遺傳性掌蹠角化症 (Inherited Palmoplantar Keratodermas) 精華筆記
總論
- 掌蹠角化症 (palmoplantar keratoderma, PPK):一群臨床與遺傳上異質性的角化異常,特徵為手掌與足底皮膚的局灶性或瀰漫性異常增厚;可為後天性或遺傳性,遺傳性者多為家族性、發病年齡早。
- 三種型態分類:(1) 瀰漫型 (diffuse) — 整個掌蹠表面均勻受侵;(2) 局灶型 (focal) — 局限於壓力點,再分為斑塊型 (areata/nummular) 與條紋型 (striate);(3) 點狀型 (punctate) — 多發、散在、1-mm 至 1-cm 圓形角化丘疹。
- 進一步特徵:transgrediens(角化向手足背側、腕屈側與足跟延伸)與假性指趾斷離 (pseudoainhum)(指趾緊縮帶導致殘毀)。
- 新分類除型態外,並納入致病基因/病因。
- 流行病學:個別罕見,但近親通婚社群盛行率高。EPPK 是瀰漫型角化症最常見型態,全球發生率每 10 萬名活產 2.2–4.4 例。
診斷與鑑別診斷
- 診斷演算法(圖 48-2)整合四組資料:皮膚發現(PPK 型態、毛髮/指甲異常)、皮膚外發現(牙周炎、耳聾)、遺傳模式、組織病理(表皮鬆解型過度角化、失黏附、角化不全),藉此將 PPK 指派至基因群再定序確診。
- 後天性鑑別(表 48-2):發炎性(乾癬 psoriasis、扁平苔癬 lichen planus、毛囊性紅糠疹、反應性關節炎)、副腫瘤性、皮膚淋巴瘤、藥物/化學暴露、甲狀腺機能低下症、更年期角化症 (keratoderma climactericum)、感染(皮癬菌、疥瘡、尋常疣、梅毒)。
- 帶 transgrediens:LK、Nagashima 型、Greither、Olmsted、Papillon-Lefevre。
- 伴感覺神經性聽力損失 (SNHL):Vohwinkel、KID 症候群與其他 GJB2 相關病、粒線體遺傳 PPK。
- 伴角化區內鱗狀細胞癌 (SCC):Huriez、Unna-Thost 症候群。
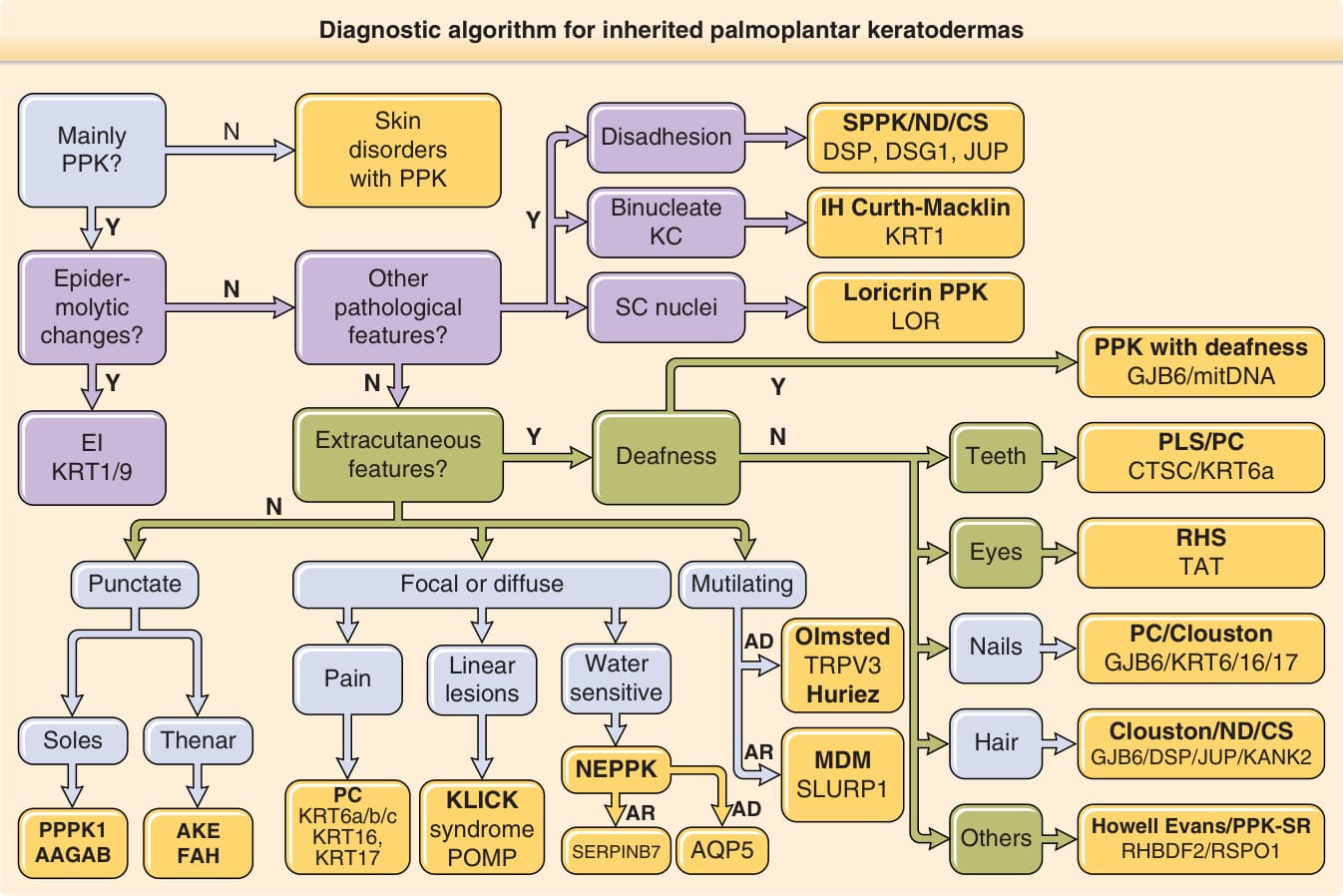
圖 48-2:遺傳性掌蹠角化症的診斷演算法
常見治療原則
- 目標為緩解表現:機械性措施、緩解疼痛、治療續發感染、助行輔具。
- 局部治療:潤膚劑、外用角質溶解劑(10% 至 20% 水楊酸 salicylic acid,或 35% 至 70% 丙二醇 propylene glycol 併厚潤膚劑,必要時封包);外用 calcipotriol 對 EPPK 有效、外用 tazarotene 對 PLS 有效;積極治療皮癬菌/細菌感染與多汗症;浸泡與機械性移除角化、矯正器/鞋墊緩解疼痛。
- 口服類視黃醇 (oral retinoids):通常有效,建議低劑量、短期或間歇治療(顧及長期副作用與皮膚脆弱風險)。對 EPPK、MDM、VS、LK、OS、Huriez、點狀型 PPK 1、PLS 有效;KID 症候群部分有效(isotretinoin 可惡化角膜新生血管)。建議低起始劑量 acitretin (0.5 mg/kg/day) 或 alitretinoin (10 mg/day),依反應與耐受性逐步增量。
- 手術:局灶型 PPK 與殘毀病例可考慮手術切除與植皮。
瀰漫型 PPK,無皮膚外特徵
表皮鬆解型 PPK (EPPK)(Vörner 型,顯性)
- 瀰漫型角化症最常見型態。黃色、瀰漫型 PPK,掌蹠邊緣具紅斑性銳利邊界;多於出生時或最初幾週出現,終生穩定(圖 48-3)。可併疼痛性裂隙、多汗症、續發感染、假性指趾斷離與屈指畸形。
- 機轉:體染色體顯性,KRT9(多數)或 KRT1(少數)雜合子突變,以顯性負向方式作用。突變熱點為 KRT9 第 163 號密碼子(p.Arg163Trp 約佔 30%)。
- 病理:緻密正過度角化、顆粒層增生、棘層肥厚、表皮鬆解型過度角化(核周空泡化+大型不規則角質透明顆粒);可能需多次切片。
- 特定治療:小鼠模型中 RNAi/shRNA 抑制突變等位基因表現有效。
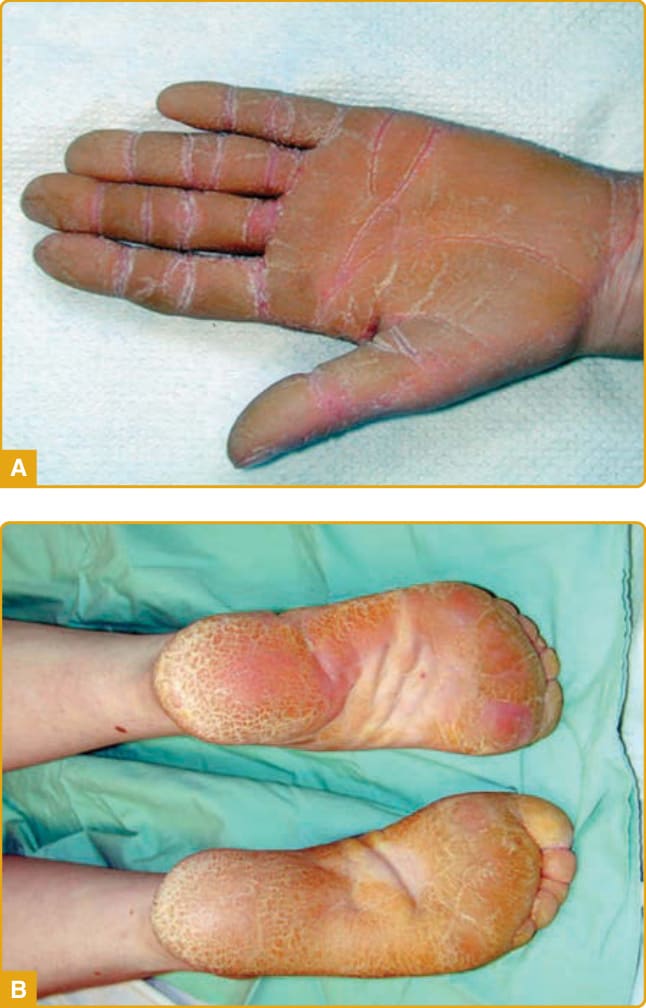
圖 48-3:表皮鬆解型掌蹠角化症,掌蹠邊緣紅斑性銳利邊界(KRT9 突變)
Unna-Thost PPK (NEPPK,顯性)
- 臨床似 EPPK 但較輕、較平滑蠟狀;多汗症、續發皮癬菌感染與凹陷性角質溶解症較 EPPK 常見。
- 機轉:KRT1 雜合子突變(V1 頭部區域錯義突變等);Unna-Thost 與 Vörner 經考證為同一疾病實體。無 KRT1 突變時可篩 KRT6c、KRT16。
- 病理:非特異(過度角化、正角化、棘層肥厚),無表皮鬆解型過度角化。
Greither 症候群(顯性)
- 瀰漫型、帶 transgrediens、阿基里斯腱與四肢延伸的角化斑塊;第五個十年後傾向消退。Sybert PPK 為較嚴重的類似型態。
- 機轉:KRT1 錯義突變(影響螺旋起始基序);Sybert 致病基因未明。
Loricrin 角化症 (LK)(顯性)
- VS 變異型,無聽力障礙但有全身性魚鱗癬。蜂窩狀 PPK、海星狀過度角化、指節墊、假性指趾斷離致自體截斷(圖 48-4A);約 35% 出生時可見膠樣膜。
- 機轉:LOR 雜合子框移突變,突變 loricrin 異常進入細胞核(功能獲得型有害效應)。
Bothnia 型 PPK (PPKB)(顯性)
- 瑞典北部高盛行(0.3%–0.55%)。瀰漫型黃色過度角化延伸至指背;接觸水後呈白色海綿狀外觀。
- 機轉:AQP5(水通道蛋白 5)功能獲得型 (GOF) 突變,角質形成細胞水分攝取增加;屬皮膚通道病變。
Mal de Meleda (MDM)(隱性)
- 體染色體隱性、進行性、殘毀性、帶 transgrediens;手套狀/長襪狀延伸(圖 48-5),紅斑性鱗狀邊界。指甲異常、假性指趾斷離、進行性功能障礙。惡性黑色素瘤 (MM) 風險升高,需定期篩檢。PPK-Gamborg-Nielsen 為其較輕等位基因變異型。
- 機轉:SLURP1 雙等位基因突變(編碼 SLURP-1,分泌型表皮神經調節因子,調節角質形成細胞分化、凋亡與免疫)。
- 病理/診斷:MDM 病人掌蹠與汗液中 SLURP-1 缺如或勉強可測;毛果芸香鹼汗液檢測可作快速篩檢。
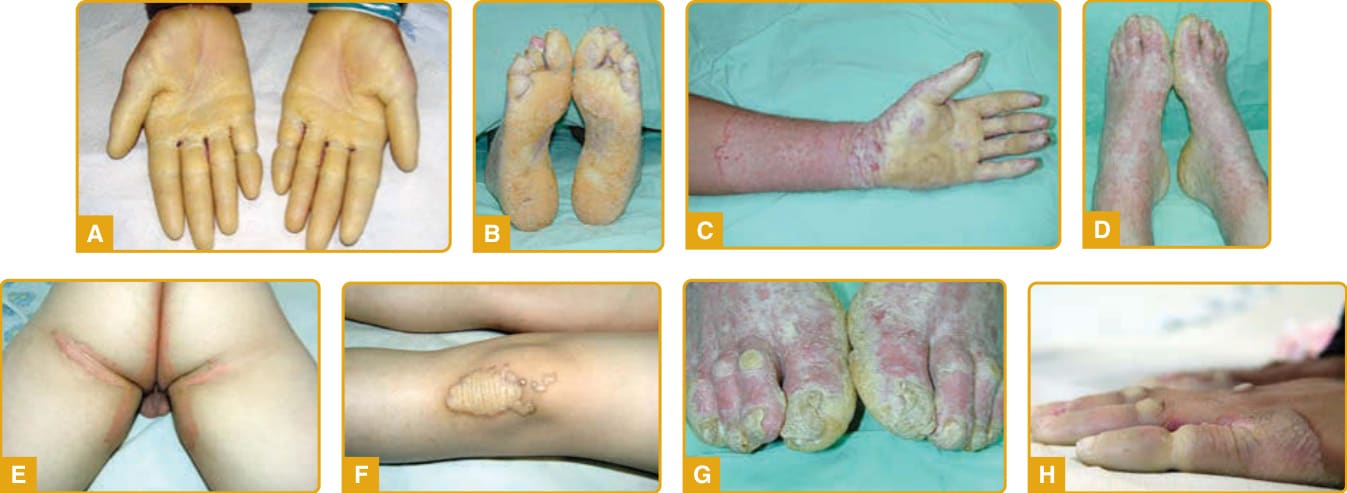
圖 48-5:Mal de Meleda,紅色鱗狀邊界勾勒的瀰漫型蠟狀過度角化,手套狀/長襪狀延伸
Nagashima 型 PPK (NPPK)(隱性)
- 亞洲族群最常見 PPK 類型(日本、中國)。瀰漫型、帶 transgrediens、青春期後非進行性、非殘毀性;接觸水 10 分鐘內呈白色海綿狀外觀。無緊縮帶或截斷。
- 機轉:SERPINB7 雙等位基因突變(編碼絲胺酸蛋白酶抑制劑);中、日復發性無義突變 c.796C>T 可作篩檢策略。
瀰漫型 PPK,伴皮膚外特徵
Olmsted 症候群 (OS)(顯性/隱性/X 連鎖)
- 瀰漫型殘毀性 PPK+開口周圍 (periorificial) 角化斑塊;疼痛性、致殘、妨礙行走。伴嚴重搔癢、甲失養、毛髮異常(捻轉發、結節性脆髮症)、禿髮、紅斑性肢痛症;PPK 內可發展 SCC、MM。
- 機轉:TRPV3 突變(顯性/隱性)或 MBTPS2 突變(X 連鎖隱性,與 IFAP 等位)。TRPV3 高表現於角質形成細胞、毛囊、感覺神經元,與 TGF-α/EGFR 訊息相關。
- 特定治療:部分/全層切除合併植皮;EGFR 抑制劑改善 PPK;TRPV3 拮抗劑對 GOF 突變者可能有效。
連接蛋白相關 PPK(GJB2/Cx26)
- Vohwinkel 症候群 (VS):三聯徵 — 蜂窩狀殘毀性 PPK(圖 48-4B)、海星狀角化斑塊、纖維性緊縮帶致自體截斷;多伴高頻 SNHL。常見 GJB2 復發突變 p.Asp66His。
- KID 症候群(角膜炎–魚鱗癬–耳聾):最嚴重的連接蛋白病。紅皮角化症、粗糙點刻顆粒狀 PPK(圖 48-6)、進行性角膜炎(可致失明)、先天性嚴重 SNHL;易黏膜皮膚感染、約 10% 發展 SCC(p53 喪失)。多攜 GJB2 p.Asp50Asn;GJB6(Cx30)突變者可伴先天性無毛症。
- Bart-Pumphrey 症候群 (BPS):SNHL、蜂窩狀 PPK、指節墊、白甲症。
- PPK 與耳聾:GJB2 第一細胞外環突變(如 p.R75Q)。
- 特定治療:VS 用交指皮瓣/Z 成形術/緊縮帶切除植皮;KID 眼部用角膜緣同種異體移植、角膜移植、免疫抑制,單例 bevacizumab 有效。
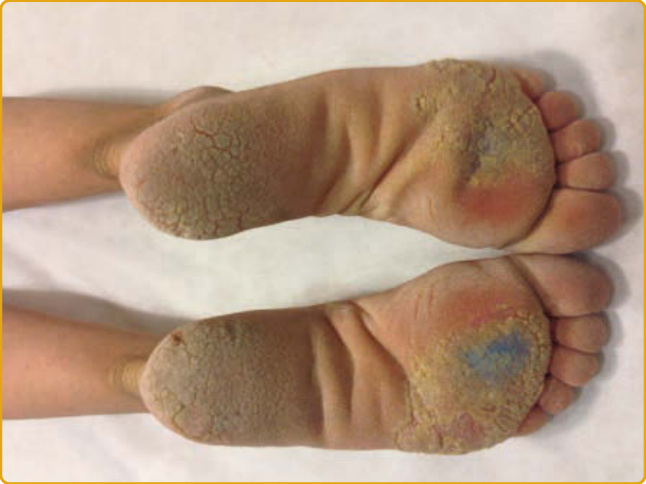
圖 48-6:KID 症候群,粗糙、點刻狀、顆粒狀的足底角化症
PPK–先天性禿髮症候群
- PPKCA1(顯性,較輕,GJA1/Cx43 GOF 突變,與 ODDD 等位);PPKCA2(隱性,伴假性指趾斷離、指端硬化、攣縮,致病基因未明)。
汗腺性外胚層發育不良(Clouston 症候群,顯性)
- 甲失養、毛髮脫落、瀰漫型鵝卵石樣 PPK,汗腺與牙齒正常;近 30% 病例僅以指甲異常表現。
- 機轉:GJB6(Cx30)雜合子突變;典型只有皮膚表現而無聽力障礙(Cx26 在內耳代償 Cx30)。
Huriez 症候群(顯性)
- 瀰漫型 PPK、手指硬化性萎縮 (scleroatrophy)、指端硬化,萎縮皮膚內發生 SCC(圖 48-4C);SCC 風險約 13%(高 100 倍以上),腫瘤分化差、轉移率高、死亡率約 5%。
- 機轉:病因未明,基因定位於 4q23;受侵皮膚蘭格漢斯細胞幾乎缺如、p53 異常。
Papillon-Lefevre 症候群 (PLS)(隱性)
- 帶 transgrediens 瀰漫型 PPK(圖 48-7A)+嚴重進行性牙周炎(乳齒與恆齒喪失);皮膚與牙齒嚴重度無相關。感染易感性增加(肝/腦膿瘍、肺炎);可伴硬腦膜鈣化。
- 機轉:CTSC(組織蛋白酶 C)LOF 突變,與 Haim-Munk 症候群等位。尿液缺乏活性 CTSC 可作可靠指標。
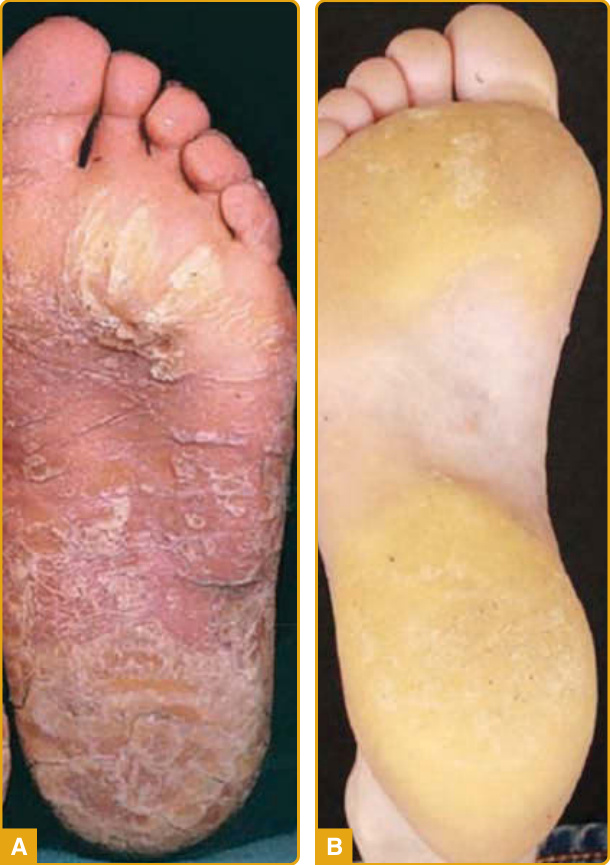
圖 48-7:Papillon-Lefevre(A,CTSC 突變瀰漫型角化症)與 Howel-Evans(B,足底壓力區局灶型厚斑塊)
Naxos 病 (ND)(隱性)
- 三聯徵:瀰漫型 PPK、心肌病變、羊毛狀髮。心律不整性右心室發育不良/心肌病變 (ARVD/C),青春期出現症狀,心室頻脈與猝死為主要死因(三分之一早逝,平均死亡年齡 32 歲)。
- 機轉:JUP(血漿斑珠蛋白 plakoglobin)雙等位基因突變。
伴皮膚 SCC 與性別逆轉症候群(隱性)
- 指端硬化、伴多發皮膚 SCC 的 PPK、早期牙齒喪失、性腺機能低下、性別逆轉(46,XX 男性表現型)。
- 機轉:RSPO1(R-spondin1)同合子突變。
伴耳聾的 PPK(粒線體遺傳)
- 母系遺傳;橙黃色瀰漫型掌蹠角化+雙側、輕至重度、可進行性的聽力損失(聽力損失外顯率 60% > 皮膚 37%)。
- 機轉:MTTS1 粒線體 tRNA 同質性點突變 c.A7445G。
局灶型 PPK
條紋型 PPK (SPPK)(顯性)
- 手掌沿指掌側面的線狀過度角化斑塊(圖 48-8A)+足底局限增厚(圖 48-8B);第一/二個十年出現,手工勞動加劇。
- 機轉:DSP、DSG1 或 KRT1 的無義/框移雜合子突變致單倍體不足;DSG1 雙等位基因突變致 SAM 症候群(圖 48-9)。
- 病理:表皮上層角質形成細胞失黏附 — 為橋粒蛋白相關 PPK 的診斷線索。
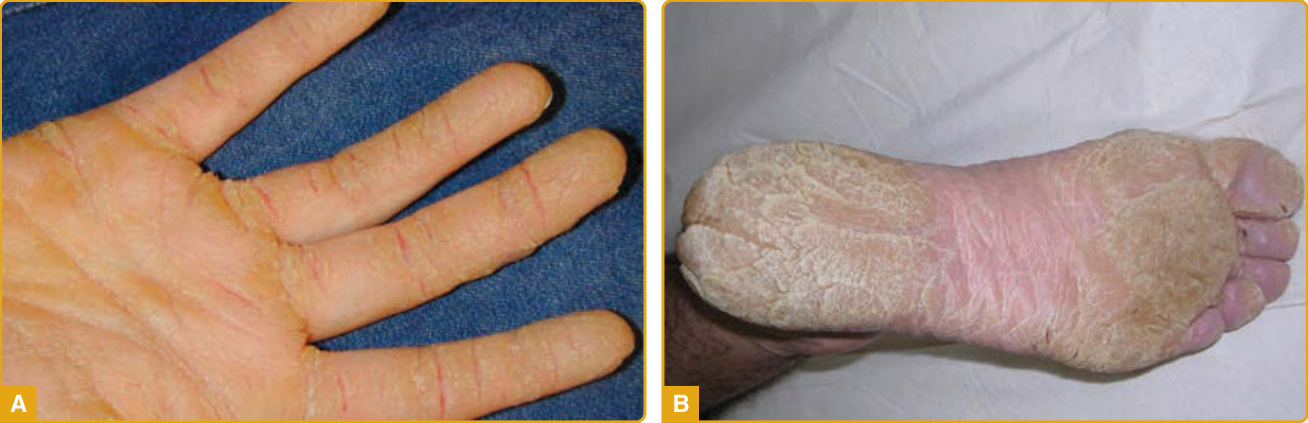
圖 48-8:DSG1 突變的局灶型 PPK,手掌線狀斑塊 (A)、足底局限斑塊 (B)
Wachters 變異型遺傳性 PPK(顯性)
- 黃色、非向外延伸、對稱、錢幣狀過度角化斑塊局限於足底壓力點;男性較多;致病基因未明。
先天性厚甲症 (Pachyonychia Congenita, PC)(顯性)
- 由五個角蛋白基因之一突變引起:KRT6A、KRT6B、KRT6C、KRT16、KRT17。
- 三大常見特徵(>90%):增厚足趾甲(V/倒 U 形甲下過度角化,圖 48-10A)、足底角化症(局灶型 PPK 伴胼胝,圖 48-10B)、足底疼痛(厚胼胝下水皰形成所致,約 60% 為神經病變性疼痛)。
- 其他:口腔白色角化症(70%)、毛囊皮脂腺囊腫(脂囊腫等,圖 48-10C,多 KRT17)、新生兒齒(15%,幾乎專屬 KRT17)、毛囊角化症。
- 基因型–表現型(表 48-3):KRT6A(40%,p.Asn172del,10 趾甲皆侵、喉部白色角化致呼吸窘迫);KRT16(25–30%,輕微 FPPK);KRT17(20%,囊腫、新生兒齒);KRT6B;KRT6C(最輕)。
- 特定治療:類視黃醇療效矛盾(可加重疼痛須停藥);甲失養以機械性治療+浸泡為主;標靶療法 — 雷帕黴素 (rapamycin) 阻斷 KRT6 mRNA 轉譯改善足底胼胝;KRT6A 特異性 siRNA 第 Ib 期試驗使胼胝消退並控制疼痛。
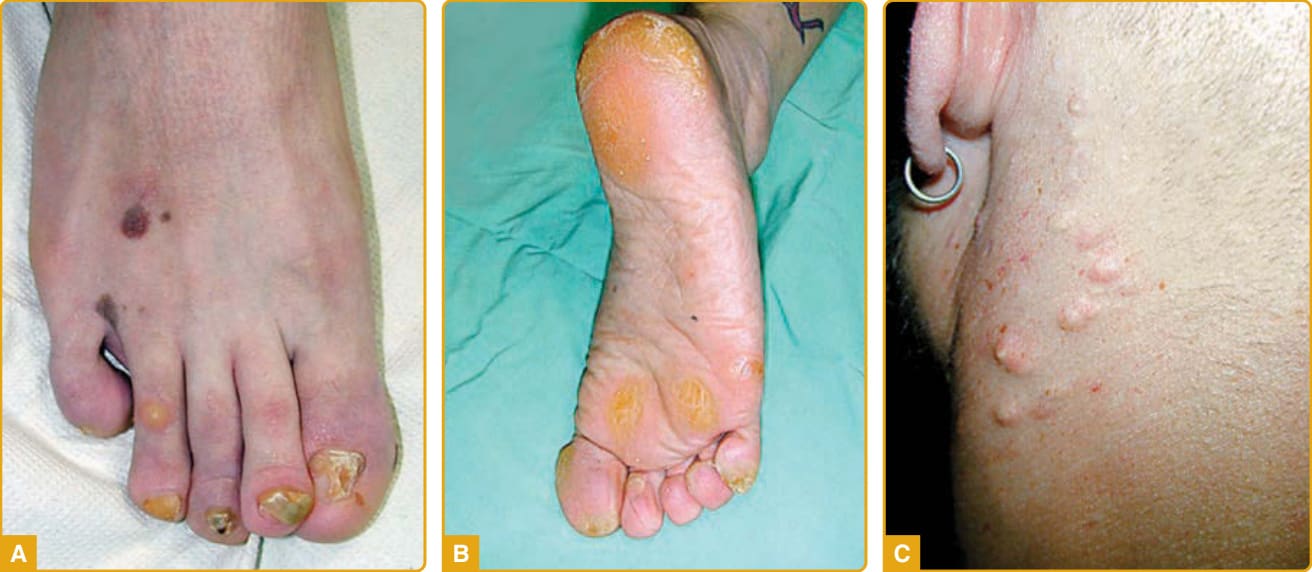
圖 48-10:先天性厚甲症。KRT6A 突變的 V 形厚甲與甲下過度角化 (A)、承重區胼胝的局灶型 PPK (B)、KRT17 突變的表皮內含囊腫 (C)
Howel-Evans 症候群(顯性)
- 局灶型 PPK(圖 48-7B)+黏膜(特別食道)SCC,伴毛囊過度角化、口腔白色角化症。約 95% 病人 65 歲時發展食道癌;建議每年胃鏡篩檢。
- 機轉:RHBDF2(iRhom2)GOF 錯義突變,調節 ADAM17/EGFR 訊息傳遞。
Richner-Hanhart 症候群 (RHS)(隱性)
- 三聯徵:疼痛性局灶型 PPK、雙側角膜炎、智能不足;源於酪胺酸結晶胞內累積。眼部表現常先於皮膚(75%)。
- 機轉:TAT(酪胺酸轉胺酶)突變致酪胺酸累積。診斷:血清酪胺酸升高(苯丙胺酸正常)+尿酪胺酸代謝物;尿琥珀醯丙酮可區分第 I 型酪胺酸血症。
- 特定治療:早期限制酪胺酸/苯丙胺酸飲食為關鍵;類視黃醇改善皮膚與眼睛但無法預防智能不足。
Carvajal 症候群 (CS)(隱性)
- 三聯徵:羊毛狀髮、條紋型 PPK (SPPK)、左心室擴張型心肌病變(>90% 第二個十年嚴重受影響,充血性心衰竭與心律不整為青春期主要死因)。
- 機轉:DSP(橋粒斑蛋白)突變。雜合子 DSP 突變另與 DCWHKTA、第 8 型 ARVD、孤立性 SPPK 相關。
點狀型 PPK
點狀型 PPK 第 I 型 (PPKP1)(顯性)
- 多發、過度角化、中央凹陷、黃至褐色丘疹,不規則分布於掌蹠(圖 48-11),可疼痛、隨年齡融合;伍氏燈下白色螢光。
- 機轉:AAGAB(p34,PPKP1A)或 COL14A1(第 XIV 型膠原蛋白 α1,PPKP1B)雜合子突變致單倍體不足。
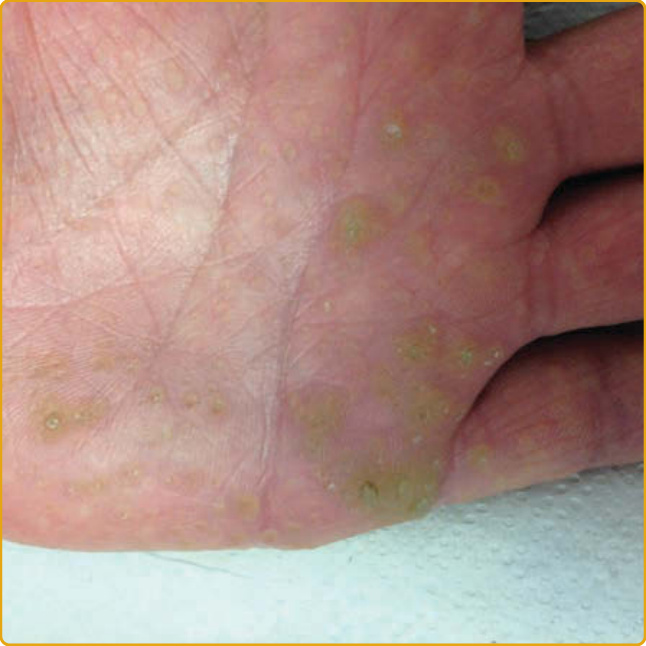
圖 48-11:AAGAB 突變的點狀型 PPK 第 I 型,多發中央凹陷的黃褐色丘疹
點狀型 PPK 第 2 型 (PPKP2)(汗孔角化型,顯性)
- 多發微小膚色至黃色角化棘狀物,伍氏燈下「月光下繁星」螢光;分子病因未明,病理為角樣板層樣柱狀角化不全。
點狀型 PPK 第 3 型 (PPKP3,AKE of Costa)(顯性)
- 圓至橢圓形白黃色半透明丘疹,好發大小魚際與壓力點,線狀/鋪石排列(圖 48-12)。病理為彈性組織減少與彈力纖維變性 (elastorrhexis);致病基因未明(可能 2p25-p12)。
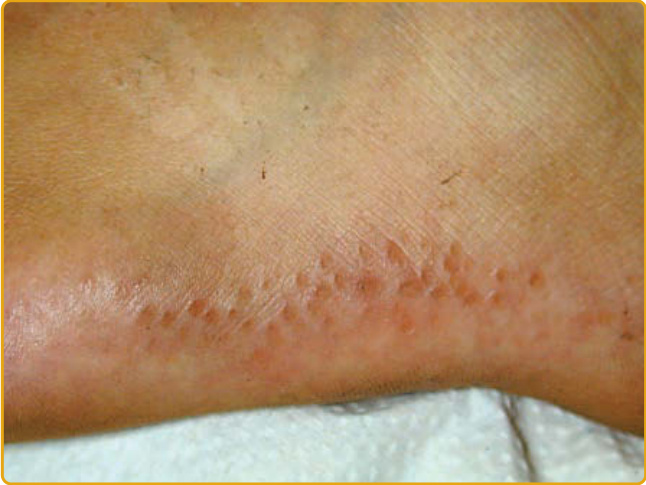
圖 48-12:點狀型 PPK 第 III 型(肢端角化彈力纖維變性症),足底外側線狀排列的黃色丘疹
Cole 病(顯性,伴皮膚外特徵)
- 點狀型 PPK+色素減少斑(近端四肢);可伴肌腱、乳房、脾臟鈣化。
- 機轉:ENPP1 雜合子突變(SMB 區域),與異常胰島素訊息傳遞相關。