The Ichthyoses
PART8
Disorders of Cornification
AT-A-GLANCE
■ The ichthyoses are a heterogeneous group of skin diseases characterized by generalized scaling, and often areas of thickened skin.
■ Most types are inherited, and these usually present at birth or appear in childhood; however, some forms are acquired.
■ Scales may vary in size, color, and body site.
■ The may be accompanied by erythema, abnormalities in adnexal structures, and palmoplantar keratoderma.
■ The may be associated with systemic findings, such as failure to thrive, increased susceptibility to infection, atopic dermatitis, neurosensory deafness, and neurologic and other disease.
■ Histopathology is usually nonspecific with few notable exceptions.
■ Early genetic testing can aid in the diagnosis and anticipation of potential systemic abnormalities.
INTRODUCTION
This chapter discusses the heterogeneous group of disorders known as the ichthyoses that share the common feature of generalized scaling and most commonly arise at birth or childhood but can be acquired later in life. Ichthyoses commonly result from genetic mutations in a diverse group of genes, including membrane transporters, lipid biosynthesis enzymes, and structural proteins, among many others. Ensuing epithelial barrier compromise, hyperproliferation, and hyperkeratosis become evident as scaling. Associated systemic abnormalities are often attributable to noncutaneous functions of mutant genes.
The hallmark of ichthyosis is scale, which reflects altered differentiation of the epidermis. Because the Greek word for horn (scale) is keras and the Latin is cornu, in this chapter, the terms epidermal differentiation, keratinization, and cornification are used synonymously. The name ichthyosis is derived from the Greek ichthys, meaning “fish,” and refers to the similarity in appearance of the skin to fish scale. Both inherited and acquired forms are found. Early reports of ichthyosis in the Indian and Chinese literature date back to several hundred years bc, and the condition was discussed by Willan in 1798.1
Ichthyosis can present at birth or develop later in life. It can occur as a disease limited to the skin or in association with abnormalities of other organ systems. A number of well-defined types of ichthyosis with characteristic features can be reliably diagnosed. However, because of the great clinical heterogeneity and the profound effect of the environment on scaling, a specific diagnosis can be challenging in certain patients and families without the aid of genetic testing.
CLASSSIFICATION OF THE ICHTHYOSES
Siemens introduced genetic concepts into the ichthyoses.2 Wells and Kerr classified the heritable ichthyoses3 and separated X-linked recessive ichthyosis from ichthyosis vulgaris (IV).4 Gassman developed the concept of retention versus hyperproliferation hyperkeratosis.5 Van Scott, Frost, and Weinstein subsequently proposed a classification of the ichthyoses based on differences in rates of epidermal turnover, characterizing them as either disorders of epidermal hyperproliferation or disorders of prolonged retention of the stratum corneum.6 Subsequently, Williams and Elias proposed a classification that lists the disorders
8
of cornification in which clinical, genetic, or biochemical data suggest a distinct disease.7
Genetic investigation of the ichthyoses has revealed causative mutations in genes relevant to structural proteins, lipid biosynthesis, cell–cell communication, desquamation, and many other pathways and has, in many cases, provided insight into previously unknown determinants of epidermal homestasis.8 The advent of whole-exome sequencing (WES) has led to phenotypic expansion, and a new classification of the ichthyoses is evolving based not only on mutant genes but also on specific mutations. Knowing which gene is mutated and what the effects of a given mutation are enables understanding of disease pathobiology. Defining these disorders on the basis of common molecular processes leads to more rational approaches to understanding their pathophysiology and treatment. Inheritance patterns and common clinical features of selected hereditary ichthyoses are shown in Tables 47-1 to 47-3. Grouping these disorders according to the function of encoded proteins (Table 47-4) facilitates understanding of the clinical phenotypes in terms of underlying mechanism. However, further work is necessary to clearly understand how specific mutations result in clinical disease and to develop targeted therapeutic interventions. In 2009, a consensus conference proposed a revised nomenclature and classification of the ichthyoses, which has replaced commonly used but varied descriptive terms with descriptors based on inheritance and pathobiology.9 Primary criteria for classification included whether the disorder is limited to the skin (nonsyndromic) or affects the skin and other organ systems (syndromic), its mode of inheritance, and disease pathobiology. Thus, disorders previously referred to as harlequin ichthyosis, lamellar ichthyosis (LI), and congenital ichthyosiform erythroderma became autosomal recessive congenital ichthyosis (ARCI) and bullous ichthyosis, bullous congenital ichthyosiform erythroderma (BCIE), epidermolytic hyperkeratosis, and ichthyosis exfoliativa became epidermolytic ichthyosis (EI), among other updates.9
Although likely to be further refined in coming years, 2009 consensus nomenclature will be used throughout this chapter.
CLINICAL PRESENTATION
Several features are useful in distinguishing different forms of ichthyosis. These include age of onset, presence of collodion membrane at birth, quality of scale, presence or absence of erythroderma, abnormalities in other parts of the skin (eg, thickened palms and soles, ectropion, eclabium) and adnexal structures (eg, alopecia or hair shaft abnormalities), and involvement of other organ systems. Among distinguishing features, the appearance of the surface of the skin can aid in diagnosis. Visible scaling may be seen in some patients, which can be distinguished by size, configuration, color, and adherence of scale, and there can be thickening of the skin with or without
776
visible scale known as keratoderma. Thickening of the stratum corneum, evident either clinically or histologically, is termed hyperkeratosis. Light microscopic features are usually diagnostic in EI and can be helpful in selected ichthyoses (eg, Refsum disease, neutral lipid storage disease, acquired ichthyosis of sarcoidosis, and mycosis fungoides), but histopathologic examination may not be useful to distinguish other ichthyoses.10 In many cases, the clinical diagnosis may be clarified by genetic analysis, although mutations are not always found. The development of ichthyosis in adulthood may be a marker of systemic disease.
GENETICS
Family history and a multigeneration pedigree may indicate the mode of inheritance. However, many autosomal dominant diseases (eg, EI) have a high frequency of spontaneous (also called de novo) mutation. Thus, the lack of a positive family history does not rule out autosomal dominant inheritance. Alternatively, the presence of parental consanguinity may suggest autosomal recessive inheritance. Distinguishing among autosomal dominant, de novo mutation, and autosomal recessive inheritance in pedigrees without prior affected individuals presents a unique clinical challenge in many cases. The advent of WES has permitted ascertainment of inheritance patterns, genetic diagnosis, and gene discovery in many cases.11,12
Prenatal molecular diagnosis has become possible. Alternative methods, including fetoscopy and fetal skin biopsy, are limited to later times in pregnancy, harbor a risk of fetal mortality, and are now rarely performed.13 When it is possible to do prenatal diagnosis by molecular analysis of a fetal sample, it is optimally performed early in pregnancy. This can be done with chorionic villous sampling in the first trimester (10 to 12 weeks after last menstrual period) or by amniocentesis in the second trimester in disorders in which the underlying genetic defect is known and the specific mutation in the family has been identified.14 Prenatal diagnosis by mutational analysis has been accomplished in a number of the ichthyoses. Preimplantation genetic diagnosis is a reasonable alternative and has been accomplished for many inherited disorders, including ARCI and EI.15 The procedure requires that the couple undergo in vitro fertilization to obtain embryos. The embryos are then screened by molecular methods to detect the mutation that is segregating in the family. Only embryos that screen negative for the mutation are selected and then can be used for implantation in the uterus to achieve pregnancy. Noninvasive methods of molecular diagnosis (evaluation of fetal DNA circulating in the mother’s blood) offer potential for the future.16,17 For autosomal recessive disorders in which the mutation is known, carrier detection may be performed for at-risk relatives.
Frequent skin infections; characteristic pungent odor KRT1, KRT10 Keratin 1 or 10 Structural protein abnormality leading to keratin intermediate filament dysfunction—epidermal fragility
Abnormal intercellular communication
Epidermolytic ichthyosis (OMIM #113800) Birth Heterogeneous; may have verrucous, firm, hyperkeratotic (hystrix) spines, often linearly arrayed in flexural creases; blisters; may have erythroderma and/or palmar/plantar keratoderma
Superficial epidermolytic ichthyosis (OMIM #146800) Birth Redness and blistering at birth; later develop hyperkeratosis, accentuated over flexures; mauserung (molting): collarette-like lesion where uppermost epidermis has been lost
Autosomal Semidominant
Autosomal Dominant
8
Abnormal intercellular communication
Abnormal intercellular
communication
gap junction channels
between cells
Follicular hyperkeratosis, scarring alopecia, dystrophic nails, susceptibility to infection
Follicular hyperkeratosis, scarring
alopecia, dystrophic nails, sus-
ceptibility to infection
Birth/ infancy Progressive corneal opacification; either mild generalized hyperkeratosis or discrete erythematous plaques, which may be symmetric; neurosensory deafness
Shortly after birth Erythematous, scaly plaques, symmetrically distributed over extremities, buttocks, and face; stabilize in early childhood; trunk tends to be spared
neurosensory deafness
Birth/
Keratitis–ichthyosis–deafness (KID) syndrome Recessive has been reported (OMIM #148210)
Keratitis–ichthyosis–deafness
has been reported (OMIM
(KID) syndrome Recessive
Progressive symmetric erythrokeratoderma (OMIM #602036)
#148210)
8
DIAGNOSIS ONSET CHARACTERISTIC CLINICAL FEATURES ASSOCIATED FEATURES GENE PROTEIN FUNCTION
X-linked recessive ichthyosis (OMIM #308100)
Birth or infancy Fine to large scales; comma-shaped corneal opacities on posterior capsule
Cryptorchidism; female carriers may have corneal opacities and delay of onset or progression of labor in affected pregnancies
Chondrodysplasia punctata X-linked recessive (CDPX; (OMIM #302950)
Birth May begin as erythroderma, linear or whorled atrophic areas or hyperkeratosis, alopecia, skeletal abnormalities, short stature
STS Steroid sulfatase Lipid metabolism— abnormal cholesterol metabolism with accumulation of cholesterol sulfate
Cataracts, deafness ARSE Arylsulfatase E Lipid metabolism— ill defined: failure of hydrolysis of sulfate ester bonds
X-linked dominant chondrodysplasia punctata (Conradi- Hünermann- Happle syndrome) (CDPX2) (OMIM #302960)
Birth CIE at birth; clears and is replaced by linear hyperkeratosis, follicular atrophoderma and pigmentary abnormalities, and stippled calcifications on radiographs
Lipid metabolism— abnormal cholesterol biosynthesis
Occurs almost exclusively in females; hair shaft abnormalities, short stature, cataracts
Birth Congenital hemidys-
CHILD syndrome
EBP EBP, also known as 3β-hydroxysteroid- ∆8,7-isomerase
NSDHL; EBP
NSDHL; EBP reported NSDHL (3 β-hydroxysteroid dehydrogenase); EBP reported
Occurs almost
CHILD syndrome (OMIM #308050) Birth Congenital hemidysplasia, ichthyosiform erythroderma, limb defects
NSDHL
Lipid metabolism—
Occurs almost exclusively in females
(OMIM #308050)
plasia, ichthyosiform erythroderma, limb defects
Lipid metabolism— postsqualene cholesterol biosynthesis
(3 β-hydroxysteroid dehydrogenase); EBP reported
exclusively in females
reported
postsqualene cholesterol biosynthesis
aThese are the predominant modes of inheritance. CHILD, congenital hemidysplasia with ichthyosiform erythroderma and limb defects; CIE, congenital ichthyosiform erythroderma; EBP, emopamil-binding protein; NSDHL,= NAD(P)H steroid dehydrogenase-like protein; OMIM, Online Mendelian Inheritance in Man.
ETIOLOGY AND PATHOGENESIS
The epidermis undergoes a regular pattern of selfrenewal. Its primary cell type, the keratinocyte, undergoes a regular pattern of differentiation to enable the skin to fulfil its role as a barrier to mechanical trauma and desiccation. The end product of differentiation is the stratum corneum, which is composed of terminally differentiated keratinocytes, corneocytes (“bricks”), surrounded by an intercellular matrix (“mortar”) (see Chaps. 5 and 14). The corneocyte bricks are protein enriched, and the intercellular mortar is composed of hydrophobic, lipid-enriched membrane bilayers.7
The keratin-laden corneocytes are thought to be primarily responsible for the resilience and water retention properties of the stratum corneum, and the matrix forms most of the permeability barrier to water loss. The normal stratum corneum undergoes desquamation in an organized and invisible manner, with individual corneocytes separating from each other and shedding as single cells. Ichthyotic skin has an abnormal quality and quantity of scale, the barrier function
778
of the stratum corneum is compromised, and there may be alterations in the kinetics of epidermal cell proliferation (see Chap. 5). The stratum corneum can be viewed as a compartment, with thickening of the stratum corneum being the result of cells entering the compartment at an increased rate, leaving (corneocyte desquamation) too slowly, or both. The process of epidermal differentiation is complex and not completely understood. Defects in many different aspects and steps of this process can lead to a similar end result: abnormal stratum corneum and scale. Genetic investigation of inherited ichthyoses has revealed pathways central to epithelial differentiation. For example, mutations in genes that encode the suprabasal epidermal keratins, keratins 1 and 10, cause EI when they affect the highly conserved encoded rod domains necessary for polymerization of keratin intermediate filaments.18-20 Mutations in the gene encoding transglutaminase-1, an enzyme that catalyzes the cross-linking of proteins and attachment of ceramides during the formation of corneocytes, are found in a large fraction of patients with autosomal recessive congenital ichthyosis.21-23 Mutations in SPINK5, encoding a serine protease inhibitor, cause
SPINK5 LEKTI (a serine protease inhibitor) Protein metabolisminhibits degradation of desmosomal proteins and perhaps filaggrin in stratum corneum.
ABCA12 ATP-binding cassette, subfamily A, member 12 Lipid metabolism— membrane transport, abnormality of lipid metabolism
Restricted respiration, feeding; neonatal sepsis; often leads to neonatal death; failure to thrive
Atopy; high serum levels of IgE; may have aminoaciduria; failure to thrive
Respiratory distress, and transient peripheral eosinophilia; the respiratory signs resolve
Large, platelike, brown scale over most of the body; accentuated on lower extremities; ectropion, eclabium, and alopecia. palmar/ plantar involvement varies Fine, white scale; generalized erythroderma; palmar/plantar involvement varies
Ichthyosis prematurity syndrome (OMIM #608649) Birth Premature delivery of infants with erythrodermic, edematous, caseous scaling skin resembling excessive vernix caseosa, evolves into dry, scaly skin with follicular accentuation with signs of atopy
Harlequin ichthyosis (OMIM #242500) Birth Markedly thickened skin with geometric, deep fissures; at birth survivors develop severe erythroderma
Netherton syndrome (OMIM #256500) Birth Ichthyosis linearis circumflexa or similar to congenital ichthyosiform erythroderma; trichorrhexis invaginata
Birth; often collodion presentation
ARCI/CIE
ARCI/LI
ARCI
8
(Continued)
Sjögren-Larsson syndrome (OMIM #270200) Ichthyosis apparent at birth Generalized fine to coarse hyperkeratosis; spastic diplegia; mental retardation; retinal glistening white dots
8
Most PAHX; PEX 7 also reported (see RCPD below) Phytanoyl-CoA hydroxylase (PhyH) Peroxisome abnormality— deficiency of phytanic acid catabolism; results in phytanic acid accumulation.
in active site of eukaryotic
sulfatases
Abnormally low sulfur content of hair. Majority have defect in ERCC2 (XPD). A few have mutations in ERCC3 (XPB), GTF2H5 (TTDA), or TTDN1.
Scarring alopecia of the scalp and eyebrows, enamel dysplasia
Retinitis pigmentosa, elevated plasma phytanic acid
Severe pruritus; neurologic abnormalities; hepatic abnormalities; lipid droplets in circulating leukocytes
skeletal abnormalities;
facial dysmorphism
Brittle hair, photosensitivity, short stature, ichthyosis, intellectual impairment, microcephaly, recurrent infections
Birth; may have collodion membrane Generalized scaling, resembles CIE; variable extracutaneous involvement: cataracts, decreased hearing, psychomotor delay.
Some have ichthyosis, which may be apparent at birth; may have collodion presentation
Multiple sulfatase deficiency
Trichothiodystrophy (Tay syndrome; PIBI(D)S, IBI(D)S, BI(D)S) (OMIM #278730, OMIM #601675, OMIM #234050)
Chanarin–Dorfman syndrome (neutral lipid storage disease; OMIM #275630)
Neonatal ichthyosis– sclerosing cholangitis syndrome (OMIM #6073718)
Selected Ichthyoses Organized by Cellular Defect
DISEASE INHERITANCE GENE/PROTEIN
Cytoskeleton
Cytoskeleton
Epidermolysis bullosa simplex AD KRT5,14/keratins 5,14
AD KRT5,14/keratins 5,14
Epidermolysis bullosa
simplex
Dowling-Degos disease AD KRT5
AD KRT5
Dowling-Degos
disease
Galli-Galli disease AD KRT5
Galli-Galli disease AD KRT5
Ichthyosis with confetti AD KRT10/KRT1
Ichthyosis with confetti AD KRT10/KRT1
Epidermolytic ichthyosis AD KRT1,10/keratins 1,10
AD KRT1,10/keratins 1,10
Epidermolytic
ichthyosis
Ichthyosis bullosa of Siemens AD KRT2/keratin 2
AD KRT2/keratin 2
Ichthyosis bullosa of
Siemens
Epidermolytic PPK AD KRT9/keratin 9
Epidermolytic PPK AD KRT9/keratin 9 9/ 9
Pachyonychia congenita AD KRT6A,6B,16,17/keratins 6A,6B,16,17
AD KRT6A,6B,16,17/keratins 7/ 7 6A,6B,16,17
Pachyonychia
congenita
Non-epidermolytic PPK AD KRT1, KRT16/keratins 1,16
Non-epidermolytic PPK AD KRT1, KRT16/keratins 1,16
Naxos AR JUP/Junctional plakoglobin
Naxos AR JUP/Junctional plakoglobin P/ P
White sponge nevus AD KRT4,13/keratins 4,13
White sponge nevus AD KRT4,13/keratins 4,13
Ichthyosis vulgaris AD FLG/filaggrin
Ichthyosis vulgaris AD FLG/filaggrin
Cornified Envelope
Cornified Envelope
Lamellar ichthyosis AR TGM1/transglutaminase 1
Lamellar ichthyosis AR TGM1/transglutaminase 1
Bathing suit ichthyosis AR TGM1 temperature-sensitive
Bathing suit ichthyosis AR TGM1 temperature-sensitive mutation
mutation
Keratoderma (Camissa) AD LOR/loricrin
Keratoderma (Camissa) AD LOR/loricrin
Generalized peeling
AR CDSN/corneodesmosin
Generalized peeling skin disease AR CDSN/corneodesmosin
skin disease
Membrane Transporters
Membrane Transporters
Darier disease AD ATP2A2/SERcalciumATPase
Darier disease AD ATP2A2/SERcalciumATPase
AD ATP2A2/SERcalciumATPase
Acrokeratosis verruciformis AD ATP2A2/SERcalciumATPase
Acrokeratosis
verruciformis
Hailey-Hailey disease AD ATP2C1
Hailey-Hailey disease AD ATP2C1
AR CLDN1/claudin-1
Ichthyosis with sclerosing cholangitis AR CLDN1/claudin-1
Ichthyosis with scle-
rosing cholangitis
Lamellar AR ABCA12
Lamellar AR ABCA12
Harlequin AR ABCA12
Harlequin AR ABCA12
AR FATP4
Ichthyosis prematurity syndrome AR FATP4
Ichthyosis prematurity
syndrome
Cell Junction Proteins
Cell Junction Proteins
AD GJB2/Connexin 26
PPK with deafness (Vohwinkel) AD GJB2/Connexin 26
PPK with deafness
(Vohwinkel)
Erythrokeratoderma variabilis AD GJB3/Connexin 31 GJB4/Connexin30.3
AD GJB3/Connexin 31 GJB4/Connexin30.3
Erythrokeratoderma
variabilis
AD GBJ2/Connexin 26
Keratitis–ichthyosis– deafness (KID) AD GBJ2/Connexin 26
Keratitis–ichthyosis–
deafness (KID)
AD DSP/desmoplakin P/ P
Nonepidermolytic PPK (striate)
Nonepidermolytic
AD DSP/desmoplakin
PPK (striate)
AR DSP/desmoplakin P/ P
AR DSP/desmoplakin
AD DSG1/desmoglein 1
AD DSG1/desmoglein 1
8
DISEASE INHERITANCE GENE/PROTEIN
Snare Protein
Snare Protein
AR SNAP29/synaptosome 9/ 9 associated protein 29
CEDNIK (cerebral
CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma)
AR SNAP29/synaptosome associated protein 29
dysgenesis, neuropathy, ichthyosis, and keratoderma)
AR VSP33B/vacuolar protein
ARC (arthrogryposis-
AR VSP33B/vacuolar protein sorting homologue B
ARC (arthrogryposisrenal dysfunctioncholestasis)
renal dysfunctioncholestasis)
sorting homologue B
Metabolic Enzymes
Metabolic Enzymes
X-linked ichthyosis X-LR STS/steroid sulfatase
X-linked ichthyosis X-LR STS/steroid sulfatase
AR TAT/tyrosine T/ T aminotransferase
Tyrosinemia II
Tyrosinemia II (Richner-Hanhart) AR TAT/tyrosine aminotransferase
(Richner-Hanhart)
Sjögren-Larsson AR ALDH3A2/fatty aldehyde
Sjögren-Larsson AR ALDH3A2/fatty aldehyde dehydrogenase
dehydrogenase
Refsum AR PAXH/phytanoyl CoA H/ H hydroxylase
Refsum AR PAXH/phytanoyl CoA hydroxylase
Conradi-Hunermann X-LD EBP/3 P/ P β-hydroxysterol δ7-isomerase
Conradi-Hunermann X-LD EBP/3β-hydroxysterol δ7-isomerase
X-LD EBP/3 P/ P β-hydroxysterol δ7-isomerase NSDHL/ NAD(P)
X-LD EBP/3β-hydroxysterol δ7-isomerase NSDHL/ NAD(P) dependent steroid dehydrogenase-like
CHILD (congenital
CHILD (congenital hemidysplasia, ichthyosiform erythroderma and limb defects)
hemidysplasia, ichthyosiform erythroderma and limb defects)
dependent steroid dehydrogenase-like
Gaucher AR GBA/glucocerebrosidase B
Gaucher AR GBA/glucocerebrosidase B
AR ABHD5/(CGI-58)
Chanarin-Dorfman
Chanarin-Dorfman (neutral lipid storage)
AR ABHD5/(CGI-58)
(neutral lipid storage)
AR PNPLA2/patatin-like
Neutral lipid storage
Neutral lipid storage with myopathy AR PNPLA2/patatin-like phospholipase domain-containing protein
with myopathy
phospholipase domain-containing protein
AR ALOXE3/lipoxygenase 3 ALOX12B/12(R)-
Autosomal recessive
Autosomal recessive congenital ichthyosis
AR ALOXE3/lipoxygenase 3 ALOX12B/12(R)- lipoxygenase E3 NIPAL4/magnesium transporter NIPA4 CYP4F22/cytochrome P450 family 4 subfamily F member 22 PNPLA1/patatin like phospholipase domain containing 1, CERS3/ ceramide synthase 3, SDR9C7/short chain dehydrogenase/ reductase family 9C, member 7, SULT2B1/ sulfotransferase family 2B member 1
congenital ichthyosis
lipoxygenase E3 NIPAL4/magnesium
transporter NIPA4 CYP4F22/cytochrome P450
family 4 subfamily F member 22 PNPLA1/patatin like
phospholipase domain containing 1, CERS3/ ceramide synthase 3, SDR9C7/short chain dehydrogenase/ reductase family 9C, member 7, SULT2B1/ sulfotransferase family 2B member 1
AR ALOX12B and ALOXE3
Self-improving
Self-improving collodion ichthyosis AR ALOX12B and ALOXE3
collodion ichthyosis
(Continued)
781
8
DISEASE INHERITANCE GENE/PROTEIN
Protease/Protease Inhibitors
Protease/Protease
Inhibitors
AR CTSC/cathepsin C C/ C
Papillion-Lefevre keratoderma AR CTSC/cathepsin C
Papillion-Lefevre
keratoderma
AR SPINK5/serine protease
Ichthyosis linearis
AR SPINK5/serine protease inhibitor 5
Ichthyosis linearis circumflexa (Netherton)
circumflexa (Netherton)
inhibitor 5
X-LR MBTPS2/membrane bound
Ichthyosis follicularis/
Ichthyosis follicularis/ photophobia (IFAP) X-LR MBTPS2/membrane bound transcription factor peptidase, site 2
photophobia (IFAP)
transcription factor peptidase, site 2
AR ST14/matriptase
ARCI with
ARCI with hypotrichosis AR ST14/matriptase
hypotrichosis
Proteasome
Proteasome
AR POMP/proteasome P/ P maturation protein
KLIK (keratosis linearis
AR POMP/proteasome maturation protein
KLIK (keratosis linearis with ichthyosis congenita and sclerosing keratoderma)
with ichthyosis congenita and sclerosing keratoderma)
DISEASE INHERITANCE GENE/PROTEIN
Secreted Proteins
Secreted Proteins
AR SLURP1/secreted Ly6/UPAR
Mal de Meleda keratoderma AR SLURP1/secreted Ly6/UPAR related protein 1
Mal de Meleda
keratoderma
related protein 1
DNA Repair and
DNA Repair and Transcription
Transcription
Trichothiodystrophy AR ERCC2/excision repair cross-
Trichothiodystrophy AR ERCC2/excision repair crosscomplementation group 2 GTF2H5/general transcription factor IIH subunit 5 GTF2E2/general transcription factor IIE subunit 2 ERCC3/excision repair crosscomplementation group 3
complementation group 2 GTF2H5/general
transcription factor IIH subunit 5 GTF2E2/general
transcription factor IIE subunit 2 ERCC3/excision repair cross-
complementation group 3
AD, autosomal dominant; AR, autosomal recessive; ARCI, autosomal recessive congenital ichthyosis; CHILD, congenital hemidysplasia, ichthyosiform erythroderma, and limb defects; PPK X-LD, X-linked dominant; X-LR, X-linked recessive.
Netherton syndrome and confirm a role for proteolysis and protease inhibitors in normal epidermal differentiation.24 Finally, mutations in FLG result in reduced or absent filaggrin and decreased moisture binding in the stratum corneum of patients with IV.25
These cardinal examples highlight the observation that mutations in a host of genes encoding proteins with highly divergent functions affect normal keratinocyte differentiation and cause ichthyosis. It is yet unknown how defects in diverse processes result in similar phenotypes, although studies suggest that defects in the barrier may result in inflammation and hyperproliferation.26,27 Furthermore, our evolving understanding of these mechanisms continues to clarify the multisystem, clinical phenotypes observed in several ichthyosiform disorders.
COMMON THERAPEUTIC APPROACHES
Current therapies for the inherited ichthyoses are symptomatic and focus on hydration, lubrication, and keratolysis.28,29 Ichthyotic skin, even if thickened, has a decreased barrier function and increased transepidermal water loss. Because pliability of the stratum corneum is a function of its water content, hydration can soften the surface of the skin. In moist, humid climates, most ichthyoses improve. Moistening the skin with, for example, long baths can hydrate it. Well-hydrated areas of hyperkeratosis can more easily be thinned with mild abrasives
782
(eg, sponges, buff puffs, pumice stones). Addition of bath oils or application of lubricants before drying can prolong the hydration and softening. Depending on the ichthyosis and environmental conditions, individual patients may prefer specific lubricating agents, which can take the form of lotions, creams, oils, ointments, or petrolatum. In dry climates and winter months, humidifiers can be used to create a more hospitable environment. Keratolytic agents are used to enhance corneocyte desquamation and thereby remove scale and thin hyperkeratotic stratum corneum. There are many commercially available keratolytic creams and lotions containing urea, salicylic acid, or α-hydroxy acids (eg, lactic acid, glycolic acid). Urea may function by its capacity to bind water. Propylene glycol (40%– 60% in water), with or without occlusion, can also be effective in scale removal. Occlusion can effectively increase skin hydration and facilitate desquamation; it can also enhance the effect of keratolytic agents. Special care should be taken when using extensive areas of occlusion with keratolytic agents and in individuals who may be heat intolerant. The markedly impaired barrier function in ichthyosis should be considered when using topical preparations over large areas of body surface. For example, widespread use of topical salicylic acid preparations can lead to significant absorption, intoxication (eg, nausea, tinnitus, dyspnea, hallucinations), and even death.30 Children are at greater risk because they have a greater body surface area per unit weight than adults, a situation that effectively heightens the possibility of developing systemic toxicity from topicals. Although
the use of topical retinoids in most of the ichthyoses appears to be safe,31 the abnormal skin barrier should be considered when treating concomitant dermatoses in patients with ichthyosis. When using topical agents such as tacrolimus, pimecrolimus, or topical steroids, when increased systemic absorption has been observed, monitoring of serum levels may be necessary.32
Another risk to children is that in several types of ichthyosis nutritional requirements may be high, and inadequate nutrition can lead to failure to thrive. This was thought to be related to the large turnover of scale; however, recent studies suggest energy loss from impaired barrier function is the cause.33
Some patients with ichthyosis have decreased sweating with heat intolerance. It is important for the parents of a newborn with ichthyosis to be aware of the possibility of decreased sweating and to be attentive for signs of heat intolerance, such as flushing and lethargy, particularly during hot weather and, as the child grows, during exercise. Avoiding hot environments, carrying spray bottles with water to moisten the skin and cool it through evaporation, and cooling vests can minimize heat stress. Systemic retinoid therapy with isotretinoin or acitretin (see Chap. 185) can induce dramatic improvement in many ichthyoses. The decision to initiate systemic retinoid therapy should be weighed carefully because after the drug is started, continued benefit usually requires chronic therapy. Retinoic acid metabolism– blocking agents, which increase endogenous retinoid levels, offer a possible alternative.34
Fungal infections are common, both of skin and nails, and are often undiagnosed because of the generalized scaling. A high index of suspicion can help diagnose tinea corporis, capitis, or versicolor when the only symptom may be localized pruritus (which is often intense) and the only sign a difference in the character of scale or a localized area of alopecia.
ICHTHYOSIS VULGARIS
Ichthyosis vulgaris (Online Mendelian Inheritance in Man [OMIM] #146700), the most common ichthyosis, is relatively mild. The disease was thought to be autosomal dominant, and a study of English school children found that 1 in 250 were affected.4 Mutations in the gene encoding profilaggrin cause IV.25 Profilaggrin contains multiple copies of filaggrin, which has various structural and biochemical functions during cornification. The inheritance pattern has been clarified to be semidominant; individuals who carry one mutated allele have a mild phenotype, but those with mutations in both profilaggrin alleles (homozygotes or compound heterozygotes for the mutations) (Fig. 47-1) manifest a severe clinical phenotype. In the Anglo- European population, the prevalence of clinical disease is as high as 1 in 80.35,36
8
CLINICAL FEATURES
CLINICAL FEATURES
Although infants usually have normal skin, the disease often manifests within the first year of life. The scale of IV is usually most prominent on the extensor surfaces of the extremities, with flexural sparing. The diaper area tends to be spared. There may be fine, white scales over large areas. Particularly on the lower extremities, which are often the most severely involved area, the scales may be centrally attached, with “cracking” (superficial fissuring through the stratum corneum) at the edges. This turning up at the edges can lead to the skin feeling rough.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Filaggrin is an epidermal protein involved in the aggregation of keratin intermediate filaments,37 is an important component of the cornified envelope,38 and helps retain moisture in the stratum corneum. Keratin filaments form a network, or cell matrix, that gives structural integrity to the epidermal keratinocytes. As keratinocytes mature into corneocytes, the keratin filaments collapse and are cross-linked to the cornified cell envelope. Filaggrin is synthesized as a high-molecular-weight precursor, profilaggrin, that contains multiple filaggrin molecules and is localized to keratohyalin granules. Biochemical studies of epidermis from patients with IV have shown absence of or decrease in filaggrin and its precursor, profilaggrin.39,40 The association between IV and atopic dermatitis has been long appreciated on clinical grounds. Null mutations in the gene encoding profilaggrin (FLG) have now been shown to be a strong predisposing factor for atopic dermatitis in the Anglo-European population. A strong association is also found with individuals who have sensitivity to common allergens, allergic rhinitis, early-onset and persistent eczema, and asthma in the presence of atopic dermatitis.35,41
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
In some cases, it is difficult to distinguish mild IV from simple dry skin (xerosis). Evolving understanding of this very common condition is beginning to clarify how a spectrum of underlying mutations can cause the diverse clinical severity of dry skin from xerosis to severe IV. In addition, on the basis of skin findings alone, males with severe IV may be difficult to differentiate from those affected with X-linked recessive ichthyosis.42,43 The histopathologic findings of IV are variable, and the characteristic orthohyperkeratosis and absent granular layer may only be seen in individuals with two abnormal alleles.
783
8
A
B
C D
E F
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
A number of other findings are commonly observed in association with IV.42 Hyperlinear palms are usually present, and some patients may have palmar/plantar thickening approaching a keratoderma. Keratosis pilaris is common, even in individuals with mild IV,
784
and usually involves the outside of the arms, extensor thighs, and buttocks. Even though IV is usually not considered a syndromic ichthyosis, atopy is also frequently observed and can manifest as hay fever, eczema, or asthma. These findings can confound an accurate diagnosis because hyperlinear palms and keratosis pilaris may be seen in atopic individuals who do not have IV. Rarely, individuals with IV may have hypohidrosis with heat intolerance. There is great variation in the severity of clinical manifestations among affected individuals in the same family. The condition
usually worsens in climates that are dry and cold and improves in warm, humid environments, where the disease may clear dramatically. Most patients respond well to regular application of emollients, and use of α-hydroxy acids can reduce scaling when present.
X-LINKED RECESSIVE ICHTHYOSIS
In the 1960s, X-linked recessive ichthyosis (OMIM #308100) was distinguished clinically from other ichthyoses.4 X-linked recessive ichthyosis occurs in approximately 1 in 1500 to 6000 males.5,44 Steroid sulfatase (arylsulfatase C) deficiency causes XLI;45,46 in 90% of cases, genetic deletion of the STS locus is causative.47
The remainder are point mutations in STS.
CLINICAL FEATURES
CLINICAL FEATURES
Scaling may begin in the newborn period and is usually most prominent on the extensor surfaces, although there is significant involvement of the flexural areas. Although the extent and degree of scaling are variable, X-linked ichthyosis can usually be distinguished from IV on clinical criteria and inheritance pattern.5,48 The latter tends to be associated with hyperlinear palms
8
and soles, keratosis pilaris, and a family history of atopy. X-linked ichthyosis tends to have more severe involvement with larger scale (Fig. 47-2), and commashaped, corneal opacities may be present in half of adult patients.49,50 Corneal opacities do not affect vision and may be present in female carriers. Affected males have an increased risk of cryptorchidism, and independently, they are at increased risk for the development of testicular cancer.51
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Steroid sulfatase hydrolyzes sulfate esters, which include cholesterol sulfate and sulfated steroid hormones.52 Sulfated fetal adrenal hormones undergo desulfation to estrogens, which are excreted in maternal urine. The absence of steroid sulfatase enzyme in the fetal placenta leads to low maternal urinary estrogens, and in some pregnancies, to a failure of labor to initiate or to progress normally. In males with X-linked recessive ichthyosis, steroid sulfatase enzyme activity is decreased or absent in many tissues, including epidermis, stratum corneum, leukocytes, and in cultured fibroblasts.53 Carrier females have been found to have leukocyte steroid sulfatase levels intermediate between those observed in normal individuals and those in affected males.
785
8
Cholesterol sulfate levels are elevated in the serum, epidermis, and scale.54 Steroid sulfatase is one of a group of arylsulfatases located on chromosome Xp22. More than 90% of the mutations in X-linked ichthyosis are deletions that can often be detected by fluorescence in situ hybridization (FISH) or array-comparative genomic hybridization (CGH), available in many clinical laboratories. Confirmation of the diagnosis can also be made by finding an elevation in serum cholesterol sulfate levels. In the epidermis, steroid sulfatase catalyzes the hydrolysis of cholesterol sulfate. Topical application of cholesterol sulfate in mice can induce a scaling disorder, further supporting the role of cholesterol sulfate hydrolysis in corneocyte desquamation. Cholesterol sulfate inhibits the proteases that degrade corneodesmosomes, resulting in inhibition of desquamation and a retention hyperkeratosis.55
DIAGNOSIS
DIAGNOSIS
Skin and eye findings, history of prolonged labor, and evidence of X-linked transmission enable diagnosis of XLI.45,56 Notably, a significant fraction of patients now are recognized in utero through maternal screening that assesses levels of α-fetoprotein, human chorionic gonadotropin, and estriol. Because steroid sulfatase is necessary for placental estrogen production, triple screen results indicating low estriol are present in XLI pregnancies.47 Because the majority of maternal urinary estrogens are derived from the fetal adrenal glands and are metabolized by the placenta, low levels can reflect fetal abnormalities or death, raising concern in parents. However, in XLI, low levels do not indicate severe fetal morbidity. Although skin biopsy is rarely done, histopathologic examination shows compact orthohyperkeratosis, acanthosis, papillomatosis, and a thickened granular layer. Many assays are available for the diagnosis of XLI, including FISH for STS, array-CGH, deletion and point mutation detection via next-generation sequencing, and enzymatic assays of steroid sulfatase function.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Although in most cases, XLI is primarily a cutaneous disorder, genetic deletions that include STS and adjacent sulfatases explain the overlap syndromes involving chondrodysplasia punctata and X-linked ichthyosis.57 The X-linked form of Kallmann syndrome, in which hypogonadotropic hypogonadism and anosmia are found, often with renal abnormalities, obesity, synkinesis (mirror image movements of the extremities), cleft palate, and spastic paraplegia, can also be seen in association with X-linked
786
recessive ichthyosis as part of a contiguous gene deletion syndrome. Because of this and because of the association with testicular carcinoma, patients with X-linked recessive ichthyosis should be queried about anosmia and have periodic testicular examination.44,58
When scaling is prominent, long bathwater soaks with or without bicarbonate can aid in desquamation.59
Emollients, including those with α-hydroxy acids or lactic acid, can aid in desquamation.
COLLODION BABY AND NEWBORN PRESENTATIONS
Most cases of ichthyosis, excluding IV and X-linked ichthyosis, present at birth with red, scaly skin. In the United States, the incidence of such cases is estimated to be 5 to 10 per 100,000 live births.60 A fraction of these present with a collodion membrane, which is shed early in life with features of congenital ichthyosis or normal skin subsequently developing. Genotype is not the sole determinant of collodion baby presentation because many with the common genotypes associated with collodion membranes present with simply red scaly skin. No differences in the pathogenesis of these two newborn presentations of ichthyosis have been identified, and management is generally similar for both.
CLINICAL FEATURES
CLINICAL FEATURES
A collodion baby is born encased in a translucent, parchment-like membrane that is taut and may impair respiration and sucking. Birth can be premature, contributing to morbidity. During the first 2 weeks of life, the membrane breaks up and peels off and can leave fissures, with impairment of the barrier to infection and water loss (Fig. 47-3).
Common
Common
■Autosomal recessive congenital ichthyosis (lamellar ichthyosis, congenital ichthyosiform erythroderma, overlap) Rare
■Autosomal recessive congenital ichthyosis (lamellar ichthyosis,
congenital ichthyosiform erythroderma, overlap) Rare
■Ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC) syndrome
■Ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC)
syndrome
■Chondrodysplasia punctata
■Chondrodysplasia punctata
■Gaucher disease
■Gaucher disease
■Loricrin keratoderma
■Loricrin keratoderma
■Neutral lipid storage disease
■Neutral lipid storage disease
■Self-healing collodion baby
■Self-healing collodion baby
■Sjögren–Larsson syndrome
■Sjögren–Larsson syndrome
■Trichothiodystrophy
■Trichothiodystrophy
■X-linked hypohidrotic ectodermal dysplasia
■X-linked hypohidrotic ectodermal dysplasia
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
The in utero aqueous environment contributes to the development of the collodion presentation, which transforms into a wide spectrum of ichthyosis phenotypes as the child grows (Table 47-5). This is the usual presentation of ARCI and is less commonly seen in several other forms of ichthyosis and rarely Gaucher disease. In addition, an autosomal recessive, selfhealing collodion phenotype has been described, in which the skin greatly clears within the first few weeks and transitions into mildly affected or normal skin.61
Eleven Swedish and four Danish patients with a selfimproving ichthyosis resulting in xerosis, hyperlinear palms, red cheeks, and anhidrosis were found to have mutations in ALOX12B, ALOX3E, or TGM1.62
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
In caring for a collodion infant or other newborn with ichthyosis, consideration of a potentially increased risk of infection, difficulties in thermal regulation, and hypernatremic dehydration caused by increased transepidermal water loss is essential.63 Newborn care should include careful monitoring for infection and of temperature, hydration, and electrolytes and measures to keep the peeling membrane soft and lubricated to facilitate flexibility and desquamation. Appropriate pain management and eye care should be used when indicated. These newborns usually benefit from a humidified incubator where the air is saturated with water; wet compresses followed by bland lubricants can be used to further hydrate the membrane and achieve maximum pliability.64 Before spontaneous peeling of the collodion membrane, the thickened
8
stratum corneum can dry and harden in areas such as the extremities and can constrict, leading to distal swelling, cyanosis, and rarely necrosis in some cases. Release of constricting bands via curettage or debridement can prevent complications.65
AUTOSOMAL RECESSIVE CONGENITAL ICHTHYOSIS
The term autosomal recessive congenital ichthyosis is used to describe a heterogeneous group of disorders that present at birth with generalized involvement of the skin. Autosomal recessive ichthyosis is rare and has been estimated to occur in about 1 in 300,000 persons.5
In older literature, nonbullous congenital ichthyosiform erythroderma (NCIE, also called lamellar ichthyosis, with autosomal recessive inheritance) was distinguished from BCIE (also called epidermolytic hyperkeratosis [EHK], with autosomal dominant inheritance) based on clinical appearance (bullae) and pattern of inheritance.6,66 That the term LI was used interchangeably with NCIE and included a spectrum of phenotypes has led to some confusion. We also now understand that genotype alone does not determine whether the phenotype will be LI or CIE. Williams and Elias distinguished LI from NCIE (usually called congenital ichthyosiform erythroderma), a milder erythrodermic form, and these descriptors remain useful clinical descriptors in clinical practice.67 In LI, one sees large, dark, platelike scales, and although infants may be red at birth, adults have little to no erythroderma (Figs. 47-4 to 47-6). In the more severe, classic presentation of LI, tautness of the facial skin leads to traction on the eyelids and lips, resulting in ectropion and eclabium. Scarring alopecia, most prominent at the periphery of the scalp, may be partly caused by traction at the hairline. In contrast, CIE has generalized redness and fine, white scales (eg, Figs. 47-7 and 47-9). Patients with classic CIE have little to no ectropion, eclabium, or alopecia. However, many patients do not fit neatly into these two clinical descriptions68 in that they have features of both LI and CIE with a clinical phenotype intermediate between both disorders. Therefore, it can be useful to consider these two distinctive presentations as ends of a spectrum, between which lie a gradation of clinical phenotypes with variable degrees of erythema and coarseness of scale. Individual features such as collodion membrane (discussed earlier), ectropion, and alopecia can occur across the spectrum. Although attempts to refine the categorization of these disorders by biochemical and ultrastructural observations have failed to yield a consistent and replicable classification scheme, identification of the spectrum of specific molecular defects underlying these conditions may aid classification.8 Identification of mutations has, so far, found ARCI to be caused by 10 different genes that are important for the formation of the intercellular lipid layer or the cornified envelope of keratinocytes, including TGM1 (OMIM #191995), ALOX12B
787
8
(OMIM #603741), ALOXE3 (OMIM #607206), NIPAL4 (OMIM #609383), CYP4F22 (OMIM #611495), ABCA12 (OMIM #607800), PNPLA1 (OMIM #612121), CERS3 (OMIM #615276), SDR9C7 (OMIM #609767), and SULT2B1 (OMIM #604125).
CLINICAL FEATURES
CLINICAL FEATURES
LAMELLAR ICHTHYOSIS PHENOTYPE
The LI phenotype of ARCI is apparent at birth, and the newborn usually presents encased in a collodion membrane (see Fig. 47-3). At this time, the skin may be red. Over time, the skin develops large, platelike scales, and most are centrally attached with raised borders. The scales tend to be largest over the lower extremities, where the large, platelike scales separated by superficial fissuring can lead to an appearance similar to that of a dry riverbed. During childhood and into adulthood, the degree of erythema may vary. Involvement of the palms and soles in LI is variable and ranges from minimal hyperlinearity to severe keratoderma. This phenotype is most often associated with mutations in TGM1, but even mutations in
788
one gene result in a highly variable phenotype (see Figs. 47-4 to 47-6). The lips and mucous membranes tend to be spared in LI, but the adnexal structures may be compromised by the adherent, firm scales. Thick stratum corneum on the scalp tends to encase hairs and in conjunction with the tautness of the skin may lead to a scarring alopecia, most marked at the periphery of the scalp. Hyperkeratosis interferes with normal sweat gland function, resulting in hypohidrosis, but the degree of impairment varies between patients. Some patients have severe heat intolerance and must be vigilant to avoid overheating. Bathing suit ichthyosis is a subtype of LI in which affected individuals develop the scaling typical of LI but limited to the bathing suit area. The distribution correlates with warmer areas of skin. Decreased transglutaminase is found in these areas, and unique, temperature-sensitive mutations in TGM1 have been identified in affected individuals.69-72
CONGENITAL ICHTHYOSIFORM ERYTHRODERMA PHENOTYPE
As with LI, the CIE phenotype of ARCI is apparent at birth, and the newborn usually presents with a taut, shiny, collodion membrane. After shedding of
8
the membrane, the skin of infants with CIE remains red, usually with a fine, white, generalized scale (see Figs. 47-7 and 47-8). On the lower legs, the scale may be larger and darker. In contrast to LI, the classic presentation of CIE may have little to no ectropion, eclabium, or alopecia. As in LI, there is a wide variation in the ability to sweat, and patients with CIE may have minimal sweating with severe heat intolerance. Mucous membranes are usually spared. Palm and sole involvement is variable. Nails may have ridging, but
they are often spared. As with all the ichthyoses, dermatophyte infection of the skin and nails is common.
HARLEQUIN ICHTHYOSIS PHENOTYPE
A dramatic, severe, and sometimes fatal presentation of ichthyosis is that of harlequin ichthyosis (OMIM #242500) (Fig. 47-9). The child is often
789
8
790
premature and born with thick, shiny plates of stratum corneum separated by deep, red fissures that tend to form geometric patterns, as seen in the patched costumes of the harlequin clowns from the Italian Commedia dell’Arte dating from the 16th and 17th centuries. There are poorly developed or absent ears and marked ectropion and eclabium. The fingertips are tapered, and there is hyperconvexity of the nails. Uniformly fatal before 1980, a substantial fraction of patients with harlequin ichthyosis now survive with a persistent and severe erythroderma (Fig. 47-10).65,73,74
8
Netherton syndrome (OMIM #256500) is an autosomal recessive disorder featuring ichthyosis, a structural hair shaft abnormality, and atopy.75 When present, a key finding is ichthyosis linearis circumflexa (ILC), a characteristic, polycyclic, serpiginous, migratory, double-edged scale at the margins of erythematous plaques (Fig. 47-11). Other patients, however, can present with a more severe erythroderma with or without ILC (Fig. 47-12). Histopathologic examination is not specific, but absent or thin stratum corneum is highly suggestive. Most patients have a specific hair shaft abnormality called trichorrhexis invaginata, in which the distal hair segment is telescoped into the proximal one, forming a ball-and-socket-like deformity on microscopic examination (Fig. 47-13). This is also known as “bamboo hair” and is caused by abnormal cornification of the internal root sheath. Hair from multiple areas should be examined because only 20% to 50% of hair may be affected; the characteristic abnormality may be more commonly observed on eyebrow hair.76-78 The hair defects may not be detectable at birth and may disappear with age. Atopy-like diathesis may occur in these patients as atopic dermatitis, asthma, or severe food allergy (particularly to nuts) and marked elevations of serum immunoglobulin E. In some patients, generalized aminoaciduria, mild developmental delay, and impaired cellular immunity may also be present.79
791
8
792
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
LAMELLAR ICHTHYOSIS AND CONGENITAL ICHTHYOSIFORM ERYTHRODERMA PHENOTYPES
The LI and CIE phenotypes represent a clinical spectrum, and mutations in many of the same genes underlie both phenotypes. A cardinal gene implicated in LI phenotypes is TGM1.21,22 Even with this gene, there is great phenotypic variability (see Figs. 47-6 to 47-8). TGM1 encodes transglutaminase 1; transglutaminases catalyze calcium-dependent cross-linking of proteins through the formation of ε-(γ-glutamyl)lysine isodipeptide bonds. During the formation of the stratum corneum, transglutaminase catalyzes the cross-linking of cellular proteins, including involucrin, loricrin, small proline-rich proteins, keratins, filaggrin, and others. The resulting protein complex is deposited on the inner side of the plasma membrane to form the cornified envelope. Transglutaminase also attaches ceramides secreted into the intercellular space by lamellar bodies to cornified envelope proteins, notably involucrin, and thereby is important in the formation of both the protein and lipid components of the stratum corneum.80
In a human skin–immunodeficient mouse xenograft model, transfer of a transglutaminase-1 gene into transglutaminase-1-deficient keratinocytes from LI patients resulted in normalization of transglutaminase expression and epidermal architecture in addition to restoration of cutaneous barrier function.81 More recently, topical application of recombinant transglutaminase-1 in liposomes has been shown to normalize histology and function in murine xenografts.82
Autosomal recessive mutations in the genes encoding lipoxygenases ALOXE3 (see Fig. 47-9) and ALOX12B (see Fig. 47-10) were found to cause CIE.83
Lipoxygenases catalyze the formation of hydroperoxides from polyunsaturated fatty acids. In the skin, the products of ALOXE3 and ALOX12B play a central role in the generated of oxidized ceramides, which are incorporated into the corneocyte lipid envelope.84
8
Altogether, mutations in 10 genes can cause ARCI phenotypes, and these include: TGM1 (OMIM #190195), ALOX12B (OMIM #603741), ALOXE3 (OMIM #∗607206), NIPAL4/Ichthyin6 (OMIM #609383), CYP4F227 (OMIM #611495), ABCA12 (OMIM #607800), PNPLA1 (OMIM #612121), CERS3 (OMIM #615276), SDR9C7 (OMIM #609767), and SULT2B1 (OMIM #604125).
HARLEQUIN ICHTHYOSIS PHENOTYPE
Harlequin ichthyosis results from autosomal recessive inheritance of mutations in ABCA12, which codes for an adenosine triphosphate (ATP)-binding cassette (ABC) transporter involved in lamellar granule secretion and epidermal lipid transport. Whereas premature termination, loss-of-function mutations most commonly underlie harlequin ichthyosis,85,86 missense mutations in ABCA12 have been identified in individuals with less severe ARCI (Fig. 47-14).87,88 In harlequin ichthyosis, normal lamellar granules are not found; instead, there are small vesicles that lack internal structure. There is also no evidence of the lipid lamellae that form between granular and cornified cells as a result of discharge of lamellar granule contents into the intercellular space.89
Netherton syndrome has been found to be caused by mutations in SPINK5, a gene encoding LEKTI (lymphoepithelial Kazal-type related inhibitor).24 LEKTI is a serine protease inhibitor that is predominantly expressed in epithelial and lymphoid tissues and may be important in the downregulation of inflammatory pathways. It inhibits the proteases that degrade the corneodesmosome, thus allowing premature desquamation. LEKTI polymorphisms are associated with common atopy and atopic dermatitis.90,91 Early reports of Netherton syndrome recognized signs of reduced cellular immunity manifesting as viral warts and negative skin testing to microbial antigens. The danger of such defective cellular immunity was reinforced with the report of Folster- Holst and coworkers, who identified oncogenic human papillomavirus in a 28-year-old woman with vulvar condyloma that rapidly transformed to an aggressive squamous cell carcinoma.92 In our experience, all adults with Netherton have papillomatous changes in intertriginous areas, but the role of papilloma viruses in the development of these lesions has not been established.
DIAGNOSIS
DIAGNOSIS
Clinical features and history are central to establishing a diagnosis in ARCI. Advances in genetic testing, either via research pathways (see section “Registry for Ichthyosis and Related Skin Disorders”) or clinical laboratories, has allowed the rapid ascertainment of genetic diagnoses. These tests use next-generation sequencing of a panel of genes or a selected subset of genes assessed via exome sequencing. Genetic testing permits confirmation of diagnosis and counseling regarding potential disease comorbidities. Clinical features supporting phenotypes include the following:
793
8
LI and CIE typically presents with a collodion membrane at birth that sheds to reveal either the thick, adherent scale of LI or the erythroderma and finer scale of CIE. Histopathologic examination rarely aids in the diagnosis of LI or CIE, with both disorders showing nonspecific acanthosis and hyperkeratosis. Harlequin ichthyosis has a classical clinical presentation, with armorlike plates of scale and deep fissures present at birth which shed to reveal severe erythroderma and scaling. Netherton syndrome presents with hair shaft abnormalities, with or without ILC or erythroderma, in the setting of atopic features. These clinical findings provide strong support for the diagnosis of Netherton syndrome.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
LAMELLAR ICHTHYOSIS AND CONGENITAL ICHTHYOSIFORM ERYTHRODERMA
Treatment with oral retinoids can improve or prevent some sequelae of these phenotypes. Patients frequently
794
notice an increase in sweating, with improved heat tolerance. Although retinoid therapy can cause blepharitis or even conjunctivitis, it is usually well tolerated by patients with LI and CIE. Moreover, the ability of systemic retinoid (and in some cases, topical retinoid) therapy to decrease thick periocular scale can decrease the tendency to develop ectropion.93 Nevertheless, patients with severe scaling and ectropion usually require careful eye maintenance. Because of the ectropion, the eyelids may fail to close fully, particularly during sleep; hydration with liquid tears during the day and ophthalmic lubricants at night can prevent exposure keratitis. Individuals with mutations in ALOXE3 and ALOX12B tend to have less scaling that those with mutations in TGM1 and, consistent with this observation, are findings in a recent study showing that ectropion is less common (35%) in CIE than in LI (57%).94
HARLEQUIN ICHTHYOSIS
Advances in neonatal intensive care, together with facilitating desquamation by judicious use of systemic retinoid therapy, have led to improvements in survival and to the use of the name “harlequin baby” rather than “harlequin fetus.” These children are at risk during the neonatal period. Abnormal water loss through the skin and poor temperature regulation lead to risk of fluid and electrolyte imbalance.
The infants are also at risk for infection beginning in the skin but at the same time (because of poor temperature regulation) do not show the usual signs of infection. Normal respiration may be restricted by the taut skin. Treatment with systemic retinoids during the newborn period can facilitate desquamation of the membrane and is associated with improved outcomes.73,95 Some infants and children have had failure to thrive and require tube feeding. After shedding of harlequin scale, erythroderma is prominent. In children and adults, topical and systemic retinoids can be helpful in the management of ectropion and hyperkeratosis.
NETHERTON SYNDROME
At birth, affected children may present with generalized erythroderma, and in some individuals, erythroderma persists throughout life with a phenotype on the LI–CIE spectrum of ARCI.96 In others, erythroderma fades, and ILC ensues. Infants and children may have feeding problems, with poor absorption and failure to thrive.97 Pruritus can be profound, and scratching can lead to lichenification at the flexures. Tacrolimus ointment, a topical immunomodulator (see Chap. 192), is effective in common atopic dermatitis with minimal systemic absorption. However, Netherton syndrome is complicated by an abnormal skin barrier, allowing increased percutaneous absorption and associated risk for systemic toxic effects. This should be considered when using topical agents such as tacrolimus because monitoring of serum levels may be necessary98 and topical steroids because iatrogenic Cushing syndrome has been reported.32 Furthermore, topical and systemic retinoids should be avoided in patients with Netherton syndrome because they can further exacerbate the condition.
KERATINOPATHIC ICHTHYOSES
Keratinopathic ichthyoses result from mutations in genes encoding keratins, components of the intermediate filament network that are central to cellular structural integrity. Phenotypes falling within the keratinopathic ichthyosis spectrum include EI, superficial epidermolytic ichthyosis (SEI, ichthyosis bullosa of Siemens), ichthyosis hysterix Curth-Macklin, annular EI, and ichthyosis with confetti (IWC).
CLINICAL FEATURES
CLINICAL FEATURES
EPIDERMOLYTIC ICHTHYOSIS
In 1902, Brocq described bullous ichthyotic erythroderma and distinguished the blistering type from the nonblistering type of CIE.99 The original description included three unrelated patients whose clinical
8
manifestations varied. However, this was probably the first description of EI (OMIM #113800). The disease is named for the distinctive histopathologic features of vacuolar degeneration of the epidermal keratinocyte (ie, epidermal lysis) and associated hyperkeratosis. EI is also known as EHK and BCIE, earlier descriptive names signifying characteristic histopathologic changes or the blistering, neonatal presentation with subsequent scaling and redness, respectively. EI is transmitted as an autosomal dominant trait with a prevalence of approximately 1 in 200,000 to 300,000 persons. There is a high frequency of spontaneous mutation, and as many as half of the patients have no family history.10 The disease usually presents at birth with blistering, redness, and peeling and can be mistaken for epidermolysis bullosa or staphylococcal scalded skin on clinical examination (Fig. 47-15A). Because there is a high frequency of new mutations, the disease may be unexpected, and the diagnosis may be unknown. The newborn may require intensive care with fluid and electrolyte monitoring.
A
B
795
8
Specialized skin care can minimize blistering and enhance healing of erosions and may include lubrication to decrease friction and mechanical trauma, protective padding, and specialized wound dressings. Newborns with extensive erosions are prone to bacterial infection and sepsis, and carefully chosen topical and systemic antibiotics can minimize the extent of infection. With time, generalized hyperkeratosis may develop, which may or may not be associated with erythroderma. EI skin usually has a characteristic odor, thought to be related to superinfection by mixed flora. Histopathology shows a thickened stratum corneum and vacuolar degeneration of the upper epidermis, leading to the histologic term EHK. The vacuolar degeneration usually involves the upper epidermis and occasionally all of the suprabasilar keratinocytes. Granular cells exhibit dense, enlarged, irregularly shaped masses that appear to be keratohyalin granules (Fig. 47-15B). All patients with EI have a characteristic corrugated scale that becomes accentuated in areas of body folds (Figs. 47-16 and 47-17). There is striking clinical heterogeneity in EI with phenotypes, including isolated, thick palmoplantar hyperkeratosis, generalized spiny scale, migratory patches of erythrokeratoderma, keratoderma with superficial peeling scale, and generalized exfoliative erythroderma.100
LINEAR ICHTHYOSIFORM ERYTHRODERMA
Linear EI, a variant of epidermal nevus, presents in a linear mosaic pattern caused by a postzygotic, spontaneous mutation during embryogenesis. Areas of hyperkeratosis alternating with normal skin are often distributed in streaks along Blaschko lines. These may be limited to a few streaks, often bounded by the midline, or there may be many stripes, with widespread, patchy involvement. Linear EI has been found to result from somatic mutations in KRT1 and KRT10 that are identical to those found in generalized disease.101,102
If the mosaicism affected gonadal tissue, individuals with linear EI can transmit the mutation to offspring with resulting generalized EI.103,104
SUPERFICIAL EPIDERMOLYTIC ICHTHYOSIS
SIE (ichthyosis bullosa of Siemens) is a rare autosomal dominant genodermatosis. Patients are born with redness and blistering. The redness subsides over the subsequent weeks to months, and the skin develops corrugated hyperkeratosis, particularly over flexural areas (Fig. 47-18). In some areas, there may be a
796
8
797
8
lichenified appearance to the skin. As with EI, the epidermis is fragile; however, the fragility is more superficial. This can result in loss of the uppermost epidermis (predominantly the stratum corneum), yielding a characteristic, collarette-like depressed area that has been described as “mauserung” (molting). Histologically, the epidermis shows hyperkeratosis and vacuolization similar to EHK but confined to the granular layer.10
Mutations have been found in the gene encoding keratin 2, a differentiation keratin of the suprabasilar epidermis that is expressed in the more superficial epidermal layers.105,106
ANNULAR EPIDERMOLYTIC ICHTHYOSIS
Annular EI is a rare autosomal dominant disorder that presents at birth or within in the first few months of life with severe, intermittent scaling and blistering that resolves during puberty.107,108 Residual, limited plaques with corrugated scale and erythema are seen primarily in flexural and intertriginous skin (Fig. 47-19). In some cases, explosive bouts of widespread erythema with blisters and pustules are seen.109 Patients subsequently develop widespread, migratory, polycyclic, and annular scaling plaques. Light and electron microscopy reveals findings of EI. Mutations in KRT10 and KRT1 have been found.109,110
ICHTHYOSIS WITH CONFETTI
This rare disorder is also known as ichthyosis variegata and congenital reticulated ichthyosiform erythroderma, descriptive terms that fail to capture its clinical evolution over the course of life. IWC presents with congenital erythroderma, malformation of the auricle, and tapering of the digits which leads to the frequent clinical diagnosis of CIE. In childhood, palmoplantar keratoderma develops, and the severity ranges from mild to severe.111 Most individuals with IWC develop small islands of normal-appearing skin beginning in childhood (Fig. 47-20), although later presentation has also been reported. Normal areas of skin have been shown to arise from loss of heterozygosity on chromosome 17q or 12q via mitotic recombination of diseasecausing mutations in the genes encoding keratin 10 (KRT10) and keratin 1 (KRT1).112-114 Histopathology shows a clear transition from affected epidermis to normal skin within white spots. In individuals with IWC caused by mutations in KRT10, bandlike parakeratosis, psoriasiform acanthosis and vacuolated, binuclear upper epidermal keratinocytes are seen, and electron microscopy shows perinuclear filament retraction and poor investment of desmosomes with intermediate filaments.114 In individuals with IWC caused by mutations in KRT1, affected skin shows psoriasiform acanthosis, prominent keratohyalin granules, perinuclear vacuolization with rare binucleate cells, and thick
798
8
hyperkeratosis, and electron microscopy shows perinuclear filament detachment.113
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Within keratinocytes, keratin intermediate filaments form an elaborate network that confers structural stability to the cells. In the suprabasilar, differentiating keratinocytes of interfollicular epidermis, this network is primarily formed by keratins 1 and 10, which polymerize to form intermediate filaments. In the granular layer, keratin 2 partially replaces keratin 1 in intermediate filaments.
EPIDERMOLYTIC ICHTHYOSIS
On electron microscopic examination, clumping of filaments is observed to begin in the first suprabasal layer. These aggregated filaments are clumps of keratin intermediate filaments that contain the suprabasal keratins 1 and 10.10,115 When expressed in keratinocytes, mutant keratins in EI and SEI aggregate and clump, with collapse of the intermediate filament network and ensuing
cytolysis. Aggregates are cytotoxic, and mutations in KRT10 have been shown to disrupt epidermal differentiation and formation of the lipid permeability barrier while inducing epidermal hyperproliferation.116,117
Mutations in genes coding keratin 1, 2, or 10 have been identified in a number of EI kindreds.118 In many cases, severe palmar/plantar involvement implies mutations in KRT1; this may reflect the “redundancy” of K9 (a keratin that occurs only in the suprabasal epidermis of palmar and plantar skin) and K10 in palmar/plantar epithelium. Mutations in KRT9 have been found in families with epidermolytic PPK (Vörner) (see Chap. 48).119
ICHTHYOSIS WITH CONFETTI
IWC does not feature marked skin fragility, and electron microscopy shows no evidence of filament aggregates, though perinuclear filament retraction is seen. Expression of KRT10 mutants in cells leads to intermediate filament network collapse and intranuclear accumulation of K10 within the nucleolus, and expression of KRT1 mutants similarly leads to intermediate filament network collapse with intranuclear accumulation of K1.112-114 Because all revertant spots arise from mitotic recombination and spots of normal skin increase in number and size over time, IWC KRT1 and
799
8
KRT10 mutations appear to affect homologous recombination and confer selective advantage to normal keratinocytes.
DIAGNOSIS
DIAGNOSIS
Diagnosis combines clinical findings, histopathology, and genetic analysis. Clinical features can inform diagnosis. For examples, the appearance of white spots of normal skin in childhood suggests IWC, but a history of blistering at birth with less frequent blistering over time and histology showing epidermolysis and hypergranulosis suggests EI or SEI. Genetic testing permits precision in diagnosis, with mutations found in KRT1, KRT10, or KRT2.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
Although the neonatal presentation of widespread blistering can be dramatic in EI and SEI, in the first few weeks of life, hyperkeratosis becomes more prominent, and blisters become more localized to areas of friction. Areas of thick hyperkeratosis, which are not pliable and have a hard, rough surface, are prone to mechanical trauma. In patients with the hystrix type of porcupine-like hyperkeratosis, the rough surface causes high traction with objects moving across the skin surface, which tend to catch on the hyperkeratotic horn and peel it off. Topical agents such as lubricants and keratolytics can reduce the thickened, rough areas and help to minimize blistering and erosion. In contrast, patients with erythroderma and peeling, who do not have the thick areas of hyperkeratotic spines, have less need for keratolytics but still need lubricants. Acute exacerbations may occur from skin infections. Bacterial infection of the skin is common, often leads to enhanced blistering, and may require frequent therapy with topical and oral antibiotics. Dilute bleach baths aid in controlling bacterial odor and colonization, and bicarbonate baths can facilitate desquamation. Topical retinoids can reduce scaling locally,120 and systemic retinoids can markedly reduce hyperkeratosis, decreasing grooming and bathing time.121,122 Dosing must be slowly titrated because fragility can increase with higher doses, particularly in EI. For reasons that are not well understood, blistering and infection are less of a problem for adults than for children with EI.
CONNEXIN DISORDERS
Mutations in genes encoding connexins lead to erythrokeratoderma, and phenotypes include keratitis ichthyosis deafness syndrome and erythrokeratodermia variabilis et progressiva (EKVP). There are
800
overlapping clinical features and phenotypic variability, even within kindreds, in these disorders.123,124
CLINICAL FEATURES
CLINICAL FEATURES
EKVP (OMIM #133200) represents a spectrum of phenotypes initially described as progressive symmetric erythrokeratodermia (PSEK) and erythrokeratodermia variabilis (EKV) but that have been found to have a common genetic basis.125 The progressive symmetric erythrokeratodermia phenotype was first definitively described by Darier in 1911126 and is characterized by well-demarcated, erythematous, hyperkeratotic plaques that are symmetrically distributed over the extremities and buttocks, and often the face127 (Fig. 47-21). The trunk tends to be spared, but the palms and soles may be involved. The plaques appear shortly after birth, progress slowly during the first few years, and then stabilize in early childhood. The plaques usually remain stable in location and appearance but may undergo partial regression at puberty. The erythrokeratodermia variabilis phenotype was described by Mendes da Costa in 1925 as a rare disorder typically presenting at birth or during the first year of life.128 Both generalized involvement (see Fig. 47-25) characterized by persistent, red-brown hyperkeratotic plaques and accentuated skin markings and localized involvement that is limited in extent and characterized by sharply demarcated, hyperkeratotic plaques that are symmetrically arrayed and remain relatively fixed for months to years have been described. Although many features are shared with PSEK, EKV also demonstrates sharply demarcated, migratory red patches that vary in size from a few to many centimeters. These geographic, figurate red patches appear or regress over minutes to hours; some individuals complain of burning at these sites, but they are asymptomatic in others. The red patches develop independently of the hyperkeratosis. Palmoplantar hyperkeratosis may be present, but hair and mucous membranes are unaffected. Histopathologic features include hyperkeratosis, acanthosis, papillomatosis, and capillary dilatation. Epidermis involved with severe papillomatosis and suprapapillary thinning may result in a “church spire” appearance. EKVP is inherited in an autosomal dominant pattern with incomplete penetrance and variable expressivity, although rare recessive cases have been described.129
KERATITIS–ICHTHYOSIS– DEAFNESS SYNDROME
KERATITIS–ICHTHYOSIS–
DEAFNESS SYNDROME
KID syndrome (OMIM #148210) is a rare disorder characterized by keratitis (with progressive corneal opacification), ichthyosis, and deafness (neurosensory). Involvement of multiple ectodermal tissues qualifies KID syndrome as an ectodermal dysplasia. Most cases are compatible with autosomal dominant inheritance.
8
The disease is characterized by discrete erythematous plaques, and there may be mild, generalized hyperkeratosis (Fig. 47-22). The distinctive plaques may have a discrete border and a verrucous appearance with crusting and may be conspicuously figurate and symmetric on the face. Furrowing about the mouth results in characteristic facies. There may be prominent follicular hyperkeratosis, which can result in a scarring alopecia of the scalp. “Leather-like” palmar/plantar keratoderma is almost always seen.130 Descriptions of nail changes vary from absent, delayed appearance after birth, atrophic, or brittle to thickened, with loss of or “rough” cuticles, subungual hyperkeratosis, and leukonychia. The teeth may be small. Auditory evoked potential studies allow detection of the hearing deficit in infancy. Keratitis may develop.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
ERYTHROKERATODERMIA VARIABILIS ET PROGRESSIVA
Mutations in GJB3, GJB4, and GJA1, the genes encoding connexin 31, 30.3, and 43, respectively, have
been identified in kindreds with EKVP.11,124,128,131,132
Connexins are a family of proteins that aggregate to form gap junctions that are important channels for intercellular communication. This intercellular signaling system is crucial for maintaining tissue homeostasis, growth control, development, and synchronized response of cells to stimuli. In the cell, connexins oligomerize to form homotypic or heterotypic hexameric connexons in the Golgi, which are transported to the cell membrane, where they can either function as nonjunctional hemichannels or dock with connexins in neighboring cells. Mutant connexins can show impaired intercellular communication through gap junctions and leakiness of hemichannels.124 The critical deficit in causing the pathology has not been established.
KERATITIS–ICHTHYOSIS–DEAFNESS SYNDROME
Dominant mutations in GJB2, the gene encoding connexin 26, have been detected in sporadic cases and familial KID syndrome.133 Functional studies of cells expressing mutated connexin 26 demonstrated failure of a fluorescent tracer to pass through gap junction channels to neighboring cells, consistent with disruption of intercellular communication.124,133 Mutant connexin 26
801
8
also forms poor gap junctions and leaky heterotypic hemichannels with connexin 43.134 Different mutations in the same gene (GJB2) encoding connexin 26 have also been found in a family with a mutilating palmoplantar keratoderma (Vohwinkel disease; see Chap. 48) and deafness (without ichthyosis).135 The identification of mutations in the genes encoding a variety of connexin proteins has highlighted the role of connexin-mediated intercellular communication through gap junctions in the development and maintenance of ectodermal tissues. Connexins 26, 30, and 31 are expressed in the stratified epithelia of the cochlea and epidermis, and abnormalities in these proteins can cause sensorineural hearing impairment and skin disorders.136
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
CLINICAL COURSE,
PROGNOSIS, AND
MANAGEMENT
ERYTHROKERATODERMIA VARIABILIS ET PROGRESSIVA
Affected individuals frequently have normal skin at birth, and initial lesions present in infancy or childhood.
802
Skin involvement can wax and wane over the course of an individual’s life. Systemic retinoid treatment improves hyperkeratotic lesions and may also clear the figurate red patches when present. The hyperkeratotic skin lesions may be triggered by trauma to the skin, and the red patches may be triggered by a change in temperature.
KERATITIS ICHTHYOSIS DEAFNESS SYNDROME
Cutaneous severity can vary significantly in KID syndrome, particularly in the neonatal period. Keratitis and deafness are variably severe; when present, they may be progressive. Developmental brain defects, often asymptomatic, are common, and some recommend routine magnetic resonance imaging.137 Affected individuals can have an increased susceptibility to bacterial, fungal, or viral infections.138 Squamous cell carcinoma of the skin and tongue has also been reported.139 In contrast to many other ichthyotic conditions, treatment of these patients with oral retinoids has been reported to be of little benefit and possibly to exacerbate the corneal neovascularization. The p.A88V and p.G45E mutations in GJB2 are associated with neonatal lethality, primarily caused by respiratory failure.140-143a
PEELING SKIN DISORDERS
CLINICAL FEATURES
CLINICAL FEATURES
The peeling skin syndromes (PSSs; OMIM #270300) were initially described in case reports and were later classified into those with acral predominance, those that were generalized and noninflammatory, and those that were generalized and inflammatory. Skin peeling was often intermittent, and patients lacked the general hyperkeratosis seen in most ichthyoses. These few early cases made it clear that this was a very heterogeneous group of diseases. In the past 12 years, five recessive gene mutations have been reported from one to four families with findings described as PSSs. The common clinical features included painless, easy peeling skin without scarring, exacerbation by friction and humidity, first appearance from the second day to mid-first decade of life. Many had itch, and some reportedly had cutaneous and systemic allergy.
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Mutations in CDSN, which encodes corneodesmosin, cause inflammatory PSS with atopy, pruritus, and marked deficits in cutaneous barrier function, termed PSS1 (OMIM #270300).144 Affected individuals have superficial exfoliation, ichthyosiform erythroderma, and frequent cutaneous infections in childhood. Corneodesmosin is a component of desmosomes and corneodesmosomes at the transition from cells of the stratum granulosum to the stratum corneum. Mutations in CDSN lead to loss of corneodesmosin expression, eliminating the adhesion between the upper epidermis and the stratum corneum and disrupting terminal differentiation. One consequence of defective differentiation is upregulation of kallikrein 5, a stratum corneum tryptic enzyme that further exacerbates the peeling skin phenotype. Mutations in the gene coding for transglutaminase 5, a ubiquitously expressed transglutaminase with widespread expression in skin, have been identified in multiple kindreds with acral peeling skin, PSS2 (OMIM #609796).145 Transglutaminase 5 is expressed in the granular layer of skin and is central to the formation of the cornified envelope, cross-linking structural proteins including involucrin, loricrin, filaggrin, and small proline-rich proteins. Failure to cross-link these proteins leads to shedding at the stratum corneum interface. Autosomal recessive mutations in CHST8, which encodes N-acetylgalactosamine-4-O-sulfotransferase, an enzyme central to the production of sulfated glycosaminoglycans in the epidermis, have been found in a single kindred with PSS3 (OMIM #616265).146
Affected individuals develop generalized white scaling after 5 years of age which is prominent on
8
the extremities and peels easily. N-acetylgalactosamine-4-O-sulfotransferase functions in glycosylation of proteins central to epidermal differentiation and desquamation. Autosomal recessive, loss of function mutations in CSTA which encodes cystatin A, a cysteine protease inhibitor expressed throughout the epidermis, have also been reported in kindreds with acral PSS, some with more generalized exfoliative ichthyosis, classified as PSS4 (OMIM #607936).147-150 Cystatin A has a central role in desmosomal adhesion in basal layers of the epidermis, and peeling skin caused by CSTA mutations accordingly shows a deeper level of cleavage than is seen in individuals with mutations in TGM5.149
Autosomal recessive mutation in SERPINB8, which belongs to a large family of protease inhibitors, causes a noninflammatory PSS, PSS5 (OMIM #617115).
SYNDROMIC ICHTHYOSES
Syndromic ichthyoses feature moderate to severe extracutaneous involvement. A selection of syndromic ichthyoses will be described.
CHANARIN-DORFMAN SYNDROME
CHANARIN-DORFMAN
SYNDROME
Chanarin-Dorfman syndrome (neutral lipid storage disease with ichthyosis; OMIM #275630) is an autosomal recessive disorder characterized by accumulation of triglycerides in the cytoplasm of keratinocytes, leukocytes, muscle, liver, fibroblasts, and other tissues with normal blood lipid levels. The resulting ichthyosis features generalized lamellar scales with variable erythema, and corrugated accentuation of hyperkeratosis in flexures is common (Fig. 47-23). An EKV-like presentation has been reported.151 Presentation is often that of a collodion baby with ectropion and eclabium. Extracutaneous involvement, which is generally not as severe as in neutral lipid storage disease without ichthyosis caused by mutations in PNPLA2, is variable and may be mild, including cataracts, decreased hearing, hepatosplenomegaly with abnormal liver enzymes and fatty liver, psychomotor delay, myopathy with elevations in serum muscle enzymes, and neurologic abnormalities. Histopathology of oil red O or Sudan III stains of frozen skin sections shows lipid droplets in dermal cells, in the basal layer (and to a lesser extent suprabasally), and in the acrosyringia of the eccrine ducts. Examination of peripheral blood smears shows lipid vacuoles within granulocytes, eosinophils, and monocytes, a feature that may also be present in carriers.152 Mutations in the ABHD5 gene have been identified.153 ABHD5 belongs to a large family of proteins, most of which are enzymes, and appears to activate adiposetriglyceride lipase in the initial step in lipolysis of triglycerides. In the absence of ABHD5, triglycerides
803
8
accumulate in cells154 and in lamellar bodies where, after secretion, they interfere with lipid lamellae formation in the stratum corneum, resulting in increased transepidermal water loss.155
CHILD SYNDROME
CHILD SYNDROME
CHILD syndrome (OMIM #308050) is a rare disorder consisting of congenital hemidysplasia, ichthyosiform erythroderma, and limb defects, which is found almost exclusively in females. It is a mosaic disorder featuring red scaly plaques intermixed with normal skin on one side of the body with minimal contralateral involvement. There may be bands of normal skin on the affected side (Fig. 47-24). Limb defects occur ipsilateral to the ichthyosis and range from digital hypoplasia to agenesis of the extremity. There may be punctate calcification of cartilage. Unilateral hypoplasia can involve the central nervous system and cardiovascular, pulmonary, renal, endocrine, and genitourinary systems. The inheritance pattern is X-linked dominant, with the condition being lethal in males. X-inactivation of the mutant allele gives rise to normal skin in affected females. Most affected individuals have loss-of-function mutations in NSDHL (NAD[P]H steroid dehydrogenase-like protein) encoding a 3β-hydroxysteroid dehydrogenase.156,157 NSDHL functions in the postsqualene cholesterol biosynthetic pathway, catalyzing
804
intermediate steps in the conversion of lanosterol to cholesterol.158 The disorder is related to X-linked dominant chondrodysplasia punctata caused by mutations in emopamil-binding protein (EBP), which also has skin and skeletal abnormalities, and one individual with a CHILD phenotype has been reported with an EBP mutation. EPB is downstream of NSDLH in the cholesterol synthetic pathway. Pathogenesis-based therapy ameliorates the cutaneous findings in CHILD syndrome. Defects in NSDHL lead to a failure to synthesize cholesterol and accumulation of 4-methyl and 4,4-dimethol sterols to toxic levels. Phenotypic improvement of the skin findings in CHILD syndrome was achieved by topical application of cholesterol and lovastatin. This supplies the end product of cholesterol biosynthesis while blocking the pathway at the level of HMG CoA reductase, proximal to NSDHL, thereby preventing synthesis of toxic intermediates.159
CHONDRODYSPLASIA PUNCTATA
CHONDRODYSPLASIA
PUNCTATA
Chondrodysplasia punctata is a clinically and genetically diverse group of rare diseases, first described by Conradi, that share the features of stippled calcification of the epiphyses and skeletal changes resulting from abnormal deposition of calcium in the areas of enchondral bone formation during fetal development and early infancy. Clinical severity ranges from severe dwarfism and death during infancy to a radiographic abnormality that resolves over time leaving minor skeletal changes. Several forms also include ichthyosiform changes. An autosomal recessive (rhizomelic) type, and both X-linked dominant160 and recessive forms161 have been described.
RHIZOMELIC CHONDRODYSPLASIA PUNCTATA
Rhizomelic chondrodysplasia punctata (RCDP; OMIM #215100) is also known as peroxisomal biogenesis disorder complementation group 11 (CG11). It is an autosomal recessive, rare, multisystem developmental disorder characterized by dwarfism caused by symmetric shortening of the proximal long bones (ie, rhizomelia), specific radiologic abnormalities (ie, the presence of stippled calcifications of cartilage, vertebral body clefting), joint contractures, congenital cataracts, ichthyosis, and severe mental retardation. Skin changes are present in approximately 25% of patients. RCDP is a disorder of peroxisomes, membrane-bound multifunctional organelles found in all nucleated cells. Their functions vary with cell type and include a variety of pathways (eg, hydrogen peroxide–based respiration, fatty acid β-oxidation, and lipid and cholesterol synthesis) involving the synthesis and degradation of various compounds.
8
Hereditary human peroxisomal disorders are subdivided into disorders of peroxisome biogenesis, in which the organelle is not formed normally, and those involving a single peroxisomal enzyme. RCDP is caused by mutations in PEX7, a gene that encodes peroxin 7, a receptor required for targeting a subset of enzymes to peroxisomes.162 Thus, it is a peroxisome biogenesis disorder characterized by loss of multiple peroxisomal metabolic functions.
X-LINKED RECESSIVE CHONDRODYSPLASIA PUNCTATA
X-linked recessive chondrodysplasia punctata (CDPX; OMIM #302950) can involve skin (linear or whorled atrophic or ichthyosiform hyperkeratosis, follicular atrophoderma, may begin as erythroderma), hair (coarse, lusterless, cicatricial alopecia), short stature and skeletal abnormalities, cataracts, and deafness. Curry and associates studied a family that had atypical ichthyosis and elevated cholesterol sulfate in two affected males and identified an X chromosomal deletion (Xp22) that included the gene for steroid sulfatase.161 There is a cluster of arylsulfatase genes at this location. Mutations in ARSE, the gene encoding the enzyme arylsulfatase E, were found in five patients; however, it is possible that the disorder may also be caused by mutations in adjacent arylsulfatase genes.163 The similarity to warfarin embryopathy suggests that warfarin embryopathy may be caused by drug-induced inhibition of the same enzyme.
X-LINKED DOMINANT CHONDRODYSPLASIA PUNCTATA
X-linked dominant chondrodysplasia punctata (CDPX2; Conradi-Hünermann-Happle syndrome, OMIM #302960) is characterized by a mosaic pattern of skin involvement along Blaschko lines caused by mosaic X-chromosome inactivation (lyonization). It occurs almost exclusively in females, with loss of the gene function hypothesized to be lethal to males.164
Affected females have a normal life expectancy, and there may be increased disease expression in successive generations (anticipation). Occurrence in a male has been observed in association with a 47, XXY karyotype. Conradi-Hünermann-Happle syndrome presents at birth as a congenital ichthyosiform erythroderma that clears over months and is replaced by linear hyperkeratosis, follicular atrophoderma, and pigmentary abnormalities. Hair shaft abnormalities and cicatricial alopecia can also occur. Stippled calcifications are seen in radiographs of areas of endochondral bone formation during childhood but may no longer be visible after puberty. Stature may be short, with asymmetric shortening of the legs. Cataracts occur, usually asymmetrically, in about two thirds of patients. Histochemical staining for calcium may show calcifications within the epidermis, especially
805
8
within hair follicles in young children, which may not be present in older children.165 Mutations in the gene encoding the EBP cause the syndrome. EBP was first identified as a binding target for the drug emopamil, a calcium channel blocker. It was later found to be 3β-hydroxysteroid-δ8-δ7-isomerase that catalyses an intermediate step in the conversion of lanosterol to cholesterol.160 Although this is another ichthyosis caused by abnormality in cholesterol synthesis, the pathophysiology of how this defect causes clinical manifestations is unclear.
ICHTHYOSIS FOLLICULARIS, ALOPECIA, AND PHOTOPHOBIA SYNDROME
ICHTHYOSIS FOLLICULARIS,
ALOPECIA, AND
PHOTOPHOBIA SYNDROME
Noninflammatory follicular hyperkeratosis, nonscarring alopecia, photophobia, and characteristic facies are seen in the X-linked recessive ichthyosis follicularis, alopecia, and photophobia syndrome (IFAP) syndrome (OMIM #308205, see Chap. 49). Less constant features include recurrent respiratory infections, nail abnormalities, angular cheilitis, keratotic plaques on the extensor surface of the extremities, inguinal hernia, cryptorchidism, short stature, seizures, and psychomotor developmental delay.166 Mutations in the MBTPS2 gene cause this syndrome presumably by resulting in impaired cholesterol homeostasis and endoplasmic reticulum stress.167
ICHTHYOSIS PREMATURITY SYNDROME
ICHTHYOSIS PREMATURITY
SYNDROME
Complications such as polyhydramnios in the second trimester of pregnancy occur in the ichthyosis prematurity syndrome (OMIM #608649), resulting in premature delivery of infants with erythrodermic, edematous, caseous scaling skin resembling excessive vernix caseosa, respiratory distress, and transient peripheral eosinophilia.168 The respiratory signs, erythema, and skin changes resolve, leaving signs of atopy and dry, scaly skin with follicular accentuation more pronounced than is seen in keratosis pilaris. Mutations in the fatty acid transport protein 4 gene FATP4 (SLC27) cause this disorder.169,170
MULTIPLE SULFATASE DEFICIENCY
MULTIPLE SULFATASE
DEFICIENCY
Multiple sulfatase deficiency (OMIM #272200) is a rare autosomal recessive disorder. Clinical features include neurologic deterioration, skeletal abnormalities, facial dysmorphism, and ichthyosis resembling that seen in
806
X-linked steroid sulfatase deficiency. The disorder is a composite of the clinical features of both metachromatic leukodystrophy and a mucopolysaccharidosis. It is characterized by a deficiency of both lysosomal (arylsulfatases A and B) and microsomal (arylsulfatase C/steroid sulfatase of X-linked ichthyosis) arylsulfatases, resulting in the accumulation of sulfatides, glycosaminoglycans, sphingolipids, and steroid sulfates in tissues and body fluids.171 The gene coding for sulfatase modifying factor 1 (SUMF1), which generates a unique amino acid derivative, Cα-formylglycine, necessary for catalytic activity of all sulfatases, is mutated in individuals with multiple sulfatase deficiency.172,173
REFSUM DISEASE
REFSUM DISEASE
Refsum disease (heredopathia atactica polyneuritiformis; OMIM #266500) is a rare, progressive, degenerative disorder of lipid metabolism resulting from the failure to break down dietary phytanic acid and its subsequent accumulation in tissues. This autosomal recessive condition affects mostly Scandinavians and populations originating from Northern Europe. Clinical manifestations include retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, cranial nerve dysfunction (neural deafness, anosmia), miosis, electrocardiographic abnormalities, cardiomyopathy, renal tubular dysfunction, and skeletal abnormalities (epiphyseal dysplasia). Ichthyosis, which is variable, generally develops after the neurologic and ophthalmologic manifestations. Often there are small white scales over the trunk and extremities resembling IV. Routine hematoxylin and eosin histologic examination shows variably sized vacuoles in the epidermal basal and suprabasilar cells, which correspond to lipid accumulation seen with lipid stains of frozen sections.174
Phytanic acid (a 20-carbon, branched-chain fatty acid) is derived from a variety of dietary sources including dairy products, ruminant fats, and chlorophyllcontaining foods, although chlorophyll-bound phytol cannot be absorbed in humans. The disease is caused by a deficiency of PhyH, a peroxisomal protein that catalyzes the α-oxidation of phytanic acid. PhyH deficiency leads to the accumulation of phytanic acid in the serum and tissues, where it substitutes for the fatty acids normally present. Although mutations in PAHX, the gene encoding PhyH, are responsible for most cases of Refsum disease, there is genetic heterogeneity.175 The Refsum phenotype has been described in patients with mutations PEX7, the affected gene in RCDP (see earlier). Phytanic acid and other chlorophyll metabolites bind the retinoid X receptor (RXR), as does its natural ligand, 9-cis-retinoic acid, and they may be physiologically active in coordinating cellular metabolism through RXR-dependent signaling pathways. However, the role of RXR in the pathogenesis of Refsum disease is unclear. Refsum disease is distinguished from infantile Refsum disease, a fulminant generalized peroxisomal biogenesis disorder in which young children present with severe neurologic abnormalities,
mental retardation, hepatomegaly, and dysmorphic features in addition to the other signs of adult Refsum disease.176
Refsum disease is a rare example of an ichthyosis whose pathophysiology was understood well enough before identification of the causative gene to make rational treatment recommendations. The diagnosis can be made by detection of elevated levels of plasma phytanic acid. In children who do not have elevated plasma levels of phytanic acid, the diagnosis may be made by measuring PhyH activity in cultured fibroblasts.177 Treatment includes dietary restriction of foods containing phytanic acid and its precursors. In the clinical setting of a delayed onset of ichthyosis in association with neurologic impairment, this disorder should be considered because therapy can arrest progression.
SJÖGREN-LARSSON SYNDROME
SJÖGREN-LARSSON
SYNDROME
In 1957, Sjögren and Larsson reported on 13 families from north Sweden with a syndrome of congenital ichthyosis, spastic paralysis, and mental retardation. Sjögren-Larsson syndrome (SLS; OMIM #270200) is a rare, autosomal recessive disorder that presents at birth with an ichthyosis that may range from fine scaling to
8
generalized hyperkeratosis. Erythema may be present at birth but tends to gradually clear by 1 year of age. Collodion-like membranes are rarely seen. The ichthyosis manifests as fine scale, large scale, or thickening of the stratum corneum without scale. Pruritus is common. Thickened areas may be yellow to brown in color and have a lichenified appearance with accentuated skin markings (Fig. 47-25). The most involved areas are the sides and back of the neck, lower abdomen, and flexures. Hair, nails, and the ability to sweat are generally normal.178 During the first 2 to 3 years, neurologic manifestations of spastic diplegia or tetraplegia and mental retardation develop and can be accompanied by speech defects and seizures. A characteristic ophthalmologic finding is the presence of glistening white dots in the macula of the retina. These occur after 1 year of age and may not be present in all patients. SLS is caused by fatty alcohol:NAD oxidoreductase (FAO) deficiency. FAO is a complex enzyme with two separate proteins that sequentially catalyze the oxidation of fatty alcohol to fatty aldehyde and subsequently to fatty acid. Mutations found in the ALDH3A2 gene confirmed the role for this enzyme in the cause of this disorder and the importance of this pathway for normal desquamation. ALDH3A2 is a microsomal enzyme that catalyzes the oxidation of medium- and longchain aliphatic aldehydes derived from metabolism of fatty alcohol, phytanic acid, ether glycerolipids, and
807
8
leukotriene B4.179 The identification of decreased fibroblast FAO activity in a family with atypical cutaneous findings (lack of ichthyosis or discrete plaques rather than generalized ichthyosis) has expanded the spectrum of clinical phenotypes associated with abnormal FAO activity.180
TRICHOTHIODYSTROPHY
TRICHOTHIODYSTROPHY
Trichothiodystrophy (TTD; OMIM #601675, also see Chap. 130) is an autosomal recessive disorder that includes a broad spectrum of clinical phenotypes linked by the characteristic features of sulfur deficient, brittle hair that exhibits alternating birefringence (tiger tail banding) when viewed under polarizing microscopy (Fig. 47-26).181 The spectrum of clinical involvement is broad, ranging from only hair to severe multisystem abnormalities. A survey of 112 cases reported in the literature found ichthyosis (65%) as the most common skin finding followed by photosensitivity (42%). Of the patients with ichthyosis, about one third presented with a collodion membrane at birth. Erythroderma, when present at birth, usually decreases over weeks with evolution into a generalized ichthyosis, usually without erythema, which varies from small fine,
scaling to large, dark yellow-brown hyperkeratosis (see Fig. 47-17B).182,183 There may be flexural sparing and palmoplantar keratoderma. Nail findings are found in more than half of the patients and include dystrophic nails (ridging, splitting) (see Fig. 47-17A), hypoplasia, brittle nails, and koilonychia.184 Ectropion usually does not occur. Photosensitivity can range from subtle to severe. Other common findings in TTD include intellectual impairment, short stature, microcephaly, characteristic facial features (protruding ears, micrognathia), recurrent infections, and cataracts. A series of mnemonics have been used to describe the constellation of findings as BIDS (brittle hair, intellectual impairment, decreased fertility, and short stature); patients who also have ichthyosis have been called IBIDS, and with the addition of photosensitivity, PIBI(D)S has been used.185 However, these terms do not account for the other multisystem findings that are commonly present. The majority of TTD patients with ichthyosis have a defect in the function or activity of TFIIH, a protein complex involved in DNA repair and transcription. Most mutations are in ERCC2. Other mutations affect ERCC3, GTF2H5, and GTF2E2. Two other genes causing TTD, MPLKIP and RNF113A, have not been associated with ichthyosis and their function is unknown.186-190 Although many patients with
808
TTD have photosensitivity, in contrast to xeroderma pigmentosum, these photosensitive patients have not been observed to be at high risk for the development of skin cancer. Nucleotide excision repair is one normal cellular mechanism by which structural DNA damage (eg, ultraviolet-induced cyclobutane pyrimidine dimers) is removed and repaired (see Chaps. 20 and 134).
ACQUIRED ICHTHYOSIS
The development of ichthyosis in adulthood can be a manifestation of systemic disease and has been described in association with malignancies, drugs, endocrine and metabolic disease, malnutrition, HIV and other infections, and autoimmune conditions.191-194
The granular layer is often attenuated in this disorder, and the scale often resembles to that seen in mild IV. Although Hodgkin disease is the most common malignancy reported with acquired ichthyosis, non- Hodgkin lymphomas and a variety of other malignancies have also been observed. Histology may be diagnostic in acquired ichthyosis associated with mycosis fungoides.195 Skin involvement may follow the course of malignancy and clear with effective cancer treatment. Acquired ichthyosis is commonly seen in association with AIDS; ichthyotic or xerotic skin has been observed in up to 30% of patients with AIDS.196 A study of HIV-1–positive intravenous drug users found acquired ichthyosis occurred only after profound helper T cell depletion, more frequently with coinfection with human T-cell leukemia or lymphoma virus type II, and suggested that it may be a marker for concomitant infection with both viruses.197
In acquired ichthyosis occurring in association with sarcoidosis, skin biopsy can be diagnostic, showing noncaseating granulomas in the dermis.198 Acquired ichthyosis may be a marker of autoimmune disease, occurring with systemic lupus erythematosus, dermatomyositis, mixed connective tissue disease, and eosinophilic fasciitis.199,200 It has been described in bone marrow transplant recipients, in whom it may be related to graft-versus-host disease.201
Although occurrence in association with cholesterollowering agents (nicotinic acid, triparanol) highlights the relationship between cholesterol metabolism and normal desquamation, acquired ichthyosis has been observed with a variety of drugs, including cimetidine, clofazimine, hydroxyurea, cholesterol-lowering agents, and others.202,203 Kava is a psychoactive beverage made from the root of a pepper plant and used for thousands of years by Pacific Islanders. Heavy kava drinkers can acquire a reversible ichthyosiform eruption called kava dermopathy. In Western nations, kava is sold in health food stores as a relaxant.204
PITYRIASIS ROTUNDA
PITYRIASIS ROTUNDA
Sharply demarcated, round or oval scaly patches with hypo- or hyperpigmentation are seen in
8
pityriasis rotunda. Although this uncommon disorder is usually acquired, occasional familial cases have been described.205
RETICULATED PAPILLOMATOSIS OF GOUGEROT AND CARTEAUD
RETICULATED
PAPILLOMATOSIS
OF GOUGEROT AND
CARTEAUD
Confluent and reticulated papillomatosis of Gougerot and Carteaud is an uncommon but distinctive acquired ichthyosiform dermatosis seen in young adults and characterized by persistent brown, scaly macules, papules, patches, and plaques. Lesions tend to be localized predominantly on the neck, upper trunk (intermammary and interscapular regions), and axillae, where they tend to be confluent and become reticulated towards the periphery. The lesions bear a clinical resemblance to tinea versicolor, a skin infection with Pityrosporum spp. A variety of treatment approaches have been reported, including topical (keratolytics, derivatives of vitamin A and D, antimicrobials) and systemic (antibiotics, retinoids) agents. Minocycline has been suggested as a first-line treatment; successful retreatment of recurrences supports the concept that this condition is an abnormal response to an infection or inflammation.206,207
RESOURCES
The Foundation for Ichthyosis and Related Skin Types (FIRST) (phone: 800-545-3286; http://www.firstskinfoundation.org; email: info@firstskinfoundation.org.) provides support and information for affected individuals, family members, friends, and health care provider. The Genetic Testing Registry (https://www.ncbi .nlm.nih.gov/gtr/) provides disease reviews, genetic testing resources, and educational materials. Online Mendelian Inheritance in Man, (http://www .ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM) (trademark Johns Hopkins University), a catalog of human genes and genetic disorders, is a useful reference with links to additional resources. The Registry for Ichthyosis and Related Disorders is a resource for investigators to improve diagnosis and treatment of these disorders (Yale University, Department of Dermatology, PO Box 208059, 333 Cedar Street, New Haven, CT 06510; email: ichthyosisregistry@yale. edu).
ACKNOWLEDGMENTS
The authors are grateful to Philip Fleckman and John J. DiGiovanna, who authored the prior version of this chapter and have provided text, tables, and photographs that appear here.
809
8
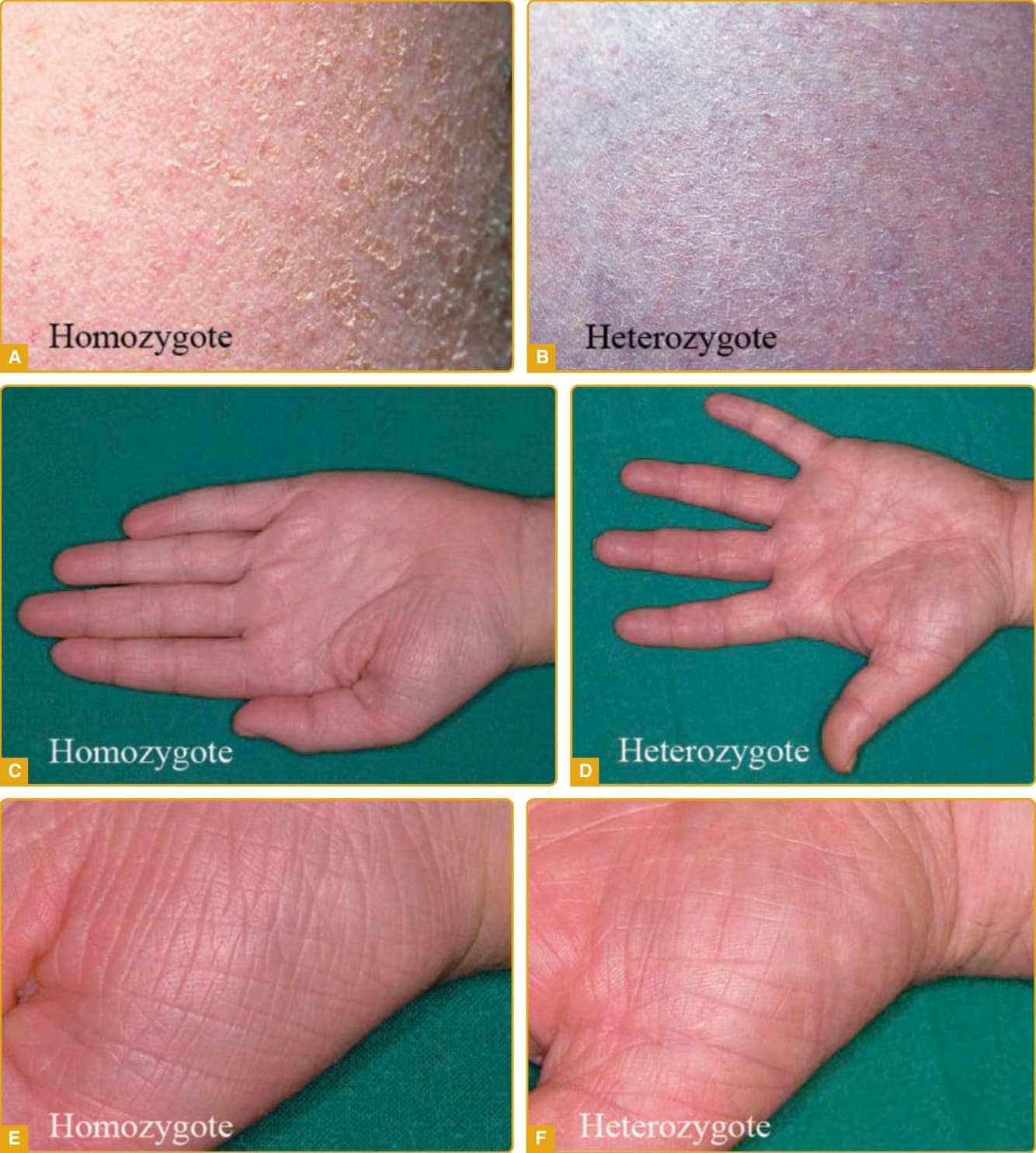
Figure 47-1 A–F, Ichthyosis vulgaris (FLG mutation). Small, centrally adherent scales generally spare intertriginous areas. Increased number and depth of palmar markings are usually noted.
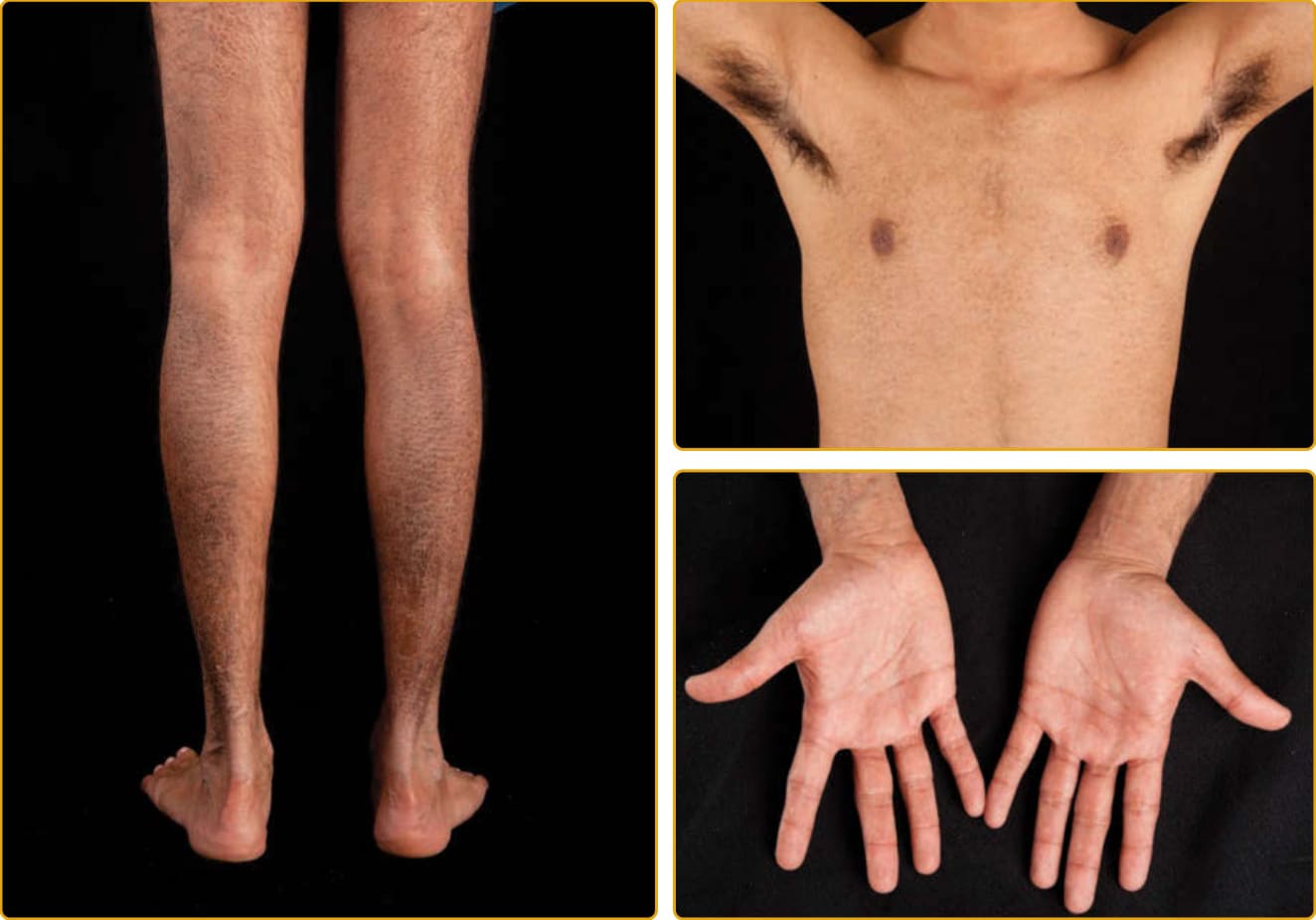
Figure 47-2 X-linked recessive ichthyosis (STS mutation). Small, centrally adherent, dark scales often involve sides of neck and face; they are less severe in flexures. Palms and soles show little to no involvement.
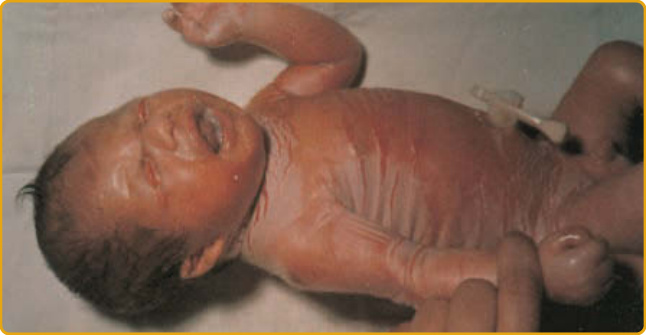
Figure 47-3 Collodion infant. The infant is 36 hours old and is covered with a macerated membrane that shows fissures; note ectropion and eclabium. The condition may develop over time into various clinical phenotypes, including autosomal recessive congenital ichthyosis and self-healing collodion baby.
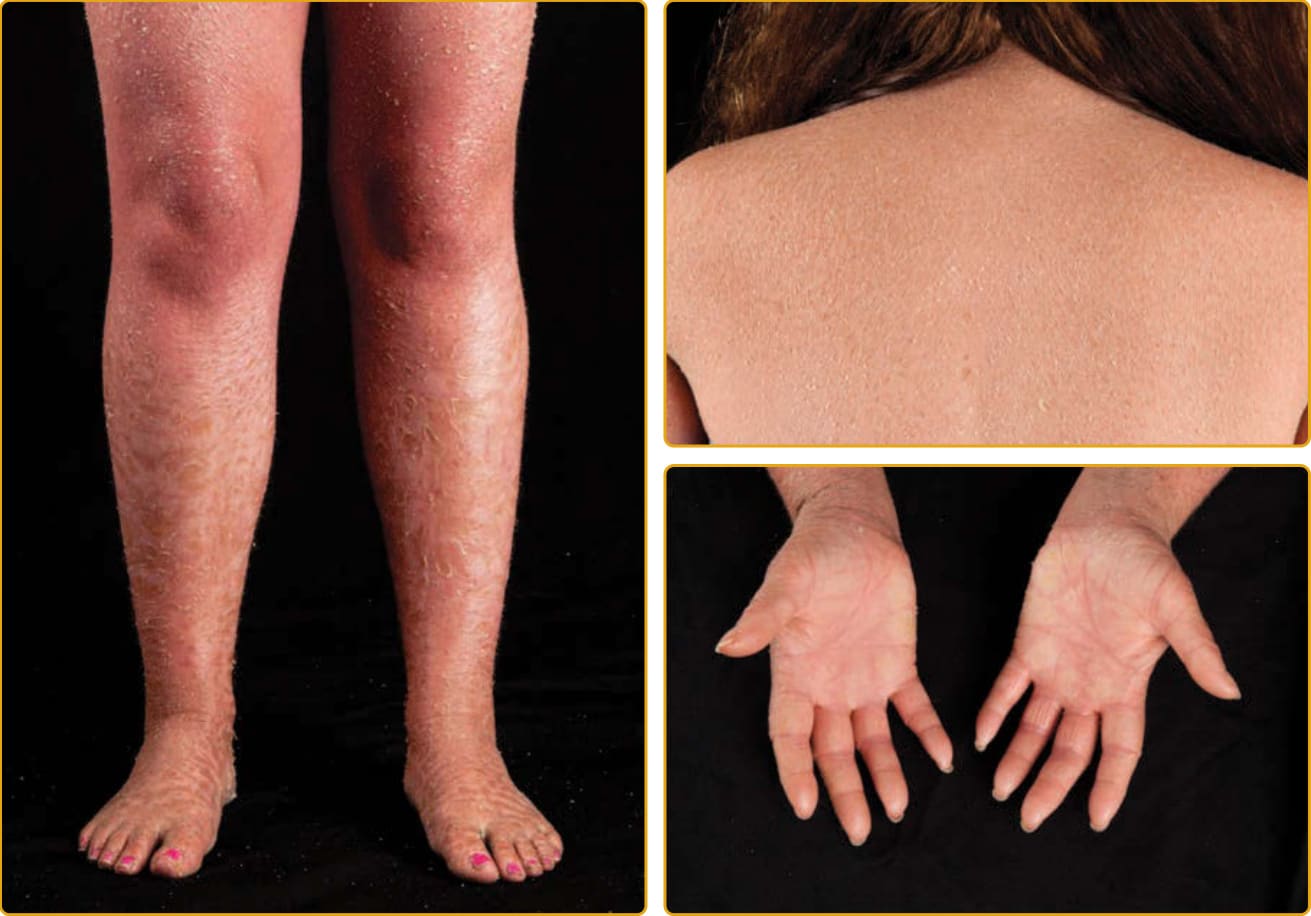
Figure 47-4 Autosomal recessive congenital ichthyosis (TGM1 mutations, classic lamellar ichthyosis, moderate). Large dark scales are often most obvious over extensor extremities and forehead. Mild to moderate erythema and palmarplantar keratoderma are usual. The severity of scale varies widely (see Fig. 47-6).
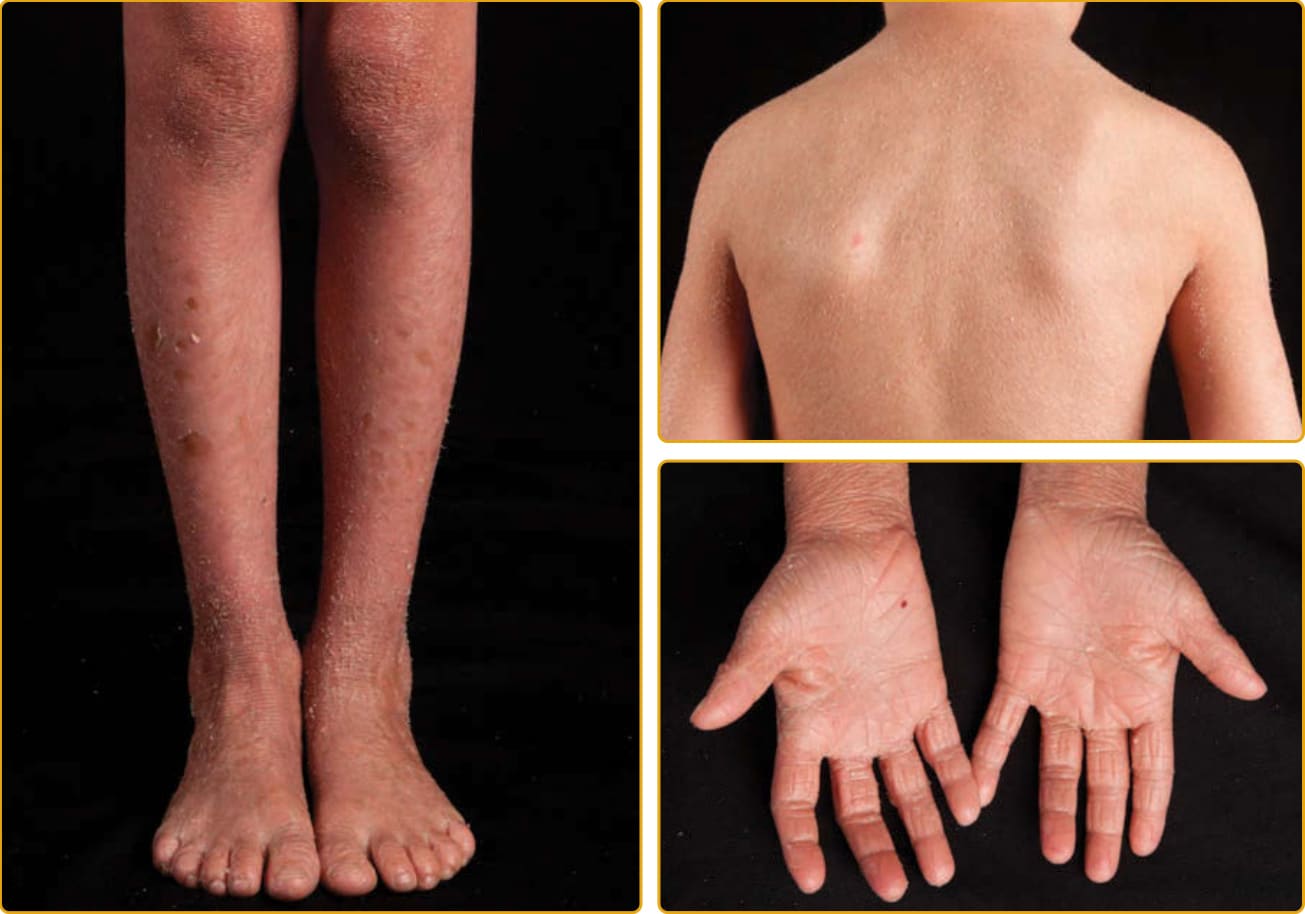
Figure 47-5 Autosomal recessive congenital ichthyosis (TGM1 mutations, classic lamellar ichthyosis, mild). Large dark scales are often most obvious over extensor extremities and forehead. Mild to moderate erythema and palmar-plantar keratoderma are usual. Severity of scale varies widely (see Fig. 47-4).
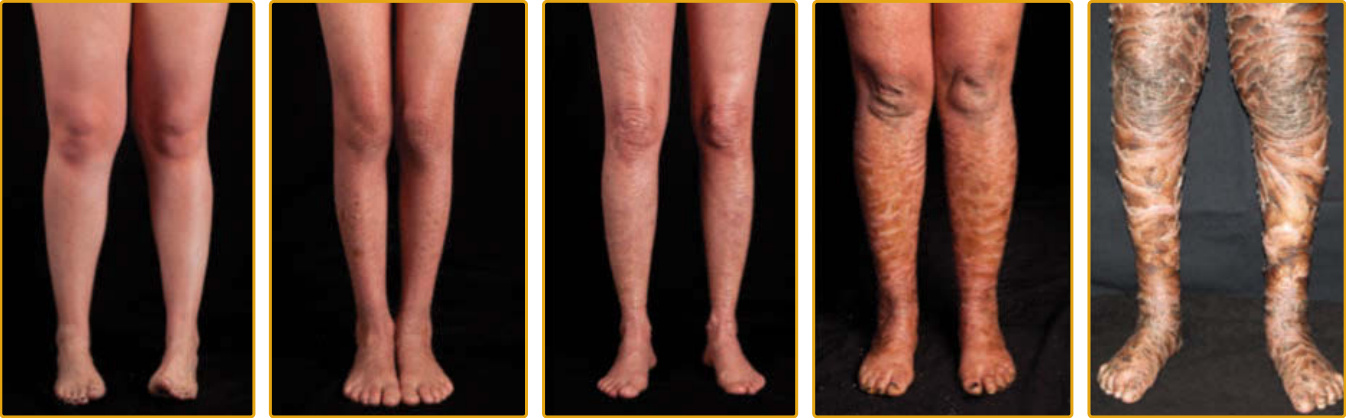
Figure 47-6 Severity spectrum for mutations in TGM1. Phenotypic variability is seen across individuals with the same mutation.
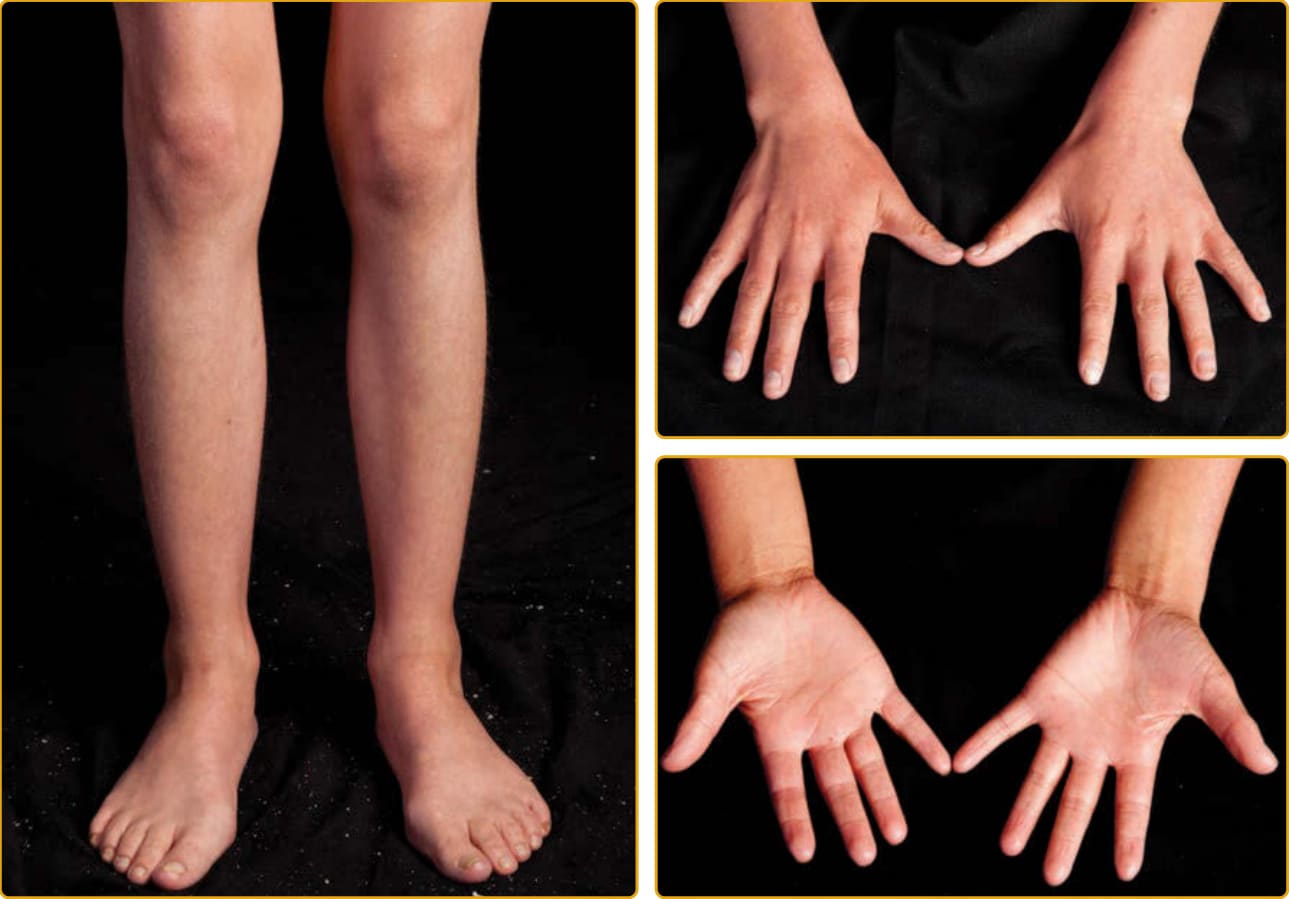
Figure 47-7 Autosomal recessive congenital ichthyosis (ALOXE3 mutations). The volar and dorsal surfaces of hands and feet show exaggerated linear markings. Scale elsewhere, when present, is fine and white and overlies mild erythema.
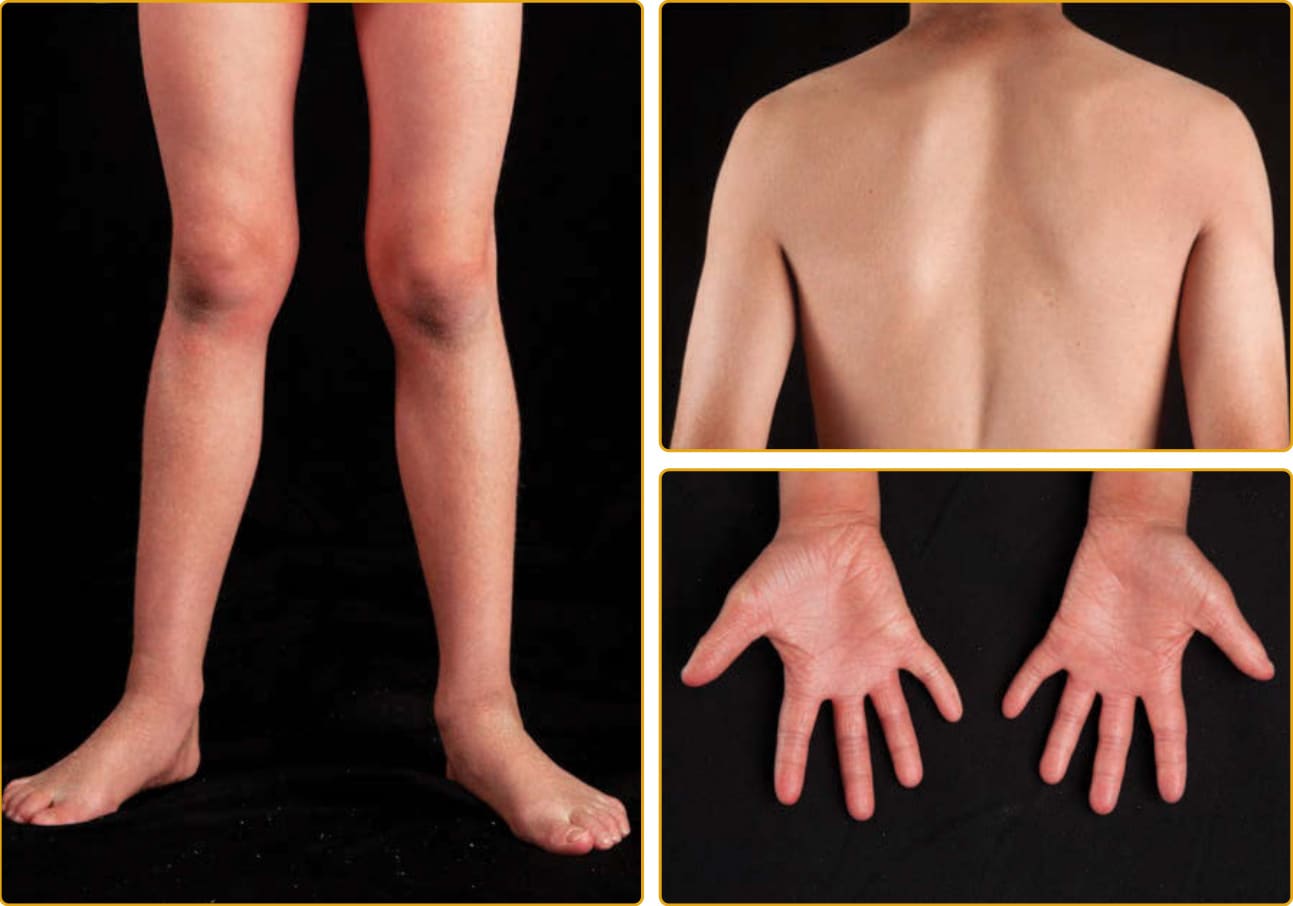
Figure 47-8 Autosomal recessive congenital ichthyosis (ALOX12B mutations). The volar and dorsal surfaces of hands and feet show increased linear markings. Scale elsewhere, when present, is fine and white and overlies mild erythema.
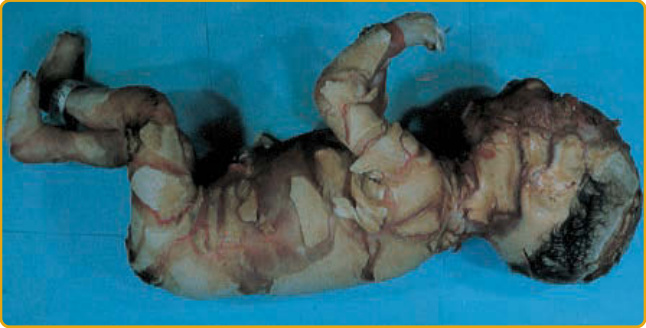
Figure 47-9 Harlequin infant. Harlequin ichthyosis. Note the rudimentary ears and the distorted appearance as a result of the thick “plates” of stratum corneum. This infant died a few days after birth.
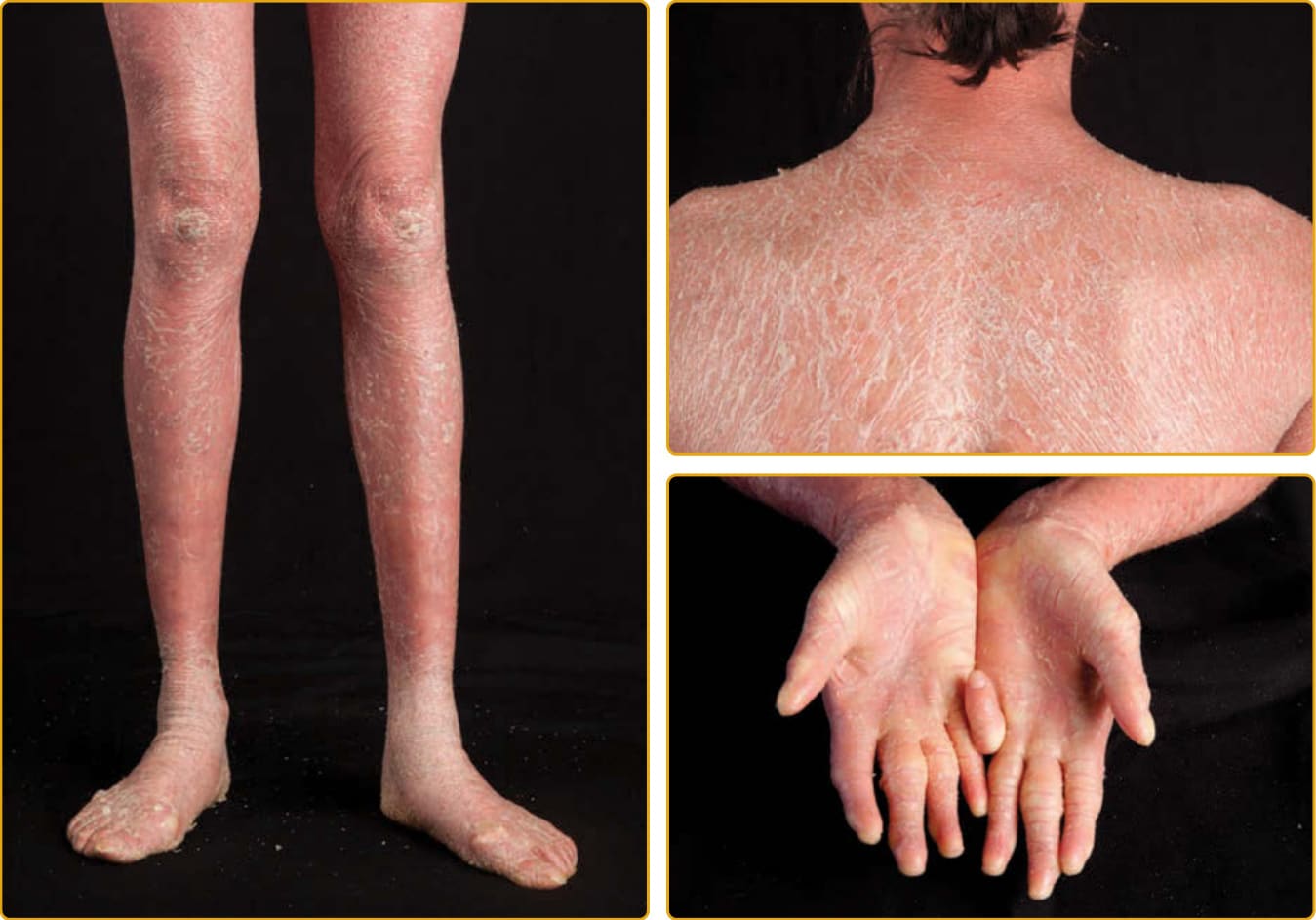
Figure 47-10 Autosomal recessive congenital ichthyosis (ABCA12 mutations, moderate). Large flat scales and moderate generalized erythema. All digits are tapered.
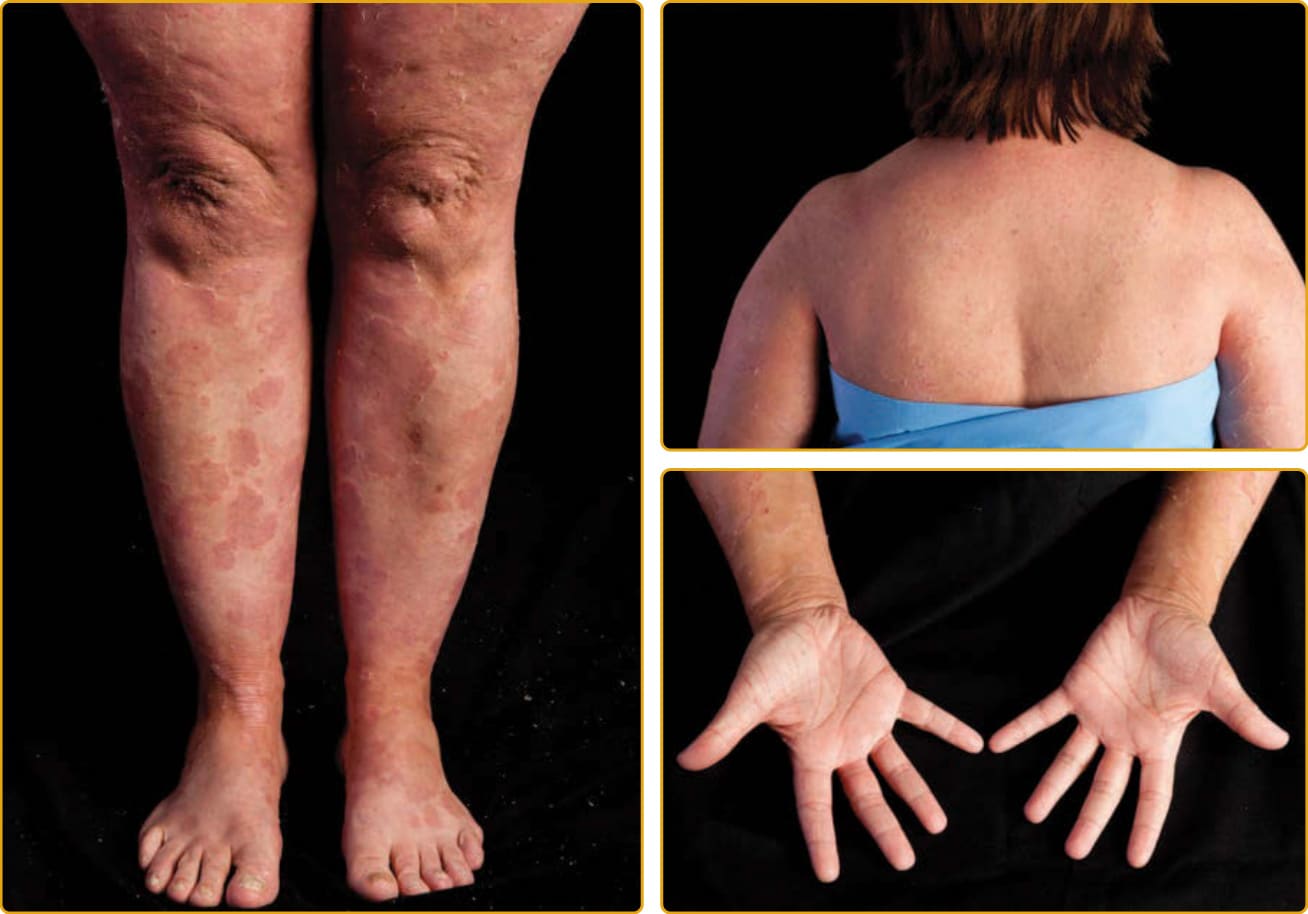
Figure 47-11 Netherton syndrome (SPINK 5 mutation, ichthyosis linearis circumflexa predominant). Serpiginous, migrating scales peel away to reveal underling erythema. The palms and soles have mild to moderate keratoderma.
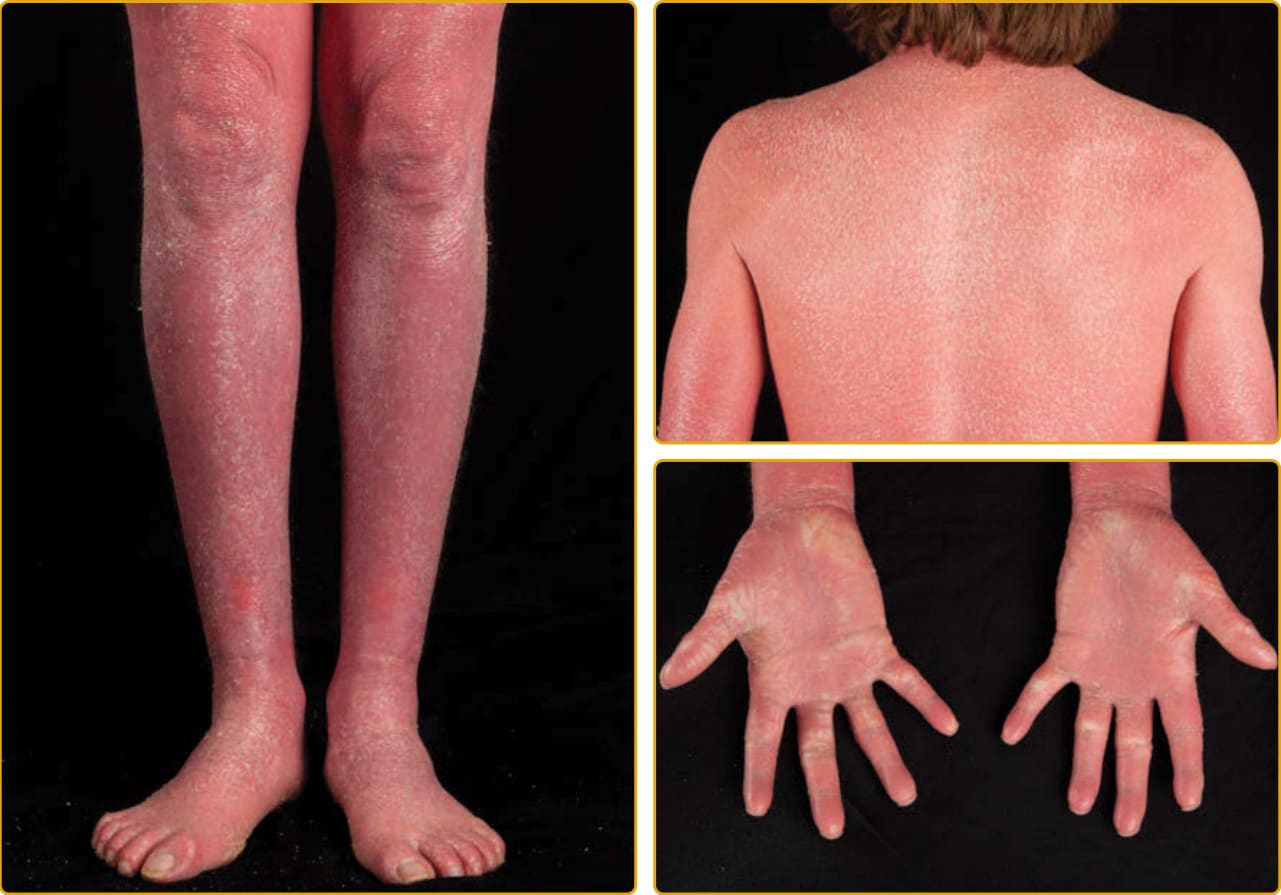
Figure 47-12 Netherton syndrome (SPINK 5 mutation, erythroderma predominant). Small white scales cover areas of mild to severe generalized erythema. Serpiginous, peeling scales are often limited to areas around the ankles and wrists. The palms and soles have mild to moderate keratoderma.
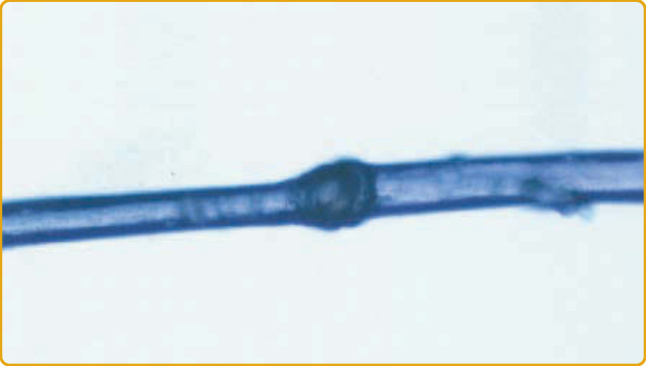
Figure 47-13 Netherton syndrome hair abnormalities. A small bamboo hair shaft shows features of trichorrhexis invaginata.
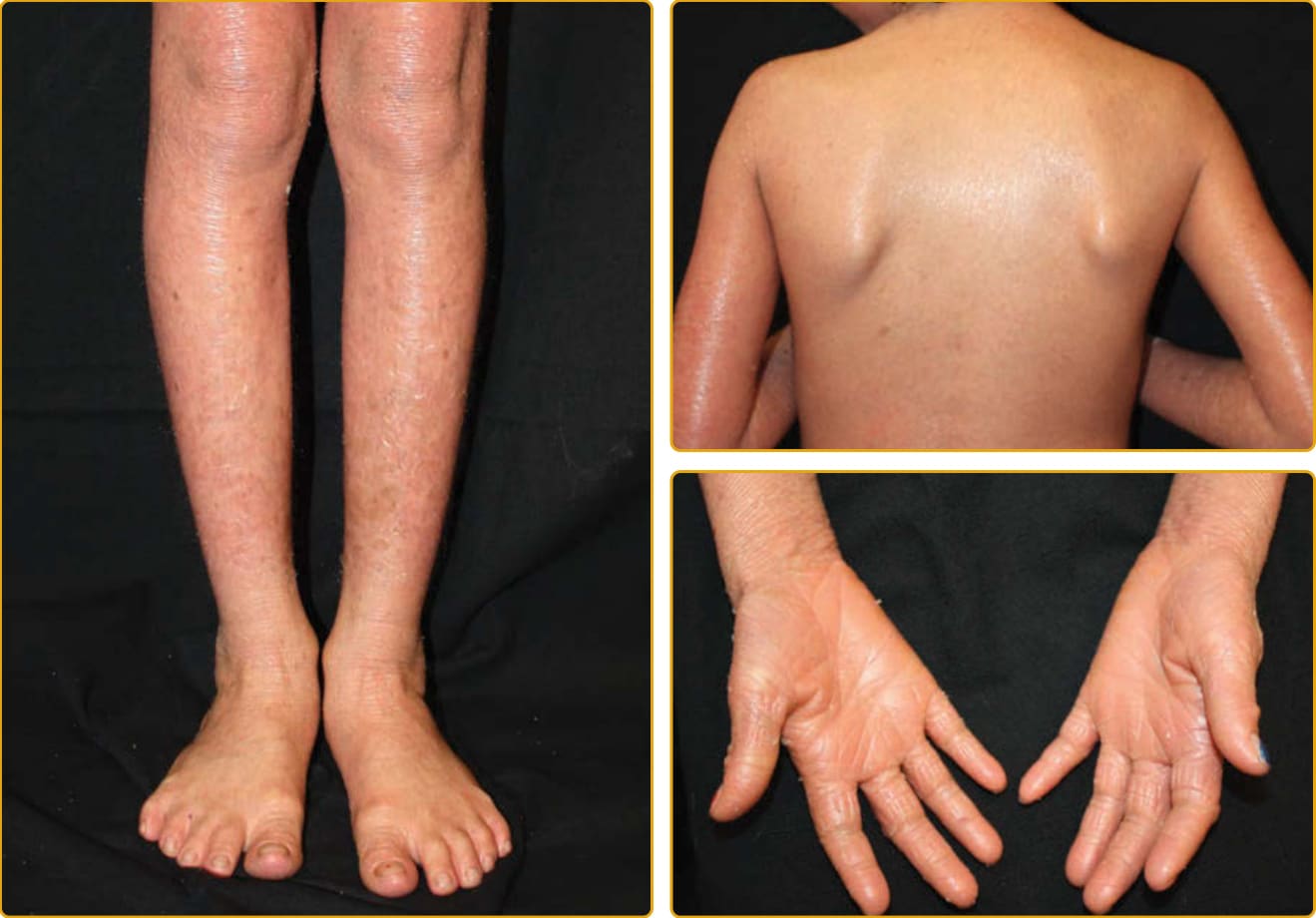
Figure 47-14 Autosomal recessive congenital ichthyosis (ABCA12 mutation, mild). Scales on the shins are large, but those elsewhere are small on background of mild erythema. Moderate palmar keratoderma with increased linear markings and tapered fifth digit.
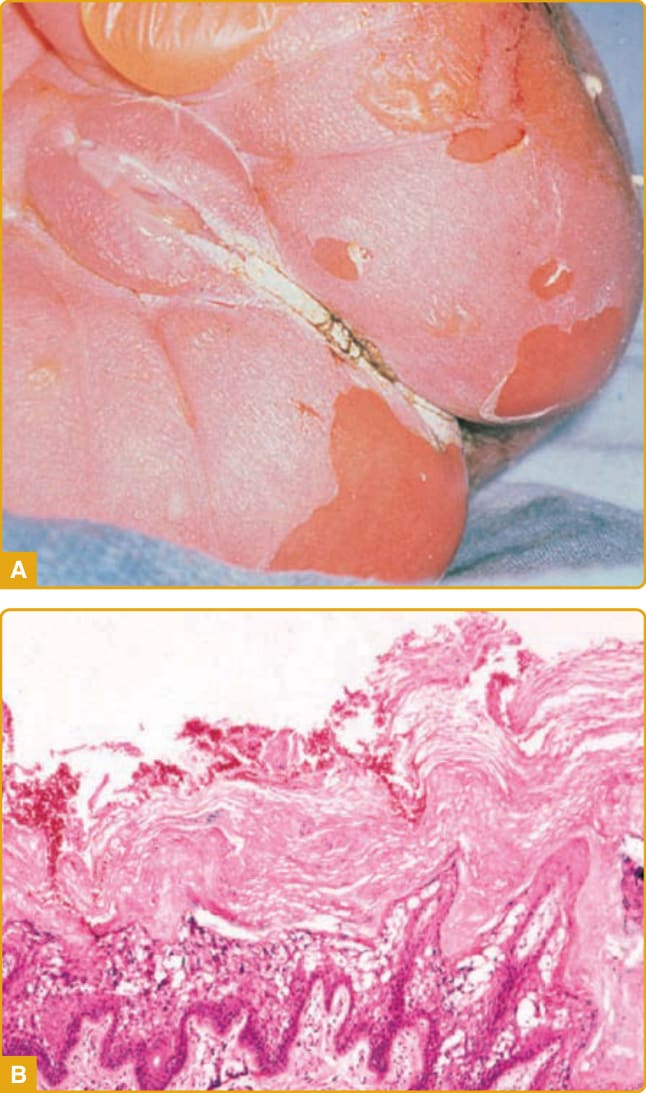
Figure 47-15 Epidermolytic ichthyosis infantile presentation. A, Newborn showing blistering and erosions. B, Epidermolytic ichthyosis histology. The stratum corneum is thickened (hyperkeratosis), and there is prominent vacuolar degeneration of suprabasilar epidermis most marked at the granular layer.
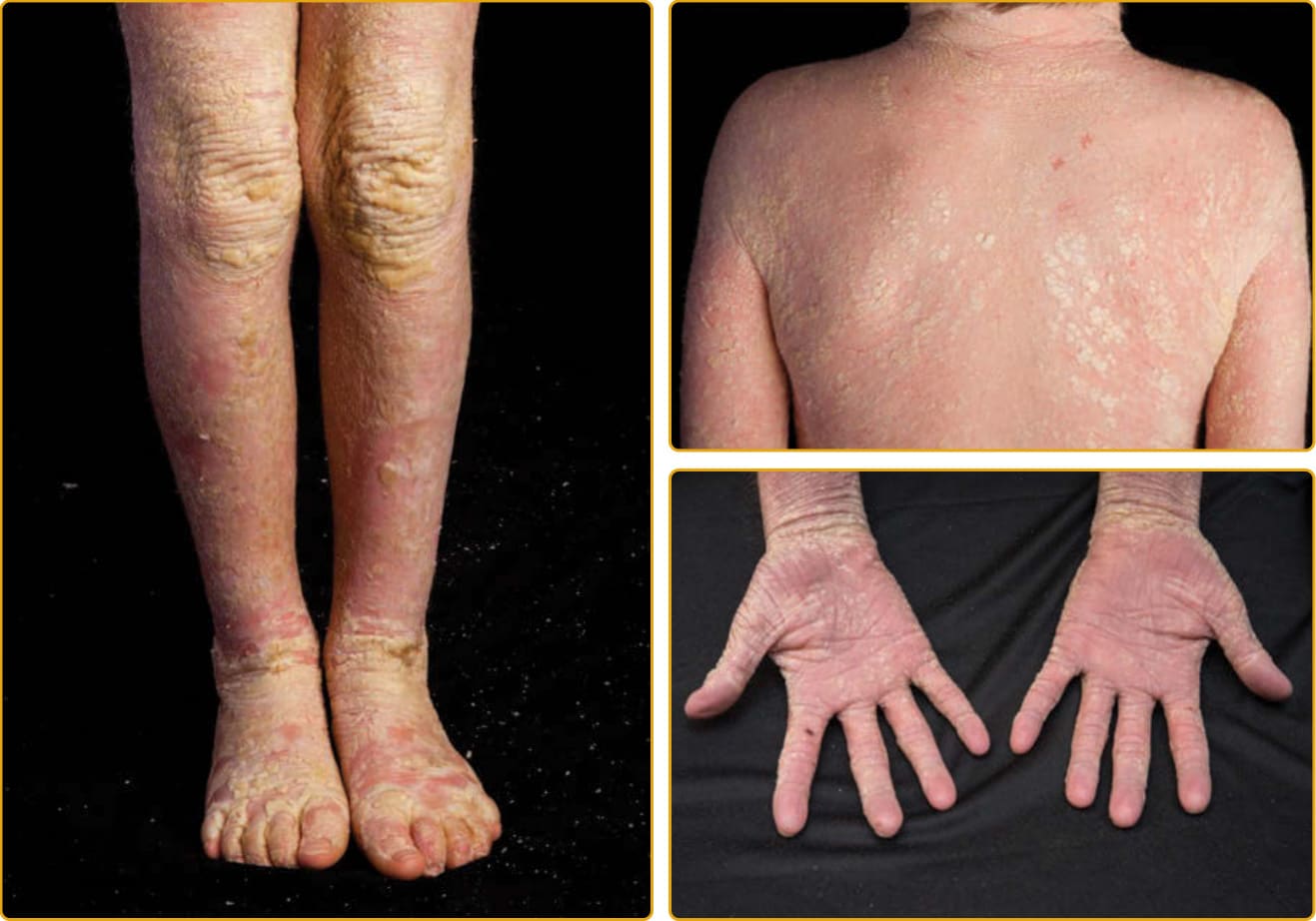
Figure 47-16 Epidermolytic ichthyosis (KRT10 mutation). Generalized coarse, columnar scales often have a corrugated pattern over flexures. Scales unevenly desquamate from the underlying epidermis to reveal mild to moderate generalized erythema. Hyperkeratosis of the palms and soles is mild to moderate, and pseudoainhum occurs.
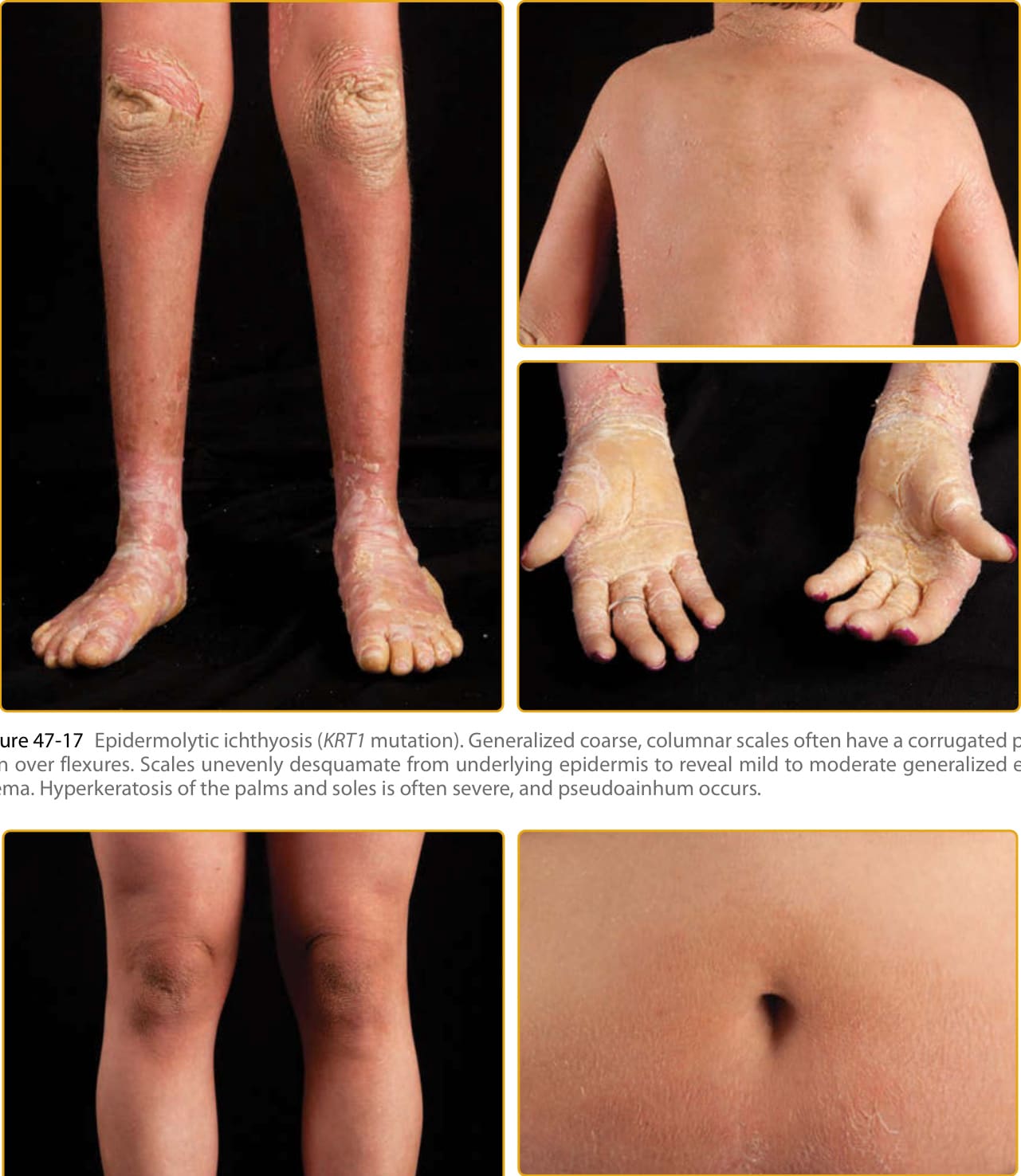
Figure 47-17 Epidermolytic ichthyosis (KRT1 mutation). Generalized coarse, columnar scales often have a corrugated pattern over flexures. Scales unevenly desquamate from underlying epidermis to reveal mild to moderate generalized erythema. Hyperkeratosis of the palms and soles is often severe, and pseudoainhum occurs.
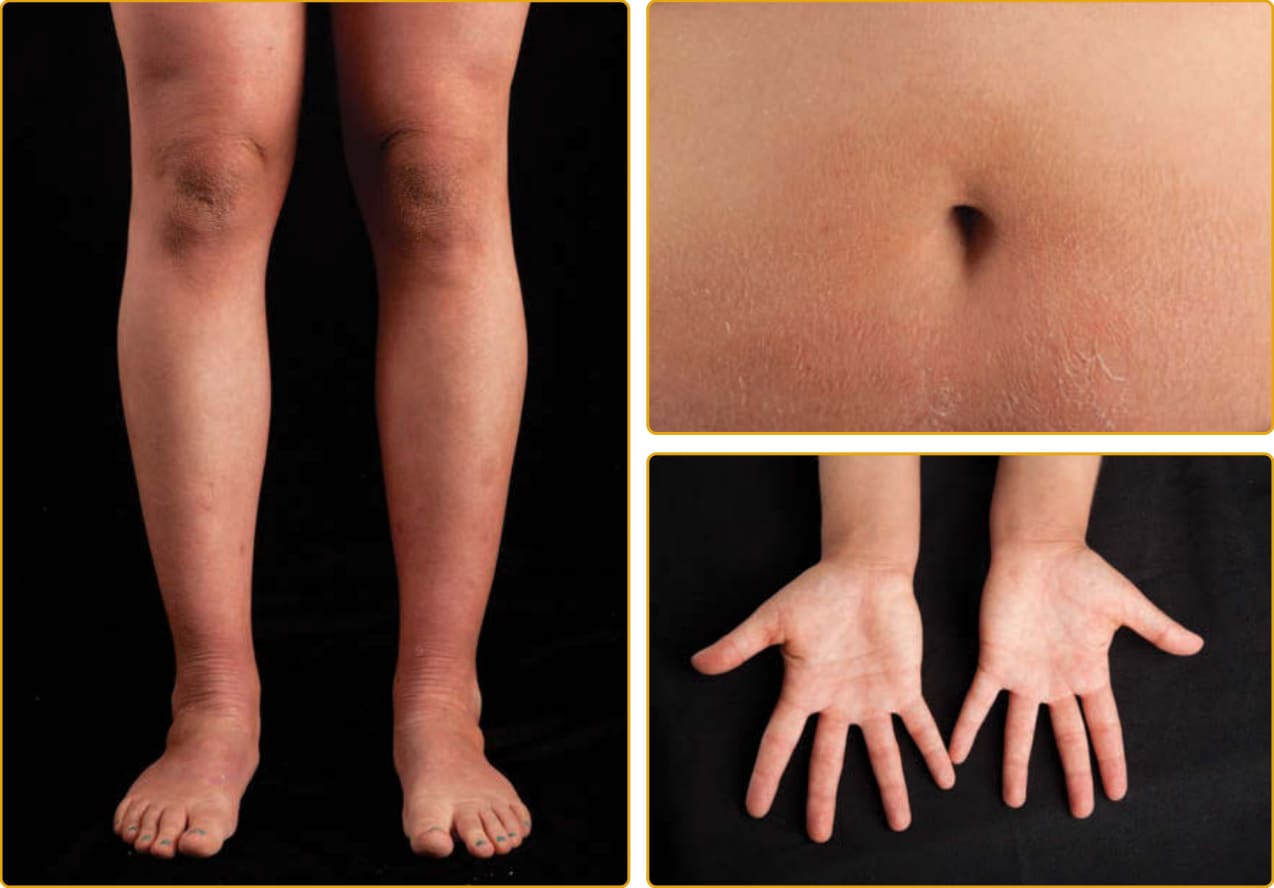
Figure 47-18 Epidermolytic ichthyosis (KRT2 mutation). Smooth, mildly corrugated scale is generally accentuated at flexures or areas of mild friction. Circular or geometric areas of stratum corneum peeling are common. Erythema is mild, and the palms and soles are spared.
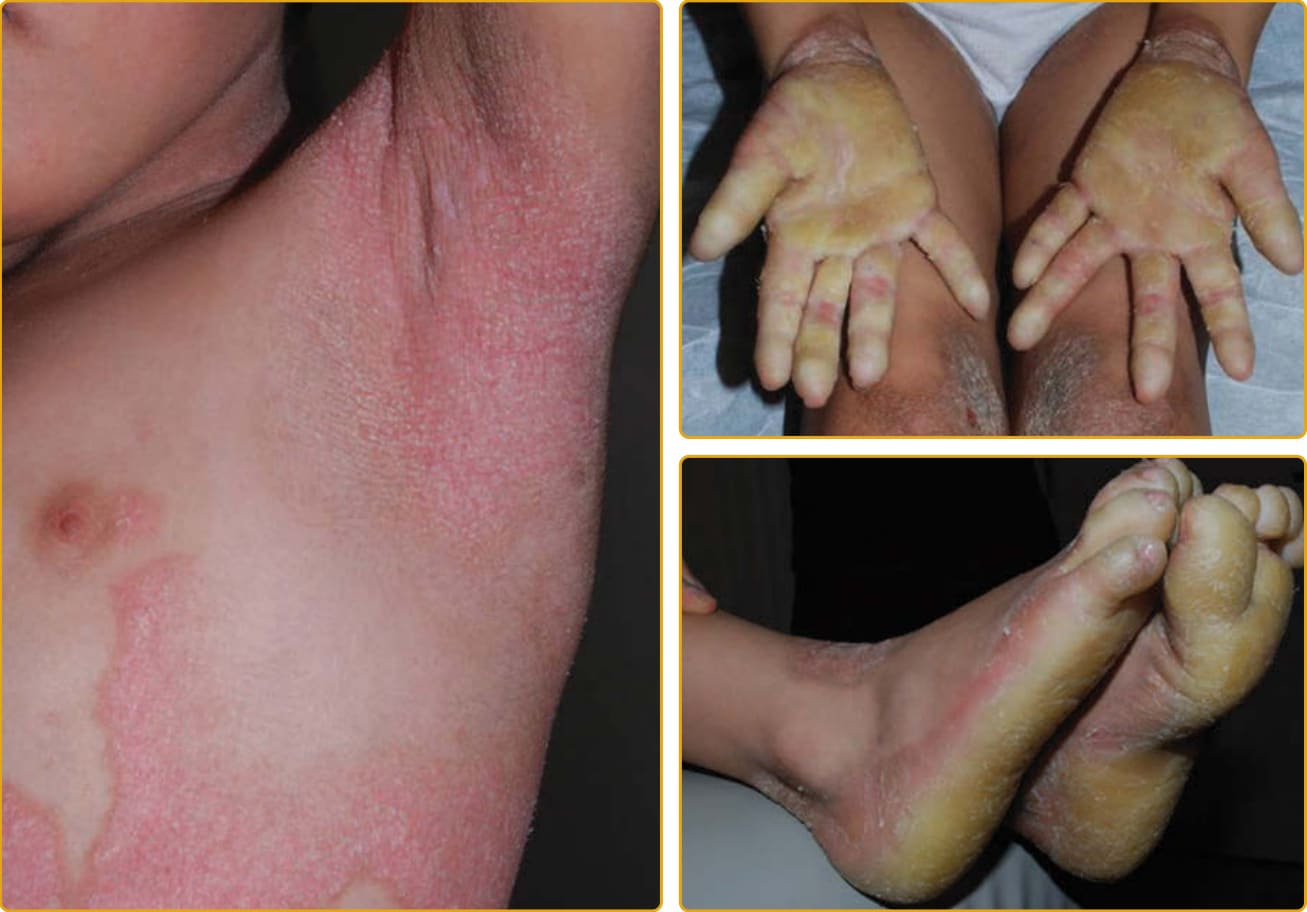
Figure 47-19 Epidermolytic ichthyosis, annular with PPK (KRT1 mutation). Circumscribed erythematous plaques with coarse scale and a corrugated pattern accentuated at flexures. The palms and soles variably involved.
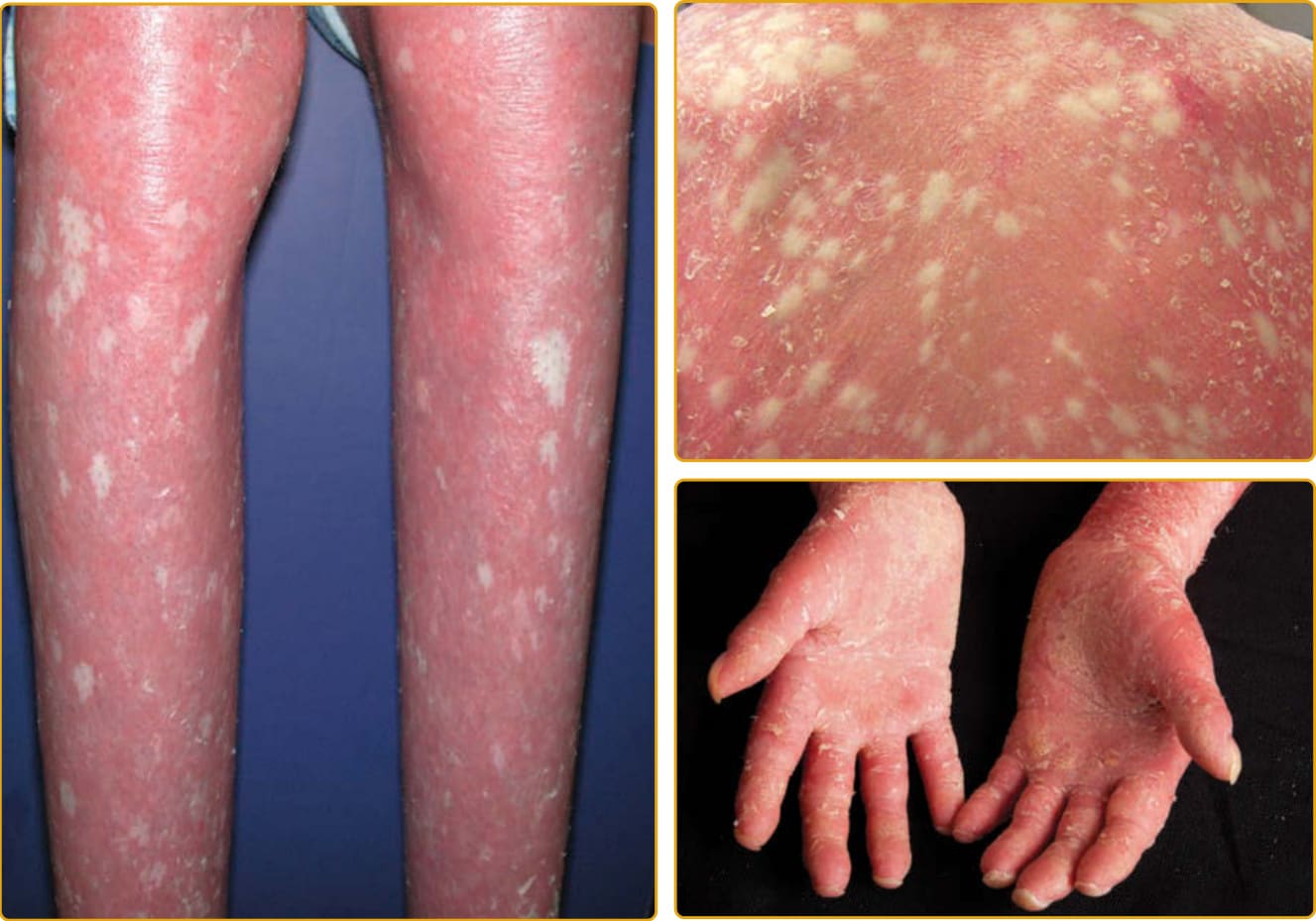
Figure 47-20 Ichthyosis with confetti (KRT10 mutation). Phylloid (leaf-like) areas of normal skin on a background of mild to severe erythema and scale increase in number and size starting in the first decade of life. Palmar and plantar erythrokeratoderma vary from mild to severe. Contractures may occur at small and large joints.
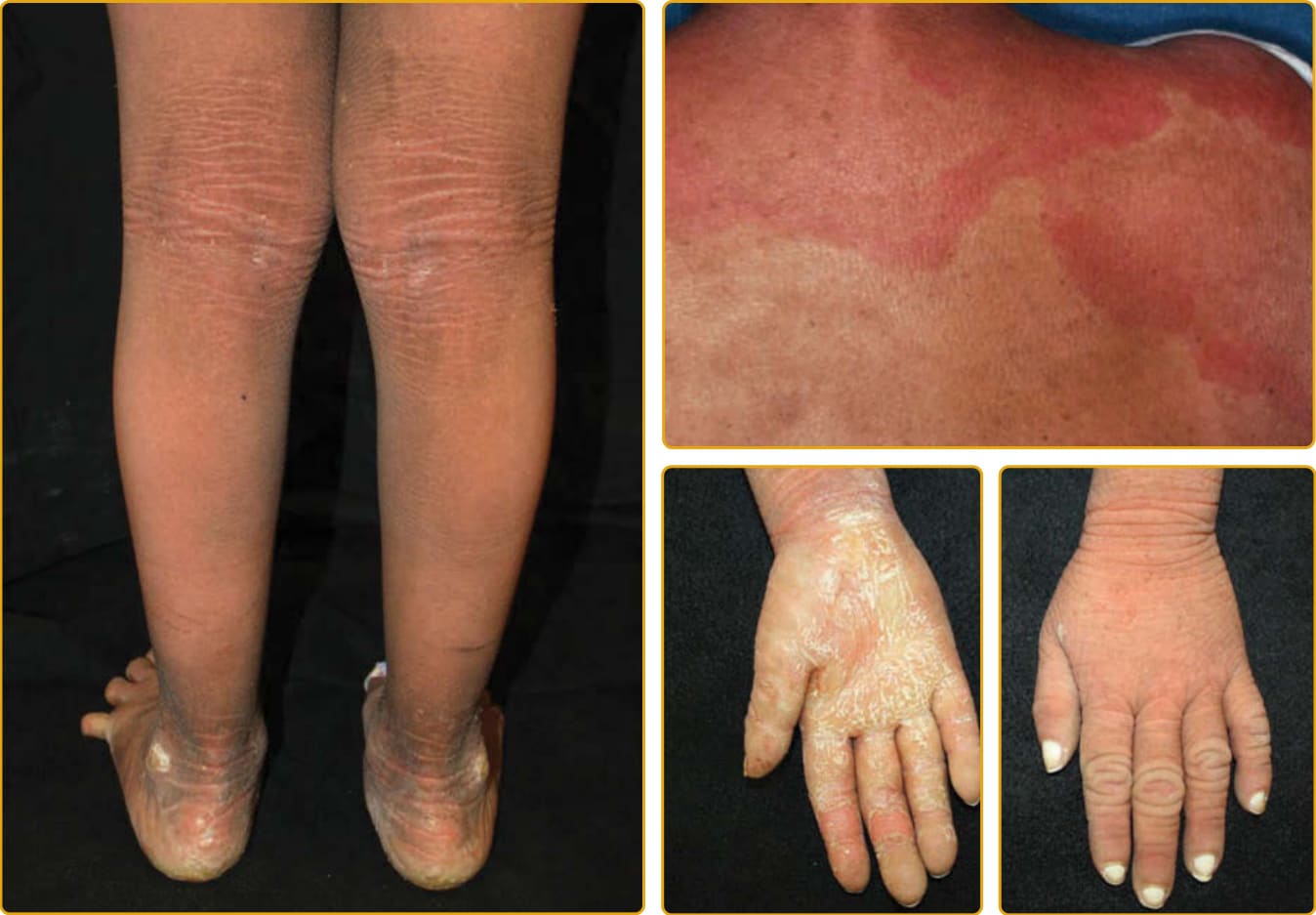
Figure 47-21 Erythrokeratoderma variabilis (GJB3; GJB4; GJA1 mutations). Corrugated coarse plaques are usually accentuated in the flexures. Palm and sole involvement may be severe. Circumscribed erythematous patches may be fixed or evanescent.
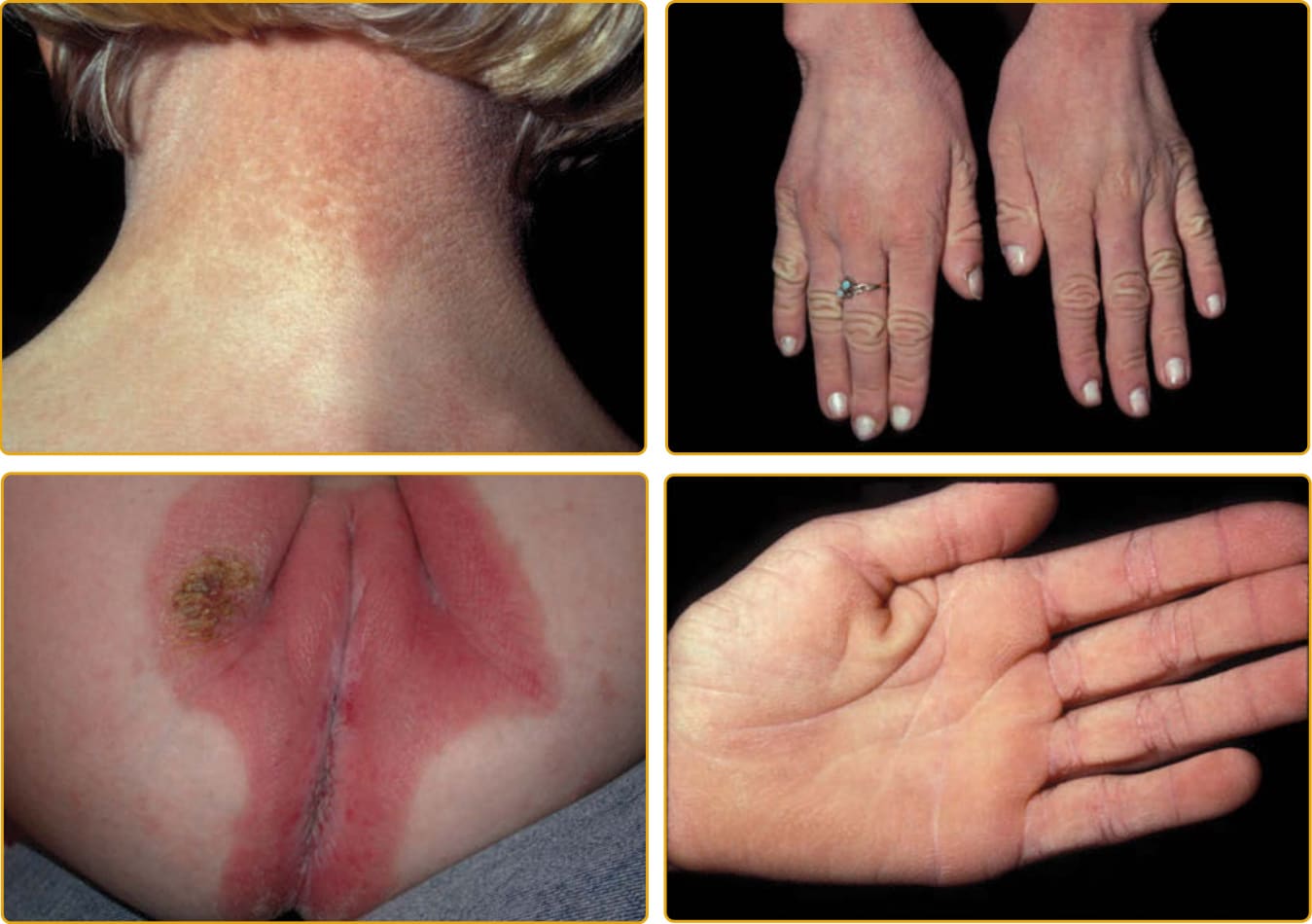
Figure 47-22 Keratitis ichthyosis deafness syndrome (GJB2 mutation). Smooth or gritty columns of hyperkeratosis give the skin a pebbly or rough appearance and are most prominent at body folds, palms, and soles and around orifices. Erythema is usually mild. Bacterial and fungal infections are common.
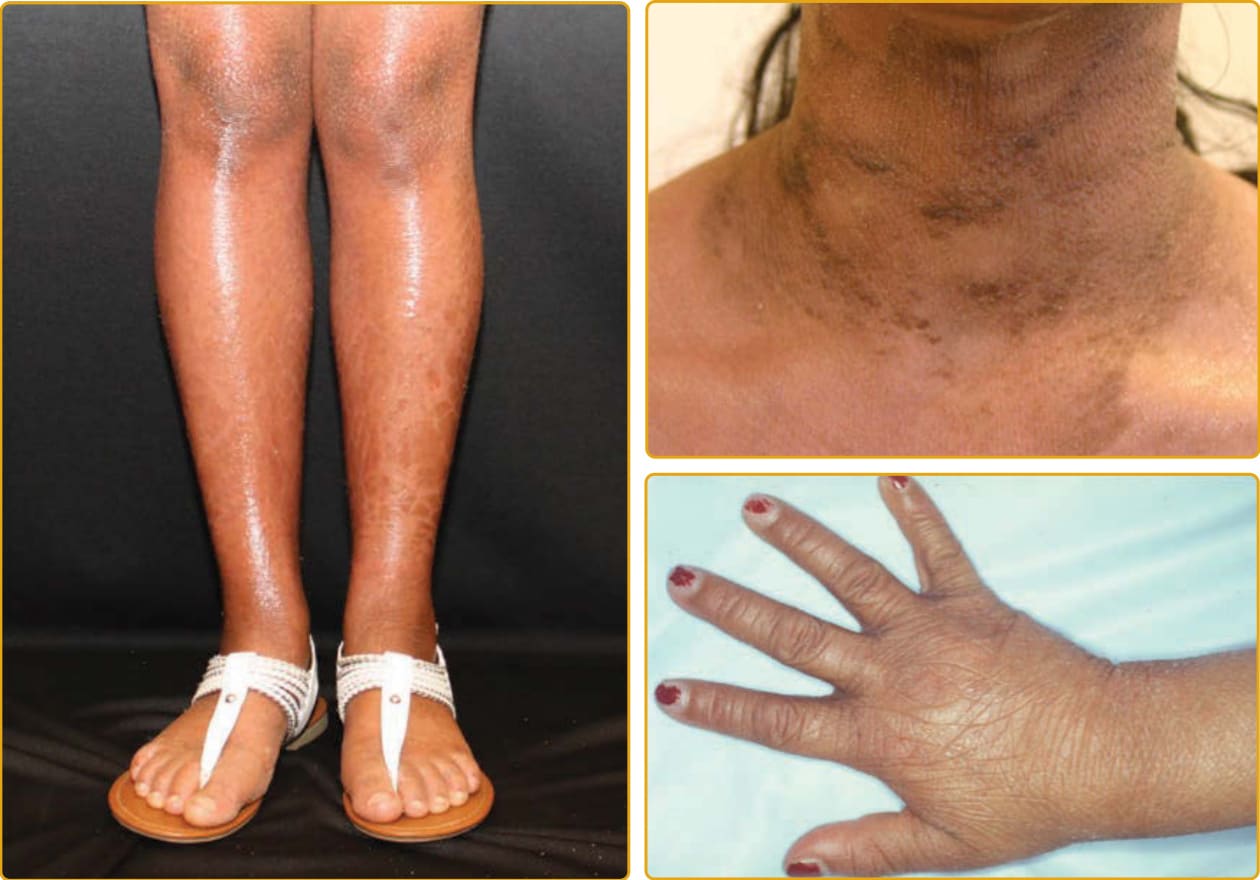
Figure 47-23 Autosomal recessive congenital ichthyosis (ABHD5 mutations, Chanarin-Dorfman disease; neutral lipid storage disease). Dark scales may vary, in the same individual, from large and flat to small and columnar. Mild to moderate erythema and exaggerated r accentuated linear markings over dorsa of hands and feet are common.
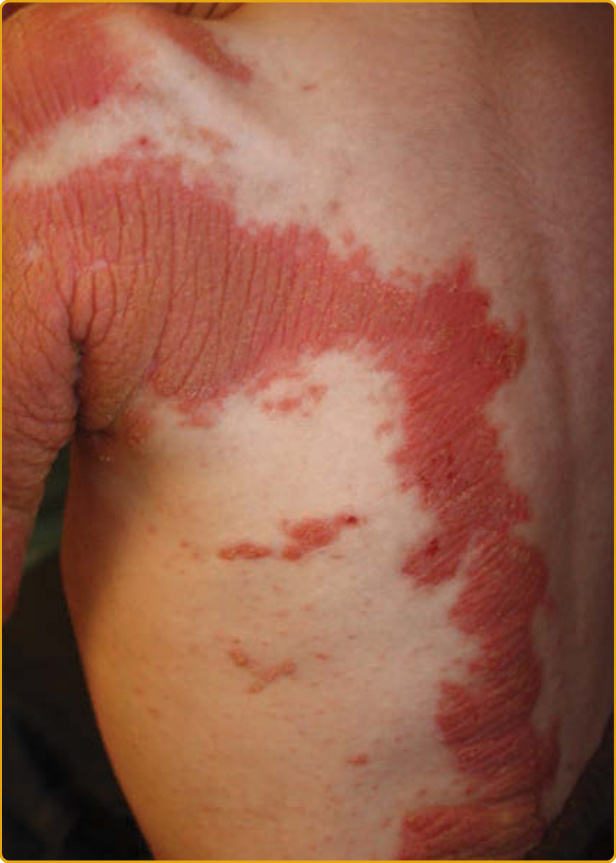
Figure 47-24 CHILD (congenital hemidysplasia, ichthyosiform erythroderma, and limb defects) syndrome. Linear stripes of erythrokeratoderma are present on the back.
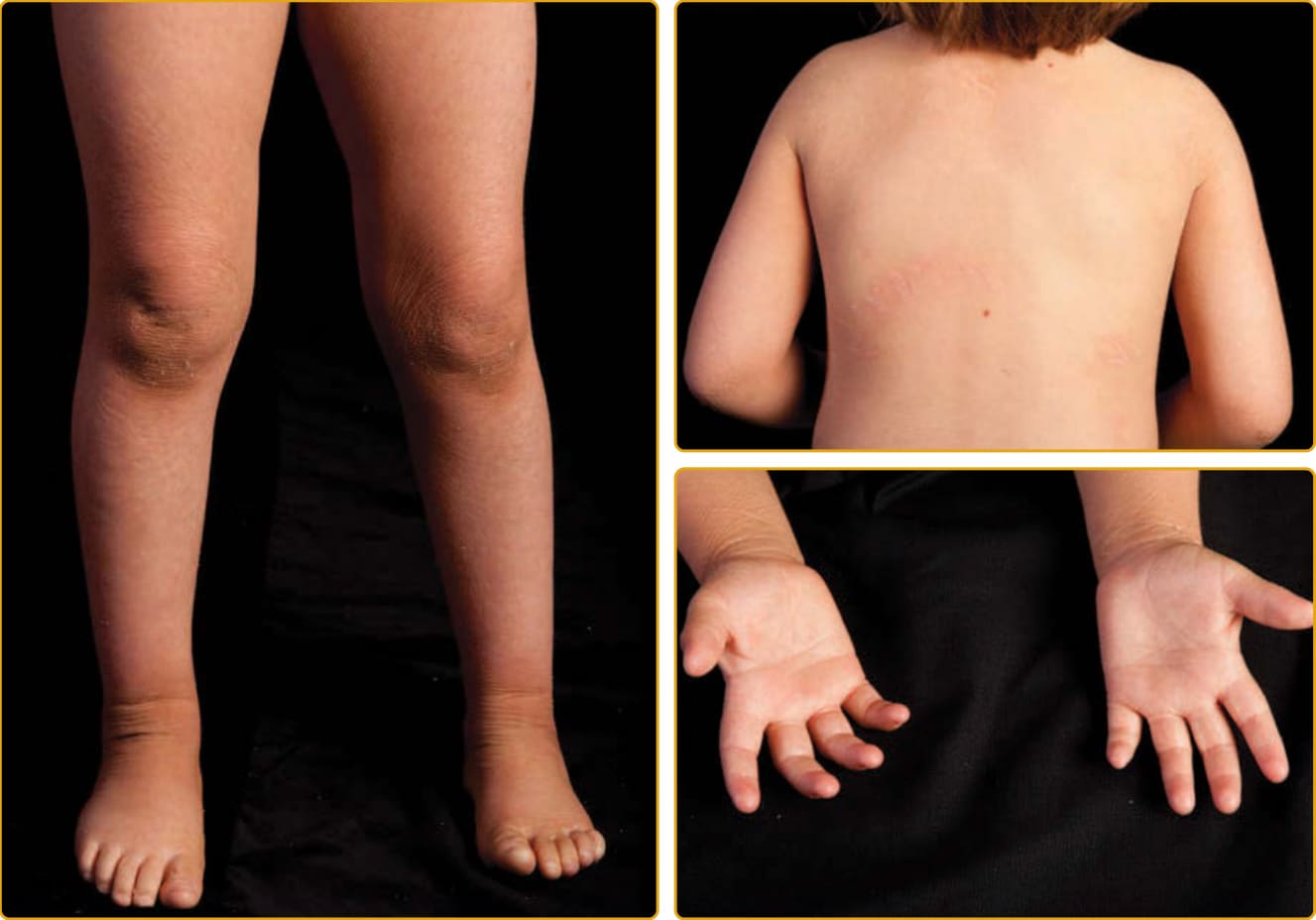
Figure 47-25 Autosomal recessive congenital ichthyosis, Sjögren-Larsson syndrome (ALDH3A2 mutations). Generalized smooth hyperkeratosis (keratoderma) results in large adherent scales on extremities and accentuated linear surface markings. There is usually little erythema, but scratching produces fine white scale and erythema.
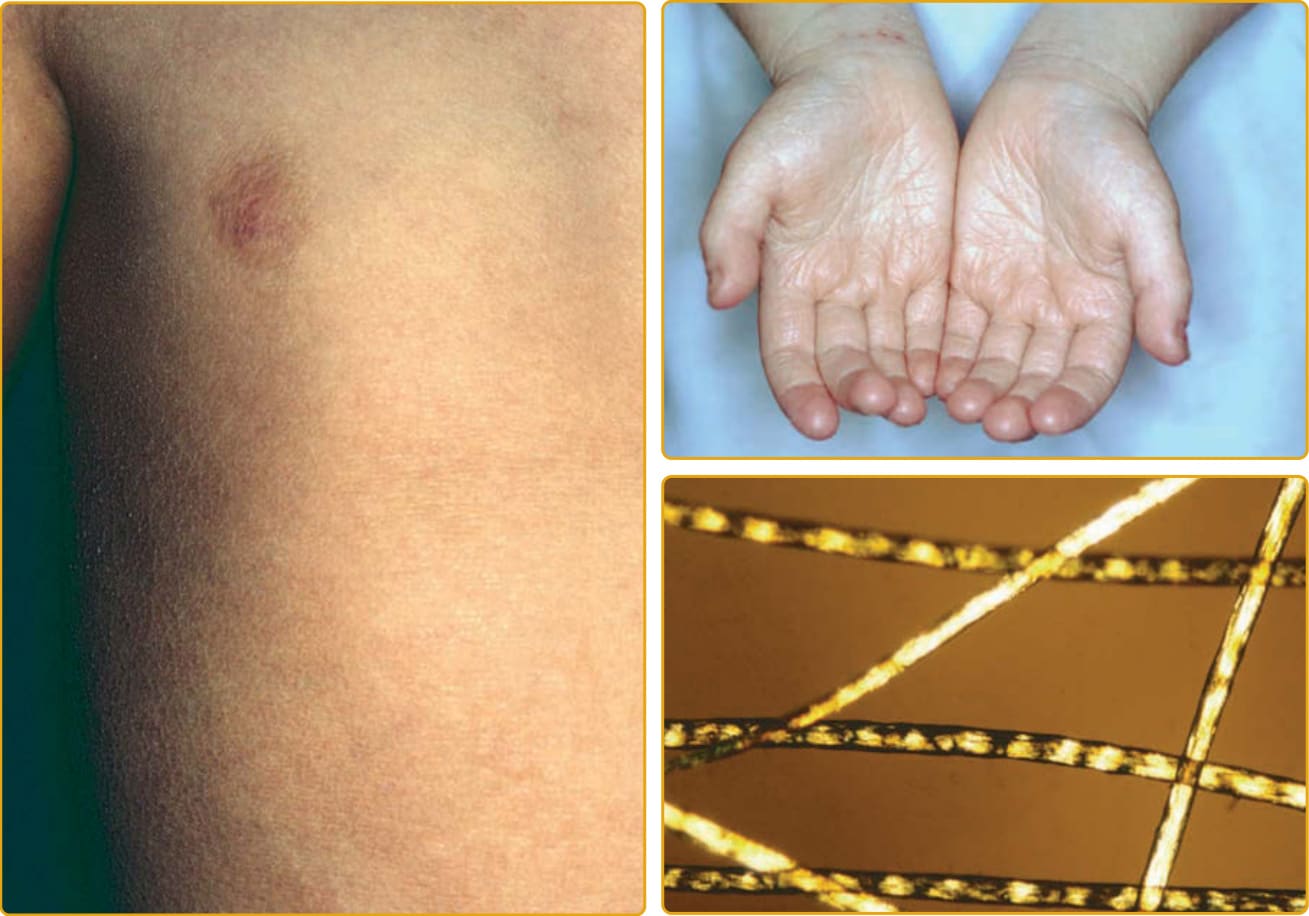
Figure 47-26 Autosomal recessive congenital ichthyosis, trichothiodystrophy (ERCC2, ERCC3, GTF2H5, GTF2E2 mutations). Beyond the newborn period, there is usually very mild generalized hyperkeratosis and minimal erythema. Hair shows characteristic alternating birefringence in polarized light.
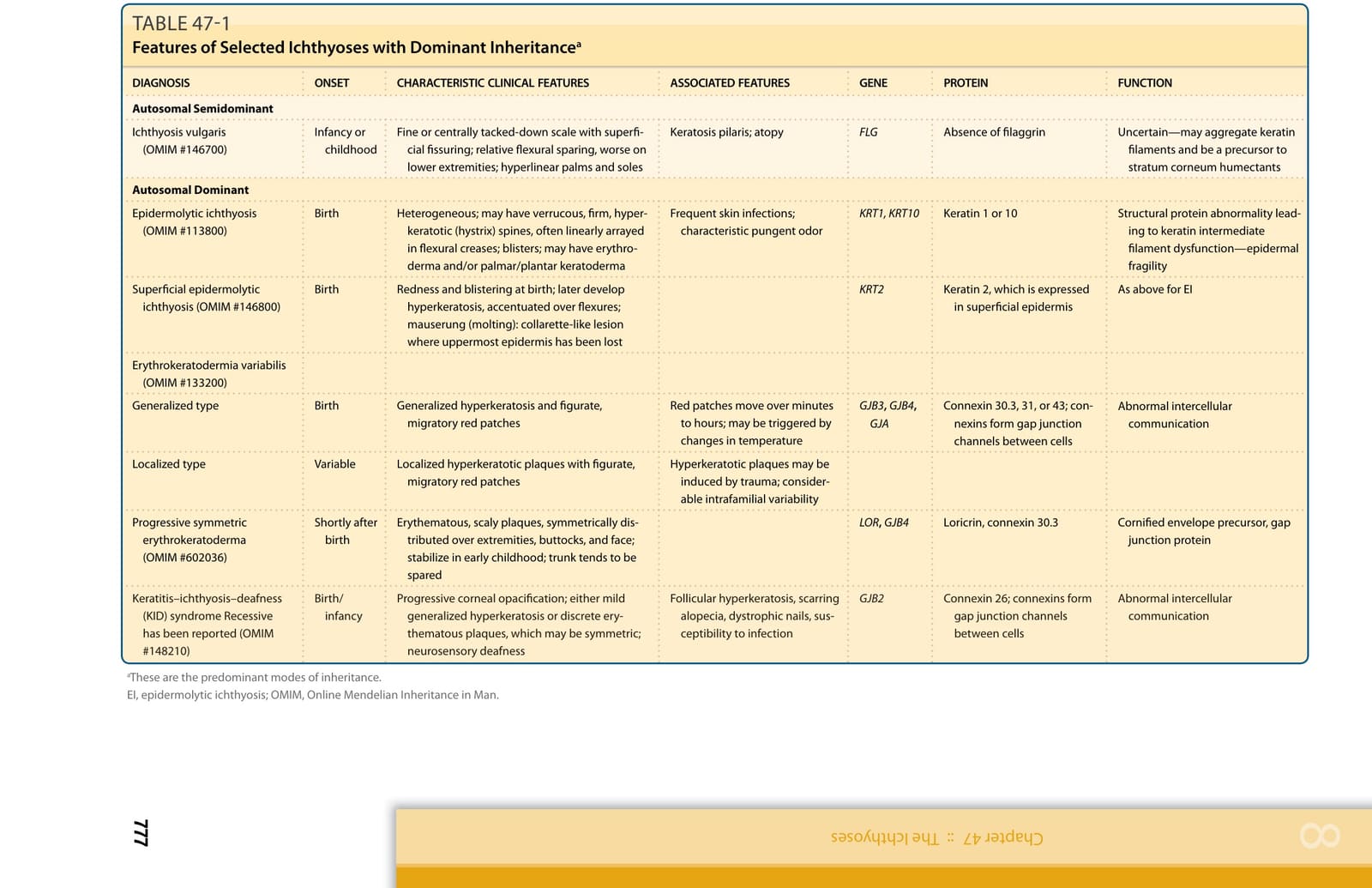
TABLE 47-1
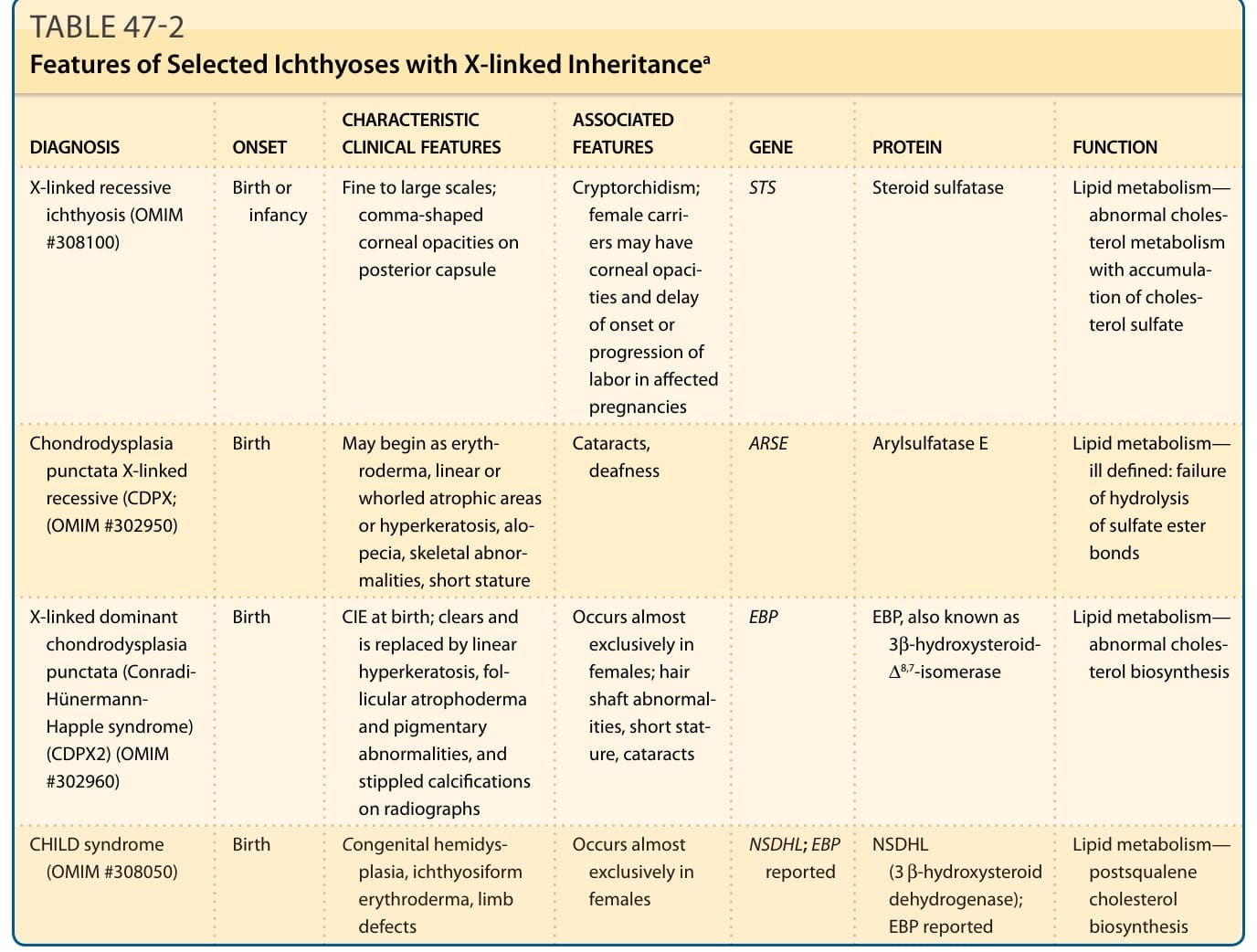
TABLE 47-2 Features of Selected Ichthyoses with X-linked Inheritancea
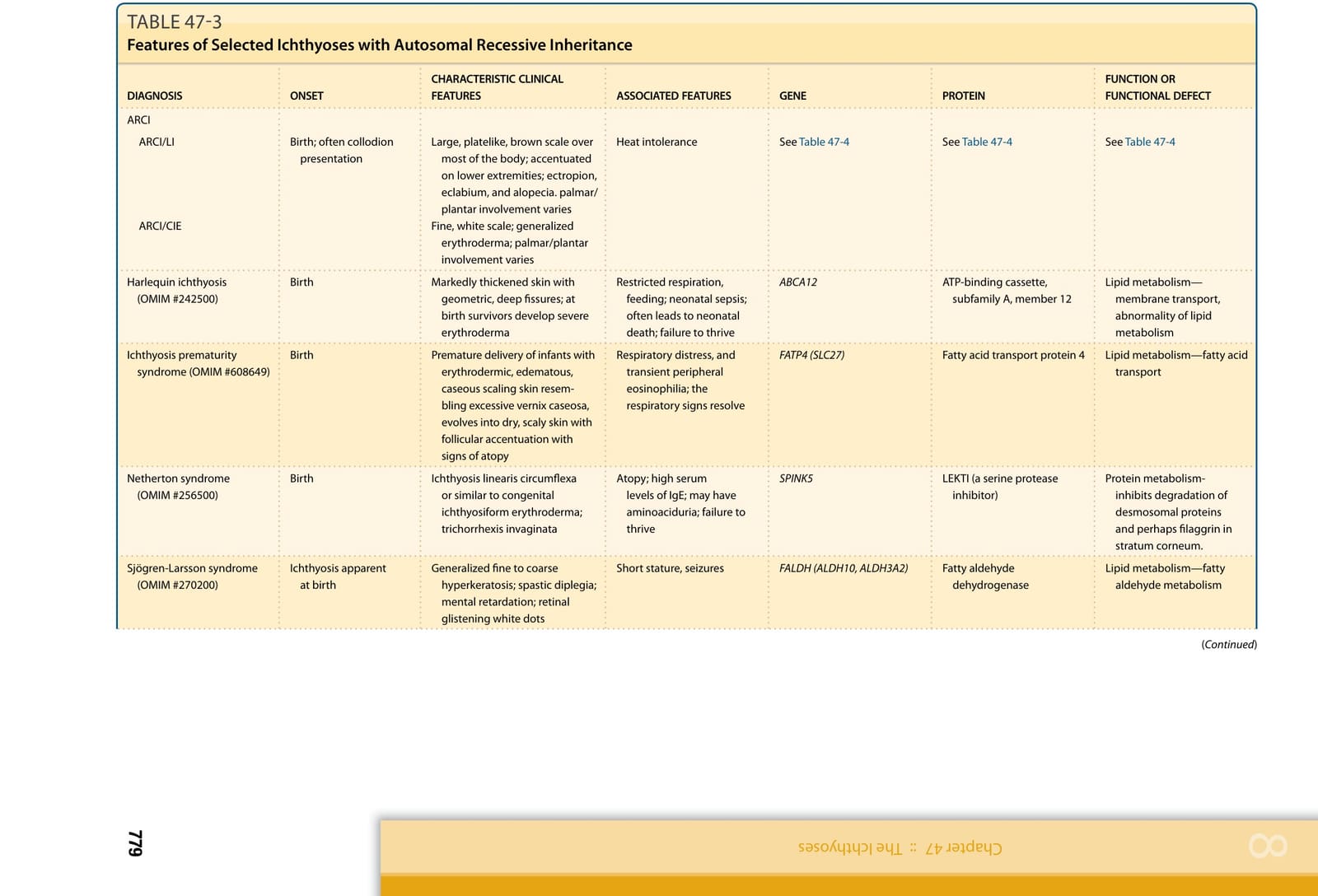
TABLE 47-3
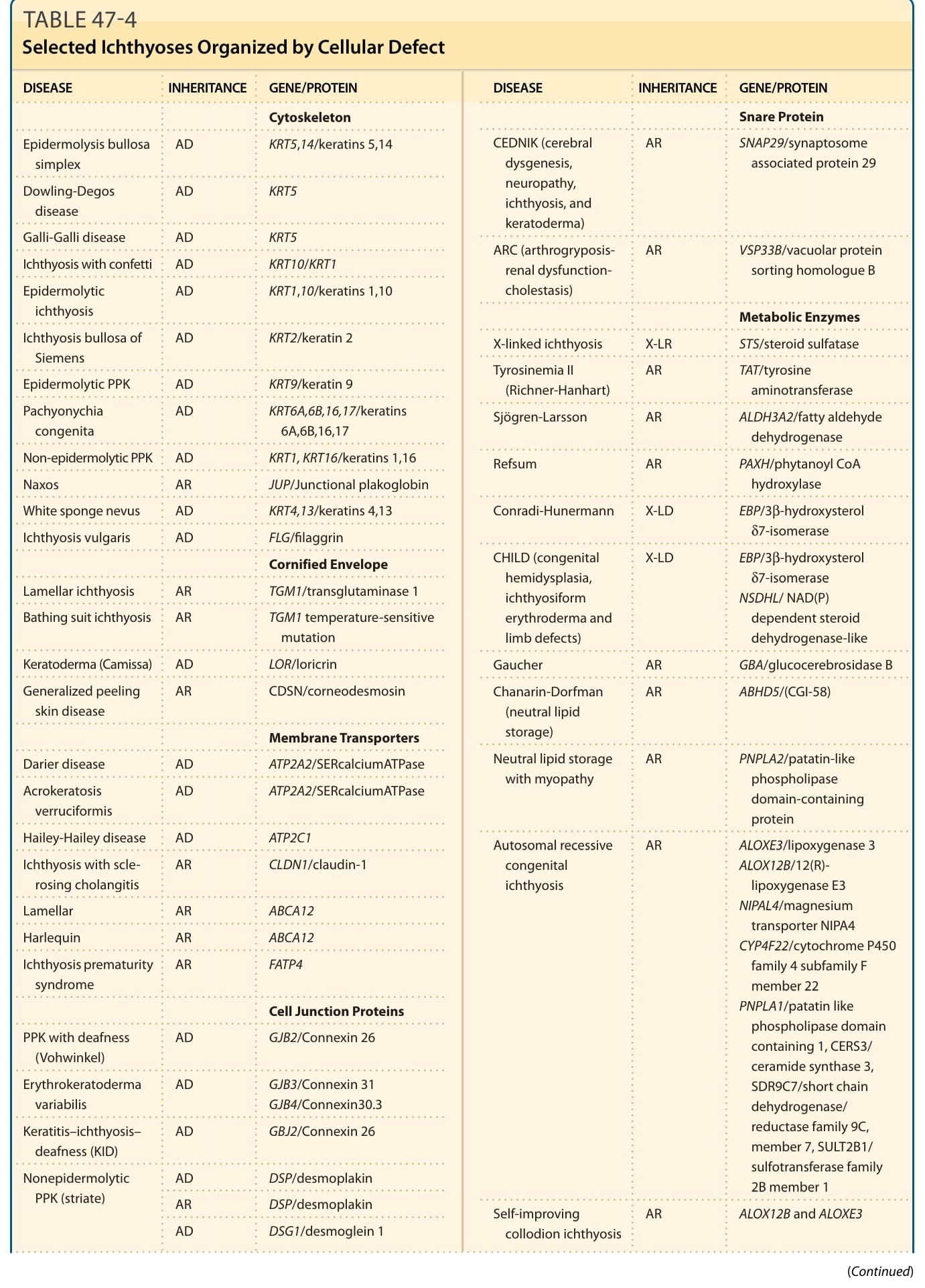
TABLE 47-4

TABLE 47-5 Disorders Associated with Collodion Membrane