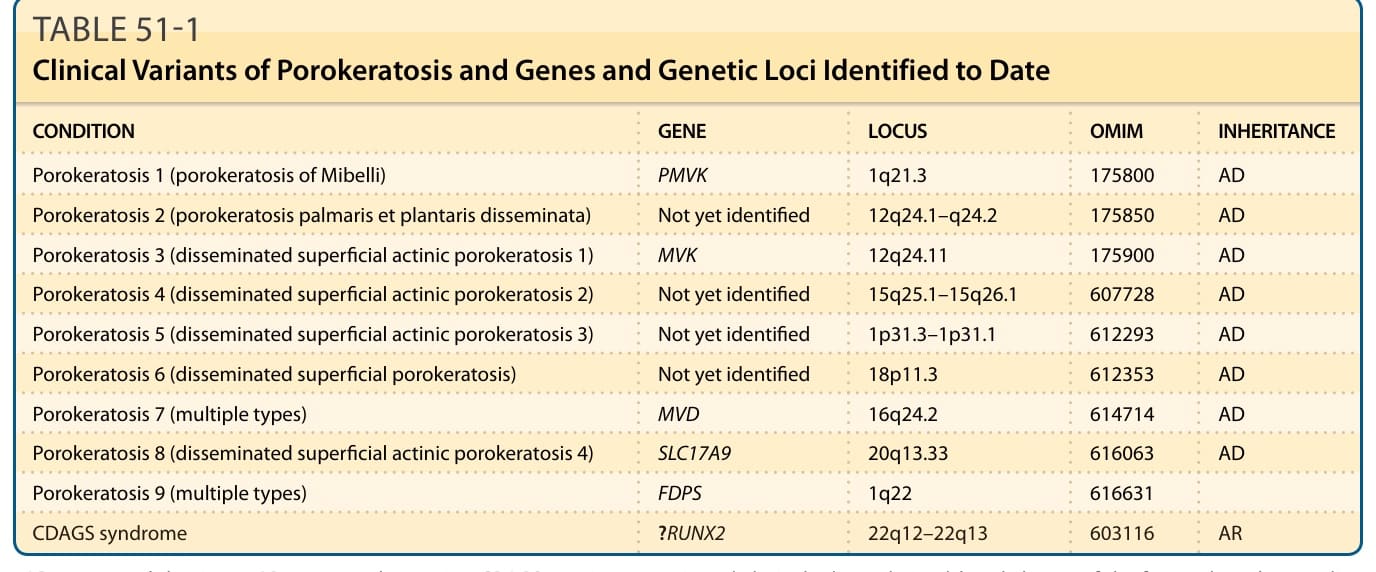
表 51-1:汗孔角化症的臨床變異型及已鑑定的基因與遺傳基因座。