Inherited Palmoplantar Keratodermas
8
AT-A-GLANCE
■ Inherited palmoplantar keratodermas (PPKs) are a heterogeneous group of genodermatoses characterized by hyperkeratosis of the palms and soles, with or without associated features. They are usually classified according to their morphology (diffuse, focal, punctate), mode of inheritance, and the presence or absence of extracutaneous features.
■ Epidermolytic PPK (EPPK) is the most common form of diffuse keratoderma. It results from heterozygous mutations in KRT9 (most cases) or KRT1 (the minority of cases) encoding keratin 9 and keratin 1, respectively. Epidermolytic hyperkeratosis is seen on histology. A similar but milder phenotype of diffuse PPK is evident in nonepidermolytic PPK (NEPPK) Unna-Thost type, caused by heterozygous mutations in KRT1.
■ Diffuse NEPPK with transgrediens may be observed in Greither syndrome and PPK Bothnia type caused by missense heterozygous mutations in KRT1 and heterozygous gain-of-function mutations in AQP5, respectively. PPK Bothnia type also manifests with a white, spongy appearance of the palms and soles upon exposure to water.
■ Mal de Meleda is a form of autosomal recessive, progressive, diffuse, mutilating PPK with transgrediens caused by biallelic mutations in SLURP1.
■ Nagashima-type PPK is the most common type of PPK in the Asian population and is characterized by diffuse, transgrediens, nonprogressive, nonmutilating PPK. The palmoplantar skin assumes a typical whitish spongy appearance after water immersion. This form of PPK is caused by biallelic mutations in SERPIN7 encoding a serine protease inhibitor.
■ Olmsted syndrome, caused by mutations in TRPV3 (autosomal dominant or autosomal recessive inheritance) or MBTPS2 (X-linked recessive inheritance), is characterized by diffuse mutilating PPK with periorificial keratotic plaques.
■ Heterozygous mutations in the gene GJB2 encoding connexin 26 result in four genodermatoses featuring PPK and hearing impairment, including Vohwinkel syndrome (mutilating honeycomblike PPK and starfish-shaped keratotic plaques), keratosis–ichthyosis–deafness (KID) syndrome (erythrokeratoderma, grainy PPK, abnormal ectodermal features, progressive keratitis, and recurrent infections), Bart-Pumphrey syndrome
(honeycomb-like PPK, knuckle pads, and leukonychia), and PPK with deafness syndrome. A form of KID syndrome associated with congenital atrichia results from mutations in GJB6 encoding Cx30.
■ Loricrin keratoderma, caused by heterozygous frameshift mutations in LOR encoding loricrin, is a variant of Vohwinkel syndrome not associated with hearing impairment but featuring generalized ichthyosis.
■ PPK with hearing impairment can also result from point mutations in the MTTS1 gene encoding a mitochondrial transfer RNA.
■ Hidrotic ectodermal dysplasia (Clouston syndrome) is a form of ectodermal dysplasia associated with diffuse PPK and nail and hair abnormalities caused by heterozygous mutations in GJB6 encoding connexin 30.
■ Huriez syndrome, an autosomal dominant inherited PPK with unknown etiology, is characterized by diffuse PPK, scleroatrophy, sclerodactyly, and occurrence of squamous cell carcinomas within atrophic skin.
■ Papillon-Lefevre syndrome is an autosomal recessive disorder caused by mutations in the gene CTSC encoding cathepsin C. It is characterized by diffuse PPK with transgrediens and severe progressive periodontitis.
■ Naxos disease (ND) and Carvajal syndrome (CS), caused by mutations in the genes JUP and DSP encoding plakoglobin and desmoplakin, respectively, are cardiocutaneous syndromes featuring PPK (diffuse in ND and striate in CS), woolly hair, and cardiomyopathy (right ventriculopathy in ND and mainly left ventricle involvement in CS).
■ Striate PPK results from heterozygous nonsense or frameshift mutations in the genes encoding desmoglein 1, desmoplakin, and to a lesser degree keratin 1.
■ Pachyonychia congenita is a group of rare autosomal dominant disorders caused by mutations in one of five keratin genes, including KRT6A, KRT6B, KRT6C, KRT16, or KRT17 encoding keratins 6a, 6b, 6c, 16, and 17, respectively, known to be expressed in differentiated epithelial tissues. It manifests with focal painful PPK, thick dystrophic nails with a characteristic appearance, and associated features such as oral leukokeratosis, pilosebaceous cysts, and natal teeth.
(Continued)
AT-A-GLANCE (Continued)
■ Howel-Evans syndrome is an autosomal dominant disorder featuring focal PPK and mucosal (particularly esophageal) squamous cell carcinomas with associated findings of follicular hyperkeratosis and oral leukokeratosis caused by heterozygous mutations in RHBDF2, encoding iRhom2, a protein known to play a role in epidermal growth factor receptor (EGFR) signaling.
■ Richner-Hanhart syndrome is an autosomal recessive disease caused by mutations in the TAT gene encoding tyrosine aminotransferase, a hepatic cytosolic enzyme important for tyrosine and phenylalanine metabolism. It is characterized by painful focal PPK, bilateral keratitis, and mental retardation.
■ Punctate PPK (PPKP) type 1 is characterized by multiple, painful, yellow-brown hyperkeratotic papules on the palms and soles that most commonly appear during the first or second decades of life. It may result from mutations in one of two genes: AAGAB encoding p34 protein with resultant upregulation of EGFR or COL14A1 encoding collagen type XIV α1. PPKP
INTRODUCTION
The term palmoplantar keratoderma (PPK) refers to a group of potentially debilitating and clinically and genetically heterogeneous disorders of cornification which are clinically characterized by abnormal focal or diffuse thickening of the skin on the palms and soles. These disorders can occur as both acquired or inherited conditions. Inherited PPKs are characterized by familial occurrence and a relatively early age of onset in most cases. Genodermatoses associated with PPK are shown in Table 48-1, and several examples are presented in Fig. 48-1. This chapter focuses on inherited disorders in which PPK is a dominant feature. Several classification systems of PPKs have been proposed. Originally, the classification was based on assessment of several clinical and histologic characteristics, including the specific morphology and pattern of palmoplantar hyperkeratosis, the extent of involvement (isolated PPK or syndromic PPK associated with ectodermal defects and/or extracutaneous manifestations), mode of inheritance (autosomal dominant, autosomal recessive, X-linked, mitochondrial), age of onset, and the presence or absence of epidermolysis on histology.1-3 Three morphological patterns are usually distinguished: (1) diffuse PPK with uniform involvement of the entire palmoplantar surface; (2) focal PPK with localized hyperkeratosis predominantly on pressure points that is further subdivided into areata or nummular type (oval lesions, mainly on the plantar surface) and striate type (longitudinal hyperkeratotic lesions extending from the palms along the volar surface of the fingers associated with focal to diffuse thickening of the
8
type 2 is characterized by skin-colored to yellow asymptomatic keratotic spines with histopathologic findings resembling cornoid lamella of porokeratosis. PPKP type 3 (acrokeratoelastoidosis of Costa) manifests with round to oval, whiteyellow translucent papules with predilection to the thenar and hypothenar areas and pressure points with histopathologic findings of decreased elastic tissue and fragmented elastic fibers. The genes responsible for PPKP type 2 and 3 are still to be elucidated.
■ Cole disease, characterized by punctate PPK, hypopigmented macules, and possible internal organs calcifications, is caused by mutations in ENPP1 leading to impaired insulin signaling.
■ Management of all forms of PPK focuses on topical treatments (mainly emollients and keratolytic agents), mechanical measures, methods to relieve pain or inhibit sweating, treatment of secondary infections, and various walking aids. Oral retinoids have been shown to be effective in several types of inherited PPK, and the development of more targeted therapeutic approaches is underway.
plantar skin); and (3) punctate PPK with multiple, discrete 1-mm to 1-cm round keratotic papules over the palms and soles. In addition, palmoplantar involvement may feature transgrediens (extension of hyperkeratosis onto the dorsal aspects of the fingers, toes, hands, feet, and flexor aspects of the wrists and heels) or mutilation caused by pseudoainhum (constricting bands around digits) formation.4 With the deciphering of the pathogenesis of most forms of inherited PPKs, novel classification schemes include the etiology of the PPK in addition to morphological features.2-6
Elucidating the molecular disease mechanisms of PPKs has led to the discovery of new disorders and syndromes, providing new insights into the biological roles of epidermal structural components and paving the way for the development of novel, disorderspecific treatment modalities.7-9
EPIDEMIOLOGY
Although inherited PPKs are rare disorders when considered individually, their actual prevalence and incidence may be underestimated given the fact that mildly affected individuals do not require medical intervention or may be incorrectly diagnosed.8,10 However, in several countries or communities in which consanguineous marriages are common, inherited PPKs may occur with high prevalence. In Northern Sweden, nonepidermolytic PPK (NEPPK) is characterized by a prevalence of 0.3% to 0.55%,11 but in Northern Ireland, the prevalence of epidermolytic PPK (EPPK) was found to be 4.4 per 100,000 people.12 In South India, a
817
8
ENTITY WITH ASSOCIATED PALMOPLANTAR KERATODERMA
Ichthyoses Autosomal recessive congenital ichthyosis (ARCI)
Epidermolytic ichthyosis (Fig. 48-10A)
Ichthyosis Curth-Macklin
Keratosis linearis–ichthyosis congenital– keratoderma (KLICK) syndrome
Trichothiodystrophy
Chanarin-Dorfman syndrome
Sjogren-Larsson syndrome
Conradi-Hunermann-Happle syndrome (Fig. 48-10B)
CEDNIK syndrome
MEDNIK syndrome
Ichthyosis prematurity syndrome
Erythrokeratodermias Erythrokeratodermia variabilis
Progressive symmetric erythrokeratodermia
Epidermolysis bullosa (EB) EB simplex (localized and generalized severe)
Recessive dystrophic epidermolysis bullosa
Kindler syndrome
Ectodermal dysplasia–skin fragility syndrome
Ectodermal dysplasias Schopf-Schulz-Passarge syndrome
Oculodentodigital dysplasia
Odontoonychodermal dysplasia
Naegeli-Franceschetti-Jadassohn syndrome
Dermatopathia pigmentosa reticularis
Autosomal recessive ectodermal dysplasia syndrome
Rasopathies Costello syndrome
Others Darier disease (see Fig. 48-10C)
Acrokeratosis verruciformis of Hopf
Cowden syndrome
Epidermodysplasia verruciformis
Pityriasis rubra pilaris
Severe dermatitis, multiple allergies and metabolic wasting (SAM) syndrome
Dyskeratosis congenita
Keratosis follicularis spinulosa decalvans (KFSD)
Ichthyosis follicularis congenital atrichia and photophobia syndrome (IFAP)
Basal cell nevus syndrome (Gorlin syndrome)
Basal cell nevus syndrome (Gorlin syndrome)
CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma; MEDNIK, mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratodermia.
prevalence rate of 5.2 in 10,000 was documented with Unna-Thost syndrome being the most prevalent entity seen in approximately 38% of cases.13 In general, EPPK
818
is the most common form of diffuse keratoderma with a worldwide incidence of 2.2 to 4.4 per 100,000 live newborns.1,14,15
DIAGNOSIS
As briefly mentioned already, the past years have led to significant progress in our understanding of the pathogenesis of inherited PPKs. These advances can in turn be harnessed to facilitate the diagnosis of these conditions using algorithms integrating both clinical and molecular features. Fig. 48-2 depicts one such algorithm based on the use of four sets of data: (1) cutaneous findings including PPK morphology (eg, diffuse, focal, punctate, mutilating), hair (eg, woolly hair), or nail abnormalities, as well as features associated with other genodermatoses (eg, skin fragility); (2) extracutaneous findings (eg, periodontitis, deafness); (3) mode of inheritance (eg, autosomal dominant, autosomal recessive, mitochondrial inheritance); and (4) histopathologic findings such as epidermolytic hyperkeratosis, disadhesion of keratinocytes, or parakeratosis. Using this algorithm, almost forms of PPK can be assigned to one set of genes which should be sequenced to lead to the final diagnosis.
DIFFERENTIAL DIAGNOSIS
Palmoplantar hyperkeratosis may represent a feature of both inherited and acquired conditions. Acquired conditions listed in Table 48-2 usually occur later in life and include inflammatory disorders (eg, psoriasis, lichen planus, pityriasis rubra pilaris, chronic dermatitis, reactive arthritis), paraneoplastic keratoderma, cutaneous lymphoma, chemical and drug exposure, malnutrition, metabolic disorders (eg, hypothyroidism), keratoderma climactericum, and infectious diseases (eg, dermatophytes, scabies, verruca vulgaris and syphilis).16
Genodermatoses that feature PPK are listed in Table 48-1. Several distinctive characteristics can be shared by some of these diseases. For example, autoamputation of digits can accompany PPK in loricrin keratoderma (LK), Vohwinkel syndrome (VS), Olmsted syndrome (OS), Sybert PPK, mal-de-Meleda (MDM), PPK-Gamborg-Nielsen, and PPK congenital alopecia syndrome 2 (PPKCA2).17,18 Other nonhereditary diseases that may cause constricting bands with or without hyperkeratosis include leprosy, tertiary syphilis, yaws, scleroderma, Raynaud syndrome, amniotic bands, ergotamine poisoning, spinal medulla tumors, syringomyelia, and scar formation from frostbite, burns, and trauma.19
PPK with transgrediens may be observed in LK, Nagashima type-PPK (NPPK), Greither disease, OS, Papillon-Lefevre syndrome (PLS). PPK with sensorineural hearing loss (SNHL) should raise the differential diagnosis of VS syndrome, keratitis–ichthyosis–deafness (KID) syndrome and other GJB2-associated diseases, or mitochondrial inherited-PPK with deafness.
A
B C
8
Several syndromes are characterized by PPK associated with squamous cell carcinoma (SCC) within areas of abnormal keratinization (eg, Huriez and Unna-Thost syndromes). Other genodermatoses including dyskeratosis congenita, Rothmund-Thomson syndrome, and
xeroderma pigmentosum are associated with elevated absolute rates of SCC. A few forms of PPKs feature gingival and dental anomalies (eg, OS, PLS, Haim-Munk syndrome, and Kindler syndromes).
819
8
Diagnostic algorithm for inherited palmoplantar keratodermas
Skin disorders with PPK
N
Mainly PPK?
SPPK/ND/CS DSP, DSG1, JUP
Disadhesion
Binucleate KC
Y Y
N Epidermolytic changes?
Other pathological features?
SC nuclei
N Y
Y
EI KRT1/9 Extracutaneous features? Deafness
N
Punctate
IH Curth-Macklin KRT1
Loricrin PPK LOR PPK with deafness GJB6/mitDNA
Y
N Teeth
PLS/PC CTSC/KRT6a
RHS TAT
Eyes
Focal or diffuse Mutilating
Water sensitive
Pain Linear lesions
Soles
Thenar
NEPPK
PC KRT6a/b/c KRT16, KRT17
KLICK syndrome POMP
AR
PPPK1 AAGAB
AKE FAH
PC/Clouston GJB6/KRT6/16/17
Olmsted TRPV3 Huriez
AD
Nails
Clouston/ND/CS GJB6/DSP/JUP/KANK2
Hair
AR
MDM SLURP1
AD
Howell Evans/PPK-SR RHBDF2/RSPO1
Others
SERPINB7 AQP5
Wrinkling of palmoplantar skin may be a feature in aquagenic wrinkling of the palms with CFTR mutations, PPK Bothnia type with AQP5 mutations, NPPK with SERPINB7 mutations, and acrokeratoelastoidosis (AKE). The differential diagnosis of punctate palmoplantar keratosis includes verruca vulgaris, corns over pressure points, porokeratosis punctate palmaris et plantaris, keratosis punctate of the palmar creases, Cole disease, Darier disease, Cowden disease, Gorlin syndrome, acrokeratosis verruciformis of Hopf, epidermodysplasia verruciformis, and arsenic exposure.20-22
PPK associated with sclerosis localized to dorsal hands and sclerodactyly may be a feature of Huriez syndrome, Unna-Thost syndrome (occasionally shows sclerodactyly or nail changes), PPKCA2, and palmoplantar hyperkeratosis with SCC of the skin and sex reversal. Systemic sclerosis may show similar features. PPK with nail changes typical for pachyonychia congenita (PC) may be observed in Clouston syndrome
820
caused by GJB6 mutations (may show similar nail changes and painful keratoderma) or homozygous mutations in FZD6, encoding frizzled 6, a Wnt-signaling pathway receptor in the nail matrix, which was recently discovered as the cause of congenital nail dystrophy.23 Dyskeratosis congenita manifest with features overlapping with PC, including nail dystrophy, PPK, hyperhidrosis, and oral leukoplakia. Onychomycosis should be excluded in cases of PPK with nail findings.
COMMON THERAPEUTIC APPROACHES
In general, the management of all forms of PPK is aimed at alleviating disease manifestations and focuses on mechanical measures, methods to relieve pain, treatment of secondary infections, and various
Inflammatory Skin Disorders Palmoplantar psoriasis Lichen planus Pityriasis rubra pilaris Chronic dermatitis Reactive arthritis
Metabolic Disorders Hypothyroidism, myxedema Chronic lymphedema
Drugs and Chemical Exposures Lithium, hydroxyurea, verapamil, bleomycin Chemotherapeutic agents Chlorinated hydrocarbon Chronic arsenic exposure
Infectious Diseases Dermatophytes Scabies Verruca vulgaris Syphilis Leprosy Tuberculosis
Malignancies Paraneoplastic keratoderma Cutaneous lymphoma
Malnutrition
Miscellaneous Keratoderma climactericum Pitted keratolysis Punctate porokeratosis
Miscellaneous Keratoderma climactericum Pitted keratolysis Punctate porokeratosis
walking aids. Topical treatment includes the regular use of emollients and topical keratolytic formulations (eg, 10% to 20% salicylic acid or 35% to 70% propylene glycol combined with thick emollient), with occlusion if necessary. In addition, topical calcipotriol and topical tazarotene have been shown to be effective in EPPK and PLS, respectively.24 Dermatophyte or bacterial secondary infections and hyperhidrosis should be diagnosed and treated aggressively to avoid disease aggravation. Soaking in water and mechanical removal of hyperkeratotic areas (eg, grooming and trimming) are additional therapeutic measures that may provide symptom relief. Pain can be relieved by orthotics or insoles, wicking socks, ventilated or cushioned footwear, and maintaining body weight to reduce repeated trauma to the feet and the tendency to develop callus and blisters.
ORAL RETINOIDS
ORAL RETINOIDS
Oral retinoids are usually effective. Low-dose, shortterm, or intermittent therapy are sometimes recommended given the possible long-term side effects, the chronic nature of the condition, and the risk of
8
aggravated skin fragility, which may exacerbate disease manifestation in several types of PPK. More specifically, systemic retinoids were shown to be effective in EPPK,25 MDM,26 VS, and LK27-30 and in cases of OS,31-34 Huriez syndrome,35 punctate PPK 1,36,37 and PLS (for both PPK and periodontopathy).38-40 Systemic retinoids have been found to be effective in KID syndrome (for hyperkeratosis, dissecting cellulitis of the scalp, and cancer chemoprophylaxis), although some reports demonstrated only partial response and exacerbation of corneal neovascularization with isotretinoin.28,41-46 Accordingly, a low starting dose of acitretin (0.5 mg/kg/day) or alitretinoin (10 mg/day) with gradual increase depending on response and tolerability is recommended.47-49
SURGICAL THERAPY
SURGICAL THERAPY
Surgical excision and grafting is an option in focal PPK and in cases associated with mutilation.4,50 Specific surgical approaches are discussed later.
INHERITED PALMOPLANTAR KERATODERMAS
DIFFUSE PALMOPLANTAR KERATODERMAS WITHOUT EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE
DIFFUSE PALMOPLANTAR
KERATODERMAS WITHOUT
EXTRACUTANEOUS
FEATURES, DOMINANT
INHERITANCE
EPIDERMOLYTIC PALMOPLANTAR KERATODERMA
Clinical Features: EPPK (Vörner type; Online Mendelian Inheritance in Man [OMIM] #144200), first described by Vörner in 1901, is an autosomal dominant disorder characterized by yellowish, diffuse PPK with erythematous sharp margins at the edge of the palms (Fig. 48-3A) and soles (Fig. 48-3B).51 Most cases present at birth or during the first weeks of life, although appearance up to the third year of life has been reported. The initial presentation is palmoplantar erythema that starts at the margins of the palms and soles, extends toward the center, and subsequently becomes covered with thick scale and remains stable throughout life.25,52 Transgrediens and hyperkeratotic lesions over the elbows, knees, and knuckle pads have been reported.53-60 It may be complicated with painful fissures, hyperhidrosis, maceration, and secondary bacterial and fungal infections with an offending odor.57 Digital mutilation with pseudoainhum and
821
8
A
B
camptodactyly has been reported.59-62 In most cases, PPK is an isolated finding, although the rare occurrence of EPPK in association with leg ulcers, scleroderma, familial multiple carcinomas, and Ehlers-Danlos syndrome type III has been reported.25,63-65
Etiology and Pathogenesis: EPPK is an autosomal dominant genodermatosis caused by heterozygous mutations in KRT9 encoding keratin 9.66
Mutations in KRT1 encoding keratin 1 have also been reported in association with EPPK in a minority of cases.67-70
Keratin 9 is a type I intermediate filament protein whose expression is confined to the suprabasal layers of the palmoplantar epidermis. Similar to all other keratins, KRT9 comprises three major domains: the head domain; the central α-helix rod domain, which is composed of four helical subsegments (1A, 1B, 2A, and 2B) that are interrupted by three nonhelical linker domains (L1, L12, and L2); and the tail domain.71,72 Most KRT9 mutations identified to date are located in the 1A or 2B segments of the α-helix rod domain, which are highly conserved in all keratin proteins and are essential for the formation of the keratin heterodimer and
822
play a pivotal role in the congregation and stabilization of keratin intermediate filaments. Most causative genetic defects are missense or frameshift (small in-frame insertion–deletion) mutations acting in a dominant-negative manner. A mutational hotspot exists at codon 163 of KRT9 (in the central α-helix rod domain) that encodes the amino acid arginine, which is often mutated to tryptophan, glutamine, or proline. p.Arg163Trp accounts for approximately 30% of all EPPK-causing mutations across different ethnic groups.73,74
Keratin 1 is a type II keratin expressed at the suprabasal cells committed to terminal differentiation that forms heterodimer with keratin 9 in palmoplantar epidermis and with keratin 10 in all other skin surfaces, including hair-bearing skin and other stratified orthokeratinized squamous epithelia.6,75-77 The reported KRT1 mutations in families with EPPK involve larger in-frame deletions affecting the helix boundary motifs of the rod domain,68 different from the missense mutations in the same domains reported in epidermolytic ichthyosis (EI; see Chap. 47) and from frameshift mutations in the V2 domain that have been found in striate keratoderma and in ichthyosis hystrix Curth- Macklin.78,79 Other mutations in KRT1 that were associated with EPPK are gain-of-function (GOF) mutations at the beginning of the helix 1B domain of the central α-helical coiled-coil rod domain and an insertion of 18 amino acids in the 2B rod domain, emphasizing that mutations located more central in the rod domain of keratin 1 do not disrupt the function of filament assembly and stability as much as those in the helix initiation and termination sequence seen in EI cases.67,70
Pathology: Skin biopsies show compact orthohyperkeratosis, hypergranulosis, acanthosis, and epidermolytic hyperkeratosis manifested by perinuclear vacuolization of the keratinocytes and large irregularly-shaped keratohyaline granules located in the granular layers of the epidermis. Moreover, dyskeratosis manifested by intracytoplasmic and perinuclear eosinophilic homogenizations with round-oval eosinophilic inclusions were identified in involved epidermis corresponding to the intracytoplasmic aggregates and clumps of tonofilaments seen ultrastructurally.3,80,81
More than one biopsy may be required to demonstrate epidermolytic changes and the typical histologic alterations of epidermolytic hyperkeratosis may be noticed in the acrosyringium.25
Ultrastructural findings include intracytoplasmic vacuolization in the upper spinous and granular cell layers; disruption and dispersion of the cellular organelles with indistinct cellular borders; intracytoplasmic dense aggregates of tonofilaments surrounding the nuclei; dense clumps of tonofilaments; abundant glycogen and ribosomes; detachment of tonofibrils from desmosomal plaques; large keratohyalin granules present from the mid malpighian layers; “composite” keratohyalin granules consisting of a homogeneous, relatively electron-lucent core; and dense peripheral deposits.65,80,82-86
Specific Treatments: Inhibition of Krt9 mutant allele expression in a mouse model of EPPK using ribonucleic acid interference (RNAi)–based therapy has been demonstrated.73 In addition, a transgenic mouse model of EPPK was treated with a mutant-specific short hairpin RNA (shRNA) that resulted in knockdown of the mutant protein with restoration of normal morphology and function of the skin.87
UNNA-THOST PALMOPLANTAR KERATODERMA Clinical Features: Diffuse nonepidermolytic PPK (NEPPK, Unna-Thost type; OMIM #600962) is an autosomal dominant genodermatosis first described by Unna and Thost in 1880. Clinical features are similar to those of diffuse EPPK, although they are often milder, with presentation in the first few months of life. Onset during the third decade of life has been reported. NEPPK is characterized by diffuse, welldemarcated, yellowish, thick hyperkeratosis with an erythematous rim overlying the palms and soles. It tends to be smoother and waxier than EPPK, which is thicker and fissured.4 Hyperhidrosis, as well as refractory secondary dermatophyte infections and pitted keratolysis leading to maceration and desquamation, are common findings seen more frequently in NEPPK than in EPPK.88-91 In addition, the presence of hyperkeratosis around the umbilicus and the nipple with mild thickening and dryness of the knees and elbows has been reported.92 Cases associated with atopic dermatitis93 and verrucous carcinoma94 have been described. Associations with acrocyanosis of the distal third of the extremities, clinodactyly of the fifth finger, and perianal or pericrural involvement have been reported.95-97
In general, EPPK and NEPPK show substantial clinical overlap, and histopathologic evaluation is often required to distinguish between them.
Etiology and Pathogenesis: Originally, Unna-Thost PPK was recognized as a nonepidermolytic form of diffuse PPK clinically resembling Vörner PPK. However, Kuster and coworkers investigated the original family reported by Unna and Thost and found epidermolytic hyperkeratosis on histology and identified a mutation located in the coil-1A domain at the beginning of the central rod domain of KRT9. This mutations is in close proximity to the mutation found in the original family reported by Vörner, suggesting that Unna-Thost and Vörner PPKs are the same entity.57,98
In a single family with diffuse NEPPK, a missense mutation was identified in the terminal V1 variable subdomain (nonhelical head domain) of keratin 1. In contrast with other reported KRT1 mutations involving central domains important for filament assembly and stability that result in epidermolysis, this mutation is likely to be important for interactions of keratin filaments with other cellular components including the desmosomes.92 This mutation results in distortion in
8
keratinocytes shape and prevention of efficient distribution of lamellar body lipid material with impaired barrier formation.99
Another missense mutation in KRT1 resulting in a mild phenotype has been identified in the 2B rod domain, again underscoring that mutations affecting the central part of the keratin 1 rod domain do not disrupt filament assembly to the same extent as mutations located at the initiation or termination motif of the helix sequence.100
Diffuse NEPPK, without other clinical features of PC, has been reported with heterozygous mutations in KRT6c and KRT16. The phenotype consists of diffuse PPK on the soles and focal hyperkeratosis on the palms. In cases of NEPPK when no pathogenic mutation is identified in KRT1, screening for KRT6c and KRT16 mutations may be valuable.101,102
Pathology: Diffuse NEPPK shows nonspecific histopathologic findings, including hyperkeratosis, orthokeratosis, acanthosis, and either hyper- or hypogranulosis with no evidence of epidermolytic hyperkeratosis.92,93 Close inspection and repeated biopsies are needed to exclude small foci of epidermolytic hyperkeratosis compatible with EPPK.
GREITHER SYNDROME Clinical Features: Transgrediens et progrediens PPK (Greither syndrome; OMIM #133200) was originally described in 1952 by Greither.103 Greither syndrome is an autosomal dominant disease with marked intra- and interfamilial variability. It characteristically starts to develop after the second year of life (although appearance soon after birth or later in childhood and adolescence has been documented). It is characterized by diffuse, thickened, scaly, yellowish PPK with an erythematous rim and transgrediens. The involvement of the skin over the Achilles tendon and gradual extension of patchy hyperkeratotic, erythematous, and hyperpigmented papules and plaques towards the shins, knees, thighs, knuckles, wrists, elbows, and flexural areas are typical.95,103-107 Greither PPK tends to involute after the fifth decade of life. Hyperhidrosis is a common feature, and pitted keratolysis may be an associated finding. A case presenting as neonatal blistering and erythroderma, initially diagnosed as EI, has been reported.77 An association with atopic dermatitis has been described.108 A case of malignant melanoma arising on the hyperkeratotic sole of a patient with Greither PPK has been documented.109 Severe cases of PPK may be complicated by spontaneous autoamputation and digit deformities.110 Greither syndrome has been described in association with incontinentia pigmenti, acrocyanosis, and erythrokeratoderma variabilis in isolated cases.104,111-113
Sybert PPK is another form of progressive, diffuse, autosomal dominant PPK with transgrediens and autoamputation, however it manifests with more severe hyperkeratosis as compared to Greither PPK.110
823
8
Etiology and Pathogenesis: Greither syndrome is inherited in an autosomal dominant fashion and was shown to result from a missense mutation in KRT1 affecting the helix initiation motif at the amino terminal end of the central rod domain of keratin 1,108
a region known to be essential for effective filament assembly.114 The causative gene for Sybert PPK has not been identified yet.
Pathology: The characteristic findings in Greither syndrome are acanthosis, marked hyperkeratosis, and hypergranulosis. A case with striking vacuolation of the superficial keratinocytes and numerous keratohyaline granules in the granular layer consistent with epidermolytic hyperkeratosis has been reported.108
Moreover, focal depressions of the epidermis occupied by round foci of a compact orthokeratotic horny layer have been reported.106 The described histopathologic findings in Sybert PPK are epidermal hyperplasia with hypergranulosis, parakeratosis, and orthokeratosis, as well as a sparse lymphocytic infiltrate in the papillary dermis. Ultrastructural studies revealed normal keratin filaments with abnormal structure and distribution of keratohyaline granules.110
LORICRIN KERATODERMA Clinical Features: LK (mutilating keratoderma with ichthyosis, VS Camisa type; OMIM #604117) is a rare, autosomal dominant disorder. It features honeycomb-like PPK, starfish-like hyperkeratoses, prominent knuckle pads on the dorsal aspects of the hands, and pseudoainhum leading to autoamputation of the digits. These signs, when associated with hearing impairment, are referred to as VS (see later).115 LK is a variant of VS reported by Camisa and Rossana in 1984 with no hearing impairment but with prominent generalized ichthyosis. The two essential clinical features for establishing the diagnosis are the characteristic honeycomb-like keratoderma and generalized ichthyosis, although the phenotype of LK is truly heterogeneous. LK manifests at birth or in early childhood and progresses gradually throughout adulthood. The clinical findings include moderate, generalized ichthyosiform dermatitis with dryness and fine scales affecting the trunk and
extremities and hyperkeratosis localized to body folds with no evidence of erythema. Palmoplantar involvement is characterized by diffuse, symmetrical, well-demarcated transgrediens honeycomb-like PPK with an erythematous border (Fig. 48-4A),27,116,117
although diffuse hyperkeratosis with no evidence of a honeycomb-like pattern may be seen. Knuckle pads, hyperkeratotic plaques on the dorsal parts of the hands, and pseudoainhum have been reported in LK pedigrees in varying frequencies,117-124 and a collodion membrane may be seen at birth in approximately 35% of cases.120,121,125 Association of LK with vitiligo, atopic dermatitis, neurodevelopmental delay, and microcephaly has been reported.121,123
Etiology and Pathogenesis: LK results from heterozygous frameshift insertion or deletion mutations in LOR which encodes loricrin. Loricrin is synthetized in the granular layer of the epidermis and then migrates to the cell periphery, where it is located beneath the plasma membrane and is crosslinked to other cytosolic proteins including involucrin, forming the cornified cell envelope (CCE).30,117,119-121,123,125-130 Most mutations in LOR implicated in LK lead to a frameshift, delayed termination, and aberrant protein elongation. Experiments in transgenic mice suggest that LOR mutations exert a gain of function deleterious effect. The mutant loricrin forms arginine-rich nuclear localization sequences that “tag” a protein exposed on the cell surface for import into the cell nucleus by nuclear transport. The abnormal localization of loricrin in the nucleus alters nuclear/nucleolar functions, including nonribosomal RNA processing and growth factor signal transduction, and disrupts apoptotic processes in terminally differentiated keratinocytes.124,126,131-134
No clear genotype–phenotype correlation has been reported and variable intrafamilial disease severity has been reported.117,120,121
LK skin demonstrates abnormal epidermal permeability barrier function130,131 similar to the skin barrier defects seen in various ichthyoses, including lamellar ichthyosis with transglutaminase-1 mutations.135 Cell lines overexpressing mutant form of LOR demonstrated increased proliferation rate through activation of AKT-kinase with phosphorylation of ERK1/2, epidermal growth factor receptor (EGFR), and signal
A B C
824
transducer and activator of transcription 3 (STAT3) compared with cell lines expressing the wild-type loricrin.136
Pathology: Histopathologic features include hyperkeratosis, significant parakeratosis, acanthosis, hypergranulosis (although the granular later may appear normal), diffuse vacuolar changes, focal cytolytic changes, and necrotic suprabasal keratinocytes.118
Ultrastructural findings consist of abundant keratohyaline granules, vacuolar changes and disruption of suprabasal keratinocytes, and deposition of electrondense intranuclear inclusions within the granular layer that contain loricrin as shown by immunogold studies.117,118
PALMOPLANTAR KERATODERMA BOTHNIA TYPE Clinical Features: PPK Bothnia type (PPKB; OMIM #600231) was first described in 1994 as an autosomal dominant form of diffuse NEPPK, which has a high prevalence (0.3% to 0.55%) in two provinces of the West and Northwest Gulf of Bothnia in Northern Sweden.11 PPKB has been described also in a large pedigree of Chinese Han descent,137 and a common founder mutation in the British population has been suggested.138 PPKB clinically resembles Nagashima PPK (see later) and usually manifests during infancy or childhood with a diffuse, homogenous hyperkeratosis with a yellowish hue over the palms and soles that extends to the dorsal digits.11
These patients, even with a barely detectable mild phenotype, demonstrate a typical white spongy appearance of affected areas upon exposure to water. Hyperhidrosis, maceration, and secondary fungal infections are common, as are abnormal nails (curved with ragged cuticles). In contrast to this condition, aquagenic wrinkling of the palms, which is often found in conjunction with cystic fibrosis (CF), is characterized by translucent whitish papules, excessive wrinkling, and palmar edema induced by brief exposure to water.137,139,140
Etiology and Pathogenesis: PPKB is an autosomal dominant disorder caused by GOF mutations in the gene AQP5, encoding water-channel protein aquaporin 5 (AQP5), first identified in 2013 by Blaydon and coworkers.141
Aquaporins are cell-membrane transporters that enable the osmotic movement of water across the cell membrane in many cell types. AQP5 is mostly found in the apical plasma membrane and is involved in the excretion of water from exocrine glands (salivary gland, lacrimal gland, and sweat gland); it is also expressed in epithelial cells of the lung and the cornea.141 Of note, hypohidrosis in Sjogren syndrome has been associated with decreased expression of AQP5.142 In normal palmoplantar skin, AQP5 is localized to the plasma membrane of keratinocytes of the stratum granulosum, and in PPKB lesions, AQP5
8
expression is retained. However, affected keratinocytes are subject to increased water uptake through the plasma membrane.141
It has been suggested from protein modeling studies that PPKB-causing mutations increase the diameter of the constriction point of the AQP5 water channel with a direct influence on AQP5 gating or water flow through the channel.141 Aquagenic wrinkling of palms (transient edematous whitish plaques on the palms upon exposure to water and hyperhidrosis) has been associated with aberrant expression of AQP5 in sweat glands.143 Activation of transient receptor potential vanilloid 4 (TRPV4) results in increased cytosolic calcium concentration essential for sweat secretion, although its overactivation in keratinocytes may result in apoptosis and hyperkeratosis as demonstrated in OS caused by TRPV3 mutations.144,145
Because TRPV4 and AQP5 are both expressed in eccrine sweat glands and keratinocytes,141 it has been speculated that palmoplantar hyperhidrosis and hyperkeratosis in PPKB are a result of GOF effect of the AQP5–TRPV4 complex, indicating that PPKB is a skin channelopathy.137 A possible disease mechanism for aquagenic wrinkling of the palms in patients with CF is dysfunction of the TRPV4 channels in keratinocytes, resulting in dysregulated water influx through eccrine ducts.139
Histopathology: Histopathologic findings are nonspecific and include orthohyperkeratosis and a mild lymphocytic infiltrate in the upper dermis.137
DIFFUSE PALMOPLANTAR KERATODERMA WITHOUT EXTRACUTANEOUS FEATURES, RECESSIVE INHERITANCE
DIFFUSE PALMOPLANTAR
KERATODERMA WITHOUT
EXTRACUTANEOUS
FEATURES, RECESSIVE
INHERITANCE
MAL DE MELEDA Clinical Features: MDM (OMIM #248300) is a rare autosomal recessive disorder initially described in patients native to the Croatian island of Meleda (Mljet). The diagnostic criteria for the disease were presented in 1969.146 Since then, the disease has been described in other parts of the world, including the Middle East, Western Europe, and North Africa with higher prevalence in certain populations, mainly in the Mediterranean and Adriatic regions, because of a founder effect. A few cases have been reported in Taiwan, China, Japan, Indonesia, and India.147-157
The onset of the disease is during infancy or early childhood with diffuse transgressive yellowish waxy hyperkeratotic plaques outlined by a red scaly border over the palms (Fig. 48-5A) and soles
825
8
A
B
E
F
D
C
G
H
(Fig. 48-5B) that are preceded by prominent and persistent erythema. The palmoplantar hyperkeratosis progresses with age and extends onto the dorsal surface of the hands and feet in a glove (Fig. 48-5C) and stocking (Fig. 48-5D) distribution, the flexor aspects of the wrists and ankles and over the Achilles tendon, forearms, elbows, and knees.147,154 Involvement of the inguinal and groin regions (Fig. 48-5E) has been described.147,148
Additional features are knuckle pads, lichenoid or keratotic plaques over joints (Fig. 48-5F), perioral erythema, hyperhidrosis with superinfection, malodorous maceration and painful fissures, nail abnormalities (eg, koilonychia, clubbing, onycholysis, nail thickening, dystrophy and subungual hyperkeratosis) (Fig. 48-5G), sclerodactyly, and brachydactyly. Circumscribed hyperkeratotic plaques over the fingers and toes result in a cone-shaped appearance (Fig. 48-5H), digital constrictions, pseudoainhum, and progressive functional impairment with reduced mobility.158-160
Although penetrance is complete, phenotype varies with geographic origin and ethnic background. In addition, no clear genotype–phenotype correlation has been identified, and environmental factors (mechanical trauma and heat) seem to influence the disease course.147,148,150,154 A case with no plantar involvement, a milder phenotype with slightly erythematous palmoplantar keratotic plaques, and an atypical appearance with multiple 2- to 5-mm keratolytic pits outlined by brownish red erythema over the palmoplantar surface (mainly plantar skin) have been described.26,147,154 Cases with no evidence of transgrediens have been described.151 Heterozygous carriers may manifest with mild diffuse PPK or smooth skin on the palms and soles with keratotic papules.161
Several cases of malignant melanoma (MM) arising in the hyperkeratotic lesions of patients with MDM have been described in the literature.149,162,163 A case
826
complicated by irregular hyperpigmented spots on palmoplantar skin and the appearance of Bowen disease on the sole in a patient have been reported.164,165
Accordingly, periodic screening for MM and other neoplasms is warranted. PPK Gamborg-Nielsen (PPK-GN; OMIM #244850), an autosomal recessive disorder first described in patients from the northernmost county of Sweden by Gamborg Nielsen in 1985, is considered by some as a milder form of MDM. It manifests with a similar phenotype but less severe diffuse palmoplantar hyperkeratosis and no nail deformities or distant keratosis, although knuckle pads and tapered fingers may be observed.166-168
Etiology and Pathogenesis: MDM is caused by biallelic mutations in the SLURP1 gene (previously known as ARS component B), which contains three translated exons and encodes the lymphocyte antigen 6 /urokinase-type plasminogen activator receptor related protein-1 (SLURP-1), a member of the Ly-Upar superfamily of proteins that play a role in transmembrane signal transduction, cell activation, and cell adhesion.148,169 SLURP-1 is considered a secreted epidermal neuromodulator that is likely to be essential for both epidermal homeostasis and inhibition of tumor necrosis factor (TNF)-α release by macrophages during wound healing.170
Genetic heterogeneity is suggested in MDM because families without a SLURP1 mutation have been reported.171,172
PPK-GN has recently been shown to be an allelic variant of MDM. A homozygous mutation in SLURP1, c.43T>C, was identified in 14 individuals from northern Sweden, and compound heterozygous mutations, c.280T>A and c.43T>C, were identified in one individual from southern Sweden. The c.43T>C mutation had been previously reported in MDM,168 supporting that PPK-GN is a mild variant of MDM and not a separate entity.173
SLURP-1 has been shown to be expressed in human skin, mainly in keratinocytes underlying the upper epidermal layers, and it has a role in maintaining the skin’s physiologic and structural integrity. SLURP-1 upregulates the expression of transglutaminase 1, cytokeratin 10, and caspases 3 and 8 with resultant increased apoptotic activity which exceeds that of TNF-α. In addition, it regulates epidermal differentiation through cholinergic pathways.174
Moreover, SLURP-1 expression correlates with abnormal immune responses. Mutations in this gene resulted in defective T-cell activation responses,149 and SLURP-1 increased acetylcholine synthesis in T cells and attenuated T-cell proliferation. These effects were abolished by a α7-nAChR antagonist, indicating that SLURP-1 modulates the functional development of T cells via α7-nAChR-mediated pathways.175 Accordingly, patients with homozygous SLURP1 mutations are prone to viral infections and to the development of MM.176 The higher incidence of MM in patients with MDM is not only attributable to defective T-cell activation and prolonged inflammation in hyperkeratotic skin162,163 but may also result from defective apoptotic activity177,178 and defective regulation of TNF-α release from macrophages.179,180
It has been suggested that SLURP-1 deficiency influences the efficiency of triglyceride hydrolysis in keratinocytes, an essential process for the formation of acylceramides important for the epidermal barrier.181,182 It has been hypothesized that leakage of interstitial fluids into the stratum corneum and accumulation of triglyceride droplets in SLURP1- deficient skin are conditions favoring microorganism growth responsible for the malodorous skin of MDM.181
SLURP-1 may play a role in the pathogenesis of psoriasis. Its expression was upregulated in the skin of imiquimod-induced psoriasis in mice, and SLURP1 mRNA expression was significantly upregulated after stimulation with interleukin (IL)-22 which was completely suppressed by STAT3 inhibition. In addition, SLURP-1 significantly suppressed the growth of Staphylococcus aureus,183 and it has been shown to regulate epithelialization and wound healing in both cutaneous and oral wounds.184
Pathology: The histologic features of MDM are hyperkeratosis, orthokeratosis, foci of parakeratosis, marked acanthosis, a more pronounced stratum lucidum, and a perivascular lymphohistiocytic infiltrate.185 Electron microscopy findings are a lessabrupt-than-normal transition between the stratum granulosum and stratum corneum; normal intermediate filaments and corneodesmosomes; and nonspecific, irregularly shaped keratohyalin granules with a spongy appearance.26 In palmoplantar sections from patients with MDM, as well as in their sweat, SLURP-1 was either absent or barely detectable. It has been suggested that SLURP-1 assessment in sweat collected by the standard pilocarpine procedure used for the diagnosis of CF may serve as a rapid screening test for MDM.186
8
NAGASHIMA-TYPE PALMOPLANTAR KERATODERMA Clinical Features: Nagashima-type PPK (NPPK; OMIM #615598), is an autosomal recessive disorder first described by Nagashima in 1977. The disease was initially named Meleda-type PPK, but in 1989 the term NPPK (or keratosis palmoplantaris Nagashima) was coined to distinguish this form of PPK from true MDM, given its milder phenotype and on-progressive nature after puberty.187,188 Most cases have been reported in Japan and China, and the estimated prevalence in those countries is 1.2 in 10,000 and 2.3 to 3.1 in 10,000, respectively,189,190 making it the most common type of PPK in Asian populations. Several cases have been described outside East Asia with estimated prevalence rates of NPPK in non-Asian populations as low as 0.5 in 100,000,000.189,191,192
NPPK typically begins in the first years of life (from birth through the fourth year) and gradually progresses until puberty with no further progression thereafter. It is characterized by a diffuse, well-demarcated, erythematous palmoplantar hyperkeratosis that extends to the dorsal surfaces of the hands, feet, inner wrists, ankles, and Achilles tendon area. Involvement of the elbows and knees is common. A single case with involvement of the central lumbar area, cubital fossae, forearms, thighs, and popliteal fossae has been described.193 Hyperkeratosis on the ears and toenail dystrophy (atrophic nail plates with fragile distal edges and spontaneous peeling) are rare features that have been reported in isolated cases.194,195 It is frequently complicated by hyperhidrosis, superinfection by dermatophytes, a distinct odor, and maceration.187,188,190-192
There is no evidence of constricting bands, spontaneous amputation, or flexion contractures in contrast to other more severe forms of autosomal recessive transgressive diffuse PPKs, such as MDM and PPK- GN.188,192 In addition, patients with NPPK display a whitish spongy appearance within 10 minutes of water exposure specifically in the erythematous hyperkeratotic areas, suggesting enhanced water permeation into the stratum corneum in NPPK lesional skin.189,190
A case of MM arising within the lesions of NPPK has been reported.196
Etiology and Pathogenesis: NPPK is caused by biallelic mutations in the gene SERPINB7, encoding serpin family B member 7 (SERPIN7), a cytoplasmic member of the serine protease inhibitor (serpin) superfamily. Most cases of NPPK are caused by lossof-function (LOF) mutations resulting from aberrant splicing or premature stop codons with nonsense or frameshift mutations.197 Compound heterozygosity for a missense founder mutation c.830C>T, resulting in proline to leucine in the highly conserved residue 277, resulted in NPPK phenotype with mislocalized SER- PIN7 within corneocytes.198
A recurrent nonsense mutation in SERPINB7, c.796C>T, has been found to be prevalent in both Chinese and Japanese NPPK patients, probably reflecting
827
8
a founder effect. Accordingly, it has been suggested that mutation screening for c.796C>T in SERPINB7 can serve as a diagnostic strategy in the setting of Chinese and Japanese patients with diffuse, nonmutilating PPKs.190,193,199
Epidermal protease inhibitors, such as LEKTI, LEKTI-2, elafin, serpins, and cystatins, play a crucial role in maintaining epidermal homeostasis by inhibiting both endogenous proteases residing in the stratum granulosum and the stratum corneum and exogenous proteases from bacteria, fungi, viruses, pollen, and house dust mites that attack the epidermis.200,201 Mutations affecting serpins can cause symptoms either by loss of protease inhibitory activity and uncontrolled protease activity or by aggregation of mutant serpins polymers in serpin-synthesizing cells with cell death and tissue damage (the “serpinopathies”). SERPINB7 has been shown to be distributed in the epidermis, especially in the stratum granulosum and upper part of the stratum corneum, with predominant cytoplasmic expression. In NPPK skin, SERPINB7 immunoreactivity is markedly diminished.189 Although SERPINB7 is ubiquitously expressed in human epidermis, NPPK is limited to the hands, feet, knees, and elbows, suggesting that chronic exposure to mechanical stress may have a role in the development of NPPK and that SERPINB7 might inhibit mechanical stressinduced proteases and protect keratinocytes or corneocytes from protease-mediated cellular damage.189
The whitish spongy change upon water exposure in NPPK, similar to PPKB with AQP5 mutations and aquagenic keratoderma with CFTR mutations has been suggested to result from enhanced water permeation into the damaged stratum corneum.
Pathology: Histopathologic findings in NPPK include orthohyperkeratosis with acanthosis, hypergranulosis, and a mild to moderate perivascular lymphocytic infiltrate in the upper dermis.191,192,199 A predominance of CD4+ T cells with barely detectable CD20+ B cells was observed in dermal infiltrates.202
DIFFUSE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE
DIFFUSE INHERITED
PALMOPLANTAR
KERATODERMA WITH
EXTRACUTANEOUS
FEATURES, DOMINANT
INHERITANCE
OLMSTED SYNDROME Clinical Features: OS (OMIM #614594), first reported in 1927 by Olmsted,203 is a rare disorder with a prevalence of less than 1 in 1,000,000. It usually appears at birth, during the neonatal period, or in early childhood, although appearance later in childhood has been described,204 and progressively worsen over time. Both
828
sexes are affected, although male patients comprise 60% of reported cases.205 It is characterized by symmetric, sharply demarcated, diffuse PPK with yellowish-brown hyperkeratosis, painful fissures, and erythematous borders in association with periorificial keratotic plaques. These plaques may be restricted to the areas around the mouth, nostrils, ear meatus, anus, and perigenital region or may extend to involve nonperiorificial sites such as the neck, upper thorax, lower abdomen, arms, elbows, knees, thighs, and inguinal folds. The PPK typically starts focally and is distributed on the pressure points. It gradually extends to most of the surface of the palms and soles with thick hyperkeratotic plaques that eventually result in flexion deformity of the fingers, pseudoainhum, and spontaneous amputation of the digits or the hands. The PPK is typically painful and disabling, interfering with walking and daily activities. Cases with more focal or punctuate keratoderma, lacking pseudoainhum or significant periorificial keratotic lesions, have been reported.204,206-209 Additional findings are severe pruritus with sleep disturbances, onychodystrophy (ridged and rough nails, onychogryphosis, leukonychia, onycholysis, paronychia, subungual hyperkeratosis, absence of nails), abnormal dentition (absence of premolar teeth, periodontal disease with premature teeth loss),210,211 sweating abnormalities, oral leukokeratosis, hyperhidrosis, hyperkeratotic linear streaks, follicular keratosis, cheilitis, and ichthyotic lesions.208,212-220 Hair abnormalities are common, although rare cases have normal hair, and include alopecia (diffuse, universal or patchy); hypotrichosis; and thinning, curly, woolly, coarse, and dry or easily broken hair. Microscopic findings of affected hair include pili torti, trichorrhexis nodosa, reduced pigment, longitudinal ridges, and transverse fractures of the hair shaft.33,205,213,217-219 Sparse and thin eyebrows and eyelashes, madarosis, as well as trichomegaly were observed in OS.206,214,221 Secondary bacterial and candidal infections,216,222,223 SCC (verrucous carcinoma or its variant epithelioma cuniculatum), and MM can develop within the PPK.224-227 A case associated with cone-shaped fingers and a sclerodactyly-like appearance overlapping with clinical features of Huriez syndrome has been described.228 Extracutaneous manifestations are uncommon and include hearing loss,211 ocular abnormalities (corneal dystrophy, inflamed lacrimal glands or ducts or meibomian gland dysfunction, chronic blepharitis),214,216 short stature,218,226 primary sclerosing cholangitis,229 mental retardation,203,230 joint laxity, ankyloses, osteopenia, and osteolysis.145,203-205,211,218,223,226,231,232 Erythromelalgia206,228,233 has been reported in several cases.
Etiology and Pathogenesis: Although most cases reported to date have been sporadic, familial cases with different modes of inheritance, including autosomal dominant, autosomal recessive and X-linked inheritance, have been reported. Autosomal dominant, semidominant, and autosomal recessive OS have been shown to be cause by mutations in the gene TRPV3 encoding transient receptor potential vanilloid 3 (TRPV3),145,206,221,232,233 and mutations in the gene MBTPS2 encoding the membrane-bound transcription factor protease site 2 have been identified in X-linked
recessive OS.234 There is no clear genotype–phenotype correlation in OS. Mutations in TRPV3 are associated with persistence of basal keratins208,235 and increased expression of Ki-67.219,220 Keratinocytes expressing mutant TRPV3 are prone to cell death, and the epidermis of OS patients displays increased apoptotic cells compared with control participants.145
TRPV3 is highly expressed in keratinocytes and in the hair follicles (HFs), spinal cord, sensory neurons, brain, and cornea. It has an important role in epidermal barrier formation, regulation of hair growth, and modulation of pain sensation and pruritus, explaining the manifestations of OS.236-240 TRPV3 activation increases intracellular Ca2+ concentration and is associated with transforming growth factor (TGF)-α/ EGFR signaling, which is known to regulate epidermal differentiation.241 The role of TRPV3 in mediating EGFR signaling in hair and skin barrier function was demonstrated using a Trpv3 knockout mouse model that developed a wavy hair coat and curly whiskers in addition to red, dry, scaly skin at birth.239 In addition, mice or rats carrying heterozygous mutations in Trpv3 exhibited hair paucity and dermatitis.242
Mutations in MBTPS2, encoding a zinc metalloprotease essential for cholesterol homeostasis and endoplasmic reticulum (ER) stress response, have been reported in cases of OS with X-linked recessive inheritance.234 Mutations in MBTPS2 were first identified in ichthyosis follicularis atrichia photophobia (IFAP) syndrome243 (see Chap. 49), which is allelic to X-linked recessive OS. A case with features of both OS and IFAP syndrome has been reported.244
There is no obvious clinical difference between OS patients carrying either TRPV3 or MBTPS2 mutations, and clinical variability is observed within the same family or among different patients harboring the same mutations, supporting a role for modifier genes, environmental effects, or epigenetic factors. Moreover, genetic heterogeneity is likely given the fact that some patients do not carry mutations in either TRPV3 or MBTPS2.
Pathology: The histopathologic findings of OS are nonspecific and include psoriasiform epidermal hyperplasia, orthohyperkeratosis, focal parakeratosis, hypo- or hypergranulosis, acanthosis, and inflammatory infiltrates in the upper dermis, which may contain mast cells.145,205,232 Epidermal vesicular degeneration has been reported.228 Electron microscopy findings include large, coarse, densely packed bundles of tonofilaments in the keratinocytes of the mid Malpighian layer and increased numbers of coarse keratohyaline granules in the granular layer. Decreased number of pigment granules and absence of Langerhans cells were reported.228
Specific Treatments: Partial excision of palmoplantar keratosis or full-thickness excision with skin grafting has been performed with clinical improvement.245 EGFR inhibitors have been shown to improve PPK,246 and TRPV3 antagonists may be an effective treatment for patients harboring GOF mutations in the TRPV3 gene.
8
CONNEXIN-ASSOCIATED PALMOPLANTAR KERATODERMAS: VOHWINKEL SYNDROME, KERATITIS–ICHTHYOSIS–DEAFNESS SYNDROME, BART-PUMPHREY SYNDROME, AND PALMOPLANTAR KERATODERMA WITH DEAFNESS Clinical Features: VS (keratoderma hereditaria mutilans; OMIM #124500) was first reported in 1929 by Vohwinkel115 and Wigley.247 VS is characterized by a triad of diffuse, mutilating PPK with a “honeycomblike” appearance (see Fig. 48-4B); “starfish-shaped” keratotic plaques on the dorsal hands, feet, and extensor surfaces; and fibrous constricting bands (pseudoainhum) at the interphalangeal joints of the hands and feet that result in autoamputation.248 Most cases are also associated with high-frequency SNHL.27,29,249-252 The disease is more common among white women. It tends to manifest in the neonatal period and progresses throughout life. Cases with PPK characterized by callosities at pressure points or striate lesions at sites of injury, suggesting an isomorphic phenomenon, have been reported.250 The disease may be associated with generalized ichthyosis,28 acanthosis nigricans,253 cicatricial or nonscarring alopecia, nail anomalies, knuckle pads, bullous lesions on the soles,249 and craniofacial anomalies (cleft lip and palate, microcephaly, and facial asymmetry).27,29,249,250,254-257 The appearance of SCC and basal cell carcinoma (BCC) in the hyperkeratotic lesions has been reported.28 Cases associated with deafmutism,18 mental retardation,258 spastic paraplegia with myopathy,29 psychomotor developmental retardation, and epileptic seizures259,260 have been reported. KID syndrome (OMIM #148210; see Chap. 47) is the most severe cutaneous connexin disorder, involving epithelia of ectodermal origin (skin, appendages, nail, teeth) along with significant inner ear and cornea involvement.261,262 Disease manifestations were first described in 1915, although the name KID syndrome was coined by Skinner and coworkers in 1981.262,263
The disease manifests at birth or during infancy. Its cutaneous features are erythrokeratoderma with symmetrical, well-circumscribed hyperkeratotic plaques with a reticulated pattern, leather-like appearance, or diffuse furfuraceous scaling with underlying erythema and a relative predilection for the axillae and neck. Other manifestations include thickened skin with coarse-grained appearance or follicular hyperkeratosis without erythema. The characteristic PPK is diffuse and has a rough, stippled, or grainy appearance (Fig. 48-6). Chronic cheilitis and perleche are common. Associated findings are nail dystrophy, oral manifestations (leukokeratosis, deep fissures of the tongue, dental abnormalities including caries and delayed eruption of teeth, persistent oral mucosal papules), hypohidrosis, and heat intolerance. Numerous KID syndrome patients have sparse hair, and 10% to 23% have congenital atrichia.261,264 Most patients develop
829
8
progressive keratitis that begins during childhood and may first manifest as photophobia. The keratitis is characterized by corneal inflammation, pain, and corneal neovascularization and may be associated with chronic blepharitis, conjunctivitis, keratoconjuctivitis sicca, and limbal defects and may cause progressive visual decline and blindness. Associated ophthalmologic features are loss of eyebrows and eyelashes, hyperkeratosis of the eyelids, trichiasis, early-onset cataract, and bilateral lacrimal punctal agenesis.261,265-272
Conversely, cases with no evidence of ophthalmologic manifestations have been described.273,274 In addition, patients with KID have congenital, bilateral, and severe SNHL, which is not progressive, compared with the corneal findings.275 Patients are prone to mucocutaneous infections, mostly bacterial and candidal, which can be fatal in the neonatal period.276,277 Viral infections (molluscum contagiosum and cytomegalovirus), recurrent pulmonary infiltrates, and recurrent otitis externa have been reported.278,279 Patients are prone to developing benign cutaneous tumors, mainly trichilemmoma,280-282 although isolated cases of multiple poromas and porokeratotic eccrine ostial and dermal duct nevus have been described.283 Approximately 10% of affected individuals are reported to develop SCC of the skin (mainly acral sites and areas of chronic infection or inflammation) and tongue, apparently caused by p53 loss in the lesions.41,261,268,269,278,280,284-287
SCCs may cause death in patients with KID given their aggressive phenotype and the tendency to develop multiple tumors.280 In addition, single cases of an association with sebaceous carcinoma, peripheral T-cell non-Hodgkin lymphoma, malignant histiocytoma, and metastatic malignant pilar tumors have been rep orted.271,281,288-290 An association between KID syndrome and hidradenitis suppurativa and dissecting cellulitis of the scalp has been described.47,281,291,292 KID syndrome may result in death in infancy from severe infections or respiratory compromise.276,293-295
Bart-Pumphrey syndrome (BPS; OMIM #149200) is a rare, autosomal dominant disorder first described by Bart and Pumphrey in 1967. It is characterized by
830
severe SNHL, PPK with a honeycomb-like appearance, knuckle pads, and leukonychia.296-298 Hearing loss and knuckle pads are the most common findings; leukonychia and PPK are seen less frequently.296,297,299-302
The PPK features diffuse, sharply demarcated thickening of the palmoplantar skin with a punctate, grainy surface reminiscent of VS. Striate appearance has been described as well.302
PPK and deafness (OMIM #148350) is an autosomal dominant condition in which PPK may be diffuse, transgrediens with fissures and underlying erythema or with a milder phenotype with skin fold accentuation over pressure points. Knuckle pads may be present. Hearing impairment may become apparent during infancy; is bilateral, prelingual, and slowly progressive; and affects high-frequency tones.303-309
Etiology and Pathogenesis: Gap junctions, which are formed by connexons, are intercellular junctions that facilitate and regulate the passage of water and small molecules between adjacent cells. The oligomerization of six connexins leads to the formation of a hemichannel called a connexon, and connexons of two opposing cells interact with each other through their extracellular portions to form a channel that is the basic unit of the gap junction. Connexins are expressed in a tissue and differentiation specific manner.310-315
The epidermis, its appendages, and other ectodermderived epithelia of the inner ear and cornea share the expression of several connexin proteins, including Cx26, Cx30, Cx31, and Cx43. Cx43 is ubiquitously expressed in all epithelial cells of ectodermal origin, and similar to Cx26, it has been shown to play a role in wound healing.316 The expression of Cx26 is limited to cochlear cells, corneal limbal cells, palmoplantar epidermis, around the openings of eccrine sweat glands and ducts, and in the inner and outer root sheath of human HFs. Cx26 expression is widely paralleled by the expression pattern of Cx30 (both share 76% amino acid identity).317,318 In contrast to Cx30, which is prevalent in the upper differentiated layers of interfollicular epidermis, Cx26 is only expressed at low levels in palmoplantar epidermis but is strongly induced in response to wounding and in hyperproliferative diseases.318-320 Cx26 is also expressed in epithelial supporting cells surrounding the sensory hair cells of the cochlea and in the fibrocytes lining the cochlear duct, where it mediates the recycling of potassium ions passing through the hair cells back to the endolymph during auditory transduction.321,322
GJB2 encodes Cx26. Recessive mutations producing complete loss of expression or function are the single most common cause of nonsyndromic SNHL in humans.323 In addition, mutations in connexin genes are known to cause several inherited human disorders not associated with PPK or other dermatological abnormalities, including X-linked Charcot-Marie-Tooth disease (Cx32),324 zonular pulverent cataract (Cx50),325 and occulodentodigitaldysplasia (Cx43).326 Moreover, several inherited genodermatoses result from heterozygous mutations in connexin genes (see Table 48-1 and Fig. 48-2).250,327-329
Connexins consist of four transmembrane domains, three cytoplasmic domains (the amino-terminus, a cytoplasmic loop and the carboxy terminus domains), and two extracellular loops. Both the N-terminal and C-terminal parts of the proteins are located in the cytoplasm. The membrane spanning and extracellular domains are highly conserved, and the main differences between connexins are found in their C-terminal tails.311,330 Most dominant-negative GJB2 mutations associated with hearing impairment and cutaneous involvement reported so far are located in the cytoplasmic N-terminal or in the first extracellular loop of Cx26, which is highly conserved among the connexins and is involved in the control of voltage gating, channel permeability, multimer assembly, and the interactions between connexons. Accordingly, mutations affecting the first extracellular domain may affect both protein transport and channel permeability.311,317 In addition, different mutations in this domain result in different pathophysiological effects, such as blockage of conductance, dominant effect on wild-type Cx26, protein folding, and altered hemichannel structural stability, leading to a variable clinical phenotype. Moreover, co-oligomerization of different connexins into heteromeric connexons plays a role in the pathogenesis and in the dynamic nature of connexin-associated phenotypes, which are mostly caused by a dominant negative effect on intercellular communication.311,331
Most reported cases of VS result from the recurrent heterozygous missense mutation, p.Asp66His, in GJB2 which results in an amino acid substitution from aspartic acid to histidine in a highly conserved residue of the first extracellular domain.330 This mutation could selectively impair the ability of Cx26 to form heteromeric and homomeric connexons,332 resulting in a change in charge or conformation of Cx26 with disruption of gating properties for certain molecules or ions. Generalized knockout of Gjb2 in mice is embryonic lethal (caused by placental insufficiency), but conditional Gjb2 knockout in the mouse inner ear resulted in deafness.333 Transgenic expression of a dominantnegative Gjb2 mutation revealed a progressive degeneration of the sensory hair cells with the loss of the tunnel of Corti as a result of disturbed cortilymph homeostasis,334 illustrating the essential role played by Cx26 in the auditory function in the inner ear. Transgenic mice expressing the p.Asp66His mutation exclusively in the suprabasal epidermis exhibited keratoderma with constriction bands on the tail, marked thickening of the epidermal cornified layers, and increased epidermal TUNEL staining, indicative of either excess apoptosis or premature terminal differentiation.335 Premature keratinocyte death might induce compensatory basal cell proliferation, leading to the massive thickening of the stratum corneum. Both autosomal dominant41,43 and autosomal recessive336 forms of KID syndrome have been described, although autosomal dominant inheritance is more common.42,337 Cases with suggested parental germline mosaicism have been reported.286,294,338 Most affected individuals harbor a recurrent missense mutation in GJB2, p.Asp50Asn, leading to replacement of aspartic
8
acid in codon 50 with asparagine.298,317,339-342 In addition, hystrix-like ichthyosis-deafness (HID) syndrome, which is allelic to KID syndrome,343 has been reported to result from the p.Asp50Asn mutation and other GJB2 missense mutations.298 Asp50 is a pore-lining residue that is highly conserved among connexins and is crucial for gap junction formation and function; p.Asp50Asn results in intracellular expression of the mutant protein, suggesting altered trafficking to the plasma membrane and absence of gap junction plaques.344,345 The mutation p.Gly45Glu has been identified in a fatal forms of KID syndrome.294 All KID syndrome–causing mutations cluster in regions coding for the first extracellular domain and the cytoplasmic amino terminal domain of Cx26 and are predicted to alter the charge and structure of this domain, in contrast to nonsyndromic Cx26-associated mutations that are located along the protein.314,317 Recent experiments in Xenopus laevis oocytes demonstrate that hemichannels formed by several Cx26 KID mutants are inhibited by mefloquine; mefloquine attenuated increased macroscopic membrane currents in primary mouse keratinocytes expressing the human Cx26 p.Gly45Glu mutation.346
A heterozygous mutation in GJB6, encoding Cx30, has been shown to result in KID syndrome as well. This mutation is predicted to alter the sequence and charge of the first transmembrane helix of Cx30 and was reported in a child with typical characteristics of KID syndrome, including follicular, spiny hyperkeratosis, congenital atrichia, and nail abnormalities.264 A similar mutation had been implicated in Clouston syndrome (see later) without evidence for abnormal sweating, hearing, photophobia, and keratitis,347 underscoring the profound influence of other genetic and epigenetic factors in modifying the clinical outcome of connexin disorders and emphasizing the phenotypic variability of GJB6 mutations. It has been speculated that the unique phenotype of this patient is related to the presence of a homozygous polymorphism in GJB2.264
In BPS, two mutations involving the first extracellular domain of Cx26, p.N54K, and p.G59S have been reported, underscoring the importance of this domain in docking of connexin hemichannels and voltage gating.299,302
PPK with deafness results from heterozygous mutations in the GJB2 gene clustered in or at the border of the first extracellular loop domain.303-306 One of these dominant mutations, p.R75Q (c.224G>A), was described for the first time by Uyguner and coworkers in a Turkish family305 and was reported in isolated deafness and in association with PPK.348
Pathology: Histopathologic manifestations of VS include compact hyperkeratosis; orthokeratosis; acanthosis; significant hypergranulosis with large, irregularly shaped keratohyaline granules; papillomatosis of the epidermis; and dermal fibrosis with a sparse perivascular lymphocytic infiltrate.256,298,349 Ultrastructural findings are marked swollen mitochondria and increased numbers of desmosomes in the spinous and granular layers with corneocytes containing many membrane coating granules and lipid-like vacuoles.349
831
8
Skin biopsies obtain from PPK in KID syndrome may display epidermal hyperplasia, compact orthokeratotic hyperkeratosis, and focal parakeratosis. Hypogranulosis may be observed, although cases with a prominent granular layer have been reported, and swollen keratinocytes with slightly vacuolated cytoplasm are described. Keratotic plugging is a common feature. Inflammatory cells in the upper dermis may be evident, especially in cases of infection.264,294,350
Histopathologic findings in BPS are massive orthokeratotic hyperkeratosis, hypergranulosis, acanthosis, and papillomatosis. Epidermal gap junctions appear normal on electron microscopy.302
Specific Treatments: In VS, cross finger flap,248
treatment with Z-plasty,351 full-thickness excision of the constriction band with a full-thickness skin graft,352
and a distant abdominal skin flap for fifth digit constriction bands353 have been described. Reported treatments for the ocular manifestations of KID syndrome are keratolimbal allograft, keratoplasty and immunosuppression,354 keratectomy,43 topical corticosteroids and cyclosporine,355 and bevacizumab has been shown to be effective in a single case.356
PALMOPLANTAR KERATODERMA– CONGENITAL ALOPECIA SYNDROME Clinical Features: Two forms of PPK with congenital alopecia have been described. An autosomal dominant form has a milder phenotype (PPKCA1, Stevanovic type; OMIM #104100),357-360 and the recessively inherited form is associated with pseudo-ainhum, sclerodactyly, contractures, and sometimes cataracts (PPKCA2, Wallis type; OMIM #212360).257,361-363 Hair is normal or sparse at birth, and noncicatricial alopecia involving the scalp, body, or facial hair becomes apparent in early infancy. Trichorrhexis nodosa may be evident on hair microscopy. PPK develops in late infancy and is well-defined, focal or linear, nonmutilating, and transgrediens in the dominant cases. In the recessive cases, it features progressive thickening of the lateral and medial aspects of palms and soles with an erythematous rim and skin cracks that subsequently involve the dorsal fingers, resulting in contractures, pseudoainhum, and sclerodactyly. Associated findings are follicular plugging with ulerythema ophryogenes– like features; keratosis pilaris; and hyperkeratotic plaques over the ankles, elbows, and popliteal fossae, with multiple spiky, horn-like lesions reminiscent of ichthyosis hystrix and nail abnormalities (eukonychia, nail dystrophy).364 Cataracts, meningocele, and unilateral deafness were reported in single cases.257,358,361
Etiology and Pathogenesis: PPKCA1 has been shown to result from a heterozygous missense mutation in the gene GJA1, encoding Cx43, which exerts a GOF effect on the Cx43 hemichannel. Cx43 is ubiquitously expressed in various organs, including the epidermis and HFs.365,366
832
PPKCA1 is allelic to oculodentodigital dysplasia (ODDD), which is characterized by craniofacial dysmorphism; dental, ophthalmologic, and limb abnormalities; and neurodegeneration, and is occasionally associated with PPK and hair and nail anomalies. In ODDD, most mutations result in retention of the mutant protein in the ER or decreased permeability of the channels with nonfunctional gap junctions.326,367-369
The responsible gene for PPKCA2 has not been identified yet.
Pathology: Hyperkeratotic plaques show orthohyperkeratosis with follicular plugging and perivascular lymphocytic infiltration in the papillary dermis.364
Scanning electron microscopy of the hair shafts reveals multiple pits with cuticular weathering364 or longitudinal grooves.360
HIDROTIC ECTODERMAL DYSPLASIA (CLOUSTON SYNDROME; SEE CHAP. 131) Clinical Features: Hidrotic ectodermal dysplasia (HED, Clouston syndrome; OMIM #129500) was first described in 1895370 and later by Clouston in families from Quebec.371,372 HED is an autosomal dominant ectodermal dysplasia particularly common among the French-Canadian population because of a founder effect,328 although it has been reported in several ethnic groups.347,373-380 The main features of this condition include nail dystrophy, hair loss, and palmoplantar hyperkeratosis with normal sweat glands and teeth.378,381,382
Nail abnormalities are usually present; in nearly 30%, they are the only manifestation of the syndrome. They range from almost normal-appearing nails to short nails and anonychia.371 Nail plate changes include thickening, brittleness, ridging, discoloration, splitting, onycholysis, and 20-nail dystrophy.347,371,372,381
Paronychia and nail infections are common and may result in nail matrix destruction.371,374,383 Hair abnormalities may be progressive and involve the scalp, facial, and body hair and include atrichia or hypotrichosis with brittle, fine, pale, or slow-growing hair.384 PPK is diffuse with a velvet-like or cobblestoned appearance extending onto the fingertips and knuckles with fissures. In a large Han Chinese family, the hyperkeratosis of the palms and soles tended to worsen with time.379 Cases of HED with nail changes resembling PC, either as a solitary finding or in association with alopecia but with no evidence of PPK have been reported.385-387 Keratotic papules and plaques over the upper and lower extremities and discrete hyperpigmentation over digital joints have been described. Rare associations include strabismus, cataract and photophobia, hearing impairment, mental deficiency, and bone abnormalities (thickening of the skull, abnormal phalanges).371,372,382,383,388-391
Etiology and Pathogenesis: HED is caused by autosomal dominant heterozygous mutations in GJB6,
encoding Cx30.328 Cx30 is a 261–amino acid polypeptide consisting of four transmembrane domains, two extracellular domains, and three cytoplasmic domains similar to other connexins.392,393 Cx30 is expressed in the epidermis (middle and upper spinous layers), HFs (outer root sheath, hair matrix), nails (nail matrix and nail bed), brain, and inner ear.328,394-399
A mouse model for HED carrying the p.A88V mutation in GJB6 demonstrates mild hyperkeratosis of palmoplantar skin, enlarged and hyperproliferative sebaceous glands, and altered hearing (both are not common features in HED).400 In addition, this mutation resulted in significant apoptosis, possibly through leaky hemichannels.401 It has been suggested that the leaky hemichannels resulting from the p.A88V mutation could stimulate proliferation through activation of Ca2+-dependent kinases with altered gene expression or cell cycle reentry.400
Similar to other connexin disorders, HED is characterized by extensive clinical and genetic heterogeneity. A patient with clinical features of HED and ODDD and extensive hyperkeratosis of the skin was found to have an heterozygous mutation, p.V41L, in GJA1 encoding Cx43, as well as a heterozygous sequence variant (P.R127H) in GJB2.402 A Chinese patient with HED was found to carry two missense mutations, p.N14S in GJB6 and p.F191L in GJB2,389 and a Japanese patient with HED associated with hearing impairment and photophobia was found to harbor a heterozygous missense mutations in GJB6 (p.Ala88Val) and a polymorphism in GJB2 (p.Val27Ile).390
In contrast to GJB2 mutations that result in skin symptoms and deafness, mutations in GJB6 typically result in skin manifestations with no evidence of hearing impairment. It is possible that Cx26 compensates for the absence of Cx30 in the inner ear and not in the skin, but Cx30 cannot completely compensate for the loss of Cx26 at both sites because mutant Cx26 exerts a dominant negative effect on other co-expressed connexins. However, a case of KID syndrome caused by a mutation in GJB6 and a case of HED with SNHL and photophobia have been reported; in addition, a p.T5M mutation in GJB6 results in dominant nonsyndromic hearing loss, suggesting that functional redundancy may be dependent on additional factors such as the location and nature of the causative mutations.264,390,403
Pathology: Hypotrichotic skin reveals normal epidermis, a normal distribution of eccrine and sebaceous glands, and absence or remnants of HFs.347,379,384 PPK lesions reveal hyperkeratosis with irregular and reticulated acanthosis.389
HURIEZ SYNDROME Clinical Features: Huriez syndrome (sclerotylosis; OMIM #181600) was first described by Huriez and coworkers in 1969 in two families from Northern France.404 Since then, kindred from different ethnicities have been described, including Tunisian, Indian, German, English, Italian, and Japanese families.405-411 Most cases are inherited in an autosomal
8
dominant fashion, and several sporadic cases have been reported.409,412 Huriez syndrome is characterized by diffuse PPK, scleroatrophy of the hands and fingers, occurrence of SCC within atrophic skin, and sclerodactyly leading to contractures. The disease is present at birth or appears in the first years of life and persists with no further progression. The PPK manifests as diffuse, yellowish-grey, nonerythematous, well-confined hyperkeratotic plaques mainly involving the palms with extension towards the fingers and accentuation of palmar creases (Fig. 48-4C). The soles are less frequently involved and usually show accentuation over pressure sites. A porokeratotic appearance has been described. Scleroatrophic changes include a pseudosclerodermoid appearance of the hands and digits with absence of dermatoglyphs and erythematous, atrophic appearance of the dorsal aspect of the hands, fingers, and tips of the fingers and toes with no evidence of Raynaud phenomenon. Associated findings are hypohidrosis and nail changes including hypoplasia, curving, onychorrhexis, koilonychia, longitudinal ridging, and clubbing. Manifestations reported in isolated cases are poikiloderma-like changes, distinctive small nodules on the fingers, and facial telangiectasia. Malignant degeneration starts with the occurrence of skin ulcers on the atrophic skin of the hands and development of SCC by the third to fourth decade of life. The estimated risk of SCC is 13%, a greater than 100-fold higher risk. These tumors tend to show poor differentiation with high rates of metastasis and a calculated mortality rate of 5%.35,404,406-408,413,414 Cases associated with internal malignancies (gastric carcinoma, pharyngeal carcinoma) have been described, but internal malignancies do not seem to be more frequent in Huriez syndrome than in the general population.35,404
Etiology and Pathogenesis: The pathogenesis of Huriez syndrome and the mechanism involved in tumor formation are unknown. Previously, a linkage between Huriez syndrome and the certain blood groups has been reported415; however subsequent studies failed to confirm this finding.35,407 Lee and coworkers mapped the gene to chromosome 4q23.416
Loss of heterozygosity of 4q has been reported in the majority of SCCs originating from head and neck and in almost 50% of cases of cervical carcinoma. However, in the former, the region involved extends distal to the locus of Huriez syndrome with possible overlap on its centromeric end.416
Immunohistochemical and ultrastructural studies revealed an almost complete absence of epidermal Langerhans cells within involved skin.405,406 Moreover, positive p53 staining (indicative of abnormal function of p53) was observed in atypical keratinocytes, suggesting that p53 mutations may be responsible for the development of actinic keratoses and SCC in Huriez syndrome.409
Pathology: Skin biopsies obtained from PPK skin reveal marked acanthosis and papillomatosis, hypergranulosis with increased number of keratohyaline granules, and orthokeratotic hyperkeratosis. No
833
8
abnormalities are observed in the dermis and sweat glands. In scleroatrophic regions, the epidermis shows hypergranulosis with orthokeratotic hyperkeratosis, dense collagen fibers in the reticular dermis, and thin elastic fibers. Sparse dermal mononuclear cell infiltrates may be seen.35,405,409,412,413 A case with vacuolar degeneration of keratinocytes in the upper spinous layers has been reported.411 Ultrastructural findings are dense bundles of tonofilaments throughout epidermal layers with abundant keratohyaline distributed in large clumps of irregular density. Areas of scleroatrophic skin show similar changes in tonofilaments and keratohyaline but with addition features of thinned oxytalan and elaunin fibers at the dermal– epidermal junction, irregular borders, and nonhomogeneous appearance of elastic fibers with evidence of elastic fibers engulfed by macrophages.35
DIFFUSE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, RECESSIVE INHERITANCE
DIFFUSE INHERITED
PALMOPLANTAR
KERATODERMA WITH
EXTRACUTANEOUS
FEATURES, RECESSIVE
INHERITANCE
PAPILLON-LEFEVRE SYNDROME Clinical Features: PLS (OMIM #245000) is a rare, autosomal recessive disorder characterized by palmoplantar hyperkeratosis and severe progressive periodontitis, causing loss of primary and permanent teeth with periosteal changes of the alveolar bone.417-420
Periodontal disease severity has been shown to peak in the teens and declines with age.421 The estimated prevalence of PLS is 1 to 4 cases per 1 million people, and the carrier rate is 2 to 4 per 1000. There is no gender predilection, but consanguinity has been observed in approximately half of cases.422,423 PPK may appear at birth or during the first months of life, although in most cases, both PPK and periodontitis develop between the sixth month and fourth year of life, often beginning with the eruption of the first teeth.424 Of note, there is no correlation between the severity of the cutaneous and dental manifestations.425 The PPK is characterized by diffuse, erythematous, sharply demarcated hyperkeratotic and scaly plaques on both palms and soles with transgrediens features extending onto the dorsal aspect of the hands and feet (Fig. 48-7A). It may be associated with hyperhidrosis, foul-smelling odor, and nail abnormalities (transverse grooving and fissuring). Atypical cases with late-onset periodontitis and palmoplantar lesions or cases with isolated PPK or periodontitis have been described.426-429 Sharply demarcated, psoriasiform, hyperkeratotic plaques can be observed on the knees, elbows, and ankles, and a more widespread distribution resembling psoriasis
834
A B
has also been described.38,423,425 Increased susceptibility to infections is typical; pyogenic skin infections as well as hepatic or cerebral abscesses and pneumonia have been reported.423,430 PLS may be associated with intellectual disability and calcification of the dura mater. Isolated cases have been reported in association with growth retardation, infantile voice, and hypothyroidism, although these may not be directly related to the PLS phenotype.38,420,423,431,432 An association with atopic diathesis and elevated immunoglobulin E (IgE) levels has been reported.38,421,433,434 Acral lentiginous melanoma may be more common in PLS, at least among Japanese patients.435
Etiology and Pathogenesis: PLS results from LOF homozygous or compound heterozygous mutations in the gene CTSC, encoding cathepsin C (CTSC), also known as dipeptidyl peptidase I. More than 70 mutations have been reported from ethnically diverse populations; approximately half are homozygous missense mutations altering protein folding and function, 25% are nonsense, and nearly 25% are frameshift mutations.38,420,436 PLS is allelic to Haim-Munk syndrome (HMS; OMIM #245010), a disorder described mostly in individuals from Cochin, India, characterized by the presence of pes planus, arachnodactyly, acroosteolysis, and onychogryphosis in addition to
PPK and periodontitis. Individuals with HMS do not have evidence of cerebral calcification and bacterial infections. Sulák and coworkers recently reported two Hungarian patients, one with PLS and one with HMS, who carry the same homozygous nonsense mutation in the CTSC gene,437 supporting the notion that PLS and HMS are phenotypic variants of the same disease. PLS is also allelic to prepubertal periodontitis without skin manifestations.438-440
CTSC belongs to the papain superfamily of cysteine peptidases and is a tetrameric enzyme consisting of four identical subunits linked together by noncovalent bonds. It plays an important role in intracellular degradation of proteins and coordinates the zymogen activation of serine proteases in cells with immune or inflammatory function including neutrophils, mast cells, cytotoxic T cells, and natural killer cells.441-446 The protein is expressed as a pro-proteinase in epithelial and myeloid cells, and a multistep process leads to its activation.418,447 The CTSC mutations in patients with PLS are associated with loss of CTSC enzymatic protease activity; carriers of the mutations demonstrate reduced activity compared with control participants.448
In has been shown that the absence of active CTSC in the urine serves as a strong and reliable indicator for PLS and allows screening for the disease in populations with a high frequency of consanguinity.417
Analysis of PLS patient neutrophils show that proteins normally found in the azurophilic granules (elastase, cathepsin G, proteinase 3) are completely absent in mature neutrophils but not in progenitor cells, indicating that CTSC mutations promotes protease degradation in mature immune cells. Because significant immunodeficiency is not evident in patients with PLS, neutrophil serine proteases are probably dispensable for human immunoprotection.449
Elevated IgE levels have been demonstrated in several cases of PLS, and the delayed resorption of the roots of primary teeth in hyper-IgE syndrome patients led to the suggestion that serum IgE may influence periodontal metabolism. This is supported by improvement in periodontal inflammation in PLS after a decline in IgE levels.421 Moreover, a T helper (Th) 2 phenotype is evident in PLS with reduced levels of Th1 cells and interferon-γ and increased levels of IL-4.434
Pathology: Histopathology analysis in PLS reveal irregular hyperkeratosis, hypergranulosis, marked acanthosis, and perivascular lymphocytic and histiocytic infiltrate in the dermis.39,428,434 Lipid-like vacuoles in corneocytes and granulocytes in addition to irregular keratohyalin granules and reduction in tonofilaments39 are evident on electron microscopy.
NAXOS DISEASE Clinical Features: Naxos disease (ND; OMIM #601214) is an autosomal recessive disease caused by mutations in JUP encoding plakoglobin (PG).450
The disease was originally described in families from the Greek island of Naxos by Protonotarios and
8
coworkers. Since then it has been reported in various ethnicities.451-455
The disease is characterized by a triad of diffuse PPK, cardiomyopathy, and woolly hair.450,456,457 The woolly hair appears at birth and features sparse and brittle, sometimes hypopigmented hair. Hair abnormalities are seen in scalp, eyebrows, and axillary and pubic hair. PPK develops during the first year of life and is characterized by diffuse, well-demarcated, nontransgredient hyperkeratotic plaques over the palms and soles, which may be surrounded by an erythematous border. Other cutaneous features are hyperhidrosis and nail abnormalities. Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) usually becomes symptomatic by adolescence with syncope being its first manifestation in most cases. Ventricular tachycardia and sudden death as a result of arrhythmia, which is a major cause of death in Naxos disease (one third of patients die prematurely with a mean age at death of 32 years), are common complications, and symptoms of right heart failure usually appear in the final stages. Left ventricular involvement may also be seen with further disease progression.452,455,456,458-461
Etiology and Pathogenesis: ND is an autosomal recessive disorder caused by biallelic mutations in JUP encoding PG. The most common reported mutation to date is a homozygous 2-bp deletion which results in a premature stop codon and expression of a truncated PG lacking 56 residues from the C-terminal domain of the protein.450,454,456,462 Heterozygous carriers of that mutation usually have no skin or hair abnormalities, although woolly hair may be evident, and minor heart involvement has been documented in approximately 25% of cases.456 Carriers of other mutations that result in residual PG expression display a milder phenotype of PPK, woolly hair, and skin fragility with no evidence of cardiomyopathy.463 Interestingly, an autosomal dominant mutation with one amino acid insertion in the PG N-terminal domain has been described in cases of ARVC/C with no evidence of PPK or hair abnormalities.464 Moreover, biallelic missense mutations in the JUP gene have been reported in association with PPK and ARVC/C similar to ND with universal alopecia instead of wooly hair.465
PG is a member of the armadillo protein family and a constituent protein in adherens and desmosomal junctions in many tissues, including heart muscle and epidermis. The intracellular tail of desmosomal cadherins (desmogleins and desmocollins) is connected to PG and plakophilins through armadillo repeat domains and amino-terminal head domains, respectively. These two armadillo proteins interact with desmoplakin, a plakin protein that links the desmosomal plaque to the keratin cytoskeleton.466-471
A Naxos-like phenotype (ARVC associated with severe left ventricular involvement, PPK, and woolly hair) is also associated with homozygous mutations in the gene DSC2, encoding the desmosomal cadherin desmocollin 2.472 In addition, woolly hair and PPK similar to ND but with no evidence of cardiomyopathy are associated with a homozygous missense mutation
835
8
in KANK2, encoding the steroid receptor coactivator interacting protein that controls activation of steroid receptors.473
Pathology: Palmoplantar skin reveals findings compatible with NEPPK.450,457
PALMOPLANTAR HYPERKERATOSIS WITH SQUAMOUS CELL CARCINOMA OF SKIN AND SEX REVERSAL SYNDROME Clinical Features: Palmoplantar hyperkeratosis with SCC of skin and sex reversal (OMIM #610644) is an autosomal recessive disorder described in patients from Southern Italy. It is characterized by sclerodactyly, PPK associated with multiple cutaneous SCCs, early teeth loss caused by chronic periodontal disease, nail hypoplasia with longitudinal ridging, hypogenitalism (ambiguity of external genitalia), gynecomastia, hypospadias with altered plasma sex hormone levels (low testosterone and high follicle-stimulating hormone), and sex reversal (46,XX karyotype in a male phenotype). Associated findings are hypertriglyceridemia, laryngeal cancer, nodular testicular hyperplasia, bilateral cataracts, and bilateral optic nerve coloboma.411,474,475 A similar presentation has been described in a case of true hermaphroditism with the presence of both ovarian and testicular tissue in a 46,XX woman.476
Etiology and Pathogenesis: This disorder results from homozygous mutations in the gene RSPO1, encoding R-spondin1. R-spondins are ligands interacting with Fzd–LRP receptor complexes important for β-catenin stabilization.477 Keratinocytes isolated from the PPK revealed fibroblast-like morphology and larger intercellular spaces with inability to form stratified epidermal layers in organotypic cultures.478
Pathology: Palmoplantar skin reveals orthokeratotic hyperkeratosis compatible with NEPPK.474
DIFFUSE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, MITOCHONDRIAL INHERITANCE
DIFFUSE INHERITED
PALMOPLANTAR
KERATODERMA WITH
EXTRACUTANEOUS
FEATURES,
MITOCHONDRIAL
INHERITANCE
PALMOPLANTAR KERATODERMA WITH DEAFNESS Clinical Features: The association of PPK and SNHL with a pattern of maternal inheritance caused
836
by a mutation in the extrachromosomal mitochondrial genome (mtDNA) has been identified in several families originating from New Zealand, Japan, France, and Portugal. PPK appears during childhood (5 to 15 years of age) and is characterized by orange-yellow diffuse, well-demarcated, palmoplantar hyperkeratotic plaques with minimal to no erythema. It predominantly involves pressure points and may progress gradually. Although less common, extension onto the dorsal aspects of the hands and feet with a honeycomblike pattern has been reported, and keratotic plaques over the knees, elbows, and Achilles tendon may be observed. PPK may also be rather focal, resembling calluses, and may involve only plantar skin. Clearing of PPK during pregnancy has been described. Hearing impairment may start in infancy but usually appears by 5 years of age and is bilateral, mild to severe, and may be progressive. Incomplete penetrance is common and is higher for hearing loss (60%) than for skin manifestations (37%). Both sexes are equally affected.
Etiology and Pathogenesis: All affected individuals harbor a common homoplasmic point mutation (c.A7445G) in the mitochondrial transfer RNA (tRNA) encoding the MTTS1 gene.479-481 This mutation results in a significant decrease in serine tRNA level with abnormal processing of mitochondrial mRNA and proteins.482
Pathology: PPK shows orthokeratotic hyperkeratosis, focal parakeratosis, acanthosis, and focal hypogranulosis.479-481 Ultrastructural findings are large keratohyaline granules with bundles of tonofilaments not attached to desmosomes and located at the periphery of the nucleus.479,481
FOCAL INHERITED PALMOPLANTAR KERATODERMA WITHOUT EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE
FOCAL INHERITED
PALMOPLANTAR
KERATODERMA WITHOUT
EXTRACUTANEOUS
FEATURES, DOMINANT
INHERITANCE
STRIATE PALMOPLANTAR KERATODERMA Clinical Features: Striate palmoplantar keratoderma (SPPK) is a rare autosomal dominant disorder that may result from mutations in DSP, DSG1, or KRT1 encoding desmoplakin, desmoglein 1, or keratin 1, respectively. SPPK is characterized by linear, thickened, hyperkeratotic plaques over the palms that extend along the volar aspect of the digits (Fig. 48-8A) and by circumscribed areas of skin thickening on the soles (Fig. 48-8B). The disease initially presents during the first or second decade of life and is typically exacerbated by manual labor. Nail abnormalities may
A B
8
rarely be observed (onycholysis, discoloration) and hyperkeratotic plaques over the knees, ankles, knuckle and toe pads, and dorsal aspect of the digits and toes have been reported.483-486 There are no other skin, hair, or extracutaneous manifestations.461,485,487-491
Mutations in DSG1 have been found to also result in diffuse and focal hyperkeratosis of the palms and soles with no evidence of striate lesions.483,486,492
Etiology and Pathogenesis: SPPK is caused by nonsense and frameshift heterozygous mutations in the DSG1 or DSP genes, demarcating two SPPK subtypes: type I (OMIM #148700) and type II (OMIM #125647), respectively.489,493,494 The mutations result in haploinsufficiency indicating that decreased expression of these proteins by 50% is sufficient for epidermal function in nonpalmoplantar skin but not at sites subjected to major mechanical trauma such as the palms and soles.461,493-495
Of note, SPPK-causing mutations in DSG1 have recently been shown to be inherited in a semidominant, rather than dominant, fashion. Biallelic mutations in DSG1 cause skin dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome (OMIM #615508)496 (Fig. 48-9). Thus, genetic counseling of any patient affected with SPPK should take into consideration these data. Desmoglein 1 and desmoplakin are important components of the desmosomal plaque. Desmoglein 1, a transmembrane protein that belongs to the family of desmosomal cadherins, is expressed in the upper epidermal layers and plays a critical role in cell–cell adhesion and signal transduction pathways regulating epidermal proliferation and differentiation.497 Loss of desmoglein 1 results in downregulation of proapoptotic signaling, promotes keratinocytes proliferation, and inhibits epidermal differentiation as a result of unopposed ERK signaling, explaining the hyperkeratotic phenotype seen in SPPK.498-501 Moreover, it has been suggested that desmoglein 1 deficiency results in SPPK via elevated Ras activity,502 which is supported by the high rate of PPK in disorders of the Ras–MAPK (mitogen-activated protein kinase) pathway.501
Similarly, loss of desmoplakin, a plaque protein that plays a crucial role in anchoring intermediate filaments to desmosomal cadherins, has been shown to result
in increased cell proliferation and enhanced G1-to-Sphase entry in the cell cycle associated with elevated phospho-ERK1/2 and phospho-Akt levels.503
A heterozygous frameshift mutation affecting the keratin 1 tail domain was found to underlie SPPK type III (OMIM #607654).79 This mutation leads to the partial loss of the glycine loop motif in the V2 domain and affects the function of the desmosomal plaque during cornification.78
Pathology: Histopathology findings in SPPK are orthohyperkeratosis, acanthosis, papillomatosis, widening of the intercellular spaces, and disadhesion of
837
8
keratinocytes in the upper epidermal layers. Although not entirely specific or sensitive for PPK, the latter two findings serve as a clue for the diagnosis of PPK caused by mutations in genes encoding desmosomal proteins.484,490,492,504 Immunohistochemical studies demonstrate abnormal perinuclear aggregation of keratin filaments associated with upregulation of KRT16 and abnormal involucrin expression.493-495 Ultrastructural findings are loosening of intercellular connections and disruption of desmosome–keratin intermediate filament interactions with clumped keratin filaments in a perinuclear distribution. Diminished abnormalappearing desmosomal structures may be observed.495
HEREDITARY KERATOSIS PALMOPLANTARIS VARIANT OF WACHTERS Clinical Features: Hereditary keratosis palmoplantaris variant of Wachters (PPK varians, Brünauer- Fohs-Siemens syndrome, Siemens syndrome) is a rare autosomal dominant disease with complete penetrance, more common in males, which normally appears in the first or second decade of life. It is characterized by yellowish, nontransgredient, symmetric, nummular, hyperkeratotic plaques localized to pressure points on the soles but that may become more confluent with time. Palmar hyperkeratotic lesions are described as either linear, nummular, membranous, fissured, or periungual. Nummular hyperkeratotic plaques over the elbows, knees, and Achilles tendon were described. Palmoplantar hyperhidrosis, painful transverse fissures, and nail changes (ridging and cuticle hyperkeratosis) may be associated.505-507 A single case of malignant melanoma arising in the hyperkeratotic lesions on the foot has been described.508 Varied phenotypic expression with inter- and intrafamilial variability resulted in the description of distinct subtypes and led to the term keratosis palmoplantaris varians that was introduced by Wachters in 1963.509
Etiology and Pathogenesis: Hereditary keratosis palmoplantaris variant of Wachters is an autosomal dominant disorder, although sporadic cases have been reported. The causative gene is yet to be identified. Immunohistochemical studies show early expression of both filaggrin and involucrin.505,510
It is worth mentioning that it is not yet clear whether hereditary keratosis palmoplantaris variant of Wachters and hereditary painful callosities are legitimate distinct entities or are in fact outstanding cases of more common forms of PPK such as PC (KRT16 mutations) or EPPK (KRT1 mutations).
Pathology: Histopathology findings include hyperkeratosis, hypergranulosis, possible focal parakeratosis, and acanthosis with no epidermolytic changes or dermal inflammatory cell infiltrate. Ultrastructural findings are tightly packed tonofibrils and large masses of keratohyaline granules with abnormal configuration.505,510
838
FOCAL INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE
FOCAL INHERITED
PALMOPLANTAR
KERATODERMA WITH
EXTRACUTANEOUS
FEATURES, DOMINANT
INHERITANCE
PACHYONYCHIA CONGENITA Clinical Features: PC (OMIM #167200 and
167210) is a group of rare autosomal dominant disorders of keratinization caused by mutations in one of five keratin genes, including KRT6A, KRT6B, KRT6C, KRT16, or KRT17. PC was first described in the beginning of the 20th century,511,512 although the causative genes were only identified in the late 1990s.513-515 PC prevalence in Western countries is 0.9 cases per million with a worldwide PC population estimated to be between 5000 and 10,000.516-518 The severity of the clinical features varies among and within families as the phenotype may be influenced by modifier genes or environmental factors.519
Historically, PC has been divided into two subtypes: PC-1 (Jadassohn-Lewandowski), featuring oral leukokeratosis and caused by mutations in KRT6A or KRT16 and PC-2 (Jackson-Lawler) caused by mutations in KRT6B or KRT17 and featuring cysts and natal teeth. However, phenotypic characterization of more than 1000 mutation-verified PC patients enrolled in the International PC Research Registry showed considerable overlap between these subtypes and lead to a new classification scheme based on the mutant gene. Cases of clinically suspected PC with no identified mutation in the known PC-related genes are termed PC-U (unknown).518,519
The three clinical features that are reported in more than 90% of PC patients are thickened toenails, plantar keratoderma, and plantar pain. Thickened toenails, the phenotypic feature that gave its name to the condition, appear during early childhood but may occur also in the first weeks of life (especially with mutations in KRT6A). Pachyonychia is characterized by significant subungual hyperkeratosis and very thick nails with a characteristic inverted U or V shape that grow to full length or terminate prematurely (Fig. 48-10A). Paronychia with staphylococcal and candida superinfection may be seen. On average, 9 toenails are involved, and mutations in KRT6A carry the highest likelihood of having 10 toenails affected. Nail involvement is variable, even within family members carrying the same mutations. Fingernails, although reported to be involved in many patients (100% of patients with KRT6A mutations), may be spared, especially with mutations in KRT6B.518 The most common manifestation of plantar keratoderma is focal PPK (FPPK) with calluses over weight-bearing areas (Fig. 48-10B), although diffuse keratoderma may be observed. Blistering, fissures,
8
A B C
and open sores are common. Palmar lesions are less prominent and usually occur in response to occupational mechanical trauma. The average age of onset of PPK is 4 years, although it may appear at birth or have delayed onset (30 years of age). It is very typical that FPPK starts when children begin to walk. Patients with KRT16 and KRT6A mutations develop keratoderma earlier than patients with KRT6B and KRT17 mutations.518 Although PC classically causes a nontransgredient FPPK, several cases with transgrediens and involvement of the dorsal feet have been described (mostly with KRT6A mutations).520 Plantar pain occurs in all subtypes, may be more severe in warmer weather, and is considered the most important and debilitating feature that affects patients’ quality of life. The severe pain is secondary to blister formation deep underneath the thick callus as demonstrated by high-frequency ultrasound.521 Approximately 60% of PC patients display neuropathic pain features with the existence of nociceptive pain in the remainder.517
Additional features are mucosal involvement, cyst formation, and natal teeth. Oral leukokeratosis was reported in 70% of patients. It tends to appear by 5 years of age, although onset at birth was reported in approximately half of the patients and was most characteristic with mutations in KRT6A or KRT17. It may be the first manifestation of PC in newborns, is often mistaken for candidiasis, and may alter sucking or feeding, causing failure to thrive. About 40% of patients (mostly with KRT17 mutations) exhibit various types of epidermal inclusion cysts. Pilosebaceous cysts, such as steatocystomas (Fig. 48-10C), and vellus hair cysts are most common and normally develop during puberty, continue throughout adulthood, and may require surgical removal. The face and trunk are affected. Comedone-like cysts may be found in the axillae, mainly in children.522 Follicular keratosis on the trunk and extremities, mostly in areas of friction such as the knees and elbows, may be observed in approximately
50% of patients and usually present by early childhood. The phenomenon of natal teeth (erupted teeth present at birth or by 1 month) was reported in 15% of patients with PC, almost exclusively with KRT17 mutations.518,519 Primary and secondary dentition is normal. Less commonly associated features are palmoplantar hyperhidrosis, angular cheilitis, and corneal dystrophy.523 Alopecia was described in two cases of homozygous, semidominant missense mutations in KRT17.524 Hoarseness with life-threatening respiratory distress secondary to laryngeal leukokeratosis has been seen in infants and children with KRT6A mutations. Moreover, infants and children with KRT6A mutations may demonstrate severe pain anterior to the ear on first sucking or eating that lasts a few seconds.525,526
In PC resulting from mutations in KRT16, mild FPPK is a universal finding; however, many mutations in KRT16 produce a less pronounced phenotype with FPPK restricted to weight-bearing areas of the soles, limited or absent nail involvement, and no evidence of oral leukokeratosis, leading to the diagnosis of focal NEPPK rather than PC.527-531
Mutations in KRT6C have been reported to be associated with a milder phenotype similar to some of the KRT16 mutations. These cases are characterized by plantar keratoderma (FPPK over pressure sites or diffuse PPK) and callosities over the palms with absent or minor nail changes, such as hypertrophy of the fifth toenail, and plantar blisters. Leukokeratosis may be seen but with no additional clinical findings.532,533 Key clinical features of PC with associated mutations are summarized in Table 48-3.
Etiology and Pathogenesis: PC is caused by heterozygous mutations in one of five keratin genes expressed in differentiated epithelial tissues: KRT6A, KRT6B, KRT6C, KRT16, and KRT17, encoding keratins 6a, 6b, and 6c (type II keratins) and keratins 16 and 17 (type I keratins). The expression of keratins
839
8
GENE FREQUENCY (%) COMMON MUTATION (S) KEY CLINICAL FEATURES
KRT6A 40 p. Asn172del Thickened toenails and fingernails since infancy Highest likelihood of having 10 toenails affected; Earlier onset of FPPK; transgrediens and involvement of the dorsal feet may occur Oral leukokeratosis Laryngeal leukokeratosis with hoarseness and life-threatening respiratory distress
KRT16 25–30 p. Arg127Cys (mild) p. Arg127Pro (severe) p. Leu128Gln (mild) p .Leu128Pro (severe)
Mild FPPK with limited or absent nail involvement Earlier onset FPPK is possible Coinheritance of FLG mutations aggravates the phenotype
KRT17 20 p. Asn92Ser Delayed onset of FPPK Oral leukokeratosis Epidermal inclusion cysts (steatocystomas, vellus hair cysts) Natal teeth Alopeciaa
KRT6B 5–9
Fingernails may be spared Delayed onset of FPPK
KRT6C 3 Mostly mild FPPK and callosities over the palms with absent or minor nail changesb
KRT6C 3
Mostly mild FPPK and callosities over the palms with absent or minor nail changesb
Leukokeratosis with no additional clinical features may be seen
Leukokeratosis with no additional clinical features may be seen
aIn a single case of homozygous semidominant inheritance.
bA milder phenotype may be the consequence of more restricted expression of keratin 6c in palmoplantar skin and nails compared with keratins 6a and 6b. KRT6C mutation carriers in the general population have been reported. FPPK, focal palmoplantar keratoderma.
6 and 16 is restricted in normal epidermis to the upper outer hair root sheath and nail bed, palmoplantar skin, and suprabasal orogenital mucosal keratinocytes.534
In addition, wounding or inflammation induces their expression in interfollicular epidermis, and they are expressed at high levels in cultured keratinocytes from both normal and psoriatic skin.535
Autosomal dominant inheritance is observed in more than half of the cases; the remaining cases are caused by spontaneous dominant mutations.536
There are a few case reports of apparent recessive PC inheritance. However, there are no such cases with confirmed genetic testing, and it is likely that most if not all of these cases represent phenocopies of PC.537
Semidominant inheritance for KRT17 mutations has been reported as mentioned earlier.524 Paternal germ cell mosaicism for a KRT6A mutation has also been described.538 Most mutations are missense mutations, and the remainder are small in-frame deletion, frameshift, nonsense, or splice-site mutations that are likely to exert a dominant-negative effect.536 The majority of reported mutations to date involve the highly conserved helix boundary domains at either end of the α-helical rod domain, being vital for the elongation phase of filament assembly.539
Skin biopsies obtained from PC-involved skin show markedly induced expression of the PC-related genes KRT6, KRT16, and KRT17, which are known to be upregulated in states of stress or injury. Mechanical stress may lead to upregulation of both wild-type and mutant forms of these keratins and perturbed intermediate filament formation caused by mutant keratin incorporation, triggering the PC phenotype. This in turn has been linked to
840
aberrant expression of additional mutant keratin as part of the wound healing response, resulting in a vicious cycle of gradual worsening.540-542 Moreover, overexpression of genes related to cell adhesion (DSC2, CDSN, and GJB2), cornified cell envelope formation (involucrin and loricrin) and desquamation (kallikrein [KLK]-5; SPINK6, and SERPINs) and non–PC-related keratins (KRT75 and KRT85) has been observed. Pain-associated genes are also upregulated in PC-involved skin, including KLK10 (catalyzes the production of bradykinin) and SPRR1A (a structural protein in keratinocytes being expressed also by neurons), which may relate to the severe pain featured by PC patients.542
Pathology: Skin biopsies obtained from PPK lesions reveal features of NEPPK. Reduced granular layer and cytolysis in the outer root sheath of HFs may be seen.513,530 Epidermolytic hyperkeratosis has been observed in a case associated with mutation in KRT16 with striate palmar hyperkeratosis and diffuse PPK of the soles.101 Ultrastructurally, suprabasal keratinocytes in PC are characterized by densely aggregated keratin filament bundles, predominantly in the perinuclear region and sparing the cell periphery. Reduced desmosome number with widened intercellular spaces and reduced keratohyaline granules are additional findings.513
Specific Treatments: Contradictory data regarding the efficacy of retinoids in PC have been published with an improvement in both PPK and nail dystrophy in some reports and no improvement in others. In addition, the treatment had to be withheld in many patients with observed improvement because
of increased pain as a result of epidermal thinning and predisposition to infections.543-546
In PC, the most effective approach to nail dystrophy consists of mechanical treatments (filing, grinding, cutting, or clipping) and soaking the nails. Surgical avulsion is sometimes ineffective because of regrowth of nails. Oral leukokeratosis may be improved by keeping good oral hygiene, gentle brushing, and oral antibiotics. Follicular hyperkeratosis is treated with oral and topical retinoids, keratolytic agents, and emollients. Steatocystoma multiplex and other pilosebaceous cysts can be treated by incision and drainage, excision, intralesional steroid injection, and oral antibiotics in the case of secondary infections.544,547-550 Recently, targeted and more specific therapies have been studied in PC. Rapamycin has been shown to result in marked improvement in painful plantar calluses and in quality of life by selectively blocking translation of mRNAs, including KRT6 mRNA. However, the use of oral rapamycin was limited by side effects, including gastrointestinal symptoms. A clinical trial with topical rapamycin is underway.526,551 Moreover, a specific siRNA that selectively silences the expression of a specific pathogenic mutation in KRT6A has been shown to lead to callus regression and pain control in a phase Ib clinical trial.552 New approaches for more efficient, safe, and practical ways to deliver siRNA into the epidermis are needed to avoid the painful injections.548,552-557
HOWEL-EVANS SYNDROME Clinical Features: Howel-Evans syndrome (tylosis with esophageal cancer; OMIM #148500) is a rare autosomal dominant disorder with complete penetrance characterized by the association of PPK and mucosal SCCs, particularly of the esophagus. It was first reported by Howel-Evans and coworkers in 1958558 and has been described in families from a range of countries.559-565 The estimated prevalence of the disorder in the general population is less than 1 in 1,000,000.566 The onset of PPK is usually during childhood or adolescence (between 5 and 15 years, although most cases are evident by 7 to 8 years of age). It is characterized by focal, yellowish, thick plaques localized to areas of pressure or friction on the palms and soles (see Fig. 48-7B) that may be associated with painful fissures and secondary infections. Sparing of the palms may be evident.567 Additional findings are follicular hyperkeratosis, cutaneous horns, and oral leukokeratosis. Cases of SCC of the oropharynx have been recorded.559-566,568,569
Esophageal lesions present as few-millimeter, white, polypoid lesions throughout the esophagus. About 95% of patients with Howel-Evans syndrome develop carcinoma by 65 years of age, similar to the onset of sporadic cases.566
In patients with Howel-Evans syndrome, screening includes annual gastroscopy. Besides surveillance, diet, and lifestyle modification to reduce risk factors for esophageal carcinoma are recommended.566
Etiology and Pathogenesis: Howel-Evans syndrome results from GOF missense mutations
8
in RHBDF2, which encodes a catalytically inactive rhomboid intramembrane serine, iRhom2. iRhom2 belongs to a family of seven transmembrane-spanning proteins, which are serine intramembrane proteases associated with EGFR signaling and mitochondrial remodeling.570,571 RHBDF2 is an iRhom that lost its protease activity during evolution but retained key nonprotease functions (regulation of EGF and TNF-α signaling pathways).572,753
Skin biopsies obtained from patients with Howel- Evans syndrome demonstrate cytoplasmic localization of iRhom2 compared with the normal membrane expression seen in normal skin. Similar cytoplasmic localization is observed in biopsies obtained from Howel-Evans syndrome esophageal carcinoma and from sporadic squamous esophageal tumors, suggesting that dysregulation of iRhom2 plays a role in these malignancies.574,575
iRhom2 has been shown to regulate the trafficking and activation of ADAM17, a membrane bound sheddase that has been shown to play a pivotal role in the proteolytic cleavage and release of substrates from the cell surface, including TNF-α, members of the EGF family of growth factors, and desmosomes.573,576 Epidermal keratinocytes from patients with Howel-Evans syndrome show upregulated ADAM17 activity, resulting in increased EGFR activity, increased desmosome processing, immature epidermal desmosomes, upregulated epidermal transglutaminase activity, and resistance to staphylococcal infection.577 Moreover, these keratinocytes demonstrate features of dysregulated wound repair in vitro.577,578
Overexpression of EGFR has been demonstrated in sporadic esophageal SCC and several other carcinomas and correlates with reduced survival. Moreover, expression of ADAM17 has been shown to correlate with progression of esophageal carcinoma.579-5815
Accordingly, precancerous esophageal lesions seen in Howel-Evans syndrome may be a result of dysregulated EGFR signaling.574
Pathology: Affected skin reveals acanthosis, orthohyperkeratosis, and hypergranulosis with no parakeratosis or spongiosis.566,567
FOCAL INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, RECESSIVE INHERITANCE
FOCAL INHERITED
PALMOPLANTAR
KERATODERMA WITH
EXTRACUTANEOUS
FEATURES, RECESSIVE
INHERITANCE
RICHNER-HANHART SYNDROME Clinical Features: Richner-Hanhart Syndrome (RHS, tyrosinemia type II, oculocutaneous tyrosinosis, keratosis palmoplantaris with corneal dystrophy;
841
8
OMIM #276600) is a rare autosomal recessive disorder named after the original reports of Richner and Hanhart. Its incidence is less than 1 in 250,000, occurring in various ethnic groups, although it seems to be particularly prevalent in Mediterranean countries and in the Arab world.582-587
Symptoms typically appear in early childhood and include a triad of painful PPK, bilateral keratitis, and mental retardation that results from intracellular accumulation of tyrosine crystals and a secondary inflammatory response in the involved tissues. Ocular manifestations include photophobia, pain, tearing, redness, and pseudoherpetiform keratitis with corneal ulcerations. Ocular manifestations occur in 75% of cases, present soon after birth or within the first year of life, and usually antedate the cutaneous manifestations. Skin manifestations usually begin after the first year of life and consist of well-demarcated, focal, palmoplantar, white-yellow, hyperkeratotic plaques surrounded by erythema on the weight-bearing areas of the soles. There is associated pain and often hyperhidrosis. The fingertips, hypothenar, and thenar eminences can also be affected. Sixty percent of cases, particularly untreated ones, present with neurologic manifestations, including mental retardation, nystagmus, tremor, ataxia, and convulsions.588-591 A case with epileptic seizures, mild mental retardation, and photophobia with no other ophthalmologic or skin manifestations has been reported.592
Etiology and Pathogenesis: RHS is an autosomal recessive disorder caused by mutations in the TAT gene encoding tyrosine aminotransferase (TAT). As a consequence of TAT deficiency, tyrosine accumulates in tissues. More than 20 mutations have been identified within the TAT gene with alteration of the activity and stability of the tyrosine aminotransferase enzyme.572,587,593-600 It was hypothesized that disease severity, particularly neurologic disabilities, correlates with the presence of a mutant protein but its total absence is associated with more favorable features.601,602
Previous reports have suggested that palmoplantar lesions in RHS result from intracellular L-tyrosine crystals that destabilize lysosomal membranes and initiate a cascade of cell injury and inflammation, resulting in the typical skin lesions. This hypothesis is consistent with the presence of crystals in tyrosine-induced lesions in the corneal epithelium of rats and in the spinous layer keratinocytes of patient epidermis.603-606 However, later studies failed to demonstrate crystals in epidermal keratinocytes and suggested that the palmoplantar hyperkeratosis results from an excessive intracellular concentration of tyrosine that leads to noncovalent cross-links among keratins and the formation of aggregated tonofilaments.607
Laboratory Findings: RHS is diagnosed by a high level of serum tyrosine (with normal phenylalanine) and accumulation of tyrosine metabolites in the
842
urine (p-hydroxyphenylpyruvate, p-phenylacetate, p-hydroxyphenylacetate).601 A static level of urinary succinylacetone distinguishes RHS from type l tyrosinemia.
Pathology: Histopathologic findings in RHS include marked acanthosis, hyperkeratosis, and significant hypergranulosis. Parakeratosis, a parakeratotic column in the acrosyringium, and multinucleated keratinocytes in the spinous cell layer are possible findings. Increased mitotic activity in the suprabasal layers and thin, elongated epidermal ridges, may be seen.586,601,608,609 Intracytoplasmic tyrosine crystals have been reported.606 Ultrastructural findings include thickening of the granular layer and increased synthesis of tonofibrils and keratohyalin, large numbers of microtubules, and tonofibrillar masses with tubular channels or inclusions of microtubules. Multinucleated keratinocytes in the spinous layer and lipid droplets in the cornified layer may be observed.586,607,609
Specific Treatments: The key to the management of RHS is early diagnosis and early initiation of a diet that restricts tyrosine and phenylalanine to reduce the risk and severity of long-term complications of hypertyrosinemia, especially in the eye and skin.601,609,610 Tandem mass spectrometry newborn screening for inborn errors of metabolism identifies RHS in the asymptomatic neonatal period.611 Retinoids improve skin and eye lesions, but they do not prevent mental retardation and should be considered discriminately in children.612 A case in which thigh skin was grafted onto a plantar lesion demonstrated hyperkeratosis that spared the graft area with the formation of a keratotic wall around it.613
CARVAJAL SYNDROME Clinical Features: Carvajal syndrome (CS; OMIM #605676), first reported by Carvajal-Huerta in 1998, is an autosomal recessive disease caused by mutations in the DSP gene (encoding desmoplakkin) and characterized by a triad of woolly hair, SPPK, and cardiomyopathy. Left ventricular dilated cardiomyopathy develops during childhood; the right ventricle may also be involved.614-616 Patients may demonstrate only skin or cardiac features or the full phenotype. Woolly hair appears at birth and may be associated with sparse eyebrows and axillary and pubic hair. SPPK develops during childhood, although cases with appearance at birth have been reported. A case featuring alopecia of the scalp, eyebrows, and eyelashes has been described.617 Additional cutaneous features are linear lichenoid keratotic papules in the flexural folds, follicular hyperkeratosis on the elbows and knees or scattered across the abdomen and lower limbs, nail abnormalities (dystrophy, onychogryphosis, clubbing), transient pruritic vesicles, and psoriasiform plaques.614-618 CS cases with dominant
inheritance are associated with hypo- or oligodontia and leukonychia or brittle nails.618,619 Co-occurrence with congenital unilateral deafness, recurrent pharyngitis, and diarrhea was reported.620 The left ventricle is severely affected by the second decade of life in more than 90% of patients, although cardiomyopathy with symptoms of progressive heart failure presenting as early as the first years of life has been reported. Congestive heart failure and ventricular arrhythmia are the most common causes of death during adolescence.617,621
Etiology and Pathogenesis: Desmoplakin is a central component of the desmosomes, which are cell-to-cell junctions in simple and stratified squamous epithelia and cardiac muscle that connect intermediate filaments to the desmosomal cadherins in the cytoplasmic membrane. The N-terminal plakin domain of DSP binds PG, plakophilins, and desmosomal cadherins, its C-terminal domain connects the cytoskeleton intermediate filaments, and its central coiled-coil rod domain is responsible for homodimerization.622 DSP is expressed in all epidermal layers, in the outer root sheath, companion layer, and Henle and Huxley layers of the HF; intercalated discs are important for cardiomyocyte binding in the heart.623-625
The first homozygous mutation in DSP causing CS was described in an Ecuadorian pedigree and resulted in a truncated protein lacking the C-domain tail region responsible for intermediate filament binding.614 Another recessive missense mutation that was associated with blisters in early childhood and ARVC during adolescence was described in Arab kindred and affected the C-terminal domain of the protein.616 Several other homozygous mutations resulting in truncated protein involving the coiledcoil rod domain and the C-terminal domain with impaired intermediate filament binding have been described.626
Dominant heterozygous mutations in DSP have been reported in cases of tooth agenesis in addition to the common triad of CS (dilated cardiomyopathy with wooly hair, keratoderma, and tooth agenesis; DCWHKTA; OMIM #615821).618,619,627,628 In these cases, the mutations affect the N-terminal domain of DSP and most involve exon 14, suggesting that this region serves as a hot spot and disrupts desmosome scaffolds in a dominant-negative fashion. The clinical feature of tooth agenesis is in line with studies in mice demonstrating a role of desmosomal proteins in teeth development.629,630
Besides DCWHKTA, heterozygous DSP mutations are linked to ARVD type 8 (OMIM #607450) and isolated SPPK (see earlier). Homozygous or compound heterozygous DSP mutations also result in lethal acantholytic epidermolysis bullosa (OMIM #609638) and skin fragility–woolly hair syndrome (OMIM #607655).628,631,632
Pathology: The histopathology findings in CS are hyperkeratosis, papillomatosis, spongiosis, epidermolytic hyperkeratosis, and dyskeratosis. Acantholysis may be seen in the spinous layer.614
8
PUNCTATE INHERITED PALMOPLANTAR KERATODERMA WITHOUT EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE
PUNCTATE INHERITED
PALMOPLANTAR
KERATODERMA WITHOUT
EXTRACUTANEOUS
FEATURES, DOMINANT
INHERITANCE
PUNCTATE PALMOPLANTAR KERATODERMA TYPE I Clinical Features: Punctate palmoplantar keratoderma (PPKP) type I (PPKP1; keratosis punctate palmoplantaris type Buschke-Fischer-Brauer; OMIM #148600 [PPKP1A] and 614936 [PPKP1B]) is a rare autosomal dominant disease. The estimated incidence of PPKP1 is approximately 1 to 3 per 100,000 individuals in various populations across Europe, the Middle East, and Asia.36,588,633-636
The disease has its onset during the first or second decades of life, although a later onset has also been described.589,591,633,637-640 It is characterized by multiple hyperkeratotic, centrally indented, yellow to brown papules distributed irregularly over the palmoplantar skin (Fig. 48-11). Lesions may be painful. They increase in size and number with advancing age and coalescence into more confluent plaques, particularly on pressure-bearing areas of plantar skin. A phenotype resembling human papilloma virus–induced papilloma-like lesions has been described.641,642
Dermoscopic findings of palmoplantar lesions are well-demarcated, structureless, yellow-orange areas
843
8
surrounded by a whitish halo with no evidence of dotted vessels commonly seen in verruca vulgaris.37
Lesions reveal white fluorescence with Wood light examination.643
Etiology and Pathogenesis: PPKP1 is an autosomal dominant disease that results from heterozygous mutations in two genes, AAGAB (PPKP1A) and COL14A1 (PPKP1B). AAGAB encodes the α-and γ-adaptin-binding protein p34,599.634,635,641 which contains an adaptin-binding domain and a Rab-like GTPase domain that might play an important role in clathrin-coated vesicle trafficking as a chaperone and in skin integrity.635,641,644,645
Inter- and intrafamilial phenotypic variability is common and may be related to the effect of modifier genes or of aging and environmental factors.36,591 Genetic anticipation (earlier disease onset in later generations) has been reported.633,639 To date, more than 30 null variants in AAGAB have been identified, and haploinsufficiency was suggested as a possible disease-causing mechanism.635
A missense mutation in COL14A1, encoding collagen type XIV α1 chain, was identified in a large Chinese family with PPKP1 and incomplete penetrance.646 The protein is mainly expressed in well-differentiated tissues and in late embryonic development, and the reported mutation involves the collagen triple helix repeat region.
Pathology: Histopathologic findings in PPPK include marked hyperkeratosis and acanthosis with epidermal invagination associated with focal parakeratosis, hyper- or hypogranulosis, and overlying orthokeratosis.634,635,641 Transmission electron microscopy of lesional plantar skin shows an abnormal abundance of small vesicles close to the cell membrane in the basal epidermal layer and prominent dilatation of the Golgi apparatus, consistent with a defect in vesicle transport.641
PUNCTATE PALMOPLANTAR KERATODERMA TYPE 2 Clinical Features: PPKP type II (PPKP2; porokeratotic type; OMIM #175860) is an autosomal dominant disease characterized by multiple, firmly attached, tiny, skin-colored to yellow, asymptomatic, keratotic spines arising on the palms and soles around puberty or in the early 20s. Lesions may increase in number over the years with possible extension onto the dorsal and lateral surfaces of the fingers.646-649 Lesions reveal white fluorescence resembling “stars under the moonlight” with Wood light examination.650
Facial sebaceous hypoplasia was reported in male patients.649 Acquired cases with a similar phenotype have been described in association with internal malignancies.651
Etiology and Pathogenesis: The molecular etiology of PPKP2 is unknown. Most described cases
844
are acquired; however, there are also familial cases with autosomal dominant inheritance. The pathogenesis involves enhanced epidermal proliferation of the basal layer under the columnar parakeratoses.652
Pathology: Histopathology findings in PPKP2 are compact vertical parakeratotic columns in the stratum corneum overlying hypogranular epidermis resembling cornoid lamellae.648 Ultrastructural findings are numerous, variable-sized, pyknotic nuclei in the stratum corneum and reduced number of keratohyaline granules in the granular layer.652
PUNCTATE PALMOPLANTAR KERATODERMA TYPE 3 Clinical Features: PPKP type III (PPKP3, AKE [of Costa]; OMIM #101850) is a rare autosomal dominant disorder that usually appears during childhood or adolescence, although onset in infancy or adulthood has also been reported. There is no racial or ethnic predilection.653 AKE is characterized by asymptomatic, round to oval, whitish to yellow and translucent papules, and more rarely nodules and plaques, with a hyperkeratotic surface or umbilication. Changes are located on the palms and soles with predilection for the thenar and hypothenar areas of the hands and pressure sites on the palms and soles. A linear pattern or “paving stones” arrangement over the radial and ulnar margins of the hands may be seen (Fig. 48-12).654,655 In severe cases, lesions may affect the dorsal aspects of hands and feet (including knuckle pads), wrists, and ankles.653,656 Aquagenic PPK can be associated,657 and unilateral involvement has been described.658,659 Nail dystrophy and hyperhidrosis may be observed.657,660 Although AKE is usually confined to the skin, a case with reduced elasticity in medium and large arteries and an association with systemic and nodular scleroderma has been reported.664-663
TRPV3 MBTPS2 (X-linked cases) TRPV3
Diffuse PPK
8
(Continued)
8
RSPO1 R-spondin1
(Continued)
Palmoplantar hyperkeratosis with SCC of skin and sex reversal syndrome
Punctate PPK
Focal PPK
tase/phosphodiesterase 1
Focal acral hyperkeratosis (FAH) is an autosomal dominant inherited disease with phenotypic resemblance to AKE, including nail abnormalities.664 However, no elastic tissue abnormalities are observed in these cases. FAH is often considered to be a variant of AKE, although this notion is not accepted by some authors.654,665-668
Etiology and Pathogenesis: PPKP3 is an autosomal dominant inherited disorder, although sporadic cases have been described.669,670 The genetic basis of the disease has yet to be elucidated, although preliminary linkage studies suggest a possible locus on 2p25-p12.671,672
Ultrastructural studies support AKE involves a defect in secretion of elastic material and a failure of elastic fiber synthesis rather than degeneration of elastic fibers.673 In addition, the formation of the keratotic papules are a result of filaggrin overproduction that accumulates over the granular layer before its incorporation into the CCE.674
Pathology: AKE lesions show orthokeratotic hyperkeratosis, acanthosis, and hypergranulosis. Both lesional and normal-appearing skin reveal decreased elastic tissue and fragmented, thickened, or thinned elastic fibers (elastorrhexis) in the reticular dermis with sparing of the papillary dermis.657 Ultrastructural findings are fibroblasts with dense granules at the cytoplasm periphery with reduced extracellular content of elastic fibers.673
PUNCTATE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE
PUNCTATE INHERITED
PALMOPLANTAR
KERATODERMA WITH
EXTRACUTANEOUS
FEATURES, DOMINANT
INHERITANCE
COLE DISEASE Clinical Features: Cole disease (OMIM #615522) is a rare autosomal dominant disorder that presents at birth or during early infancy and is characterized by punctate PPK and irregularly shaped, hypopigmented macules distributed over the proximal extremities or less typically over the trunk.674-678 Calcifications in several organs (tendons, breast, and spleen) have been described.678,680
Etiology and Pathogenesis: Cole disease results from heterozygous mutations in the gene ENPP1, encoding ectonucleotide pyrophosphatase/ phosphodiesterase 1, a cell surface protein that generates extracellular inorganic pyrophosphate by catalyzing the hydrolysis of adenosine triphosphate to adenosine monophosphate.681 ENPP1 is composed of
8
eight domains including phosphodiesterase, nuclease, and somatomedin B–like (SMB) domains.682 Biallelic mutations affecting the phosphodiesterase domain that mediates ENPP1 catalytic activity or the nuclease domain have been shown to be associated with inherited disorders featuring abnormal calcium homeostasis or ectopic calcifications,683-685 and mutations associated with Cole disease affect highly conserved cysteine residues clustered within the SMB domains.678,680 Given the notion that ENPP1 has been shown to inhibit insulin signaling through the interaction between its SMB domain and insulin receptor,684-687 it is suggested that abnormal insulin signaling plays a role in the pathogenesis of Cole disease.688-691
Pathology: Skin biopsies obtained from palmoplantar lesions show hyperkeratosis, orthokeratosis, hypergranulosis, and acanthosis. Calcified deposits in the dermis may be seen.676 Hypopigmented macules demonstrate hyperkeratosis, reduced melanin content in keratinocytes, and normal or reduced number of melanocytes.678,680,692 Ultrastructural findings of hypopigmented lesions consist of disproportionately large numbers of melanosomes within the cytoplasm and dendrites of melanocytes with paucity of melanosomes in adjacent keratinocytes, suggesting abnormal melanosome transfer as the primary pathomechanism for hypopigmentation in Cole disease.678,680
SUMMARY
See Table 48-4.
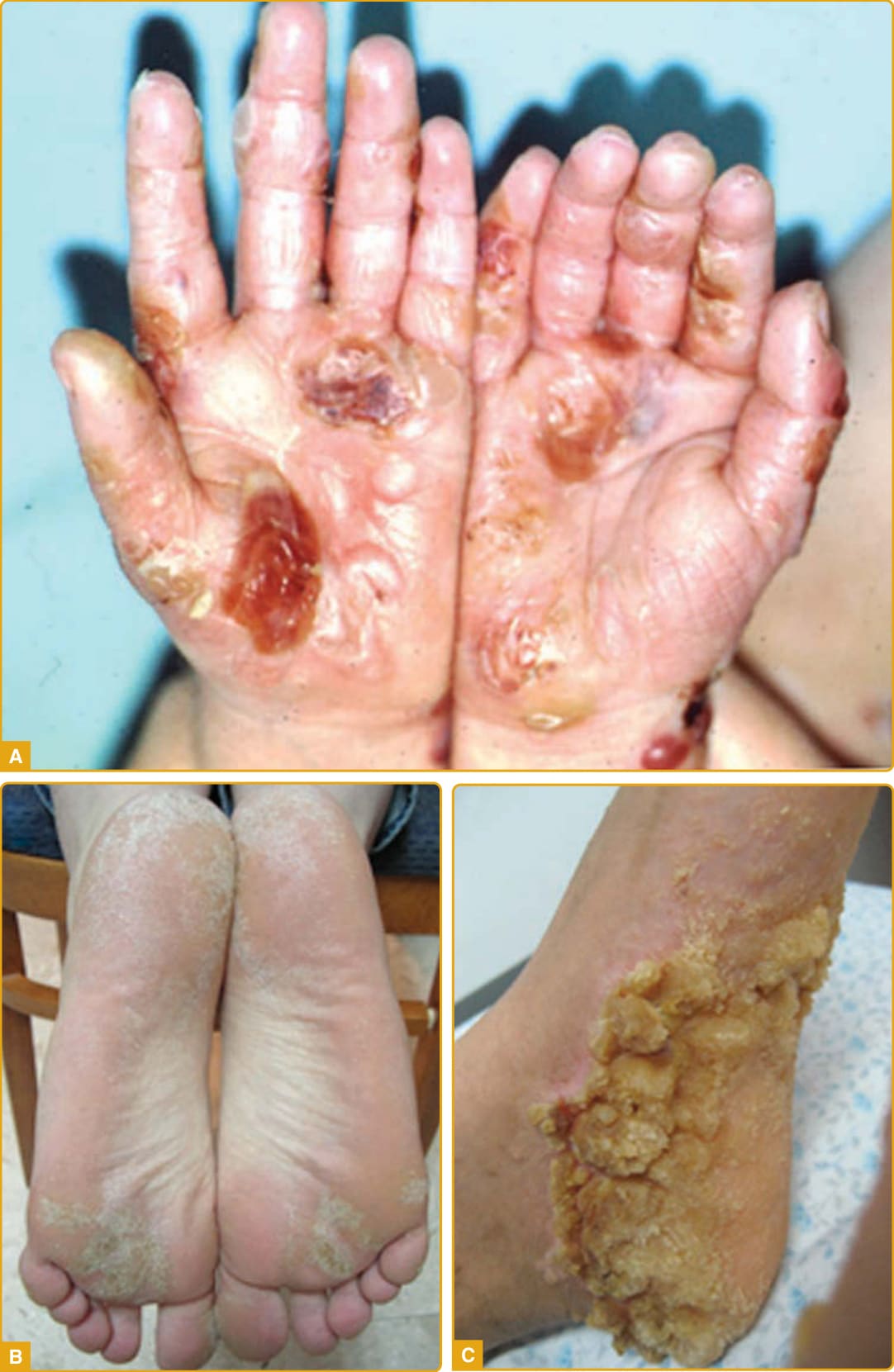
Figure 48-1 Genodermatoses with palmoplantar keratoderma as an associated feature. A, Diffuse palmar keratoderma with blisters in a patient with epidermolytic ichthyosis caused by KRT1 mutation. B, Focal hyperkeratotic plantar plaques on weight-bearing areas in a patient with Conradi-Hunermann-Happle syndrome. C, Thick, yellow, hyperkeratotic plaques on plantar skin in a patient with Darier disease.
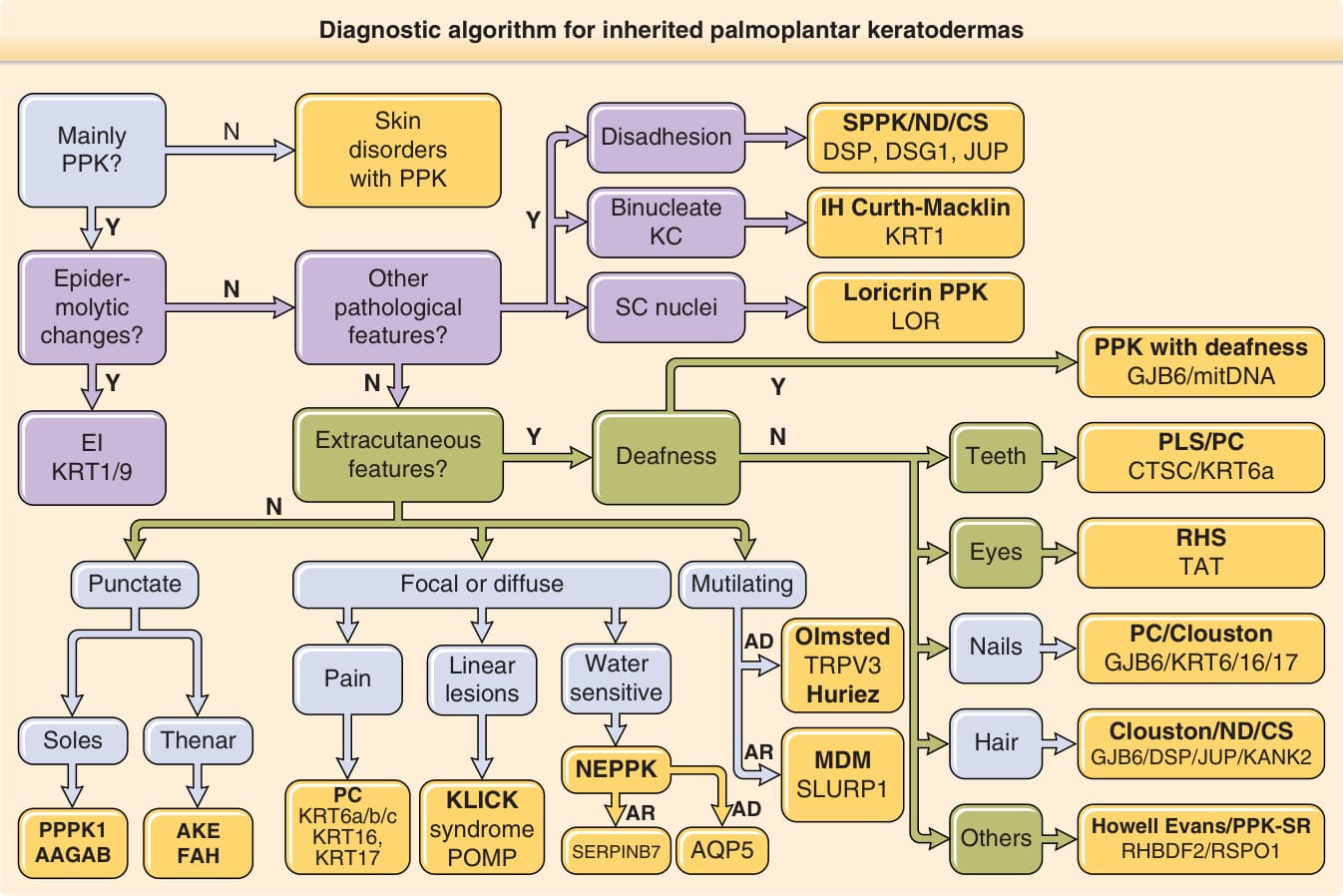
Figure 48-2 Diagnostic algorithm for inherited palmoplantar keratodermas (PPKs). Most patients with inherited PPK can be assigned to one gene based on four criteria: cutaneous findings, extracutaneous manifestations, mode of inheritance, and histopathologic findings. AAGAB, α-and γ-adaptin-binding protein P34; AKE, acrokeratoelastoidosis; AQP5, aquaporin 5; CS, Carvajal syndrome; CTSC, cathepsin C; DSG1, desmoglein 1; DSP, desmoplakin; EI, epidermolytic ichthyosis; FAH, focal acral hyperkeratosis; GJB6, gap junction protein β6; IH, ichthyosis hystrix; JUP, plakoglobin; KANK2, KN motif and ankyrin repeat domains 2; KC, keratinocytes; KLICK, Keratosis linearis–ichthyosis congenital–keratoderma; KRT, keratin; LOR, loricrin; MDM, mal de Meleda; ND, Naxos disease; NEPPK, nonepidermolytic palmoplantar keratoderma; PC, pachyonychia congenita; PLS, Papillon-Lefevre syndrome; POMP, proteasome maturation protein; PPK-SR, palmoplantar keratoderma with sex reversal; PPPK1, punctate palmoplantar keratoderma type I; RHBDF2, rhomboid 5 homolog 2; RHS, Richner-Hanhart syndrome; RSPO1, R-spondin 1; SC, stratum corneum; SERPINB7, serpin family B member 7; SLURP-1, secreted LY6/PLAUR domain containing 1; SPPK, striate palmoplantar keratoderma; TAT, tyrosine aminotransferase; TRPV3, transient receptor potential cation channel subfamily V member 3. (The authors acknowledge the contributions of Maurice van Steensel, Vinzenz Oji, Edel A. O’Toole, David Hansen, Mary Schwartz, and Frances Smith in the preparation of this figure.)
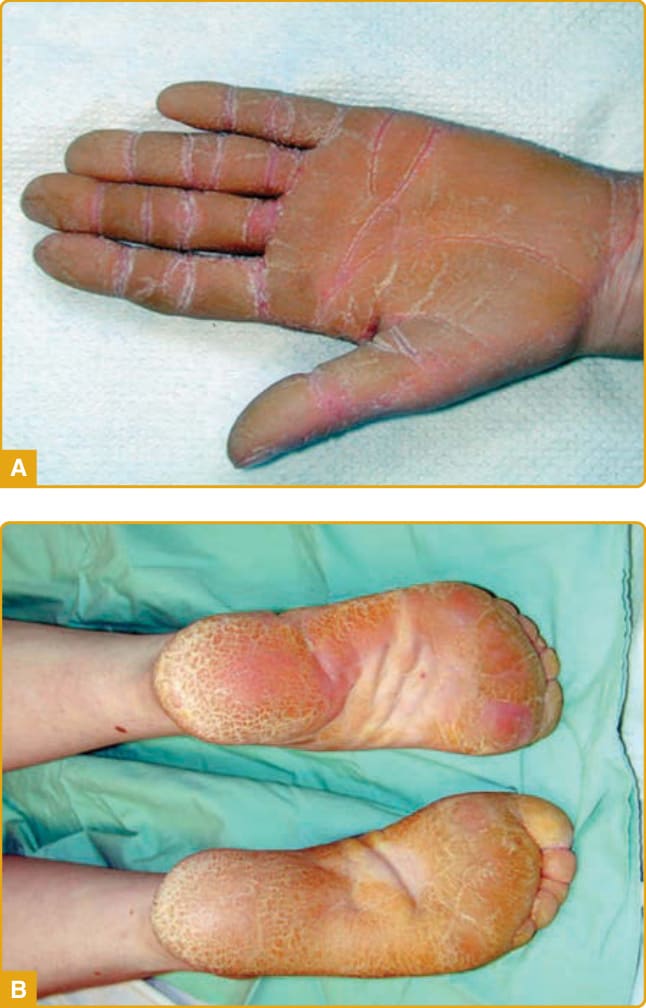
Figure 48-3 Epidermolytic palmoplantar keratoderma. Yellow, diffuse palmoplantar keratoderma with erythematous sharp margins at the edge of the palms (A) and soles (B). A heterozygous mutation in KRT9 was identified in this patient.
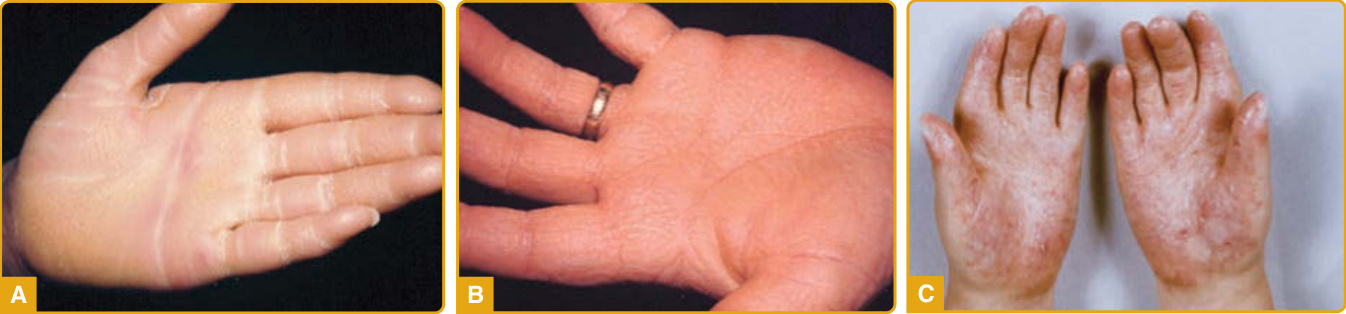
Figure 48-4 Rare forms of transgrediens palmoplantar keratoderma. A, A case of loricrin palmoplantar keratoderma with diffuse, well-demarcated, honeycomb-like palmoplantar keratoderma with an erythematous border. B, Vohwinkel syndrome with a “honeycomb-like” appearance C, Huriez syndrome. Diffuse, well-confined hyperkeratotic plaques over the palms. (Used with permission from Dr. Cameron Kennedy, Bristol Royal Infirmary, United Kingdom.)
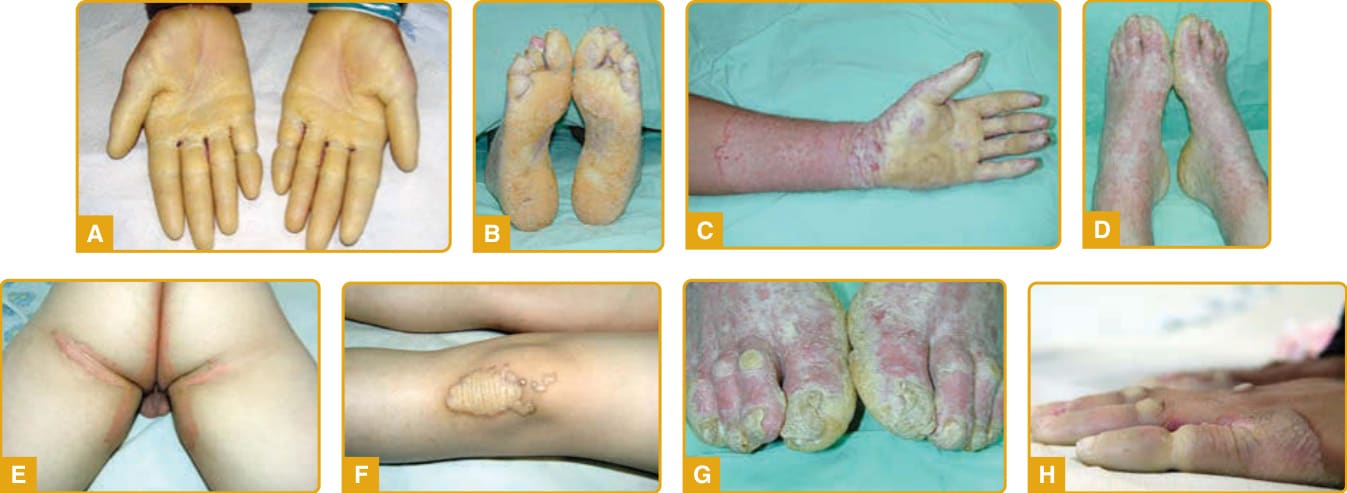
Figure 48-5 Mal de Meleda. Diffuse, yellow, waxy hyperkeratotic plaque outlined by a red, scaly border over the palms (A) and soles (B) with extension onto the dorsal surface of the hands and feet in a glove (C) and stocking (D) distribution. Involvement of the groin area (E), keratotic plaques over joints (F), nail abnormalities (G), and pseudoainhum (H) are associated findings.
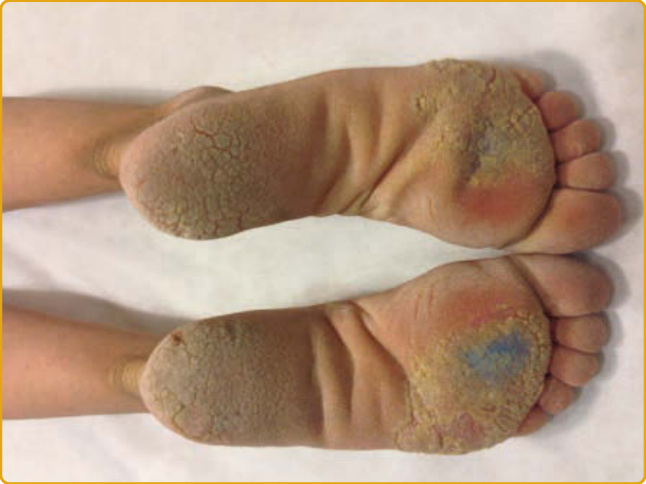
Figure 48-6 Keratitis–ichthyosis–deafness (KID) syndrome. Diffuse, hyperkeratotic, plantar keratoderma with the characteristic rough, stippled, and grainy appearance.
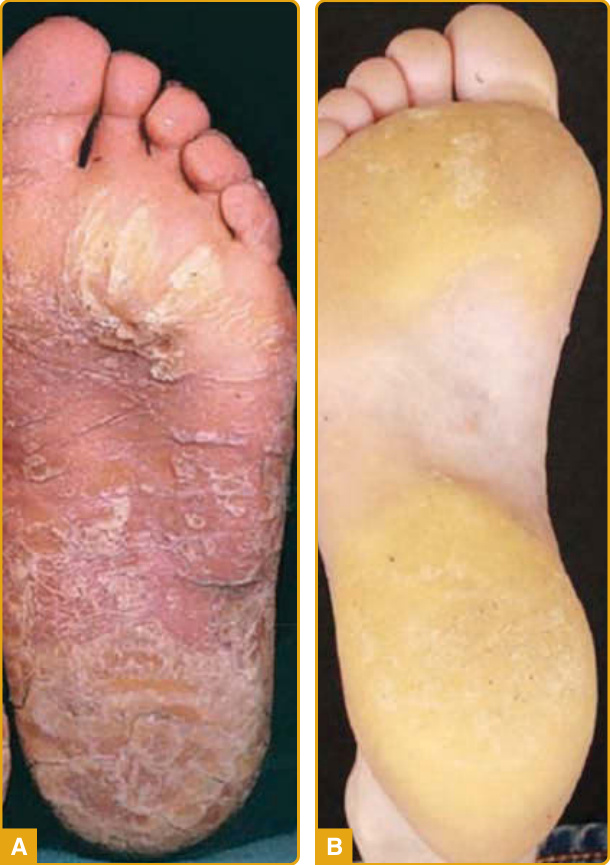
Figure 48-7 Plantar keratodermas of Papillon-Lefèvre and Howel-Evans syndromes. A, Diffuse keratoderma in Papillon-Lefèvre syndrome caused by mutation in cathepsin C. B, Howell-Evans syndrome showing focal, yellow, thick plaques localized to areas of pressure on the soles. (Part A used with permission from Barts and the London NHS Trust, United Kingdom.)
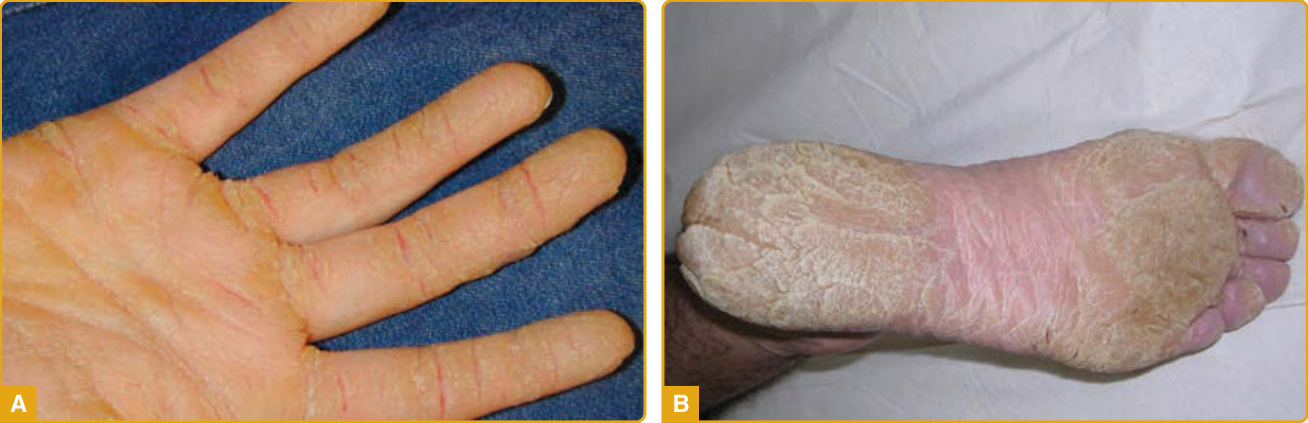
Figure 48-8 Focal palmoplantar keratoderma with DSG1 mutation. Linear, thickened, hyperkeratotic plaques on the palms which extend along the volar aspect of the digits (A) and circumscribed areas of hyperkeratotic plaques on the soles (B).
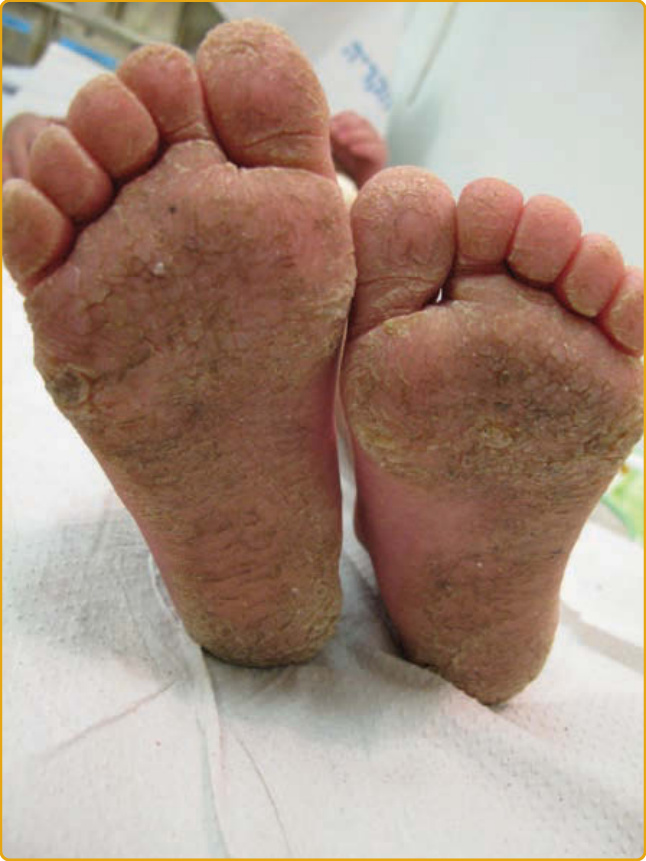
Figure 48-9 Plantar keratoderma in a patient with skin dermatitis, multiple allergies, and metabolic wasting syndrome resulting from biallelic mutations in DSG1.
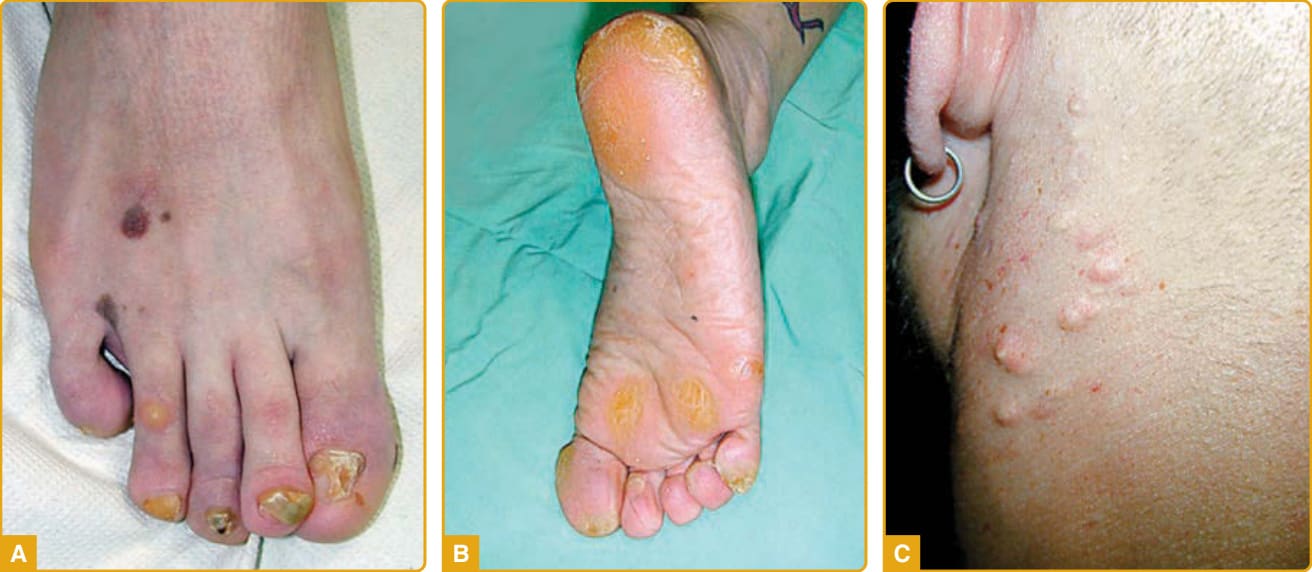
Figure 48-10 Pachyonychia congenita. KRT6A mutation with the characteristic nail abnormalities (V-shape thick nails and subungual hyperkeratosis) (A) and focal palmoplantar keratoderma with calluses over weight-bearing areas (B). Mutations in KRT17 are typically associated with various types of epidermal inclusion cysts (C).
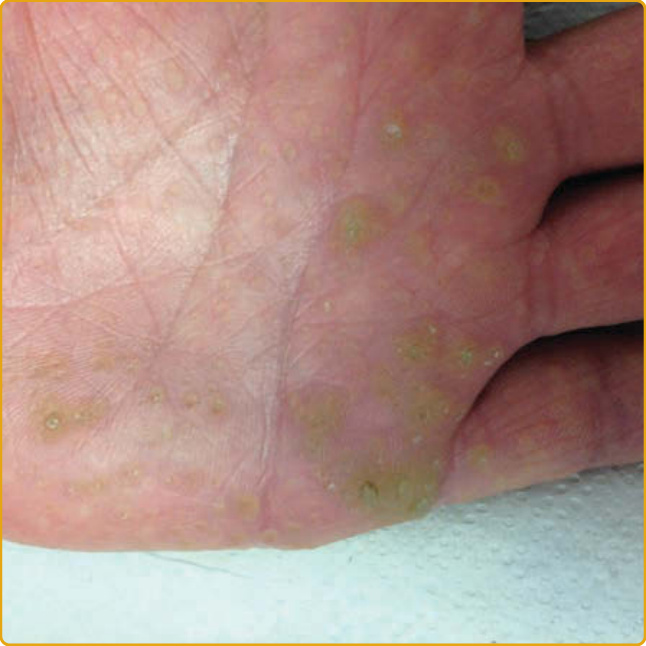
Figure 48-11 Punctate palmoplantar keratoderma type I caused by mutation in AAGAB. Multiple hyperkeratotic, centrally indented, yellow to brown papules irregularly distributed over the palmar skin.
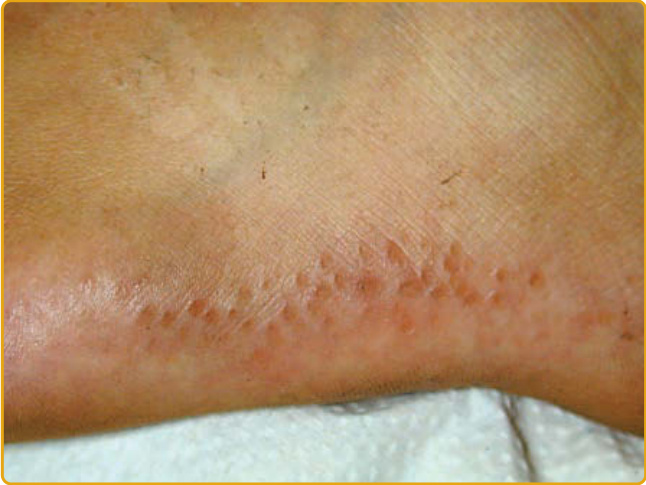
Figure 48-12 Punctate palmoplantar keratoderma type III (acrokeratoelastoidosis). Round to oval yellow papules with central umbilication and a linear pattern of arrangement over the lateral margin of the sole.
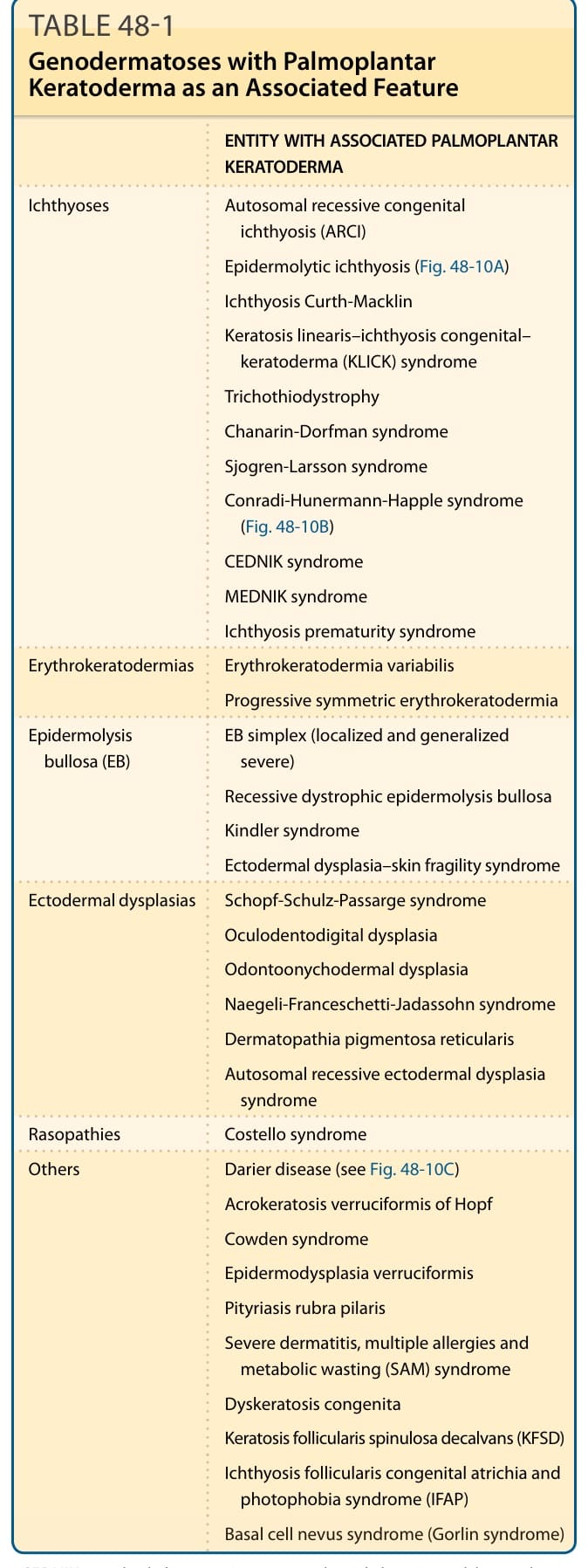
TABLE 48-1 Genodermatoses with Palmoplantar Keratoderma as an Associated Feature
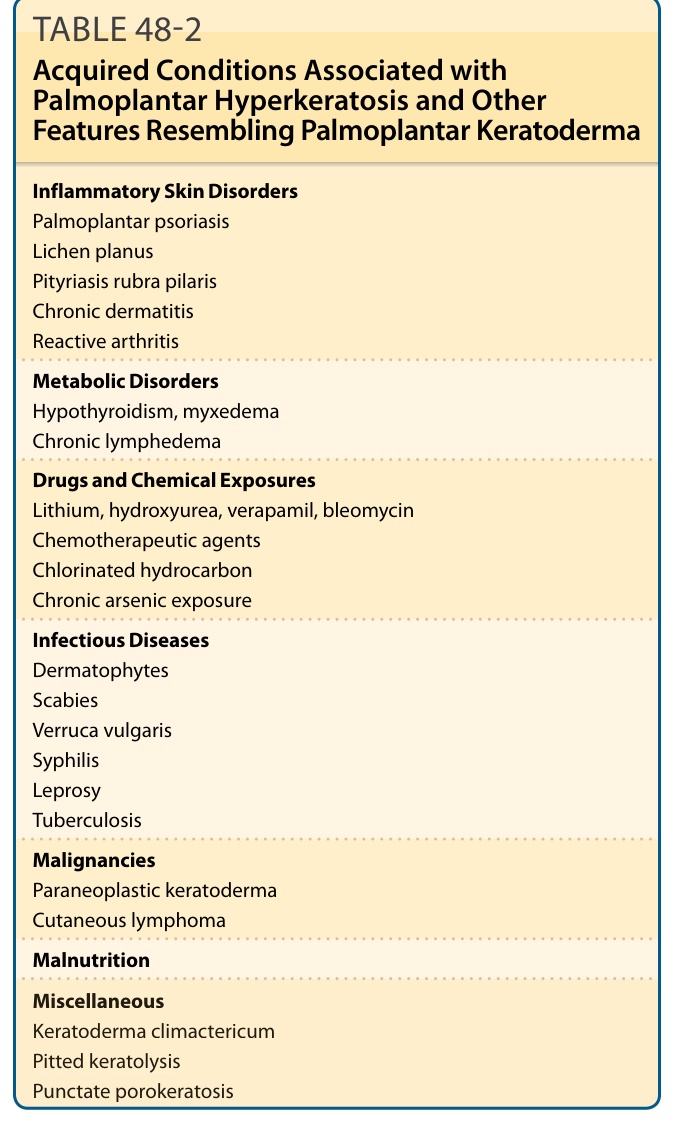
TABLE 48-2 Acquired Conditions Associated with Palmoplantar Hyperkeratosis and Other Features Resembling Palmoplantar Keratoderma
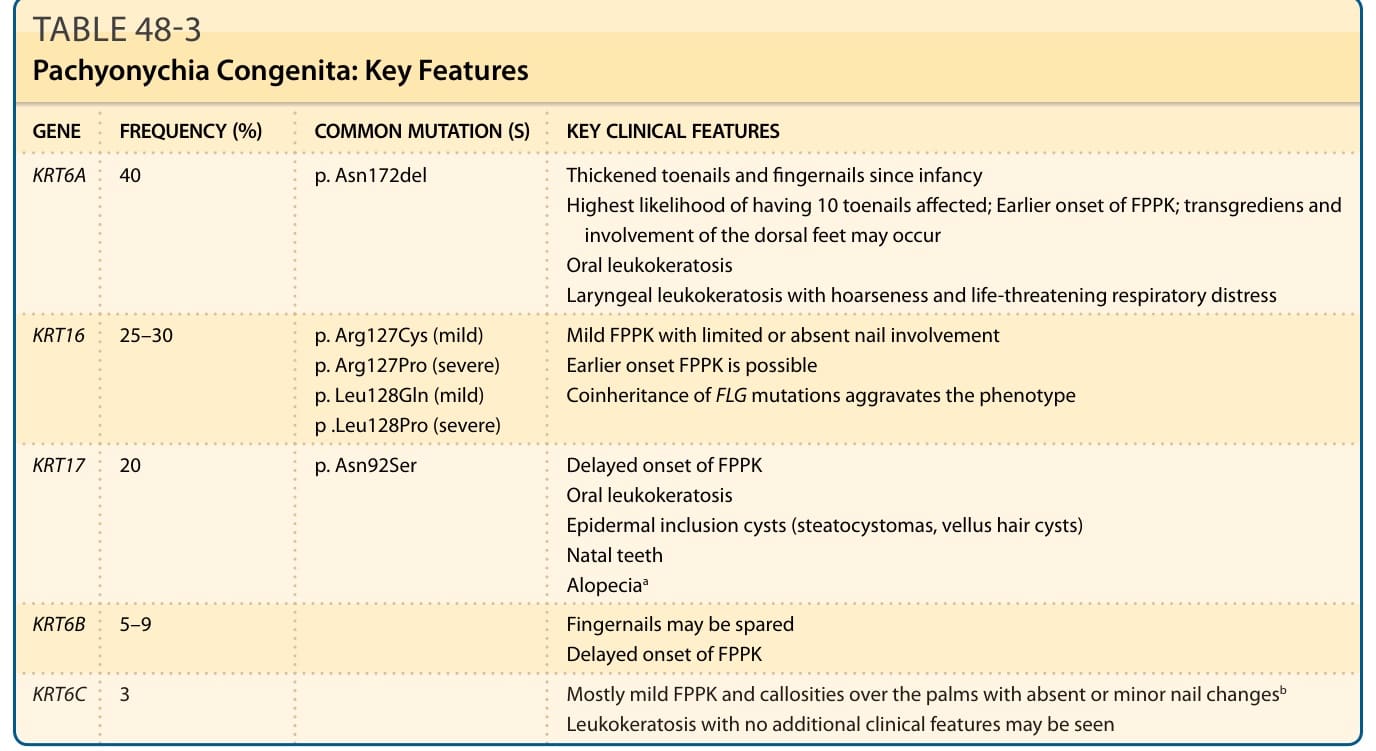
TABLE 48-3 Pachyonychia Congenita: Key Features
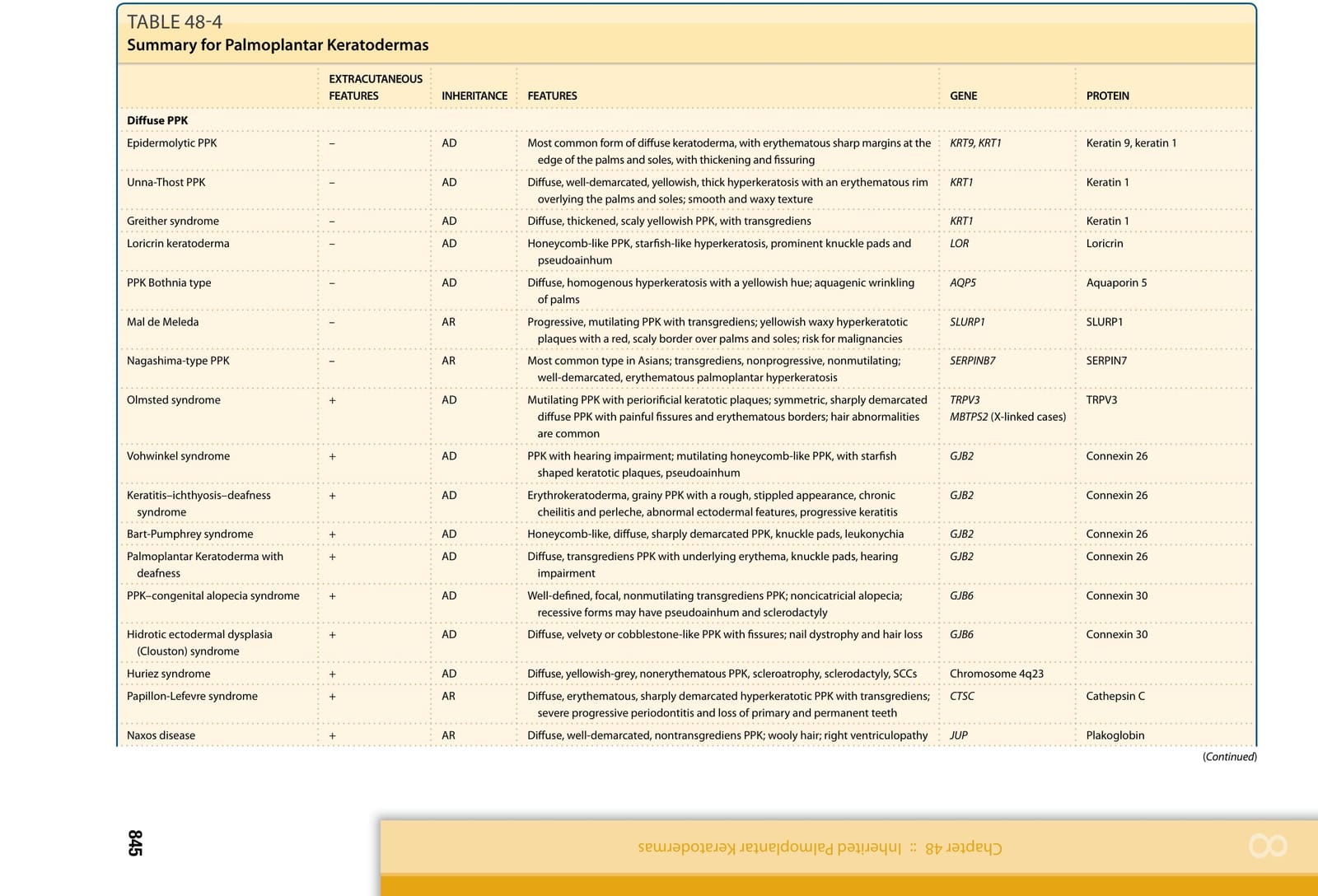
TABLE 48-4