遺傳性掌蹠角化症 (Inherited Palmoplantar Keratodermas)
PART 8
角化異常 (Disorders of Cornification)
重點一覽 (AT-A-GLANCE)
■ 遺傳性掌蹠角化症(inherited palmoplantar keratodermas, PPKs)是一群異質性的遺傳性皮膚病(genodermatoses),其特徵為手掌與足底的過度角化(hyperkeratosis),可伴隨或不伴隨相關特徵。通常依照其型態(瀰漫型 diffuse、局灶型 focal、點狀型 punctate)、遺傳模式,以及是否具有皮膚外(extracutaneous)特徵來加以分類。
■ 表皮鬆解型掌蹠角化症(epidermolytic PPK, EPPK)是瀰漫型角化症最常見的型態。它起因於 KRT9(多數病例)或 KRT1(少數病例)的雜合子突變(heterozygous mutation),分別編碼角蛋白 9(keratin 9)與角蛋白 1(keratin 1)。組織學上可見表皮鬆解型過度角化(epidermolytic hyperkeratosis)。在非表皮鬆解型掌蹠角化症(nonepidermolytic PPK, NEPPK)Unna-Thost 型中,可見類似但較輕微的瀰漫型 PPK 表現型,其由 KRT1 的雜合子突變所引起。
■ 帶有 transgrediens(角化向外延伸)的瀰漫型 NEPPK 可見於 Greither 症候群與 Bothnia 型 PPK,分別由 KRT1 的錯義雜合子突變(missense heterozygous mutation)與 AQP5 的功能獲得型雜合子突變(gain-of-function heterozygous mutation)所引起。Bothnia 型 PPK 在接觸水後,手掌與足底還會呈現白色海綿狀外觀。
■ Mal de Meleda 是一種體染色體隱性遺傳、進行性、瀰漫性、殘毀性(mutilating)、帶有 transgrediens 的 PPK,由 SLURP1 的雙等位基因突變(biallelic mutation)所引起。
■ Nagashima 型 PPK 是亞洲族群中最常見的 PPK 類型,特徵為瀰漫型、帶 transgrediens、非進行性、非殘毀性的 PPK。掌蹠皮膚在浸水後呈現典型的白色海綿狀外觀。此型 PPK 由 SERPIN7(編碼一種絲胺酸蛋白酶抑制劑 serine protease inhibitor)的雙等位基因突變所引起。
■ Olmsted 症候群由 TRPV3(體染色體顯性或體染色體隱性遺傳)或 MBTPS2(X 染色體連鎖隱性遺傳)的突變所引起,特徵為瀰漫型殘毀性 PPK,並伴有開口周圍(periorificial)角化斑塊。
■ 編碼連接蛋白 26(connexin 26)的 GJB2 基因雜合子突變,會導致四種具有 PPK 與聽力障礙特徵的遺傳性皮膚病,包括:Vohwinkel 症候群(殘毀性蜂窩狀 PPK 與海星狀角化斑塊)、角膜炎–魚鱗癬–耳聾(keratosis–ichthyosis–deafness, KID)症候群(紅皮角化症 erythrokeratoderma、顆粒狀 PPK、異常的外胚層特徵、進行性角膜炎與反覆感染)、Bart-Pumphrey 症候群(蜂窩狀 PPK、指節墊 knuckle pads 與白甲症 leukonychia),以及伴隨耳聾症候群的 PPK。一種伴隨先天性無毛症(congenital atrichia)的 KID 症候群型態,起因於編碼 Cx30 的 GJB6 突變。
■ Loricrin 角化症由編碼 loricrin 的 LOR 基因雜合子框移突變(frameshift mutation)所引起,是 Vohwinkel 症候群的一種變異型,不伴隨聽力障礙,但具有全身性魚鱗癬(generalized ichthyosis)特徵。
■ 伴隨聽力障礙的 PPK 亦可由編碼粒線體轉送 RNA(mitochondrial transfer RNA)的 MTTS1 基因點突變所引起。
■ 汗腺性外胚層發育不良(hidrotic ectodermal dysplasia,Clouston 症候群)是一種外胚層發育不良,伴隨瀰漫型 PPK 與指甲、毛髮異常,由編碼連接蛋白 30(connexin 30)的 GJB6 基因雜合子突變所引起。
■ Huriez 症候群是一種病因未明、體染色體顯性遺傳的 PPK,特徵為瀰漫型 PPK、硬化性萎縮(scleroatrophy)、指端硬化(sclerodactyly),以及在萎縮皮膚內發生鱗狀細胞癌(squamous cell carcinomas)。
■ Papillon-Lefevre 症候群是一種體染色體隱性遺傳疾病,由編碼組織蛋白酶 C(cathepsin C)的 CTSC 基因突變所引起。特徵為帶 transgrediens 的瀰漫型 PPK 與嚴重的進行性牙周炎(periodontitis)。
■ Naxos 病(Naxos disease, ND)與 Carvajal 症候群(Carvajal syndrome, CS)分別由編碼血漿斑珠蛋白(plakoglobin)與橋粒斑蛋白(desmoplakin)的 JUP 與 DSP 基因突變所引起,是一種心臟–皮膚症候群(cardiocutaneous syndromes),具有 PPK(ND 為瀰漫型、CS 為條紋型 striate)、羊毛狀髮(woolly hair)與心肌病變(cardiomyopathy)(ND 為右心室病變、CS 主要為左心室侵犯)。
■ 條紋型 PPK(striate PPK)起因於編碼橋粒糖蛋白 1(desmoglein 1)、橋粒斑蛋白(desmoplakin)以及程度較低的角蛋白 1(keratin 1)等基因的無義(nonsense)或框移雜合子突變。
■ 先天性厚甲症(pachyonychia congenita)是一群罕見的體染色體顯性遺傳疾病,由五個角蛋白基因之一的突變所引起,包括 KRT6A、KRT6B、KRT6C、KRT16 或 KRT17,分別編碼角蛋白 6a、6b、6c、16 與 17,這些角蛋白已知表現於分化的上皮組織中。其表現為局灶型疼痛性 PPK、具有特徵性外觀的厚而失養(dystrophic)的指甲,以及相關特徵如口腔白色角化症(oral leukokeratosis)、毛囊皮脂腺囊腫(pilosebaceous cysts)與新生兒齒(natal teeth)。
(續)
重點一覽(續)(AT-A-GLANCE (Continued))
■ Howel-Evans 症候群是一種體染色體顯性遺傳疾病,特徵為局灶型 PPK 與黏膜(特別是食道)鱗狀細胞癌,並伴有毛囊過度角化(follicular hyperkeratosis)與口腔白色角化症,由編碼 iRhom2 的 RHBDF2 基因雜合子突變所引起;iRhom2 是一種已知在表皮生長因子受體(epidermal growth factor receptor, EGFR)訊息傳遞中扮演角色的蛋白質。
■ Richner-Hanhart 症候群是一種體染色體隱性遺傳疾病,由編碼酪胺酸轉胺酶(tyrosine aminotransferase)的 TAT 基因突變所引起;酪胺酸轉胺酶是一種對於酪胺酸(tyrosine)與苯丙胺酸(phenylalanine)代謝重要的肝臟胞質酵素。其特徵為疼痛性局灶型 PPK、雙側角膜炎(keratitis)與智能不足(mental retardation)。
■ 點狀型 PPK(punctate PPK, PPKP)第 1 型的特徵為手掌與足底上多發、疼痛、黃褐色的過度角化丘疹,最常出現於生命的第一或第二個十年。它可能由以下兩個基因之一的突變所引起:編碼 p34 蛋白並導致 EGFR 上調的 AAGAB,或編碼第 XIV 型膠原蛋白 α1(collagen type XIV α1)的 COL14A1。PPKP
前言 (INTRODUCTION)
掌蹠角化症(palmoplantar keratoderma, PPK)一詞指的是一群可能造成功能障礙、在臨床與遺傳上具有異質性的角化異常疾病,其臨床特徵為手掌與足底皮膚異常的局灶性或瀰漫性增厚。這些疾病可以是後天性或遺傳性的。遺傳性 PPK 的特徵是家族性發生,且在多數病例中發病年齡相對較早。與 PPK 相關的遺傳性皮膚病列於表 48-1,數個範例呈現於圖 48-1。本章聚焦於以 PPK 為主要特徵的遺傳性疾病。已有數種 PPK 分類系統被提出。最初的分類基於數項臨床與組織學特徵的評估,包括掌蹠過度角化的特定型態與分布模式、侵犯範圍(孤立型 PPK 或伴隨外胚層缺陷與/或皮膚外表現的症候群型 PPK)、遺傳模式(體染色體顯性、體染色體隱性、X 染色體連鎖、粒線體遺傳)、發病年齡,以及組織學上是否有表皮鬆解(epidermolysis)。通常可區分為三種型態模式:(1)瀰漫型 PPK,整個掌蹠表面均勻受侵犯;(2)局灶型 PPK,過度角化局限於以壓力點為主的部位,並可進一步細分為斑塊型(areata 或 nummular 型;橢圓形病灶,主要位於足底表面)與條紋型(striate 型;縱向過度角化病灶,從手掌沿手指掌側面延伸,伴隨足底皮膚的局灶性至瀰漫性增厚);以及(3)點狀型 PPK,於手掌與足底上有多發、散在、1-mm 至 1-cm 的圓形角化丘疹。此外,掌蹠侵犯可能表現出 transgrediens(過度角化延伸至手指、足趾、手、足的背側面,以及手腕屈側面與足跟)或因假性指趾斷離(pseudoainhum,指趾周圍的緊縮帶 constricting bands)形成所致的殘毀。隨著大多數遺傳性 PPK 致病機轉的解析,新的分類方案除了型態特徵外,也納入 PPK 的病因。
闡明 PPK 的分子疾病機轉已促成新疾病與新症候群的發現,提供關於表皮結構成分生物學角色的新見解,並為發展新穎、疾病特異性的治療方式鋪路。
流行病學 (EPIDEMIOLOGY)
雖然個別來看,遺傳性 PPK 是罕見疾病,但鑑於輕度受影響的個體不需要醫療介入或可能被誤診,其實際盛行率與發生率可能被低估。然而,在數個近親通婚常見的國家或社群中,遺傳性 PPK 可能以高盛行率發生。在瑞典北部,非表皮鬆解型 PPK(NEPPK)的盛行率為 0.3% 至 0.55%,但在北愛爾蘭,表皮鬆解型 PPK(EPPK)的盛行率為每 10 萬人 4.4 例。在南印度,記載到的盛行率為每 1 萬人 5.2 例,其中 Unna-Thost 症候群為最常見的疾病實體,約見於 38% 的病例。一般而言,EPPK 是瀰漫型角化症最常見的型態,全球發生率為每 10 萬名活產新生兒 2.2 至 4.4 例。
與掌蹠角化症相關的疾病實體 (ENTITY WITH ASSOCIATED PALMOPLANTAR KERATODERMA)
(以下對應原文 Table 48-1 之內容,依疾病類別列舉伴隨掌蹠角化症的遺傳性皮膚病。)
魚鱗癬類(Ichthyoses)
- 體染色體隱性先天性魚鱗癬(autosomal recessive congenital ichthyosis, ARCI)
- 表皮鬆解型魚鱗癬(epidermolytic ichthyosis)(圖 48-10A)
- Curth-Macklin 魚鱗癬(Ichthyosis Curth-Macklin)
- 線狀角化–先天性魚鱗癬–角化症(keratosis linearis–ichthyosis congenital–keratoderma, KLICK)症候群
- 毛髮硫營養不良症(trichothiodystrophy)
- Chanarin-Dorfman 症候群
- Sjögren-Larsson 症候群
- Conradi-Hünermann-Happle 症候群(圖 48-10B)
- CEDNIK 症候群
- MEDNIK 症候群
- 魚鱗癬早產症候群(ichthyosis prematurity syndrome)
紅皮角化症類(Erythrokeratodermias)
- 可變性紅皮角化症(erythrokeratodermia variabilis)
- 進行性對稱性紅皮角化症(progressive symmetric erythrokeratodermia)
表皮分解性水皰症(epidermolysis bullosa, EB)
- 單純型 EB(EB simplex,局限型與全身性嚴重型)
- 隱性失養型表皮分解性水皰症(recessive dystrophic epidermolysis bullosa)
- Kindler 症候群
- 外胚層發育不良–皮膚脆弱症候群(ectodermal dysplasia–skin fragility syndrome)
外胚層發育不良類(Ectodermal dysplasias)
- Schöpf-Schulz-Passarge 症候群
- 眼–齒–指發育不良(oculodentodigital dysplasia)
- 齒–甲–皮發育不良(odontoonychodermal dysplasia)
- Naegeli-Franceschetti-Jadassohn 症候群
- 網狀色素性皮膚病(dermatopathia pigmentosa reticularis)
- 體染色體隱性外胚層發育不良症候群(autosomal recessive ectodermal dysplasia syndrome)
RAS 病變類(Rasopathies)
- Costello 症候群
其他(Others)
- 達瑞氏病(Darier disease)(見圖 48-10C)
- Hopf 疣狀肢端角化症(acrokeratosis verruciformis of Hopf)
- Cowden 症候群
- 疣狀表皮發育不良(epidermodysplasia verruciformis)
- 毛囊性紅糠疹(pityriasis rubra pilaris)
- 嚴重皮膚炎、多重過敏與代謝性耗損(severe dermatitis, multiple allergies and metabolic wasting, SAM)症候群
- 先天性角化不良症(dyskeratosis congenita)
- 脫髮性棘狀毛囊角化症(keratosis follicularis spinulosa decalvans, KFSD)
- 魚鱗癬性毛囊角化–先天性無毛–畏光症候群(ichthyosis follicularis congenital atrichia and photophobia syndrome, IFAP)
- 基底細胞母斑症候群(basal cell nevus syndrome,Gorlin 症候群)
CEDNIK:大腦發育不全、神經病變、魚鱗癬與角化症(cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma);MEDNIK:智能不足、腸病變、耳聾、神經病變、魚鱗癬與角化症(mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratodermia)。
診斷 (DIAGNOSIS)
如前簡述,過去數年我們對遺傳性 PPK 致病機轉的理解有顯著進展。這些進展反過來可用於促進這些疾病的診斷,方法是使用整合臨床與分子特徵的演算法。圖 48-2 描繪了其中一種演算法,基於四組資料的運用:(1)皮膚發現,包括 PPK 型態(如瀰漫型、局灶型、點狀型、殘毀型)、毛髮(如羊毛狀髮)或指甲異常,以及與其他遺傳性皮膚病相關的特徵(如皮膚脆弱);(2)皮膚外發現(如牙周炎、耳聾);(3)遺傳模式(如體染色體顯性、體染色體隱性、粒線體遺傳);以及(4)組織病理學發現,如表皮鬆解型過度角化、角質形成細胞失黏附(disadhesion)或角化不全(parakeratosis)。使用此演算法,幾乎所有形式的 PPK 都可被指派至一組基因,並應對其進行定序以達成最終診斷。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
掌蹠過度角化可能是遺傳性與後天性疾病的特徵。表 48-2 所列的後天性疾病通常較晚發病,包括發炎性疾病(如乾癬 psoriasis、扁平苔癬 lichen planus、毛囊性紅糠疹、慢性皮膚炎、反應性關節炎)、副腫瘤性角化症(paraneoplastic keratoderma)、皮膚淋巴瘤、化學與藥物暴露、營養不良、代謝性疾病(如甲狀腺機能低下症 hypothyroidism)、更年期角化症(keratoderma climactericum),以及感染性疾病(如皮癬菌 dermatophytes、疥瘡 scabies、尋常疣 verruca vulgaris 與梅毒 syphilis)。
具有 PPK 特徵的遺傳性皮膚病列於表 48-1。這些疾病中有些可共享數項獨特特徵。例如,指趾自體截斷(autoamputation)可伴隨於 loricrin 角化症(loricrin keratoderma, LK)、Vohwinkel 症候群(VS)、Olmsted 症候群(OS)、Sybert PPK、mal-de-Meleda(MDM)、PPK-Gamborg-Nielsen,以及 PPK 先天性禿髮症候群 2(PPK congenital alopecia syndrome 2, PPKCA2)的 PPK。其他可能造成緊縮帶(伴隨或不伴隨過度角化)的非遺傳性疾病包括痲瘋、三期梅毒、雅司病(yaws)、硬皮症、雷諾氏症候群(Raynaud syndrome)、羊膜帶(amniotic bands)、麥角胺中毒(ergotamine poisoning)、脊髓腫瘤、脊髓空洞症(syringomyelia),以及凍傷、燒傷與外傷後的疤痕形成。
帶 transgrediens 的 PPK 可見於 LK、Nagashima 型 PPK(NPPK)、Greither 病、OS、Papillon-Lefevre 症候群(PLS)。伴隨感覺神經性聽力損失(sensorineural hearing loss, SNHL)的 PPK 應引起對 VS 症候群、角膜炎–魚鱗癬–耳聾(KID)症候群與其他 GJB2 相關疾病,或粒線體遺傳伴隨耳聾之 PPK 的鑑別診斷考量。
數種症候群的特徵為 PPK 伴隨於異常角化區域內發生的鱗狀細胞癌(squamous cell carcinoma, SCC)(如 Huriez 與 Unna-Thost 症候群)。其他遺傳性皮膚病,包括先天性角化不良症、Rothmund-Thomson 症候群與著色性乾皮症(xeroderma pigmentosum),則與升高的 SCC 絕對發生率相關。少數形式的 PPK 具有牙齦與牙齒異常的特徵(如 OS、PLS、Haim-Munk 症候群與 Kindler 症候群)。
遺傳性掌蹠角化症的診斷演算法 (Diagnostic algorithm for inherited palmoplantar keratodermas)
(以下對應原文圖 48-2 的診斷演算法流程內容,以皮膚病伴隨 PPK 為起點。)
伴隨 PPK 的皮膚疾病(Skin disorders with PPK)
- 是否主要為 PPK?(Mainly PPK?)
- 是否有失黏附(disadhesion)/雙核角質形成細胞(binucleate KC)?→ 條紋型 PPK/ND/CS:DSP、DSG1、JUP
- 是否有表皮鬆解性變化(epidermolytic changes)?
- 角質層核(SC nuclei)/其他病理特徵?
- EI(表皮鬆解型魚鱗癬):KRT1/9
- Curth-Macklin 魚鱗癬(IH Curth-Macklin):KRT1
- 是否有皮膚外特徵(extracutaneous features)?耳聾(Deafness)
- Loricrin PPK:LOR
- 伴隨耳聾之 PPK(PPK with deafness):GJB6/粒線體 DNA(mitDNA)
- 牙齒(Teeth):PLS/PC:CTSC/KRT6a;RHS:TAT
- 眼睛(Eyes)
- 局灶型或瀰漫型(Focal or diffuse)/殘毀型(Mutilating)/對水敏感(Water sensitive)
- 疼痛(Pain)/線狀病灶(Linear lesions)
- 足底(Soles)/大魚際(Thenar)
- NEPPK(非表皮鬆解型 PPK)
- PC(先天性厚甲症):KRT6a/b/c、KRT16、KRT17
- KLICK 症候群:POMP
- 點狀型(Punctate):PPPK1:AAGAB
- AKE(acrokeratoelastoidosis):FAH
- PC/Clouston:GJB6/KRT6/16/17
- Olmsted:TRPV3;Huriez
- 指甲(Nails):Clouston/ND/CS:GJB6/DSP/JUP/KANK2
- 毛髮(Hair)
- MDM:SLURP1
- Howell-Evans/PPK-SR:RHBDF2/RSPO1
- 其他(Others):SERPINB7、AQP5
- 遺傳模式分支:AR(體染色體隱性)、AD(體染色體顯性)
掌蹠皮膚的皺褶(wrinkling)可能是以下疾病的特徵:伴隨 CFTR 突變的手掌水源性皺褶(aquagenic wrinkling of the palms)、伴隨 AQP5 突變的 Bothnia 型 PPK、伴隨 SERPINB7 突變的 NPPK,以及肢端角化彈力纖維變性症(acrokeratoelastoidosis, AKE)。點狀掌蹠角化症的鑑別診斷包括尋常疣、壓力點上的雞眼(corns)、掌蹠點狀汗孔角化症(porokeratosis punctate palmaris et plantaris)、掌紋點狀角化症(keratosis punctate of the palmar creases)、Cole 病、達瑞氏病、Cowden 病、Gorlin 症候群、Hopf 疣狀肢端角化症、疣狀表皮發育不良,以及砷暴露。
伴隨局限於手背的硬化(sclerosis)與指端硬化的 PPK,可能是 Huriez 症候群、Unna-Thost 症候群(偶爾呈現指端硬化或指甲變化)、PPKCA2,以及伴隨皮膚 SCC 與性別逆轉(sex reversal)的掌蹠過度角化的特徵。全身性硬化症(systemic sclerosis)可能呈現類似特徵。具有先天性厚甲症(pachyonychia congenita, PC)典型指甲變化的 PPK,可見於由 GJB6 突變引起的 Clouston 症候群(可能呈現類似的指甲變化與疼痛性角化症),或由編碼 frizzled 6 的 FZD6 同合子突變所引起;frizzled 6 是甲基質(nail matrix)中的一種 Wnt 訊息傳遞路徑受體,最近被發現為先天性指甲失養(congenital nail dystrophy)的病因。先天性角化不良症的表現與 PC 有重疊特徵,包括指甲失養、PPK、多汗症(hyperhidrosis)與口腔白斑(oral leukoplakia)。在伴隨指甲發現的 PPK 病例中,應排除甲癬(onychomycosis)。
常見治療方法 (COMMON THERAPEUTIC APPROACHES)
一般而言,所有形式 PPK 的處置目標都在緩解疾病表現,並聚焦於機械性措施、緩解疼痛的方法、續發性感染的治療,以及各種助行輔具。
(以下對應原文表 48-2 之內容,列舉與掌蹠過度角化相關、且具有類似 PPK 其他特徵的後天性疾病。)
發炎性皮膚疾病(Inflammatory Skin Disorders)
- 掌蹠乾癬(palmoplantar psoriasis)
- 扁平苔癬(lichen planus)
- 毛囊性紅糠疹(pityriasis rubra pilaris)
- 慢性皮膚炎(chronic dermatitis)
- 反應性關節炎(reactive arthritis)
代謝性疾病(Metabolic Disorders)
- 甲狀腺機能低下症、黏液水腫(hypothyroidism, myxedema)
- 慢性淋巴水腫(chronic lymphedema)
藥物與化學暴露(Drugs and Chemical Exposures)
- 鋰鹽(lithium)、羥基脲(hydroxyurea)、維拉帕米(verapamil)、博來黴素(bleomycin)
- 化療藥物(chemotherapeutic agents)
- 氯化碳氫化合物(chlorinated hydrocarbon)
- 慢性砷暴露(chronic arsenic exposure)
感染性疾病(Infectious Diseases)
- 皮癬菌(dermatophytes)
- 疥瘡(scabies)
- 尋常疣(verruca vulgaris)
- 梅毒(syphilis)
- 痲瘋(leprosy)
- 結核(tuberculosis)
惡性腫瘤(Malignancies)
- 副腫瘤性角化症(paraneoplastic keratoderma)
- 皮膚淋巴瘤(cutaneous lymphoma)
營養不良(Malnutrition)
其他(Miscellaneous)
- 更年期角化症(keratoderma climactericum)
- 凹陷性角質溶解症(pitted keratolysis)
- 點狀汗孔角化症(punctate porokeratosis)
局部治療包括規律使用潤膚劑(emollients)與外用角質溶解配方(如 10% 至 20% 水楊酸 salicylic acid,或 35% 至 70% 丙二醇 propylene glycol 與厚潤膚劑併用),必要時加以封包(occlusion)。此外,外用骨化三醇衍生物 calcipotriol 與外用 tazarotene 已分別證實對 EPPK 與 PLS 有效。皮癬菌或細菌續發性感染與多汗症應被診斷並積極治療,以避免疾病加劇。浸泡於水中與機械性移除過度角化區域(如修整 grooming 與修剪 trimming)是可提供症狀緩解的額外治療措施。疼痛可藉由矯正器(orthotics)或鞋墊(insoles)、吸濕襪(wicking socks)、通風或具緩衝的鞋具,以及維持體重以減少對足部的反覆創傷與形成胼胝(callus)與水皰的傾向來緩解。
口服類視黃醇 (ORAL RETINOIDS)
口服類視黃醇(oral retinoids)通常有效。鑑於可能的長期副作用、疾病的慢性本質,以及可能加重皮膚脆弱(這在數種 PPK 中可能使疾病表現惡化)的風險,有時建議採用低劑量、短期或間歇性治療。更具體地說,全身性類視黃醇已證實對 EPPK、MDM、VS 與 LK 有效,並對 OS、Huriez 症候群、點狀型 PPK 1 與 PLS(對 PPK 與牙周病變兩者皆然)的病例有效。全身性類視黃醇已發現對 KID 症候群有效(對於過度角化、頭皮剝離性蜂窩織炎 dissecting cellulitis of the scalp,以及癌症化學預防 cancer chemoprophylaxis),雖然部分報告顯示僅有部分反應,且使用 isotretinoin 會使角膜新生血管(corneal neovascularization)惡化。因此,建議以低起始劑量的 acitretin(0.5 mg/kg/day)或 alitretinoin(10 mg/day)開始,並依反應與耐受性逐步增量。
手術治療 (SURGICAL THERAPY)
手術切除與植皮(surgical excision and grafting)是局灶型 PPK 與伴隨殘毀病例的一種選項。具體的手術方式於後文討論。
遺傳性掌蹠角化症 (INHERITED PALMOPLANTAR KERATODERMAS)
瀰漫型掌蹠角化症,無皮膚外特徵,顯性遺傳 (DIFFUSE PALMOPLANTAR KERATODERMAS WITHOUT EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE)
表皮鬆解型掌蹠角化症 (Epidermolytic Palmoplantar Keratoderma)
臨床特徵: EPPK(Vörner 型;線上人類孟德爾遺傳資料庫 [Online Mendelian Inheritance in Man, OMIM] #144200)由 Vörner 於 1901 年首次描述,是一種體染色體顯性遺傳疾病,特徵為黃色、瀰漫型 PPK,在手掌(圖 48-3A)與足底(圖 48-3B)邊緣具有紅斑性的銳利邊界。多數病例於出生時或生命最初幾週出現,雖然亦有報告在三歲前才出現。最初的表現是掌蹠紅斑,從手掌與足底邊緣開始,向中央延伸,隨後被厚鱗屑覆蓋,並終生維持穩定。已有報告肘部、膝部與指節墊上的 transgrediens 與過度角化病灶。它可能併發疼痛性裂隙(fissures)、多汗症、浸潤糜爛(maceration),以及帶有惡臭的續發性細菌與真菌感染。已有報告伴隨假性指趾斷離(pseudoainhum)的指趾殘毀與屈指畸形(camptodactyly)。在多數病例中,PPK 是一項孤立的發現,雖然已有報告 EPPK 罕見伴隨腿部潰瘍、硬皮症、家族性多發性癌症與第 III 型 Ehlers-Danlos 症候群發生。
病因與致病機轉: EPPK 是一種體染色體顯性遺傳性皮膚病,由編碼角蛋白 9 的 KRT9 雜合子突變所引起。少數病例中也有報告編碼角蛋白 1 的 KRT1 突變與 EPPK 相關。
角蛋白 9 是一種第 I 型中間絲(intermediate filament)蛋白,其表現局限於掌蹠表皮的基底上層(suprabasal layers)。與所有其他角蛋白相似,KRT9 包含三個主要區域:頭部區域(head domain);中央 α 螺旋桿狀區域(central α-helix rod domain),由四個螺旋亞節段(1A、1B、2A 與 2B)組成,並被三個非螺旋連接區域(L1、L12 與 L2)所中斷;以及尾部區域(tail domain)。迄今所鑑定的多數 KRT9 突變位於 α 螺旋桿狀區域的 1A 或 2B 節段,這些節段在所有角蛋白中高度保守,對於角蛋白異二聚體(heterodimer)的形成至關重要,並在角蛋白中間絲的聚集與穩定中扮演關鍵角色。多數致病的遺傳缺陷為錯義或框移(小型框內插入–缺失 small in-frame insertion–deletion)突變,以顯性負向(dominant-negative)方式作用。KRT9 的第 163 號密碼子(位於中央 α 螺旋桿狀區域)存在一個突變熱點,編碼胺基酸精胺酸(arginine),常突變為色胺酸(tryptophan)、麩醯胺酸(glutamine)或脯胺酸(proline)。p.Arg163Trp 在不同族群中約佔所有致 EPPK 突變的 30%。
角蛋白 1 是一種第 II 型角蛋白,表現於致力於終末分化的基底上層細胞,在掌蹠表皮中與角蛋白 9 形成異二聚體,並在所有其他皮膚表面(包括帶毛皮膚與其他複層正角化鱗狀上皮)中與角蛋白 10 形成異二聚體。在 EPPK 家族中報告的 KRT1 突變涉及影響桿狀區域螺旋邊界基序(helix boundary motifs)的較大型框內缺失,這與表皮鬆解型魚鱗癬(epidermolytic ichthyosis, EI;見第 47 章)中報告的同一區域錯義突變不同,也與在條紋型角化症與 Curth-Macklin 豪豬狀魚鱗癬(ichthyosis hystrix Curth-Macklin)中發現的 V2 區域框移突變不同。其他與 EPPK 相關的 KRT1 突變是位於中央 α 螺旋捲曲螺旋桿狀區域之螺旋 1B 區域起始處的功能獲得型(gain-of-function, GOF)突變,以及 2B 桿狀區域中 18 個胺基酸的插入,這強調了位於角蛋白 1 桿狀區域較中央位置的突變,對絲狀體組裝與穩定功能的破壞程度,不如 EI 病例中位於螺旋起始與終止序列的突變。
病理學: 皮膚切片顯示緻密的正過度角化(compact orthohyperkeratosis)、顆粒層增生(hypergranulosis)、棘層肥厚(acanthosis),以及表皮鬆解型過度角化,後者表現為角質形成細胞的核周空泡化(perinuclear vacuolization),以及位於表皮顆粒層的大型不規則形角質透明顆粒(keratohyaline granules)。此外,在受侵犯的表皮中可鑑定出角化不良(dyskeratosis),表現為胞內與核周的嗜伊紅均質化(eosinophilic homogenizations)伴圓–橢圓形嗜伊紅內含物,對應於超微結構下所見的張力絲(tonofilaments)胞內聚集物與團塊。
可能需要多於一次的切片才能顯示表皮鬆解性變化,而表皮鬆解型過度角化的典型組織學改變可能於肢端汗管(acrosyringium)中被注意到。
超微結構發現包括:上棘層與顆粒細胞層的胞內空泡化;細胞胞器的破壞與分散,細胞邊界不清;圍繞細胞核的胞內緻密張力絲聚集物;緻密的張力絲團塊;豐富的肝糖(glycogen)與核糖體(ribosomes);張力纖維(tonofibrils)自橋粒斑(desmosomal plaques)脫離;自中馬氏層(mid malpighian layers)起出現的大型角質透明顆粒;由均質、相對電子透亮核心構成的「複合」角質透明顆粒;以及緻密的周邊沉積物。
特定治療: 在 EPPK 的小鼠模型中,已證實使用核糖核酸干擾(ribonucleic acid interference, RNAi)為基礎的療法可抑制 Krt9 突變等位基因的表現。此外,一個 EPPK 的轉殖基因小鼠模型以突變特異性短髮夾 RNA(short hairpin RNA, shRNA)治療,導致突變蛋白被剔除(knockdown),並恢復皮膚的正常型態與功能。
Unna-Thost 掌蹠角化症 (Unna-Thost Palmoplantar Keratoderma)
臨床特徵: 瀰漫型非表皮鬆解型 PPK(NEPPK,Unna-Thost 型;OMIM #600962)是一種體染色體顯性遺傳性皮膚病,由 Unna 與 Thost 於 1880 年首次描述。臨床特徵與瀰漫型 EPPK 相似,雖然往往較輕微,於生命最初幾個月出現。亦有報告於生命第三個十年發病。NEPPK 的特徵為瀰漫型、界線分明、黃色、覆蓋於手掌與足底之上、帶有紅斑性邊緣的厚過度角化。它傾向於比 EPPK 更平滑、更蠟狀,而 EPPK 則較厚且有裂隙。多汗症,以及難治性的續發性皮癬菌感染與凹陷性角質溶解症(導致浸潤糜爛與脫屑)是常見的發現,在 NEPPK 中比在 EPPK 中更常見。此外,已有報告肚臍周圍與乳頭周圍的過度角化,伴隨膝部與肘部的輕微增厚與乾燥。已描述伴隨異位性皮膚炎與疣狀癌(verrucous carcinoma)的病例。已有報告伴隨四肢遠端三分之一處的肢端發紺(acrocyanosis)、第五指的斜指(clinodactyly),以及肛門周圍或股溝周圍侵犯。
一般而言,EPPK 與 NEPPK 顯示出相當大的臨床重疊,常需要組織病理學評估以區分兩者。
病因與致病機轉: 最初,Unna-Thost PPK 被認為是臨床上類似 Vörner PPK 的一種非表皮鬆解型瀰漫型 PPK。然而,Kuster 與同僚調查了 Unna 與 Thost 最初報告的家族,在組織學上發現表皮鬆解型過度角化,並鑑定出一個位於 KRT9 中央桿狀區域起始處 coil-1A 區域的突變。此突變與 Vörner 最初報告家族中發現的突變位置十分接近,顯示 Unna-Thost 與 Vörner PPK 為同一疾病實體。
在一個患有瀰漫型 NEPPK 的單一家族中,於角蛋白 1 的末端 V1 可變亞區(非螺旋頭部區域)鑑定出一個錯義突變。與其他報告中涉及對絲狀體組裝與穩定重要之中央區域、並導致表皮鬆解的 KRT1 突變不同,此突變可能對於角蛋白絲與包括橋粒在內的其他細胞成分之間的交互作用很重要。此突變導致角質形成細胞形狀的扭曲,並妨礙板層小體(lamellar body)脂質物質的有效分布,造成屏障形成受損。
另一個導致輕微表現型的 KRT1 錯義突變,於 2B 桿狀區域被鑑定出,再次強調影響角蛋白 1 桿狀區域中央部分的突變,對絲狀體組裝的破壞程度不如位於螺旋序列起始或終止基序的突變。
瀰漫型 NEPPK,在無 PC 其他臨床特徵的情況下,已有報告伴隨 KRT6c 與 KRT16 的雜合子突變。其表現型包括足底瀰漫型 PPK 與手掌局灶型過度角化。在 NEPPK 病例中,若 KRT1 未鑑定出致病突變,篩檢 KRT6c 與 KRT16 突變可能有價值。
病理學: 瀰漫型 NEPPK 顯示非特異性的組織病理學發現,包括過度角化、正角化(orthokeratosis)、棘層肥厚,以及顆粒層增生或減少(hyper- 或 hypogranulosis),無表皮鬆解型過度角化的證據。需要仔細檢查與重複切片,以排除符合 EPPK 的小型表皮鬆解型過度角化病灶。
Greither 症候群 (Greither Syndrome)
臨床特徵: Transgrediens et progrediens PPK(Greither 症候群;OMIM #133200)最初由 Greither 於 1952 年描述。Greither 症候群是一種體染色體顯性遺傳疾病,具有顯著的家族內與家族間變異性。它典型於兩歲後開始發展(雖然亦有記載出生後不久或於兒童期與青春期較晚出現)。其特徵為瀰漫型、增厚、有鱗屑、黃色、帶有紅斑性邊緣與 transgrediens 的 PPK。阿基里斯腱(Achilles tendon)上方皮膚的侵犯,以及斑片狀過度角化、紅斑性與色素過度沉著的丘疹與斑塊向脛部、膝部、大腿、指節、手腕、肘部與屈側區域逐漸延伸是典型特徵。Greither PPK 傾向於在第五個十年後消退。多汗症是常見特徵,凹陷性角質溶解症可能是相關發現。已有報告一例以新生兒水皰與紅皮症(erythroderma)表現、最初被診斷為 EI 的病例。已描述伴隨異位性皮膚炎的關聯。已記載一例 Greither PPK 病人在過度角化足底上發生惡性黑色素瘤(malignant melanoma)的病例。嚴重的 PPK 病例可能併發自發性自體截斷與指趾畸形。Greither 症候群已在個別病例中描述伴隨色素失禁症(incontinentia pigmenti)、肢端發紺與可變性紅皮角化症。
Sybert PPK 是另一種進行性、瀰漫型、體染色體顯性遺傳、帶 transgrediens 與自體截斷的 PPK,然而與 Greither PPK 相比,它表現出更嚴重的過度角化。
病因與致病機轉: Greither 症候群以體染色體顯性方式遺傳,已證實由 KRT1 的錯義突變所致,該突變影響角蛋白 1 中央桿狀區域胺基末端的螺旋起始基序(helix initiation motif),此區域已知對有效的絲狀體組裝至關重要。Sybert PPK 的致病基因尚未被鑑定。
病理學: Greither 症候群的特徵性發現是棘層肥厚、顯著的過度角化與顆粒層增生。已有報告一例淺層角質形成細胞顯著空泡化,且顆粒層中有大量符合表皮鬆解型過度角化的角質透明顆粒。
此外,已有報告表皮的局灶性凹陷,由緻密正角化角質層的圓形病灶所佔據。Sybert PPK 中所描述的組織病理學發現為表皮增生伴顆粒層增生、角化不全與正角化,以及乳頭層真皮中稀疏的淋巴球浸潤。超微結構研究顯示正常的角蛋白絲,但角質透明顆粒的結構與分布異常。
Loricrin 角化症 (Loricrin Keratoderma)
臨床特徵: LK(伴隨魚鱗癬的殘毀性角化症,VS Camisa 型;OMIM #604117)是一種罕見的體染色體顯性遺傳疾病。它具有蜂窩狀 PPK、海星狀過度角化、手背側面顯著的指節墊,以及導致指趾自體截斷的假性指趾斷離等特徵。當這些徵象伴隨聽力障礙時,即稱為 VS(見後文)。LK 是由 Camisa 與 Rossana 於 1984 年報告的 VS 變異型,無聽力障礙,但具有顯著的全身性魚鱗癬。建立診斷的兩項必要臨床特徵是特徵性的蜂窩狀角化症與全身性魚鱗癬,雖然 LK 的表現型確實具有異質性。LK 於出生時或兒童早期表現,並在整個成年期逐漸進展。臨床發現包括中度、全身性的魚鱗癬樣皮膚炎,伴隨乾燥與細鱗屑,影響軀幹與四肢,以及局限於身體皺褶的過度角化,無紅斑證據。掌蹠侵犯的特徵為瀰漫型、對稱、界線分明、帶 transgrediens、具紅斑性邊界的蜂窩狀 PPK(圖 48-4A),雖然亦可見無蜂窩狀模式的瀰漫型過度角化。指節墊、手背側部的過度角化斑塊與假性指趾斷離,已在 LK 的家系中以不同頻率被報告,並且約 35% 的病例於出生時可見膠樣膜(collodion membrane)。已有報告 LK 伴隨白斑症(vitiligo)、異位性皮膚炎、神經發展遲緩與小頭症(microcephaly)。
病因與致病機轉: LK 起因於 LOR 的雜合子框移插入或缺失突變,該基因編碼 loricrin。Loricrin 於表皮顆粒層合成,然後遷移至細胞周邊,位於細胞質膜(plasma membrane)下方,並與其他胞質蛋白(包括兜甲蛋白 involucrin)交聯,形成角化細胞外套膜(cornified cell envelope, CCE)。多數涉及 LK 的 LOR 突變導致框移、延遲終止與異常的蛋白延長。轉殖基因小鼠的實驗顯示,LOR 突變施加一種功能獲得型的有害效應。突變的 loricrin 形成富含精胺酸的核定位序列(nuclear localization sequences),將一個暴露於細胞表面的蛋白「標記」,使其經核轉運被輸入細胞核。loricrin 在細胞核中的異常定位改變了核/核仁功能,包括非核糖體 RNA 處理與生長因子訊息傳導,並破壞終末分化角質形成細胞中的凋亡過程。
尚無明確的基因型–表現型相關性被報告,並已報告家族內疾病嚴重度的變異。
LK 皮膚顯示出異常的表皮通透性屏障功能,類似於各種魚鱗癬中所見的皮膚屏障缺陷,包括伴隨轉麩醯胺酸酶 1(transglutaminase-1)突變的板層狀魚鱗癬(lamellar ichthyosis)。過度表現突變型 LOR 的細胞株,與表現野生型 loricrin 的細胞株相比,透過 AKT 激酶(AKT-kinase)的活化,伴隨 ERK1/2、表皮生長因子受體(EGFR)與訊號轉導及轉錄活化因子 3(signal transducer and activator of transcription 3, STAT3)的磷酸化,顯示出增加的增殖率。
病理學: 組織病理學特徵包括過度角化、顯著的角化不全、棘層肥厚、顆粒層增生(雖然顆粒層可能呈現正常)、瀰漫性空泡變化、局灶性細胞溶解性變化,以及壞死的基底上層角質形成細胞。
超微結構發現包括豐富的角質透明顆粒、基底上層角質形成細胞的空泡變化與破壞,以及顆粒層內電子緻密的核內內含物沉積,免疫金(immunogold)研究顯示其中含有 loricrin。
Bothnia 型掌蹠角化症 (Palmoplantar Keratoderma Bothnia Type)
臨床特徵: Bothnia 型 PPK(PPKB;OMIM #600231)於 1994 年首次被描述為一種瀰漫型 NEPPK 的體染色體顯性型態,在瑞典北部 Bothnia 灣西部與西北部兩個省份具有高盛行率(0.3% 至 0.55%)。PPKB 亦已在一個漢族(Han)後裔的大型家系中被描述,並有人提出在英國人口中有一個共同的奠基者突變(founder mutation)。PPKB 臨床上類似 Nagashima PPK(見後文),通常於嬰兒期或兒童期表現出瀰漫型、均質、帶黃色調的過度角化,覆蓋於手掌與足底,並延伸至指趾背側。
這些病人,即使是僅有勉強可偵測的輕微表現型者,在接觸水後也會在受影響區域顯示典型的白色海綿狀外觀。多汗症、浸潤糜爛與續發性真菌感染很常見,異常指甲(彎曲且有破爛的甲小皮 ragged cuticles)亦然。與此疾病相反,常與囊腫纖維化(cystic fibrosis, CF)一同出現的手掌水源性皺褶,其特徵為半透明的白色丘疹、過度皺褶,以及由短暫接觸水所誘發的手掌水腫。
病因與致病機轉: PPKB 是一種體染色體顯性遺傳疾病,由 AQP5 基因的 GOF 突變所引起,AQP5 編碼水通道蛋白水通道蛋白 5(aquaporin 5, AQP5),由 Blaydon 與同僚於 2013 年首次鑑定。
水通道蛋白(aquaporins)是細胞膜轉運蛋白,使水能在許多細胞類型中經由滲透作用跨越細胞膜移動。AQP5 主要存在於頂端質膜(apical plasma membrane),參與外分泌腺(唾液腺、淚腺與汗腺)的水分排出;它亦表現於肺與角膜的上皮細胞。值得注意的是,乾燥症候群(Sjögren syndrome)中的少汗症(hypohidrosis)已被發現與 AQP5 表現降低相關。在正常掌蹠皮膚中,AQP5 定位於顆粒層(stratum granulosum)角質形成細胞的質膜,而在 PPKB 病灶中,AQP5 表現得以保留。然而,受影響的角質形成細胞會經由質膜增加水分攝取。
蛋白模型研究提示,致 PPKB 的突變增加了 AQP5 水通道收縮點(constriction point)的直徑,直接影響 AQP5 的閘控(gating)或水分通過通道的流動。手掌水源性皺褶(接觸水後手掌上短暫的水腫性白色斑塊與多汗症)已被發現與汗腺中 AQP5 的異常表現相關。瞬時受體電位香草素 4(transient receptor potential vanilloid 4, TRPV4)的活化導致胞質鈣濃度增加,這對汗液分泌至關重要,雖然其在角質形成細胞中的過度活化可能導致凋亡與過度角化,如在由 TRPV3 突變引起的 OS 中所示。
由於 TRPV4 與 AQP5 兩者皆表現於外分泌汗腺與角質形成細胞,因此有人推測 PPKB 中的掌蹠多汗症與過度角化是 AQP5–TRPV4 複合體 GOF 效應的結果,顯示 PPKB 是一種皮膚通道病變(skin channelopathy)。CF 病人手掌水源性皺褶的一個可能疾病機轉是角質形成細胞中 TRPV4 通道的功能障礙,導致經由外分泌導管的水分流入失調。
組織病理學: 組織病理學發現為非特異性的,包括正過度角化與上真皮中輕微的淋巴球浸潤。
瀰漫型掌蹠角化症,無皮膚外特徵,隱性遺傳 (DIFFUSE PALMOPLANTAR KERATODERMA WITHOUT EXTRACUTANEOUS FEATURES, RECESSIVE INHERITANCE)
Mal de Meleda
臨床特徵: MDM(OMIM #248300)是一種罕見的體染色體隱性遺傳疾病,最初在克羅埃西亞 Meleda 島(Mljet)原住病人身上描述。該疾病的診斷標準於 1969 年提出。自此之後,該疾病已在世界其他地區被描述,包括中東、西歐與北非,因奠基者效應(founder effect)在某些族群中具有較高的盛行率,主要在地中海與亞得里亞海地區。在台灣、中國、日本、印尼與印度已有少數病例報告。
該疾病於嬰兒期或兒童早期發病,於手掌(圖 48-5A)與足底(圖 48-5B)上出現瀰漫型、向外延伸(transgressive)、黃色、蠟狀、由紅色鱗狀邊界所勾勒的過度角化斑塊,其前有顯著且持續的紅斑。掌蹠過度角化隨年齡進展,並以手套狀(圖 48-5C)與長襪狀(圖 48-5D)分布延伸至手足背側面、手腕與足踝的屈側面,以及阿基里斯腱、前臂、肘部與膝部上方。已描述腹股溝與股溝區域的侵犯(圖 48-5E)。
其他特徵包括指節墊、關節上方的苔癬樣或角化斑塊(圖 48-5F)、口周紅斑、伴隨重複感染的多汗症、惡臭的浸潤糜爛與疼痛性裂隙、指甲異常(如匙狀甲 koilonychia、杵狀指 clubbing、甲剝離 onycholysis、指甲增厚、失養與甲下過度角化)(圖 48-5G)、指端硬化與短指(brachydactyly)。手指與足趾上的局限性過度角化斑塊導致圓錐狀外觀(圖 48-5H)、指趾緊縮、假性指趾斷離,以及伴隨活動度降低的進行性功能障礙。
雖然外顯率(penetrance)完全,但表現型隨地理來源與族裔背景而變化。此外,尚未鑑定出明確的基因型–表現型相關性,環境因素(機械性創傷與熱)似乎影響疾病病程。已描述一例無足底侵犯、表現型較輕微(具有輕微紅斑性的掌蹠角化斑塊),以及一種非典型外觀(掌蹠表面,主要為足底皮膚,上有多發 2- 至 5-mm、由棕紅色紅斑勾勒的角質溶解凹陷 keratolytic pits)的病例。已描述無 transgrediens 證據的病例。雜合子帶因者可能表現出輕微的瀰漫型 PPK,或手掌與足底皮膚平滑但帶有角化丘疹。
文獻中已描述數例 MDM 病人在過度角化病灶內發生惡性黑色素瘤(malignant melanoma, MM)。已報告一例併發掌蹠皮膚不規則色素過度沉著斑點,以及一例病人足底出現鮑文氏病(Bowen disease)。
因此,定期篩檢 MM 與其他腫瘤是必要的。PPK Gamborg-Nielsen(PPK-GN;OMIM #244850)是一種體染色體隱性遺傳疾病,由 Gamborg Nielsen 於 1985 年首次在瑞典最北部縣份的病人身上描述,部分人認為它是 MDM 的較輕微型態。它表現出類似的表現型,但瀰漫型掌蹠過度角化較不嚴重,且無指甲畸形或遠處角化,雖然可觀察到指節墊與漸尖的手指。
病因與致病機轉: MDM 由 SLURP1 基因(先前稱為 ARS component B)的雙等位基因突變所引起,該基因含有三個轉譯外顯子,編碼淋巴球抗原 6/尿激酶型纖溶酶原活化因子受體相關蛋白 1(lymphocyte antigen 6 / urokinase-type plasminogen activator receptor related protein-1, SLURP-1),是 Ly-Upar 蛋白超家族的成員之一,這些蛋白在跨膜訊號轉導、細胞活化與細胞黏附中扮演角色。SLURP-1 被認為是一種分泌型表皮神經調節因子(neuromodulator),可能對於表皮恆定,以及在傷口癒合期間抑制巨噬細胞釋放腫瘤壞死因子(tumor necrosis factor, TNF)-α 兩者皆至關重要。
MDM 中提示存在遺傳異質性,因為已有報告無 SLURP1 突變的家族。
PPK-GN 最近已被證實為 MDM 的等位基因變異型。在來自瑞典北部的 14 名個體中鑑定出 SLURP1 的同合子突變 c.43T>C,並在一名來自瑞典南部的個體中鑑定出複合雜合子突變 c.280T>A 與 c.43T>C。c.43T>C 突變先前曾在 MDM 中被報告,支持 PPK-GN 是 MDM 的輕微變異型而非獨立的疾病實體。
SLURP-1 已被證實表現於人類皮膚,主要在上表皮層下方的角質形成細胞中,並在維持皮膚的生理與結構完整性中扮演角色。SLURP-1 上調轉麩醯胺酸酶 1、細胞角蛋白 10(cytokeratin 10)與胱天蛋白酶 3 與 8(caspases 3 and 8)的表現,從而增加凋亡活性,其程度超過 TNF-α。此外,它透過膽鹼性途徑(cholinergic pathways)調節表皮分化。
此外,SLURP-1 表現與異常的免疫反應相關。此基因的突變導致 T 細胞活化反應缺陷,且 SLURP-1 增加 T 細胞中乙醯膽鹼(acetylcholine)的合成並減弱 T 細胞增殖。這些效應被 α7-nAChR 拮抗劑所廢除,顯示 SLURP-1 經由 α7-nAChR 媒介的途徑調節 T 細胞的功能性發展。因此,具有同合子 SLURP1 突變的病人易發生病毒感染並發展 MM。MDM 病人中 MM 較高的發生率不僅可歸因於 T 細胞活化缺陷與過度角化皮膚中延長的發炎,也可能源於凋亡活性缺陷,以及巨噬細胞 TNF-α 釋放調節的缺陷。
有人提出,SLURP-1 缺乏會影響角質形成細胞中三酸甘油酯水解(triglyceride hydrolysis)的效率,而此過程對於形成對表皮屏障重要的醯基神經醯胺(acylceramides)至關重要。有人假設,組織間液滲漏進入角質層,以及在 SLURP1 缺乏皮膚中三酸甘油酯小滴的累積,是有利於微生物生長的條件,這負責了 MDM 皮膚的惡臭。
SLURP-1 可能在乾癬的致病機轉中扮演角色。在咪喹莫特(imiquimod)誘發的小鼠乾癬皮膚中其表現上調,且在介白素(interleukin, IL)-22 刺激後 SLURP1 mRNA 表現顯著上調,這完全被 STAT3 抑制所壓制。此外,SLURP-1 顯著抑制金黃色葡萄球菌(Staphylococcus aureus)的生長,並已被證實調節皮膚與口腔傷口中的上皮化(epithelialization)與傷口癒合。
病理學: MDM 的組織學特徵為過度角化、正角化、局灶性角化不全、顯著的棘層肥厚、更明顯的透明層(stratum lucidum),以及血管周圍的淋巴組織球浸潤。電子顯微鏡發現包括:顆粒層與角質層之間比正常更不突然的轉變;正常的中間絲與角質橋粒(corneodesmosomes);以及具有海綿狀外觀、非特異性、不規則形的角質透明顆粒。在 MDM 病人的掌蹠切片中,以及在其汗液中,SLURP-1 不是缺如就是勉強可偵測。有人提出,在用於診斷 CF 的標準毛果芸香鹼(pilocarpine)程序所收集的汗液中評估 SLURP-1,可作為 MDM 的快速篩檢試驗。
Nagashima 型掌蹠角化症 (Nagashima-Type Palmoplantar Keratoderma)
臨床特徵: Nagashima 型 PPK(NPPK;OMIM #615598)是一種體染色體隱性遺傳疾病,由 Nagashima 於 1977 年首次描述。該疾病最初被命名為 Meleda 型 PPK,但於 1989 年創造了 NPPK(或 keratosis palmoplantaris Nagashima)一詞,以區分此型 PPK 與真正的 MDM,因為其表現型較輕微且青春期後具非進行性本質。多數病例報告於日本與中國,這些國家的估計盛行率分別為每 1 萬人 1.2 例與每 1 萬人 2.3 至 3.1 例,使其成為亞洲族群中最常見的 PPK 類型。已有數例在東亞以外被描述,NPPK 在非亞洲族群中的估計盛行率低至每 1 億人 0.5 例。
NPPK 典型於生命最初幾年開始(從出生至四歲),並逐漸進展至青春期,此後不再進展。其特徵為瀰漫型、界線分明、紅斑性的掌蹠過度角化,延伸至手足背側面、內側手腕、足踝與阿基里斯腱區域。肘部與膝部的侵犯很常見。已描述一例侵犯中央腰部區域、肘窩、前臂、大腿與膕窩的病例。耳朵上的過度角化與足趾甲失養(trophy 萎縮性甲板伴脆弱的遠端邊緣與自發性剝離)是罕見特徵,已在個別病例中被報告。它經常併發多汗症、皮癬菌重複感染、特殊氣味與浸潤糜爛。
與其他更嚴重形式的體染色體隱性向外延伸瀰漫型 PPK(如 MDM 與 PPK-GN)相反,無緊縮帶、自發性截斷或屈曲攣縮(flexion contractures)的證據。此外,NPPK 病人在接觸水 10 分鐘內,特別在紅斑性過度角化區域顯示白色海綿狀外觀,提示 NPPK 病灶皮膚的角質層水分滲透增強。
已報告一例在 NPPK 病灶內發生 MM 的病例。
病因與致病機轉: NPPK 由 SERPINB7 基因的雙等位基因突變所引起,該基因編碼絲胺酸蛋白酶抑制劑家族 B 成員 7(serpin family B member 7, SERPIN7),是絲胺酸蛋白酶抑制劑(serpin)超家族的一個胞質成員。多數 NPPK 病例由功能喪失型(loss-of-function, LOF)突變所引起,源於異常剪接或具有無義或框移突變的早熟終止密碼子。一個錯義奠基者突變 c.830C>T 的複合雜合性(導致高度保守的第 277 號殘基由脯胺酸變為白胺酸 leucine),造成 NPPK 表現型,並使 SERPIN7 在角質細胞(corneocytes)內錯誤定位。
SERPINB7 中一個復發性無義突變 c.796C>T,已被發現在中國與日本 NPPK 病人中皆普遍存在,可能反映奠基者效應。因此,有人提出在中國與日本患有瀰漫型、非殘毀性 PPK 的病人情境中,篩檢 SERPINB7 的 c.796C>T 突變可作為一種診斷策略。
表皮蛋白酶抑制劑(如 LEKTI、LEKTI-2、elafin、serpins 與 cystatins)在維持表皮恆定中扮演關鍵角色,方法是抑制位於顆粒層與角質層的內源性蛋白酶,以及來自攻擊表皮的細菌、真菌、病毒、花粉與屋塵蟎(house dust mites)的外源性蛋白酶。影響 serpins 的突變可經由兩種方式造成症狀:喪失蛋白酶抑制活性與不受控制的蛋白酶活性,或突變 serpin 多聚體在合成 serpin 的細胞中聚集,伴隨細胞死亡與組織損傷(「serpin 病變 serpinopathies」)。SERPINB7 已被證實分布於表皮,特別是顆粒層與角質層上部,以胞質表現為主。在 NPPK 皮膚中,SERPINB7 免疫反應性顯著減少。雖然 SERPINB7 在人類表皮中普遍表現,但 NPPK 局限於手、足、膝與肘,提示慢性暴露於機械性壓力可能在 NPPK 的發展中扮演角色,且 SERPINB7 可能抑制機械性壓力誘導的蛋白酶,並保護角質形成細胞或角質細胞免於蛋白酶媒介的細胞損傷。
NPPK 接觸水後的白色海綿狀變化,與伴隨 AQP5 突變的 PPKB 及伴隨 CFTR 突變的水源性角化症相似,有人提出這源於受損角質層水分滲透的增強。
病理學: NPPK 的組織病理學發現包括正過度角化伴棘層肥厚、顆粒層增生,以及上真皮中輕至中度的血管周圍淋巴球浸潤。在真皮浸潤中觀察到以 CD4+ T 細胞為主,CD20+ B 細胞勉強可偵測。
瀰漫型遺傳性掌蹠角化症,伴皮膚外特徵,顯性遺傳 (DIFFUSE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE)
Olmsted 症候群 (Olmsted Syndrome)
臨床特徵: OS(OMIM #614594)由 Olmsted 於 1927 年首次報告,是一種罕見疾病,盛行率低於每 100 萬人 1 例。它通常於出生時、新生兒期或兒童早期出現,雖然已描述較晚於兒童期出現,並隨時間進行性惡化。兩性皆受影響,雖然男性病人佔報告病例的 60%。其特徵為對稱、界線銳利、瀰漫型 PPK,伴黃褐色過度角化、疼痛性裂隙與紅斑性邊界,並伴隨開口周圍角化斑塊。這些斑塊可能局限於口、鼻孔、耳道、肛門與生殖器周圍區域,或可能延伸侵犯非開口周圍部位,如頸部、上胸部、下腹部、手臂、肘部、膝部、大腿與腹股溝皺褶。PPK 典型於局灶開始,分布於壓力點。它逐漸延伸至手掌與足底大部分表面,伴厚過度角化斑塊,最終導致手指屈曲畸形、假性指趾斷離,以及指趾或手的自發性截斷。PPK 典型為疼痛性與致殘性,妨礙行走與日常活動。已報告一些角化症較局灶或點狀、缺乏假性指趾斷離或顯著開口周圍角化病灶的病例。其他發現包括伴睡眠障礙的嚴重搔癢、甲失養(嵴狀與粗糙的指甲、甲彎曲 onychogryphosis、白甲症、甲剝離、甲溝炎 paronychia、甲下過度角化、無甲)、異常齒列(前臼齒缺如、伴隨早期牙齒喪失的牙周病)、出汗異常、口腔白色角化症、多汗症、過度角化線狀條紋、毛囊角化症、唇炎(cheilitis)與魚鱗癬樣病灶。毛髮異常很常見,雖然罕見病例毛髮正常,包括禿髮(瀰漫型、全身性或斑塊狀)、毛髮稀少(hypotrichosis),以及稀疏、捲曲、羊毛狀、粗糙、乾燥或易斷的毛髮。受影響毛髮的顯微發現包括捻轉發(pili torti)、結節性脆髮症(trichorrhexis nodosa)、色素減少、縱向嵴狀與毛幹的橫向斷裂。OS 中觀察到稀疏細薄的眉毛與睫毛、睫毛脫落(madarosis),以及長睫症(trichomegaly)。續發性細菌與念珠菌感染、SCC(疣狀癌或其變異型上皮瘤 epithelioma cuniculatum)與 MM 可在 PPK 內發展。已描述一例伴隨圓錐狀手指與類指端硬化外觀、與 Huriez 症候群臨床特徵重疊的病例。皮膚外表現不常見,包括聽力損失、眼部異常(角膜失養、發炎的淚腺或淚管或瞼板腺功能障礙、慢性瞼緣炎 blepharitis)、身材矮小、原發性硬化性膽管炎(primary sclerosing cholangitis)、智能不足、關節鬆弛、關節僵直(ankyloses)、骨質減少(osteopenia)與骨溶解(osteolysis)。已有數例報告紅斑性肢痛症(erythromelalgia)。
病因與致病機轉: 雖然迄今報告的多數病例為散發性,但已報告具有不同遺傳模式的家族性病例,包括體染色體顯性、體染色體隱性與 X 染色體連鎖遺傳。體染色體顯性、半顯性(semidominant)與體染色體隱性 OS 已被證實由 TRPV3 基因的突變所引起,該基因編碼瞬時受體電位香草素 3(transient receptor potential vanilloid 3, TRPV3),而 MBTPS2 基因(編碼膜結合轉錄因子蛋白酶位點 2 membrane-bound transcription factor protease site 2)的突變已在 X 染色體連鎖隱性 OS 中被鑑定。OS 中無明確的基因型–表現型相關性。TRPV3 突變與基底角蛋白的持續存在以及 Ki-67 表現增加相關。表現突變型 TRPV3 的角質形成細胞易發生細胞死亡,且 OS 病人的表皮與對照參與者相比顯示出增加的凋亡細胞。
TRPV3 高度表現於角質形成細胞與毛囊(hair follicles, HFs)、脊髓、感覺神經元、腦與角膜。它在表皮屏障形成、毛髮生長調節,以及疼痛感覺與搔癢調節中具有重要角色,可解釋 OS 的表現。TRPV3 活化增加胞內 Ca2+ 濃度,並與轉化生長因子(transforming growth factor, TGF)-α/EGFR 訊息傳遞相關,後者已知調節表皮分化。使用發展出波浪狀毛被與捲曲鬍鬚、並於出生時出現紅色乾燥脫屑皮膚的 Trpv3 基因剔除小鼠模型,證實了 TRPV3 在媒介毛髮與皮膚屏障功能之 EGFR 訊息傳遞中的角色。此外,攜帶 Trpv3 雜合子突變的小鼠或大鼠表現出毛髮稀少與皮膚炎。
編碼一種對膽固醇恆定與內質網(endoplasmic reticulum, ER)壓力反應至關重要的鋅金屬蛋白酶(zinc metalloprotease)的 MBTPS2 突變,已在 X 染色體連鎖隱性遺傳的 OS 病例中被報告。MBTPS2 突變最初在魚鱗癬性毛囊角化–無毛–畏光(ichthyosis follicularis atrichia photophobia, IFAP)症候群中被鑑定(見第 49 章),該症候群與 X 染色體連鎖隱性 OS 為等位基因關係。已報告一例同時具有 OS 與 IFAP 症候群特徵的病例。
攜帶 TRPV3 或 MBTPS2 突變的 OS 病人之間無明顯臨床差異,且在同一家族內或攜帶相同突變的不同病人之間觀察到臨床變異性,支持修飾基因(modifier genes)、環境效應或表觀遺傳因素的角色。此外,鑑於部分病人不攜帶 TRPV3 或 MBTPS2 任一者的突變,遺傳異質性是可能的。
病理學: OS 的組織病理學發現為非特異性的,包括乾癬樣表皮增生(psoriasiform epidermal hyperplasia)、正過度角化、局灶性角化不全、顆粒層減少或增生、棘層肥厚,以及上真皮中可能含有肥大細胞(mast cells)的發炎性浸潤。已報告表皮水皰性退化(vesicular degeneration)。電子顯微鏡發現包括中馬氏層角質形成細胞中大型、粗糙、緻密堆積的張力絲束,以及顆粒層中粗糙角質透明顆粒數量增加。已報告色素顆粒數量減少與蘭格漢斯細胞(Langerhans cells)缺如。
特定治療: 已執行掌蹠角化症的部分切除或全層切除合併植皮,並有臨床改善。EGFR 抑制劑已被證實可改善 PPK,而 TRPV3 拮抗劑對於攜帶 TRPV3 基因 GOF 突變的病人可能是有效的治療。
連接蛋白相關掌蹠角化症:Vohwinkel 症候群、角膜炎–魚鱗癬–耳聾症候群、Bart-Pumphrey 症候群與伴隨耳聾的掌蹠角化症 (Connexin-Associated Palmoplantar Keratodermas: Vohwinkel Syndrome, Keratitis–Ichthyosis–Deafness Syndrome, Bart-Pumphrey Syndrome, and Palmoplantar Keratoderma with Deafness)
臨床特徵: VS(殘毀性遺傳性角化症 keratoderma hereditaria mutilans;OMIM #124500)於 1929 年由 Vohwinkel 與 Wigley 首次報告。VS 的特徵為三聯徵:瀰漫型、殘毀性、具「蜂窩狀」外觀的 PPK(見圖 48-4B);手背、足背與伸側面上的「海星狀」角化斑塊;以及手足指趾間關節(interphalangeal joints)上導致自體截斷的纖維性緊縮帶(假性指趾斷離)。多數病例亦伴隨高頻感覺神經性聽力損失(SNHL)。該疾病在白人女性中較常見。它傾向於在新生兒期表現並終生進展。已報告以壓力點上的胼胝或損傷部位的條紋病灶(提示同形現象 isomorphic phenomenon)為特徵之 PPK 的病例。該疾病可能伴隨全身性魚鱗癬、黑色棘皮症(acanthosis nigricans)、瘢痕性或非瘢痕性禿髮、指甲異常、指節墊、足底水皰性病灶,以及顱面異常(唇顎裂、小頭症與面部不對稱)。已報告過度角化病灶中出現 SCC 與基底細胞癌(basal cell carcinoma, BCC)。已報告伴隨聾啞(deaf-mutism)、智能不足、伴肌病變的痙攣性下肢癱瘓(spastic paraplegia with myopathy)、精神運動發展遲緩與癲癇發作的病例。KID 症候群(OMIM #148210;見第 47 章)是最嚴重的皮膚連接蛋白疾病,侵犯外胚層來源的上皮(皮膚、附屬器、指甲、牙齒)以及顯著的內耳與角膜侵犯。疾病表現於 1915 年首次被描述,雖然 KID 症候群這個名稱由 Skinner 與同僚於 1981 年所創。
該疾病於出生時或嬰兒期表現。其皮膚特徵為紅皮角化症,伴對稱、界線分明、具網狀模式的過度角化斑塊、皮革樣外觀,或瀰漫型糠狀鱗屑(furfuraceous scaling)伴底層紅斑,並相對好發於腋窩與頸部。其他表現包括增厚、具粗顆粒外觀的皮膚,或無紅斑的毛囊過度角化。特徵性的 PPK 為瀰漫型,具有粗糙、點刻狀(stippled)或顆粒狀外觀(圖 48-6)。慢性唇炎與口角炎(perleche)很常見。相關發現包括甲失養、口腔表現(白色角化症、舌頭深裂、牙齒異常包括齲齒與牙齒延遲萌發、持續性口腔黏膜丘疹)、少汗症與熱不耐受。許多 KID 症候群病人毛髮稀疏,10% 至 23% 具有先天性無毛症。多數病人發展出進行性角膜炎,於兒童期開始,並可能首先表現為畏光(photophobia)。角膜炎的特徵為角膜發炎、疼痛與角膜新生血管,並可能伴隨慢性瞼緣炎、結膜炎、乾性角結膜炎(keratoconjunctivitis sicca)與角膜緣缺陷(limbal defects),並可能造成進行性視力下降與失明。相關的眼科特徵為眉毛與睫毛脫落、眼瞼過度角化、倒睫(trichiasis)、早發性白內障與雙側淚點發育不全(lacrimal punctal agenesis)。
相反地,已描述無眼科表現證據的病例。此外,KID 病人具有先天性、雙側、嚴重的 SNHL,與角膜發現相比,其為非進行性。病人易發生黏膜皮膚感染,多為細菌與念珠菌感染,在新生兒期可能致命。已報告病毒感染(傳染性軟疣 molluscum contagiosum 與巨細胞病毒 cytomegalovirus)、反覆肺部浸潤與反覆外耳炎。病人易發展良性皮膚腫瘤,主要為毛根鞘瘤(trichilemmoma),雖然已描述多發性汗孔瘤(poromas)與汗孔角化性外分泌孔及真皮導管痣(porokeratotic eccrine ostial and dermal duct nevus)的個別病例。據報告約 10% 的受影響個體發展皮膚(主要為肢端部位與慢性感染或發炎區域)與舌頭的 SCC,顯然由病灶中 p53 喪失所致。
鑑於 SCC 的侵襲性表現型與發展多發腫瘤的傾向,其可能導致 KID 病人死亡。此外,已報告伴隨皮脂腺癌(sebaceous carcinoma)、周邊 T 細胞非何杰金氏淋巴瘤、惡性組織球瘤(malignant histiocytoma)與轉移性惡性毛髮腫瘤(pilar tumors)的個別病例。已描述 KID 症候群與化膿性汗腺炎(hidradenitis suppurativa)及頭皮剝離性蜂窩織炎之間的關聯。KID 症候群可能因嚴重感染或呼吸窘迫而導致嬰兒期死亡。
Bart-Pumphrey 症候群(BPS;OMIM #149200)是一種罕見的體染色體顯性遺傳疾病,由 Bart 與 Pumphrey 於 1967 年首次描述。其特徵為嚴重的 SNHL、具蜂窩狀外觀的 PPK、指節墊與白甲症。聽力損失與指節墊是最常見的發現;白甲症與 PPK 較不常見。
該 PPK 具有瀰漫型、界線銳利的掌蹠皮膚增厚,伴點狀、顆粒狀表面,令人聯想到 VS。亦已描述條紋狀外觀。
PPK 與耳聾(OMIM #148350)是一種體染色體顯性遺傳狀況,其中 PPK 可能為瀰漫型、帶 transgrediens 伴裂隙與底層紅斑,或為較輕微的表現型,於壓力點上有皮膚皺褶加重。可能存在指節墊。聽力障礙可能於嬰兒期變得明顯;為雙側、語言發展前(prelingual)、緩慢進行性,並影響高頻音調。
病因與致病機轉: 間隙連接(gap junctions)由連接子(connexons)形成,是促進並調節水與小分子在相鄰細胞之間通過的細胞間連接。六個連接蛋白的寡聚化導致形成一個稱為連接子的半通道(hemichannel),而兩個相對細胞的連接子透過其細胞外部分彼此交互作用,形成一個通道,即間隙連接的基本單位。連接蛋白以組織與分化特異性的方式表現。
表皮、其附屬器,以及內耳與角膜的其他外胚層來源上皮,共享數種連接蛋白的表現,包括 Cx26、Cx30、Cx31 與 Cx43。Cx43 普遍表現於所有外胚層來源的上皮細胞,並與 Cx26 相似,已被證實在傷口癒合中扮演角色。Cx26 的表現局限於耳蝸細胞、角膜緣細胞、掌蹠表皮、外分泌汗腺與汗管開口周圍,以及人類 HFs 的內外根鞘(root sheath)。Cx26 的表現大致與 Cx30 的表現模式平行(兩者共享 76% 的胺基酸同一性)。與普遍存在於毛囊間表皮上層分化層的 Cx30 相反,Cx26 在掌蹠表皮中僅以低量表現,但在受傷反應與過度增生疾病中強烈被誘導。Cx26 也表現於圍繞耳蝸感覺毛細胞的上皮支持細胞,以及襯於耳蝸管的纖維細胞中,在此處它媒介聽覺轉導期間通過毛細胞的鉀離子回收至內淋巴(endolymph)。
GJB2 編碼 Cx26。產生完全表現喪失或功能喪失的隱性突變,是人類非症候群性 SNHL 的單一最常見原因。此外,已知連接蛋白基因的突變會造成數種與 PPK 或其他皮膚異常無關的遺傳性人類疾病,包括 X 染色體連鎖 Charcot-Marie-Tooth 病(Cx32)、帶狀粉末性白內障(zonular pulverent cataract,Cx50)與眼–齒–指發育不良(Cx43)。此外,數種遺傳性皮膚病源於連接蛋白基因的雜合子突變(見表 48-1 與圖 48-2)。
連接蛋白由四個跨膜區域、三個胞質區域(胺基末端、一個胞質環與羧基末端區域)與兩個細胞外環構成。蛋白的 N 末端與 C 末端部分皆位於胞質中。跨膜與細胞外區域高度保守,連接蛋白之間的主要差異存在於其 C 末端尾部。迄今報告的多數與聽力障礙及皮膚侵犯相關的顯性負向 GJB2 突變,位於 Cx26 的胞質 N 末端或第一個細胞外環,後者在連接蛋白之間高度保守,並參與電壓閘控、通道通透性、多聚體組裝,以及連接子之間交互作用的控制。因此,影響第一個細胞外區域的突變可能同時影響蛋白運輸與通道通透性。此外,此區域的不同突變導致不同的病理生理效應,例如傳導阻斷、對野生型 Cx26 的顯性效應、蛋白摺疊,以及半通道結構穩定性的改變,導致可變的臨床表現型。此外,不同連接蛋白共寡聚化(co-oligomerization)成異聚連接子(heteromeric connexons),在致病機轉與連接蛋白相關表現型的動態本質中扮演角色,這些表現型多由對細胞間溝通的顯性負向效應所引起。
多數報告的 VS 病例源於 GJB2 中復發性雜合子錯義突變 p.Asp66His,導致在第一個細胞外區域一個高度保守殘基處由天門冬胺酸(aspartic acid)取代為組胺酸(histidine)的胺基酸取代。此突變可能選擇性地損害 Cx26 形成異聚與同聚連接子的能力,導致 Cx26 電荷或構象的改變,破壞對特定分子或離子的閘控特性。Gjb2 在小鼠中的全身性基因剔除是胚胎致死的(由胎盤功能不全所致),但小鼠內耳的條件性 Gjb2 基因剔除導致耳聾。顯性負向 Gjb2 突變的轉殖基因表現,揭示了感覺毛細胞的進行性退化,並因 cortilymph 恆定受擾而喪失 Corti 隧道(tunnel of Corti),說明 Cx26 在內耳聽覺功能中扮演的必要角色。專一在基底上層表皮表現 p.Asp66His 突變的轉殖基因小鼠,表現出尾部具緊縮帶的角化症、表皮角化層的顯著增厚,以及表皮 TUNEL 染色增加,指示過度凋亡或早熟終末分化。早熟的角質形成細胞死亡可能誘導代償性的基底細胞增殖,導致角質層的大量增厚。KID 症候群的體染色體顯性與體染色體隱性型態皆已被描述,雖然體染色體顯性遺傳較常見。已報告提示親代生殖細胞鑲嵌(germline mosaicism)的病例。多數受影響個體攜帶 GJB2 中復發性錯義突變 p.Asp50Asn,導致第 50 號密碼子的天門冬胺酸被天門冬醯胺(asparagine)取代。此外,與 KID 症候群為等位基因關係的豪豬狀魚鱗癬–耳聾(hystrix-like ichthyosis-deafness, HID)症候群,已被報告源於 p.Asp50Asn 突變與其他 GJB2 錯義突變。Asp50 是一個襯於孔道(pore-lining)的殘基,在連接蛋白之間高度保守,對間隙連接的形成與功能至關重要;p.Asp50Asn 導致突變蛋白的胞內表現,提示其向質膜的運送改變與間隙連接斑的缺如。p.Gly45Glu 突變已在致命型態的 KID 症候群中被鑑定。所有致 KID 症候群的突變聚集於編碼 Cx26 第一個細胞外區域與胞質胺基末端區域的區段,並預測會改變此區域的電荷與結構,這與沿著蛋白分布的非症候群性 Cx26 相關突變不同。最近在非洲爪蟾(Xenopus laevis)卵母細胞中的實驗證實,由數個 Cx26 KID 突變體形成的半通道被甲氟喹(mefloquine)所抑制;甲氟喹減弱了表現人類 Cx26 p.Gly45Glu 突變之原代小鼠角質形成細胞中增加的巨觀膜電流。
編碼 Cx30 的 GJB6 中一個雜合子突變,已被證實也會導致 KID 症候群。此突變預測會改變 Cx30 第一個跨膜螺旋的序列與電荷,並在一名具有 KID 症候群典型特徵(包括毛囊性、棘狀過度角化、先天性無毛症與指甲異常)的兒童中被報告。一個類似的突變先前曾被牽涉於 Clouston 症候群(見後文)中,無異常出汗、聽力、畏光與角膜炎的證據,強調其他遺傳與表觀遺傳因素在修飾連接蛋白疾病臨床結果中的深遠影響,並強調 GJB6 突變的表現型變異性。有人推測此病人的獨特表現型與 GJB2 中一個同合子多型性(polymorphism)的存在有關。
在 BPS 中,已報告兩個涉及 Cx26 第一個細胞外區域的突變 p.N54K 與 p.G59S,強調此區域在連接蛋白半通道對接(docking)與電壓閘控中的重要性。
PPK 與耳聾源於 GJB2 基因中聚集於第一個細胞外環區域內或邊界的雜合子突變。其中一個顯性突變 p.R75Q(c.224G>A),由 Uyguner 與同僚在一個土耳其家族中首次描述,並被報告於孤立性耳聾以及伴隨 PPK 的情況中。
病理學: VS 的組織病理學表現包括緻密的過度角化;正角化;棘層肥厚;伴大型不規則形角質透明顆粒的顯著顆粒層增生;表皮的乳頭瘤病(papillomatosis);以及伴稀疏血管周圍淋巴球浸潤的真皮纖維化。超微結構發現為棘層與顆粒層中顯著腫脹的粒線體與增加數量的橋粒,角質細胞含有許多膜被覆顆粒(membrane coating granules)與脂質樣空泡。
KID 症候群中由 PPK 取得的皮膚切片可能顯示表皮增生、緻密的正角化過度角化與局灶性角化不全。可能觀察到顆粒層減少,雖然已報告具有顯著顆粒層的病例,並描述了胞質略微空泡化的腫脹角質形成細胞。角化栓塞(keratotic plugging)是常見特徵。上真皮中的發炎細胞可能明顯,尤其在感染病例中。
BPS 的組織病理學發現為大量正角化過度角化、顆粒層增生、棘層肥厚與乳頭瘤病。電子顯微鏡下表皮間隙連接呈現正常。
特定治療: 在 VS 中,已描述交指皮瓣(cross finger flap)、Z 成形術(Z-plasty)治療、緊縮帶全層切除合併全層植皮,以及用於第五指緊縮帶的遠處腹部皮瓣。KID 症候群眼部表現的已報告治療有角膜緣同種異體移植(keratolimbal allograft)、角膜移植與免疫抑制、角膜切除術(keratectomy)、外用皮質類固醇與環孢素(cyclosporine),且 bevacizumab 已在單一病例中被證實有效。
掌蹠角化症–先天性禿髮症候群 (Palmoplantar Keratoderma–Congenital Alopecia Syndrome)
臨床特徵: 已描述兩種形式的伴隨先天性禿髮之 PPK。一種體染色體顯性型態具有較輕微的表現型(PPKCA1,Stevanovic 型;OMIM #104100),而隱性遺傳型態則伴隨假性指趾斷離、指端硬化、攣縮,有時伴白內障(PPKCA2,Wallis 型;OMIM #212360)。毛髮於出生時正常或稀疏,而侵犯頭皮、身體或面部毛髮的非瘢痕性禿髮於嬰兒早期變得明顯。毛髮顯微鏡下可能可見結節性脆髮症。PPK 於嬰兒後期發展,在顯性病例中為界線分明、局灶型或線狀、非殘毀性且帶 transgrediens。在隱性病例中,其特徵為手掌與足底外側與內側面的進行性增厚,伴紅斑性邊緣與皮膚裂痕,隨後侵犯手指背側,導致攣縮、假性指趾斷離與指端硬化。相關發現包括伴隨類眉部潰瘍性紅斑(ulerythema ophryogenes-like features)的毛囊栓塞、毛孔角化症(keratosis pilaris),以及足踝、肘部與膕窩上方的過度角化斑塊,伴隨多發、尖刺、角狀的病灶,令人聯想到豪豬狀魚鱗癬與指甲異常(白甲症、甲失養)。在個別病例中報告了白內障、腦膜膨出(meningocele)與單側耳聾。
病因與致病機轉: PPKCA1 已被證實源於編碼 Cx43 的 GJA1 基因雜合子錯義突變,該突變對 Cx43 半通道施加 GOF 效應。Cx43 普遍表現於各種器官,包括表皮與 HFs。
PPKCA1 與眼–齒–指發育不良(oculodentodigital dysplasia, ODDD)為等位基因關係,後者的特徵為顱面畸形;牙齒、眼科與肢體異常;以及神經退化,並偶爾伴隨 PPK 與毛髮、指甲異常。在 ODDD 中,多數突變導致突變蛋白滯留於 ER 或通道通透性降低,間隙連接無功能。
PPKCA2 的致病基因尚未被鑑定。
病理學: 過度角化斑塊顯示正過度角化伴毛囊栓塞,以及乳頭層真皮中的血管周圍淋巴球浸潤。
毛幹的掃描電子顯微鏡顯示多發凹陷伴甲小皮風化(cuticular weathering)或縱向溝槽。
汗腺性外胚層發育不良(Clouston 症候群;見第 131 章)(Hidrotic Ectodermal Dysplasia [Clouston Syndrome; See Chap. 131])
臨床特徵: 汗腺性外胚層發育不良(hidrotic ectodermal dysplasia, HED,Clouston 症候群;OMIM #129500)於 1895 年首次被描述,後來由 Clouston 在魁北克(Quebec)的家族中描述。HED 是一種體染色體顯性遺傳的外胚層發育不良,因奠基者效應在法裔加拿大人口中特別常見,雖然已在數個族群中被報告。此狀況的主要特徵包括甲失養、毛髮脫落與掌蹠過度角化,伴正常的汗腺與牙齒。
指甲異常通常存在;在將近 30% 的病例中,它們是該症候群的唯一表現。它們範圍從幾乎正常外觀的指甲到短指甲與無甲症(anonychia)。甲板變化包括增厚、脆性、嵴狀、變色、裂開、甲剝離與 20 甲失養(20-nail dystrophy)。甲溝炎與指甲感染很常見,並可能導致甲基質破壞。毛髮異常可能為進行性,侵犯頭皮、面部與身體毛髮,包括無毛症或毛髮稀少,伴脆、細、淡或生長緩慢的毛髮。PPK 為瀰漫型,具天鵝絨樣或鵝卵石樣(cobblestoned)外觀,延伸至指尖與指節,伴裂隙。在一個漢族大型家族中,手掌與足底的過度角化傾向於隨時間惡化。已報告具有類似 PC 之指甲變化的 HED 病例,可為單獨發現或伴隨禿髮,但無 PPK 證據。已描述上下肢上的角化丘疹與斑塊,以及指節關節上的散在色素過度沉著。罕見的關聯包括斜視、白內障與畏光、聽力障礙、智能缺陷與骨骼異常(顱骨增厚、異常指骨)。
病因與致病機轉: HED 由 GJB6 的體染色體顯性雜合子突變所引起,該基因編碼 Cx30。Cx30 是一個 261 個胺基酸的多胜肽,由四個跨膜區域、兩個細胞外區域與三個胞質區域構成,與其他連接蛋白相似。Cx30 表現於表皮(中與上棘層)、HFs(外根鞘、毛基質)、指甲(甲基質與甲床)、腦與內耳。
一個攜帶 GJB6 中 p.A88V 突變的 HED 小鼠模型,顯示掌蹠皮膚輕微的過度角化、增大且過度增生的皮脂腺,以及聽力改變(兩者在 HED 中皆非常見特徵)。此外,此突變導致顯著的凋亡,可能透過滲漏的半通道(leaky hemichannels)。有人提出,由 p.A88V 突變所致的滲漏半通道可能透過活化 Ca2+ 依賴性激酶(伴隨基因表現改變或細胞週期重新進入)來刺激增殖。
與其他連接蛋白疾病相似,HED 的特徵為廣泛的臨床與遺傳異質性。一名具有 HED 與 ODDD 臨床特徵及廣泛皮膚過度角化的病人,被發現在編碼 Cx43 的 GJA1 中有雜合子突變 p.V41L,並在 GJB2 中有雜合子序列變異(p.R127H)。一名患有 HED 的中國病人被發現攜帶兩個錯義突變,GJB6 中的 p.N14S 與 GJB2 中的 p.F191L,而一名患有伴隨聽力障礙與畏光之 HED 的日本病人,被發現攜帶 GJB6 中的雜合子錯義突變(p.Ala88Val)與 GJB2 中的多型性(p.Val27Ile)。
與導致皮膚症狀與耳聾的 GJB2 突變相反,GJB6 突變典型導致皮膚表現而無聽力障礙證據。Cx26 可能在內耳中代償 Cx30 的缺如,而在皮膚中則不然,但 Cx30 無法在兩個部位完全代償 Cx26 的喪失,因為突變的 Cx26 對其他共表現的連接蛋白施加顯性負向效應。然而,已報告一例由 GJB6 突變引起的 KID 症候群,以及一例伴隨 SNHL 與畏光的 HED;此外,GJB6 中的 p.T5M 突變導致顯性非症候群性聽力損失,提示功能冗餘性(functional redundancy)可能依賴於額外因素,如致病突變的位置與性質。
病理學: 毛髮稀少的皮膚顯示正常表皮、外分泌腺與皮脂腺的正常分布,以及 HFs 的缺如或殘餘。PPK 病灶顯示過度角化伴不規則與網狀的棘層肥厚。
Huriez 症候群 (Huriez Syndrome)
臨床特徵: Huriez 症候群(硬化性胼胝病 sclerotylosis;OMIM #181600)由 Huriez 與同僚於 1969 年在來自北法的兩個家族中首次描述。自此之後,已描述來自不同族裔的家系,包括突尼西亞、印度、德國、英國、義大利與日本家族。多數病例以體染色體顯性方式遺傳,並已報告數例散發性病例。Huriez 症候群的特徵為瀰漫型 PPK、手與手指的硬化性萎縮、萎縮皮膚內發生 SCC,以及導致攣縮的指端硬化。該疾病於出生時存在或於生命最初幾年出現,並持續而無進一步進展。PPK 表現為瀰漫型、黃灰色、非紅斑性、界線分明的過度角化斑塊,主要侵犯手掌並延伸至手指,伴掌紋加重(圖 48-4C)。足底較少受侵犯,通常於壓力部位顯示加重。已描述汗孔角化樣(porokeratotic)外觀。硬化性萎縮變化包括手與指趾的假性硬皮樣(pseudosclerodermoid)外觀,伴皮紋(dermatoglyphs)缺如,以及手、手指與指趾尖背側面的紅斑性、萎縮外觀,無雷諾現象(Raynaud phenomenon)證據。相關發現為少汗症與指甲變化,包括發育不全、彎曲、甲脆裂(onychorrhexis)、匙狀甲、縱向嵴狀與杵狀指。在個別病例中報告的表現有異色症樣(poikiloderma-like)變化、手指上獨特的小結節,以及面部毛細血管擴張。惡性退化從手部萎縮皮膚上的皮膚潰瘍開始,並於第三至第四個十年發展 SCC。估計 SCC 風險為 13%,是高出 100 倍以上的風險。這些腫瘤傾向於顯示分化不良,伴高轉移率與計算出 5% 的死亡率。已描述伴隨內臟惡性腫瘤(胃癌、咽癌)的病例,但內臟惡性腫瘤在 Huriez 症候群中似乎不比一般人群更頻繁。
病因與致病機轉: Huriez 症候群的致病機轉與腫瘤形成所涉及的機轉尚未明瞭。先前曾報告 Huriez 症候群與某些血型之間的連鎖;然而後續研究未能證實此發現。Lee 與同僚將該基因定位於第 4q23 號染色體。
第 4q 的雜合性喪失(loss of heterozygosity)已在源於頭頸部的多數 SCC,以及將近 50% 的子宮頸癌病例中被報告。然而,在前者中,所涉及的區域延伸至 Huriez 症候群基因座的遠端,在其著絲點端可能有重疊。
免疫組織化學與超微結構研究顯示,受侵犯皮膚內表皮蘭格漢斯細胞幾乎完全缺如。此外,在非典型角質形成細胞中觀察到陽性的 p53 染色(指示 p53 功能異常),提示 p53 突變可能負責 Huriez 症候群中光化性角化症(actinic keratoses)與 SCC 的發展。
病理學: 由 PPK 皮膚取得的皮膚切片顯示顯著的棘層肥厚與乳頭瘤病、伴角質透明顆粒數量增加的顆粒層增生,以及正角化過度角化。真皮與汗腺未觀察到異常。在硬化性萎縮區域,表皮顯示顆粒層增生伴正角化過度角化、網狀真皮中緻密的膠原纖維,以及細薄的彈性纖維。可見稀疏的真皮單核細胞浸潤。已報告一例上棘層角質形成細胞空泡退化的病例。超微結構發現為整個表皮層中緻密的張力絲束,伴豐富的角質透明物質分布為大型且密度不規則的團塊。硬化性萎縮皮膚區域顯示張力絲與角質透明物質的類似變化,但具有額外特徵:真皮–表皮交界處變薄的 oxytalan 與 elaunin 纖維、不規則邊界,以及彈性纖維非均質的外觀,並有彈性纖維被巨噬細胞吞噬的證據。
瀰漫型遺傳性掌蹠角化症,伴皮膚外特徵,隱性遺傳 (DIFFUSE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, RECESSIVE INHERITANCE)
Papillon-Lefevre 症候群 (Papillon-Lefevre Syndrome)
臨床特徵: PLS(OMIM #245000)是一種罕見的體染色體隱性遺傳疾病,特徵為掌蹠過度角化與嚴重的進行性牙周炎,造成乳齒與恆齒喪失,伴齒槽骨(alveolar bone)的骨膜變化。
牙周病嚴重度已被證實於青少年期達到高峰,並隨年齡下降。PLS 的估計盛行率為每 100 萬人 1 至 4 例,帶因率為每 1000 人 2 至 4 例。無性別偏好,但約半數病例觀察到近親血緣。PPK 可能於出生時或生命最初幾個月出現,雖然在多數病例中,PPK 與牙周炎在第六個月至四歲之間發展,常於第一顆牙萌發時開始。值得注意的是,皮膚與牙齒表現的嚴重度之間無相關性。PPK 的特徵為瀰漫型、紅斑性、界線銳利的過度角化與鱗狀斑塊,覆蓋於兩側手掌與足底,具有延伸至手足背側面的 transgrediens 特徵(圖 48-7A)。它可能伴隨多汗症、惡臭氣味與指甲異常(橫向溝紋與裂隙)。已描述晚發性牙周炎與掌蹠病灶的非典型病例,或孤立性 PPK 或牙周炎的病例。可在膝部、肘部與足踝上觀察到界線銳利、乾癬樣的過度角化斑塊,並已描述類似乾癬之更廣泛分布。對感染的易感性增加是典型的;已報告化膿性皮膚感染以及肝或腦膿瘍與肺炎。PLS 可能伴隨智能障礙與硬腦膜(dura mater)鈣化。已報告個別病例伴隨生長遲緩、嬰兒型聲音與甲狀腺機能低下症,雖然這些可能與 PLS 表現型無直接關係。已報告伴隨異位性體質(atopic diathesis)與升高的免疫球蛋白 E(immunoglobulin E, IgE)濃度的關聯。肢端雀斑樣黑色素瘤(acral lentiginous melanoma)在 PLS 中可能更常見,至少在日本病人中如此。
病因與致病機轉: PLS 源於編碼組織蛋白酶 C(cathepsin C, CTSC,亦稱二肽基肽酶 I dipeptidyl peptidase I)的 CTSC 基因 LOF 同合子或複合雜合子突變。已從族裔多樣的族群中報告超過 70 個突變;約半數為改變蛋白摺疊與功能的同合子錯義突變,25% 為無義突變,將近 25% 為框移突變。PLS 與 Haim-Munk 症候群(Haim-Munk syndrome, HMS;OMIM #245010)為等位基因關係,後者主要在來自印度科欽(Cochin)的個體中描述,除了 PPK 與牙周炎外,其特徵為扁平足(pes planus)、蜘蛛指(arachnodactyly)、肢端骨溶解(acroosteolysis)與甲彎曲。HMS 個體無腦鈣化與細菌感染證據。Sulák 與同僚最近報告兩名匈牙利病人,一名患 PLS、一名患 HMS,他們攜帶 CTSC 基因中相同的同合子無義突變,支持 PLS 與 HMS 是同一疾病的表現型變異型這一概念。PLS 也與無皮膚表現的青春期前牙周炎(prepubertal periodontitis)為等位基因關係。
CTSC 屬於半胱胺酸肽酶(cysteine peptidases)的木瓜蛋白酶(papain)超家族,是一種四聚體酵素,由四個透過非共價鍵連接在一起的相同亞基構成。它在蛋白的胞內降解中扮演重要角色,並協調具有免疫或發炎功能之細胞(包括嗜中性球、肥大細胞、細胞毒性 T 細胞與自然殺手細胞)中絲胺酸蛋白酶的酶原(zymogen)活化。該蛋白在上皮與骨髓細胞中以前蛋白酶(pro-proteinase)形式表現,並經由多步驟過程導致其活化。PLS 病人中的 CTSC 突變與 CTSC 酵素蛋白酶活性的喪失相關;突變的帶因者與對照參與者相比顯示出降低的活性。
已證實尿液中缺乏活性 CTSC,可作為 PLS 強而可靠的指標,並允許在近親血緣高頻率的族群中篩檢該疾病。
PLS 病人嗜中性球的分析顯示,正常存在於嗜天青顆粒(azurophilic granules)中的蛋白(彈性蛋白酶 elastase、組織蛋白酶 G、蛋白酶 3)在成熟嗜中性球中完全缺如,但在前驅細胞中則否,指示 CTSC 突變促進成熟免疫細胞中的蛋白酶降解。由於 PLS 病人中無明顯的免疫缺陷,嗜中性球絲胺酸蛋白酶對人類免疫保護可能是非必需的。
數例 PLS 中已證實升高的 IgE 濃度,且高 IgE 症候群(hyper-IgE syndrome)病人乳齒牙根的延遲吸收,導致有人提出血清 IgE 可能影響牙周代謝。這由 PLS 中 IgE 濃度下降後牙周發炎的改善所支持。此外,PLS 中明顯具有 T 輔助(T helper, Th)2 表現型,伴 Th1 細胞與干擾素-γ 濃度降低,以及 IL-4 濃度增加。
病理學: PLS 的組織病理學分析顯示不規則的過度角化、顆粒層增生、顯著的棘層肥厚,以及真皮中血管周圍的淋巴球與組織球浸潤。電子顯微鏡下明顯可見角質細胞與顆粒球中的脂質樣空泡,以及不規則的角質透明顆粒與張力絲的減少。
Naxos 病 (Naxos Disease)
臨床特徵: Naxos 病(Naxos disease, ND;OMIM #601214)是一種體染色體隱性遺傳疾病,由編碼血漿斑珠蛋白(plakoglobin, PG)的 JUP 突變所引起。
該疾病最初由 Protonotarios 與同僚在來自希臘 Naxos 島的家族中描述。自此之後,已在各種族裔中被報告。
該疾病的特徵為三聯徵:瀰漫型 PPK、心肌病變與羊毛狀髮。羊毛狀髮於出生時出現,具有稀疏、脆、有時色素減少的毛髮。毛髮異常見於頭皮、眉毛、腋毛與陰毛。PPK 於生命第一年發展,特徵為手掌與足底上瀰漫型、界線分明、非向外延伸(nontransgredient)的過度角化斑塊,可能被紅斑性邊界所環繞。其他皮膚特徵為多汗症與指甲異常。心律不整性右心室發育不良/心肌病變(arrhythmogenic right ventricular dysplasia/cardiomyopathy, ARVD/C)通常於青春期變得有症狀,暈厥(syncope)是多數病例的首發表現。心室頻脈與因心律不整所致的猝死(這是 Naxos 病的主要死因,三分之一的病人早逝,平均死亡年齡為 32 歲)是常見併發症,右心衰竭的症狀通常出現於末期。隨著疾病進一步進展,亦可見左心室侵犯。
病因與致病機轉: ND 是一種體染色體隱性遺傳疾病,由編碼 PG 的 JUP 雙等位基因突變所引起。迄今最常報告的突變是一個同合子的 2-bp 缺失,導致早熟終止密碼子並表現出一個缺乏該蛋白 C 末端區域 56 個殘基的截短 PG。該突變的雜合子帶因者通常無皮膚或毛髮異常,雖然可能可見羊毛狀髮,且約 25% 的病例記載到輕微的心臟侵犯。攜帶其他導致殘餘 PG 表現之突變的帶因者,顯示出較輕微的 PPK、羊毛狀髮與皮膚脆弱表現型,無心肌病變證據。有趣的是,已在無 PPK 或毛髮異常證據的 ARVC/C 病例中,描述一個 PG N 末端區域有一個胺基酸插入的體染色體顯性突變。此外,已報告 JUP 基因的雙等位基因錯義突變伴隨類似 ND 之 PPK 與 ARVC/C,但以全身性禿髮取代羊毛狀髮。
PG 是 armadillo 蛋白家族的成員,是許多組織(包括心肌與表皮)中黏附連接(adherens)與橋粒連接的組成蛋白。橋粒鈣黏蛋白(desmosomal cadherins,橋粒糖蛋白 desmogleins 與橋粒膠蛋白 desmocollins)的胞內尾部,分別透過 armadillo 重複區域與胺基末端頭部區域與 PG 及斑菲素蛋白(plakophilins)連接。這兩個 armadillo 蛋白與橋粒斑蛋白(desmoplakin)交互作用,後者是一種將橋粒斑連接至角蛋白細胞骨架的 plakin 蛋白。
類 Naxos 表現型(ARVC 伴隨嚴重的左心室侵犯、PPK 與羊毛狀髮)也與編碼橋粒鈣黏蛋白橋粒膠蛋白 2(desmocollin 2)的 DSC2 基因同合子突變相關。此外,類似 ND 但無心肌病變證據的羊毛狀髮與 PPK,與 KANK2 的同合子錯義突變相關,KANK2 編碼控制類固醇受體活化的類固醇受體共活化因子交互作用蛋白(steroid receptor coactivator interacting protein)。
病理學: 掌蹠皮膚顯示符合 NEPPK 的發現。
伴隨皮膚鱗狀細胞癌與性別逆轉症候群之掌蹠過度角化症 (Palmoplantar Hyperkeratosis with Squamous Cell Carcinoma of Skin and Sex Reversal Syndrome)
臨床特徵: 伴隨皮膚 SCC 與性別逆轉的掌蹠過度角化(OMIM #610644)是一種體染色體隱性遺傳疾病,在來自南義大利的病人中描述。其特徵為指端硬化、伴多發皮膚 SCC 的 PPK、因慢性牙周病所致的早期牙齒喪失、伴縱向嵴狀的指甲發育不全、性腺機能低下(hypogenitalism,外生殖器模糊不清)、男性女乳症(gynecomastia)、伴隨血漿性激素濃度改變(低睪固酮與高濾泡刺激素 follicle-stimulating hormone)的尿道下裂(hypospadias),以及性別逆轉(男性表現型中的 46,XX 核型)。相關發現為高三酸甘油酯血症、喉癌、結節性睪丸增生、雙側白內障與雙側視神經缺損(optic nerve coloboma)。已在一例真兩性畸形(true hermaphroditism)的病例中描述類似的表現,該 46,XX 女性同時存在卵巢與睪丸組織。
病因與致病機轉: 此疾病源於編碼 R-spondin1 的 RSPO1 基因同合子突變。R-spondins 是與 Fzd–LRP 受體複合體交互作用的配體,對 β-catenin 穩定化很重要。由 PPK 分離出的角質形成細胞顯示纖維母細胞樣(fibroblast-like)型態與較大的細胞間隙,在器官型培養(organotypic cultures)中無法形成複層表皮層。
病理學: 掌蹠皮膚顯示符合 NEPPK 的正角化過度角化。
瀰漫型遺傳性掌蹠角化症,伴皮膚外特徵,粒線體遺傳 (DIFFUSE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, MITOCHONDRIAL INHERITANCE)
伴隨耳聾的掌蹠角化症 (Palmoplantar Keratoderma with Deafness)
臨床特徵: PPK 與 SNHL 的關聯,伴隨母系遺傳模式、由染色體外粒線體基因組(mitochondrial genome, mtDNA)突變所引起,已在源於紐西蘭、日本、法國與葡萄牙的數個家族中被鑑定。PPK 於兒童期出現(5 至 15 歲),特徵為橙黃色、瀰漫型、界線分明的掌蹠過度角化斑塊,伴極少至無紅斑。它主要侵犯壓力點,並可能逐漸進展。雖然較不常見,但已報告以蜂窩狀模式延伸至手足背側面,並可觀察到膝部、肘部與阿基里斯腱上方的角化斑塊。PPK 亦可能相當局灶,類似胼胝,並可能僅侵犯足底皮膚。已描述懷孕期間 PPK 的清除。聽力障礙可能於嬰兒期開始,但通常於 5 歲時出現,為雙側、輕至重度,並可能為進行性。不完全外顯率(incomplete penetrance)很常見,且對於聽力損失(60%)高於皮膚表現(37%)。兩性受影響程度相當。
病因與致病機轉: 所有受影響個體攜帶編碼 MTTS1 基因之粒線體轉送 RNA(transfer RNA, tRNA)中一個共同的同質性點突變(homoplasmic point mutation)(c.A7445G)。此突變導致絲胺酸 tRNA 濃度顯著下降,伴粒線體 mRNA 與蛋白的異常處理。
病理學: PPK 顯示正角化過度角化、局灶性角化不全、棘層肥厚與局灶性顆粒層減少。超微結構發現為大型角質透明顆粒,伴未附著於橋粒、位於細胞核周邊的張力絲束。
局灶型遺傳性掌蹠角化症,無皮膚外特徵,顯性遺傳 (FOCAL INHERITED PALMOPLANTAR KERATODERMA WITHOUT EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE)
條紋型掌蹠角化症 (Striate Palmoplantar Keratoderma)
臨床特徵: 條紋型掌蹠角化症(striate palmoplantar keratoderma, SPPK)是一種罕見的體染色體顯性遺傳疾病,可能源於 DSP、DSG1 或 KRT1 的突變,分別編碼橋粒斑蛋白、橋粒糖蛋白 1 或角蛋白 1。SPPK 的特徵為手掌上線狀、增厚的過度角化斑塊,沿指部掌側面延伸(圖 48-8A),以及足底上局限性的皮膚增厚區域(圖 48-8B)。該疾病最初於生命第一或第二個十年表現,典型因手工勞動而加劇。罕見可觀察到指甲異常(甲剝離、變色),並已報告膝部、足踝、指節與趾墊,以及指趾背側面的過度角化斑塊。無其他皮膚、毛髮或皮膚外表現。
DSG1 突變已被發現也會導致手掌與足底的瀰漫型與局灶型過度角化,無條紋病灶證據。
病因與致病機轉: SPPK 由 DSG1 或 DSP 基因的無義與框移雜合子突變所引起,劃分出兩種 SPPK 亞型:第 I 型(OMIM #148700)與第 II 型(OMIM #125647)。這些突變導致單倍體不足(haploinsufficiency),指示這些蛋白表現減少 50% 對於非掌蹠皮膚的表皮功能已足夠,但對於承受重大機械性創傷的部位(如手掌與足底)則不足。值得注意的是,致 SPPK 的 DSG1 突變最近已被證實以半顯性而非顯性的方式遺傳。DSG1 的雙等位基因突變造成皮膚炎、多重過敏與代謝性耗損(SAM)症候群(OMIM #615508)(圖 48-9)。因此,任何受 SPPK 影響的病人的遺傳諮詢都應考量這些資料。橋粒糖蛋白 1 與橋粒斑蛋白是橋粒斑的重要組成。橋粒糖蛋白 1 是一種屬於橋粒鈣黏蛋白家族的跨膜蛋白,表現於表皮上層,並在細胞間黏附與調節表皮增殖與分化的訊號轉導路徑中扮演關鍵角色。橋粒糖蛋白 1 的喪失導致促凋亡(proapoptotic)訊息傳遞下調、促進角質形成細胞增殖,並因無對抗的 ERK 訊息傳遞而抑制表皮分化,這可解釋 SPPK 中所見的過度角化表現型。此外,有人提出橋粒糖蛋白 1 缺乏經由升高的 Ras 活性導致 SPPK,這由 Ras–MAPK(絲裂原活化蛋白激酶 mitogen-activated protein kinase)路徑疾病中高比率的 PPK 所支持。
類似地,橋粒斑蛋白(一種在將中間絲錨定至橋粒鈣黏蛋白中扮演關鍵角色的斑蛋白)的喪失,已被證實導致細胞增殖增加,並增強細胞週期中由 G1 期至 S 期的進入,伴隨升高的磷酸化-ERK1/2 與磷酸化-Akt 濃度。
一個影響角蛋白 1 尾部區域的雜合子框移突變,被發現是 SPPK 第 III 型(OMIM #607654)的基礎。此突變導致 V2 區域中甘胺酸環基序(glycine loop motif)的部分喪失,並影響角化過程中橋粒斑的功能。
病理學: SPPK 的組織病理學發現為正過度角化、棘層肥厚、乳頭瘤病、細胞間隙加寬,以及表皮上層角質形成細胞的失黏附。雖然對 PPK 不完全特異或敏感,但後兩項發現作為診斷由編碼橋粒蛋白基因突變所致 PPK 的線索。免疫組織化學研究顯示角蛋白絲的異常核周聚集,伴隨 KRT16 上調與兜甲蛋白表現異常。超微結構發現為細胞間連接的鬆動,以及橋粒–角蛋白中間絲交互作用的破壞,伴核周分布的角蛋白絲團塊。可觀察到減少、外觀異常的橋粒結構。
Wachters 變異型遺傳性掌蹠角化症 (Hereditary Keratosis Palmoplantaris Variant of Wachters)
臨床特徵: Wachters 變異型遺傳性掌蹠角化症(PPK varians,Brünauer-Fohs-Siemens 症候群,Siemens 症候群)是一種罕見、具完全外顯率的體染色體顯性遺傳疾病,在男性中較常見,通常於生命第一或第二個十年出現。其特徵為黃色、非向外延伸、對稱、錢幣狀(nummular)的過度角化斑塊,局限於足底的壓力點,但可能隨時間變得更融合。手掌過度角化病灶被描述為線狀、錢幣狀、膜狀、有裂隙或甲周(periungual)。已描述肘部、膝部與阿基里斯腱上方的錢幣狀過度角化斑塊。可能伴隨掌蹠多汗症、疼痛性橫向裂隙與指甲變化(嵴狀與甲小皮過度角化)。已描述一例在足部過度角化病灶中發生惡性黑色素瘤的病例。伴有家族間與家族內變異性的不同表現型表達,導致不同亞型的描述,並引出 keratosis palmoplantaris varians 一詞,由 Wachters 於 1963 年所引入。
病因與致病機轉: Wachters 變異型遺傳性掌蹠角化症是一種體染色體顯性遺傳疾病,雖然已報告散發性病例。致病基因尚待鑑定。免疫組織化學研究顯示絲聚蛋白(filaggrin)與兜甲蛋白兩者的早期表現。
值得一提的是,目前尚不清楚 Wachters 變異型遺傳性掌蹠角化症與遺傳性疼痛性胼胝(hereditary painful callosities)是否為合理的不同疾病實體,或實際上是更常見形式 PPK(如 PC(KRT16 突變)或 EPPK(KRT1 突變))的突出病例。
病理學: 組織病理學發現包括過度角化、顆粒層增生、可能的局灶性角化不全與棘層肥厚,無表皮鬆解性變化或真皮發炎細胞浸潤。超微結構發現為緊密堆積的張力纖維與具異常構型的大團塊角質透明顆粒。
局灶型遺傳性掌蹠角化症,伴皮膚外特徵,顯性遺傳 (FOCAL INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE)
先天性厚甲症 (Pachyonychia Congenita)
臨床特徵: PC(OMIM #167200 與 167210)是一群罕見的體染色體顯性遺傳角化異常疾病,由五個角蛋白基因之一的突變所引起,包括 KRT6A、KRT6B、KRT6C、KRT16 或 KRT17。PC 於 20 世紀初首次被描述,雖然致病基因直至 1990 年代後期才被鑑定。PC 在西方國家的盛行率為每百萬人 0.9 例,全球 PC 人口估計在 5000 至 10,000 人之間。臨床特徵的嚴重度在家族之間與家族內部各異,因為表現型可能受修飾基因或環境因素影響。
歷史上,PC 被分為兩種亞型:PC-1(Jadassohn-Lewandowski),具有口腔白色角化症,由 KRT6A 或 KRT16 突變所引起;以及 PC-2(Jackson-Lawler),由 KRT6B 或 KRT17 突變所引起,具有囊腫與新生兒齒。然而,對國際 PC 研究登錄(International PC Research Registry)中超過 1000 名突變確認 PC 病人的表現型特徵研究,顯示這些亞型之間有相當大的重疊,並導致一個基於突變基因的新分類方案。臨床上懷疑 PC 但在已知 PC 相關基因中未鑑定出突變的病例,稱為 PC-U(unknown,未知)。
在超過 90% 的 PC 病人中報告的三項臨床特徵是增厚的足趾甲、足底角化症與足底疼痛。增厚的足趾甲(賦予該疾病名稱的表現型特徵)於兒童早期出現,但也可能於生命最初幾週發生(尤其是 KRT6A 突變)。厚甲(pachyonychia)的特徵為顯著的甲下過度角化與非常厚的指甲,具有特徵性的倒 U 形或 V 形,生長至全長或早熟終止(圖 48-10A)。可見伴隨葡萄球菌與念珠菌重複感染的甲溝炎。平均而言,9 個足趾甲受侵犯,而 KRT6A 突變具有 10 個足趾甲皆受影響的最高可能性。指甲侵犯各異,即使在攜帶相同突變的家族成員之間亦然。手指甲雖然據報告在許多病人中受侵犯(KRT6A 突變病人中 100%),但可能倖免,尤其是 KRT6B 突變。足底角化症最常見的表現是局灶型 PPK(focal PPK, FPPK),於承重區域上有胼胝(圖 48-10B),雖然可觀察到瀰漫型角化症。水皰、裂隙與開放性瘡(open sores)很常見。手掌病灶較不顯著,通常因職業性機械性創傷而發生。PPK 的平均發病年齡為 4 歲,雖然它可能於出生時出現或延遲發病(30 歲)。FPPK 在孩童開始行走時開始是非常典型的。KRT16 與 KRT6A 突變病人比 KRT6B 與 KRT17 突變病人更早發展角化症。雖然 PC 典型造成非向外延伸的 FPPK,但已描述數例帶 transgrediens 與足背侵犯的病例(多為 KRT6A 突變)。足底疼痛發生於所有亞型,在較溫暖的天氣可能更嚴重,並被認為是影響病人生活品質最重要且最致殘的特徵。此嚴重疼痛繼發於厚胼胝深處下方的水皰形成,如高頻超音波所示。約 60% 的 PC 病人顯示神經病變性疼痛(neuropathic pain)特徵,其餘則存在傷害感受性疼痛(nociceptive pain)。
其他特徵為黏膜侵犯、囊腫形成與新生兒齒。口腔白色角化症在 70% 的病人中被報告。它傾向於 5 歲時出現,雖然約半數病人報告出生時發病,且最典型於 KRT6A 或 KRT17 突變。它可能是新生兒 PC 的首發表現,常被誤認為念珠菌症,並可能改變吸吮或進食,造成生長遲滯(failure to thrive)。約 40% 的病人(多為 KRT17 突變)表現出各種類型的表皮內含囊腫(epidermal inclusion cysts)。毛囊皮脂腺囊腫(如脂囊腫 steatocystomas(圖 48-10C)與毳毛囊腫 vellus hair cysts)最常見,通常於青春期發展,持續至整個成年期,並可能需要手術移除。面部與軀幹受影響。粉刺樣囊腫(comedone-like cysts)可能見於腋窩,主要在兒童中。軀幹與四肢上的毛囊角化症(主要在摩擦區域如膝部與肘部)可在約 50% 的病人中觀察到,通常於兒童早期出現。新生兒齒現象(出生時或 1 個月時已萌發的牙齒)在 15% 的 PC 病人中被報告,幾乎專屬於 KRT17 突變。乳齒與恆齒列正常。較不常見的相關特徵為掌蹠多汗症、口角炎(angular cheilitis)與角膜失養。在兩例 KRT17 同合子半顯性錯義突變的病例中描述了禿髮。在 KRT6A 突變的嬰兒與兒童中,已見繼發於喉部白色角化症的聲音嘶啞,伴危及生命的呼吸窘迫。此外,KRT6A 突變的嬰兒與兒童可能在首次吸吮或進食時表現出耳前的嚴重疼痛,持續數秒。
在源於 KRT16 突變的 PC 中,輕微的 FPPK 是普遍的發現;然而,許多 KRT16 突變產生較不顯著的表現型,FPPK 局限於足底的承重區域,指甲侵犯有限或缺如,且無口腔白色角化症證據,導致診斷為局灶型 NEPPK 而非 PC。
KRT6C 突變已被報告與類似某些 KRT16 突變的較輕微表現型相關。這些病例的特徵為足底角化症(壓力部位的 FPPK 或瀰漫型 PPK)與手掌上的胼胝,伴缺如或輕微的指甲變化,如第五足趾甲的肥大與足底水皰。可見白色角化症,但無額外的臨床發現。PC 與相關突變的主要臨床特徵總結於表 48-3。
(以下對應原文表 48-3 之內容。)
| 基因 (GENE) | 頻率 (%) (FREQUENCY) | 常見突變 (COMMON MUTATION(S)) | 主要臨床特徵 (KEY CLINICAL FEATURES) |
|---|---|---|---|
| KRT6A | 40 | p.Asn172del | 自嬰兒期起增厚的足趾甲與手指甲;具 10 個足趾甲皆受影響的最高可能性;FPPK 較早發病;可能發生 transgrediens 與足背侵犯;口腔白色角化症;喉部白色角化症伴聲音嘶啞與危及生命的呼吸窘迫 |
| KRT16 | 25–30 | p.Arg127Cys(輕微)、p.Arg127Pro(嚴重)、p.Leu128Gln(輕微)、p.Leu128Pro(嚴重) | 輕微 FPPK,指甲侵犯有限或缺如;可能較早發病的 FPPK;共同遺傳 FLG 突變會加重表現型 |
| KRT17 | 20 | p.Asn92Ser | FPPK 延遲發病;口腔白色角化症;表皮內含囊腫(脂囊腫、毳毛囊腫);新生兒齒;禿髮ᵃ |
| KRT6B | 5–9 | 手指甲可能倖免;FPPK 延遲發病 | |
| KRT6C | 3 | 多為輕微 FPPK 與手掌上的胼胝,伴缺如或輕微的指甲變化ᵇ;可見白色角化症而無額外臨床特徵 |
ᵃ僅見於一例同合子半顯性遺傳的病例。
ᵇ較輕微的表現型可能是角蛋白 6c 在掌蹠皮膚與指甲中的表現,相較於角蛋白 6a 與 6b 更為受限的結果。已報告一般人群中的 KRT6C 突變帶因者。FPPK:局灶型掌蹠角化症(focal palmoplantar keratoderma)。
病因與致病機轉: PC 由五個表現於分化上皮組織之角蛋白基因之一的雜合子突變所引起:KRT6A、KRT6B、KRT6C、KRT16 與 KRT17,分別編碼角蛋白 6a、6b 與 6c(第 II 型角蛋白)以及角蛋白 16 與 17(第 I 型角蛋白)。角蛋白 6 與 16 的表現在正常表皮中局限於上外側毛根鞘與甲床、掌蹠皮膚,以及基底上層的口腔生殖器黏膜角質形成細胞。
此外,受傷或發炎會誘導它們在毛囊間表皮中的表現,且它們在來自正常與乾癬皮膚的培養角質形成細胞中以高量表現。
超過半數的病例觀察到體染色體顯性遺傳;其餘病例由自發性顯性突變所引起。
有少數明顯隱性 PC 遺傳的病例報告。然而,沒有經基因檢測確認的此類病例,且這些病例中即使不是全部、大多數也可能代表 PC 的表型擬態(phenocopies)。
如前所述,已報告 KRT17 突變的半顯性遺傳。亦已描述 KRT6A 突變的父系生殖細胞鑲嵌。多數突變為錯義突變,其餘為可能施加顯性負向效應的小型框內缺失、框移、無義或剪接位點突變。迄今報告的多數突變涉及位於 α 螺旋桿狀區域兩端的高度保守螺旋邊界區域,這對絲狀體組裝的延長階段至關重要。
由 PC 受侵犯皮膚取得的皮膚切片顯示 PC 相關基因 KRT6、KRT16 與 KRT17 顯著被誘導的表現,這些基因已知在壓力或損傷狀態下被上調。機械性壓力可能導致這些角蛋白的野生型與突變型兩者皆被上調,並因突變角蛋白的併入而擾亂中間絲形成,觸發 PC 表現型。這反過來與額外突變角蛋白作為傷口癒合反應一部分的異常表現相關,導致逐漸惡化的惡性循環。此外,已觀察到與細胞黏附(DSC2、CDSN 與 GJB2)、角化細胞外套膜形成(兜甲蛋白與 loricrin)與脫屑(激肽釋放酶 kallikrein [KLK]-5、SPINK6 與 SERPINs)相關基因,以及非 PC 相關角蛋白(KRT75 與 KRT85)的過度表現。疼痛相關基因在 PC 受侵犯皮膚中也被上調,包括 KLK10(催化緩激肽 bradykinin 的產生)與 SPRR1A(角質形成細胞中的一種結構蛋白,也由神經元表現),這可能與 PC 病人所具有的嚴重疼痛有關。
病理學: 由 PPK 病灶取得的皮膚切片顯示 NEPPK 的特徵。可見 HFs 外根鞘的顆粒層減少與細胞溶解。在一例伴隨 KRT16 突變、具有條紋狀手掌過度角化與瀰漫型足底 PPK 的病例中,觀察到表皮鬆解型過度角化。超微結構上,PC 中基底上層角質形成細胞的特徵為緻密聚集的角蛋白絲束,主要在核周區域,並倖免細胞周邊。橋粒數量減少伴細胞間隙加寬,以及角質透明顆粒減少,是額外的發現。
特定治療: 關於類視黃醇在 PC 中療效的矛盾資料已被發表,部分報告顯示 PPK 與甲失養兩者皆改善,其他則無改善。此外,在許多觀察到改善的病人中,治療不得不停止,因為表皮變薄與感染易感性所致的疼痛增加。
在 PC 中,處理甲失養最有效的方法包括機械性治療(銼磨、研磨、切割或修剪)與浸泡指甲。手術拔除(surgical avulsion)有時無效,因為指甲會再生長。口腔白色角化症可能因保持良好口腔衛生、輕柔刷牙與口服抗生素而改善。毛囊過度角化以口服與外用類視黃醇、角質溶解劑與潤膚劑治療。多發性脂囊腫(steatocystoma multiplex)與其他毛囊皮脂腺囊腫可藉由切開與引流、切除、病灶內類固醇注射,以及在續發性感染情況下使用口服抗生素來治療。最近,已在 PC 中研究標靶與更具特異性的療法。雷帕黴素(rapamycin)已被證實可選擇性阻斷包括 KRT6 mRNA 在內的 mRNA 轉譯,從而顯著改善疼痛性足底胼胝與生活品質。然而,口服雷帕黴素的使用受副作用所限,包括腸胃道症狀。一項使用外用雷帕黴素的臨床試驗正在進行中。此外,一種選擇性靜默 KRT6A 中特定致病突變表現的特異性 siRNA,已在第 Ib 期臨床試驗中被證實導致胼胝消退與疼痛控制。需要更有效、安全且實用的方式將 siRNA 遞送至表皮,以避免疼痛的注射。
Howel-Evans 症候群 (Howel-Evans Syndrome)
臨床特徵: Howel-Evans 症候群(伴食道癌的胼胝病 tylosis with esophageal cancer;OMIM #148500)是一種罕見、具完全外顯率的體染色體顯性遺傳疾病,特徵為 PPK 與黏膜(特別是食道)SCC 的關聯。它由 Howel-Evans 與同僚於 1958 年首次報告,並已在一系列國家的家族中被描述。該疾病在一般人群中的估計盛行率低於每 100 萬人 1 例。PPK 的發病通常於兒童期或青春期(5 至 15 歲之間,雖然多數病例於 7 至 8 歲時明顯)。其特徵為局灶型、黃色、厚斑塊,局限於手掌與足底的壓力或摩擦區域(見圖 48-7B),可能伴隨疼痛性裂隙與續發性感染。可能明顯倖免手掌。其他發現為毛囊過度角化、皮角(cutaneous horns)與口腔白色角化症。已記載口咽 SCC 的病例。
食道病灶表現為遍布食道、幾毫米大小、白色、息肉樣的病灶。約 95% 的 Howel-Evans 症候群病人於 65 歲時發展癌症,與散發性病例的發病相似。
在 Howel-Evans 症候群病人中,篩檢包括每年的胃鏡檢查。除了監測外,建議調整飲食與生活方式以降低食道癌的危險因子。
病因與致病機轉: Howel-Evans 症候群源於 RHBDF2 的 GOF 錯義突變,該基因編碼一種催化無活性的菱形跨膜絲胺酸蛋白(rhomboid intramembrane serine),iRhom2。iRhom2 屬於一個七次跨膜蛋白家族,這些蛋白是與 EGFR 訊息傳遞及粒線體重塑相關的絲胺酸跨膜蛋白酶(intramembrane proteases)。RHBDF2 是一種在演化過程中喪失其蛋白酶活性、但保留關鍵非蛋白酶功能(調節 EGF 與 TNF-α 訊息傳遞路徑)的 iRhom。
由 Howel-Evans 症候群病人取得的皮膚切片,相較於正常皮膚所見的正常膜表現,顯示 iRhom2 的胞質定位。在 Howel-Evans 症候群食道癌與散發性鱗狀食道腫瘤取得的切片中,觀察到類似的胞質定位,提示 iRhom2 的失調在這些惡性腫瘤中扮演角色。
iRhom2 已被證實調節 ADAM17 的運送與活化,ADAM17 是一種膜結合脫落酶(sheddase),已被證實在受質自細胞表面的蛋白水解切割與釋放中扮演關鍵角色,包括 TNF-α、EGF 生長因子家族成員與橋粒。來自 Howel-Evans 症候群病人的表皮角質形成細胞顯示上調的 ADAM17 活性,導致 EGFR 活性增加、橋粒處理增加、不成熟的表皮橋粒、上調的表皮轉麩醯胺酸酶活性,以及對葡萄球菌感染的抗性。此外,這些角質形成細胞在體外顯示傷口修復失調的特徵。
EGFR 的過度表現已在散發性食道 SCC 與數種其他癌症中被證實,並與較低的存活率相關。此外,ADAM17 的表現已被證實與食道癌的進展相關。
因此,Howel-Evans 症候群中所見的癌前食道病灶,可能是 EGFR 訊息傳遞失調的結果。
病理學: 受影響皮膚顯示棘層肥厚、正過度角化與顆粒層增生,無角化不全或海綿狀水腫(spongiosis)。
局灶型遺傳性掌蹠角化症,伴皮膚外特徵,隱性遺傳 (FOCAL INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, RECESSIVE INHERITANCE)
Richner-Hanhart 症候群 (Richner-Hanhart Syndrome)
臨床特徵: Richner-Hanhart 症候群(Richner-Hanhart Syndrome, RHS,第 II 型酪胺酸血症 tyrosinemia type II、眼皮膚酪胺酸沉著症 oculocutaneous tyrosinosis、伴角膜失養的掌蹠角化症;OMIM #276600)是一種罕見的體染色體隱性遺傳疾病,以 Richner 與 Hanhart 的最初報告命名。其發生率低於每 25 萬人 1 例,發生於各種族群中,雖然似乎在地中海國家與阿拉伯世界特別盛行。
症狀典型於兒童早期出現,包括三聯徵:疼痛性 PPK、雙側角膜炎與智能不足,這源於酪胺酸結晶的胞內累積與受侵犯組織中的續發性發炎反應。眼部表現包括畏光、疼痛、流淚、發紅與假皰疹樣角膜炎(pseudoherpetiform keratitis)伴角膜潰瘍。眼部表現發生於 75% 的病例,於出生後不久或生命第一年內出現,通常先於皮膚表現。皮膚表現通常於生命第一年後開始,包括界線分明、局灶型、掌蹠、白黃色的過度角化斑塊,圍繞著紅斑,位於足底的承重區域。有相關的疼痛且常伴多汗症。指尖、小魚際(hypothenar)與大魚際隆起也可能受影響。60% 的病例,特別是未治療者,呈現神經學表現,包括智能不足、眼球震顫(nystagmus)、震顫、運動失調(ataxia)與抽搐。已報告一例伴隨癲癇發作、輕度智能不足與畏光,無其他眼科或皮膚表現的病例。
病因與致病機轉: RHS 是一種體染色體隱性遺傳疾病,由編碼酪胺酸轉胺酶(tyrosine aminotransferase, TAT)的 TAT 基因突變所引起。由於 TAT 缺乏,酪胺酸在組織中累積。已在 TAT 基因內鑑定出超過 20 個突變,改變酪胺酸轉胺酶酵素的活性與穩定性。有人假設疾病嚴重度(特別是神經學障礙)與突變蛋白的存在相關,但其完全缺如則與較有利的特徵相關。
先前的報告提示 RHS 中的掌蹠病灶源於胞內 L-酪胺酸結晶,這些結晶使溶酶體膜(lysosomal membranes)不穩定並啟動一連串細胞損傷與發炎,導致典型的皮膚病灶。此假說與大鼠角膜上皮的酪胺酸誘導病灶以及病人表皮棘層角質形成細胞中存在結晶的情況一致。然而,後來的研究未能證實表皮角質形成細胞中的結晶,並提示掌蹠過度角化源於酪胺酸過高的胞內濃度,導致角蛋白之間的非共價交聯與聚集張力絲的形成。
實驗室發現: RHS 藉由高濃度的血清酪胺酸(伴正常的苯丙胺酸)與尿液中酪胺酸代謝物(對羥基苯丙酮酸 p-hydroxyphenylpyruvate、對苯乙酸 p-phenylacetate、對羥基苯乙酸 p-hydroxyphenylacetate)的累積來診斷。穩定濃度的尿液琥珀醯丙酮(succinylacetone)可區分 RHS 與第 I 型酪胺酸血症。
病理學: RHS 的組織病理學發現包括顯著的棘層肥厚、過度角化與顯著的顆粒層增生。角化不全、肢端汗管中的角化不全柱,以及棘層中的多核角質形成細胞是可能的發現。可見基底上層的有絲分裂活性增加,以及細薄、延長的表皮嵴。已報告胞內酪胺酸結晶。超微結構發現包括顆粒層的增厚與張力纖維及角質透明物質的合成增加、大量微管(microtubules),以及具有管狀通道或微管內含物的張力纖維團塊。可觀察到棘層中的多核角質形成細胞與角化層中的脂質小滴。
特定治療: RHS 處置的關鍵是早期診斷與早期開始限制酪胺酸與苯丙胺酸的飲食,以降低高酪胺酸血症(hypertyrosinemia)長期併發症的風險與嚴重度,特別在眼睛與皮膚。針對先天性代謝異常的串聯質譜(tandem mass spectrometry)新生兒篩檢,可在無症狀的新生兒期鑑定 RHS。類視黃醇改善皮膚與眼睛病灶,但它們無法預防智能不足,並應在兒童中審慎考量。在一例將大腿皮膚移植至足底病灶的病例中,移植區域倖免過度角化,並在其周圍形成角化壁。
Carvajal 症候群 (Carvajal Syndrome)
臨床特徵: Carvajal 症候群(Carvajal syndrome, CS;OMIM #605676)由 Carvajal-Huerta 於 1998 年首次報告,是一種體染色體隱性遺傳疾病,由 DSP 基因(編碼橋粒斑蛋白)的突變所引起,特徵為三聯徵:羊毛狀髮、SPPK 與心肌病變。左心室擴張型心肌病變(left ventricular dilated cardiomyopathy)於兒童期發展;右心室亦可能受侵犯。病人可能僅表現皮膚或心臟特徵,或完整的表現型。羊毛狀髮於出生時出現,並可能伴隨稀疏的眉毛與腋毛及陰毛。SPPK 於兒童期發展,雖然已報告出生時出現的病例。已描述一例具有頭皮、眉毛與睫毛禿髮的病例。其他皮膚特徵為屈側皺褶中的線狀苔癬樣角化丘疹、肘部與膝部上方或散布於腹部與下肢的毛囊過度角化、指甲異常(失養、甲彎曲、杵狀指)、短暫的搔癢性水皰,以及乾癬樣斑塊。具顯性遺傳的 CS 病例伴隨齒少(hypo- 或 oligodontia)與白甲症或脆甲。已報告與先天性單側耳聾、反覆咽炎與腹瀉的共同發生。在超過 90% 的病人中,左心室於生命第二個十年嚴重受影響,雖然已報告早至生命最初幾年表現進行性心衰竭症狀的心肌病變。充血性心衰竭與心室心律不整是青春期最常見的死因。
病因與致病機轉: 橋粒斑蛋白是橋粒的核心組成,橋粒是單純與複層鱗狀上皮及心肌中的細胞間連接,將中間絲連接至胞質膜中的橋粒鈣黏蛋白。DSP 的 N 末端 plakin 區域結合 PG、斑菲素蛋白與橋粒鈣黏蛋白,其 C 末端區域連接細胞骨架中間絲,而其中央的捲曲螺旋桿狀區域負責同二聚化(homodimerization)。DSP 表現於所有表皮層、外根鞘、伴隨層(companion layer)以及 HF 的 Henle 層與 Huxley 層;間盤(intercalated discs)對心臟中心肌細胞的結合很重要。
導致 CS 的第一個 DSP 同合子突變在一個厄瓜多家系中被描述,導致一個缺乏負責中間絲結合之 C 區域尾部的截短蛋白。另一個與兒童早期水皰及青春期 ARVC 相關的隱性錯義突變,在阿拉伯家系中被描述,並影響該蛋白的 C 末端區域。已描述數個其他導致涉及捲曲螺旋桿狀區域與 C 末端區域之截短蛋白、伴中間絲結合受損的同合子突變。
除了 CS 常見的三聯徵外,已在牙齒缺如(tooth agenesis)的病例中報告 DSP 的顯性雜合子突變(擴張型心肌病變伴羊毛狀髮、角化症與牙齒缺如;dilated cardiomyopathy with wooly hair, keratoderma, and tooth agenesis, DCWHKTA;OMIM #615821)。在這些病例中,突變影響 DSP 的 N 末端區域,且多數涉及第 14 外顯子,提示此區域作為熱點並以顯性負向方式破壞橋粒支架。牙齒缺如的臨床特徵與小鼠研究一致,後者證實橋粒蛋白在牙齒發育中的角色。
除了 DCWHKTA 外,雜合子 DSP 突變與第 8 型 ARVD(OMIM #607450)及孤立性 SPPK(見前文)有關。同合子或複合雜合子 DSP 突變也導致致命性棘層鬆解性表皮分解性水皰症(lethal acantholytic epidermolysis bullosa;OMIM #609638)與皮膚脆弱–羊毛狀髮症候群(skin fragility–woolly hair syndrome;OMIM #607655)。
病理學: CS 的組織病理學發現為過度角化、乳頭瘤病、海綿狀水腫、表皮鬆解型過度角化與角化不良。棘層中可見棘層鬆解(acantholysis)。
點狀型遺傳性掌蹠角化症,無皮膚外特徵,顯性遺傳 (PUNCTATE INHERITED PALMOPLANTAR KERATODERMA WITHOUT EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE)
點狀型掌蹠角化症第 I 型 (Punctate Palmoplantar Keratoderma Type I)
臨床特徵: 點狀型掌蹠角化症(punctate palmoplantar keratoderma, PPKP)第 I 型(PPKP1;Buschke-Fischer-Brauer 型點狀掌蹠角化症 keratosis punctate palmoplantaris type Buschke-Fischer-Brauer;OMIM #148600 [PPKP1A] 與 614936 [PPKP1B])是一種罕見的體染色體顯性遺傳疾病。PPKP1 的估計發生率在歐洲、中東與亞洲各族群中約為每 10 萬人 1 至 3 例。
該疾病於生命第一或第二個十年發病,雖然亦已描述較晚的發病。其特徵為多發、過度角化、中央凹陷、黃至褐色的丘疹,不規則地分布於掌蹠皮膚(圖 48-11)。病灶可能疼痛。它們隨年齡增長在大小與數量上增加,並融合成更融合的斑塊,特別在足底皮膚的承壓區域。已描述一種類似人類乳突病毒(human papilloma virus)誘導之乳突瘤樣病灶的表現型。
掌蹠病灶的皮膚鏡(dermoscopic)發現為界線分明、無結構、黃橙色的區域,被白色光暈所環繞,無尋常疣中常見的點狀血管(dotted vessels)證據。
病灶在伍氏燈(Wood light)檢查下顯示白色螢光。
病因與致病機轉: PPKP1 是一種體染色體顯性遺傳疾病,源於兩個基因的雜合子突變:AAGAB(PPKP1A)與 COL14A1(PPKP1B)。AAGAB 編碼 α- 與 γ-接合素結合蛋白 p34(α-and γ-adaptin-binding protein p34),其含有一個接合素結合區域與一個類 Rab GTP 酶(Rab-like GTPase)區域,後者作為伴護蛋白(chaperone)可能在網格蛋白被覆囊泡(clathrin-coated vesicle)運送與皮膚完整性中扮演重要角色。
家族間與家族內的表現型變異性很常見,可能與修飾基因或老化與環境因素的效應有關。已報告遺傳預期(genetic anticipation,後代世代更早發病)。迄今,已鑑定出超過 30 個 AAGAB 的無效變異(null variants),並提出單倍體不足作為可能的致病機轉。
在一個患有 PPKP1 且具不完全外顯率的大型中國家族中,鑑定出編碼第 XIV 型膠原蛋白 α1 鏈的 COL14A1 中一個錯義突變。該蛋白主要表現於分化良好的組織與胚胎發育後期,且所報告的突變涉及膠原蛋白三股螺旋重複區域(collagen triple helix repeat region)。
病理學: PPPK 的組織病理學發現包括顯著的過度角化與棘層肥厚,伴表皮內陷(invagination),並伴隨局灶性角化不全、顆粒層增生或減少,以及上覆的正角化。病灶足底皮膚的穿透式電子顯微鏡顯示基底表皮層中接近細胞膜處異常豐富的小囊泡,以及高基氏體(Golgi apparatus)的顯著擴張,符合囊泡運輸的缺陷。
點狀型掌蹠角化症第 2 型 (Punctate Palmoplantar Keratoderma Type 2)
臨床特徵: PPKP 第 II 型(PPKP2;汗孔角化型 porokeratotic type;OMIM #175860)是一種體染色體顯性遺傳疾病,特徵為多發、緊密附著、微小、膚色至黃色、無症狀的角化棘狀物(keratotic spines),於青春期前後或 20 歲出頭時出現於手掌與足底。病灶可能隨數年增加數量,並可能延伸至手指的背側與外側面。病灶在伍氏燈檢查下顯示類似「月光下繁星」的白色螢光。
男性病人報告了面部皮脂腺發育不全。已描述後天性、具有類似表現型、伴隨內臟惡性腫瘤的病例。
病因與致病機轉: PPKP2 的分子病因未明。多數所描述的病例為後天性;然而,亦有具體染色體顯性遺傳的家族性病例。其致病機轉涉及柱狀角化不全(columnar parakeratoses)下方基底層的表皮增生增強。
病理學: PPKP2 的組織病理學發現為角質層中緻密的垂直角化不全柱,上覆於顆粒層減少的表皮,類似角樣板層(cornoid lamellae)。超微結構發現為角質層中眾多、大小不一的固縮核(pyknotic nuclei),以及顆粒層中角質透明顆粒數量的減少。
點狀型掌蹠角化症第 3 型 (Punctate Palmoplantar Keratoderma Type 3)
臨床特徵: PPKP 第 III 型(PPKP3,AKE [of Costa];OMIM #101850)是一種罕見的體染色體顯性遺傳疾病,通常於兒童期或青春期出現,雖然亦已報告嬰兒期或成年期發病。無種族或族裔偏好。AKE 的特徵為無症狀、圓至橢圓形、白至黃色且半透明的丘疹,較罕見為結節與斑塊,具有過度角化表面或臍凹(umbilication)。變化位於手掌與足底,好發於手的大魚際與小魚際區域以及手掌與足底的壓力部位。可見手部橈側與尺側邊緣上的線狀模式或「鋪石」排列(圖 48-12)。在嚴重病例中,病灶可能影響手足背側面(包括指節墊)、手腕與足踝。可伴隨水源性 PPK,並已描述單側侵犯。可觀察到甲失養與多汗症。雖然 AKE 通常局限於皮膚,但已報告一例中型與大型動脈彈性降低,並伴隨全身性與結節性硬皮症的病例。
(以下對應原文圖中嵌入的演算法/表格文字片段。)
TRPV3、MBTPS2(X 染色體連鎖病例)、TRPV3
瀰漫型 PPK(Diffuse PPK)
(續)
RSPO1、R-spondin1
(續)
伴隨皮膚 SCC 與性別逆轉症候群之掌蹠過度角化症(Palmoplantar hyperkeratosis with SCC of skin and sex reversal syndrome)
點狀型 PPK(Punctate PPK)
局灶型 PPK(Focal PPK)
焦磷酸酶/磷酸二酯酶 1(tase/phosphodiesterase 1)
局灶型肢端過度角化(focal acral hyperkeratosis, FAH)是一種體染色體顯性遺傳疾病,表現型與 AKE 相似,包括指甲異常。然而,這些病例中未觀察到彈性組織異常。FAH 常被認為是 AKE 的變異型,雖然此概念不被部分作者所接受。
病因與致病機轉: PPKP3 是一種體染色體顯性遺傳疾病,雖然已描述散發性病例。該疾病的遺傳基礎尚待闡明,雖然初步的連鎖研究提示在 2p25-p12 上可能有一個基因座。
超微結構研究支持 AKE 涉及彈性物質分泌的缺陷與彈性纖維合成的失敗,而非彈性纖維的退化。此外,角化丘疹的形成是絲聚蛋白過度產生的結果,其在併入 CCE 之前累積於顆粒層之上。
病理學: AKE 病灶顯示正角化過度角化、棘層肥厚與顆粒層增生。病灶與外觀正常的皮膚兩者皆顯示網狀真皮中彈性組織減少,以及破碎、增厚或變薄的彈性纖維(彈力纖維變性 elastorrhexis),並倖免乳頭層真皮。超微結構發現為胞質周邊具緻密顆粒的纖維母細胞,伴彈性纖維的細胞外含量減少。
點狀型遺傳性掌蹠角化症,伴皮膚外特徵,顯性遺傳 (PUNCTATE INHERITED PALMOPLANTAR KERATODERMA WITH EXTRACUTANEOUS FEATURES, DOMINANT INHERITANCE)
Cole 病 (Cole Disease)
臨床特徵: Cole 病(OMIM #615522)是一種罕見的體染色體顯性遺傳疾病,於出生時或嬰兒早期表現,特徵為點狀型 PPK 與不規則形、色素減少的斑(macules),分布於近端四肢或較不典型地分布於軀幹。已描述數個器官(肌腱、乳房與脾臟)的鈣化。
病因與致病機轉: Cole 病源於 ENPP1 基因的雜合子突變,該基因編碼外核苷酸焦磷酸酶/磷酸二酯酶 1(ectonucleotide pyrophosphatase/phosphodiesterase 1),這是一種細胞表面蛋白,藉由催化三磷酸腺苷(ATP)水解為單磷酸腺苷(AMP)來生成細胞外無機焦磷酸(inorganic pyrophosphate)。ENPP1 由八個區域組成,包括磷酸二酯酶、核酸酶與類生長調節素 B(somatomedin B–like, SMB)區域。影響媒介 ENPP1 催化活性之磷酸二酯酶區域或核酸酶區域的雙等位基因突變,已被證實與具有異常鈣恆定或異位鈣化特徵的遺傳性疾病相關,而與 Cole 病相關的突變影響聚集於 SMB 區域內的高度保守半胱胺酸殘基。鑑於 ENPP1 已被證實透過其 SMB 區域與胰島素受體之間的交互作用抑制胰島素訊息傳遞,有人提出異常的胰島素訊息傳遞在 Cole 病的致病機轉中扮演角色。
病理學: 由掌蹠病灶取得的皮膚切片顯示過度角化、正角化、顆粒層增生與棘層肥厚。可見真皮中的鈣化沉積。色素減少斑顯示過度角化、角質形成細胞中黑色素含量減少,以及正常或減少的黑色素細胞數量。色素減少病灶的超微結構發現包括黑色素細胞胞質與樹突中數量不成比例龐大的黑色素體(melanosomes),而相鄰角質形成細胞中黑色素體稀少,提示異常的黑色素體轉移是 Cole 病色素減少的主要病理機轉。
總結 (SUMMARY)
見表 48-4。
圖表 (Figures and Tables)
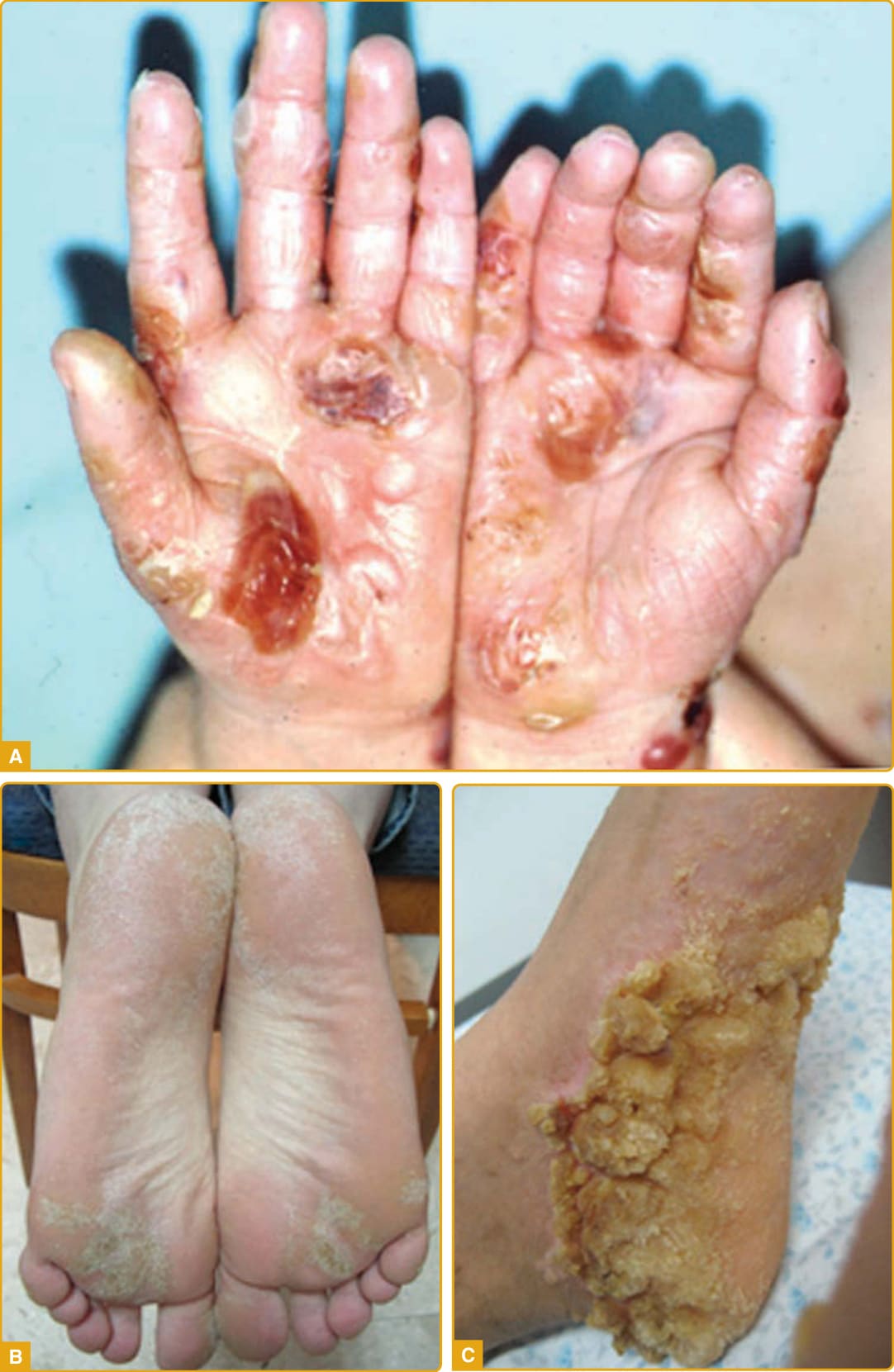
圖 48-1:以掌蹠角化症為相關特徵的遺傳性皮膚病 (Genodermatoses with palmoplantar keratoderma as an associated feature)。A,一名由 KRT1 突變所致表皮鬆解型魚鱗癬 (epidermolytic ichthyosis) 病人,呈現伴水皰的瀰漫型手掌角化症。B,一名 Conradi-Hünermann-Happle 症候群病人,於承重區域上呈現局灶型過度角化足底斑塊。C,一名達瑞氏病 (Darier disease) 病人,足底皮膚上呈現厚、黃、過度角化斑塊。
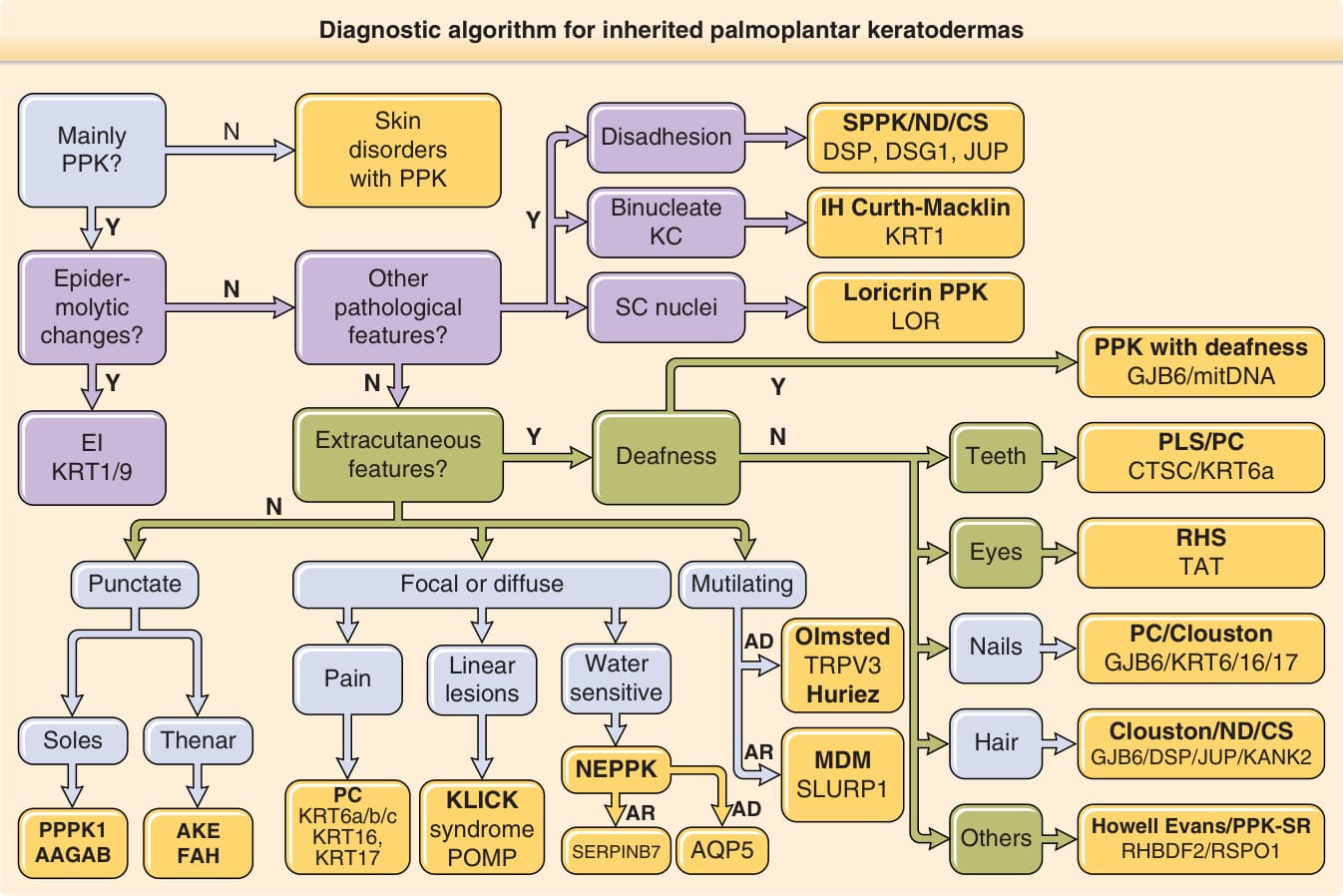
圖 48-2:遺傳性掌蹠角化症 (palmoplantar keratodermas, PPKs) 的診斷演算法。多數遺傳性 PPK 病人可基於四項標準被指派至一個基因:皮膚發現、皮膚外表現、遺傳模式與組織病理學發現。AAGAB,α- 與 γ-接合素結合蛋白 P34;AKE,肢端角化彈力纖維變性症 (acrokeratoelastoidosis);AQP5,水通道蛋白 5 (aquaporin 5);CS,Carvajal 症候群;CTSC,組織蛋白酶 C (cathepsin C);DSG1,橋粒糖蛋白 1 (desmoglein 1);DSP,橋粒斑蛋白 (desmoplakin);EI,表皮鬆解型魚鱗癬 (epidermolytic ichthyosis);FAH,局灶型肢端過度角化 (focal acral hyperkeratosis);GJB6,間隙連接蛋白 β6 (gap junction protein β6);IH,豪豬狀魚鱗癬 (ichthyosis hystrix);JUP,血漿斑珠蛋白 (plakoglobin);KANK2,KN motif and ankyrin repeat domains 2;KC,角質形成細胞 (keratinocytes);KLICK,線狀角化–先天性魚鱗癬–角化症 (Keratosis linearis–ichthyosis congenital–keratoderma);KRT,角蛋白 (keratin);LOR,loricrin;MDM,mal de Meleda;ND,Naxos 病 (Naxos disease);NEPPK,非表皮鬆解型掌蹠角化症 (nonepidermolytic palmoplantar keratoderma);PC,先天性厚甲症 (pachyonychia congenita);PLS,Papillon-Lefevre 症候群;POMP,蛋白酶體成熟蛋白 (proteasome maturation protein);PPK-SR,伴性別逆轉之掌蹠角化症 (palmoplantar keratoderma with sex reversal);PPPK1,點狀型掌蹠角化症第 I 型 (punctate palmoplantar keratoderma type I);RHBDF2,rhomboid 5 homolog 2;RHS,Richner-Hanhart 症候群;RSPO1,R-spondin 1;SC,角質層 (stratum corneum);SERPINB7,serpin family B member 7;SLURP-1,secreted LY6/PLAUR domain containing 1;SPPK,條紋型掌蹠角化症 (striate palmoplantar keratoderma);TAT,酪胺酸轉胺酶 (tyrosine aminotransferase);TRPV3,瞬時受體電位陽離子通道亞家族 V 成員 3 (transient receptor potential cation channel subfamily V member 3)。(作者感謝 Maurice van Steensel、Vinzenz Oji、Edel A. O’Toole、David Hansen、Mary Schwartz 與 Frances Smith 對本圖製作的貢獻。)
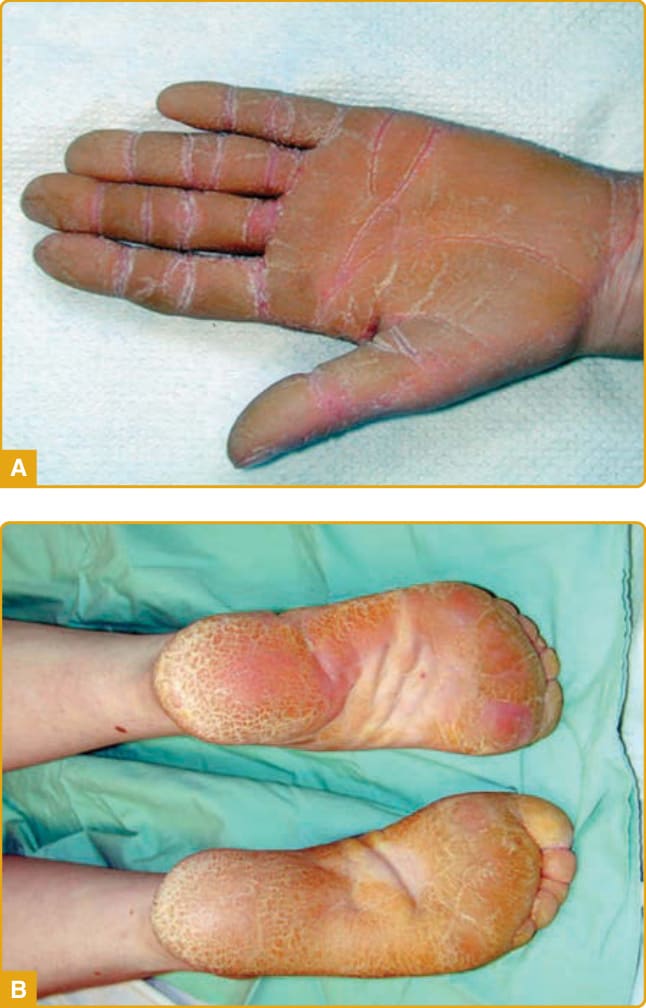
圖 48-3:表皮鬆解型掌蹠角化症 (epidermolytic palmoplantar keratoderma)。黃色、瀰漫型掌蹠角化症,在手掌 (A) 與足底 (B) 邊緣具紅斑性銳利邊界。此病人鑑定出 KRT9 的雜合子突變。
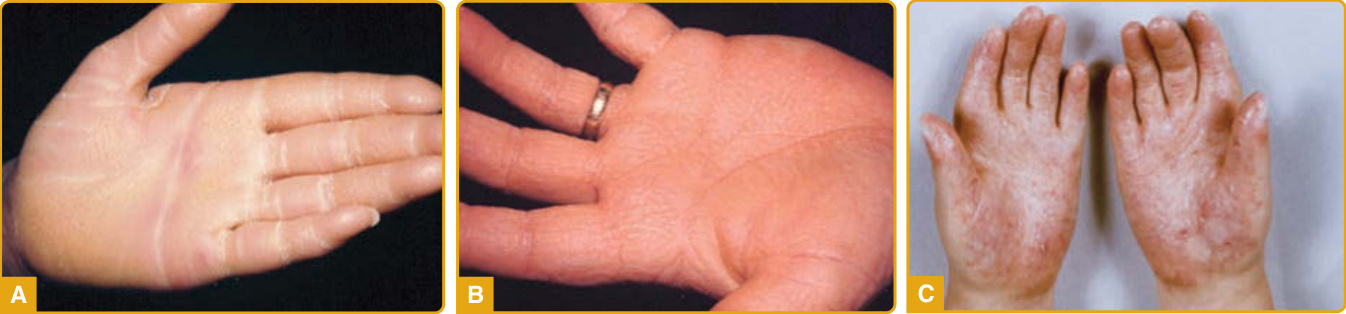
圖 48-4:罕見形式的帶 transgrediens 掌蹠角化症 (rare forms of transgrediens palmoplantar keratoderma)。A,一例 loricrin 掌蹠角化症,呈現瀰漫型、界線分明、蜂窩狀、帶紅斑性邊界的掌蹠角化症。B,Vohwinkel 症候群,呈現「蜂窩狀」外觀。C,Huriez 症候群。手掌上瀰漫型、界線分明的過度角化斑塊。(經 Dr. Cameron Kennedy 許可使用,Bristol Royal Infirmary,英國。)
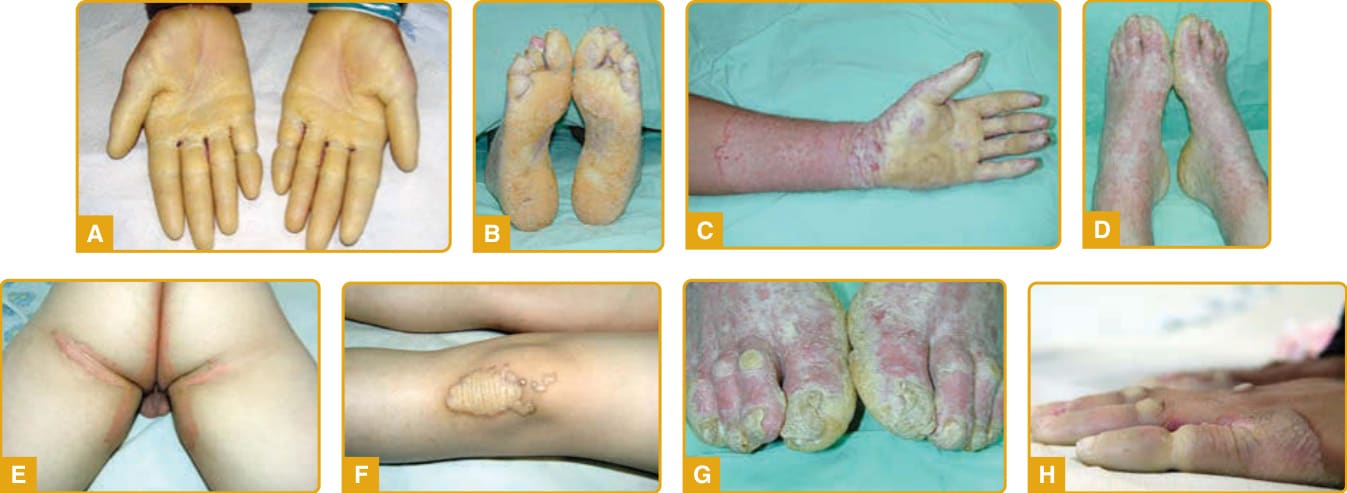
圖 48-5:Mal de Meleda。瀰漫型、黃色、蠟狀的過度角化斑塊,由紅色鱗狀邊界勾勒於手掌 (A) 與足底 (B),並以手套狀 (C) 與長襪狀 (D) 分布延伸至手足背側面。腹股溝區域的侵犯 (E)、關節上方的角化斑塊 (F)、指甲異常 (G) 與假性指趾斷離 (H) 為相關發現。
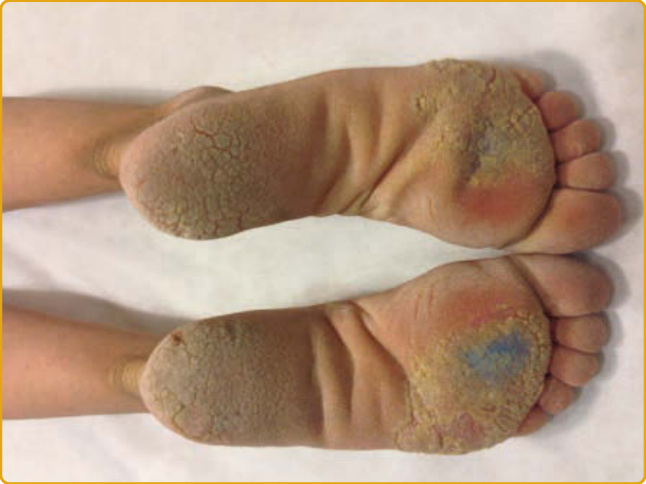
圖 48-6:角膜炎–魚鱗癬–耳聾 (keratitis–ichthyosis–deafness, KID) 症候群。瀰漫型、過度角化的足底角化症,具有特徵性的粗糙、點刻狀與顆粒狀外觀。
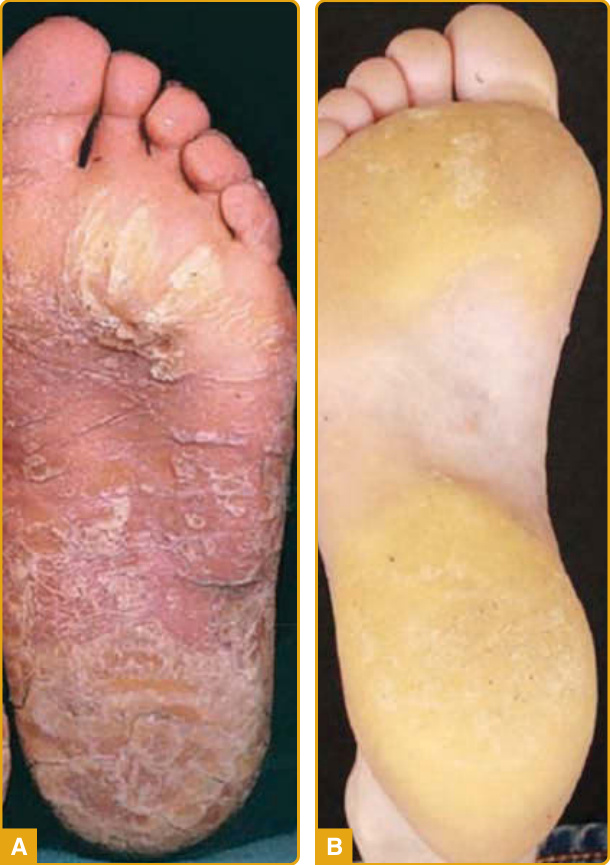
圖 48-7:Papillon-Lefèvre 與 Howel-Evans 症候群的足底角化症。A,由組織蛋白酶 C (cathepsin C) 突變所致 Papillon-Lefèvre 症候群中的瀰漫型角化症。B,Howell-Evans 症候群,呈現局限於足底壓力區域的局灶型、黃色、厚斑塊。(A 部分經許可使用,Barts and the London NHS Trust,英國。)
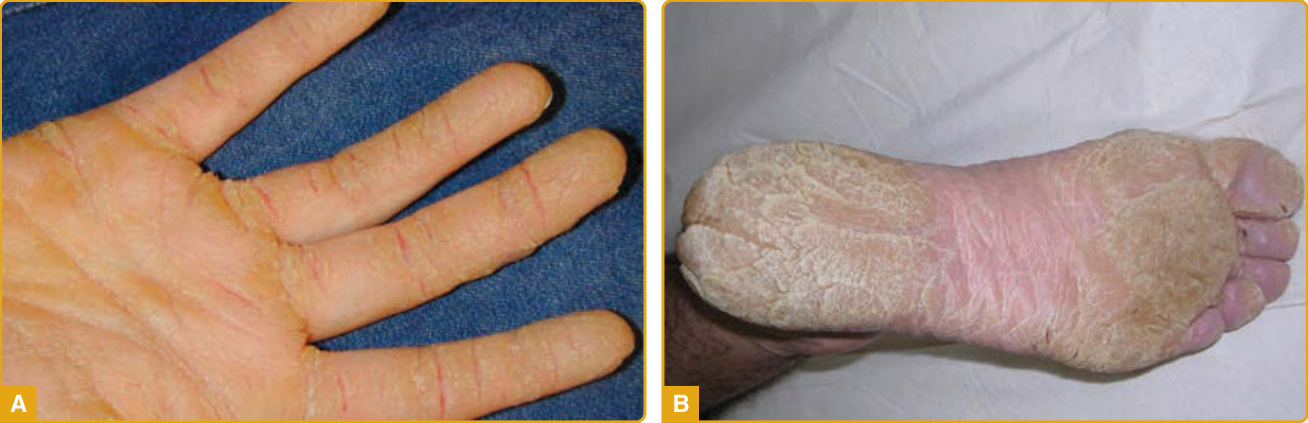
圖 48-8:伴 DSG1 突變的局灶型掌蹠角化症 (focal palmoplantar keratoderma)。手掌上線狀、增厚的過度角化斑塊,沿指部掌側面延伸 (A),以及足底上局限性的過度角化斑塊區域 (B)。
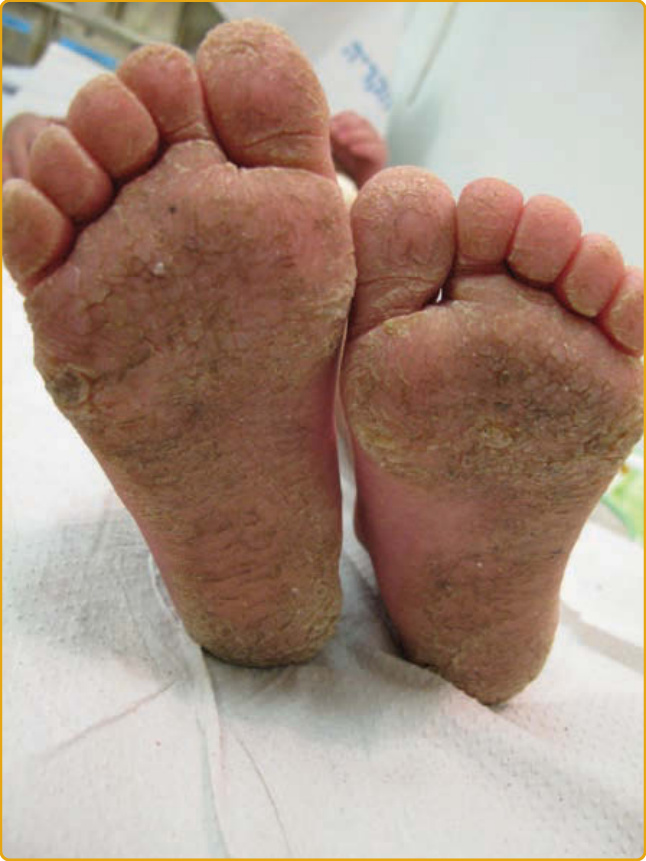
圖 48-9:一名由 DSG1 雙等位基因突變所致皮膚炎、多重過敏與代謝性耗損症候群 (skin dermatitis, multiple allergies, and metabolic wasting syndrome) 病人的足底角化症。
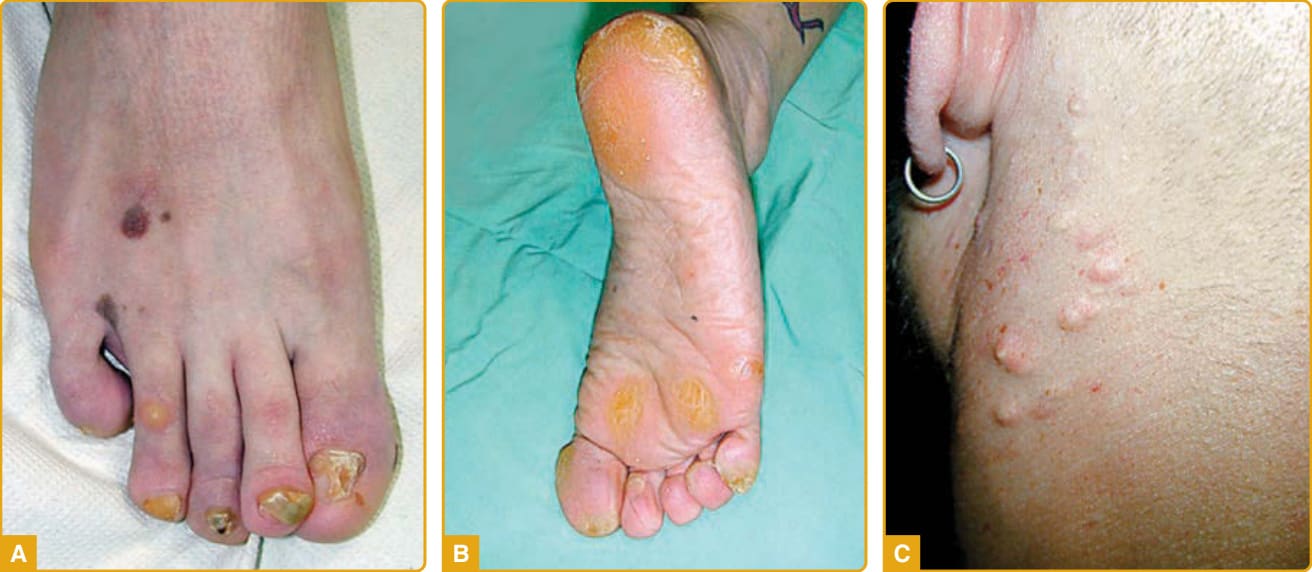
圖 48-10:先天性厚甲症 (pachyonychia congenita)。KRT6A 突變伴特徵性的指甲異常(V 形厚甲與甲下過度角化)(A),以及承重區域上有胼胝的局灶型掌蹠角化症 (B)。KRT17 突變典型伴隨各種類型的表皮內含囊腫 (C)。
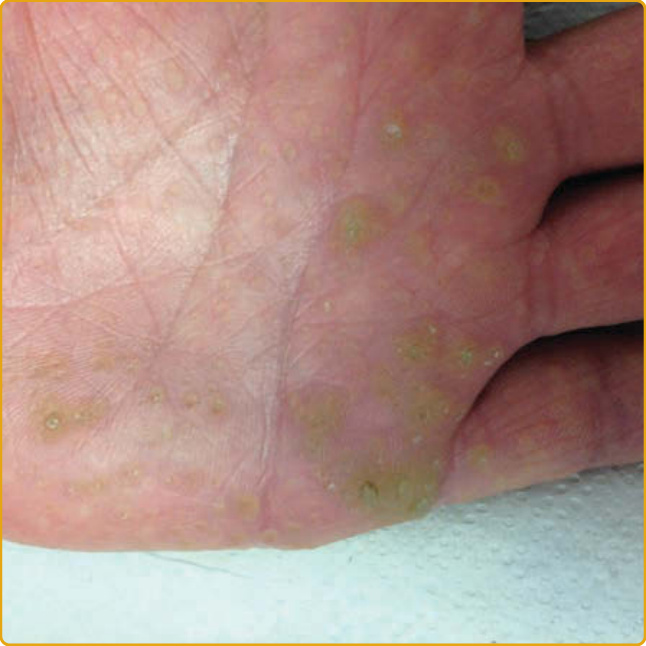
圖 48-11:由 AAGAB 突變所致點狀型掌蹠角化症第 I 型 (punctate palmoplantar keratoderma type I)。多發、過度角化、中央凹陷、黃至褐色的丘疹,不規則地分布於手掌皮膚。
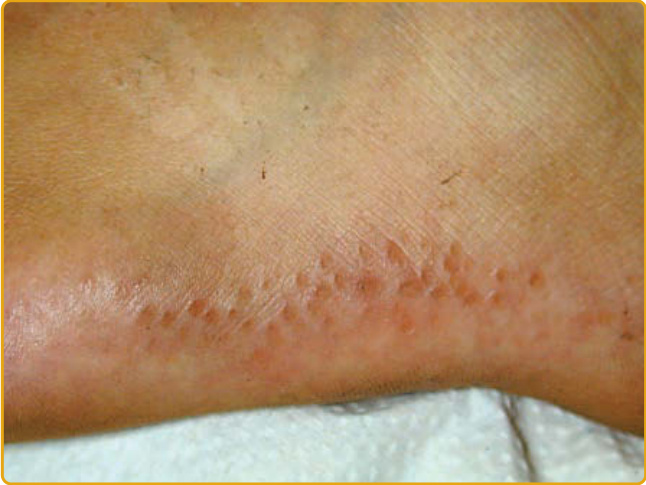
圖 48-12:點狀型掌蹠角化症第 III 型(肢端角化彈力纖維變性症 acrokeratoelastoidosis)。圓至橢圓形黃色丘疹,伴中央臍凹,以線狀模式排列於足底外側邊緣。
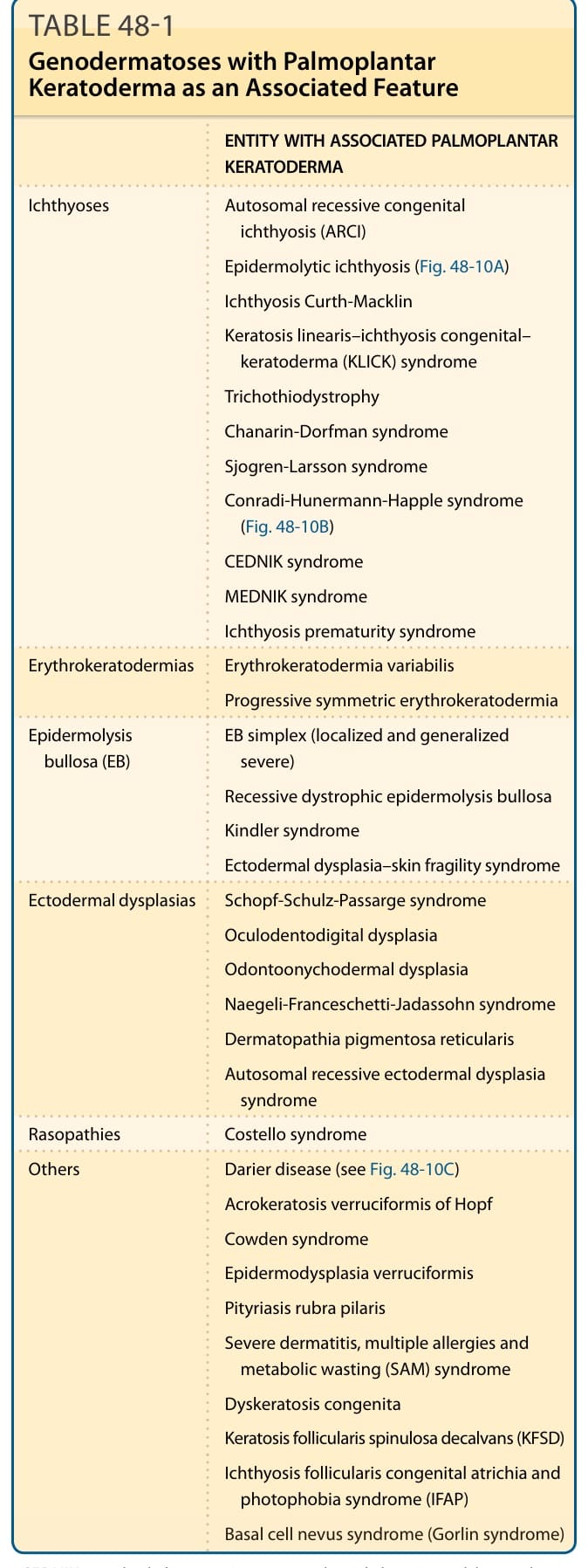
表 48-1:以掌蹠角化症為相關特徵的遺傳性皮膚病 (Genodermatoses with Palmoplantar Keratoderma as an Associated Feature)
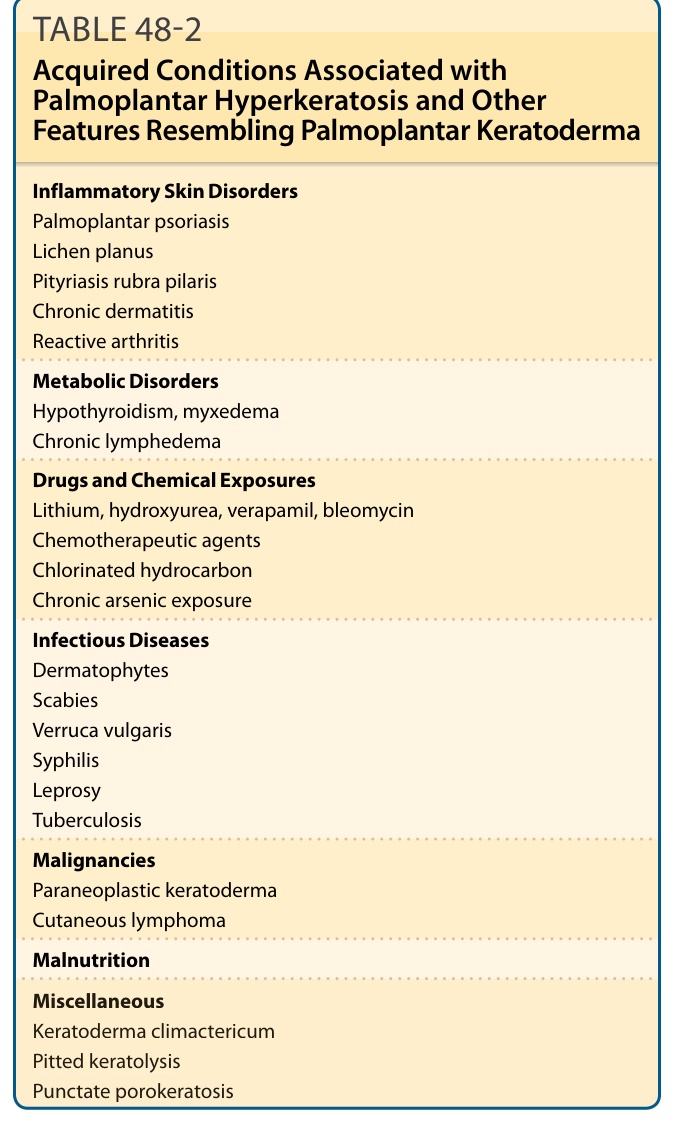
表 48-2:與掌蹠過度角化及其他類似掌蹠角化症特徵相關的後天性疾病 (Acquired Conditions Associated with Palmoplantar Hyperkeratosis and Other Features Resembling Palmoplantar Keratoderma)
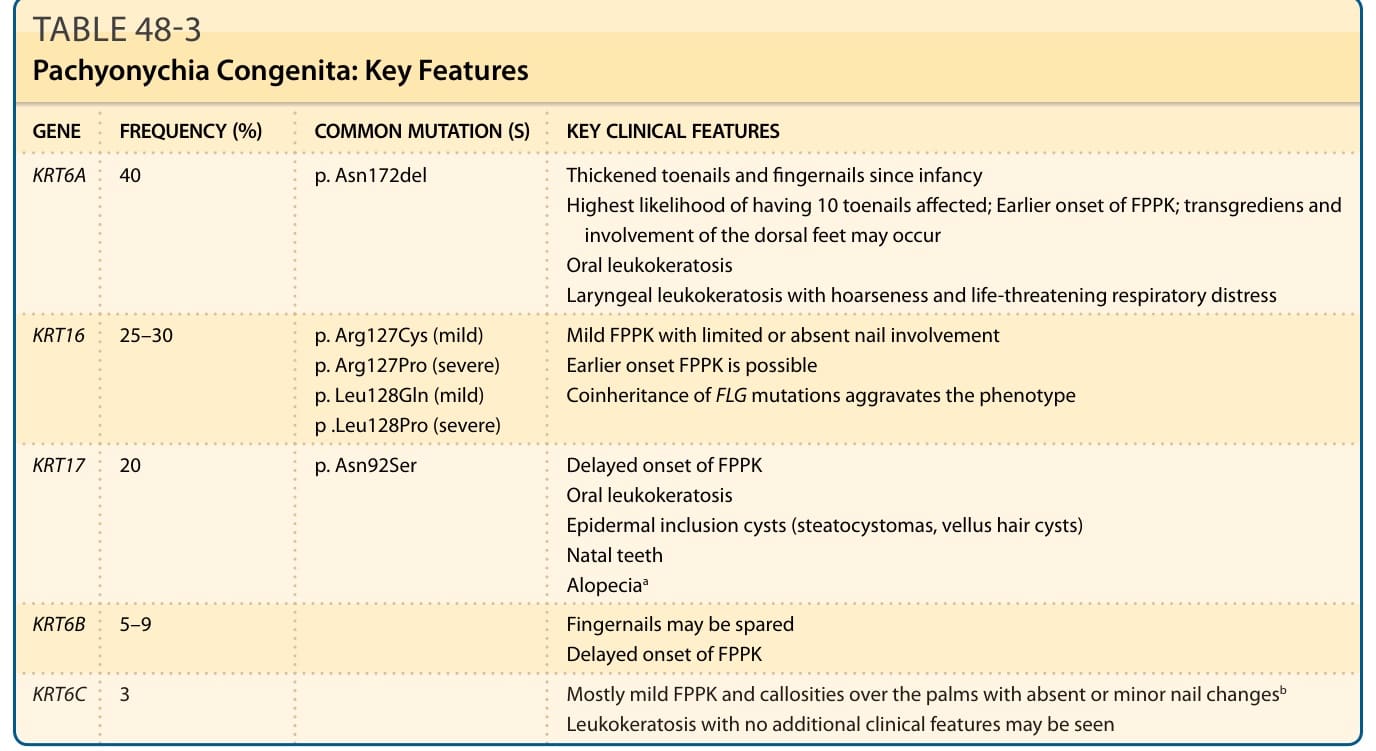
表 48-3:先天性厚甲症:主要特徵 (Pachyonychia Congenita: Key Features)
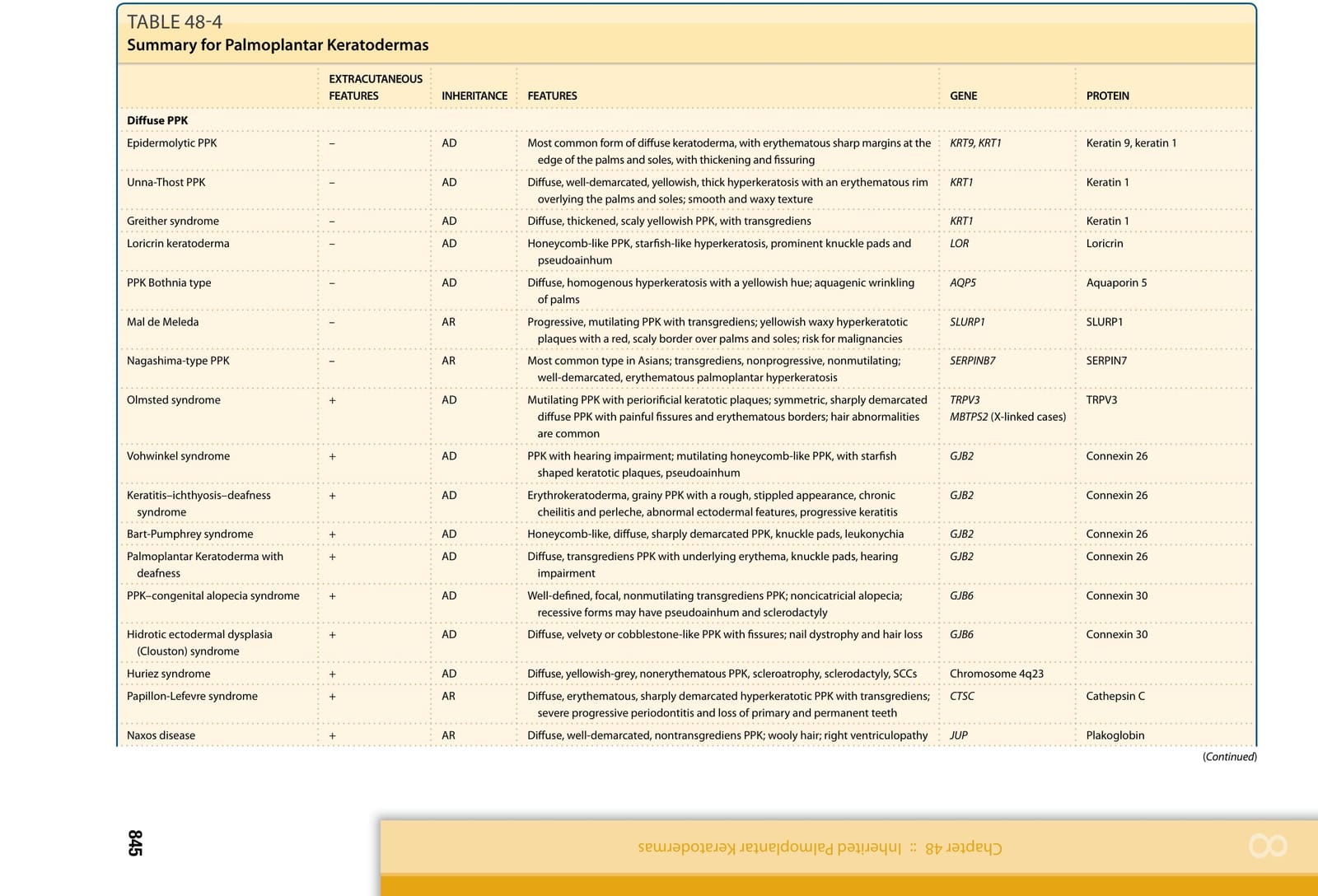
表 48-4:(總結表 Summary)