魚鱗癬 (The Ichthyoses) 精華筆記
總論
- 定義:魚鱗癬 (ichthyoses) 是一群異質性皮膚疾病,共同特徵為全身性脫屑 (generalized scaling),常合併皮膚增厚。多數為遺傳性(出生時或兒童期出現),少數為後天性 (acquired)。
- 致病機轉:突變基因多樣(膜轉運蛋白、脂質生合成酶、結構蛋白等),導致上皮屏障受損、過度增生與過度角化而表現為脫屑。表皮分化終產物為角質層(角質形成細胞「磚塊」+富含脂質的細胞間「灰泥」);角質層增厚可因細胞進入過快、脫屑過慢或兩者所致。
- 分類:2009 年共識命名以遺傳模式與病理生物學取代描述性名稱。主要標準:僅限皮膚(非症候群性 nonsyndromic)或合併他系統(症候群性 syndromic)、遺傳模式、病理生物學。原小丑魚鱗癬 (harlequin)、板層狀魚鱗癬 (LI)、先天性魚鱗癬樣紅皮症 (CIE) 歸入體染色體隱性先天性魚鱗癬 (ARCI);水疱型相關歸入表皮鬆解性魚鱗癬 (EI)。
- 臨床鑑別特徵:發病年齡、出生時有無膠樣膜 (collodion membrane)、鱗屑性質、有無紅皮症 (erythroderma)、瞼外翻 (ectropion)/唇外翻 (eclabium)、附屬器異常(禿髮、毛幹異常)、他系統侵犯。組織病理學多為非特異性(EI 為可診斷的例外)。成年期才出現魚鱗癬可能是全身性疾病的標誌。
- 遺傳/診斷:許多體染色體顯性病(如 EI)有高頻率新生 (de novo) 突變,故無家族史不能排除顯性遺傳。全外顯子定序 (WES) 有助確認遺傳模式與基因診斷。可行產前分子診斷(第一孕期絨毛膜絨毛取樣、第二孕期羊膜穿刺)與胚胎著床前基因診斷。
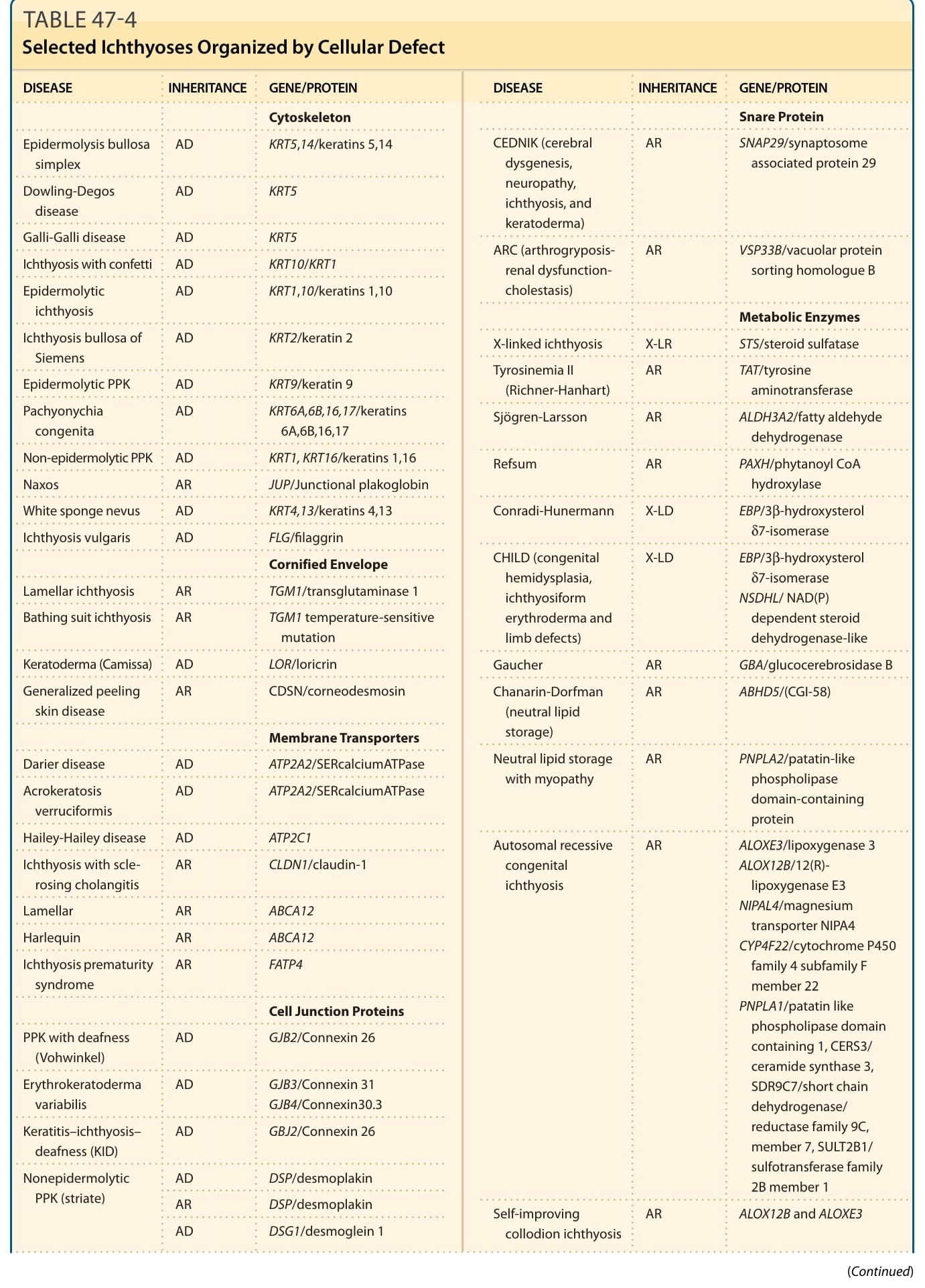
表 47-4:依細胞缺陷組織的選定魚鱗癬
常見治療途徑
- 治療為症狀性,重點:保濕 (hydration)、潤滑 (lubrication)、角質溶解 (keratolysis)。潮濕氣候多會改善;長時間泡澡+沐浴油/潤滑劑(乳液、乳霜、油、軟膏、凡士林)。
- 角質溶解劑:含尿素 (urea)、水楊酸 (salicylic acid)、α-羥基酸(乳酸、甘醇酸)。丙二醇 (propylene glycol,水中 40%–60%) 亦有效。封閉可增強保濕、脫屑與角質溶解效果。
- 屏障受損安全警示:大範圍外用水楊酸可顯著吸收致中毒(噁心、耳鳴、呼吸困難、幻覺)甚至死亡,兒童風險更高。使用 tacrolimus、pimecrolimus 或外用類固醇時可能需監測血清濃度。
- 營養需求可能高,能量流失(屏障受損)可致生長遲滯 (failure to thrive)。部分病人出汗減少 (decreased sweating) 合併熱不耐受,須注意潮紅、嗜睡等徵象,可用降溫背心。
- 全身性類視黃醇 (isotretinoin 或 acitretin) 可使許多魚鱗癬戲劇性改善,但益處須長期治療維持。黴菌感染常見且常被脫屑掩蓋。
尋常性魚鱗癬 (Ichthyosis Vulgaris, IV, OMIM #146700)
- 最常見、相對輕微。FLG(編碼前絲聚蛋白 profilaggrin)突變所致,遺傳為半顯性 (semidominant):單一突變等位基因表現輕微,雙等位基因突變則嚴重。英-歐族群盛行率高達每 80 人 1 人。
- 臨床:嬰兒多正常,第一年內顯現;鱗屑在四肢伸側 (extensor) 最明顯,屈側與尿布區倖免;細小白色鱗屑,下肢鱗屑可中央附著、邊緣龜裂。常合併手掌過度線狀紋路 (hyperlinear palms)、毛孔角化症 (keratosis pilaris) 與異位性 (atopy)。
- 致病機轉:絲聚蛋白參與角蛋白絲聚集、為角質化外膜成分並保留水分。FLG 無效突變 (null mutations) 為異位性皮膚炎 (atopic dermatitis) 的強烈易感因子。
- 鑑別/病程:輕微 IV 與單純乾燥皮膚 (xerosis) 難分;嚴重 IV 男性與 X 性聯隱性魚鱗癬難分。乾冷惡化、溫暖潮濕改善。多對潤膚劑反應良好,脫屑時用 α-羥基酸。
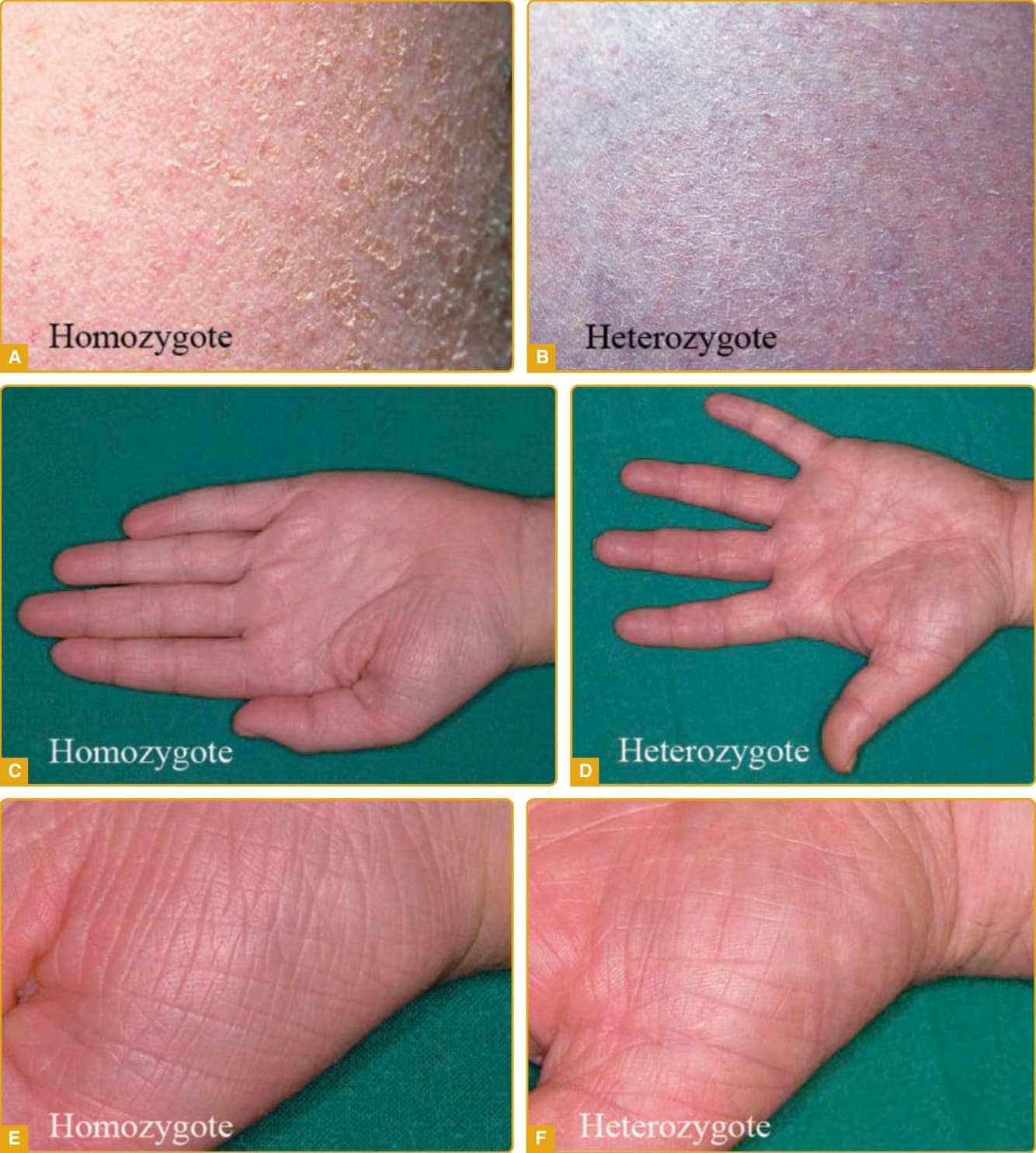
圖 47-1:尋常性魚鱗癬(FLG 突變),小型中央附著鱗屑、倖免皺褶間區。
X 性聯隱性魚鱗癬 (X-Linked Recessive Ichthyosis, XLI, OMIM #308100)
- 約每 1500–6000 名男性 1 人。類固醇硫酸酯酶 (steroid sulfatase) 缺乏所致;90% 病例為 STS 基因座缺失,其餘為點突變。
- 臨床:脫屑可始於新生兒期,伸側最明顯但屈側也顯著侵犯,鱗屑較大、深色;半數成年病人有逗點狀 (comma-shaped) 角膜混濁(不影響視力,女性帶因者亦可見)。受影響男性隱睪症 (cryptorchidism) 與睪丸癌 (testicular cancer) 風險增加。
- 致病機轉:類固醇硫酸酯酶水解硫酸膽固醇 (cholesterol sulfate);血清/表皮/鱗屑中硫酸膽固醇升高,抑制降解角質橋粒的蛋白酶,導致脫屑受抑與滯留性過度角化 (retention hyperkeratosis)。胎盤酵素缺失致母體尿雌激素低下,可致分娩異常。
- 診斷:可藉 FISH、array-CGH、次世代定序、血清硫酸膽固醇升高、酵素檢測。母體三指標篩檢 (triple screen) 顯示雌三醇 (estriol) 偏低。
- 重疊症候群:連續基因缺失可合併點狀軟骨發育異常或 X 性聯 Kallmann 症候群(低促性腺素性腺低能症、嗅覺缺失);故應詢問嗅覺缺失並定期睪丸檢查。
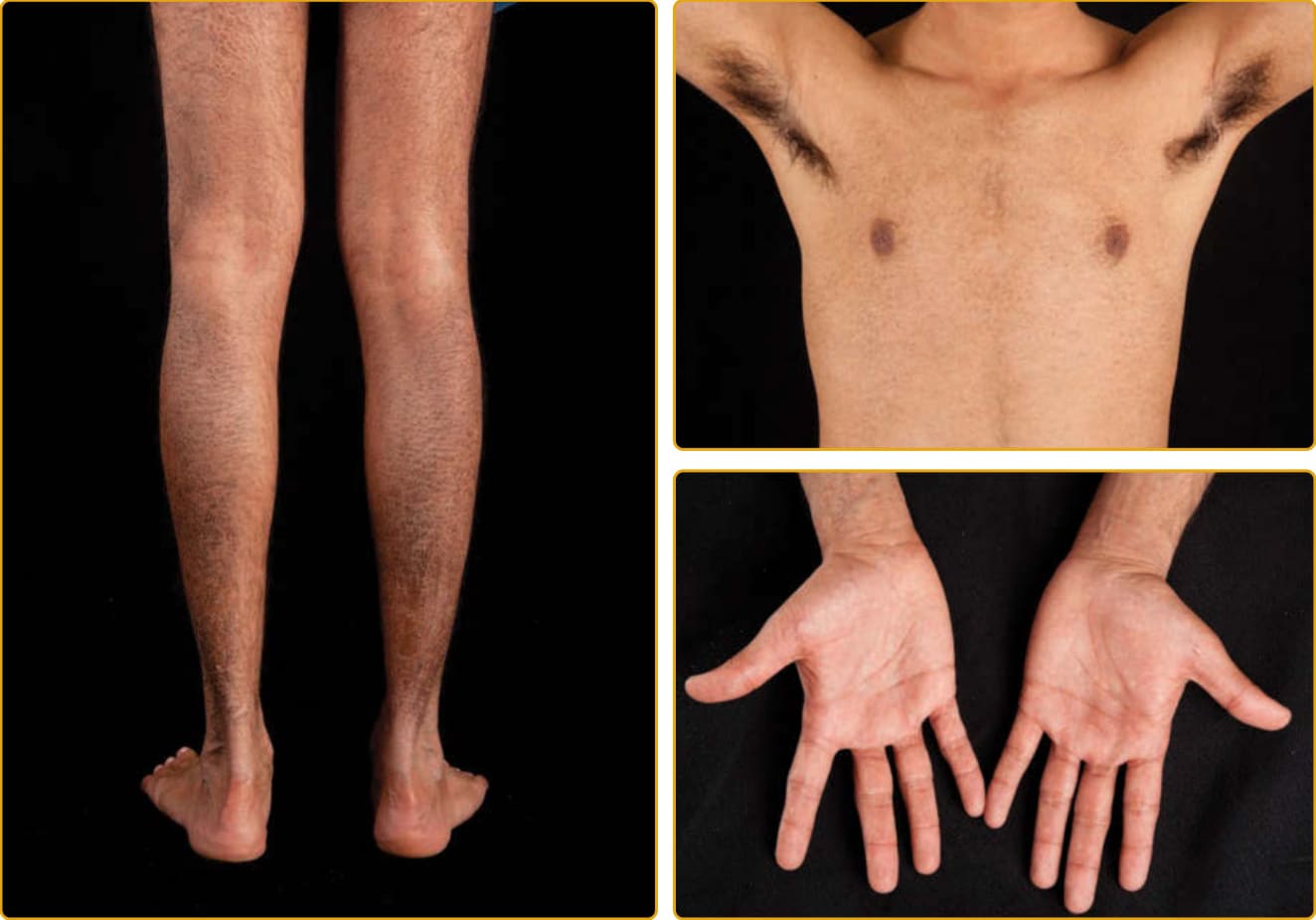
圖 47-2:X 性聯隱性魚鱗癬(STS 突變),深色中央附著鱗屑侵犯頸與臉兩側。
膠樣嬰兒與新生兒表現 (Collodion Baby)
- 多數魚鱗癬(不含 IV 與 XLI)出生時即以紅色脫屑皮膚表現,美國發生率約每 10 萬活產 5–10 例。
- 膠樣嬰兒出生時被半透明、羊皮紙樣緊繃膜包裹,可妨礙呼吸與吸吮、可能早產;膜於前 2 週破裂剝落,留下裂隙、屏障受損(圖 47-3)。基因型非唯一決定因子。
- 最常見的相關疾病為 ARCI;少見於其他型別,罕見於高雪氏病 (Gaucher disease)。另有自癒型膠樣嬰兒(皮膚數週內大幅清除;帶 ALOX12B、ALOXE3 或 TGM1 突變)。
- 新生兒處置:注意感染風險、體溫調節困難、經表皮水分流失致高血鈉性脫水;用水分飽和加濕保溫箱、濕敷後塗潤滑劑。收縮帶可致遠端腫脹、發紺,必要時以刮除/清創釋放。
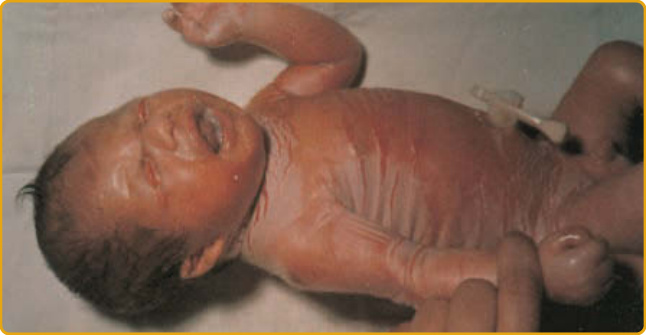
圖 47-3:膠樣嬰兒,浸軟膜伴裂隙、瞼外翻與唇外翻。
體染色體隱性先天性魚鱗癬 (ARCI)
- 一群異質性疾病,出生時即全身性侵犯,罕見(約每 30 萬人 1 人)。由 10 個基因突變所致:TGM1、ALOX12B、ALOXE3、NIPAL4、CYP4F22、ABCA12、PNPLA1、CERS3、SDR9C7、SULT2B1。LI 與 CIE 為一臨床譜系的兩端,基因型不決定表現型。
- 板層狀魚鱗癬 (LI) 表現型:常以膠樣膜起始,後發展大型深色板片狀鱗屑(下肢如乾涸河床),臉部緊繃致瞼外翻、唇外翻,周邊瘢痕性禿髮 (scarring alopecia),出汗不足與熱不耐受。最常與 TGM1 突變相關。泳衣型魚鱗癬 (bathing suit ichthyosis) 為亞型,僅泳衣覆蓋(較溫暖)區脫屑,由 TGM1 溫度敏感性突變所致。
- 先天性魚鱗癬樣紅皮症 (CIE) 表現型:出生時膠樣膜,膜脫落後持續發紅伴細小白色鱗屑,少有瞼外翻/唇外翻/禿髮;由脂氧合酶 (lipoxygenase) ALOXE3 與 ALOX12B 突變等所致。
- 小丑魚鱗癬 (harlequin ichthyosis, OMIM #242500):戲劇性、嚴重、有時致命;厚發亮角質板塊被深紅裂隙分隔成幾何圖案,耳發育不良、明顯瞼外翻與唇外翻、指尖錐形。由 ABCA12(ATP 結合卡匣轉運蛋白,層板顆粒分泌與脂質運輸)功能喪失突變所致;錯義突變則致較輕的 ARCI。1980 年前一律致命,今多數存活但伴嚴重持續紅皮症。
- Netherton 症候群 (OMIM #256500):體染色體隱性,三聯為魚鱗癬+毛幹異常+異位性。關鍵發現為環狀線狀魚鱗癬 (ichthyosis linearis circumflexa, ILC)(雙邊緣、蛇行、遷移性鱗屑);毛幹異常為套疊性脆髮症 (trichorrhexis invaginata,竹節狀毛髮)(僅 20%–50% 毛髮受影響,眉毛較易見)。IgE 顯著升高、類異位性體質、可有胺基酸尿症與細胞免疫受損。由 SPINK5(編碼蛋白酶抑制劑 LEKTI)突變所致。
- TGM1/轉麩醯胺酸酶 1:催化角質化外膜蛋白(兜甲蛋白 involucrin、loricrin、絲聚蛋白等)之鈣依賴交聯,並將神經醯胺附著至外膜。外用微脂體包裹之重組轉麩醯胺酸酶-1 可使小鼠異種移植正常化。
- 治療:
- LI/CIE:口服類視黃醇 (oral retinoids) 可改善出汗與熱耐受、減少眼周厚鱗屑以降低瞼外翻傾向;瞼外翻者白天用人工淚液、夜間用眼用潤滑劑防暴露性角膜炎。瞼外翻在 CIE(35%)較 LI(57%)少見。
- 小丑魚鱗癬:新生兒重症照護+審慎全身性類視黃醇促進脫屑改善存活;外用與全身性類視黃醇處置瞼外翻與過度角化。
- Netherton 症候群:tacrolimus 軟膏有效但屏障異常致全身吸收增加(曾有醫源性庫欣症候群報告),須監測;外用與全身性類視黃醇應避免(可能惡化)。
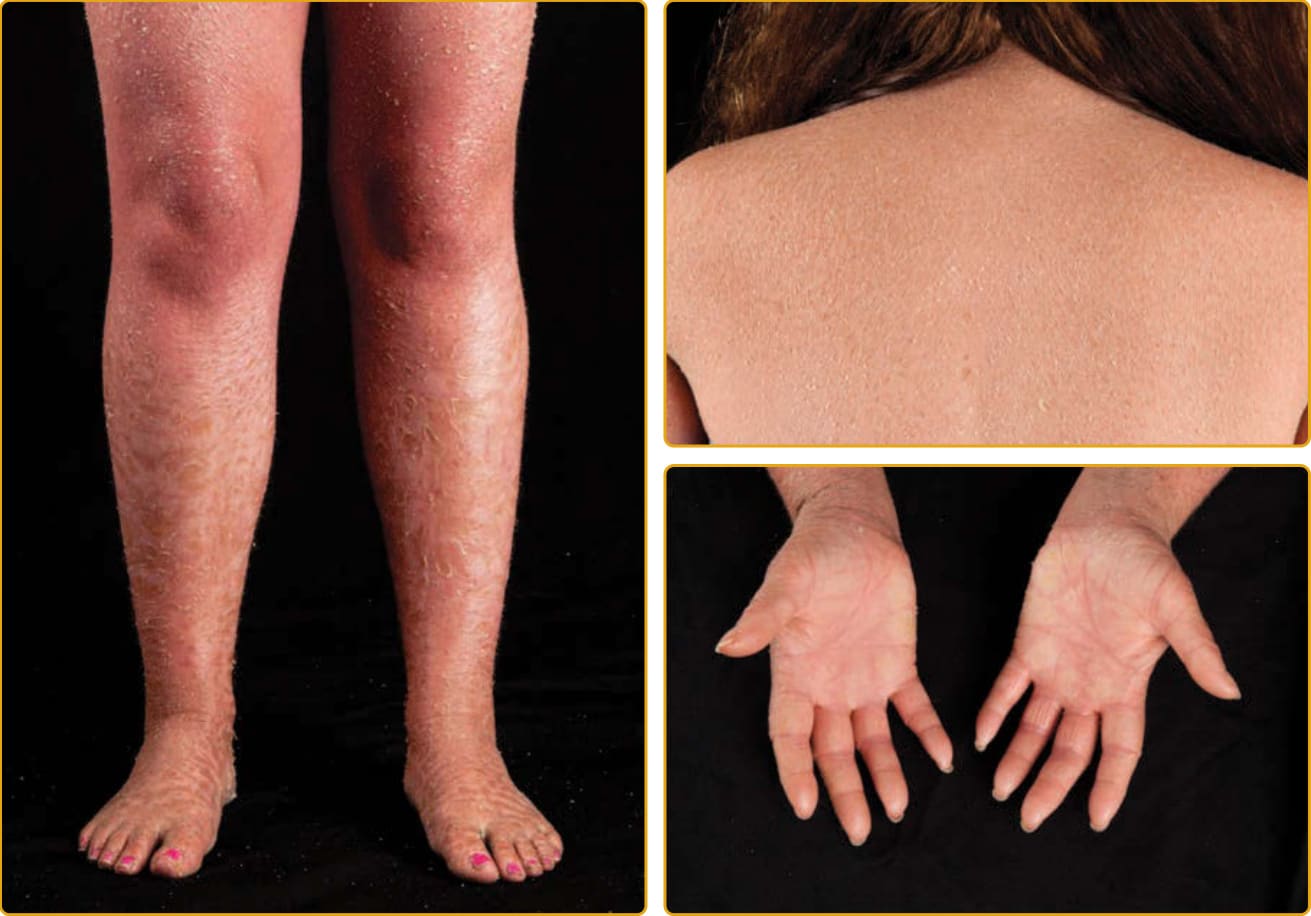
圖 47-4:ARCI(TGM1 突變,經典板層狀魚鱗癬),大型深色鱗屑於伸側與前額。
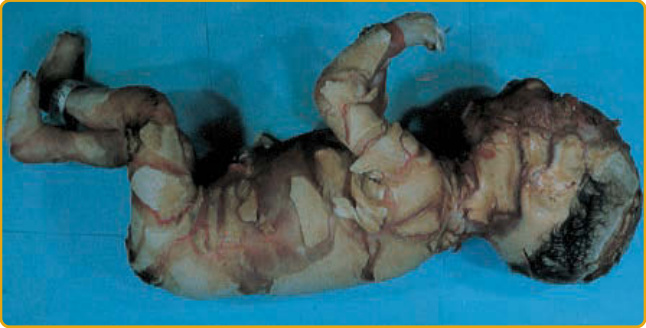
圖 47-9:小丑嬰兒,發育不全的耳朵與厚板塊狀角質層。
角蛋白病性魚鱗癬 (Keratinopathic Ichthyoses)
- 由角蛋白 (keratin) 基因突變所致。譜系含 EI、淺表性表皮鬆解性魚鱗癬 (SEI)、Curth-Macklin 豪豬狀魚鱗癬、環狀 EI、五彩碎紙樣魚鱗癬 (IWC)。
- 表皮鬆解性魚鱗癬 (EI, OMIM #113800):體染色體顯性(高新生突變率,半數無家族史),盛行率約每 20–30 萬人 1 人。由 KRT1/KRT10(影響角蛋白桿狀區)突變所致。出生時水疱、發紅、剝皮,易誤認為表皮鬆解水疱症或葡萄球菌性燙傷樣皮膚;後發展全身性過度角化與特徵性波紋狀 (corrugated) 鱗屑(皺褶區明顯),常有特徵性氣味(繼發感染)。組織學:上表皮空泡變性+過度角化(即 EHK),顆粒細胞有似透明角質顆粒的緻密團塊。
- 線狀 EI:表皮母斑變異,合子後體細胞突變,沿 Blaschko 線鑲嵌;若及性腺可遺傳致全身性 EI。
- 淺表性表皮鬆解性魚鱗癬 (SEI,Siemens 水疱型魚鱗癬, OMIM #146800):體染色體顯性;脆弱性較淺表,最上層角質層脫失形成項圈樣凹陷「mauserung(換羽)」。由 KRT2 突變所致;組織學空泡化侷限於顆粒層。
- 環狀 EI:罕見體染色體顯性,出生或最初數月嚴重間歇脫屑與水疱,青春期消退,殘留波紋狀鱗屑斑塊;KRT10 與 KRT1 突變。
- 五彩碎紙樣魚鱗癬 (IWC):先天性紅皮症、耳廓畸形、手指錐形;兒童期起出現小島狀正常皮膚(源自 KRT10/KRT1 致病突變的雜合性喪失、有絲分裂重組)。無明顯皮膚脆弱性。
- 治療:潤滑劑與角質溶解劑減少增厚粗糙區並降低水疱糜爛。細菌感染常致水疱增多,稀釋漂白水浴控菌、碳酸氫鹽浴促脫屑。全身性類視黃醇顯著減少過度角化,但劑量須緩慢滴定(EI 脆弱性可隨高劑量增加)。
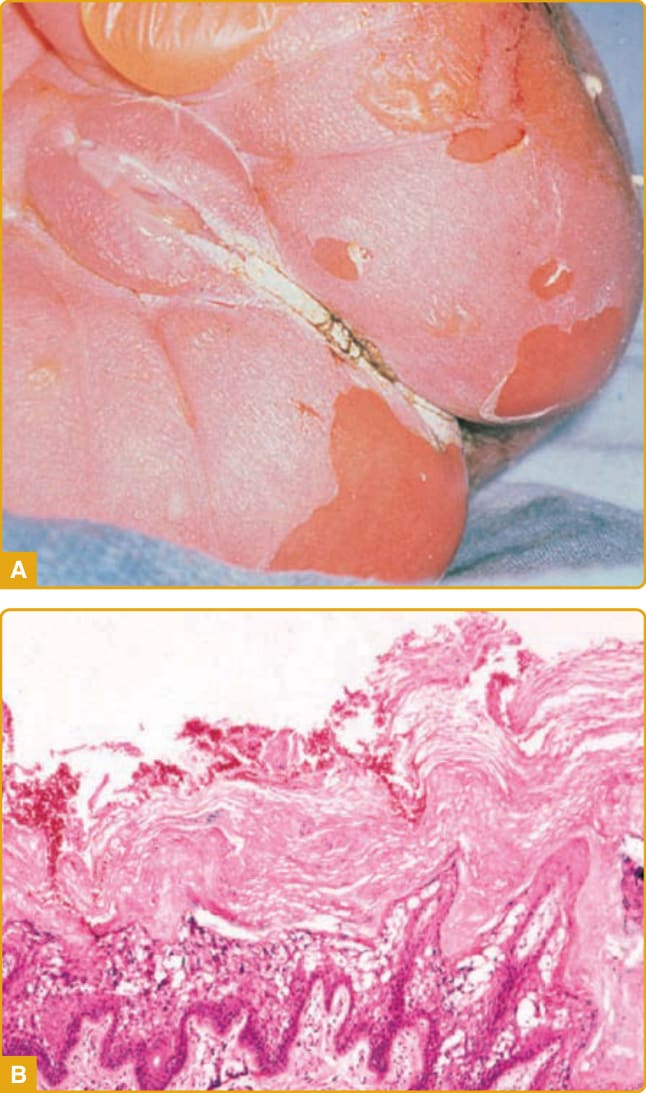
圖 47-15:EI 嬰兒期表現。A,新生兒水疱與糜爛;B,組織學示上表皮空泡變性與過度角化。
接合蛋白疾病 (Connexin Disorders)
- 由接合蛋白 (connexins,形成間隙連接) 基因突變所致,表現型重疊。
- 可變性與進行性紅斑角皮症 (EKVP, OMIM #133200):含 PSEK 與 EKV。界限分明、紅斑、過度角化斑塊對稱分布;EKV 另有遷移性、地圖樣紅斑塊(數分鐘至數小時內生滅)。由 GJB3、GJB4、GJA1(接合蛋白 31、30.3、43)突變所致。全身性類視黃醇改善過度角化。
- 角膜炎-魚鱗癬-聾症候群 (KID, OMIM #148210):角膜炎+魚鱗癬+神經感覺性聾,屬外胚層發育不良,多為體染色體顯性。離散紅斑斑塊、皮革樣掌蹠角皮症、特徵性面容;由 GJB2(接合蛋白 26)突變所致。可有皮膚/舌頭鱗狀細胞癌。口服類視黃醇益處甚少且可能加劇角膜新生血管。GJB2 之 p.A88V 與 p.G45E 突變與新生兒致死相關。
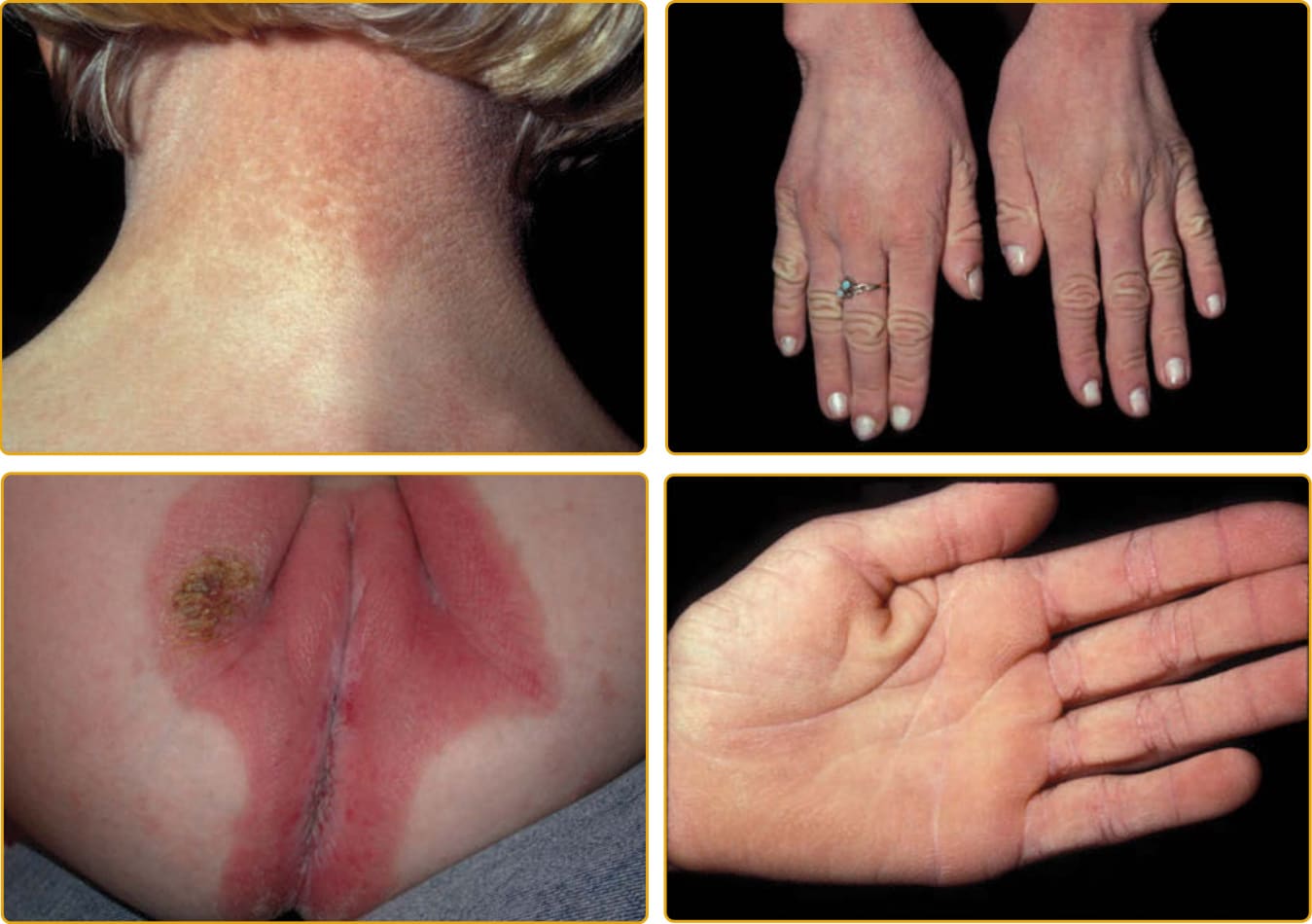
圖 47-22:KID 症候群(GJB2 突變),過度角化柱狀物致卵石樣外觀。
剝皮性皮膚疾病 (Peeling Skin Disorders)
- 剝皮症候群 (PSSs, OMIM #270300) 為異質性,皮膚剝落常間歇且缺乏全身性過度角化;無痛、易剝落、不留疤,因摩擦與濕度加劇。
- PSS1(CDSN/角質橋粒蛋白):發炎性,伴異位性、搔癢與屏障缺損。PSS2(TGM5/轉麩醯胺酸酶 5):肢端剝皮。PSS3(CHST8)。PSS4(CSTA/cystatin A,裂解層次較深)。PSS5(SERPINB8,非發炎性)。
症候群性魚鱗癬 (Syndromic Ichthyoses)
- Chanarin-Dorfman 症候群 (中性脂質儲積病, OMIM #275630):體染色體隱性,三酸甘油酯在多組織累積(血脂正常);全身性板層狀鱗屑伴屈側波紋狀加強,常以膠樣嬰兒表現。周邊血抹片可見顆粒球內脂質空泡。由 ABHD5 突變所致。
- CHILD 症候群 (OMIM #308050):先天性半側發育不良+魚鱗癬樣紅皮症+肢體缺損,幾乎僅見女性,X 性聯顯性(男性致死),鑲嵌性、單側侵犯。多數為 NSDHL(角鯊烯後膽固醇生合成)功能喪失突變,少數 EBP。外用膽固醇+lovastatin 可基於機轉改善皮膚(補終產物並阻斷毒性中間產物)。
- 點狀軟骨發育異常 (chondrodysplasia punctata):骨骺點狀鈣化。根肢型 (RCDP, OMIM #215100):體染色體隱性過氧化體生成障礙,PEX7 突變,根肢化侏儒、白內障、魚鱗癬、智能障礙。X 性聯隱性 (CDPX, OMIM #302950):ARSE(芳基硫酸酯酶 E)等突變,類似 warfarin 胚胎病變。X 性聯顯性 (CDPX2, Conradi-Hünermann-Happle, OMIM #302960):幾乎僅女性(男性致死),沿 Blaschko 線鑲嵌,EBP(3β-羥基類固醇-δ8-δ7-異構酶)突變。
- IFAP 症候群 (OMIM #308205):X 性聯隱性,魚鱗癬性毛囊角化+禿髮+畏光,由 MBTPS2 突變所致。
- 魚鱗癬早產症候群 (OMIM #608649):羊水過多致早產,紅皮症、水腫、乾酪樣脫屑皮膚,由 FATP4 (SLC27) 突變所致。
- 多發性硫酸酯酶缺乏 (OMIM #272200):體染色體隱性,溶體與微粒體芳基硫酸酯酶皆缺乏,由 SUMF1 突變所致。
- Refsum 病 (OMIM #266500):植烷酸 (phytanic acid) 分解障礙累積;色素性視網膜炎、周邊神經病變、小腦共濟失調、神經性聾/嗅覺缺失;魚鱗癬類似 IV。由 PhyH (PAHX) 缺乏所致(部分為 PEX7)。膳食限制植烷酸可阻止進展。
- Sjögren-Larsson 症候群 (SLS, OMIM #270200):體染色體隱性,先天性魚鱗癬+痙攣性雙下肢癱 (spastic diplegia)+智能障礙;視網膜黃斑閃亮白點為特徵。由脂肪醇:NAD 氧化還原酶缺乏(ALDH3A2 突變)所致。
- 毛髮硫營養不良 (TTD, OMIM #601675):體染色體隱性,硫缺乏脆弱毛髮(偏光下虎尾條紋 tiger tail banding);魚鱗癬最常見(65%)。記憶法 PIBI(D)S(光敏、智能障礙、生育力降低、魚鱗癬、矮小)。多數由 ERCC2 等 TFIIH 相關基因突變所致;與著色性乾皮症不同,這些光敏病人皮膚癌風險未升高。
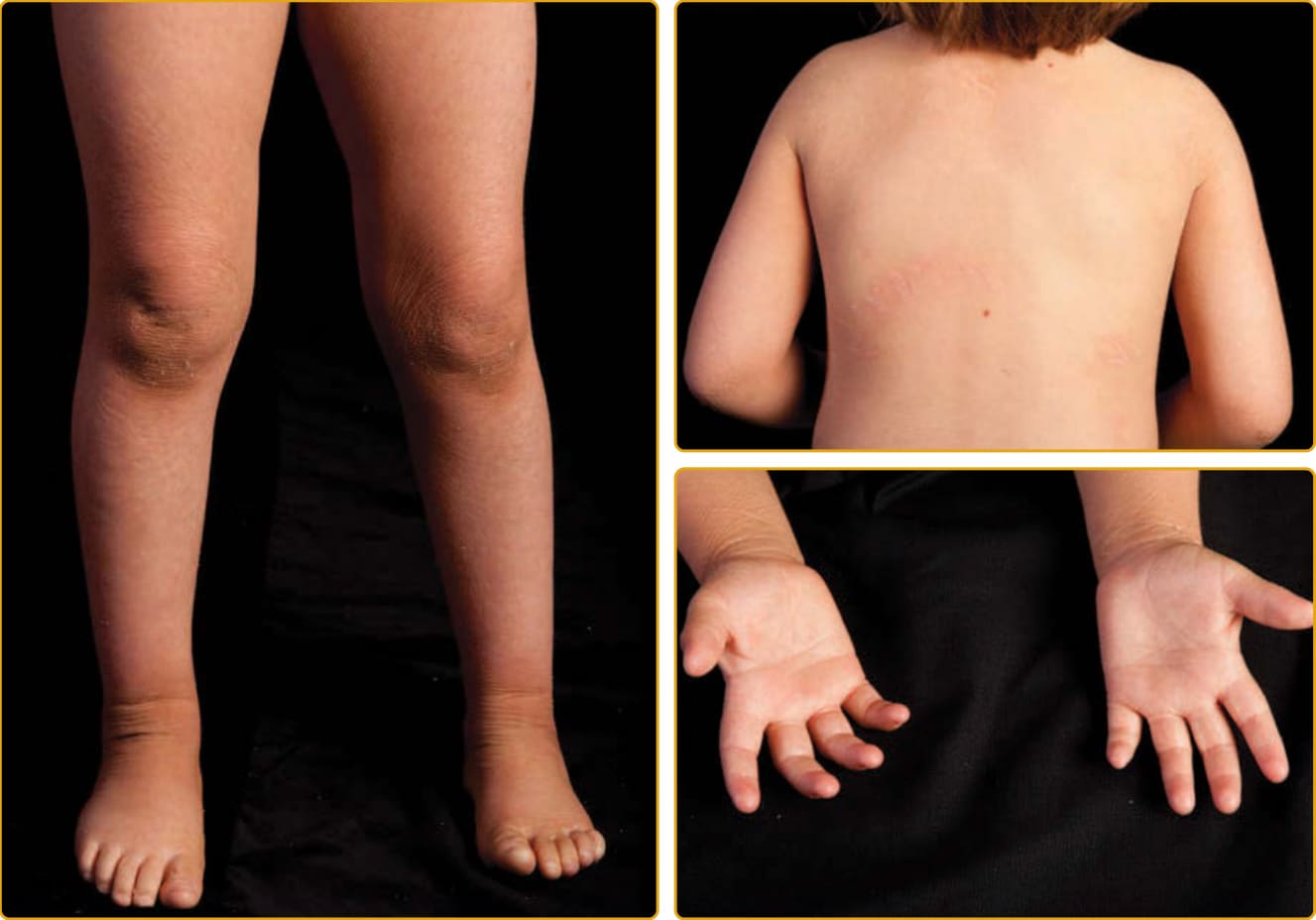
圖 47-25:SLS(ALDH3A2 突變),全身性平滑過度角化與加強的線狀表面紋路。
後天性魚鱗癬與相關病 (Acquired Ichthyosis)
- 成年期發病可為全身性疾病表現,與惡性腫瘤、藥物、內分泌代謝病、營養不良、HIV/感染、自體免疫疾病相關。顆粒層常變薄,鱗屑似輕微 IV。
- 何杰金氏病 (Hodgkin disease) 為最常報告的相關惡性腫瘤;與蕈狀肉芽腫 (mycosis fungoides) 相關者組織學可具診斷性。多達 30% AIDS 病人有魚鱗癬樣/乾燥皮膚。類肉瘤病 (sarcoidosis) 相關者切片可見非乾酪性肉芽腫。藥物:cimetidine、clofazimine、hydroxyurea、降膽固醇藥(菸鹼酸、triparanol)等;kava 致可逆 kava 皮膚病變。
- 圓形糠疹 (pityriasis rotunda):界限分明圓/卵圓形脫屑斑片伴色素改變,多為後天性。
- Gougerot 與 Carteaud 融合性與網狀乳頭瘤病:年輕成人持續棕色脫屑性病灶,分布頸、上軀幹、腋窩,臨床似汗斑;Minocycline 為建議第一線治療。