魚鱗癬 (The Ichthyoses)
PART 8
角化障礙 (Disorders of Cornification)
重點一覽 (AT-A-GLANCE)
■ 魚鱗癬 (ichthyoses) 是一群異質性 (heterogeneous) 的皮膚疾病,特徵為全身性脫屑 (generalized scaling),且常合併皮膚增厚的區域。
■ 大多數型別為遺傳性,這些型別通常在出生時出現或在兒童期顯現;然而部分型別為後天性 (acquired)。
■ 鱗屑 (scales) 的大小、顏色與分布部位可有差異。
■ 可能合併紅斑 (erythema)、附屬器構造異常,以及掌蹠角皮症 (palmoplantar keratoderma)。
■ 可能伴隨全身性表現,如生長遲滯 (failure to thrive)、感染易感性增加、異位性皮膚炎 (atopic dermatitis)、神經感覺性聾 (neurosensory deafness),以及神經學與其他疾病。
■ 組織病理學 (histopathology) 通常非特異性,僅少數值得注意的例外。
■ 早期基因檢測 (genetic testing) 有助於診斷與預期潛在的全身性異常。
前言 (INTRODUCTION)
本章討論一群被稱為魚鱗癬 (ichthyoses) 的異質性疾病,這群疾病共有全身性脫屑 (generalized scaling) 的共同特徵,最常在出生或兒童期出現,但也可在生命較晚期才後天獲得。魚鱗癬常起因於一群多樣化基因的突變,包括膜轉運蛋白 (membrane transporters)、脂質生合成酶 (lipid biosynthesis enzymes),以及結構蛋白 (structural proteins) 等多種。隨之而來的上皮屏障受損 (epithelial barrier compromise)、過度增生 (hyperproliferation) 與過度角化 (hyperkeratosis) 表現為脫屑。相關的全身性異常通常可歸因於突變基因的非皮膚功能。
魚鱗癬的標誌是鱗屑 (scale),它反映了表皮分化 (differentiation) 的改變。由於希臘文「角(鱗)」為 keras、拉丁文為 cornu,本章中表皮分化 (epidermal differentiation)、角質化 (keratinization) 與角化 (cornification) 三詞作同義使用。ichthyosis 一名源自希臘文 ichthys(意為「魚」),指皮膚外觀與魚鱗相似。遺傳性與後天性型別皆存在。在印度與中國文獻中,魚鱗癬的早期記載可追溯至西元前數百年,而此病曾於 1798 年由 Willan 討論。
魚鱗癬可在出生時出現或在生命較晚期發展。它可表現為僅限於皮膚的疾病,或合併其他器官系統的異常。許多具有特徵性表現、定義明確的魚鱗癬型別可被可靠地診斷。然而,由於極大的臨床異質性,以及環境對脫屑的深遠影響,在某些病人與家族中,若無基因檢測的協助,特定診斷可能具挑戰性。
魚鱗癬的分類 (CLASSIFICATION OF THE ICHTHYOSES)
Siemens 將遺傳學概念引入魚鱗癬研究。Wells 與 Kerr 對遺傳性魚鱗癬進行分類,並將 X 性聯隱性魚鱗癬 (X-linked recessive ichthyosis) 與尋常性魚鱗癬 (ichthyosis vulgaris, IV) 區分開來。Gassman 發展出滯留性 (retention) 與過度增生性 (hyperproliferation) 過度角化的概念。隨後 Van Scott、Frost 與 Weinstein 依據表皮更新速率 (rates of epidermal turnover) 的差異提出魚鱗癬分類,將其歸類為表皮過度增生疾病 (disorders of epidermal hyperproliferation) 或角質層延長滯留疾病 (disorders of prolonged retention of the stratum corneum)。其後 Williams 與 Elias 提出一套分類,列出在臨床、遺傳或生化資料上提示為獨立疾病的角化障礙。
魚鱗癬的基因研究已揭示與結構蛋白、脂質生合成、細胞間通訊 (cell–cell communication)、脫屑 (desquamation) 及許多其他路徑相關之基因的致病突變,並在許多情況下提供了過去未知之表皮恆定 (epidermal homeostasis) 決定因子的洞見。全外顯子定序 (whole-exome sequencing, WES) 的出現帶來了表現型擴展 (phenotypic expansion),一套不僅基於突變基因、亦基於特定突變 (specific mutations) 的新魚鱗癬分類正在演進。得知哪個基因發生突變、以及特定突變的效應為何,能讓人理解疾病病理生物學 (pathobiology)。以共同的分子過程來定義這些疾病,能更合理地理解其病理生理與治療。選定遺傳性魚鱗癬的遺傳模式與常見臨床特徵列於表 47-1 至表 47-3。依據所編碼蛋白的功能對這些疾病進行分組(表 47-4),有助於以潛在機轉理解臨床表現型。然而,仍需進一步研究以清楚理解特定突變如何導致臨床疾病,並發展標靶治療介入。2009 年,一場共識會議提出修訂後的命名與分類,以基於遺傳與病理生物學的描述詞,取代常用但分歧的描述性術語。分類的主要標準包括:疾病是否僅限於皮膚(非症候群性 nonsyndromic)或同時侵犯皮膚與其他器官系統(症候群性 syndromic)、其遺傳模式,以及疾病病理生物學。因此,過去稱為小丑魚鱗癬 (harlequin ichthyosis)、板層狀魚鱗癬 (lamellar ichthyosis, LI) 與先天性魚鱗癬樣紅皮症 (congenital ichthyosiform erythroderma) 者,成為體染色體隱性先天性魚鱗癬 (autosomal recessive congenital ichthyosis, ARCI);而水疱型魚鱗癬 (bullous ichthyosis)、水疱型先天性魚鱗癬樣紅皮症 (bullous congenital ichthyosiform erythroderma, BCIE)、表皮鬆解性過度角化 (epidermolytic hyperkeratosis) 與剝脫性魚鱗癬 (ichthyosis exfoliativa) 則成為表皮鬆解性魚鱗癬 (epidermolytic ichthyosis, EI),此外尚有其他更新。
雖然未來數年內可能會進一步修訂,本章將通篇使用 2009 年共識命名。
臨床表現 (CLINICAL PRESENTATION)
數項特徵有助於區分不同型別的魚鱗癬。這些特徵包括發病年齡、出生時是否有膠樣膜 (collodion membrane)、鱗屑的性質、是否有紅皮症 (erythroderma)、皮膚其他部位的異常(如手掌與足底增厚、瞼外翻 ectropion、唇外翻 eclabium)與附屬器構造的異常(如禿髮 alopecia 或毛幹異常),以及其他器官系統的侵犯。在這些鑑別特徵中,皮膚表面的外觀有助於診斷。部分病人可見明顯脫屑,可依鱗屑的大小、構型 (configuration)、顏色與附著程度加以區分;皮膚也可能增厚而有或無可見鱗屑,後者稱為角皮症 (keratoderma)。角質層的增厚(無論臨床或組織學上明顯)稱為過度角化 (hyperkeratosis)。光學顯微鏡特徵通常可診斷 EI,並在選定的魚鱗癬中有幫助(如 Refsum 病、中性脂質儲積病 neutral lipid storage disease、類肉瘤病 sarcoidosis 之後天性魚鱗癬,以及蕈狀肉芽腫 mycosis fungoides),但組織病理學檢查對於區分其他魚鱗癬可能無用。在許多情況下,臨床診斷可藉由基因分析釐清,雖然並非總能找到突變。成年期才發展出魚鱗癬,可能是全身性疾病的標誌。
遺傳學 (GENETICS)
家族史與多世代家系圖 (multigeneration pedigree) 可指出遺傳模式。然而,許多體染色體顯性疾病(如 EI)有高頻率的自發性(亦稱新生 de novo)突變。因此,缺乏陽性家族史並不能排除體染色體顯性遺傳。另一方面,父母近親婚配 (consanguinity) 的存在可能提示體染色體隱性遺傳。在無先前受影響個體的家系中,要區分體染色體顯性、新生突變與體染色體隱性遺傳,在許多情況下構成獨特的臨床挑戰。WES 的出現使許多病例得以確認遺傳模式、進行基因診斷與基因發現。
產前分子診斷已成為可能。其他方法,包括胎兒鏡 (fetoscopy) 與胎兒皮膚切片 (fetal skin biopsy),僅限於妊娠較晚期進行,且帶有胎兒死亡的風險,現已甚少施行。當可藉由胎兒檢體的分子分析進行產前診斷時,最理想是在妊娠早期進行。在已知潛在遺傳缺陷且家族特定突變已被鑑定的疾病中,可於第一孕期(末次月經後 10 至 12 週)行絨毛膜絨毛取樣 (chorionic villous sampling),或於第二孕期行羊膜穿刺 (amniocentesis)。已有多種魚鱗癬以突變分析完成產前診斷。胚胎著床前基因診斷 (preimplantation genetic diagnosis) 是合理的替代方案,並已對許多遺傳性疾病完成,包括 ARCI 與 EI。此程序需要夫婦接受體外受精 (in vitro fertilization) 以取得胚胎,再以分子方法篩檢胚胎以偵測家族中分離的突變。僅選擇對該突變篩檢陰性的胚胎,再用於植入子宮以達成妊娠。非侵入性分子診斷方法(評估母血中循環之胎兒 DNA)為未來提供了潛力。對於已知突變的體染色體隱性疾病,可對高風險親屬進行帶因者檢測 (carrier detection)。
註:本章將數種選定魚鱗癬的遺傳模式、特徵性臨床表現、相關特徵、基因與蛋白功能整理於表 47-1 至表 47-3。各表以圖檔形式呈現(內容摘要:表皮鬆解性魚鱗癬 KRT1/KRT10、淺表性表皮鬆解性魚鱗癬、X 性聯隱性魚鱗癬 STS、X 性聯隱性點狀軟骨發育異常 ARSE、X 性聯顯性點狀軟骨發育異常/Conradi-Hünermann-Happle 症候群 EBP、CHILD 症候群 NSDHL/EBP、Netherton 症候群 SPINK5/LEKTI、小丑魚鱗癬與板層狀魚鱗癬 ABCA12、魚鱗癬早產症候群、Sjögren-Larsson 症候群、毛髮硫營養不良 trichothiodystrophy、Chanarin-Dorfman 症候群、新生兒魚鱗癬-硬化性膽管炎症候群等之發病時間、臨床特徵、相關特徵、基因及蛋白功能與遺傳模式)。縮寫:CHILD,先天性半側發育不良合併魚鱗癬樣紅皮症與肢體缺損;CIE,先天性魚鱗癬樣紅皮症;EBP,emopamil 結合蛋白;NSDHL,NAD(P)H 類固醇去氫酶樣蛋白;OMIM,線上人類孟德爾遺傳資料庫。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
表皮會進行規律的自我更新模式。其主要細胞型別——角質細胞 (keratinocyte)——進行規律的分化模式,使皮膚得以發揮其作為對抗機械創傷與乾燥之屏障的角色。分化的最終產物是角質層 (stratum corneum),由終末分化的角質細胞——角質形成細胞 (corneocytes,「磚塊」)——所組成,並由細胞間基質(「灰泥」)所包圍(見第 5 章與第 14 章)。角質形成細胞「磚塊」富含蛋白質,而細胞間「灰泥」由疏水性、富含脂質的膜雙層 (membrane bilayers) 構成。
富含角蛋白的角質形成細胞被認為主要負責角質層的韌性與保水特性,而基質則形成大部分對抗水分流失的通透性屏障。正常角質層以有組織且不可見的方式進行脫屑,個別角質形成細胞彼此分離並以單一細胞的形式脫落。魚鱗癬皮膚的鱗屑在性質與數量上異常,角質層的屏障功能受損,且表皮細胞增生 (epidermal cell proliferation) 的動力學可能有所改變(見第 5 章)。角質層可被視為一個區室 (compartment),角質層增厚乃因細胞以增加的速率進入此區室、離開(角質形成細胞脫屑)過慢,或兩者兼有所致。表皮分化的過程複雜且尚未完全了解。此過程許多不同層面與步驟的缺陷可導致相似的最終結果:異常的角質層與鱗屑。遺傳性魚鱗癬的基因研究揭示了對上皮分化至關重要的路徑。例如,編碼基底上層表皮角蛋白(角蛋白 1 與角蛋白 10)的基因,若突變影響到角蛋白中間絲 (keratin intermediate filaments) 聚合所必需之高度保守的桿狀區 (rod domains),便會造成 EI。編碼轉麩醯胺酸酶-1 (transglutaminase-1) 的基因(此酶催化蛋白質的交聯與角質形成細胞形成過程中神經醯胺 ceramides 的附著)的突變,可見於相當大比例的 ARCI 病人。編碼絲胺酸蛋白酶抑制劑的 SPINK5 突變會造成 Netherton 症候群,並確認了蛋白質水解 (proteolysis) 與蛋白酶抑制劑在正常表皮分化中的角色。最後,FLG 突變導致絲聚蛋白 (filaggrin) 減少或缺失,使 IV 病人角質層的保濕結合減少。
這些典型範例凸顯出一項觀察:許多編碼功能高度分歧之蛋白質的基因,其突變皆會影響正常角質細胞分化並造成魚鱗癬。目前尚不清楚不同過程的缺陷如何導致相似的表現型,雖然研究提示屏障缺陷可能導致發炎與過度增生。此外,我們對這些機轉持續演進的理解,持續釐清了數種魚鱗癬樣疾病所觀察到的多系統臨床表現型。
常見治療途徑 (COMMON THERAPEUTIC APPROACHES)
目前對遺傳性魚鱗癬的治療為症狀性,著重於保濕 (hydration)、潤滑 (lubrication) 與角質溶解 (keratolysis)。魚鱗癬皮膚即使增厚,仍有降低的屏障功能與增加的經表皮水分流失 (transepidermal water loss)。由於角質層的柔軟度為其含水量的函數,保濕可軟化皮膚表面。在潮濕的氣候中,大多數魚鱗癬會改善。以例如長時間泡澡來濕潤皮膚可使其保濕。充分保濕的過度角化區域可較容易地用溫和的研磨物(如海綿、buff puff、浮石)磨薄。在乾燥前加入沐浴油或塗抹潤滑劑可延長保濕與軟化的效果。依魚鱗癬型別與環境條件不同,個別病人可能偏好特定的潤滑劑,可為乳液 (lotions)、乳霜 (creams)、油 (oils)、軟膏 (ointments) 或凡士林 (petrolatum) 形式。在乾燥氣候與冬季月份,可使用加濕器以創造較宜人的環境。角質溶解劑 (keratolytic agents) 用於增強角質形成細胞脫屑,藉此去除鱗屑並磨薄過度角化的角質層。市面上有許多含尿素 (urea)、水楊酸 (salicylic acid) 或 α-羥基酸 (α-hydroxy acids,如乳酸 lactic acid、甘醇酸 glycolic acid) 的角質溶解乳霜與乳液。尿素可能藉由其結合水分的能力發揮作用。丙二醇 (propylene glycol,水中 40%–60%),無論有無封閉 (occlusion),亦可有效去除鱗屑。封閉可有效增加皮膚保濕並促進脫屑;它也可增強角質溶解劑的效果。在大範圍使用封閉合併角質溶解劑時,以及對可能熱不耐受 (heat intolerant) 的個體,應特別小心。在大範圍體表使用外用製劑時,應考量魚鱗癬中明顯受損的屏障功能。例如,廣泛使用外用水楊酸製劑可導致顯著吸收、中毒(如噁心、耳鳴、呼吸困難、幻覺),甚至死亡。兒童風險較大,因其每單位體重的體表面積大於成人,此情況有效地提高了由外用藥產生全身性毒性的可能性。雖然在大多數魚鱗癬中使用外用類視黃醇 (topical retinoids) 似乎是安全的,但在治療魚鱗癬病人併發的皮膚病時仍應考量其異常的皮膚屏障。當使用如 tacrolimus、pimecrolimus 或外用類固醇等已觀察到全身吸收增加的藥物時,可能需要監測血清濃度。
對兒童的另一項風險是,在數種型別的魚鱗癬中,營養需求可能很高,營養不足可導致生長遲滯 (failure to thrive)。此情形過去被認為與鱗屑的大量更新有關;然而,近期研究提示,因屏障功能受損所造成的能量流失才是原因。
部分魚鱗癬病人有出汗減少 (decreased sweating) 合併熱不耐受。對患有魚鱗癬之新生兒的父母而言,重要的是要意識到出汗減少的可能性,並留意熱不耐受的徵象,如潮紅 (flushing) 與嗜睡 (lethargy),尤其是在炎熱天氣,以及隨著孩子長大、在運動時。避免炎熱環境、攜帶裝水噴瓶以濕潤皮膚並透過蒸發降溫,以及降溫背心 (cooling vests) 可將熱壓力降至最低。以 isotretinoin 或 acitretin 進行全身性類視黃醇治療(見第 185 章)可使許多魚鱗癬戲劇性地改善。啟動全身性類視黃醇治療的決定應審慎權衡,因為藥物開始後,持續的益處通常需要長期治療。視黃酸代謝阻斷劑 (retinoic acid metabolism–blocking agents) 可增加內源性類視黃醇濃度,提供了一種可能的替代方案。
黴菌感染 (fungal infections) 常見於皮膚與指甲,且常因全身性脫屑而未被診斷出。高度的懷疑指數有助於診斷體癬 (tinea corporis)、頭癬 (tinea capitis) 或汗斑 (versicolor),此時唯一症狀可能僅是局部搔癢(常很劇烈),唯一徵象是鱗屑性質的差異或局部禿髮區域。
尋常性魚鱗癬 (ICHTHYOSIS VULGARIS)
尋常性魚鱗癬 (ichthyosis vulgaris,線上人類孟德爾遺傳資料庫 [OMIM] #146700) 是最常見的魚鱗癬,相對輕微。此病過去被認為是體染色體顯性,一項英國學童研究發現每 250 人中有 1 人受影響。編碼前絲聚蛋白 (profilaggrin) 的基因突變造成 IV。前絲聚蛋白含有多個絲聚蛋白 (filaggrin) 複本,後者在角化過程中具有多種結構與生化功能。其遺傳模式已釐清為半顯性 (semidominant);帶有一個突變等位基因的個體有輕微表現型,但兩個前絲聚蛋白等位基因皆突變者(突變的同型合子或複合異型合子)(圖 47-1)則表現出嚴重的臨床表現型。在英-歐 (Anglo-European) 族群中,臨床疾病的盛行率高達每 80 人中 1 人。
臨床特徵 (CLINICAL FEATURES)
雖然嬰兒通常有正常皮膚,此病常在出生後第一年內顯現。IV 的鱗屑通常在四肢伸側 (extensor surfaces) 最為明顯,而屈側 (flexural) 則倖免。尿布區 (diaper area) 傾向倖免。在大範圍區域可能有細小、白色的鱗屑。尤其在下肢(常是侵犯最嚴重的區域),鱗屑可能中央附著,邊緣有「龜裂」(穿過角質層的淺表裂隙)。這種邊緣翹起可導致皮膚摸起來粗糙。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
絲聚蛋白 (filaggrin) 是一種參與角蛋白中間絲聚集的表皮蛋白,是角質化外膜 (cornified envelope) 的重要成分,並有助於保留角質層中的水分。角蛋白絲形成一個網絡或細胞基質,賦予表皮角質細胞結構完整性。當角質細胞成熟為角質形成細胞時,角蛋白絲塌陷並交聯至角質化細胞外膜。絲聚蛋白以高分子量前驅物——前絲聚蛋白 (profilaggrin)——的形式合成,前絲聚蛋白含有多個絲聚蛋白分子,並定位於透明角質顆粒 (keratohyalin granules)。對 IV 病人表皮的生化研究顯示絲聚蛋白及其前驅物前絲聚蛋白的缺失或減少。IV 與異位性皮膚炎 (atopic dermatitis) 之間的關聯,臨床上早已被認識。編碼前絲聚蛋白的基因 (FLG) 之無效突變 (null mutations) 現已被證實是英-歐族群中異位性皮膚炎的強烈易感因子。在對常見過敏原敏感、過敏性鼻炎 (allergic rhinitis)、早發且持續性濕疹,以及在有異位性皮膚炎的情況下合併氣喘 (asthma) 的個體中,也發現強烈的關聯。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
在某些病例中,要區分輕微的 IV 與單純乾燥皮膚 (xerosis) 是困難的。對這種非常常見之疾病演進中的理解,正開始釐清潛在突變的譜系如何造成從乾燥症到嚴重 IV 的多樣化臨床嚴重度。此外,僅根據皮膚表現,患有嚴重 IV 的男性可能難以與 X 性聯隱性魚鱗癬 (X-linked recessive ichthyosis) 患者區分。IV 的組織病理學發現可變,特徵性的正角化過度 (orthohyperkeratosis) 與顆粒層缺失 (absent granular layer) 可能僅見於帶有兩個異常等位基因的個體。
臨床病程、預後與處置 (CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT)
許多其他發現常與 IV 相關被觀察到。手掌過度線狀紋路 (hyperlinear palms) 通常存在,部分病人可能有接近角皮症的掌蹠增厚。毛孔角化症 (keratosis pilaris) 常見,即使在輕微 IV 的個體中亦然,通常侵犯手臂外側、大腿伸側與臀部。儘管 IV 通常不被視為症候群性魚鱗癬,異位性 (atopy) 也常被觀察到,可表現為花粉熱 (hay fever)、濕疹 (eczema) 或氣喘。這些發現可能混淆準確診斷,因為手掌過度線狀紋路與毛孔角化症也可見於沒有 IV 的異位性個體。罕見情況下,IV 個體可能有出汗不足 (hypohidrosis) 合併熱不耐受。同一家族中受影響個體之間臨床表現的嚴重度有極大差異。此病通常在乾冷氣候中惡化,在溫暖、潮濕的環境中改善,後者可使疾病戲劇性地清除。大多數病人對規律塗抹潤膚劑 (emollients) 反應良好,而當有脫屑時,使用 α-羥基酸可減少脫屑。
X 性聯隱性魚鱗癬 (X-LINKED RECESSIVE ICHTHYOSIS)
在 1960 年代,X 性聯隱性魚鱗癬 (OMIM #308100) 在臨床上被與其他魚鱗癬區分開來。X 性聯隱性魚鱗癬發生於約每 1500 至 6000 名男性中 1 人。類固醇硫酸酯酶 (steroid sulfatase,芳基硫酸酯酶 C arylsulfatase C) 缺乏造成 XLI;在 90% 的病例中,STS 基因座的遺傳性缺失 (genetic deletion) 為致病原因。其餘為 STS 的點突變 (point mutations)。
臨床特徵 (CLINICAL FEATURES)
脫屑可能始於新生兒期,通常在伸側最為明顯,但屈側區域也有顯著侵犯。儘管脫屑的範圍與程度可變,X 性聯魚鱗癬通常可依臨床標準與遺傳模式與 IV 區分。後者傾向合併手掌與足底過度線狀紋路、毛孔角化症與異位性家族史。X 性聯魚鱗癬傾向有較嚴重的侵犯與較大的鱗屑(圖 47-2),而半數成年病人可能出現逗點狀 (comma-shaped) 角膜混濁。角膜混濁不影響視力,且可見於女性帶因者。受影響男性的隱睪症 (cryptorchidism) 風險增加,且獨立地,他們發生睪丸癌 (testicular cancer) 的風險亦增加。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
類固醇硫酸酯酶水解硫酸酯 (sulfate esters),包括硫酸膽固醇 (cholesterol sulfate) 與硫酸化類固醇激素。硫酸化的胎兒腎上腺激素去硫酸化為雌激素 (estrogens),並排泄至母體尿液中。胎兒胎盤中類固醇硫酸酯酶的缺失導致母體尿液雌激素低下,在某些妊娠中,導致分娩無法正常啟動或進展。在患有 X 性聯隱性魚鱗癬的男性中,類固醇硫酸酯酶活性在許多組織中減少或缺失,包括表皮、角質層、白血球,以及培養的纖維母細胞。已發現帶因女性的白血球類固醇硫酸酯酶濃度介於正常個體與受影響男性之間。
硫酸膽固醇濃度在血清、表皮與鱗屑中升高。類固醇硫酸酯酶是位於第 X 染色體 Xp22 之一群芳基硫酸酯酶中的一員。X 性聯魚鱗癬中超過 90% 的突變是缺失,常可藉由螢光原位雜交 (fluorescence in situ hybridization, FISH) 或陣列比較基因組雜交 (array-comparative genomic hybridization, CGH) 偵測,許多臨床實驗室皆有提供。診斷的確認亦可藉由發現血清硫酸膽固醇濃度升高來達成。在表皮中,類固醇硫酸酯酶催化硫酸膽固醇的水解。在小鼠中外用硫酸膽固醇可誘發脫屑性疾病,進一步支持硫酸膽固醇水解在角質形成細胞脫屑中的角色。硫酸膽固醇抑制降解角質橋粒 (corneodesmosomes) 的蛋白酶,導致脫屑受抑制與滯留性過度角化 (retention hyperkeratosis)。
診斷 (DIAGNOSIS)
皮膚與眼部發現、分娩延長的病史,以及 X 性聯傳遞的證據,可診斷 XLI。值得注意的是,現有相當比例的病人透過評估 α-胎兒蛋白 (α-fetoprotein)、人類絨毛膜促性腺激素 (human chorionic gonadotropin) 與雌三醇 (estriol) 濃度的母體篩檢,在子宮內即被辨識出來。由於類固醇硫酸酯酶為胎盤雌激素產生所必需,XLI 妊娠的三指標篩檢 (triple screen) 結果顯示雌三醇偏低。由於母體尿液雌激素大多源自胎兒腎上腺並經胎盤代謝,低濃度可反映胎兒異常或死亡,引起父母的擔憂。然而在 XLI 中,低濃度並不代表嚴重的胎兒病態。雖然皮膚切片甚少進行,組織病理學檢查顯示緻密的正角化過度 (compact orthohyperkeratosis)、棘層肥厚 (acanthosis)、乳頭瘤病 (papillomatosis) 與增厚的顆粒層。診斷 XLI 有許多可用的檢測,包括 STS 的 FISH、array-CGH、透過次世代定序 (next-generation sequencing) 偵測缺失與點突變,以及類固醇硫酸酯酶功能的酵素檢測。
臨床病程、預後與處置 (CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT)
雖然在大多數病例中 XLI 主要是皮膚疾病,但包含 STS 及相鄰硫酸酯酶的遺傳性缺失,可解釋涉及點狀軟骨發育異常 (chondrodysplasia punctata) 與 X 性聯魚鱗癬的重疊症候群。Kallmann 症候群的 X 性聯型(其中可見低促性腺素性腺低能症 hypogonadotropic hypogonadism 與嗅覺缺失 anosmia,常合併腎臟異常、肥胖、聯帶運動 synkinesis〔四肢的鏡像動作〕、顎裂 cleft palate 與痙攣性下肢輕癱 spastic paraplegia),亦可作為連續基因缺失症候群 (contiguous gene deletion syndrome) 的一部分與 X 性聯隱性魚鱗癬一同出現。因為這一點,以及因為與睪丸癌的關聯,X 性聯隱性魚鱗癬病人應被詢問是否有嗅覺缺失,並定期接受睪丸檢查。
當脫屑明顯時,長時間泡澡(無論有無碳酸氫鹽 bicarbonate)可協助脫屑。潤膚劑,包括含 α-羥基酸或乳酸者,可協助脫屑。
膠樣嬰兒與新生兒表現 (COLLODION BABY AND NEWBORN PRESENTATIONS)
大多數魚鱗癬病例(不含 IV 與 X 性聯魚鱗癬)在出生時即以紅色、脫屑的皮膚表現。在美國,此類病例的發生率估計為每 100,000 名活產中 5 至 10 例。其中一部分以膠樣膜 (collodion membrane) 表現,此膜在生命早期脫落,隨後發展出先天性魚鱗癬的特徵或正常皮膚。基因型並非膠樣嬰兒表現的唯一決定因子,因為許多帶有與膠樣膜相關之常見基因型者,僅以紅色脫屑皮膚表現。這兩種魚鱗癬新生兒表現之間,尚未辨識出致病機轉的差異,且兩者的處置大致相似。
臨床特徵 (CLINICAL FEATURES)
膠樣嬰兒 (collodion baby) 出生時被包裹在一層半透明、類羊皮紙樣的膜中,此膜緊繃並可能妨礙呼吸與吸吮。可能早產,造成病態。在生命的前 2 週,此膜破裂並剝落,可留下裂隙,伴隨對感染與水分流失之屏障的受損(圖 47-3)。
與膠樣膜相關的疾病:
常見 (Common)
■ 體染色體隱性先天性魚鱗癬(板層狀魚鱗癬、先天性魚鱗癬樣紅皮症、重疊型)
罕見 (Rare)
■ 瞼緣黏連-外胚層發育不良-唇/顎裂 (Ankyloblepharon-ectodermal dysplasia-cleft lip/palate, AEC) 症候群
■ 點狀軟骨發育異常 (Chondrodysplasia punctata)
■ 高雪氏病 (Gaucher disease)
■ Loricrin 角皮症 (Loricrin keratoderma)
■ 中性脂質儲積病 (Neutral lipid storage disease)
■ 自癒型膠樣嬰兒 (Self-healing collodion baby)
■ Sjögren–Larsson 症候群
■ 毛髮硫營養不良 (Trichothiodystrophy)
■ X 性聯出汗不足型外胚層發育不良 (X-linked hypohidrotic ectodermal dysplasia)
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
子宮內的水性環境促成膠樣表現的發展,此表現隨著孩子長大而轉變為廣泛的魚鱗癬表現型譜系(表 47-5)。這是 ARCI 的常見表現,較少見於數種其他型別的魚鱗癬,罕見於高雪氏病。此外,已有描述一種體染色體隱性、自癒型膠樣表現型,其皮膚在最初數週內大幅清除,並轉變為輕微受影響或正常的皮膚。
11 名瑞典與 4 名丹麥病人,患有導致乾燥症、手掌過度線狀紋路、紅頰 (red cheeks) 與無汗症 (anhidrosis) 的自我改善型魚鱗癬,被發現帶有 ALOX12B、ALOX3E 或 TGM1 的突變。
臨床病程、預後與處置 (CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT)
在照護膠樣嬰兒或其他患有魚鱗癬的新生兒時,考量感染風險可能增加、體溫調節困難,以及因經表皮水分流失增加所造成的高血鈉性脫水 (hypernatremic dehydration) 是必要的。新生兒照護應包括對感染與體溫、水分及電解質的仔細監測,以及保持脫落膜柔軟與潤滑以促進柔韌性與脫屑的措施。在需要時應使用適當的疼痛處置與眼部照護。這些新生兒通常受益於空氣為水分飽和的加濕保溫箱;其後可用濕敷接著塗抹溫和潤滑劑以進一步濕潤膜並達到最大柔韌度。在膠樣膜自發剝落之前,增厚的角質層可能在四肢等區域乾燥變硬並收縮,導致遠端腫脹、發紺 (cyanosis),少數情況下甚至壞死。藉由刮除術 (curettage) 或清創 (debridement) 釋放收縮帶可預防併發症。
體染色體隱性先天性魚鱗癬 (AUTOSOMAL RECESSIVE CONGENITAL ICHTHYOSIS)
體染色體隱性先天性魚鱗癬 (autosomal recessive congenital ichthyosis) 一詞用於描述一群異質性疾病,這些疾病在出生時即以皮膚的全身性侵犯表現。體染色體隱性魚鱗癬罕見,估計發生於約每 300,000 人中 1 人。
在較舊的文獻中,非水疱型先天性魚鱗癬樣紅皮症 (nonbullous congenital ichthyosiform erythroderma, NCIE,亦稱板層狀魚鱗癬,體染色體隱性遺傳) 被依臨床外觀(水疱 bullae)與遺傳模式,與 BCIE(亦稱表皮鬆解性過度角化 epidermolytic hyperkeratosis, EHK,體染色體顯性遺傳)區分開來。LI 一詞與 NCIE 交替使用並涵蓋一系列表現型,已導致某些混淆。我們現也了解到基因型本身並不決定表現型將是 LI 或 CIE。Williams 與 Elias 將 LI 與 NCIE(通常稱為先天性魚鱗癬樣紅皮症,一種較輕微的紅皮症型)區分開來,而這些描述詞在臨床實務中仍是有用的臨床描述詞。在 LI 中,可見大型、深色、板片狀的鱗屑,雖然嬰兒在出生時可能發紅,成人則少有或沒有紅皮症(圖 47-4 至圖 47-6)。在較嚴重、典型的 LI 表現中,臉部皮膚的緊繃導致對眼瞼與嘴唇的牽引,造成瞼外翻 (ectropion) 與唇外翻 (eclabium)。瘢痕性禿髮 (scarring alopecia),以頭皮周邊最為明顯,可能部分由髮際線的牽引所致。相對地,CIE 有全身性發紅與細小、白色的鱗屑(如圖 47-7 與圖 47-9)。典型 CIE 病人少有或沒有瞼外翻、唇外翻或禿髮。然而,許多病人並不能整齊地歸入這兩種臨床描述,因其同時具有 LI 與 CIE 的特徵,臨床表現型介於兩種疾病之間。因此,將這兩種獨特表現視為一個譜系的兩端是有用的,兩端之間存在著紅斑程度與鱗屑粗糙度不一的臨床表現型漸層。個別特徵,如膠樣膜(前述)、瞼外翻與禿髮,可跨此譜系出現。儘管以生化與超微結構觀察來精煉這些疾病分類的嘗試未能產生一致且可重現的分類方案,鑑定這些疾病潛在的特定分子缺陷譜系可能有助於分類。截至目前,突變鑑定發現 ARCI 由 10 個對角質形成細胞之細胞間脂質層或角質化外膜形成重要的不同基因所致,包括 TGM1 (OMIM #191995)、ALOX12B (OMIM #603741)、ALOXE3 (OMIM #607206)、NIPAL4 (OMIM #609383)、CYP4F22 (OMIM #611495)、ABCA12 (OMIM #607800)、PNPLA1 (OMIM #612121)、CERS3 (OMIM #615276)、SDR9C7 (OMIM #609767) 與 SULT2B1 (OMIM #604125)。
臨床特徵 (CLINICAL FEATURES)
板層狀魚鱗癬表現型 (LAMELLAR ICHTHYOSIS PHENOTYPE)
ARCI 的 LI 表現型在出生時即顯現,新生兒通常以包裹在膠樣膜中的形式表現(見圖 47-3)。此時皮膚可能發紅。隨時間經過,皮膚發展出大型、板片狀的鱗屑,大多數中央附著並有抬高的邊緣。鱗屑傾向在下肢最大,該處由淺表裂隙分隔的大型板片狀鱗屑可造成類似乾涸河床的外觀。在兒童期至成年期,紅斑的程度可能有所變化。LI 中手掌與足底的侵犯可變,範圍從輕微的過度線狀紋路到嚴重的角皮症。此表現型最常與 TGM1 突變相關,但即使是單一基因的突變也會造成高度可變的表現型(見圖 47-4 至圖 47-6)。LI 中嘴唇與黏膜傾向倖免,但附屬器構造可能被附著的堅硬鱗屑所損害。頭皮上的厚角質層傾向包覆毛髮,並合併皮膚的緊繃,可能導致瘢痕性禿髮,以頭皮周邊最為顯著。過度角化干擾正常汗腺功能,導致出汗不足 (hypohidrosis),但損害程度因病人而異。部分病人有嚴重的熱不耐受,必須警覺以避免過熱。泳衣型魚鱗癬 (bathing suit ichthyosis) 是 LI 的一個亞型,受影響個體發展出 LI 典型的脫屑但僅限於泳衣覆蓋區域。此分布與皮膚較溫暖的區域相關。在這些區域中發現轉麩醯胺酸酶減少,並在受影響個體中鑑定出 TGM1 獨特的溫度敏感性 (temperature-sensitive) 突變。
先天性魚鱗癬樣紅皮症表現型 (CONGENITAL ICHTHYOSIFORM ERYTHRODERMA PHENOTYPE)
與 LI 相同,ARCI 的 CIE 表現型在出生時即顯現,新生兒通常以緊繃、發亮的膠樣膜表現。膜脫落後,CIE 嬰兒的皮膚仍維持發紅,通常伴有細小、白色的全身性鱗屑(見圖 47-7 與圖 47-8)。在小腿上,鱗屑可能較大且較深色。與 LI 相對,典型的 CIE 表現可能少有或沒有瞼外翻、唇外翻或禿髮。如同 LI,出汗能力有很大變異,CIE 病人可能僅有極少出汗合併嚴重的熱不耐受。黏膜通常倖免。手掌與足底侵犯可變。指甲可能有脊紋 (ridging),但常倖免。如同所有魚鱗癬,皮膚與指甲的皮癬菌 (dermatophyte) 感染常見。
小丑魚鱗癬表現型 (HARLEQUIN ICHTHYOSIS PHENOTYPE)
魚鱗癬中一種戲劇性、嚴重且有時致命的表現是小丑魚鱗癬 (harlequin ichthyosis, OMIM #242500)(圖 47-9)。此類孩童常為早產,出生時帶有厚實、發亮的角質層板塊,被深的紅色裂隙分隔,這些裂隙傾向形成幾何圖案,如 16 與 17 世紀義大利即興喜劇 (Commedia dell’Arte) 中小丑服裝的補丁圖案。耳朵發育不良或缺失,並有明顯的瞼外翻與唇外翻。指尖呈錐形,指甲過度凸起。在 1980 年之前一律致命,現有相當比例的小丑魚鱗癬病人存活下來,伴隨持續且嚴重的紅皮症(圖 47-10)。
Netherton 症候群 (OMIM #256500) 是一種以魚鱗癬、毛幹結構異常與異位性為特徵的體染色體隱性疾病。當存在時,一項關鍵發現是環狀線狀魚鱗癬 (ichthyosis linearis circumflexa, ILC),這是位於紅斑性斑塊邊緣的特徵性、多環狀、蛇行狀、遷移性、雙邊緣鱗屑(圖 47-11)。然而,其他病人可能以較嚴重的紅皮症表現,無論有無 ILC(圖 47-12)。組織病理學檢查非特異性,但缺失或變薄的角質層具高度提示性。大多數病人有一種特定的毛幹異常,稱為套疊性脆髮症 (trichorrhexis invaginata),其中遠端毛段套入近端毛段,在顯微鏡檢查下形成球-臼樣 (ball-and-socket-like) 變形。這也稱為「竹節狀毛髮」(bamboo hair),由內髮根鞘 (internal root sheath) 的異常角化所致。應檢查多個區域的毛髮,因為只有 20% 至 50% 的毛髮可能受影響;特徵性異常可能更常在眉毛上被觀察到。毛髮缺陷在出生時可能無法偵測,並可能隨年齡消失。這些病人可能發生類異位性體質 (atopy-like diathesis),表現為異位性皮膚炎、氣喘或嚴重食物過敏(尤其是對堅果),以及血清免疫球蛋白 E (immunoglobulin E) 的顯著升高。在某些病人中,亦可能存在全身性胺基酸尿症 (aminoaciduria)、輕度發展遲緩,以及細胞免疫受損。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
板層狀魚鱗癬與先天性魚鱗癬樣紅皮症表現型 (LAMELLAR ICHTHYOSIS AND CONGENITAL ICHTHYOSIFORM ERYTHRODERMA PHENOTYPES)
LI 與 CIE 表現型代表一個臨床譜系,且許多相同基因的突變為兩種表現型之基礎。在 LI 表現型中牽涉的一個主要基因是 TGM1。即使是此基因,仍有極大的表現型變異(見圖 47-6 至圖 47-8)。TGM1 編碼轉麩醯胺酸酶 1 (transglutaminase 1);轉麩醯胺酸酶透過形成 ε-(γ-麩醯胺酸)賴胺酸異二肽鍵 (ε-(γ-glutamyl)lysine isodipeptide bonds) 催化蛋白質的鈣依賴性交聯。在角質層形成過程中,轉麩醯胺酸酶催化細胞蛋白的交聯,包括兜甲蛋白 (involucrin)、loricrin、小富脯胺酸蛋白 (small proline-rich proteins)、角蛋白、絲聚蛋白等。所得的蛋白複合物沉積於質膜內側以形成角質化外膜。轉麩醯胺酸酶也將層板小體 (lamellar bodies) 分泌至細胞間隙的神經醯胺附著至角質化外膜蛋白(特別是兜甲蛋白),因此對角質層之蛋白與脂質成分的形成皆很重要。
在人類皮膚-免疫缺陷小鼠異種移植模型中,將轉麩醯胺酸酶-1 基因轉入 LI 病人之轉麩醯胺酸酶-1 缺乏的角質細胞中,除了恢復皮膚屏障功能外,也使轉麩醯胺酸酶表現與表皮結構正常化。更近期,外用包裹於微脂體 (liposomes) 的重組轉麩醯胺酸酶-1,已被證實能使小鼠異種移植的組織學與功能正常化。
編碼脂氧合酶 (lipoxygenases) ALOXE3(見圖 47-9)與 ALOX12B(見圖 47-10)之基因的體染色體隱性突變,被發現會造成 CIE。脂氧合酶催化由多元不飽和脂肪酸形成氫過氧化物 (hydroperoxides)。在皮膚中,ALOXE3 與 ALOX12B 的產物在生成被併入角質形成細胞脂質外膜之氧化神經醯胺 (oxidized ceramides) 中扮演核心角色。
總計,10 個基因的突變可造成 ARCI 表現型,這些包括:TGM1 (OMIM #190195)、ALOX12B (OMIM #603741)、ALOXE3 (OMIM *607206)、NIPAL4/Ichthyin (OMIM #609383)、CYP4F22 (OMIM #611495)、ABCA12 (OMIM #607800)、PNPLA1 (OMIM #612121)、CERS3 (OMIM #615276)、SDR9C7 (OMIM #609767) 與 SULT2B1 (OMIM #604125)。
小丑魚鱗癬表現型 (HARLEQUIN ICHTHYOSIS PHENOTYPE)
小丑魚鱗癬起因於 ABCA12 突變的體染色體隱性遺傳,ABCA12 編碼一種參與層板顆粒分泌與表皮脂質運輸的三磷酸腺苷 (ATP)-結合卡匣 (ABC) 轉運蛋白。雖然提前終止、功能喪失 (loss-of-function) 突變最常為小丑魚鱗癬的基礎,ABCA12 的錯義突變 (missense mutations) 已在較不嚴重的 ARCI 個體中被鑑定出來(圖 47-14)。在小丑魚鱗癬中,找不到正常的層板顆粒;取而代之的是缺乏內部結構的小囊泡。也沒有層板顆粒內容物排放至細胞間隙所形成、介於顆粒細胞與角質化細胞之間的脂質層板 (lipid lamellae) 的證據。
Netherton 症候群被發現由 SPINK5 突變所致,此基因編碼 LEKTI(淋巴上皮 Kazal 型相關抑制劑 lymphoepithelial Kazal-type related inhibitor)。LEKTI 是一種絲胺酸蛋白酶抑制劑,主要表現於上皮與淋巴組織,可能在發炎路徑的下調 (downregulation) 中很重要。它抑制降解角質橋粒的蛋白酶,因而允許提前脫屑。LEKTI 多型性與常見的異位性及異位性皮膚炎相關。Netherton 症候群的早期報告即認識到細胞免疫降低的徵象,表現為病毒性疣 (viral warts) 與對微生物抗原的皮膚試驗陰性。此種有缺陷細胞免疫的危險,因 Folster-Holst 及其同事的報告而被強化,他們在一名 28 歲女性的外陰菜花 (vulvar condyloma) 中鑑定出致癌性人類乳突病毒 (oncogenic human papillomavirus),該病灶迅速轉化為侵襲性鱗狀細胞癌 (squamous cell carcinoma)。依我們的經驗,所有患有 Netherton 的成人在皺褶間區域都有乳突瘤樣 (papillomatous) 變化,但乳突病毒在這些病灶發展中的角色尚未確立。
診斷 (DIAGNOSIS)
臨床特徵與病史對於建立 ARCI 的診斷至為核心。基因檢測的進展,無論透過研究途徑(見「魚鱗癬及相關皮膚疾病登錄」一節)或臨床實驗室,已使遺傳診斷得以快速確定。這些檢測使用一組基因的次世代定序,或透過外顯子定序評估選定的基因子集。基因檢測可確認診斷,並針對潛在的疾病共病進行諮詢。支持各表現型的臨床特徵包括以下:
LI 與 CIE 通常在出生時以膠樣膜表現,膜脫落後顯露出 LI 的厚實、附著鱗屑,或 CIE 的紅皮症與較細鱗屑。組織病理學檢查甚少有助於 LI 或 CIE 的診斷,兩種疾病皆顯示非特異性的棘層肥厚與過度角化。小丑魚鱗癬有經典的臨床表現,出生時有盔甲狀 (armorlike) 鱗屑板塊與深裂隙,脫落後顯露出嚴重的紅皮症與脫屑。Netherton 症候群在異位性特徵的背景下,以毛幹異常表現,無論有無 ILC 或紅皮症。這些臨床發現為 Netherton 症候群的診斷提供強力支持。
臨床病程、預後與處置 (CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT)
板層狀魚鱗癬與先天性魚鱗癬樣紅皮症 (LAMELLAR ICHTHYOSIS AND CONGENITAL ICHTHYOSIFORM ERYTHRODERMA)
以口服類視黃醇 (oral retinoids) 治療可改善或預防這些表現型的某些後遺症。病人常注意到出汗增加,熱耐受性改善。儘管類視黃醇治療可造成眼瞼炎 (blepharitis) 甚至結膜炎 (conjunctivitis),LI 與 CIE 病人通常對其耐受良好。此外,全身性類視黃醇(在某些情況下為外用類視黃醇)治療減少眼周厚鱗屑的能力,可降低發展出瞼外翻的傾向。儘管如此,患有嚴重脫屑與瞼外翻的病人通常需要仔細的眼部維護。由於瞼外翻,眼瞼可能無法完全閉合,尤其在睡眠時;白天以人工淚液 (liquid tears)、夜間以眼用潤滑劑 (ophthalmic lubricants) 保濕,可預防暴露性角膜炎 (exposure keratitis)。帶有 ALOXE3 與 ALOX12B 突變的個體傾向有較少的脫屑,相較於帶有 TGM1 突變者,與此觀察一致的是近期一項研究顯示瞼外翻在 CIE(35%)中較 LI(57%)中少見。
小丑魚鱗癬 (HARLEQUIN ICHTHYOSIS)
新生兒重症照護的進展,連同藉由審慎使用全身性類視黃醇治療以促進脫屑,已帶來存活率的改善,並使「小丑胎兒 (harlequin fetus)」一名被「小丑嬰兒 (harlequin baby)」取代。這些孩童在新生兒期處於風險中。經皮膚的異常水分流失與不良的體溫調節,導致體液與電解質失衡的風險。
這些嬰兒也有感染的風險,感染始於皮膚,但同時(因為不良的體溫調節)不會顯現出感染的常見徵象。緊繃的皮膚可能限制正常呼吸。在新生兒期以全身性類視黃醇治療可促進膜的脫屑,並與改善的結果相關。部分嬰兒與孩童曾有生長遲滯,需要管灌餵食。小丑鱗屑脫落後,紅皮症明顯。在孩童與成人中,外用與全身性類視黃醇有助於瞼外翻與過度角化的處置。
Netherton 症候群 (NETHERTON SYNDROME)
出生時,受影響的孩童可能以全身性紅皮症表現,在某些個體中,紅皮症終生持續,表現型落在 ARCI 的 LI–CIE 譜系上。在其他人中,紅皮症消退,隨後出現 ILC。嬰兒與孩童可能有餵食問題,伴隨吸收不良與生長遲滯。搔癢可能很劇烈,搔抓可導致屈側的苔癬化 (lichenification)。Tacrolimus 軟膏,一種外用免疫調節劑(見第 192 章),對常見的異位性皮膚炎有效且全身吸收極少。然而,Netherton 症候群因異常的皮膚屏障而變得複雜,允許增加的經皮吸收與相關的全身性毒性風險。在使用如 tacrolimus 等外用藥時應考量此點,因為可能需要監測血清濃度;在使用外用類固醇時亦然,因為已有醫源性庫欣症候群 (iatrogenic Cushing syndrome) 的報告。此外,外用與全身性類視黃醇應在 Netherton 症候群病人中避免,因其可能進一步惡化病情。
角蛋白病性魚鱗癬 (KERATINOPATHIC ICHTHYOSES)
角蛋白病性魚鱗癬 (keratinopathic ichthyoses) 起因於編碼角蛋白的基因突變,角蛋白是對細胞結構完整性至為核心之中間絲網絡的成分。落入角蛋白病性魚鱗癬譜系內的表現型包括 EI、淺表性表皮鬆解性魚鱗癬 (superficial epidermolytic ichthyosis, SEI,Siemens 水疱型魚鱗癬 ichthyosis bullosa of Siemens)、Curth-Macklin 豪豬狀魚鱗癬 (ichthyosis hystrix Curth-Macklin)、環狀 EI (annular EI),以及五彩碎紙樣魚鱗癬 (ichthyosis with confetti, IWC)。
臨床特徵 (CLINICAL FEATURES)
表皮鬆解性魚鱗癬 (EPIDERMOLYTIC ICHTHYOSIS)
1902 年,Brocq 描述了水疱型魚鱗癬樣紅皮症,並將水疱型與非水疱型 CIE 區分開來。原始描述包括三名不相關的病人,其臨床表現各異。然而,這可能是 EI (OMIM #113800) 的首次描述。此病以表皮角質細胞空泡變性(即表皮溶解 epidermal lysis)與相關過度角化的獨特組織病理學特徵命名。EI 也稱為 EHK 與 BCIE,這些較早的描述性名稱分別代表特徵性的組織病理學變化,或水疱型、新生兒期表現伴隨後續脫屑與發紅。EI 以體染色體顯性性狀傳遞,盛行率約為每 200,000 至 300,000 人中 1 人。其自發性突變頻率高,多達半數病人沒有家族史。此病通常在出生時以水疱、發紅與剝皮表現,臨床檢查時可能被誤認為表皮鬆解水疱症 (epidermolysis bullosa) 或葡萄球菌性燙傷樣皮膚 (staphylococcal scalded skin)(圖 47-15A)。因為新生突變頻率高,此病可能出乎意料,診斷可能未知。新生兒可能需要重症照護並監測體液與電解質。
具專業性的皮膚照護可將水疱降至最低並增進糜爛 (erosions) 的癒合,可包括潤滑以減少摩擦與機械創傷、保護性墊襯,以及專業傷口敷料。有廣泛糜爛的新生兒易發生細菌感染與敗血症,謹慎選擇的外用與全身性抗生素可將感染範圍降至最低。隨時間經過,可能發展出全身性過度角化,可能伴有或不伴有紅皮症。EI 皮膚通常有特徵性氣味,被認為與混合菌叢的繼發感染有關。組織病理學顯示增厚的角質層與上表皮的空泡變性,導致組織學術語 EHK。空泡變性通常侵犯上表皮,偶爾侵犯所有基底上層角質細胞。顆粒細胞表現出緻密、增大、形狀不規則的團塊,看似透明角質顆粒(圖 47-15B)。所有 EI 病人都有特徵性的波紋狀 (corrugated) 鱗屑,在身體皺褶區域變得更明顯(圖 47-16 與圖 47-17)。EI 有顯著的臨床異質性,表現型包括:孤立的厚掌蹠過度角化、全身性棘刺狀鱗屑、紅皮症的遷移性斑塊、伴淺表剝皮鱗屑的角皮症,以及全身性剝脫性紅皮症。
線狀魚鱗癬樣紅皮症 (LINEAR ICHTHYOSIFORM ERYTHRODERMA)
線狀 EI (linear EI),表皮母斑 (epidermal nevus) 的一種變異,以線狀鑲嵌 (mosaic) 模式表現,由胚胎發生期間合子後 (postzygotic)、自發的突變所致。過度角化區與正常皮膚交替,常沿 Blaschko 線分布成條紋狀。這些可能僅限於少數條紋,常以中線為界,或可能有許多條紋,伴隨廣泛、斑片狀的侵犯。線狀 EI 已被發現起因於 KRT1 與 KRT10 的體細胞突變 (somatic mutations),與全身性疾病中所發現者相同。
若鑲嵌現象影響到性腺組織,患有線狀 EI 的個體可將突變傳給後代,導致後代發生全身性 EI。
淺表性表皮鬆解性魚鱗癬 (SUPERFICIAL EPIDERMOLYTIC ICHTHYOSIS)
SEI(Siemens 水疱型魚鱗癬)是一種罕見的體染色體顯性遺傳性皮膚病。病人出生時有發紅與水疱。發紅在隨後數週至數月內消退,皮膚發展出波紋狀過度角化,特別是在屈側區域(圖 47-18)。在某些區域,皮膚可能有苔癬化外觀。如同 EI,表皮脆弱;然而其脆弱性較為淺表。這可導致最上層表皮(主要是角質層)的脫失,產生一種特徵性、項圈樣 (collarette-like) 的凹陷區域,被描述為「mauserung」(換羽 molting)。組織學上,表皮顯示類似 EHK 的過度角化與空泡化,但侷限於顆粒層。
在編碼角蛋白 2 的基因中已發現突變,角蛋白 2 是基底上層表皮的一種分化角蛋白,表現於較淺表的表皮層。
環狀表皮鬆解性魚鱗癬 (ANNULAR EPIDERMOLYTIC ICHTHYOSIS)
環狀 EI 是一種罕見的體染色體顯性疾病,在出生時或生命最初數月內以嚴重、間歇性的脫屑與水疱表現,並在青春期消退。殘留、有限的斑塊伴隨波紋狀鱗屑與紅斑,主要見於屈側與皺褶間皮膚(圖 47-19)。在某些病例中,可見廣泛紅斑伴水疱與膿疱的爆發性發作。病人隨後發展出廣泛、遷移性、多環狀與環狀的脫屑性斑塊。光學與電子顯微鏡顯示 EI 的發現。已發現 KRT10 與 KRT1 的突變。
五彩碎紙樣魚鱗癬 (ICHTHYOSIS WITH CONFETTI)
此罕見疾病也稱為雜色魚鱗癬 (ichthyosis variegata) 與先天性網狀魚鱗癬樣紅皮症 (congenital reticulated ichthyosiform erythroderma),這些描述性術語未能捕捉其一生中的臨床演變。IWC 以先天性紅皮症、耳廓畸形與手指錐形化表現,這導致常見的 CIE 臨床診斷。在兒童期,發展出掌蹠角皮症,嚴重度從輕微到嚴重不等。大多數 IWC 個體從兒童期開始發展出小島狀的正常外觀皮膚(圖 47-20),雖然也有較晚發病的報告。正常皮膚區域已被證實源自第 17q 或 12q 染色體之雜合性喪失 (loss of heterozygosity),透過編碼角蛋白 10 (KRT10) 與角蛋白 1 (KRT1) 之致病突變的有絲分裂重組 (mitotic recombination)。組織病理學顯示在白點內從受影響的表皮到正常皮膚有清楚的轉變。在由 KRT10 突變所致的 IWC 個體中,可見帶狀角化不全 (bandlike parakeratosis)、乾癬樣棘層肥厚 (psoriasiform acanthosis) 與空泡化、雙核的上表皮角質細胞,而電子顯微鏡顯示核周絲收縮與橋粒上中間絲的不良包覆。在由 KRT1 突變所致的 IWC 個體中,受影響皮膚顯示乾癬樣棘層肥厚、明顯的透明角質顆粒、伴罕見雙核細胞的核周空泡化,以及厚的過度角化,而電子顯微鏡顯示核周絲分離。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
在角質細胞內,角蛋白中間絲形成一個精密的網絡,賦予細胞結構穩定性。在毛囊間表皮 (interfollicular epidermis) 的基底上層、分化中的角質細胞中,此網絡主要由角蛋白 1 與 10 形成,兩者聚合形成中間絲。在顆粒層中,角蛋白 2 部分取代中間絲中的角蛋白 1。
表皮鬆解性魚鱗癬 (EPIDERMOLYTIC ICHTHYOSIS)
在電子顯微鏡檢查中,可觀察到絲的成簇 (clumping) 始於第一個基底上層。這些聚集的絲是含有基底上層角蛋白 1 與 10 的角蛋白中間絲團塊。當在角質細胞中表現時,EI 與 SEI 中的突變型角蛋白聚集並成簇,伴隨中間絲網絡的塌陷與隨之而來的細胞溶解 (cytolysis)。聚集物具細胞毒性,且 KRT10 突變已被證實會破壞表皮分化與脂質通透性屏障的形成,同時誘發表皮過度增生。
在許多 EI 家系中已鑑定出編碼角蛋白 1、2 或 10 之基因的突變。在許多病例中,嚴重的掌蹠侵犯暗示 KRT1 突變;這可能反映了 K9(一種僅出現於掌蹠皮膚之基底上層表皮的角蛋白)與 K10 在掌蹠上皮中的「冗餘性 (redundancy)」。在患有表皮鬆解性掌蹠角皮症 (epidermolytic PPK, Vörner) 的家族中已發現 KRT9 突變(見第 48 章)。
五彩碎紙樣魚鱗癬 (ICHTHYOSIS WITH CONFETTI)
IWC 不以明顯的皮膚脆弱性為特徵,電子顯微鏡顯示無絲聚集的證據,雖然可見核周絲收縮。在細胞中表現 KRT10 突變導致中間絲網絡塌陷與 K10 在核仁 (nucleolus) 內的核內累積 (intranuclear accumulation),而表現 KRT1 突變同樣導致中間絲網絡塌陷與 K1 的核內累積。由於所有回復突變 (revertant) 斑點皆源自有絲分裂重組,且正常皮膚斑點隨時間在數量與大小上增加,IWC 的 KRT1 與 KRT10 突變似乎影響同源重組 (homologous recombination),並賦予正常角質細胞選擇優勢。
診斷 (DIAGNOSIS)
診斷結合臨床發現、組織病理學與基因分析。臨床特徵可指引診斷。例如,兒童期出現正常皮膚的白點提示 IWC,但出生時水疱、隨時間水疱頻率減少的病史,以及顯示表皮溶解與顆粒層增厚的組織學,則提示 EI 或 SEI。基因檢測能使診斷精確,可在 KRT1、KRT10 或 KRT2 中找到突變。
臨床病程、預後與處置 (CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT)
雖然 EI 與 SEI 中廣泛水疱的新生兒表現可能很戲劇性,但在生命最初數週,過度角化變得更明顯,水疱變得更侷限於摩擦區域。厚過度角化的區域,不柔韌且有堅硬、粗糙的表面,易受機械創傷。在患有豪豬狀 (hystrix type)、類豪豬針般過度角化的病人中,粗糙表面在物體掃過皮膚表面時造成高度牽引,這些物體傾向勾住過度角化的角質並將其剝離。外用藥如潤滑劑與角質溶解劑可減少增厚、粗糙的區域,並有助於將水疱與糜爛降至最低。相對地,有紅皮症與剝皮、沒有過度角化棘刺厚區的病人,對角質溶解劑的需求較少,但仍需要潤滑劑。急性惡化可能由皮膚感染引起。皮膚的細菌感染常見,常導致水疱增多,可能需要頻繁以外用與口服抗生素治療。稀釋漂白水浴 (dilute bleach baths) 有助於控制細菌氣味與菌落定植,而碳酸氫鹽浴 (bicarbonate baths) 可促進脫屑。外用類視黃醇可局部減少脫屑,全身性類視黃醇可顯著減少過度角化,減少梳理與沐浴時間。劑量必須緩慢滴定,因為脆弱性可能隨較高劑量增加,尤其在 EI 中。基於尚未充分了解的原因,對成人而言,水疱與感染比兒童 EI 病人來得不成問題。
接合蛋白疾病 (CONNEXIN DISORDERS)
編碼接合蛋白 (connexins) 之基因的突變導致紅斑角皮症 (erythrokeratoderma),表現型包括角膜炎-魚鱗癬-聾症候群 (keratitis ichthyosis deafness syndrome) 與可變性與進行性紅斑角皮症 (erythrokeratodermia variabilis et progressiva, EKVP)。這些疾病有重疊的臨床特徵與表現型變異,即使在同一家系內亦然。
臨床特徵 (CLINICAL FEATURES)
EKVP (OMIM #133200) 代表一系列表現型,最初被描述為進行性對稱性紅斑角皮症 (progressive symmetric erythrokeratodermia, PSEK) 與可變性紅斑角皮症 (erythrokeratodermia variabilis, EKV),但被發現有共同的遺傳基礎。進行性對稱性紅斑角皮症表現型於 1911 年首次由 Darier 明確描述,特徵為界限分明、紅斑性、過度角化的斑塊,對稱分布於四肢與臀部,常及於臉部(圖 47-21)。軀幹傾向倖免,但手掌與足底可能受侵犯。斑塊在出生後不久出現,在最初數年緩慢進展,然後在兒童期早期穩定。斑塊通常在位置與外觀上維持穩定,但可能在青春期部分消退。可變性紅斑角皮症表現型於 1925 年由 Mendes da Costa 描述為一種罕見疾病,通常在出生時或生命第一年內表現。已有描述全身性侵犯(見圖 47-25,特徵為持續的紅棕色過度角化斑塊與加強的皮膚紋路)與局部侵犯(範圍有限,特徵為界限清楚、過度角化的斑塊,對稱排列並維持相對固定數月至數年)兩種型態。雖然許多特徵與 PSEK 相同,EKV 也表現出界限清楚、遷移性的紅斑塊,大小從數公分到許多公分不等。這些地圖樣、有形的紅斑塊在數分鐘至數小時內出現或消退;部分個體抱怨這些部位有灼熱感,但其他人則無症狀。紅斑塊獨立於過度角化而發展。可能存在掌蹠過度角化,但毛髮與黏膜不受影響。組織病理學特徵包括過度角化、棘層肥厚、乳頭瘤病與微血管擴張。被嚴重乳頭瘤病與乳頭上方變薄所侵犯的表皮,可能造成「教堂尖塔 (church spire)」外觀。EKVP 以體染色體顯性模式遺傳,伴不完全外顯率 (incomplete penetrance) 與可變的表現度 (variable expressivity),雖然已有罕見的隱性病例被描述。
角膜炎-魚鱗癬-聾症候群 (KERATITIS–ICHTHYOSIS–DEAFNESS SYNDROME)
KID 症候群 (OMIM #148210) 是一種罕見疾病,特徵為角膜炎(伴進行性角膜混濁 progressive corneal opacification)、魚鱗癬與聾(神經感覺性 neurosensory)。多種外胚層組織的侵犯使 KID 症候群符合外胚層發育不良 (ectodermal dysplasia) 的資格。大多數病例符合體染色體顯性遺傳。
此病以離散的紅斑性斑塊為特徵,可能有輕度、全身性的過度角化(圖 47-22)。這些獨特的斑塊可能有清楚的邊界與疣狀外觀伴結痂,在臉部可能顯著地呈有形與對稱。嘴部周圍的溝紋造成特徵性面容。可能有明顯的毛囊性過度角化 (follicular hyperkeratosis),可導致頭皮的瘢痕性禿髮。幾乎總是可見「皮革樣 (leather-like)」掌蹠角皮症。指甲變化的描述各異,從缺失、出生後延遲出現、萎縮或脆弱,到增厚伴角質層 (cuticle) 喪失或「粗糙」、甲下過度角化與白甲 (leukonychia)。牙齒可能很小。聽覺誘發電位 (auditory evoked potential) 研究允許在嬰兒期偵測聽力缺損。角膜炎可能發展。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
可變性與進行性紅斑角皮症 (ERYTHROKERATODERMIA VARIABILIS ET PROGRESSIVA)
在 EKVP 家系中,已鑑定出 GJB3、GJB4 與 GJA1(分別編碼接合蛋白 31、30.3 與 43)的突變。接合蛋白 (connexins) 是一群蛋白質,聚集形成間隙連接 (gap junctions),這是細胞間通訊的重要通道。此細胞間訊息系統對於維持組織恆定、生長控制、發育,以及細胞對刺激的同步反應至為關鍵。在細胞中,接合蛋白於高基氏體 (Golgi) 中寡聚化形成同型或異型的六聚體連接子 (hexameric connexons),被運送至細胞膜,在該處可作為非連接半通道 (nonjunctional hemichannels) 運作,或與鄰近細胞中的接合蛋白對接。突變型接合蛋白可表現出透過間隙連接之細胞間通訊的受損,以及半通道的滲漏。造成此病理的關鍵缺陷尚未確立。
角膜炎-魚鱗癬-聾症候群 (KERATITIS–ICHTHYOSIS–DEAFNESS SYNDROME)
在偶發病例與家族性 KID 症候群中已偵測到編碼接合蛋白 26 之基因 GJB2 的顯性突變。表現突變型接合蛋白 26 之細胞的功能研究顯示,螢光示蹤劑無法透過間隙連接通道傳至鄰近細胞,與細胞間通訊的破壞一致。突變型接合蛋白 26 也與接合蛋白 43 形成不良的間隙連接與滲漏的異型半通道。在編碼接合蛋白 26 的同一基因 (GJB2) 中,不同的突變也在一個患有殘毀性掌蹠角皮症(Vohwinkel 病;見第 48 章)與聾(無魚鱗癬)的家族中被發現。對編碼多種接合蛋白之基因的突變鑑定,凸顯了接合蛋白介導之透過間隙連接的細胞間通訊在外胚層組織發育與維持中的角色。接合蛋白 26、30 與 31 表現於耳蝸 (cochlea) 與表皮的複層上皮,這些蛋白的異常可造成感覺神經性聽力障礙 (sensorineural hearing impairment) 與皮膚疾病。
臨床病程、預後與處置 (CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT)
可變性與進行性紅斑角皮症 (ERYTHROKERATODERMIA VARIABILIS ET PROGRESSIVA)
受影響個體在出生時常有正常皮膚,初始病灶於嬰兒期或兒童期出現。皮膚侵犯可在個體一生中起伏消長。全身性類視黃醇治療改善過度角化病灶,當有有形紅斑塊存在時,也可能使其清除。過度角化的皮膚病灶可能由皮膚創傷觸發,而紅斑塊可能由溫度變化觸發。
角膜炎-魚鱗癬-聾症候群 (KERATITIS ICHTHYOSIS DEAFNESS SYNDROME)
KID 症候群中皮膚的嚴重度可有顯著差異,尤其在新生兒期。角膜炎與聾的嚴重度可變;當存在時,可能呈進行性。發展性腦部缺陷常見且常無症狀,有人建議常規磁振造影 (magnetic resonance imaging)。受影響個體可能對細菌、黴菌或病毒感染易感性增加。皮膚與舌頭的鱗狀細胞癌亦曾被報告。與許多其他魚鱗癬疾病相對,以口服類視黃醇治療這些病人被報告益處甚少,且可能加劇角膜新生血管形成。GJB2 中的 p.A88V 與 p.G45E 突變與新生兒致死性相關,主要由呼吸衰竭所致。
剝皮性皮膚疾病 (PEELING SKIN DISORDERS)
臨床特徵 (CLINICAL FEATURES)
剝皮症候群 (peeling skin syndromes, PSSs; OMIM #270300) 最初在病例報告中被描述,後來被分類為以肢端為主者、全身性且非發炎性者,以及全身性且發炎性者。皮膚剝落常為間歇性,病人缺乏大多數魚鱗癬所見的全身性過度角化。這少數早期病例清楚顯示這是一群非常異質性的疾病。在過去 12 年中,已從一至四個家族報告了五個隱性基因突變,其發現被描述為 PSSs。共同的臨床特徵包括無痛、易剝落且不留疤的皮膚,因摩擦與濕度而加劇,初次出現於生命第二天至第一個十年中期之間。許多人有搔癢,部分據報有皮膚與全身性過敏。
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
CDSN(編碼角質橋粒蛋白 corneodesmosin)的突變造成伴異位性、搔癢與顯著皮膚屏障功能缺損的發炎性 PSS,稱為 PSS1 (OMIM #270300)。受影響個體有淺表脫屑、魚鱗癬樣紅皮症,以及兒童期頻繁的皮膚感染。角質橋粒蛋白是橋粒 (desmosomes) 與角質橋粒 (corneodesmosomes) 在顆粒層細胞至角質層之過渡處的成分。CDSN 突變導致角質橋粒蛋白表現喪失,消除了上表皮與角質層之間的黏附並破壞終末分化。分化缺陷的一個後果是激肽釋放酶 5 (kallikrein 5) 的上調,這是一種角質層胰蛋白酶樣酵素,進一步加劇剝皮表現型。在多個患有肢端剝皮的家系中,已鑑定出編碼轉麩醯胺酸酶 5 (transglutaminase 5) 之基因的突變,此為 PSS2 (OMIM #609796),轉麩醯胺酸酶 5 是一種在皮膚中廣泛表現的泛在表現轉麩醯胺酸酶。轉麩醯胺酸酶 5 表現於皮膚顆粒層,對角質化外膜的形成至為核心,交聯結構蛋白,包括兜甲蛋白、loricrin、絲聚蛋白與小富脯胺酸蛋白。無法交聯這些蛋白導致在角質層界面的脫落。在單一家系中發現 CHST8(編碼 N-乙醯半乳糖胺-4-O-硫酸轉移酶 N-acetylgalactosamine-4-O-sulfotransferase,一種對表皮中硫酸化糖胺聚醣 sulfated glycosaminoglycans 產生至為核心的酵素)的體染色體隱性突變,此為 PSS3 (OMIM #616265)。
受影響個體在 5 歲後發展出全身性白色脫屑,在四肢明顯且易剝落。N-乙醯半乳糖胺-4-O-硫酸轉移酶在對表皮分化與脫屑至為核心之蛋白的糖基化 (glycosylation) 中發揮功能。在患有肢端 PSS(部分伴隨較全身性的剝脫性魚鱗癬)的家系中,也已報告 CSTA(編碼半胱胺酸蛋白酶抑制劑 cystatin A,一種表現於整個表皮的半胱胺酸蛋白酶抑制劑)的體染色體隱性、功能喪失突變,分類為 PSS4 (OMIM #607936)。Cystatin A 在表皮基底層的橋粒黏附中扮演核心角色,因此由 CSTA 突變所致的剝皮顯示較 TGM5 突變個體所見更深的裂解層次。
SERPINB8(屬於一個龐大的蛋白酶抑制劑家族)的體染色體隱性突變造成一種非發炎性 PSS,即 PSS5 (OMIM #617115)。
症候群性魚鱗癬 (SYNDROMIC ICHTHYOSES)
症候群性魚鱗癬以中度至重度的皮膚外侵犯為特徵。以下將描述一部分症候群性魚鱗癬。
Chanarin-Dorfman 症候群 (CHANARIN-DORFMAN SYNDROME)
Chanarin-Dorfman 症候群(伴魚鱗癬的中性脂質儲積病 neutral lipid storage disease with ichthyosis; OMIM #275630)是一種體染色體隱性疾病,特徵為三酸甘油酯 (triglycerides) 在角質細胞、白血球、肌肉、肝臟、纖維母細胞與其他組織的細胞質中累積,但血脂濃度正常。所致的魚鱗癬以全身性板層狀鱗屑伴可變的紅斑為特徵,屈側過度角化的波紋狀加強常見(圖 47-23)。已有 EKV 樣表現的報告。表現常為膠樣嬰兒伴瞼外翻與唇外翻。皮膚外侵犯(一般不如由 PNPLA2 突變所致、無魚鱗癬之中性脂質儲積病嚴重)可變且可能輕微,包括白內障、聽力降低、肝脾腫大伴肝臟酵素異常與脂肪肝、精神運動遲緩、伴血清肌肉酵素升高的肌病變,以及神經學異常。冷凍皮膚切片的油紅 O (oil red O) 或蘇丹 III (Sudan III) 染色之組織病理學顯示脂質滴 (lipid droplets) 位於真皮細胞、基底層(以及程度較低的基底上層),以及外分泌汗管的肢端汗管 (acrosyringia) 中。周邊血液抹片檢查顯示顆粒球、嗜酸性球與單核球內的脂質空泡,此特徵也可能存在於帶因者中。已鑑定出 ABHD5 基因的突變。ABHD5 屬於一個龐大的蛋白家族,其中大多數為酵素,似乎在三酸甘油酯脂解 (lipolysis) 的初始步驟中活化脂肪三酸甘油酯脂解酶 (adipose triglyceride lipase)。在 ABHD5 缺失的情況下,三酸甘油酯在細胞中與層板小體中累積,在分泌後干擾角質層中脂質層板的形成,導致經表皮水分流失增加。
CHILD 症候群 (CHILD SYNDROME)
CHILD 症候群 (OMIM #308050) 是一種罕見疾病,由先天性半側發育不良 (congenital hemidysplasia)、魚鱗癬樣紅皮症與肢體缺損 (limb defects) 組成,幾乎只見於女性。它是一種鑲嵌性疾病,特徵為身體一側的紅色脫屑斑塊與正常皮膚交錯,對側侵犯極少。受影響側可能有正常皮膚的條帶(圖 47-24)。肢體缺損發生在魚鱗癬的同側,範圍從手指發育不全到肢端發育不全。可能有軟骨的點狀鈣化。單側發育不全可侵犯中樞神經系統與心血管、肺、腎、內分泌與泌尿生殖系統。遺傳模式為 X 性聯顯性,此病在男性中致死。突變等位基因的 X 去活化 (X-inactivation) 使受影響女性產生正常皮膚。大多數受影響個體在 NSDHL(NAD[P]H 類固醇去氫酶樣蛋白,編碼一種 3β-羥基類固醇去氫酶)中有功能喪失突變。NSDHL 在角鯊烯後 (postsqualene) 膽固醇生合成路徑中發揮功能,催化羊毛固醇 (lanosterol) 轉化為膽固醇的中間步驟。此病與由 emopamil 結合蛋白 (EBP) 突變所致的 X 性聯顯性點狀軟骨發育異常相關,後者也有皮膚與骨骼異常,且已有一名具 CHILD 表現型的個體被報告帶有 EBP 突變。EBP 在膽固醇合成路徑中位於 NSDHL 下游。基於致病機轉的治療可改善 CHILD 症候群的皮膚發現。NSDHL 缺陷導致無法合成膽固醇,並使 4-甲基與 4,4-二甲基固醇 (4-methyl and 4,4-dimethyl sterols) 累積至毒性濃度。CHILD 症候群皮膚發現的表現型改善,可藉由外用膽固醇與 lovastatin 達成。這供應了膽固醇生合成的最終產物,同時在 HMG CoA 還原酶(位於 NSDHL 近端)的層次阻斷路徑,藉此預防毒性中間產物的合成。
點狀軟骨發育異常 (CHONDRODYSPLASIA PUNCTATA)
點狀軟骨發育異常 (chondrodysplasia punctata) 是一群臨床與遺傳上多樣化的罕見疾病,首次由 Conradi 描述,共有骨骺 (epiphyses) 點狀鈣化的特徵,以及由胎兒發育與嬰兒早期軟骨內成骨 (enchondral bone formation) 區域之異常鈣質沉積所致的骨骼變化。臨床嚴重度範圍從嚴重侏儒症與嬰兒期死亡,到隨時間消退僅留下輕微骨骼變化的影像學異常。數種型別也包括魚鱗癬樣變化。已有描述體染色體隱性(根肢型 rhizomelic)型,以及 X 性聯顯性與隱性型。
根肢型點狀軟骨發育異常 (RHIZOMELIC CHONDRODYSPLASIA PUNCTATA)
根肢型點狀軟骨發育異常 (rhizomelic chondrodysplasia punctata, RCDP; OMIM #215100) 也稱為過氧化體生成障礙互補群 11 (peroxisomal biogenesis disorder complementation group 11, CG11)。它是一種體染色體隱性、罕見的多系統發育性疾病,特徵為由近端長骨對稱性縮短(即根肢化 rhizomelia)所致的侏儒症、特定的放射學異常(即軟骨的點狀鈣化、椎體裂 vertebral body clefting)、關節攣縮、先天性白內障、魚鱗癬與嚴重智能障礙。皮膚變化存在於約 25% 的病人中。RCDP 是一種過氧化體 (peroxisomes) 的疾病,過氧化體是存在於所有有核細胞中之膜結合的多功能胞器。其功能隨細胞型別而異,包括多種路徑(如基於過氧化氫的呼吸、脂肪酸 β-氧化,以及脂質與膽固醇合成),涉及多種化合物的合成與降解。
遺傳性人類過氧化體疾病細分為過氧化體生成障礙(胞器無法正常形成)與涉及單一過氧化體酵素者。RCDP 由 PEX7 突變所致,PEX7 編碼過氧化體蛋白 7 (peroxin 7),這是將一部分酵素標靶至過氧化體所需的受體。因此,它是一種以多種過氧化體代謝功能喪失為特徵的過氧化體生成障礙。
X 性聯隱性點狀軟骨發育異常 (X-LINKED RECESSIVE CHONDRODYSPLASIA PUNCTATA)
X 性聯隱性點狀軟骨發育異常 (X-linked recessive chondrodysplasia punctata, CDPX; OMIM #302950) 可侵犯皮膚(線狀或漩渦狀萎縮性或魚鱗癬樣過度角化、毛囊性萎縮性皮膚病 follicular atrophoderma,可能始於紅皮症)、毛髮(粗糙、無光澤、瘢痕性禿髮)、矮小身材與骨骼異常、白內障與聾。Curry 及其同事研究了一個家族,其中兩名受影響男性有非典型魚鱗癬與硫酸膽固醇升高,並鑑定出一個包含類固醇硫酸酯酶基因的 X 染色體缺失 (Xp22)。在此位置有一群芳基硫酸酯酶基因。在五名病人中發現 ARSE(編碼芳基硫酸酯酶 E arylsulfatase E 的基因)的突變;然而,此病也可能由相鄰的芳基硫酸酯酶基因突變所致。與 warfarin 胚胎病變 (warfarin embryopathy) 的相似性提示,warfarin 胚胎病變可能由藥物誘發對同一酵素的抑制所致。
X 性聯顯性點狀軟骨發育異常 (X-LINKED DOMINANT CHONDRODYSPLASIA PUNCTATA)
X 性聯顯性點狀軟骨發育異常 (CDPX2; Conradi-Hünermann-Happle 症候群, OMIM #302960) 的特徵為沿 Blaschko 線的鑲嵌性皮膚侵犯,由鑲嵌性 X 染色體去活化 (lyonization) 所致。它幾乎只發生於女性,據推測基因功能喪失對男性致死。受影響女性有正常的預期壽命,且在連續世代中可能有疾病表現增加(早現現象 anticipation)。已觀察到一名男性發病合併 47, XXY 核型。Conradi-Hünermann-Happle 症候群在出生時以先天性魚鱗癬樣紅皮症表現,數月內清除並被線狀過度角化、毛囊性萎縮性皮膚病與色素異常所取代。也可發生毛幹異常與瘢痕性禿髮。在兒童期軟骨內成骨區域的放射線檢查中可見點狀鈣化,但青春期後可能不再可見。身材可能矮小,伴腿部的不對稱縮短。白內障發生於約三分之二的病人,通常不對稱。鈣的組織化學染色可能顯示表皮內的鈣化,尤其在幼童的毛囊內,在較大兒童中可能不存在。編碼 EBP 之基因的突變造成此症候群。EBP 最初被鑑定為藥物 emopamil(一種鈣通道阻斷劑)的結合標靶。後來發現它是 3β-羥基類固醇-δ8-δ7-異構酶 (3β-hydroxysteroid-δ8-δ7-isomerase),催化羊毛固醇轉化為膽固醇的一個中間步驟。雖然這是另一種由膽固醇合成異常所致的魚鱗癬,此缺陷如何造成臨床表現的病理生理尚不清楚。
魚鱗癬性毛囊角化、禿髮與畏光症候群 (ICHTHYOSIS FOLLICULARIS, ALOPECIA, AND PHOTOPHOBIA SYNDROME)
在 X 性聯隱性的魚鱗癬性毛囊角化、禿髮與畏光症候群 (ichthyosis follicularis, alopecia, and photophobia syndrome, IFAP; OMIM #308205,見第 49 章) 中,可見非發炎性毛囊性過度角化、非瘢痕性禿髮、畏光 (photophobia) 與特徵性面容。較不固定的特徵包括反覆呼吸道感染、指甲異常、口角炎 (angular cheilitis)、四肢伸側的角化斑塊、腹股溝疝氣、隱睪症、矮小身材、癲癇發作與精神運動發展遲緩。MBTPS2 基因的突變造成此症候群,推測是藉由導致膽固醇恆定受損與內質網壓力 (endoplasmic reticulum stress) 所致。
魚鱗癬早產症候群 (ICHTHYOSIS PREMATURITY SYNDROME)
在魚鱗癬早產症候群 (ichthyosis prematurity syndrome, OMIM #608649) 中發生如妊娠第二孕期羊水過多 (polyhydramnios) 等併發症,導致早產,產下帶有紅皮症、水腫性、乾酪樣 (caseous) 脫屑皮膚(類似過多胎脂 vernix caseosa)、呼吸窘迫與短暫性周邊嗜酸性球增多 (peripheral eosinophilia) 的嬰兒。呼吸徵象、紅斑與皮膚變化消退,留下異位性徵象與乾燥、脫屑的皮膚,伴隨較毛孔角化症所見更明顯的毛囊性加強。脂肪酸轉運蛋白 4 基因 FATP4 (SLC27) 的突變造成此病。
多發性硫酸酯酶缺乏 (MULTIPLE SULFATASE DEFICIENCY)
多發性硫酸酯酶缺乏 (multiple sulfatase deficiency, OMIM #272200) 是一種罕見的體染色體隱性疾病。臨床特徵包括神經學退化、骨骼異常、面部畸形,以及類似 X 性聯類固醇硫酸酯酶缺乏所見的魚鱗癬。此病是異染性腦白質失養症 (metachromatic leukodystrophy) 與黏多醣症 (mucopolysaccharidosis) 兩者臨床特徵的綜合體。它以溶體 (lysosomal) 性(芳基硫酸酯酶 A 與 B)與微粒體 (microsomal) 性(芳基硫酸酯酶 C/X 性聯魚鱗癬的類固醇硫酸酯酶)芳基硫酸酯酶皆缺乏為特徵,導致硫脂 (sulfatides)、糖胺聚醣、神經鞘脂 (sphingolipids) 與類固醇硫酸鹽在組織與體液中累積。編碼硫酸酯酶修飾因子 1 (sulfatase modifying factor 1, SUMF1) 的基因在多發性硫酸酯酶缺乏個體中發生突變,SUMF1 生成一種獨特的胺基酸衍生物 Cα-甲醯甘胺酸 (Cα-formylglycine),這是所有硫酸酯酶催化活性所必需的。
Refsum 病 (REFSUM DISEASE)
Refsum 病(遺傳性共濟失調性多發性神經炎 heredopathia atactica polyneuritiformis; OMIM #266500)是一種罕見、進行性、退化性的脂質代謝疾病,起因於無法分解膳食植烷酸 (phytanic acid) 及其後續在組織中的累積。此體染色體隱性疾病主要影響斯堪地那維亞人與源自北歐的族群。臨床表現包括色素性視網膜炎 (retinitis pigmentosa)、周邊神經病變、小腦共濟失調、腦神經功能障礙(神經性聾、嗅覺缺失)、瞳孔縮小、心電圖異常、心肌病變、腎小管功能障礙與骨骼異常(骨骺發育不良)。魚鱗癬可變,通常在神經學與眼科表現之後發展。常在軀幹與四肢有類似 IV 的小白色鱗屑。常規蘇木精-伊紅 (hematoxylin and eosin) 組織學檢查顯示表皮基底與基底上層細胞中大小不一的空泡,對應於冷凍切片脂質染色所見的脂質累積。
植烷酸(一種 20 碳、支鏈脂肪酸)源自多種膳食來源,包括乳製品、反芻動物脂肪與含葉綠素的食物,雖然與葉綠素結合的植醇 (phytol) 在人體中無法被吸收。此病由 PhyH 缺乏所致,PhyH 是一種催化植烷酸 α-氧化的過氧化體蛋白。PhyH 缺乏導致植烷酸在血清與組織中累積,在該處取代正常存在的脂肪酸。雖然編碼 PhyH 之基因 PAHX 的突變為大多數 Refsum 病病例的原因,但存在遺傳異質性。已有在帶有 PEX7(RCDP 中的受累基因,見前述)突變的病人中描述 Refsum 表現型。植烷酸與其他葉綠素代謝物結合視黃醇 X 受體 (retinoid X receptor, RXR),如同其天然配體 9-順式視黃酸 (9-cis-retinoic acid),且它們可能透過 RXR 依賴性訊息路徑在協調細胞代謝中具生理活性。然而,RXR 在 Refsum 病致病機轉中的角色尚不清楚。Refsum 病與嬰兒型 Refsum 病 (infantile Refsum disease) 區分,後者是一種猛爆性的全身性過氧化體生成障礙,幼童除了成人 Refsum 病的其他徵象外,還以嚴重神經學異常、智能障礙、肝腫大與畸形特徵表現。
Refsum 病是一個罕見的範例,其病理生理在致病基因鑑定之前即已被充分了解,足以做出合理的治療建議。診斷可藉由偵測血漿植烷酸濃度升高來達成。在血漿植烷酸濃度未升高的兒童中,診斷可藉由測量培養纖維母細胞中的 PhyH 活性來達成。治療包括膳食限制含植烷酸及其前驅物的食物。在魚鱗癬延遲發病合併神經學損害的臨床情境中,應考慮此疾病,因為治療可阻止其進展。
Sjögren-Larsson 症候群 (SJÖGREN-LARSSON SYNDROME)
1957 年,Sjögren 與 Larsson 報告了北瑞典的 13 個家族,患有先天性魚鱗癬、痙攣性麻痺與智能障礙的症候群。Sjögren-Larsson 症候群 (SLS; OMIM #270200) 是一種罕見的體染色體隱性疾病,在出生時以魚鱗癬表現,可從細小脫屑到全身性過度角化不等。出生時可能有紅斑,但傾向在 1 歲時逐漸清除。罕見膠樣樣膜。魚鱗癬表現為細鱗屑、大鱗屑,或無鱗屑的角質層增厚。搔癢常見。增厚區域可能呈黃至棕色,並有苔癬化外觀伴加強的皮膚紋路(圖 47-25)。侵犯最嚴重的區域是頸部的兩側與後方、下腹部與屈側。毛髮、指甲與出汗能力一般正常。在最初 2 至 3 年,發展出痙攣性雙下肢癱 (spastic diplegia) 或四肢癱 (tetraplegia) 與智能障礙的神經學表現,可伴隨言語缺陷與癲癇發作。一項特徵性的眼科發現是視網膜黃斑部有閃亮的白點。這些在 1 歲後出現,可能並非所有病人都存在。SLS 由脂肪醇:NAD 氧化還原酶 (fatty alcohol:NAD oxidoreductase, FAO) 缺乏所致。FAO 是一種複合酵素,有兩個獨立的蛋白,依序催化脂肪醇氧化為脂肪醛,隨後氧化為脂肪酸。在 ALDH3A2 基因中發現的突變確認了此酵素在此病病因中的角色,以及此路徑對正常脫屑的重要性。ALDH3A2 是一種微粒體酵素,催化源自脂肪醇、植烷酸、醚甘油脂 (ether glycerolipids) 與白三烯 B4 (leukotriene B4) 代謝之中鏈與長鏈脂肪族醛的氧化。在一個有非典型皮膚發現(缺乏魚鱗癬或為離散斑塊而非全身性魚鱗癬)的家族中,鑑定出纖維母細胞 FAO 活性降低,這擴展了與異常 FAO 活性相關的臨床表現型譜系。
毛髮硫營養不良 (TRICHOTHIODYSTROPHY)
毛髮硫營養不良 (trichothiodystrophy, TTD; OMIM #601675,亦見第 130 章) 是一種體染色體隱性疾病,包括廣泛的臨床表現型譜系,以硫缺乏、脆弱毛髮的特徵性表現相連結,這種毛髮在偏光顯微鏡下觀察時呈現交替的雙折射(虎尾條紋 tiger tail banding)(圖 47-26)。臨床侵犯的譜系廣泛,從僅有毛髮到嚴重的多系統異常。一項對文獻中報告的 112 個病例的調查發現,魚鱗癬(65%)是最常見的皮膚發現,其次是光敏感性(42%)。在患有魚鱗癬的病人中,約三分之一在出生時以膠樣膜表現。當出生時存在紅皮症時,通常在數週內減輕,並演變為全身性魚鱗癬(通常無紅斑),從細小脫屑到大型、深黃棕色的過度角化不等(見圖 47-17B)。可能有屈側倖免與掌蹠角皮症。指甲發現見於超過半數的病人,包括營養不良性指甲(脊紋、裂開)(見圖 47-17A)、發育不全、脆弱指甲與匙狀甲 (koilonychia)。瞼外翻通常不發生。光敏感性的範圍從輕微到嚴重。TTD 的其他常見發現包括智能障礙、矮小身材、小頭症 (microcephaly)、特徵性面部特徵(耳朵突出、小頷 micrognathia)、反覆感染與白內障。已使用一系列記憶法 (mnemonics) 來描述這一系列發現,如 BIDS(脆弱毛髮 brittle hair、智能障礙、生育力降低、矮小身材);同時也有魚鱗癬的病人被稱為 IBIDS,再加上光敏感性,則使用 PIBI(D)S。然而,這些術語並未涵蓋常存在的其他多系統發現。大多數患有魚鱗癬的 TTD 病人在 TFIIH(一種參與 DNA 修復與轉錄的蛋白複合物)的功能或活性上有缺陷。大多數突變位於 ERCC2。其他突變影響 ERCC3、GTF2H5 與 GTF2E2。另兩個造成 TTD 的基因 MPLKIP 與 RNF113A 未與魚鱗癬相關,其功能未知。雖然許多 TTD 病人有光敏感性,但與著色性乾皮症 (xeroderma pigmentosum) 相對,這些光敏感性病人未被觀察到有發展皮膚癌的高風險。核苷酸切除修復 (nucleotide excision repair) 是清除與修復結構性 DNA 損傷(如紫外線誘發的環丁烷嘧啶二聚體 cyclobutane pyrimidine dimers)的正常細胞機制之一(見第 20 章與第 134 章)。
後天性魚鱗癬 (ACQUIRED ICHTHYOSIS)
成年期才發展出魚鱗癬,可為全身性疾病的表現,已有描述其與惡性腫瘤、藥物、內分泌與代謝疾病、營養不良、HIV 與其他感染,以及自體免疫疾病相關。
此病的顆粒層常變薄,鱗屑常類似輕微 IV 所見。雖然何杰金氏病 (Hodgkin disease) 是後天性魚鱗癬最常報告的惡性腫瘤,但非何杰金氏淋巴瘤與多種其他惡性腫瘤也曾被觀察到。在與蕈狀肉芽腫 (mycosis fungoides) 相關的後天性魚鱗癬中,組織學可能具診斷性。皮膚侵犯可能隨惡性腫瘤的病程,並隨有效的癌症治療而清除。後天性魚鱗癬常見與 AIDS 相關;多達 30% 的 AIDS 病人曾被觀察到有魚鱗癬樣或乾燥的皮膚。一項對 HIV-1 陽性靜脈藥物使用者的研究發現,後天性魚鱗癬僅在輔助 T 細胞嚴重耗竭後發生,且更常見於合併感染人類 T 細胞白血病或淋巴瘤病毒第 II 型者,並提示它可能是同時感染這兩種病毒的標誌。
在與類肉瘤病 (sarcoidosis) 相關發生的後天性魚鱗癬中,皮膚切片可能具診斷性,顯示真皮中的非乾酪性肉芽腫 (noncaseating granulomas)。後天性魚鱗癬可能是自體免疫疾病的標誌,與全身性紅斑性狼瘡 (systemic lupus erythematosus)、皮肌炎 (dermatomyositis)、混合性結締組織病與嗜酸性球性筋膜炎 (eosinophilic fasciitis) 一同發生。它已被描述見於骨髓移植受者,在這些人中可能與移植物對抗宿主疾病 (graft-versus-host disease) 有關。
雖然與降膽固醇藥物(菸鹼酸 nicotinic acid、triparanol)相關的發生凸顯了膽固醇代謝與正常脫屑之間的關係,後天性魚鱗癬也已被觀察到與多種藥物相關,包括 cimetidine、clofazimine、hydroxyurea、降膽固醇藥物等。Kava 是一種由胡椒科植物根部製成的精神活性飲料,太平洋島民使用已有數千年。大量飲用 kava 者可獲得一種可逆的魚鱗癬樣疹,稱為 kava 皮膚病變 (kava dermopathy)。在西方國家,kava 在健康食品店作為鬆弛劑販售。
圓形糠疹 (PITYRIASIS ROTUNDA)
在圓形糠疹 (pityriasis rotunda) 中,可見界限分明、圓形或卵圓形的脫屑斑片,伴低色素或高色素沉著。雖然此罕見疾病通常為後天性,但偶有家族性病例被描述。
Gougerot 與 Carteaud 網狀乳頭瘤病 (RETICULATED PAPILLOMATOSIS OF GOUGEROT AND CARTEAUD)
Gougerot 與 Carteaud 融合性與網狀乳頭瘤病 (confluent and reticulated papillomatosis of Gougerot and Carteaud) 是一種少見但獨特的後天性魚鱗癬樣皮膚病,見於年輕成人,特徵為持續的棕色、脫屑性斑點、丘疹、斑片與斑塊。病灶傾向主要分布於頸部、上軀幹(乳房間與肩胛間區域)與腋窩,在這些部位傾向融合並朝周邊變成網狀。病灶在臨床上類似汗斑 (tinea versicolor),後者為一種由 Pityrosporum 屬引起的皮膚感染。已報告多種治療途徑,包括外用(角質溶解劑、維生素 A 與 D 衍生物、抗微生物劑)與全身性(抗生素、類視黃醇)藥物。Minocycline 已被建議為第一線治療;成功的復發再治療支持此病為對感染或發炎之異常反應的概念。
資源 (RESOURCES)
魚鱗癬及相關皮膚型別基金會 (The Foundation for Ichthyosis and Related Skin Types, FIRST)(電話:800-545-3286;http://www.firstskinfoundation.org;電郵:info@firstskinfoundation.org)為受影響個體、家庭成員、朋友與醫療提供者提供支持與資訊。基因檢測登錄 (Genetic Testing Registry, https://www.ncbi.nlm.nih.gov/gtr/) 提供疾病回顧、基因檢測資源與教育材料。線上人類孟德爾遺傳資料庫 (Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM)(約翰霍普金斯大學商標)是人類基因與遺傳疾病的目錄,是一個有用的參考資源並有連往額外資源的連結。魚鱗癬及相關疾病登錄 (The Registry for Ichthyosis and Related Disorders) 是供研究者改善這些疾病診斷與治療的資源(耶魯大學皮膚科,PO Box 208059, 333 Cedar Street, New Haven, CT 06510;電郵:ichthyosisregistry@yale.edu)。
誌謝 (ACKNOWLEDGMENTS)
作者感謝 Philip Fleckman 與 John J. DiGiovanna,他們撰寫了本章的前一版,並提供了出現於此處的文字、表格與照片。
圖表 (FIGURES AND TABLES)
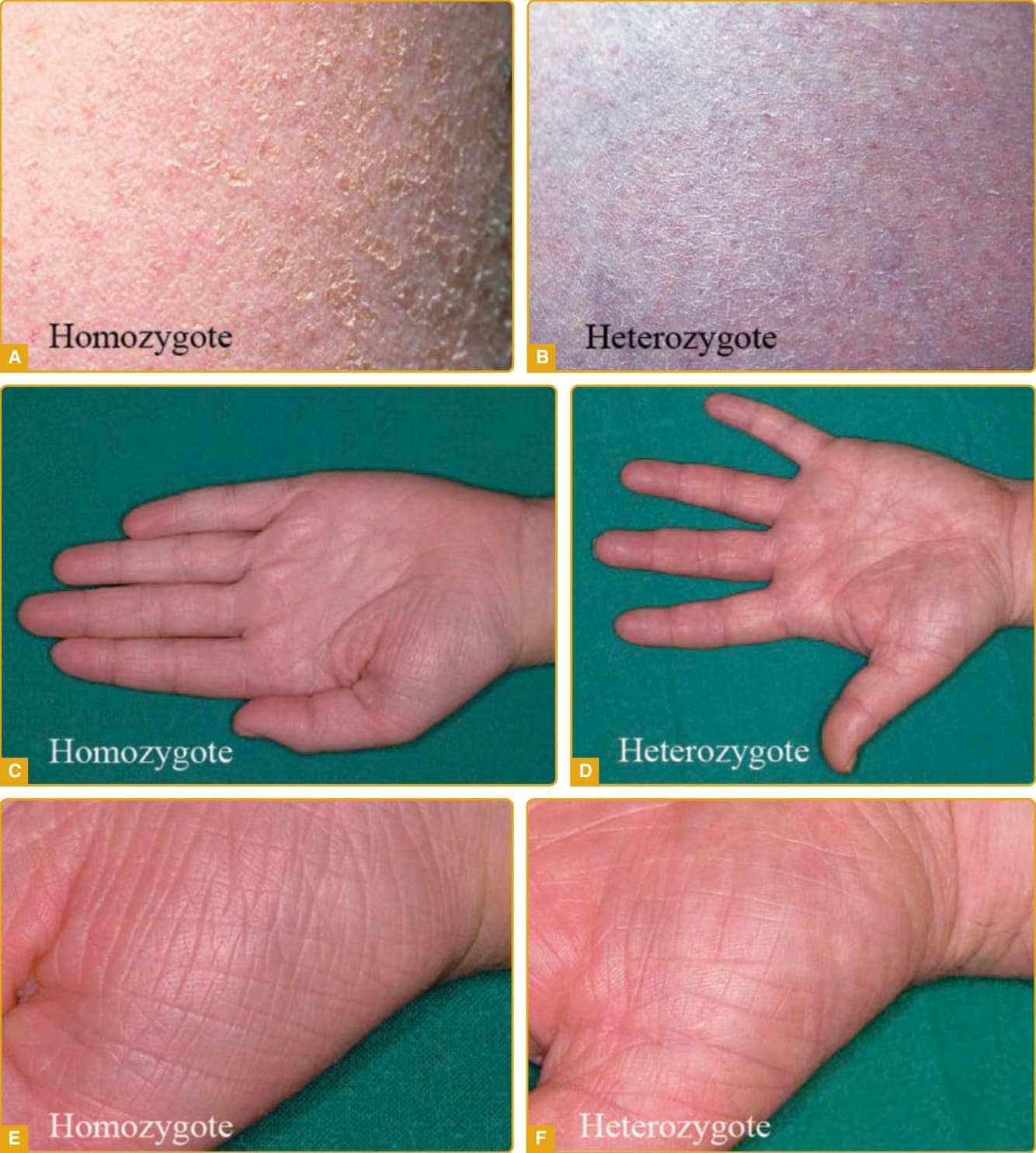
圖 47-1 A–F,尋常性魚鱗癬 (ichthyosis vulgaris)(FLG 突變)。小型、中央附著的鱗屑一般倖免皺褶間區域。通常注意到手掌紋路的數目與深度增加。
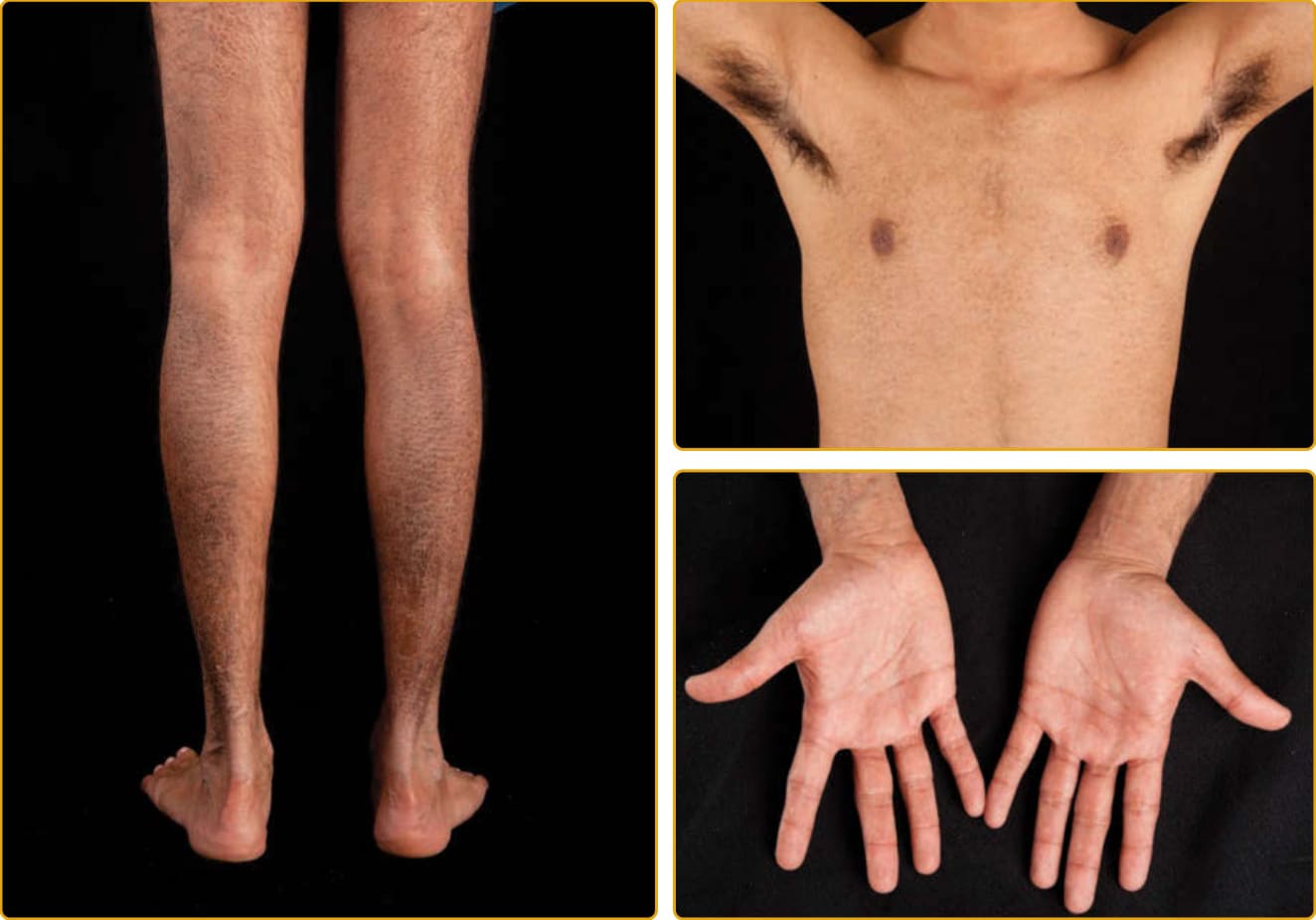
圖 47-2 X 性聯隱性魚鱗癬 (X-linked recessive ichthyosis)(STS 突變)。小型、中央附著的深色鱗屑常侵犯頸部與臉部兩側;在屈側較不嚴重。手掌與足底顯示少有或沒有侵犯。
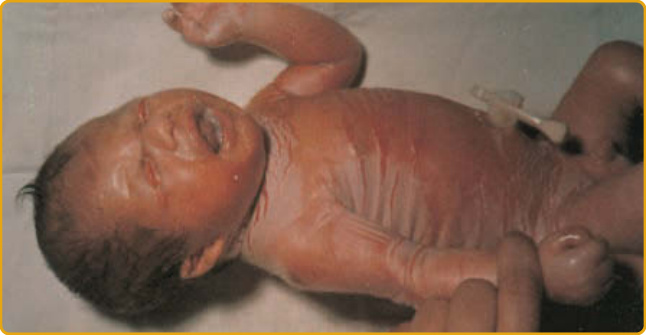
圖 47-3 膠樣嬰兒 (collodion infant)。此嬰兒出生 36 小時,覆蓋著顯示裂隙的浸軟膜;注意瞼外翻與唇外翻。此情況可能隨時間發展為各種臨床表現型,包括體染色體隱性先天性魚鱗癬與自癒型膠樣嬰兒。
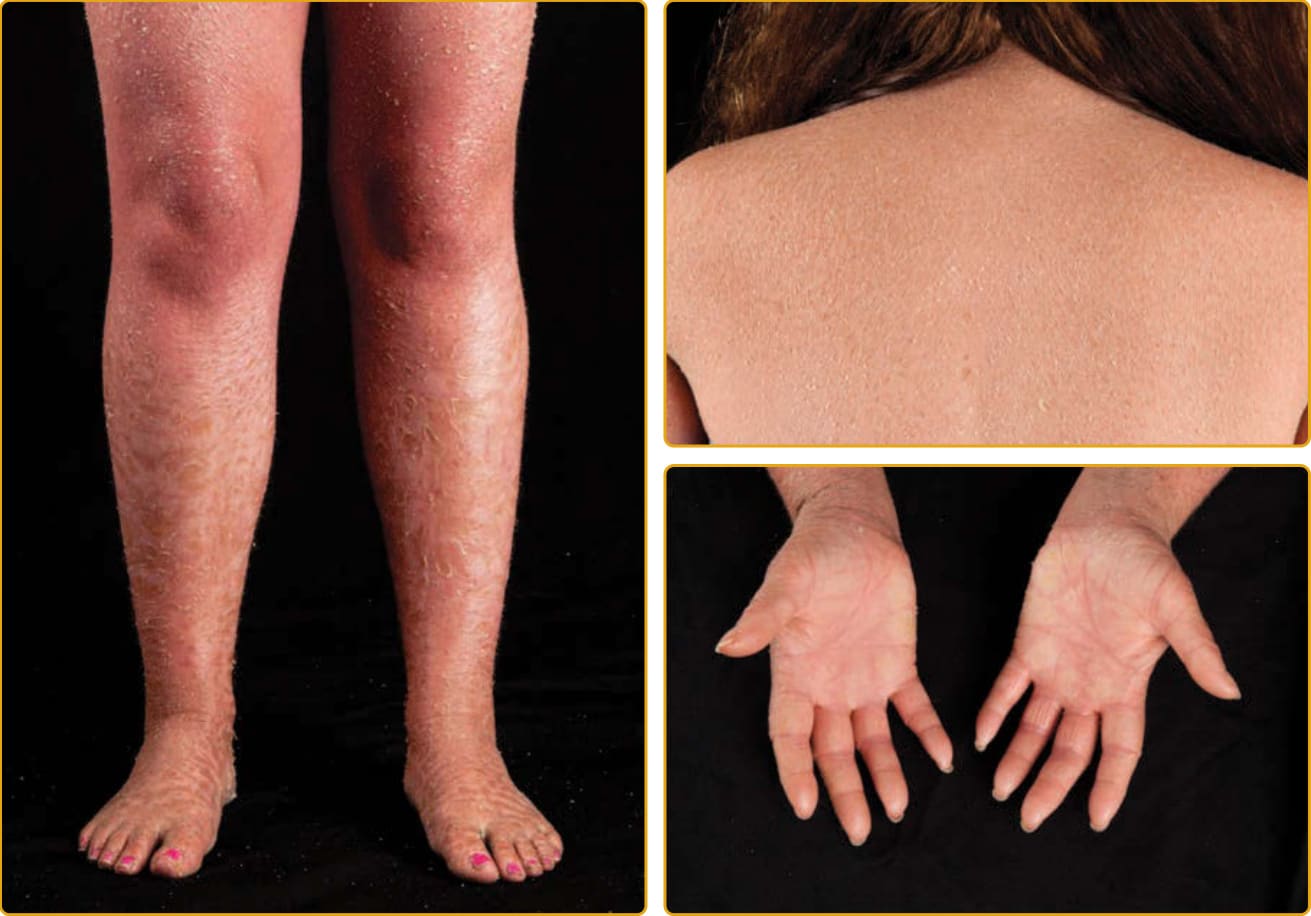
圖 47-4 體染色體隱性先天性魚鱗癬 (autosomal recessive congenital ichthyosis)(TGM1 突變,經典板層狀魚鱗癬,中度)。大型深色鱗屑常在四肢伸側與前額最為明顯。輕至中度紅斑與掌蹠角皮症為常見。鱗屑嚴重度差異很大(見圖 47-6)。
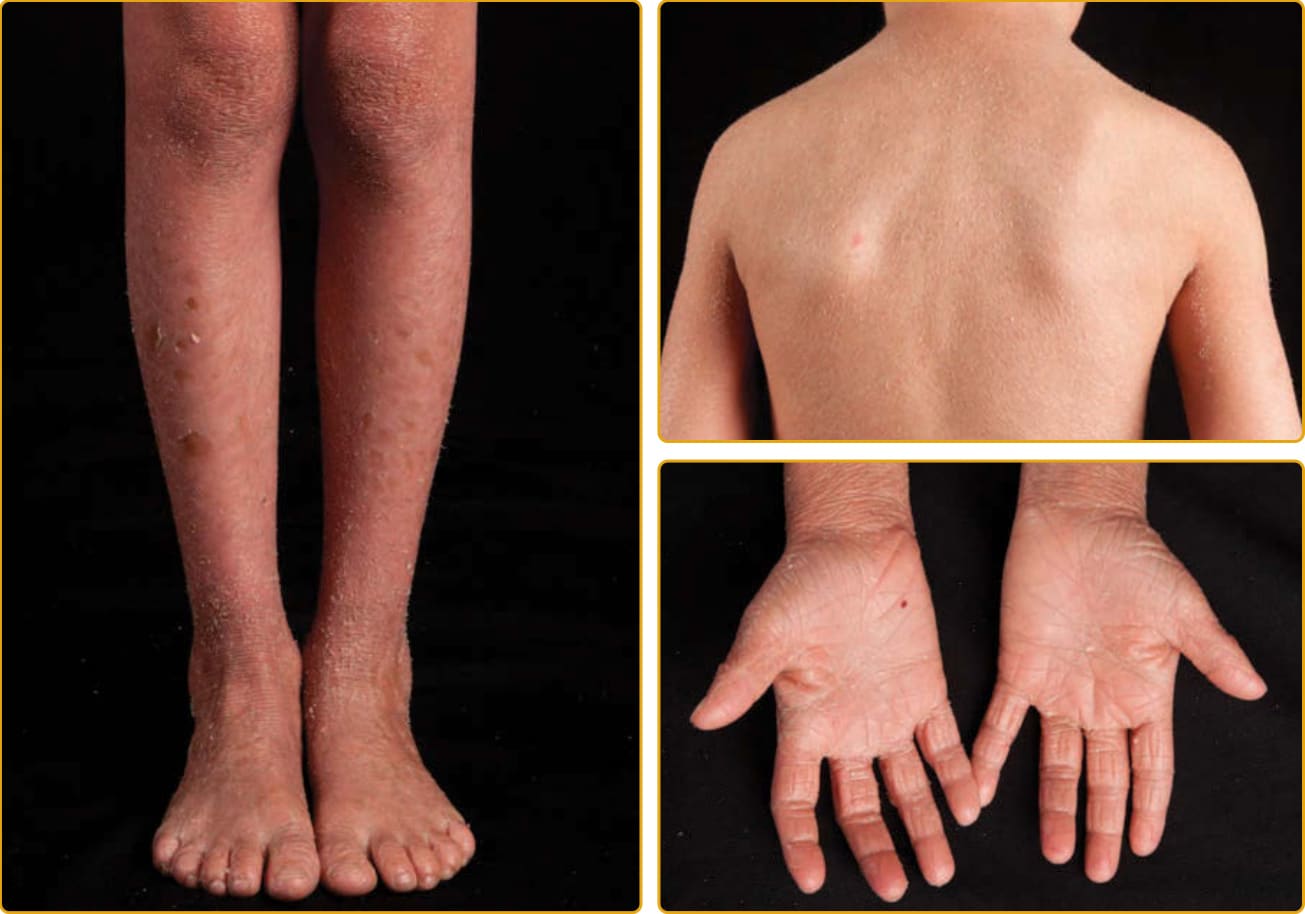
圖 47-5 體染色體隱性先天性魚鱗癬(TGM1 突變,經典板層狀魚鱗癬,輕度)。大型深色鱗屑常在四肢伸側與前額最為明顯。輕至中度紅斑與掌蹠角皮症為常見。鱗屑嚴重度差異很大(見圖 47-4)。
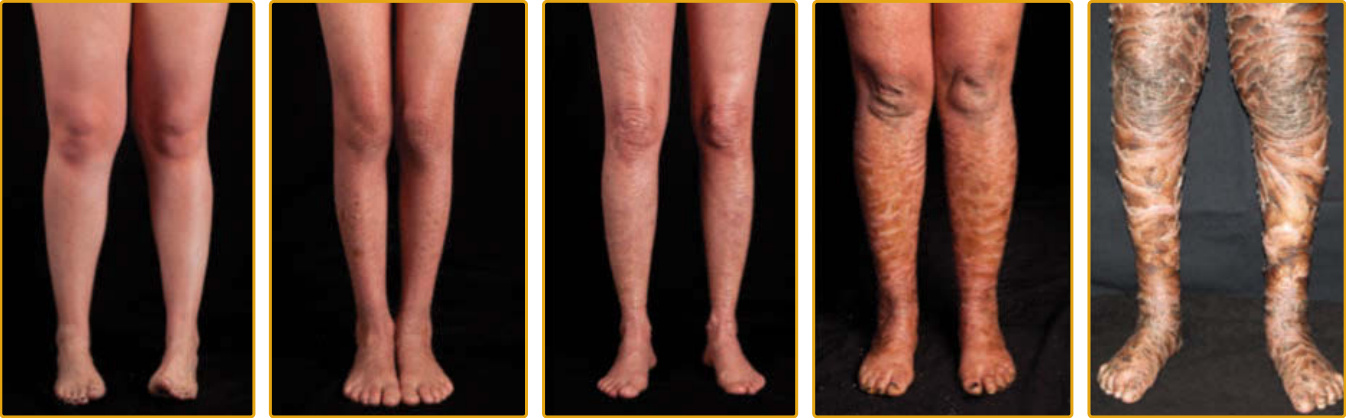
圖 47-6 TGM1 突變的嚴重度譜系。在帶有相同突變的個體之間可見表現型變異。
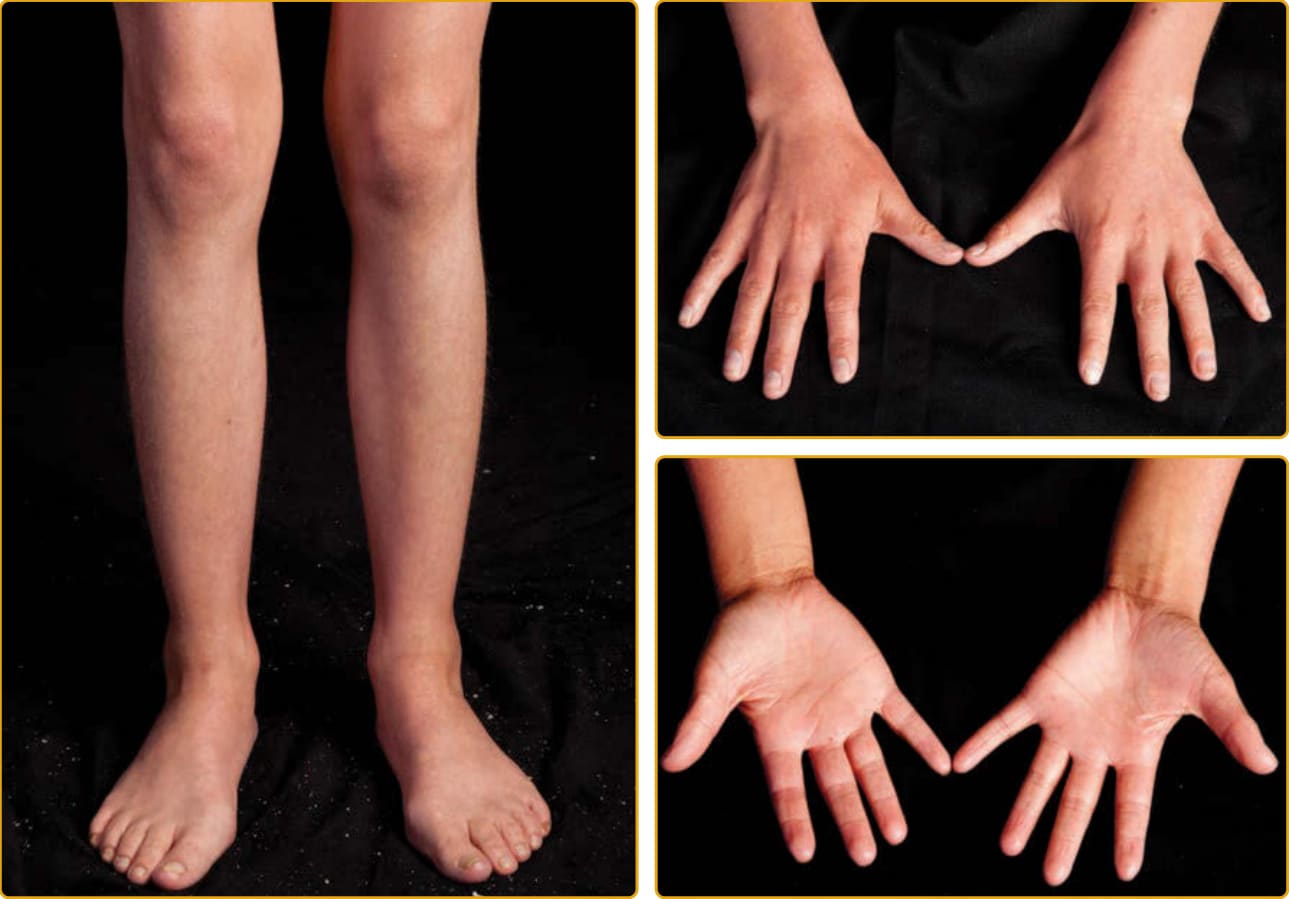
圖 47-7 體染色體隱性先天性魚鱗癬(ALOXE3 突變)。手與足的掌側 (volar) 與背側 (dorsal) 表面顯示誇大的線狀紋路。當其他部位有鱗屑時,鱗屑細小、白色,覆蓋於輕度紅斑之上。
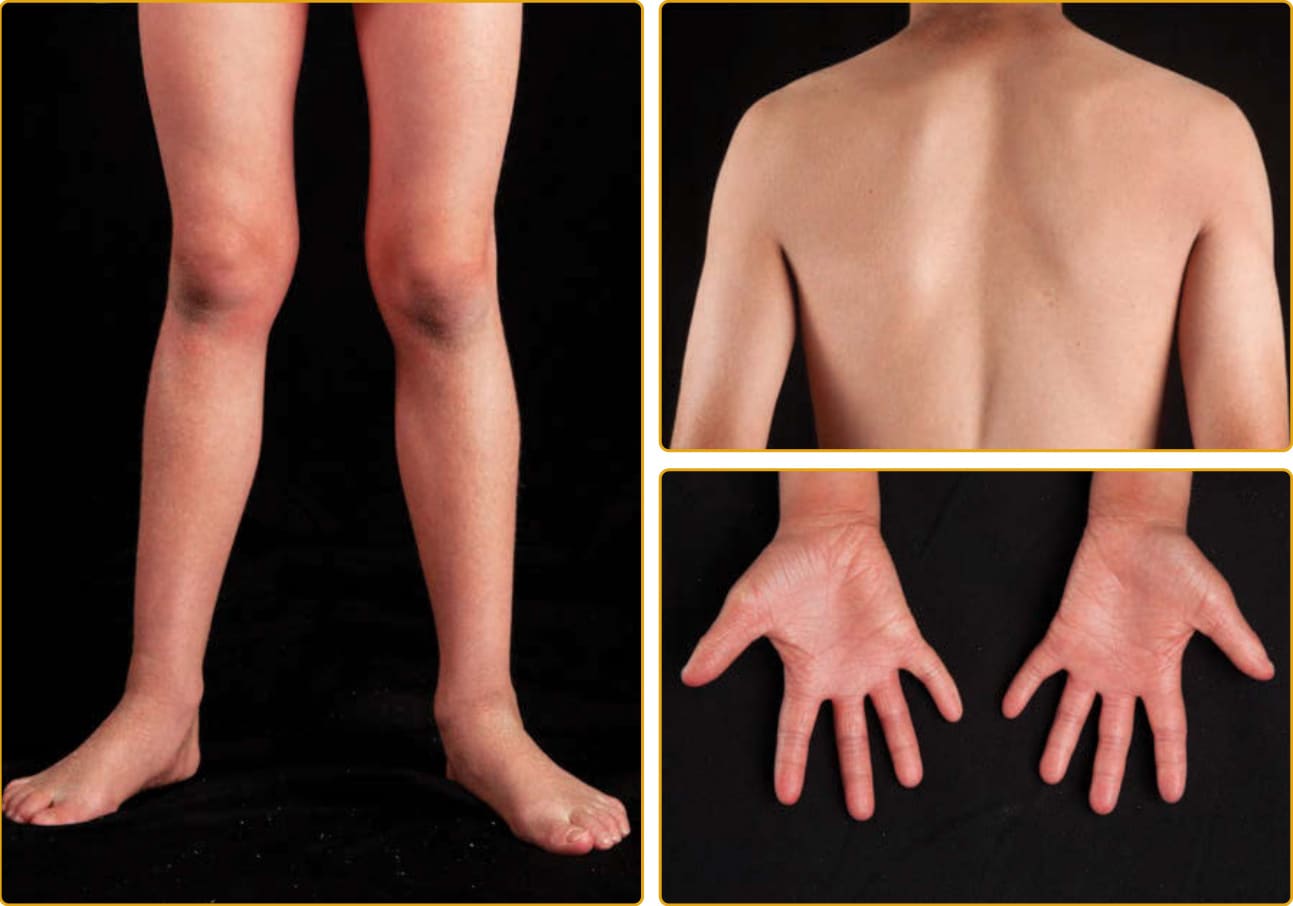
圖 47-8 體染色體隱性先天性魚鱗癬(ALOX12B 突變)。手與足的掌側與背側表面顯示增加的線狀紋路。當其他部位有鱗屑時,鱗屑細小、白色,覆蓋於輕度紅斑之上。
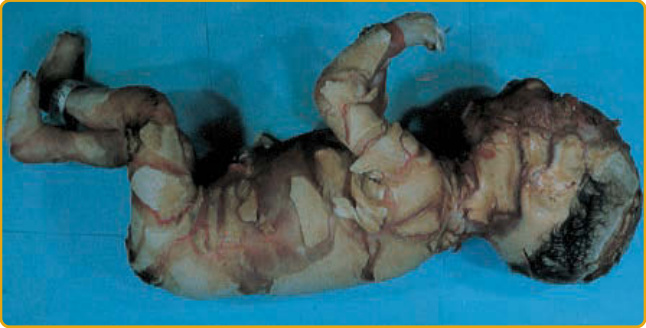
圖 47-9 小丑嬰兒 (harlequin infant)。小丑魚鱗癬。注意發育不全的耳朵,以及因厚「板塊」狀角質層所致的扭曲外觀。此嬰兒出生後數天死亡。
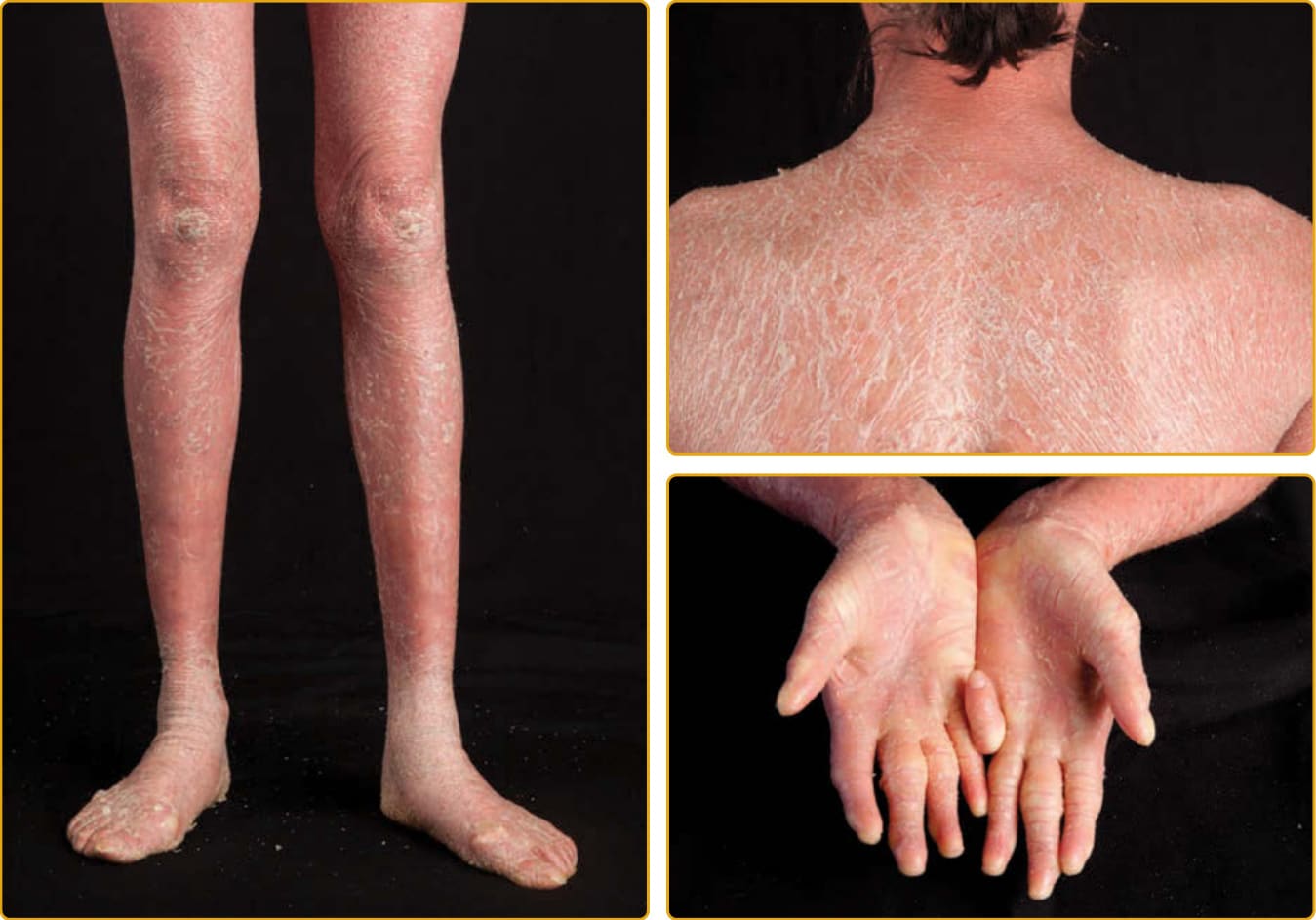
圖 47-10 體染色體隱性先天性魚鱗癬(ABCA12 突變,中度)。大型扁平鱗屑與中度全身性紅斑。所有手指皆呈錐形。
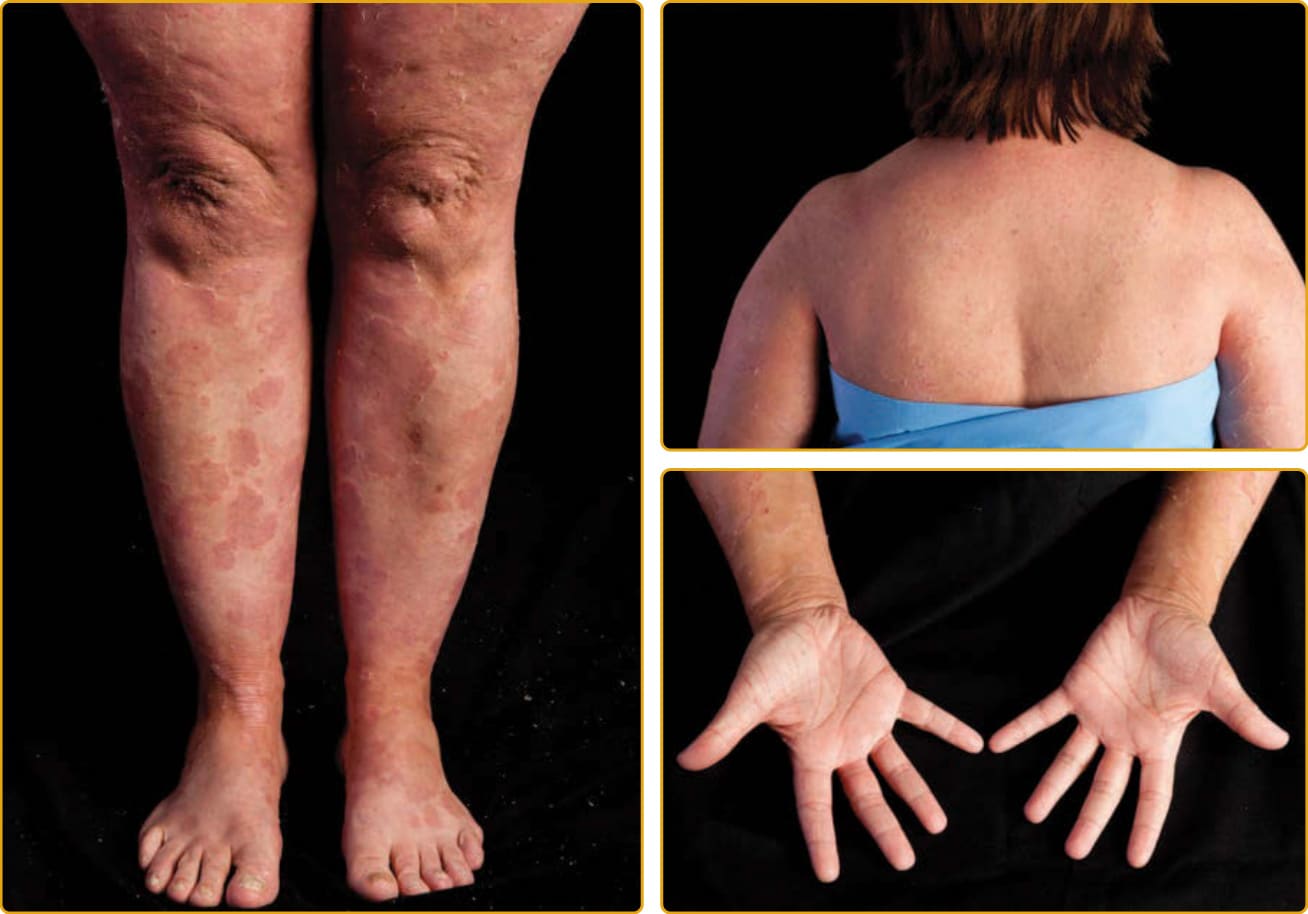
圖 47-11 Netherton 症候群(SPINK5 突變,以環狀線狀魚鱗癬 ichthyosis linearis circumflexa 為主)。蛇行狀、遷移性的鱗屑剝離以顯露下方的紅斑。手掌與足底有輕至中度角皮症。
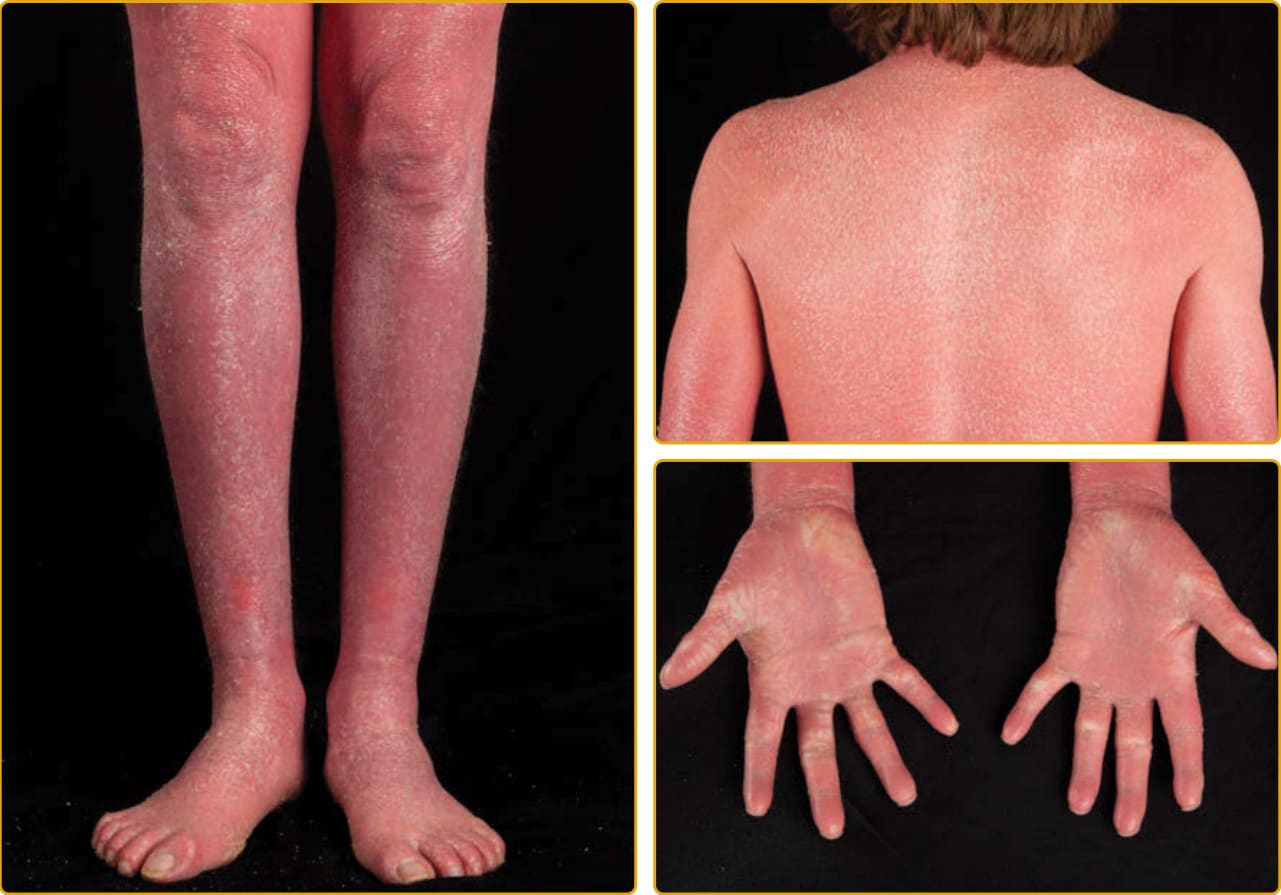
圖 47-12 Netherton 症候群(SPINK5 突變,以紅皮症為主)。小型白色鱗屑覆蓋著輕至重度全身性紅斑的區域。蛇行狀、剝離的鱗屑常侷限於踝部與腕部周圍區域。手掌與足底有輕至中度角皮症。
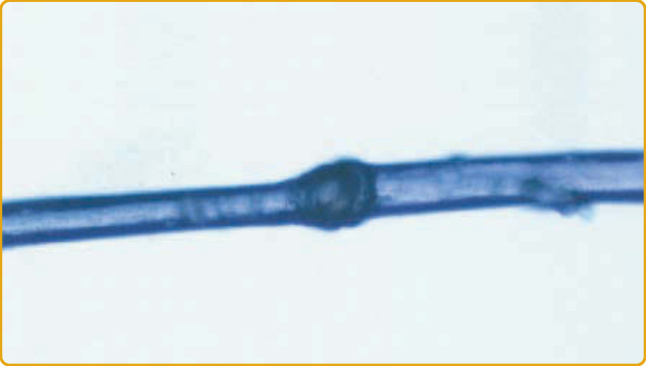
圖 47-13 Netherton 症候群毛髮異常。一小段竹節狀毛幹顯示套疊性脆髮症 (trichorrhexis invaginata) 的特徵。
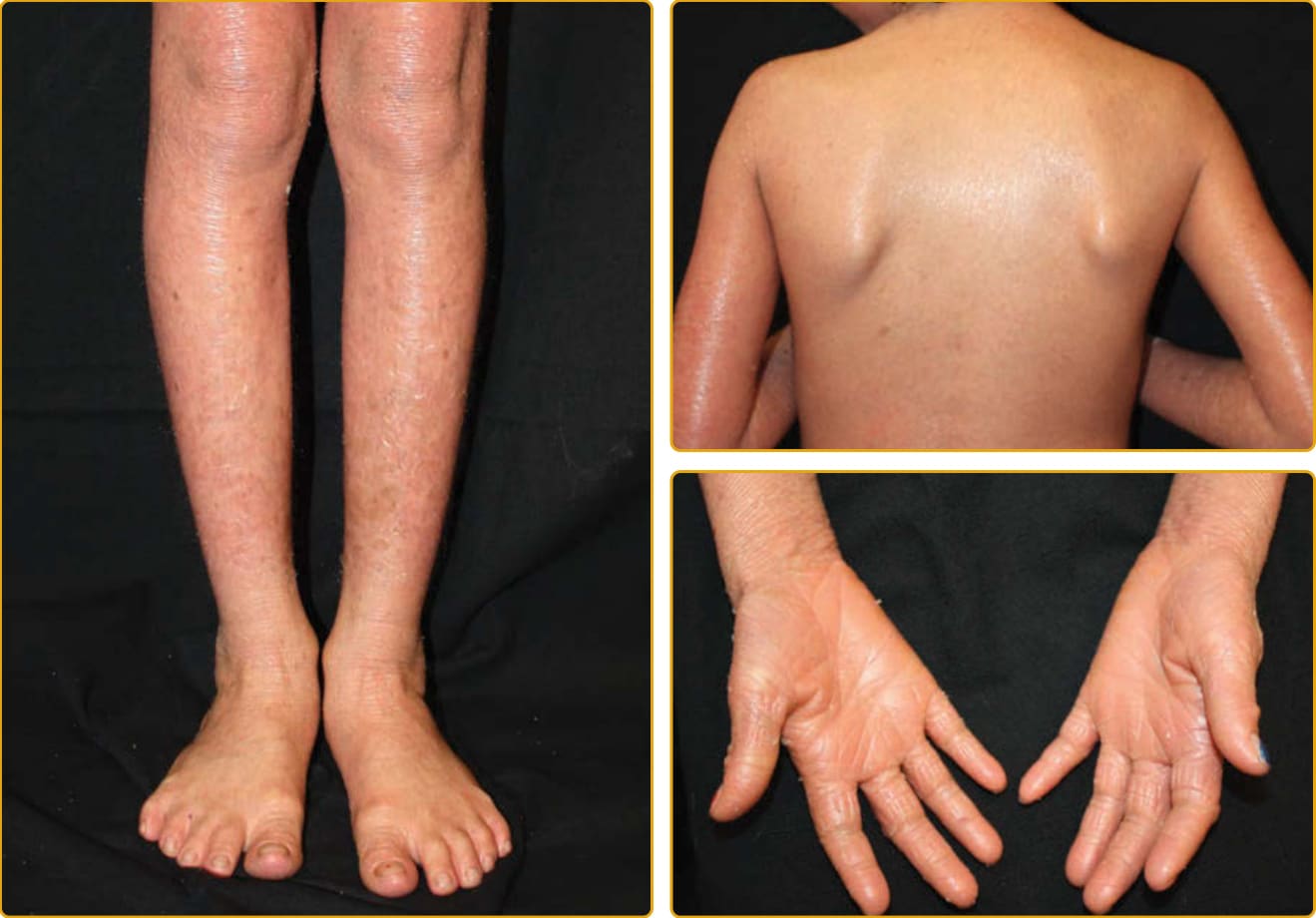
圖 47-14 體染色體隱性先天性魚鱗癬(ABCA12 突變,輕度)。脛部的鱗屑很大,但其他部位的鱗屑在輕度紅斑背景上為小型。中度手掌角皮症伴增加的線狀紋路與錐形第五指。
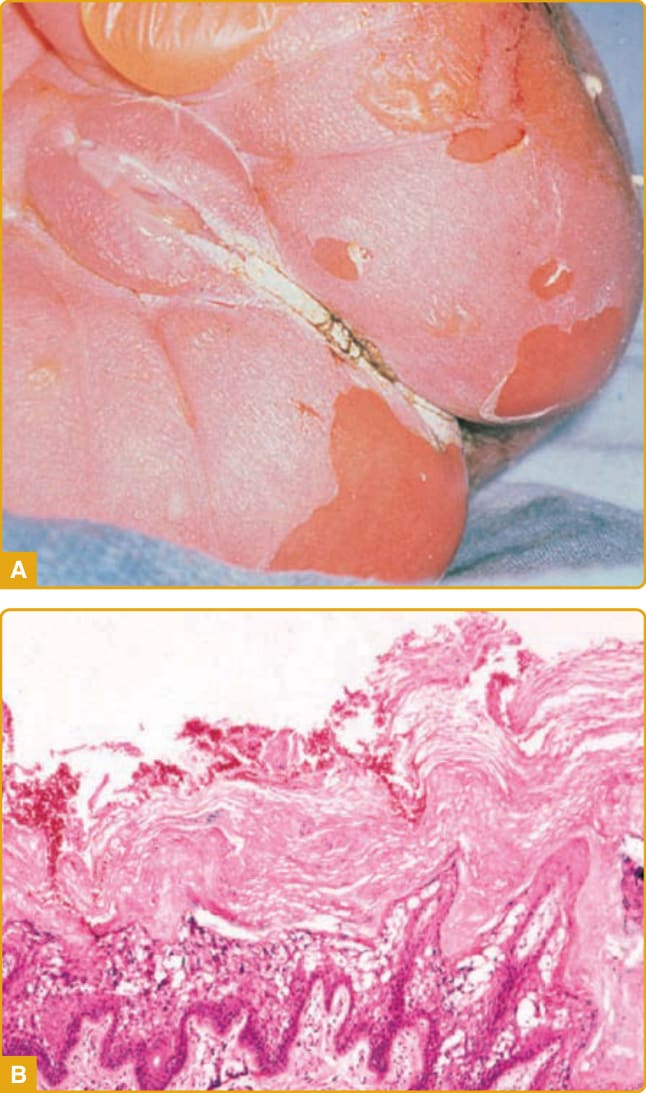
圖 47-15 表皮鬆解性魚鱗癬 (epidermolytic ichthyosis) 嬰兒期表現。A,新生兒顯示水疱與糜爛。B,表皮鬆解性魚鱗癬組織學。角質層增厚(過度角化),基底上層表皮有明顯的空泡變性,以顆粒層最為顯著。
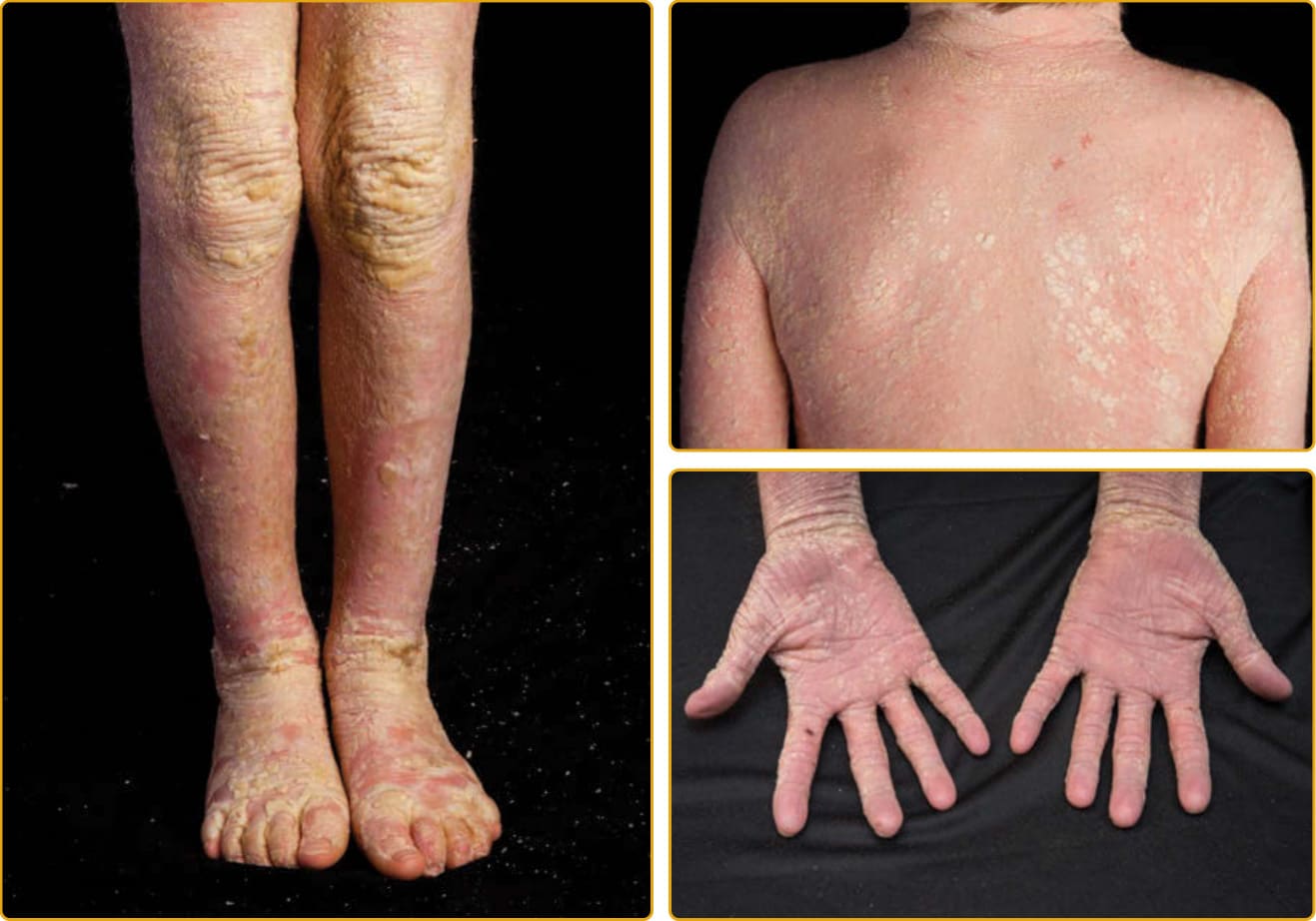
圖 47-16 表皮鬆解性魚鱗癬(KRT10 突變)。全身性粗糙、柱狀鱗屑常在屈側有波紋狀模式。鱗屑從下方表皮不均勻地脫屑,顯露出輕至中度全身性紅斑。手掌與足底的過度角化為輕至中度,並發生假性指趾斷裂症 (pseudoainhum)。
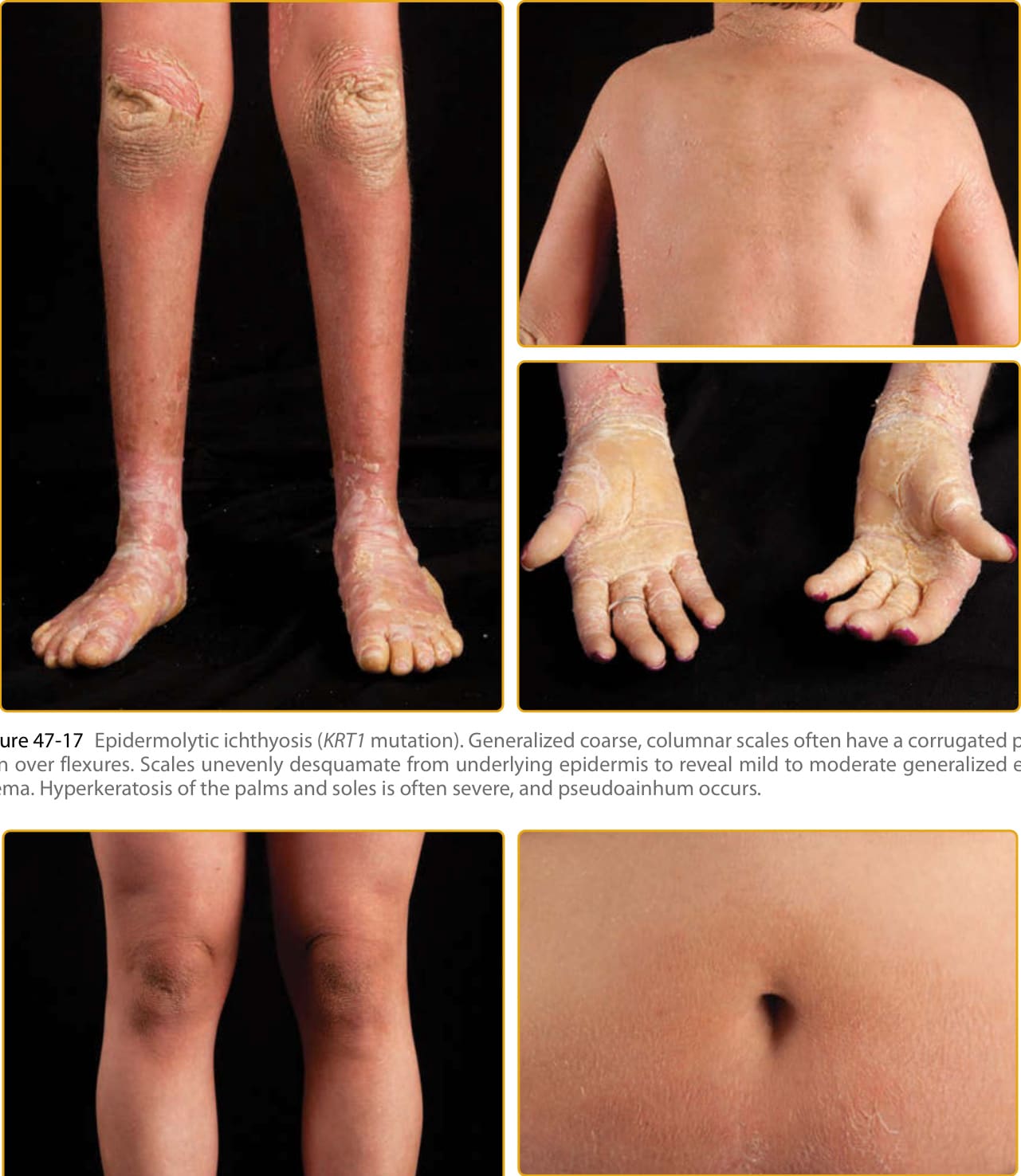
圖 47-17 表皮鬆解性魚鱗癬(KRT1 突變)。全身性粗糙、柱狀鱗屑常在屈側有波紋狀模式。鱗屑從下方表皮不均勻地脫屑,顯露出輕至中度全身性紅斑。手掌與足底的過度角化常為嚴重,並發生假性指趾斷裂症。
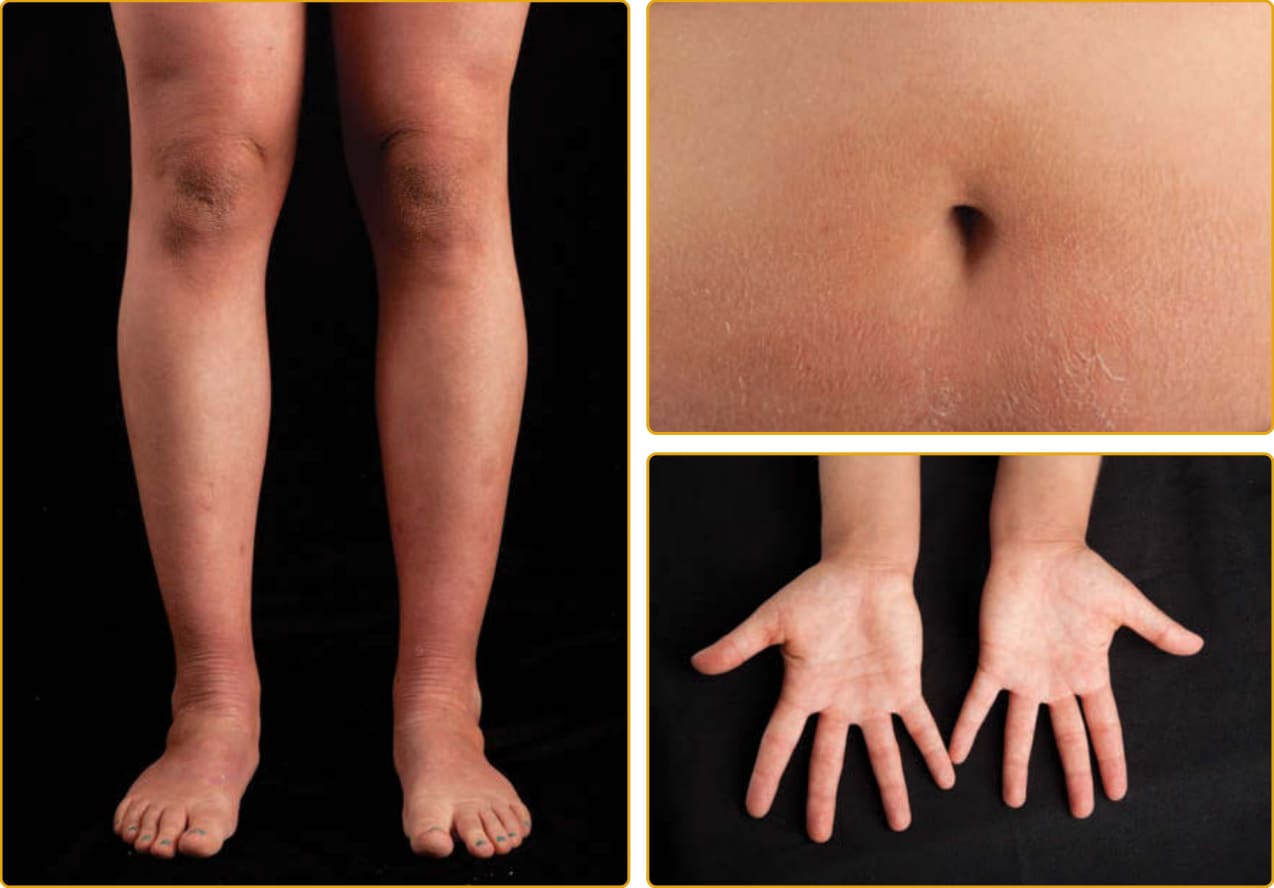
圖 47-18 表皮鬆解性魚鱗癬(KRT2 突變)。平滑、輕度波紋狀的鱗屑一般在屈側或輕度摩擦區域加強。圓形或幾何形的角質層剝離區域常見。紅斑輕微,手掌與足底倖免。
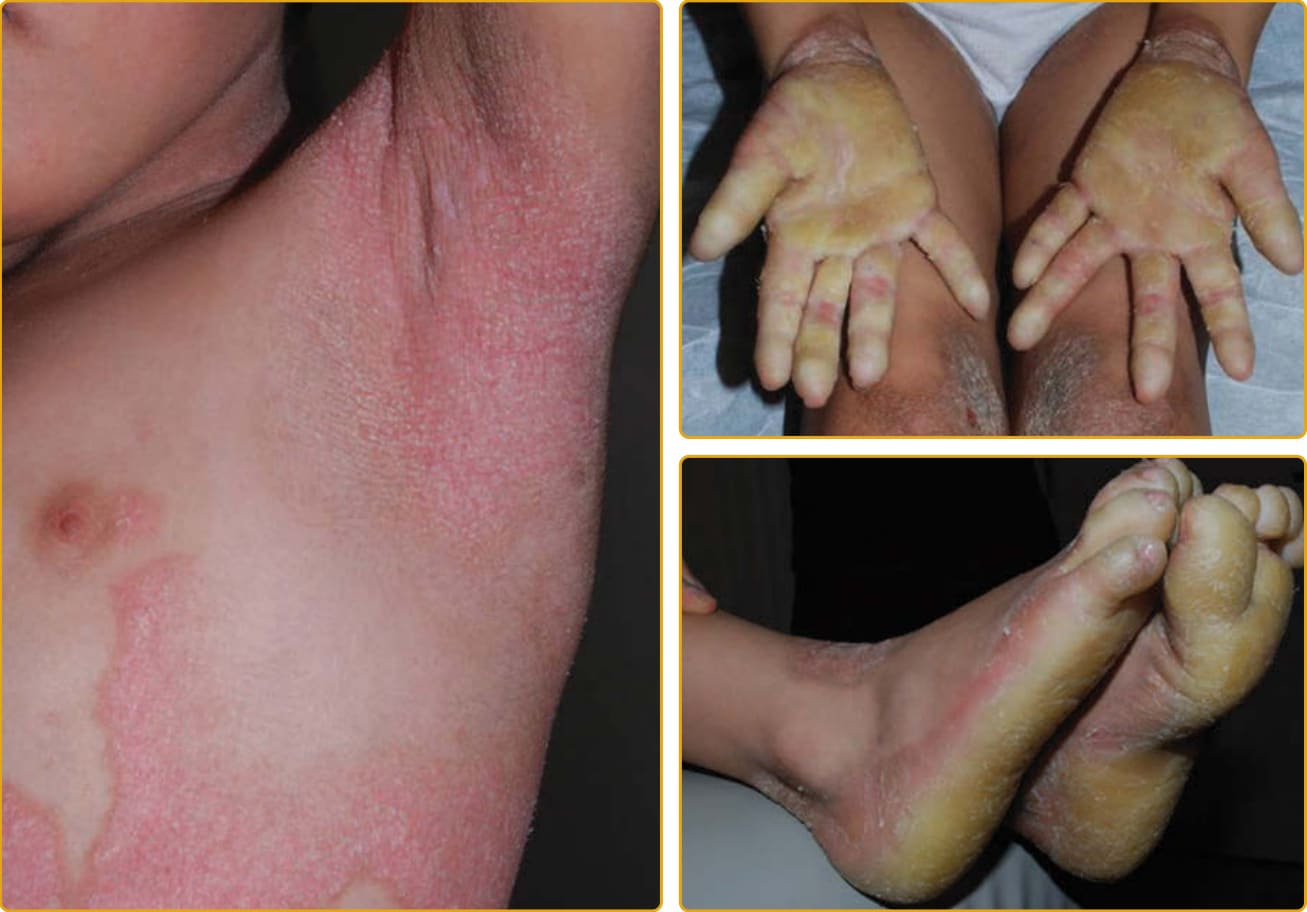
圖 47-19 表皮鬆解性魚鱗癬,環狀伴掌蹠角皮症 (PPK)(KRT1 突變)。界限分明的紅斑性斑塊伴粗糙鱗屑與在屈側加強的波紋狀模式。手掌與足底受侵犯程度不一。
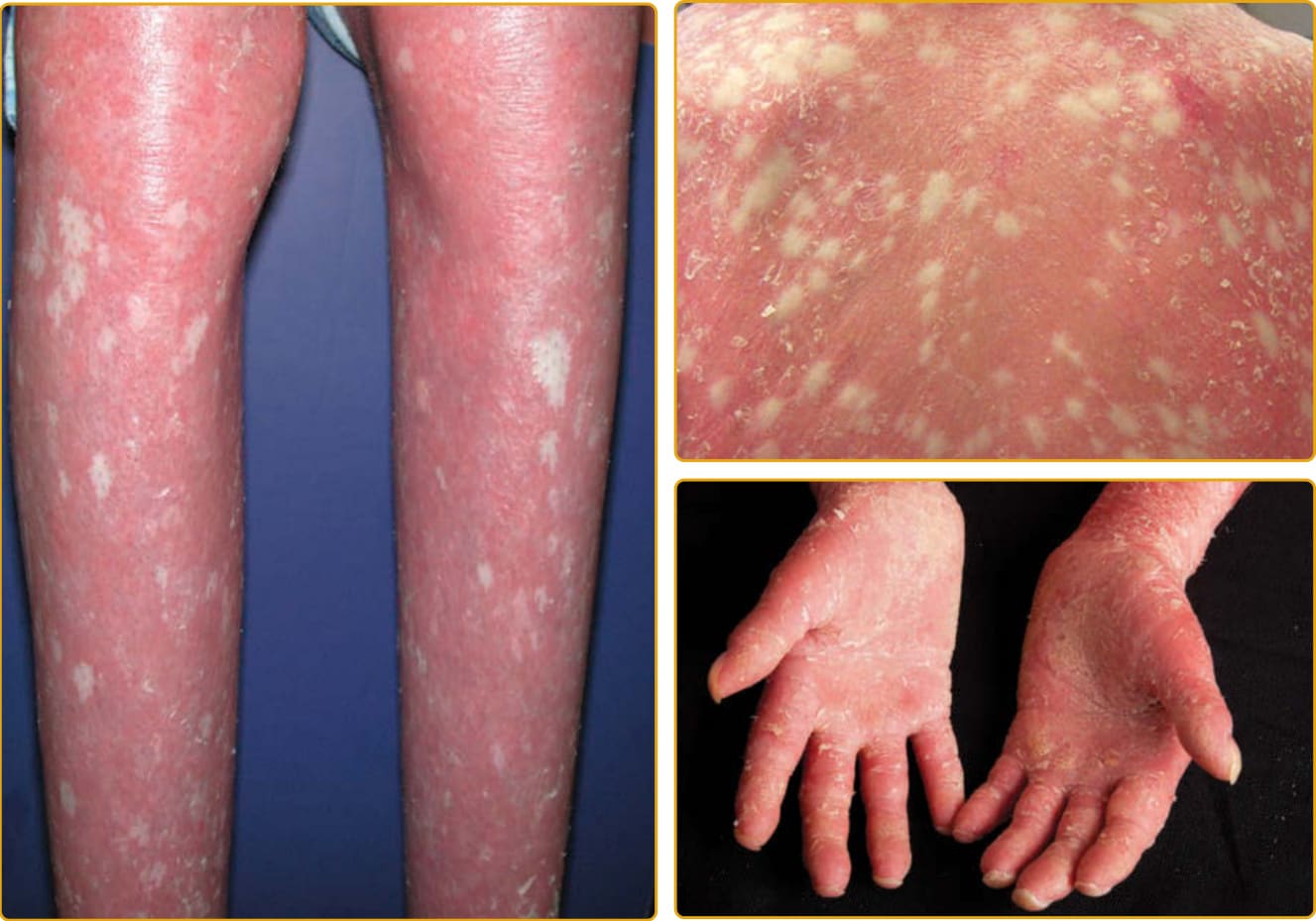
圖 47-20 五彩碎紙樣魚鱗癬 (ichthyosis with confetti)(KRT10 突變)。葉狀 (phylloid,葉片樣) 的正常皮膚區域在輕至重度紅斑與鱗屑的背景上,從生命第一個十年開始在數目與大小上增加。掌蹠紅斑角皮症從輕微到嚴重不等。小與大關節可能發生攣縮。
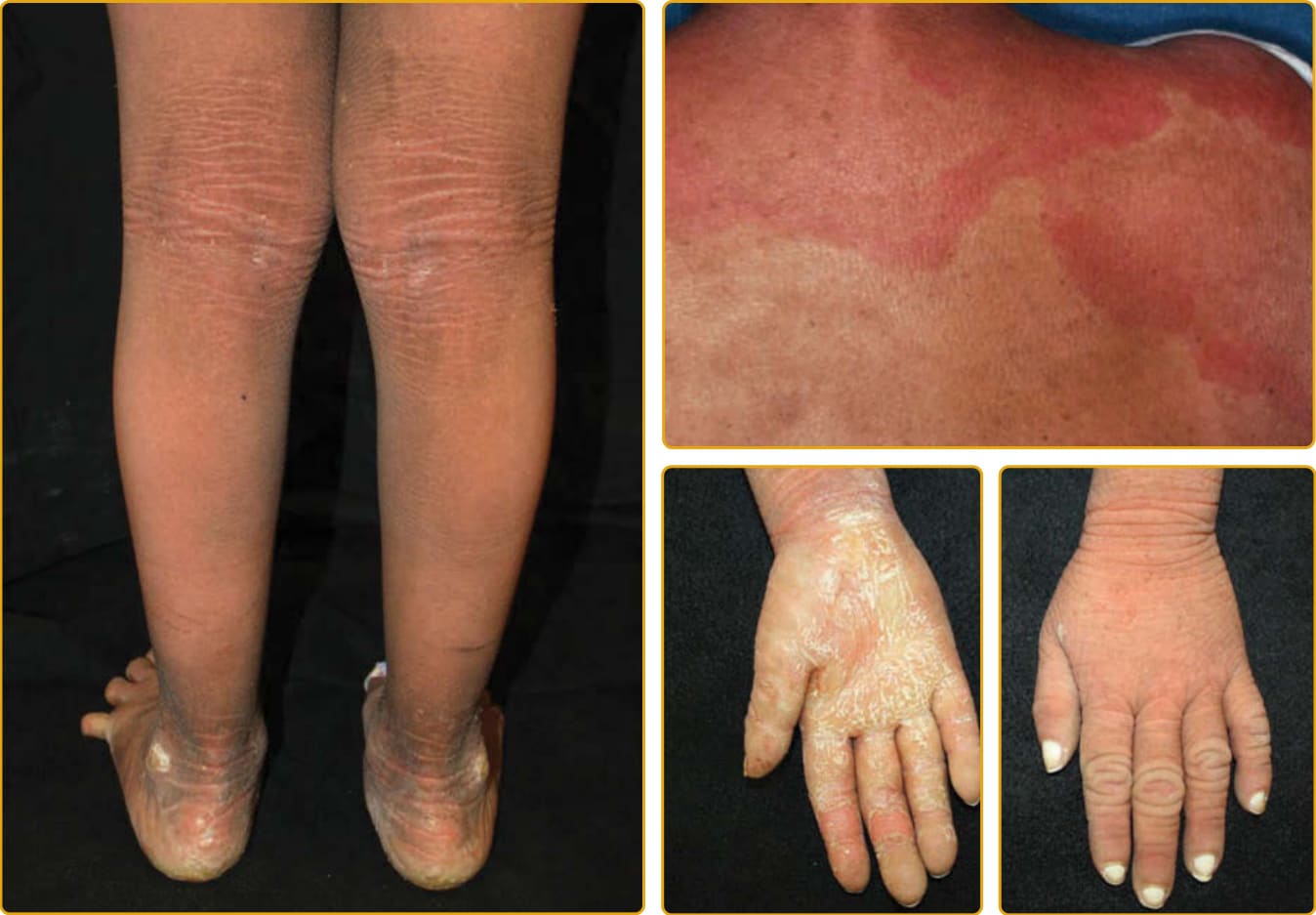
圖 47-21 可變性紅斑角皮症 (erythrokeratoderma variabilis)(GJB3;GJB4;GJA1 突變)。波紋狀粗糙斑塊通常在屈側加強。手掌與足底侵犯可能嚴重。界限分明的紅斑性斑片可為固定或一過性 (evanescent)。
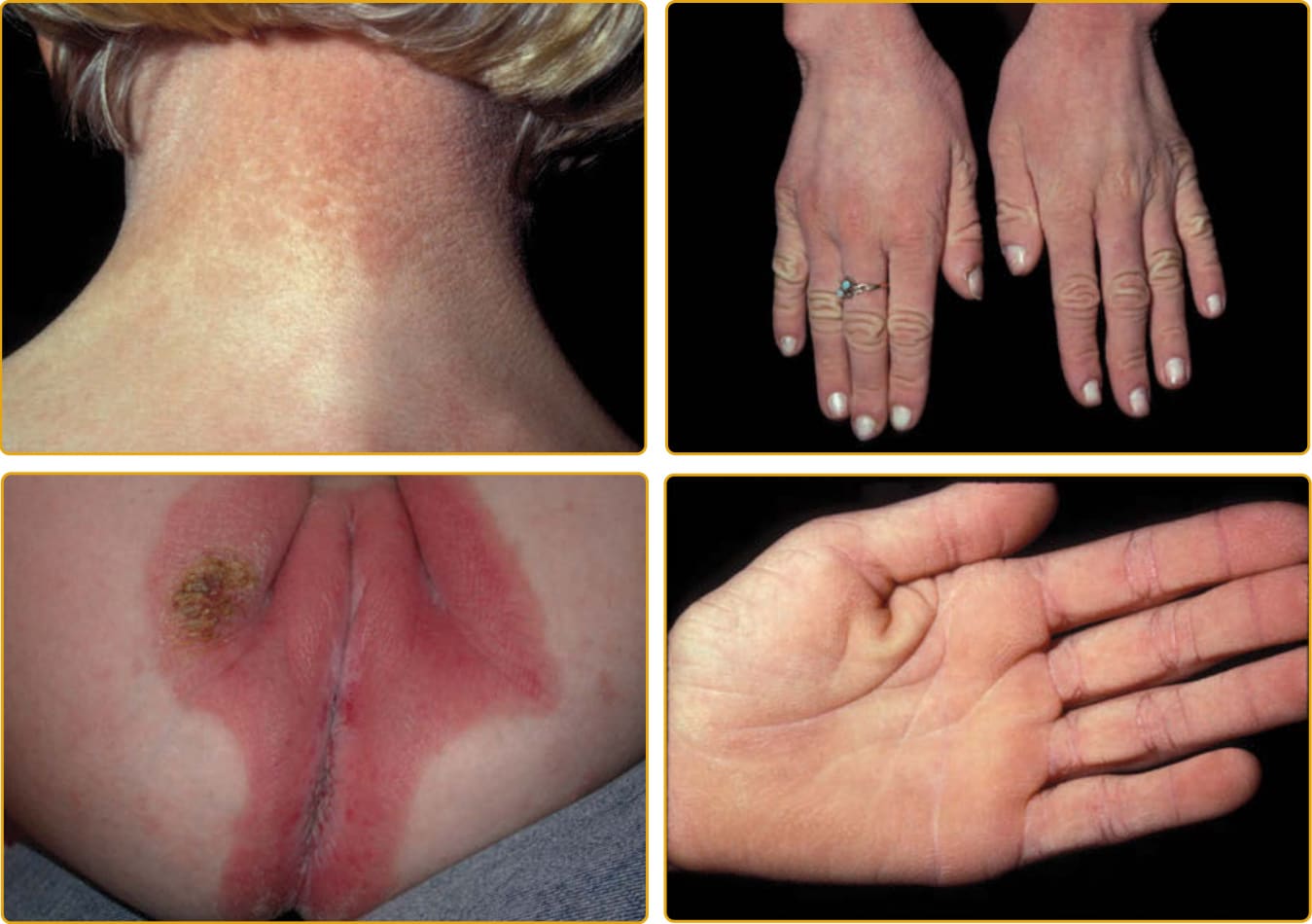
圖 47-22 角膜炎-魚鱗癬-聾症候群 (keratitis ichthyosis deafness syndrome)(GJB2 突變)。平滑或粗砂狀的過度角化柱狀物使皮膚呈卵石樣或粗糙外觀,在身體皺褶、手掌、足底與孔口周圍最為明顯。紅斑通常輕微。細菌與黴菌感染常見。
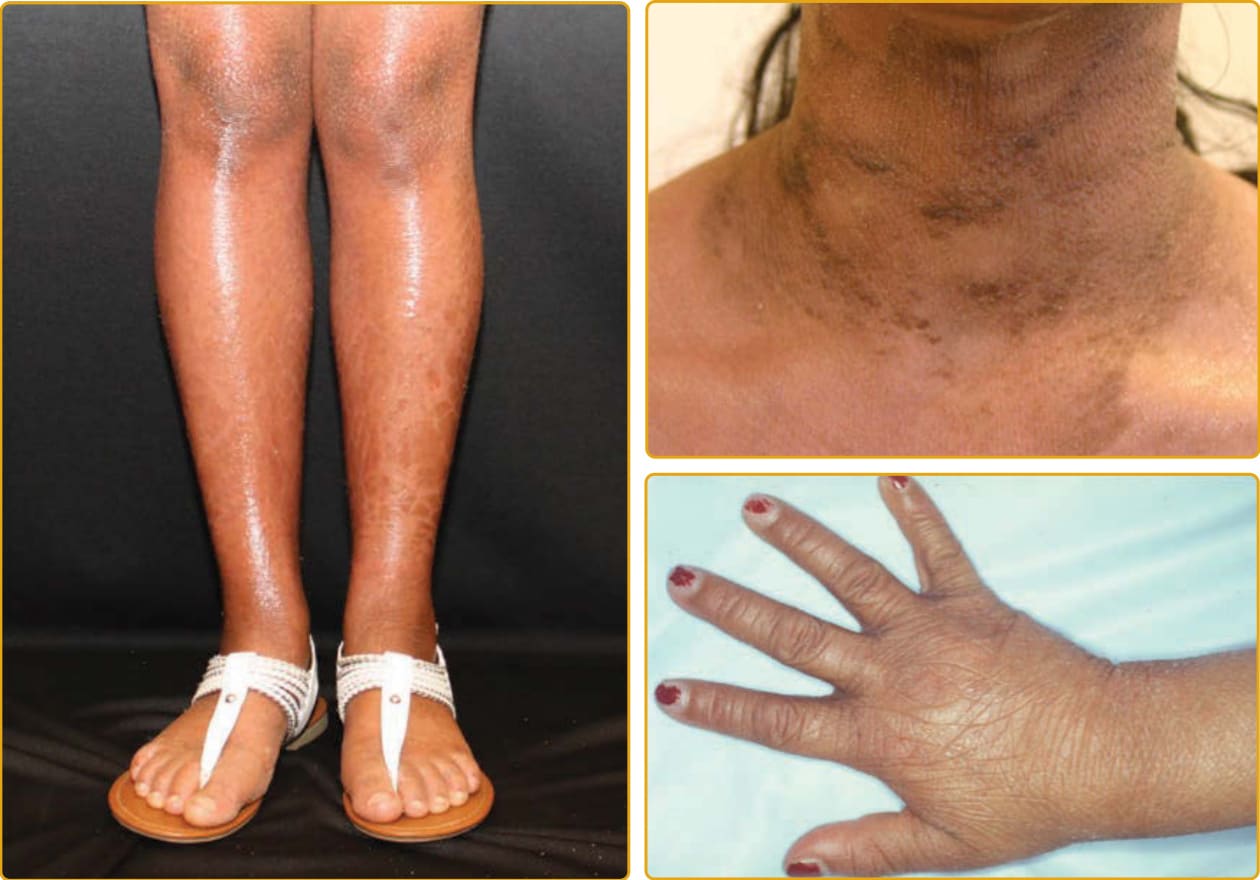
圖 47-23 體染色體隱性先天性魚鱗癬(ABHD5 突變,Chanarin-Dorfman 病;中性脂質儲積病)。深色鱗屑在同一個體中可有差異,從大型扁平到小型柱狀。手與足背側的輕至中度紅斑與誇大、加強的線狀紋路為常見。
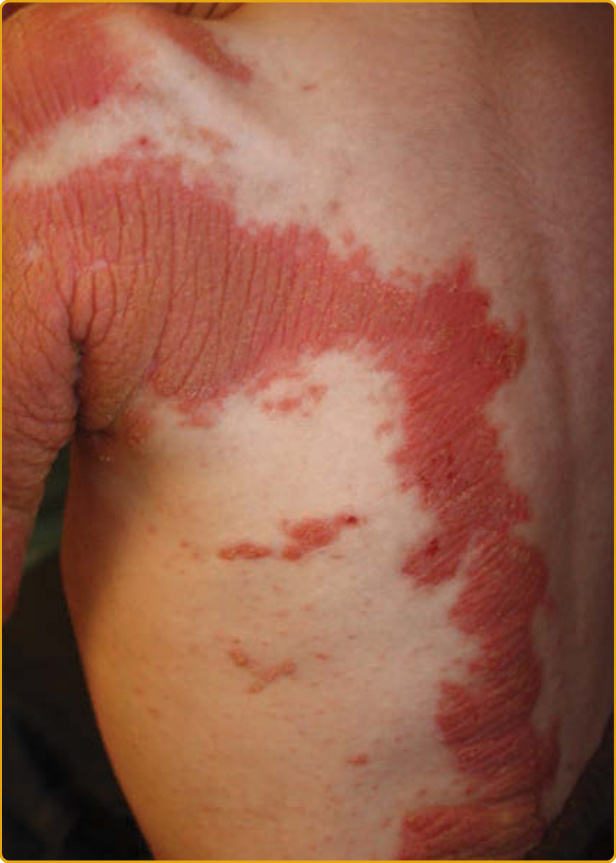
圖 47-24 CHILD(先天性半側發育不良、魚鱗癬樣紅皮症與肢體缺損)症候群。背部出現線狀條紋的紅斑角皮症。
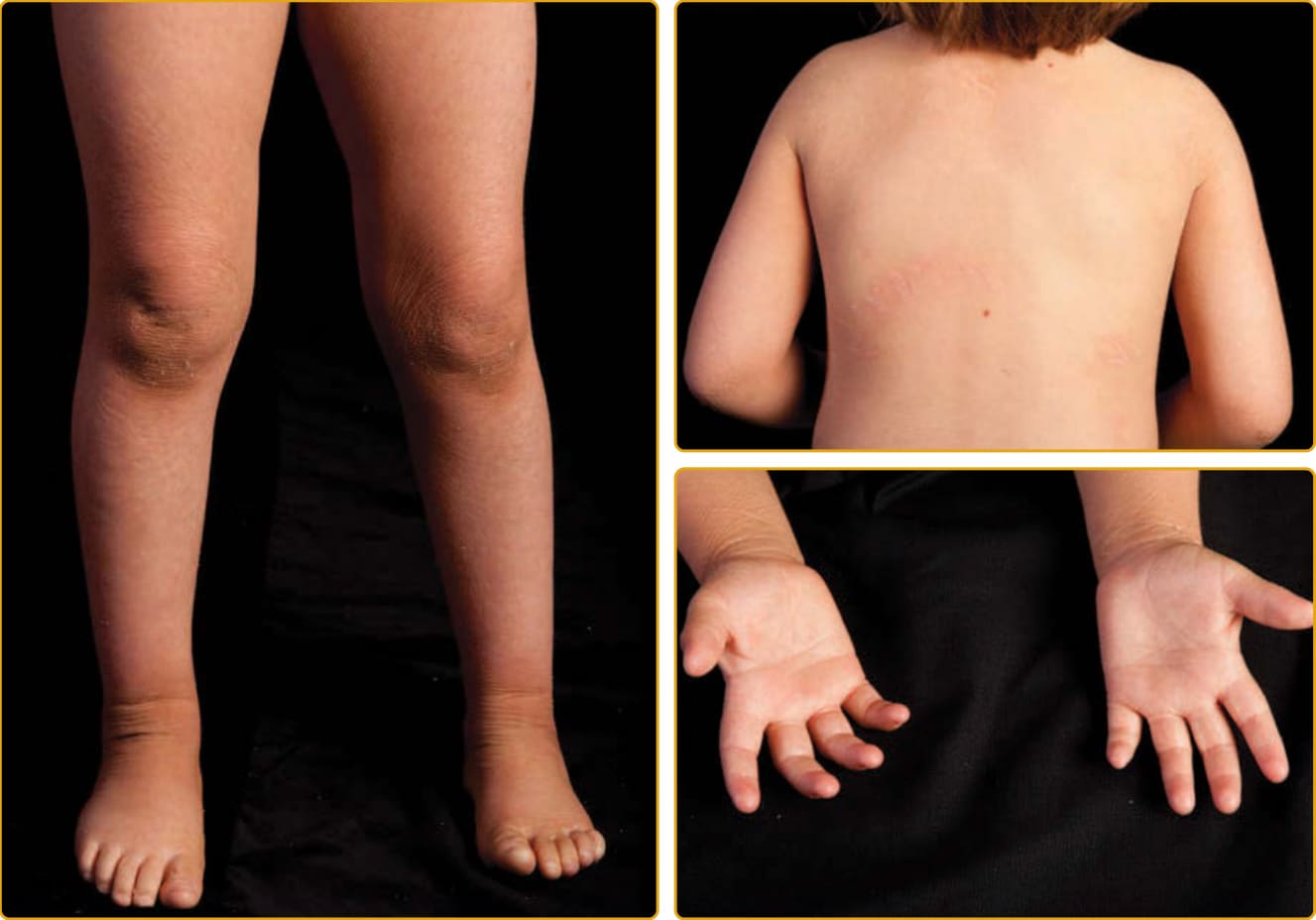
圖 47-25 體染色體隱性先天性魚鱗癬,Sjögren-Larsson 症候群(ALDH3A2 突變)。全身性平滑過度角化(角皮症)導致四肢上大型附著的鱗屑與加強的線狀表面紋路。通常少有紅斑,但搔抓會產生細白色鱗屑與紅斑。
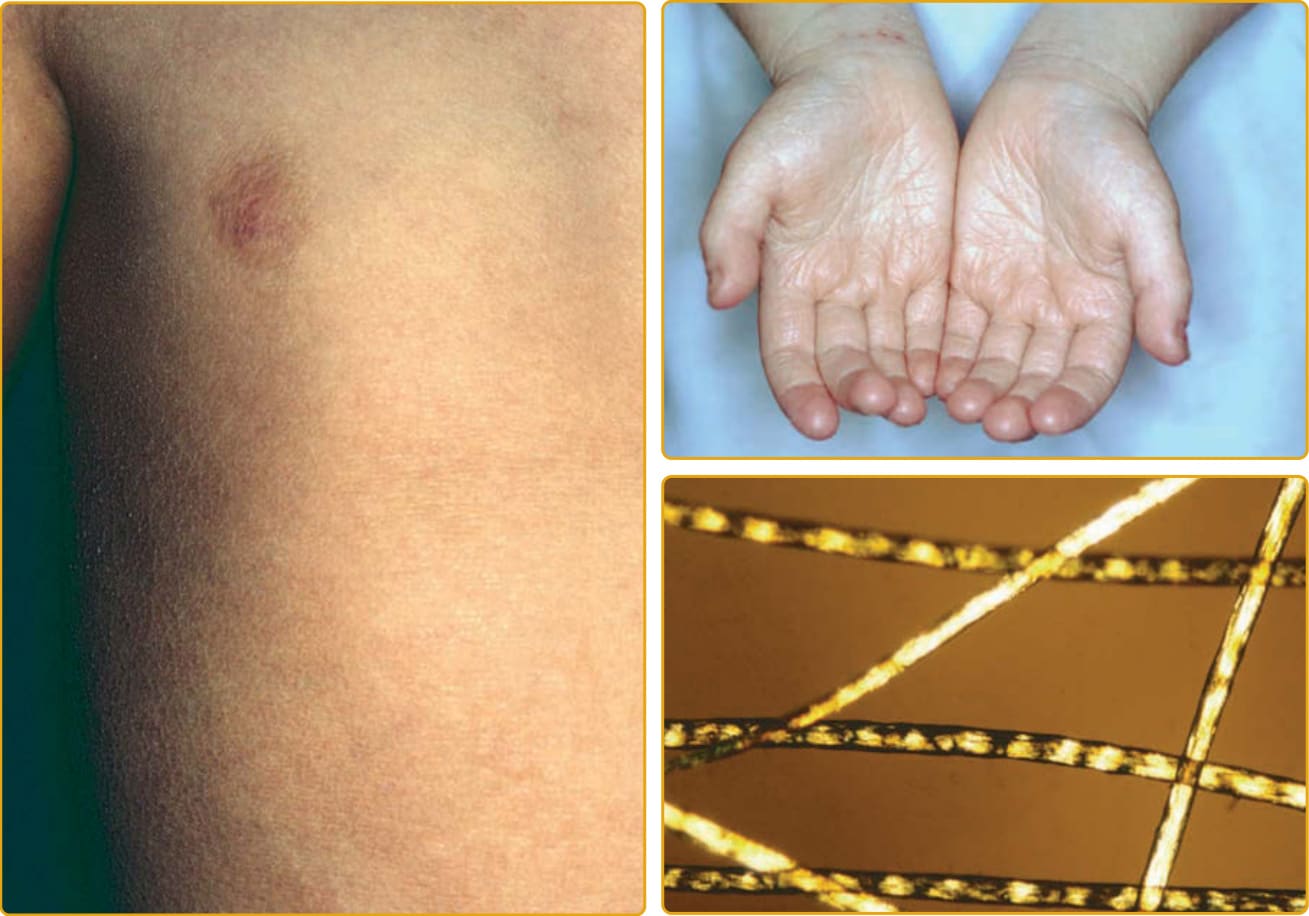
圖 47-26 體染色體隱性先天性魚鱗癬,毛髮硫營養不良 (trichothiodystrophy)(ERCC2、ERCC3、GTF2H5、GTF2E2 突變)。新生兒期之後,通常有非常輕度的全身性過度角化與極少紅斑。毛髮在偏振光下顯示特徵性的交替雙折射。
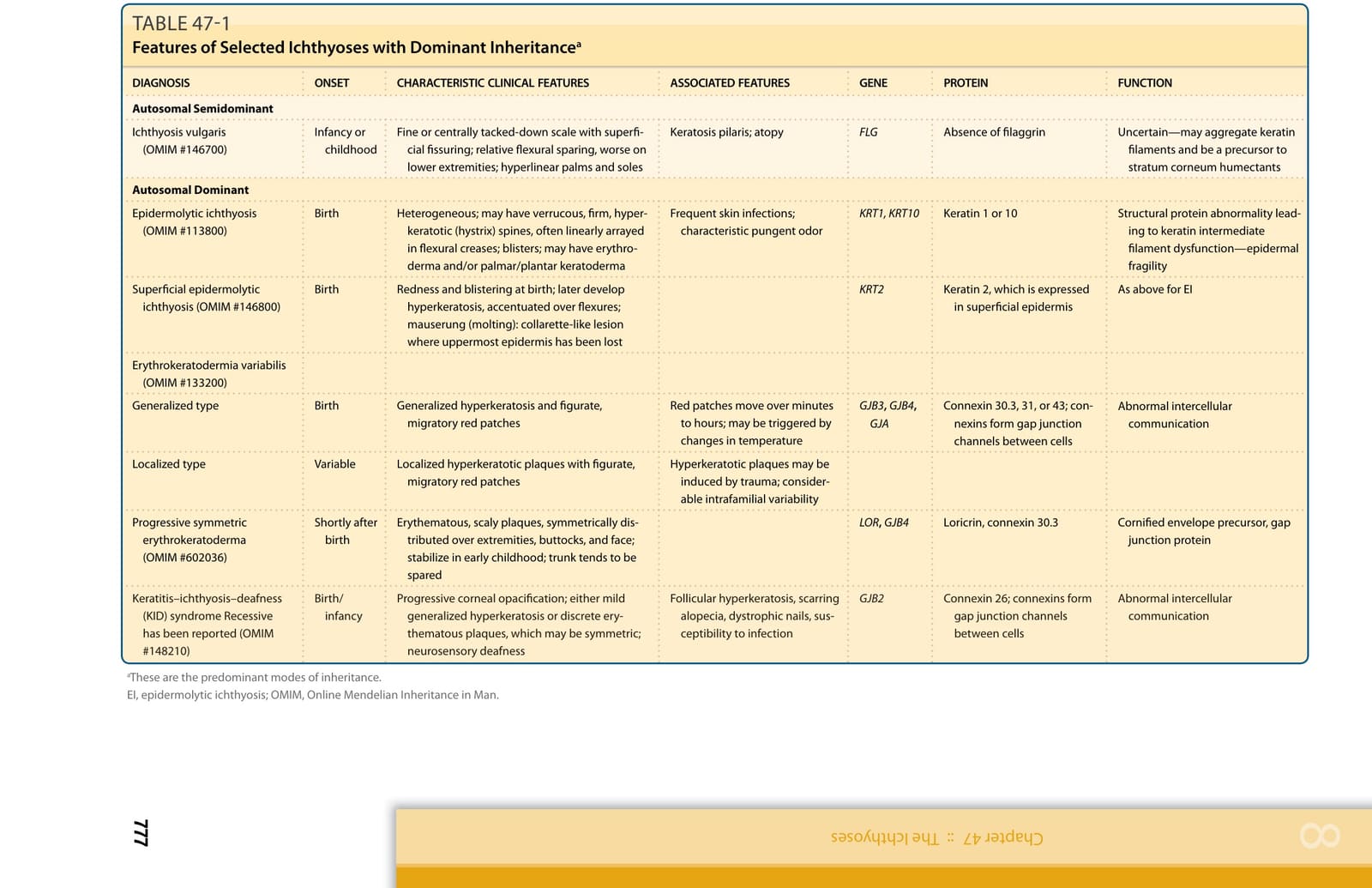
表 47-1:選定魚鱗癬的遺傳模式與常見臨床特徵(一)(Inheritance Patterns and Common Clinical Features of Selected Hereditary Ichthyoses)
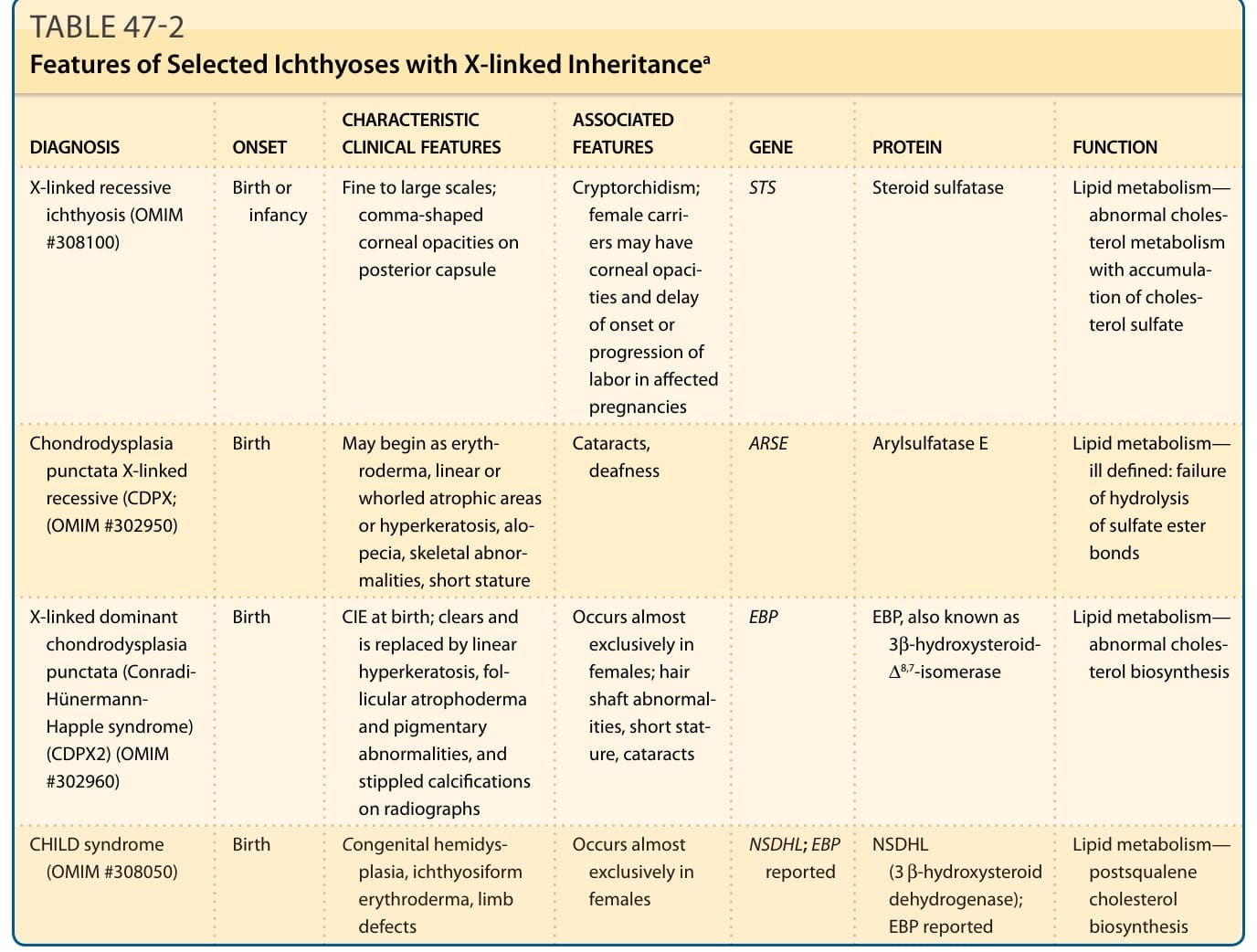
表 47-2:選定 X 性聯遺傳魚鱗癬的特徵 (Features of Selected Ichthyoses with X-linked Inheritance)
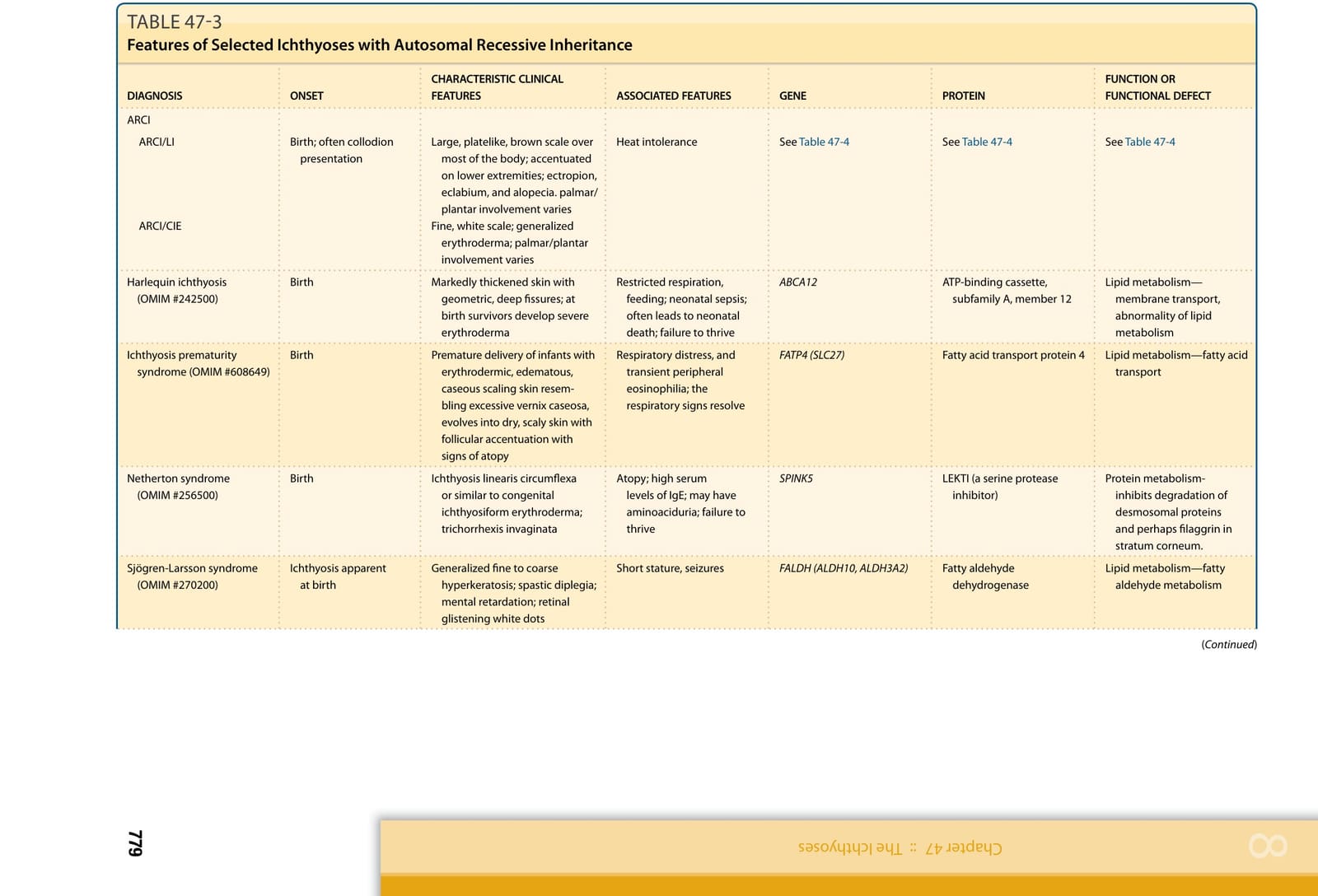
表 47-3:選定魚鱗癬的遺傳模式與常見臨床特徵(二)(Inheritance Patterns and Common Clinical Features of Selected Hereditary Ichthyoses)
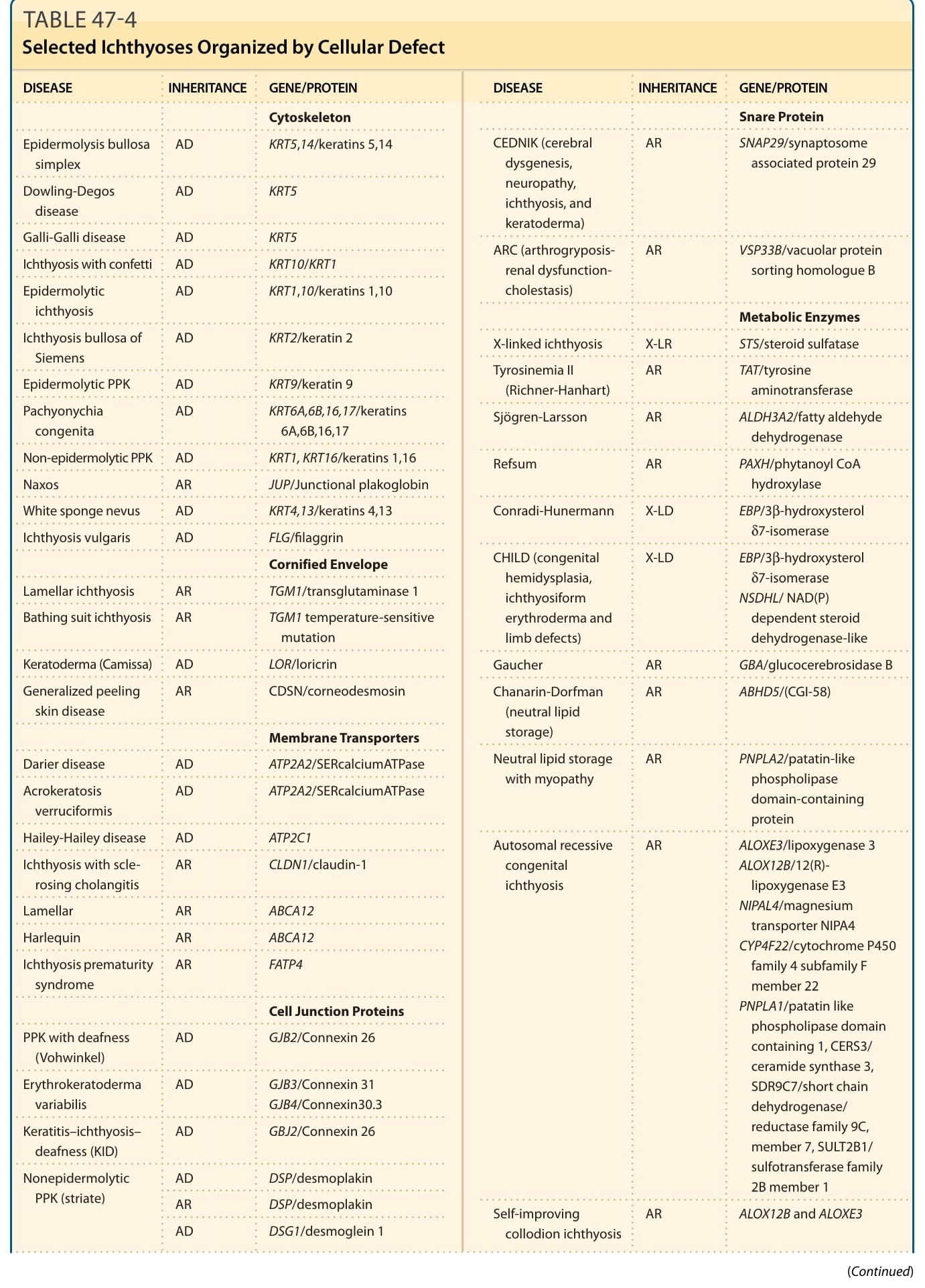
表 47-4:依細胞缺陷組織的選定魚鱗癬 (Selected Ichthyoses Organized by Cellular Defect)

表 47-5:與膠樣膜相關的疾病 (Disorders Associated with Collodion Membrane)