Porokeratosis
8
AT-A-GLANCE
■ Porokeratosis is a chronic progressive disorders of keratinization characterized clinically by hyperkeratotic papules or plaques surrounded by a threadlike, elevated border that expands centrifugally.
■ Porokeratosis is genetically heterogeneous; the majority are inherited as autosomal dominant traits.
■ At least six clinical variants of porokeratosis have been described.
■ The classic form, porokeratosis of Mibelli, presents in infancy or childhood as asymptomatic, small, brown to skin-colored, annular papules with a characteristic raised border.
■ The most common type, disseminated superficial actinic porokeratosis, presents with multiple papules distributed symmetrically on sun-exposed areas.
■ Linear porokeratosis presents at birth or in childhood with lesions distributed along Blaschko lines.
■ Punctate porokeratosis appears during or after adolescence as 1- to 2-mm papules on the palms or soles.
■ In all variants, a thin column of parakeratotic cells (cornoid lamella) corresponds to the hyperkeratotic border and extends throughout the stratum corneum in histologic sections.
■ Malignant epithelial neoplasms are reported in all subtypes except the punctate variety.
INTRODUCTION
Porokeratosis is a morphologically distinct disorder of keratinization characterized clinically by hyperkeratotic papules or plaques surrounded by a threadlike elevated border that expands centrifugally. Histologically, a thin column of parakeratotic cells extends throughout the stratum corneum and is seen in all variants. This distinctive histopathologic feature, known as the cornoid lamella, corresponds to the raised hyperkeratotic border evident clinically. At least six clinical variants of porokeratosis are recognized, and recent genetic progress has additionally allowed classification according to the underlying mutation (Table 51-1). Reports of one type of porokeratosis coexisting with other forms and different types developing in multiple members of an affected family suggest more similarities than disparities,
particularly in the disseminated forms. Thus, it is important to allow for some flexibility around clinical subcategorization.1-3
EPIDEMIOLOGY
The most common variants are disseminated superficial actinic porokeratosis and porokeratosis of Mibelli. The other forms are rare. Porokeratosis usually occurs in fair-skinned individuals and is rare in darker-skinned people. Porokeratosis of Mibelli and porokeratosis palmaris et plantaris disseminata are twice as likely to affect men. Disseminated superficial actinic porokeratosis is twice as likely to occur in women.4 Linear porokeratosis is seen with equal incidence in men and women. Porokeratosis of Mibelli usually develops in childhood. Disseminated superficial porokeratosis generally develops in the third or fourth decade of life. Porokeratosis palmaris et plantaris disseminata and linear porokeratosis can be seen at any age from birth to adulthood.
CLINICAL FINDINGS
POROKERATOSIS OF MIBELLI
POROKERATOSIS OF MIBELLI
Classic porokeratosis of Mibelli (PM) begins during infancy or childhood as asymptomatic, small, brown to skin-colored, annular papules with a characteristic annular border (Fig. 51-1). The well-demarcated hyperkeratotic border is usually more than 1 mm in height, with a characteristic longitudinal furrow. The center of the lesion may be hyperpigmented, hypopigmented, depressed, atrophic, or anhidrotic. Lesions range in diameter from millimeters to several centimeters, but giant lesions measuring up to 20 cm may occur. Such giant porokeratoses are rare and occur predominantly on the lower leg and foot. Large lesions are associated with a higher malignant potential.5 Multiple lesions may arise; however, they are usually regionally localized and unilateral. The condition is inherited as an autosomal dominant trait. Lesions persist indefinitely.
DISSEMINATED SUPERFICIAL ACTINIC POROKERATOSIS
DISSEMINATED
SUPERFICIAL ACTINIC
POROKERATOSIS
Disseminated superficial actinic porokeratosis (DSAP) is the most common of the porokeratoses. Lesions are
8
CONDITION GENE LOCUS OMIM INHERITANCE
Porokeratosis 1 (porokeratosis of Mibelli) PMVK 1q21.3 175800 AD
Porokeratosis 2 (porokeratosis palmaris et plantaris disseminata) Not yet identified 12q24.1–q24.2 175850 AD
Porokeratosis 3 (disseminated superficial actinic porokeratosis 1) MVK 12q24.11 175900 AD
Porokeratosis 4 (disseminated superficial actinic porokeratosis 2) Not yet identified 15q25.1–15q26.1 607728 AD
Porokeratosis 5 (disseminated superficial actinic porokeratosis 3) Not yet identified 1p31.3–1p31.1 612293 AD
Porokeratosis 6 (disseminated superficial porokeratosis) Not yet identified 18p11.3 612353 AD
Porokeratosis 7 (multiple types) MVD 16q24.2 614714 AD
Porokeratosis 8 (disseminated superficial actinic porokeratosis 4) SLC17A9 20q13.33 616063 AD
Porokeratosis 9 (multiple types) FDPS 1q22 616631
CDAGS syndrome ?RUNX2 22q12–22q13 603116 AR
CDAGS syndrome ?RUNX2 22q12–22q13 603116 AR
AD, autosomal dominant; AR, autosomal recessive; CDAGS, craniosynostosis and clavicular hypoplasia, delayed closure of the fontanel, anal anomalies, genitourinary malformations, skin eruption; OMIM, Online Mendelian Inheritance in Man.
characteristically uniformly small, annular, asymptomatic, or mildly pruritic papules ranging from 2 to 5 mm in diameter, distributed symmetrically on the extremities. Lesions are more generalized than other forms of porokeratosis, with typically more than 50 lesions located predominantly in sun-exposed sites (Fig. 51-2). Although widespread, lesions typically spare the palms, soles, and mucous membranes. Compared with porokeratosis of Mibelli, the hyperkeratotic border is subtler. As the lesions progress, the older, central area becomes atrophic and anhidrotic. DSAP is inherited as an autosomal dominant disorder, with the earliest reported age of onset at 7 years, and is usually
902
fully penetrant by the third or fourth decade of life.6
Initial reports of induction of lesions by exposure to ultraviolet (UV) light and hypersensitivity of DSAPderived fibroblasts to x-rays have not been consistently reproduced, and the pathogenesis of DSAP remains unknown.7-10
DISSEMINATED SUPERFICIAL POROKERATOSIS
DISSEMINATED
SUPERFICIAL
POROKERATOSIS
Disseminated superficial porokeratosis (DSP) also shows an autosomal dominant pattern of inheritance and has its onset in the third or fourth decade of life. Lesions primarily are morphologically identical to those of DSAP, occur on the extremities, and are typically distributed symmetrically but do not spare sun-protected areas as in DSAP. Similar to DSAP, more than 100 lesions may be present, with a predilection for the extensor surfaces of the extremities. Notably, involvement of the face is rare in both DSAP and DSP. In both disseminated forms, there is a reported female predominance, with a female-tomale ratio of 2 to 1.
DISSEMINATED SUPERFICIAL POROKERATOSIS OF IMMUNOSUPPRESSION
DISSEMINATED
SUPERFICIAL
POROKERATOSIS OF
IMMUNOSUPPRESSION
Disseminated superficial porokeratosis in the context of immunosuppression is recognized after renal,
A B
8
hepatic, and cardiac transplantation; electron-beam irradiation;11 immunosuppressive chemotherapy; monoclonal antibodies;12 systemic corticosteroids with hematopoietic malignancies;13 and in the setting of HIV infection.14 It has recently been described in association with type 2 diabetes mellitus as well.15 Porokeratosis has also been reported after bone marrow transplantation in the absence of ongoing immunosuppressive therapy, which suggests a more complex association than immunosuppression alone.16 The distribution and morphology of DSP of immunosuppression are similar to those of DSAP, but a history of sun exposure is less evident.
LINEAR POROKERATOSIS
LINEAR POROKERATOSIS
Linear porokeratosis is an uncommon variant, traditionally categorized as a separate entity, but is increasingly recognized as a mosaic manifestation of one of the other types of porokeratosis.17 Typically, it presents in early childhood, although congenital presentations have been reported.18 Two distinct clinical variants have been described. The more common presentation consists of a unilateral lesion confined to an extremity following Blaschko lines (Fig. 51-3). In the rare generalized form, multiple lesions affect several extremities and may involve the trunk. Linear variants have the highest potential for malignant degeneration of all the porokeratoses.
It has been postulated that this association can be attributed to allelic loss caused by a postzygotic mutation.19 The proband in Mibelli’s original publication most likely had coexistent linear porokeratosis and DSAP, with several subsequent reports confirming this phenomenon.3,17,20,21 These findings may be explained by the loss of heterozygosity for the DSAP allele and provides an example of the type 2 segmental manifestations of an autosomal dominant disorder.22,23
903
8
POROKERATOSIS PALMARIS ET PLANTARIS DISSEMINATA
POROKERATOSIS
PALMARIS ET PLANTARIS
DISSEMINATA
Porokeratosis palmaris et plantaris disseminata (porokeratosis punctata palmaris et plantaris) is a genodermatosis with an autosomal dominant inheritance pattern characterized by small, relatively uniform lesions (Fig. 51-4) that initially appear on the palms and soles. Subsequently, lesions spread to involve other parts of the body, including the mucous membranes and non–sun-exposed sites. The palmar and plantar lesions are generally more hyperkeratotic, and the characteristic longitudinal furrow along this ridge may be quite pronounced (Fig. 51-5). Typically, the lesions appear during adolescence or early adulthood and are bilateral and distributed symmetrically. Porokeratosis palmaris et plantaris disseminata affects males twice as often as females.
PUNCTATE POROKERATOSIS
PUNCTATE POROKERATOSIS
Punctate porokeratosis usually appears during adolescence or adulthood and may be seen concomitantly with other types of porokeratosis. Multiple minute and discrete punctate, hyperkeratotic lesions surrounded by a thin, raised margin are present on the palms and
904
soles. Lesions may occur in a linear arrangement, or they may aggregate to form plaques. Punctate porokeratosis must be differentiated clinically and histologically from punctate keratoderma, also referred to as punctate porokeratotic keratoderma or as porokeratosis punctata palmaris et plantaris (see Chap. 48).
CDAGS SYNDROME
CDAGS SYNDROME
CDAGS syndrome (craniosynostosis and clavicular hypoplasia, delayed closure of the fontanel, anal anomalies, genitourinary malformations, skin eruption), also referred to as CAP syndrome (craniosynostosis, anal anomalies, and porokeratosis), is a rare genodermatosis reported in four ethnically diverse families to date.24,25 The main phenotypic features consist of craniosynostosis and clavicular hypoplasia, anal anomalies, and porokeratosis. It appears to segregate as an autosomal recessive trait, with possible linkage to chromosome band 22q12–13.25 The cutaneous manifestations are strikingly consistent, with the development of small, widespread porokeratotic papules from 1 month of age in affected individuals, predominantly affecting the face and extremities, with reported photoaggravation of lesions.
ETIOLOGY AND PATHOGENESIS
Porokeratosis is a genetically heterogeneous disorder with multiple loci identified to date. A splicing mutation in phosphomevalonate kinase (PMVK), involved in the cholesterol biosynthetic pathway, has been shown to cause PM.26 The mevalonate kinase (MVK) gene on
chromosome 12q24 has recently been identified as a causative gene in DSAP.27 The product of this gene has a role in regulating calcium-induced keratinocyte differentiation and may confer protection against apoptosis induced by ultraviolet A radiation. Loci at chromosome bands 12q23.2–24.1 and 15q25 (DSAP1 and DSAP2) have been reported in other forms of familial disseminated superficial actinic porokeratoses; a further locus has been identified for disseminated superficial porokeratosis (DSP) at 18p11.3.28,29 The locus at DSAP1 includes several candidate genes, including SART3 (squamous cell antigen recognized by T cells 3), SSH1 (slingshot 1), and ARPC3.30,31 Porokeratosis punctata palmaris et plantaris maps to a region at chromosome band 12q24.1–24.2, overlapping with the region identified for DSAP1, suggesting that the two forms may be allelic.32 The centrifugal expansion of lesions is postulated to reflect the migration of a mutant clone of keratinocytes.33,34 The tumor suppressor proteins p53 and pRb are overexpressed in keratinocytes immediately beneath and adjacent to the cornoid lamella, although to date p53 mutations have not been identified, and there is no significant expression of p53 at the mRNA level.7,35-38 The increased prevalence of porokeratosis in immunosuppressed patients suggests that impaired immunity may be permissive in genetically predisposed individuals.39-42 Other reported triggering factors such as exposure to UV light, together with the increased potential for malignant transformation, highlight the dysplastic potential of affected keratinocytes. Malignant degeneration has been described in all variants of porokeratosis, except for the punctate variety.5,43,44
HISTOPATHOLOGY
Histopathologic patterns are similar in all forms of porokeratosis, with the characteristic changes evident at the raised and advancing edge of the lesion. The stratum corneum is hyperkeratotic, with a thin column of poorly staining parakeratotic cells, the cornoid lamella, running through the surrounding normal-staining cells (Fig. 51-6). The underlying keratinocytes are edematous
8
with spongiosis and shrunken nuclei, and a striking dermal lymphocytic pattern may be evident. Underlying the cornoid lamella, the granular layer is either absent or markedly reduced but is of normal thickness in other areas of the lesion. The epidermis in the central portion of porokeratosis may be normal, hyperplastic, or atrophic. Although characteristic of porokeratosis, the cornoid lamella is not pathognomonic and may also be found in other conditions, such as viral warts, some ichthyoses, and nevoid hyperkeratoses.
DIFFERENTIAL DIAGNOSIS
The classic lesions of porokeratosis are clinically distinctive, and the diagnosis is usually clinically apparent. However, atypical lesions may require differentiation (Table 51-2). Actinic keratoses, for example, may show cornoid lamellae but also show cytologic atypia. Verruca vulgaris often shows mounds of parakeratosis that are sometimes identical to cornoid lamellae, but koilocytosis is usually present. Linear porokeratosis may be clinically confused with other linear lesions (see Table 51-2), none of which has a cornoid lamella.
TREATMENT
Lesions of porokeratosis are chronic, slowly progressive, and relatively asymptomatic, although intense pruritus has been reported.45 Intervention is usually unnecessary, and disease surveillance is standard. If the lesions are problematic or cosmetically unacceptable, treatment with potent topical steroids, keratolytics, topical retinoids, topical 5-fluorouracil,46
imiquimod 5%, calcipotriol,47 anthralin,48 cryotherapy,49
carbon dioxide laser,50 pulsed dye laser,51 or Nd:YAG (neodymium-doped yttrium aluminum garnet) laser may be considered (Table 51-3). Tacrolimus 0.1% has been shown to be useful in treating patients with linear porokeratosis.52 Electrochemotherapy with intralesional bleomycin has been successful in treating
Localized Lesions Most Likely
■Granuloma annulare
■Tinea corporis
■Actinic keratosis
■Viral warts Consider
■Elastosis perforans
■Lichen planus
■Focal dermal hypoplasia (Goltz syndrome)
Linear Consider
Linear Consider
■Linear inflammatory verrucous epidermal nevus
■Linear inflammatory verrucous epidermal nevus
■Incontinentia pigmenti (stage II)
■Incontinentia pigmenti (stage II)
■Linear lichen planus
■Linear lichen planus
905
■Ichthyosis linearis circumflexa
■Ichthyosis linearis circumflexa
8
TOPICAL SURGICAL SYSTEMIC
First line
Photoprotection 5-Fluorouracil Cryotherapy
CO2 laser vaporization
Second line
Calcipotriol Imiquimod Topical corticosteroids Topical retinoids
Oral retinoids
Third line Dermabrasion Nd:YAG laser Grenz ray
Surgical
Third line
Dermabrasion Nd:YAG laser Grenz ray
Surgical excision
excision
Nd:YAG, neodymium-doped yttrium aluminum garnet.
patients with squamous cell carcinomas (SCCs) associated with linear porokeratosis.53 To prevent residual lesions or recurrence, ablative measures that reach the middermis are required. Techniques such as curettage, excision, and dermabrasion54 have been used with variable degrees of success. Oral retinoids have been shown to give the most reproducible results, although the disease typically recurs after their discontinuation. Given the association with malignancy, closer disease surveillance and a lower threshold for biopsy of
suspicious lesions may be warranted in cases of giant porokeratoses or linear lesions and in immunosuppressed individuals.
COURSE AND PROGNOSIS
The porokeratoses are generally chronic and progressive, with lesions increasing in size and number with time. Typically, this process occurs over decades in PM, but may be rapid in DSAP, particularly after sun exposure. In cases of immune compromise, fluctuations in severity may parallel the state of immune competence, and there are reports of remission after removal of primary malignancy.42 The disease is generally considered a benign process; however, malignant degeneration may occur. Malignancy is thought to arise in 7% to 11% of individuals, although these figures are likely overestimated. SCC is the most frequently associated tumor and may be invasive. Bowen disease and basal cell carcinoma have also been reported. Spontaneous resolution of lesions has been reported, although it is exceptionally rare.55
SUMMARY
A summary of the porokeratosis subtypes can be found in Table 51-4.
INHERITANCE AND RISK FACTORS EPIDEMIOLOGY CLINICAL FINDINGS SPECIAL CONCERNS
Porokeratosis of Mibelli Autosomal dominant Onset in infancy or childhood; male predominance Asymptomatic, small, brown to skin-colored annular papules with an annular border
Disseminated superficial actinic porokeratosis (DSAP)
Large lesions have malignant potential
Autosomal dominant Most common porokeratosis; as early as childhood; often present by third to fourth decade of life; female predominance
Disseminated superficial porokeratosis
Uniform, small, annular, papules from 2-5 mm, with symmetrical distribution on photoexposed extremities; asymptomatic or mildly pruritic
—
Autosomal dominant Onset by third to fourth decade of life; female predominance
Disseminated superfi- cial porokeratosis of immunosuppression
Immunosuppression after organ transplantation, drugs, or infections (see text)
Similar to above but with distribution to photoprotected sites
—
Dependent on exposure (see risk factors) Similar to DSAP —
Linear porokeratosis Mosaic manifestation of other porokeratosis variants Uncommon; presents in early childhood Primary manifestation dependent on particular variant, however configuration follows lines of Blaschko
Porokeratosis palmaris et plantaris disseminata
Highest potential for malignant degeneration
Autosomal dominant Appear during adolescence or early adulthood; male predominance
Punctate porokeratosis Concomitant occurrence with
Small, uniform lesions appearing on palms and soles with involvement to other sites; bilateral, symmetric involvement
—
Appears during adolescence
Multiple discrete punctate hyper-
—
Punctate porokeratosis Concomitant occurrence with other forms of porokeratosis Appears during adolescence or adulthood Multiple discrete punctate hyperkeratotic lesions; may aggregate to form plaques
other forms of porokeratosis
or adulthood
906
—
keratotic lesions; may aggregate to form plaques
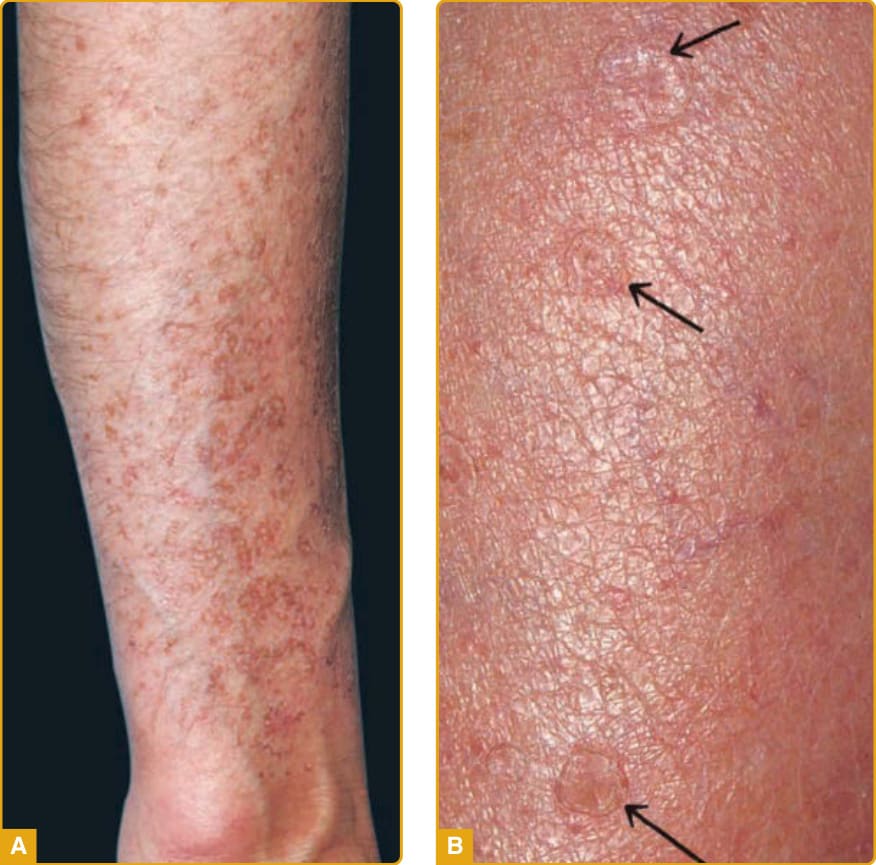
Figure 51-2 A, Disseminated superficial actinic porokeratosis with multiple lesions on the forearm in this severely affected individual. B, The characteristic furrowing (arrows) is clearly demonstrated on the forearm of this patient.
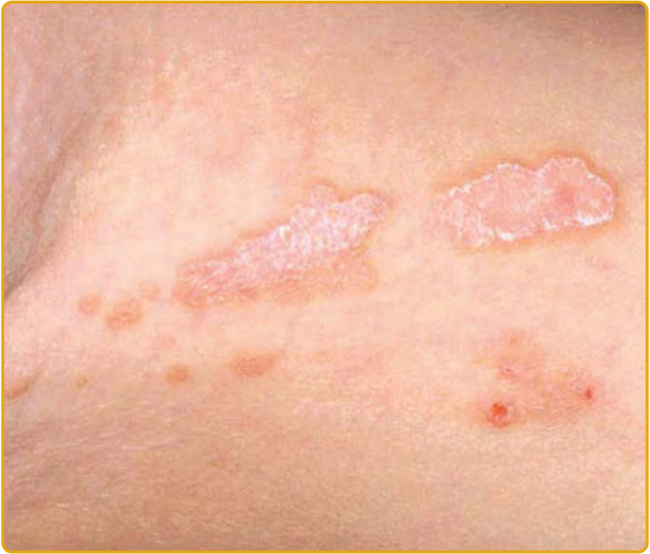
Figure 51-3 Linear porokeratosis showing the characteristic distribution along Blaschko lines.

Figure 51-4 Porokeratosis palmaris et plantaris disseminata showing multiple superficial lesions on the calf. Note the similarity to disseminated superficial actinic porokeratosis shown in Fig. 51-2A.
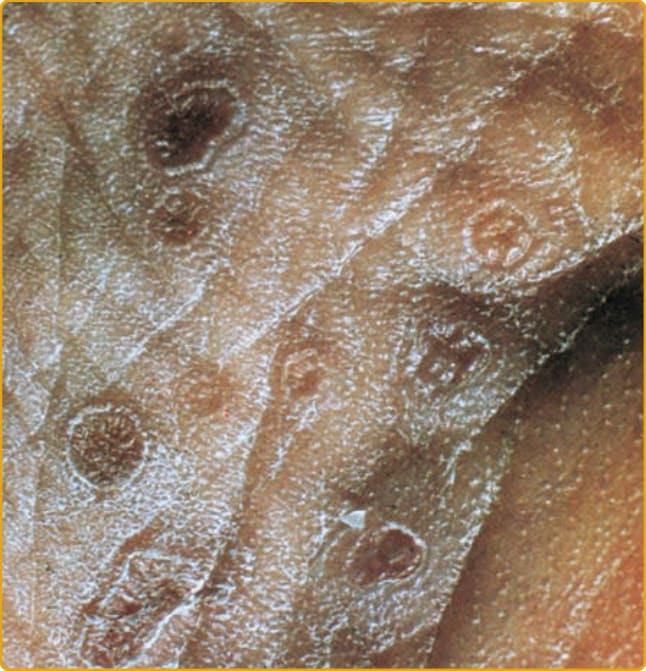
Figure 51-5 Sole of the foot in a patient with porokeratosis palmaris et plantaris disseminata. The ridge and furrow are clearly seen.
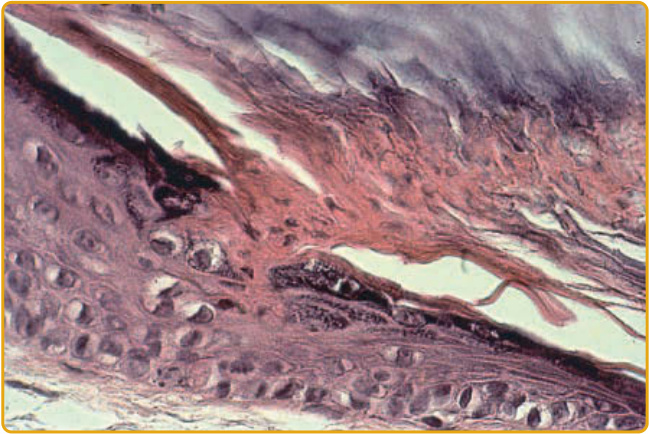
Figure 51-6 The cornoid lamella arises from an indentation of the epidermis and extends as a thin column throughout the stratum corneum. The underlying granular layer is either absent or reduced.
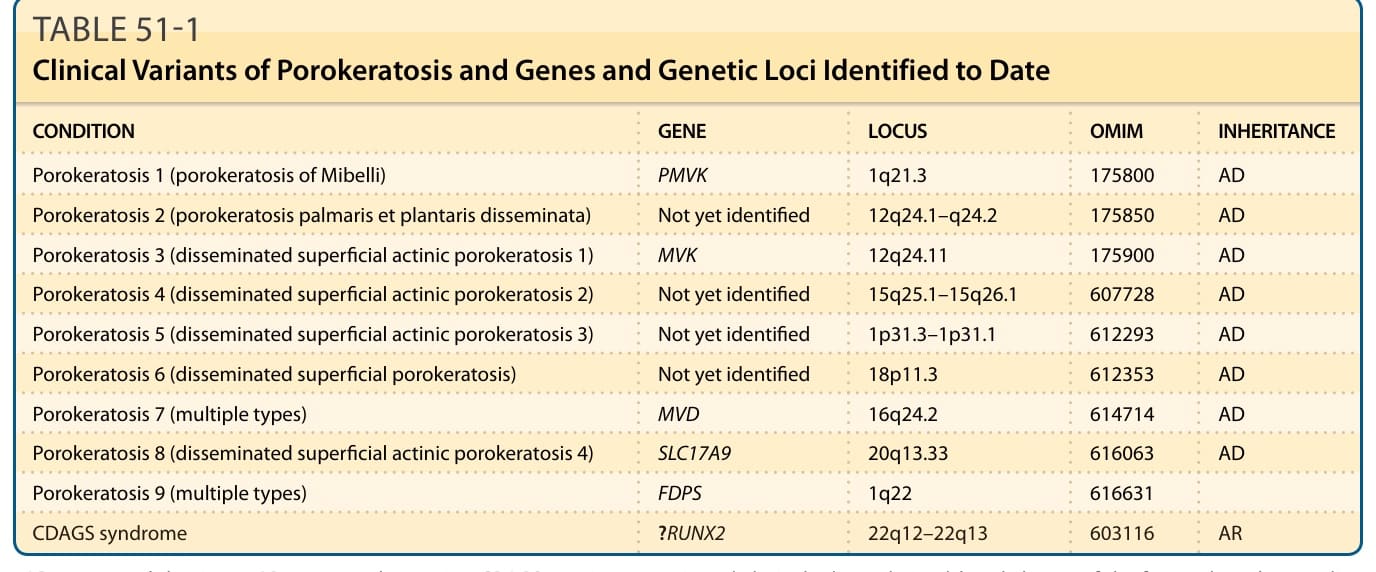
TABLE 51-1 Clinical Variants of Porokeratosis and Genes and Genetic Loci Identified to Date
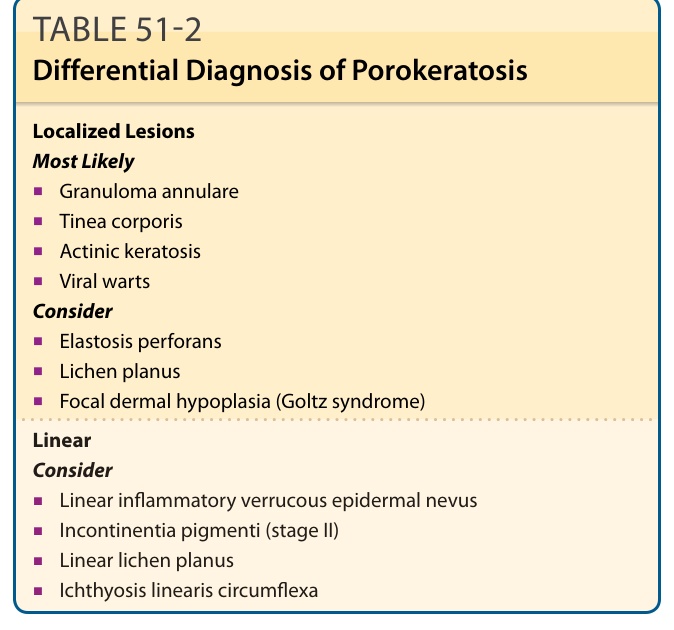
TABLE 51-2 Differential Diagnosis of Porokeratosis
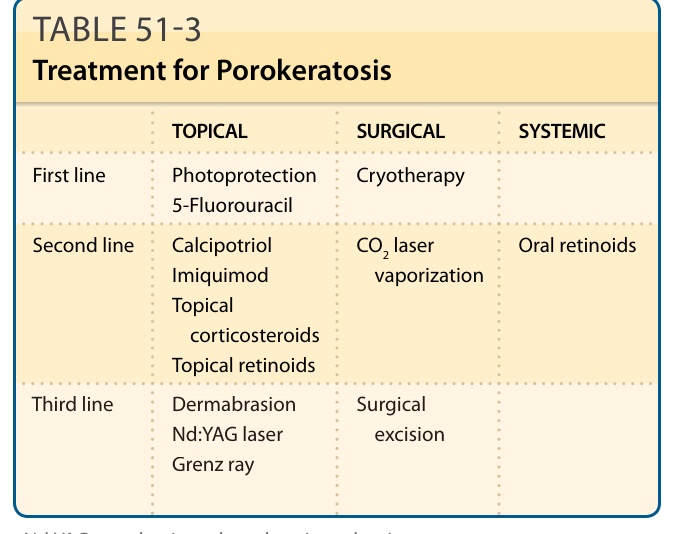
TABLE 51-3 Treatment for Porokeratosis
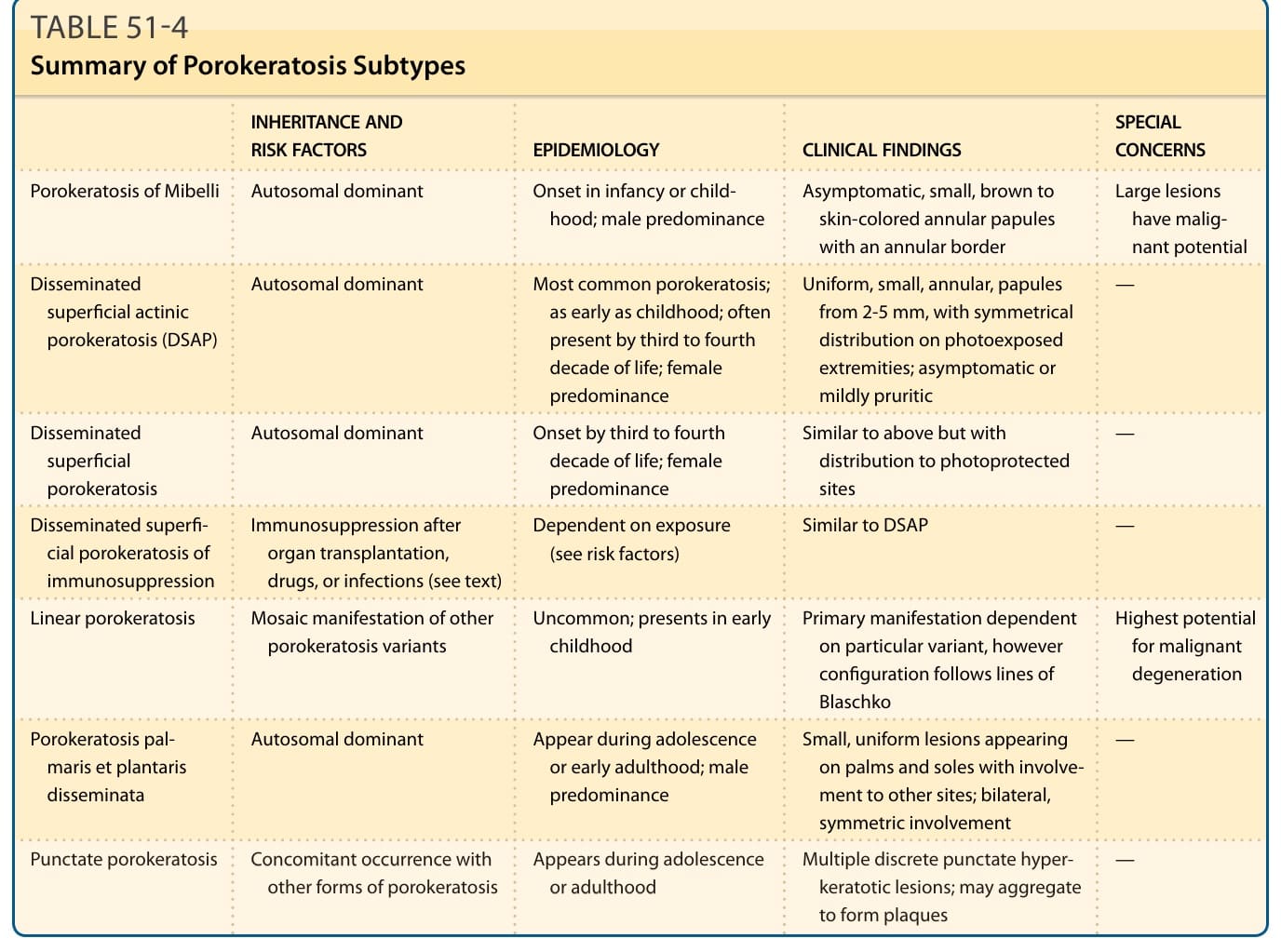
TABLE 51-4 Summary of Porokeratosis Subtypes