嗜酸性球疾病 (Eosinophilic Diseases)
嗜酸性球之生成與活化的調控 (Regulation of the Production and Activation of Eosinophils)
基本生物學
- 嗜酸性球 (eosinophils) 為骨髓衍生細胞,正常占循環白血球最多 6%(最多 600/mm³);血中僅短暫存在 8 至 18 小時,主要為組織駐留細胞,組織內平均壽命 2 至 5 天,存活因子可延長至 14 天。
- 由 CD34+ 骨髓系前驅細胞發育而來;關鍵生長因子為 IL-3、GM-CSF 與 IL-5。IL-5 最具選擇性,且高親和力 IL-5R 表現為嗜酸性球發育的先決條件。
- 人類僅骨髓、脾、淋巴結、胸腺與胃腸道(胃至結腸、不含食道)正常駐留嗜酸性球;胃腸道為骨髓外唯一在恆定狀態即有顆粒蛋白沉積之器官。CCL11 控制嗜酸性球歸巢至胃腸道、胸腺、子宮與乳腺。
超微結構與顆粒蛋白
- 成熟嗜酸性球直徑 12 至 17 µm,具雙葉核;次級(特異性)顆粒含核心,每橫切面約 30 個。
- 五種高度鹼性顆粒蛋白:主要鹼性蛋白 (MBP)-1、MBP-2、嗜酸性球衍生神經毒素 (EDN/RNase2)、嗜酸性球陽離子蛋白 (ECP/RNase3)、嗜酸性球過氧化酶 (EPO)。
- 沉積後組織中持續時間:EPO 1 週、ECP 2 週、EDN 2.5 週、MBP-1 6 週。
- MBP 占顆粒蛋白主要部分(天竺鼠約 55%),等電點 >pH 11;EPO 按重量最豐富,約占特異性顆粒總蛋白質量的 25%,pI 10.8。
- 初級顆粒含夏科-雷登結晶蛋白(Charcot-Leyden crystal protein,即 galectin-10);夏科-雷登結晶見於氣喘痰液與蠕蟲感染/嗜酸性球性胃腸炎糞便。
免疫功能與致病角色
- 對大型無法吞噬的蠕蟲具細胞毒性(抗體/補體依賴性);ECP 致碎裂、MBP-1 致外皮膜氣球狀剝離、EDN 高濃度致皺縮。然小鼠模型中中和 IL-5 後寄生蟲感染強度不變,提示其在寄生蟲防禦角色有限。
- 透過 DNA 陷阱 (DNA trap) 釋放粒線體 DNA 參與抗菌先天免疫;可作為抗原呈現細胞,並促 Th2 極化(促進 Th1 凋亡)。
- 顆粒蛋白具血管舒張作用,促成 NERDS、Gleich 症候群、蕁麻疹、Wells 症候群與蟲咬反應中的水腫;MBP 注射致劑量依賴性風疹塊與紅暈反應,ECP、EDN 致潰瘍。
- 在塞扎里症候群 (Sézary syndrome) 中腫瘤細胞產 IL-5 致嗜酸性球增多,反映腫瘤負擔。
組織運輸與活化
- VLA-4 與內皮 VCAM-1 交互作用為關鍵;CCR3 及其配體(CCL5、CCL11、CCL13、CCL24、CCL26)為嗜酸性球選擇性趨化的重大突破。
- 活化後出現顆粒減少、空泡化與密度降低之低密度 (hypodense) 細胞,其數量預示過敏性疾病嚴重度。
- 組織中經 3 種機轉釋放顆粒:零散脫顆粒、調節性分泌與溶細胞性脫顆粒。
藥理學操控
- 非選擇性減少嗜酸性球:糖皮質素、鈣調神經磷酸酶抑制劑、IFN-α/γ、LT 拮抗劑、骨髓抑制/細胞毒性藥物等,無一具特異性。
- 糖皮質素透過隔離至淋巴組織、誘導凋亡、減少骨髓生成、改變趨化激素產生而起效;部分患者出現類固醇抗性。
- 骨髓增生性 HES(FIP1L1-PDGFRA)對酪胺酸激酶抑制劑伊馬替尼 (imatinib mesylate) 反應佳。
- 標靶療法:美泊利單抗 (mepolizumab,抗 IL-5)、貝那利單抗 (benralizumab,抗 IL-5Rα)、阿崙單抗 (alemtuzumab,抗 CD52)。
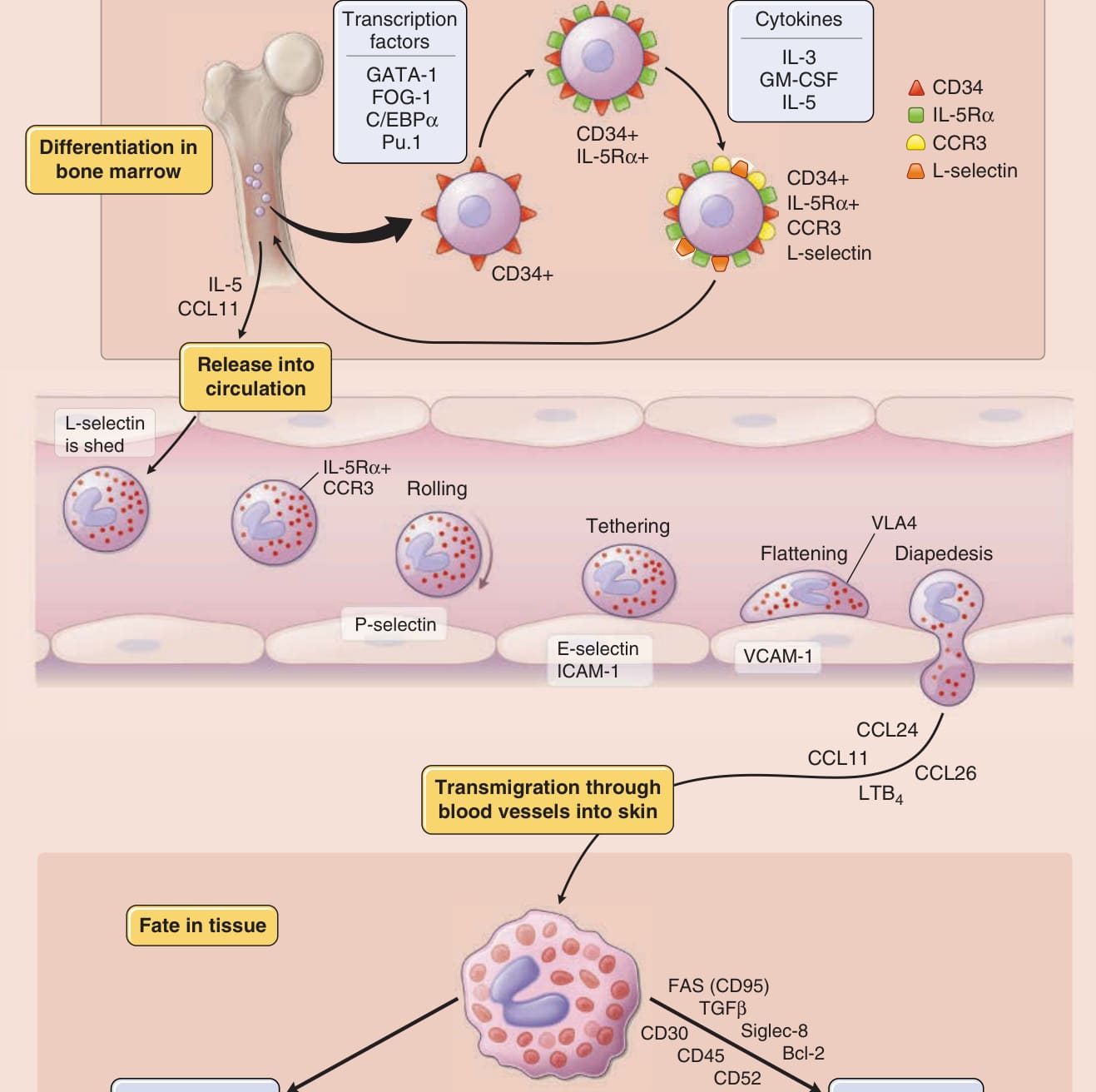
圖 40-1:嗜酸性球從未分化造血細胞到組織中命運的進程(分化、跨血管遷移與關鍵因子)。
嗜酸性球在皮膚疾病中 (Eosinophils in Cutaneous Diseases)
- 嗜酸性球可見於廣泛皮膚病切片,但對任何皮膚病皆不具病理特徵性 (pathognomonic)。
- 常見伴嗜酸性球之皮膚病:節肢動物咬傷、藥物疹、寄生蟲感染、自體免疫水疱性疾病(大疱性類天疱瘡與天疱瘡,可呈嗜酸性球性海綿狀水腫 eosinophilic spongiosis)。
- 嗜酸性球的有無或數量對鑑別診斷(如藥物反應對上急性移植物抗宿主病)價值有限;其致病影響可能與組織內細胞數量無關,因顆粒蛋白沉積可在無完整細胞下造成組織效應。

表 40-1:嗜酸性球在皮膚疾病中——以組織嗜酸性球為特徵、典型相關、由嗜酸性球定義之組織學型態,及診斷價值有限者之分類。
高嗜酸性球症候群 (Hypereosinophilic Syndromes, HES)
概述與分類
- 由標準定義的一系列實體;皮膚病灶常見並可能為表現徵象。
- 兩大亞型:淋巴球性 HES(產 IL-5 的 T 細胞克隆,性別分布相等,可達 25% 患者)與骨髓增生性 HES(>90% 為男性,與第 4q12 染色體缺失產生 FIP1L1-PDGFRA 酪胺酸激酶融合基因相關,又稱慢性嗜酸性球白血病)。
- 嗜酸性球性食道炎為器官局限性重疊型 HES,盛行率兒童高達 1:2500、成人高達 1:4000。
臨床特徵
- 搔癢性紅斑性斑疹、丘疹、斑塊、風疹塊或結節見於 >50% 患者;CD3⁻ CD4⁺ 淋巴球性 HES 皮膚表現盛行率高達 94%。
- 蕁麻疹與血管性水腫見於所有亞型;可見離心性環狀紅斑、大疱性類天疱瘡、網狀青斑、Wells 症候群等。
- 骨髓增生性 HES:發燒、體重減輕、疲倦、皮膚病灶、肝脾腫大,並可見口咽或肛門生殖器黏膜潰瘍。
- 心臟疾病常見:嗜酸性球黏附心內膜致血栓形成與心內膜下纖維化、限制型心肌病變;指甲下出血/甲褶梗塞可預示血栓栓塞。
病因與致病機轉
- 嗜酸性球為所有亞型大多數終末器官損傷之原因。淋巴球性 HES 之 T 細胞克隆表型異常(CD3⁺CD4⁻CD8⁻、CD3⁻CD4⁺),分泌高量 IL-5。
- FIP1L1-PDGFRA 突變者血清胰蛋白酶升高、骨髓非典型紡錘狀肥大細胞增加,符合肥大細胞增生症標準;PDGFRB 與 FGFR1 異常者具進展為侵襲性骨髓惡性腫瘤潛力。
診斷
- 修訂標準:至少 2 次測定血液嗜酸性球 >1500 eosinophils/mm³(或顯著組織嗜酸性球增多伴症狀與明顯血液增多),並排除繼發性原因(寄生蟲/病毒感染、過敏、藥物、腎上腺功能低下、腫瘤)。
- Chic2 螢光原位雜交或 PCR 偵測 FIP1L1-PDGFRA(陽性者對伊馬替尼有反應);應做心臟超音波(因可發生嗜酸性球性心內膜心肌疾病)、肝酵素、IgE、B12、胰蛋白酶與 T 細胞克隆性檢測。
鑑別診斷
- 寄生蟲感染(蠕蟲病常見總 IgE >500 IU/mL)、蕁麻疹、遺傳性血管性水腫、異位性/接觸性皮膚炎、藥物反應、蕈狀肉芽腫、塞扎里症候群;口腔生殖器潰瘍須鑑別貝賽特症候群、克隆氏病、潰瘍性結腸炎、賴特症候群、梅毒等。
病程、預後與處置
- 伴黏膜病灶之骨髓增生性 HES 預示侵襲性病程,未治療者多在表現後 2 年內死亡;淋巴球性 HES 一般良性,但可能演變為淋巴瘤(尤 CD3⁻CD4⁺)。
- 整體 5 年存活率 80%;主要死因為限制型心肌病變所致充血性心衰竭,其次為敗血症。
- 治療目標:緩解症狀、改善器官功能,將周邊嗜酸性球維持於 1000 至 2000/mm³。
- 排除糞小桿線蟲 (Strongyloides) 感染後,無基因突變者第一線為皮質類固醇(約 70% 反應)。
- FIP1L1-PDGFRA 陽性者給予伊馬替尼甲磺酸鹽(足以根除 FIP1L1-PDGFRA 之劑量為 100-400 mg/day),心臟侵犯者合併糖皮質素;治療前後須監測肌鈣蛋白。未定義型/複雜型可用全身性糖皮質素 0.5-1 mg/kg/day。
- 其他:IFN-α、體外光分離置換術、羥基脲、氨苯碸、英夫利昔單抗、阿崙單抗、骨髓移植,及抗 IL-5 單株抗體(美泊利單抗、瑞利珠單抗)。
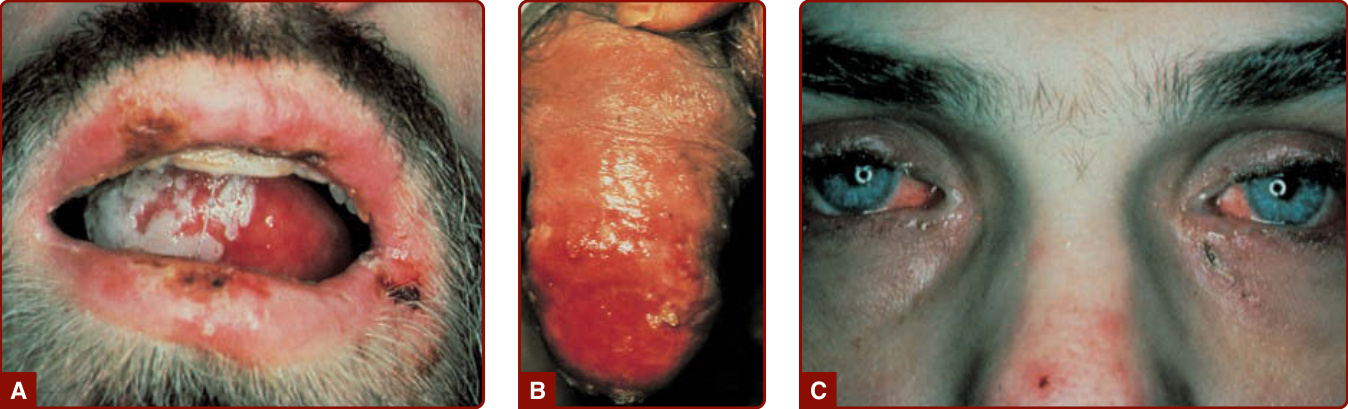
圖 40-3:高嗜酸性球症候群。口腔 (A) 與龜頭 (B) 的黏膜糜爛與潰瘍;結膜刺激 (C)。
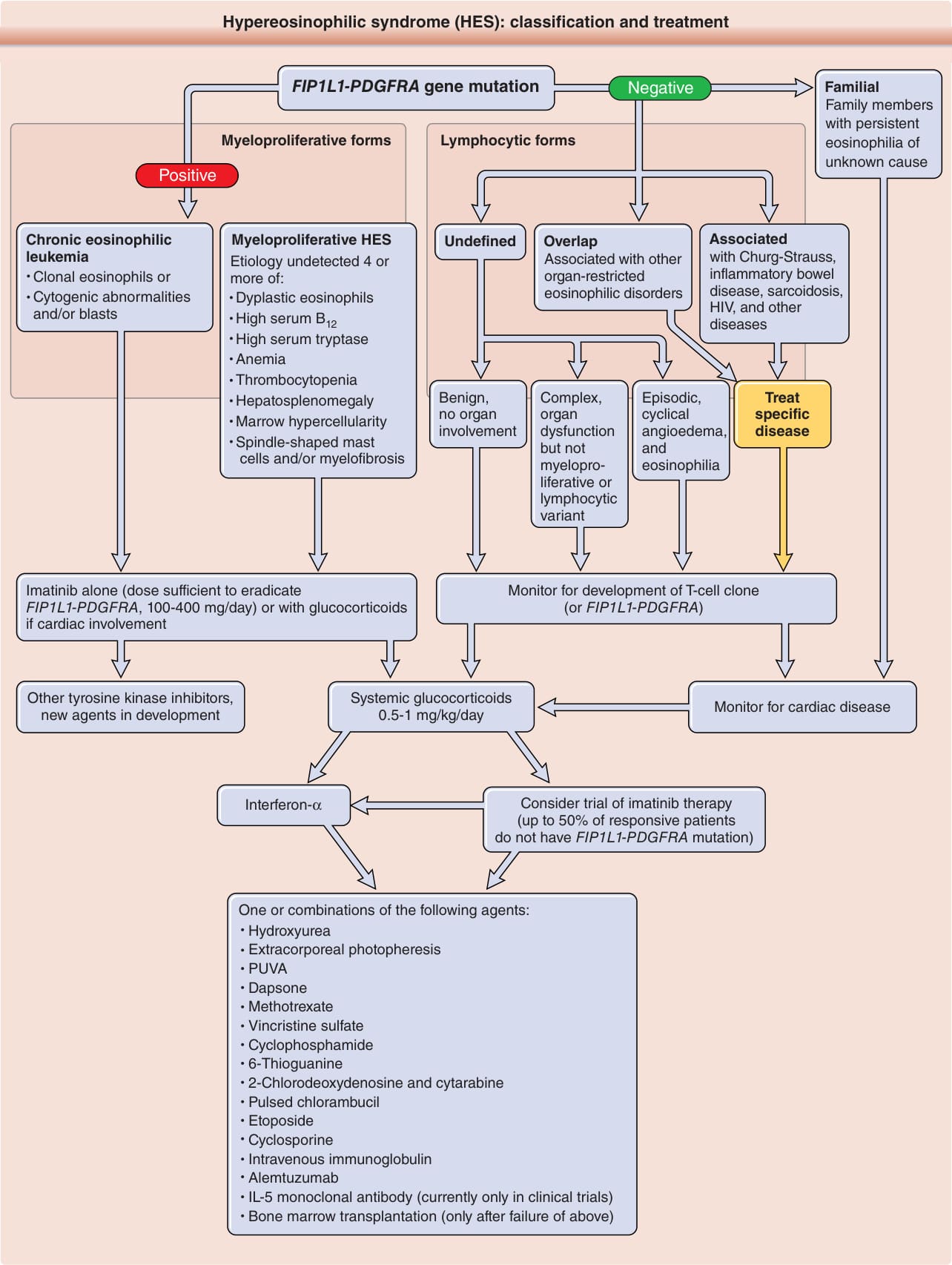
圖 40-4:高嗜酸性球症候群:分類與治療。
Wells 症候群 (Wells Syndrome,嗜酸性球性蜂窩組織炎)
臨床特徵
- 前驅性灼熱或搔癢後,以紅斑與水腫起始,數天內演變為帶紫紅色邊緣的大型水腫性斑塊;可發展水疱。病灶逐漸由鮮紅變棕紅、再變藍灰/綠灰色,類似硬斑病 (morphea)。
- 病灶單發或多發,典型侵犯四肢、較少軀幹;最常見全身症狀為不適,少數發燒。多次復發。
病因與診斷
- 病因不明,視為對外源/內源刺激的非特異性過敏反應;觸發因子包括蟲咬、感染、藥物與疫苗。
- 約 50% 患者有周邊血液嗜酸性球增多;組織學特徵為瀰漫性真皮浸潤伴嗜酸性球、組織球與火焰狀圖像 (flame figures)(後期組織球柵欄狀圍繞),通常無血管炎。火焰狀圖像為特徵性但非診斷性(亦見於其他疾病),免疫螢光顯示 MBP 廣泛細胞外染色。
鑑別診斷與處置
- 早期須鑑別蕁麻疹、丹毒 (erysipelas) 與急性蜂窩組織炎;後期類似硬斑病;有水疱時提示類天疱瘡。
- 通常數週至數月內無瘢痕消退但常復發。全身性糖皮質素通常戲劇性改善,多數患者 1 個月內逐漸減量耐受良好;復發或持續性病例可用低劑量 prednisone 5 mg 隔日維持。其他選項:美諾四環素、氨苯碸、灰黴素、抗組織胺、環孢素與 IFN-α;輕度疾病可用局部糖皮質素。
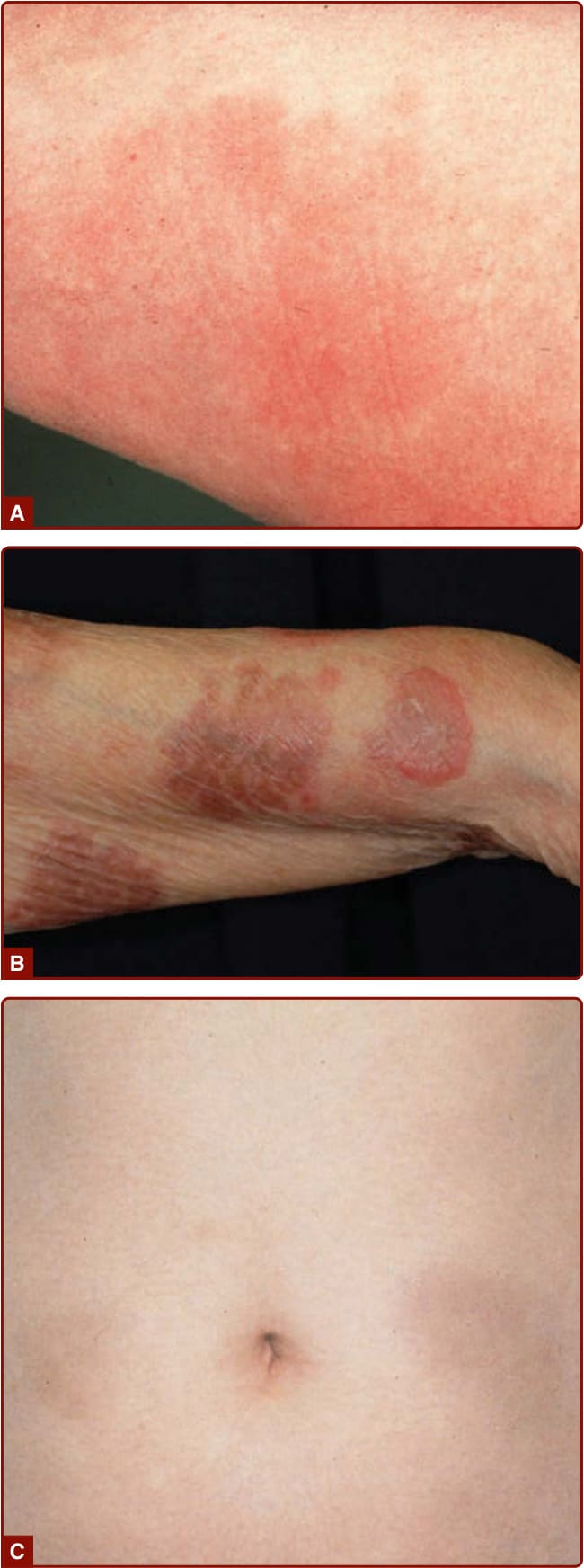
圖 40-5:Wells 症候群。A,早期紅斑與水腫病灶;B,弧形斑塊;C,類似硬斑病的後期病灶。
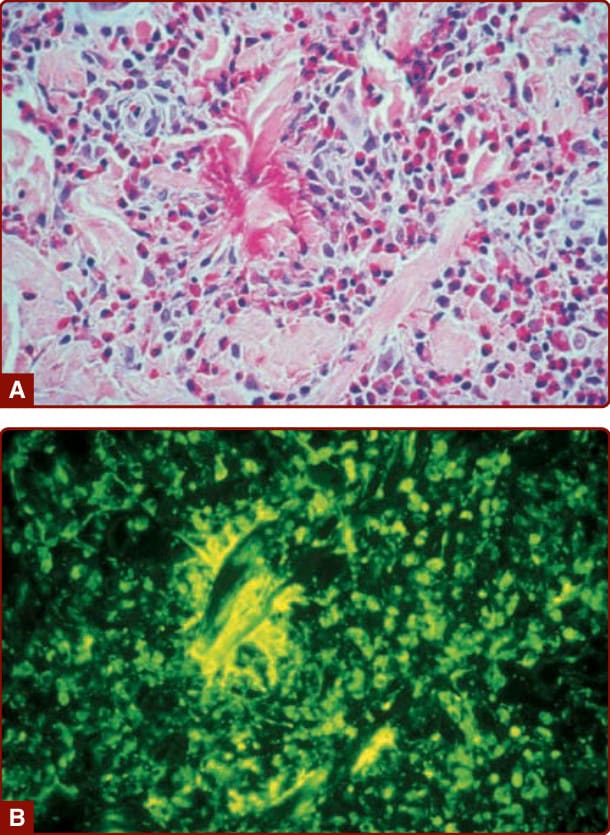
圖 40-7:火焰狀圖像。A,H&E 染色;B,MBP 免疫染色顯示顆粒蛋白沉積定位於火焰狀圖像。
嗜酸性球增多性血管淋巴樣增生 (ALHE,上皮樣血管瘤) 與木村病 (Kimura Disease, KD)
特徵與比較
- ALHE 發生於所有種族、女性為主,多介於 30 至 50 歲;KD 主要發生於亞洲男性、年齡較輕。兩者皆以頭頸部復發性真皮/皮下病灶與周邊血液嗜酸性球增多為特徵。
- ALHE 病灶較小、表淺、數量多,可能疼痛/搔癢/搏動;KD 病灶較大、侵犯皮下組織、區域淋巴結與唾液腺,一般無症狀。
- IgE 升高與腎臟疾病(10% 至 20%)僅見於 KD。
- 組織學:ALHE 以血管增生伴大型上皮樣/組織球樣內皮細胞為主、纖維化有限;KD 以淋巴樣增生伴生發中心、顯著纖維化為主,可見嗜酸性球膿瘍。
診斷與處置
- ALHE 約 20% 有周邊嗜酸性球增多、IgE 正常、無腎病變;基質典型為黏液樣 (myxoid)。鑑別含 KD、化膿性肉芽腫、上皮樣血管內皮瘤/血管肉瘤、卡波西氏肉瘤。
- ALHE 病程慢性不緩解;單發或少數小病灶可切除或莫氏手術(術部可復發),其他:全身性/病灶內糖皮質素、IFN-α、冷凍療法、雷射、局部他克莫司。
- KD 首選手術切除(可復發),其他:全身性糖皮質素、環孢素、放射治療;組織中 PDGF-α 與 c-kit 提示伊馬替尼可能有效。
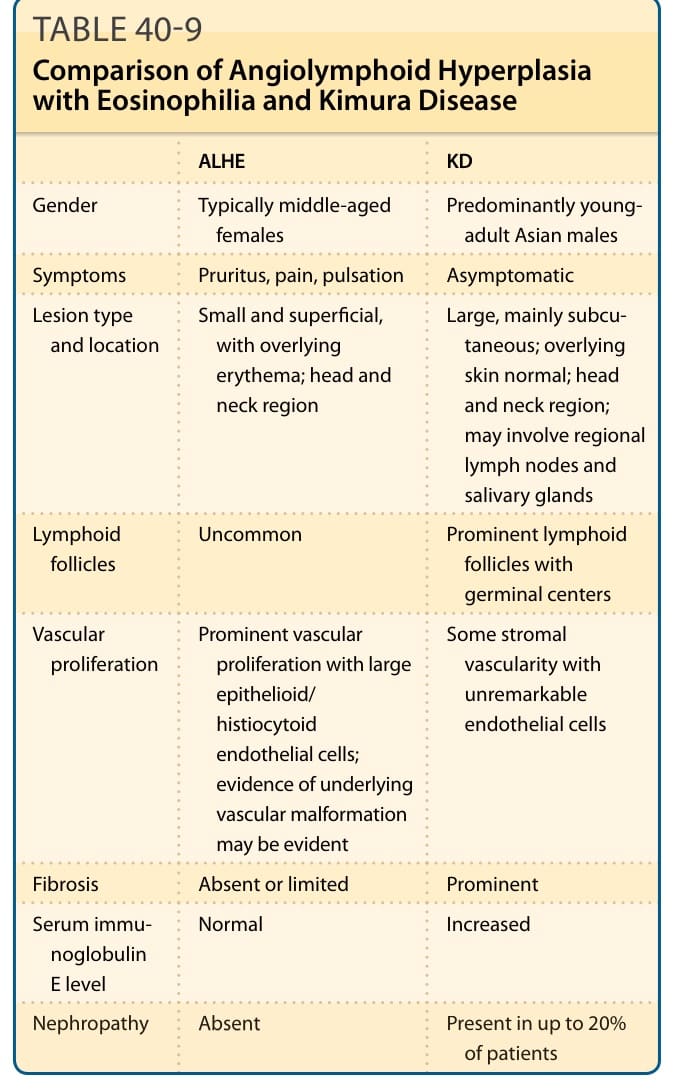
表 40-9:ALHE 與木村病的比較(性別、症狀、病灶位置、淋巴濾泡、血管增生、纖維化、IgE 與腎病變)。
嗜酸性球性膿疱性毛囊炎 (Eosinophilic Pustular Folliculitis, EPF)
- 三種變異型:經典型(Ofuji 病)——日本患者慢性復發性毛囊性膿疱、傾向環狀斑塊、脂漏性分布、約 20% 有掌蹠侵犯;免疫抑制相關型——最常見於 HIV 感染者,臉部與上軀幹極度搔癢丘疹;嬰兒/新生兒型——頭皮毛囊性膿疱。
- 病因不明;部分 HIV 相關型在開始 HAART 後出現,提示為免疫重建而非缺陷之結果。
- 診斷:應評估潛在免疫缺陷(尤 HIV);周邊血液嗜酸性球增多為所有型別組成成分。HIV 陽性者常伴淋巴球減少、低 CD4 與高 IgE。組織學以嗜酸性球浸潤毛囊與毛囊周圍為特徵。
- 鑑別:細菌/黴菌性毛囊炎、脂漏性皮膚炎、掌蹠膿疱性乾癬、痤瘡、新生兒毒性紅斑、毛囊性黏蛋白沉積症;最重要者為皮膚 T 細胞淋巴瘤。
- 處置:第一線為局部糖皮質素與局部鈣調神經磷酸酶抑制劑(局部他克莫司利於臉部病灶);非類固醇抗發炎藥(尤吲哚美辛 indomethacin)亦為第一線。其他:紫外線光療、全身性類視黃醇、環孢素、伊曲康唑、美諾四環素、氨苯碸、IFNs;HIV 相關型隨抗反轉錄病毒治療改善。嬰兒型預後良好,傾向數年內自發消退。
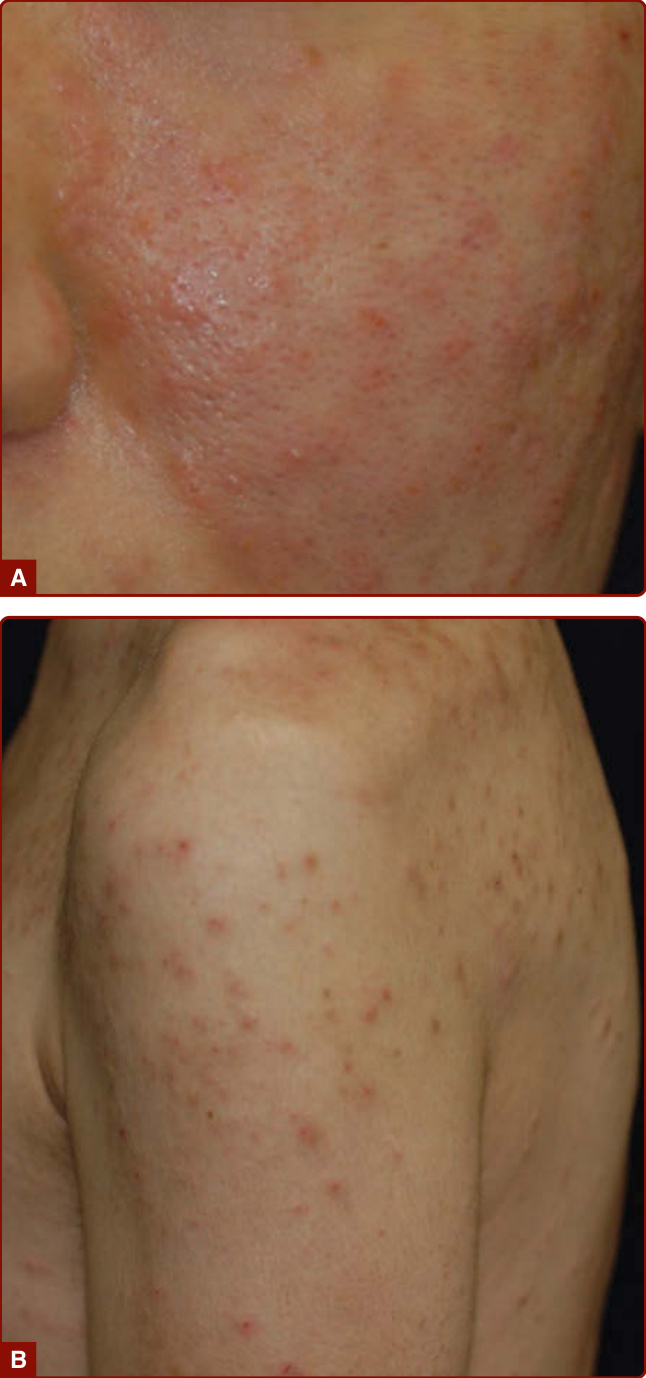
圖 40-11:嗜酸性球性膿疱性毛囊炎。A,臉頰搔癢紅色丘疹與膿疱;B,HIV 相關病例,上臂、肩、背搔癢紅色丘疹。
Ofuji 丘疹性紅皮症 (Papuloerythroderma of Ofuji)
- 好發老年男性(70 至 90 歲),病灶通常搔癢;由平頂、紅至棕色、具鋪石狀外觀的融合丘疹形成紅皮症樣疹,影響四肢與軀幹、臉部與屈側通常倖免。
- 特徵性腹部皮膚皺褶倖免稱躺椅徵 (deck-chair sign);黏膜、毛髮、指甲總是倖免。
- 約 20% 與血液性(非何杰金淋巴瘤、白血病)或內臟惡性腫瘤(胃癌、結腸癌、前列腺癌)相關;可能進展為皮膚 T 細胞淋巴瘤。
- 診斷:超過 80% 偵測到周邊嗜酸性球增多;組織學為非特異性海綿狀皮膚炎樣型態。
- 處置:口服 prednisolone 多有效;紫外線治療(單獨或合併口服與局部皮質類固醇)非常有效;依曲替酯、環孢素、IFN 亦曾報告有效,但常對治療難治。
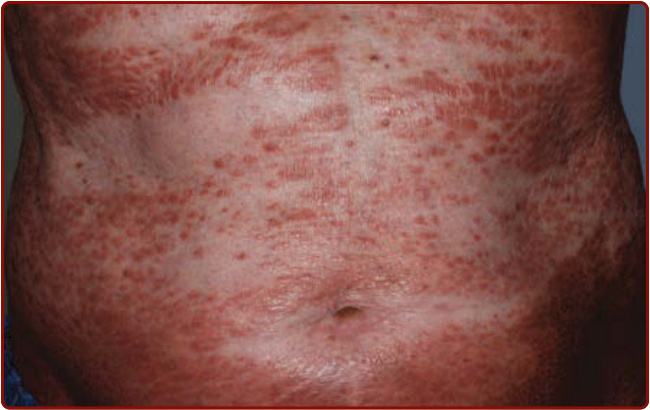
圖 40-12:Ofuji 丘疹性紅皮症。腹部鋪石狀平頂紅至棕色丘疹,可見皮膚皺褶倖免(躺椅徵)。
顏面肉芽腫 (Granuloma Faciale)
- 不常見的發炎性皮膚病,主要侵犯臉部的紅棕色丘疹與斑塊;好發成年男性(平均 52 歲)、白人較常見。
- 臨床:單發無症狀紅/棕/紫紅色斑塊,常顯示毛囊強化與毛細血管擴張,顯著毛囊開口呈「橘皮樣 (peau d’orange)」;好發鼻、耳前、臉頰、前額、眼瞼與耳朵;偶有觸痛、灼熱或搔癢。
- 病因不明,視為局部慢性纖維化血管炎;免疫螢光示血管壁免疫球蛋白與補體沉積(第 III 型免疫反應)。
- 診斷:建議含真皮全層之打孔切片;組織學示外觀正常表皮、與浸潤間隔一狹窄無浸潤帶 (grenz zone),真皮緻密瀰漫浸潤淋巴細胞、漿細胞、嗜酸性球與嗜中性球並有白血球破碎;後期血管周圍纖維蛋白沉積廣泛、含鐵血黃素致棕色。
- 鑑別:主要為持久性隆起性紅斑 (erythema elevatum diutinum, EED)(嗜酸性球與漿細胞在顏面肉芽腫更突出、嗜中性球在 EED 更常見、無浸潤帶在 EED 不典型);亦含圓盤狀紅斑性狼瘡、類肉瘤病、Jessner 浸潤、ALHE 等。
- 病程慢性、罕自發消退、常對治療有抗性並易復發。處置(表 40-13):局部與病灶內類固醇(中度改善)、冷凍手術;因屬慢性白血球破碎性血管炎變異型,氨苯碸 25 至 100 mg/day 多有益;局部他克莫司軟膏 0.1% 亦成功;手術切除適小病灶;585 至 595 nm 脈衝染料雷射可改善。
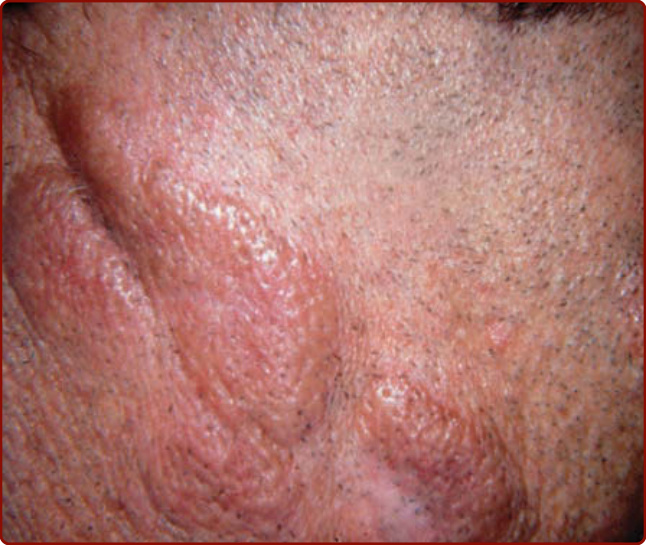
圖 40-13:顏面肉芽腫。臉頰隆起水腫性斑塊,顯著毛囊孔。
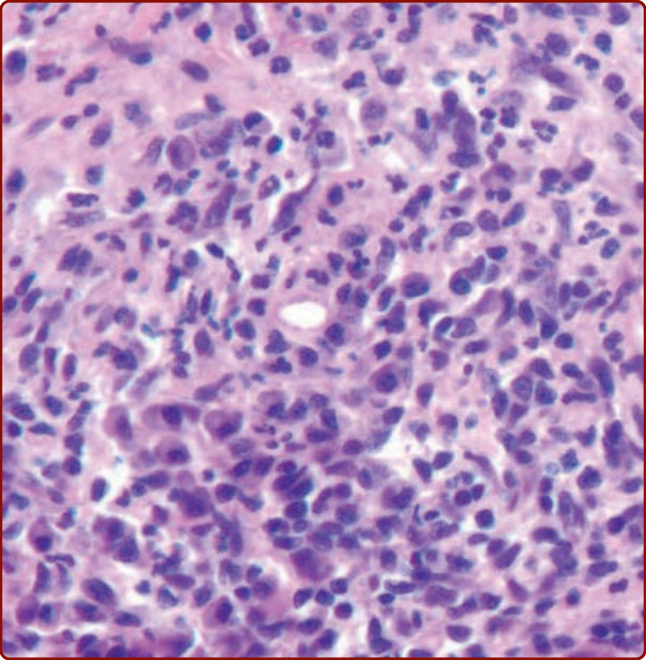
圖 40-16:顏面肉芽腫。血管周圍纖維蛋白沉積與淋巴細胞、嗜中性球、嗜酸性球混合性浸潤。
有嗜酸性球參與的其他臨床反應型態
- 嗜酸性球常於破裂後失去形態完整性而組織學上無法辨識,但顆粒蛋白沉積持續並造成組織效應。反應型態含水腫、慢性皮膚炎/搔癢(異位性皮膚炎、結節性癢疹)、藥物反應、水疱(類天疱瘡)、纖維化與血管炎。
嗜酸性球性筋膜炎 (Eosinophilic Fasciitis,Shulman 症候群)
- 以四肢疼痛、紅斑、水腫與硬結,伴周邊血液嗜酸性球增多與高伽瑪球蛋白血症表現;可發展攣縮與皮膚波紋。溝槽徵 (groove sign) 為特徵(淺表靜脈走向凹陷,患肢抬高更明顯);筋膜增厚並有淋巴細胞、漿細胞、肥大細胞與嗜酸性球浸潤。
嗜酸性球增多-肌痛症候群 (Eosinophilia-Myalgia Syndrome)
- 歷史上與攝入某些批次 L-色胺酸 (L-tryptophan) 相關;以明顯周邊嗜酸性球增多、失能性全身肌痛、肺炎、心肌炎、神經病變、腦病變與纖維化為特徵。皮膚異常含水腫、搔癢、淡紅斑疹、毛髮脫落與橘皮樣/硬斑病樣病灶;肌束膜與筋膜有顯著發炎浸潤及顆粒蛋白沉積。