壞疽性膿皮症 (Pyoderma Gangrenosum) 精華筆記
定義與分類
- 壞疽性膿皮症 (pyoderma gangrenosum, PG) 是一種不常見、慢性的嗜中性球發炎性皮膚病況。典型潰瘍型初期表現為疼痛的結節、斑塊或膿疱,逐漸擴大崩解,形成進行性擴大的潰瘍,邊緣隆起、潛掘 (undermined)、呈紫紅色 (violaceous),並圍繞一圈紅斑帶;癒合時發展出篩狀 (cribriform) 外觀。
- 因疼痛激惹現象 (pathergic phenomenon),外傷或刺激可誘發 PG 惡化,傾向發生與復發於外傷部位(高達 50% 病例)。
- 四種型態學型別:(a) 潰瘍型 (ulcerative,最常見且最早被描述)、(b) 大疱型 (bullous,又稱非典型 PG)、(c) 膿疱型 (pustular)、(d) 增殖型 (vegetative,又稱淺表肉芽腫性膿皮症 superficial granulomatous pyoderma)。通常一種型別主導臨床表現。
流行病學與疾病關聯
- 估計每年每百萬人影響高達 10 例,占慢性腿部潰瘍高達 3%;多數發生於第二至第六個十年,可能女性偏多。
- 高達 70% 病例與潛在全身性疾病相關,PG 可能先於這些疾病出現:
- 將近三分之一與發炎性腸道疾病 (inflammatory bowel disease, IBD) 相關(克隆氏症 Crohn disease 與潰瘍性結腸炎 ulcerative colitis 頻率相近)。
- 副蛋白血症(單株免疫球蛋白病 monoclonal gammopathy)見於 10% 病例,最常見為 IgA 型。
- 血液惡性腫瘤見於高達 7%,最常見為急性骨髓性白血病 (acute myeloid leukemia),多由骨髓增生不良症候群 (myelodysplastic syndrome) 等前驅。
- 其他:骨髓增生性疾病、淋巴瘤、實體腫瘤、化膿性汗腺炎 (hidradenitis suppurativa);藥物誘發者包括 G-CSF(顆粒球群落刺激因子)。
臨床表現
- 潰瘍型:初始為劇痛的紅斑性丘疹、膿疱或結節性癤腫,好發於腿部;中央壞死糜爛形成潰瘍,藍紫色潛掘邊緣、基底覆膿性壞死物質,可深及肌肉或肌腱。
- 大疱型:疼痛、快速擴大的淺表水疱,迅速糜爛;常與血液疾病相關,好發於上肢;可與 Sweet 症候群重疊。
- 膿疱型(IBD 的膿疱性疹):幾乎只見於急性 IBD(通常為 UC)惡化,軀幹多發大型膿疱。
- 增殖型:單一癤腫樣結節/膿瘍/斑塊/淺表潰瘍,多在軀幹;發作緩慢、不適輕微、通常不伴全身疾病。
- 術後 (postoperative) 與造口旁 (parastomal) PG 為潰瘍型展現疼痛激惹的例子。
- 非皮膚表現:內臟器官(肺、骨關節、CNS、心血管、腹腔內臟、眼)可發生無菌性嗜中性球浸潤。
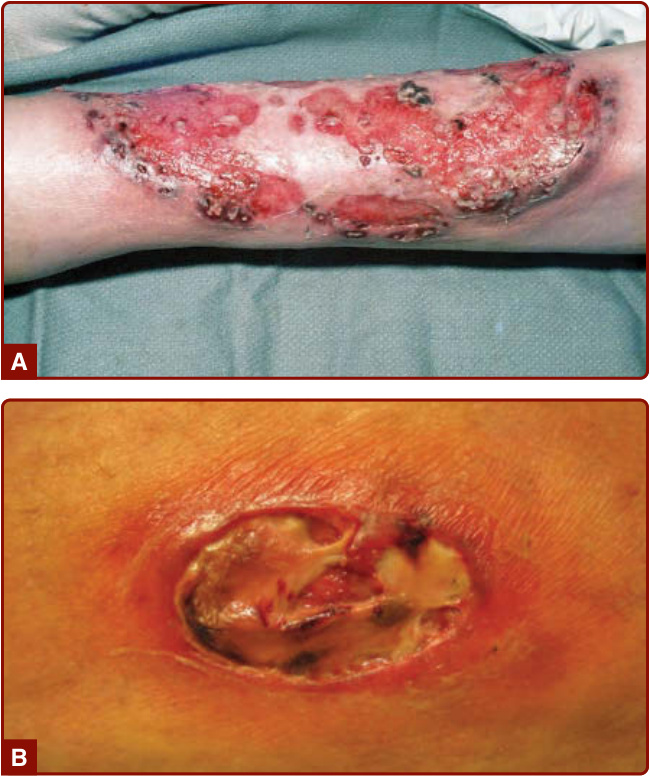
圖 37-1:典型潰瘍型壞疽性膿皮症已形成病灶,界線分明的潰瘍、潛掘與周圍紅斑帶。
致病機轉
- 病因不明,機轉所知甚少,可能依潛在疾病而異;多因子,含基因傾向、副免疫與副腫瘤現象。假說:多種相關疾病匯聚於導致異常嗜中性球活性的發炎途徑。
- 傷口邊緣以 CD3+ T 細胞與 CD163+ 巨噬細胞為主;傷口床 TNF-α、IL-8(嗜中性球趨化)、IL-17、MMP-2/MMP-9 增加。嗜中性球趨化、訊號傳遞與遷移異常。
- TNF-α 促發炎並增強嗜中性球脫顆粒;IL-23 升高驅動 IL-17 相關發炎。
- 遺傳:PAPA 症候群(化膿性關節炎、PG、痤瘡)為體染色體顯性,由第 15q 染色體 PSTPIP1(CD2BP1)基因突變所致,導致發炎體抑制減少、IL-1β 與 IL-18 增加。變異型含 PAPASH(PSTPIP1 E277D 錯義突變)、PASH(啟動子 CCTG 微衛星重複增加)。部分 IBD 易感基因座(如 IL8RA)亦與 PG 相關。
診斷
- 為臨床診斷,必須先排除其他病因(尤其感染,如非典型分枝桿菌、深部黴菌感染);無特異性血清、組織或基因標記。
- 診斷標準:需同時符合兩項主要標準 + 至少 2 項次要標準。
- 主要:(a) 無發燒/毒血症/相關藥物下,突發具特徵型態的疼痛病灶;(b) 組織病理排除血管炎、惡性腫瘤、感染微生物(特殊染色+陰性培養),並排除血管鬱滯/阻塞。
- 次要:(a) 合併全身性疾病;(b) 典型組織學發現(曾用全身性類固醇者嗜中性球可能被鈍化、改以單核細胞為主);(c) 高劑量全身性皮質類固醇下疼痛與發炎迅速減輕,1 個月內潰瘍縮小 50%;(d) 疼痛激惹病史或篩狀瘢痕。
- 組織病理(非診斷性但高度提示):應於病灶邊緣取切開楔形切片,一部分送細菌/分枝桿菌/黴菌培養,一部分送組織學(H&E+PAS、Giemsa、Fite、Gram)。中央區可見強烈真皮嗜中性球浸潤、膿瘍形成;邊緣紅斑帶可見血管周圍淋巴球浸潤(淋巴球性血管炎)。
- 實驗室:CBC+白血球分類、完整代謝套組、ESR、自體抗體(ANA、anti-Ro/La)、類風濕因子、抗磷脂質抗體、ANCA、血清蛋白電泳與免疫固定法、胸部 X 光。
- 血管檢查:腿部潰瘍者做靜脈逆流檢查(排除靜脈功能不全與深部靜脈血栓)及踝/趾肱指數(排除缺血)。
- 其他:內視鏡排除 IBD、骨髓抽吸、腹部超音波、CT。
鑑別診斷
- 範圍廣泛,依型別與部位不同(見表 37-1),主要須排除血管疾病、感染、惡性腫瘤、其他嗜中性球皮膚病與人為性疾患。
病程與預後
- 典型潰瘍型為慢性復發性疾病,具顯著病態與死亡率;大於 65 歲與男性預後較差。
- 增殖型預後佳,較可能不靠口服皮質類固醇而癒合;造口旁 PG 預後佳,常對外用/病灶內類固醇有反應;膿疱型若控制 IBD 常完全緩解;大疱型伴血液疾病者預後不佳(可能為白血病性轉變前兆)。
治療
- 全身性類固醇及/或環孢素 (cyclosporine) 不論潛在疾病皆可能有效,但常需合併治療;同時治療潛在疾病(如 IBD 用 infliximab、類風濕關節炎用 methotrexate)。
- 輕度 PG/增殖型斑塊:外用第 I 級皮質類固醇(clobetasol propionate 0.05%)、病灶內皮質類固醇,或鈣調神經磷酸酶抑制劑(tacrolimus 0.1%)每日兩次。
- 較嚴重/廣泛 PG 第一線:全身性糖皮質素。通常以口服 prednisone 每日 1 至 2 mg/kg 開始,最大劑量 150 mg/day。非常侵襲性疾病可用靜脈脈衝式 methylprednisone 1 g/day 連續 3 至 5 天,接續口服。反應通常迅速(1 至 2 週內停止進展),進展停止後緩慢減量,並加類固醇節省輔助劑(cyclosporine、dapsone、infliximab)。
- 環孢素:無法耐受糖皮質素者的另一第一線,亦作輔助。隨機對照試驗顯示口服 prednisolone 0.75 mg/kg/day 與 cyclosporine 4 mg/kg/day 療效相當;不良反應為腎毒性與高血壓,建議療程少於 1 年。
- 難治性 PG:重新評估未受控共病、新合併感染、是否需更導向藥物;可考慮基因分析(如 PAPA/IL-1β 增加者用 canakinumab、anakinra、TNF-α 抑制劑;JAK2 突變真性紅血球增多症用 ruxolitinib;MTHFR 突變者用 B 群維生素)。
- 傷口照護:每日以微溫無菌生理食鹽水或溫和消毒液清潔;1% 磺胺嘧啶銀 (silver sulfadiazine) 乳膏舒緩並促進肉芽組織;非黏性敷料+縐布彈性繃帶;淺表病灶可用水膠體敷料留置 2 至 3 天。避免刺激物(化學去腐劑、硝酸銀、黏附性紗布)。
- 處置:清創不可積極進行,盡量避免植皮(疼痛激惹、供皮區誘發新病灶風險);疾病控制但再上皮化不完全者可用培養組織同種異體/自體移植或牛膠原蛋白基質。
- 預防:有 PG 病史者避免皮膚外傷;必須手術者由皮膚科密切監督,侵襲性 PG 病史者可於術中及術後(2 週或更久)給予一療程全身性類固醇,盡量用皮內縫合。
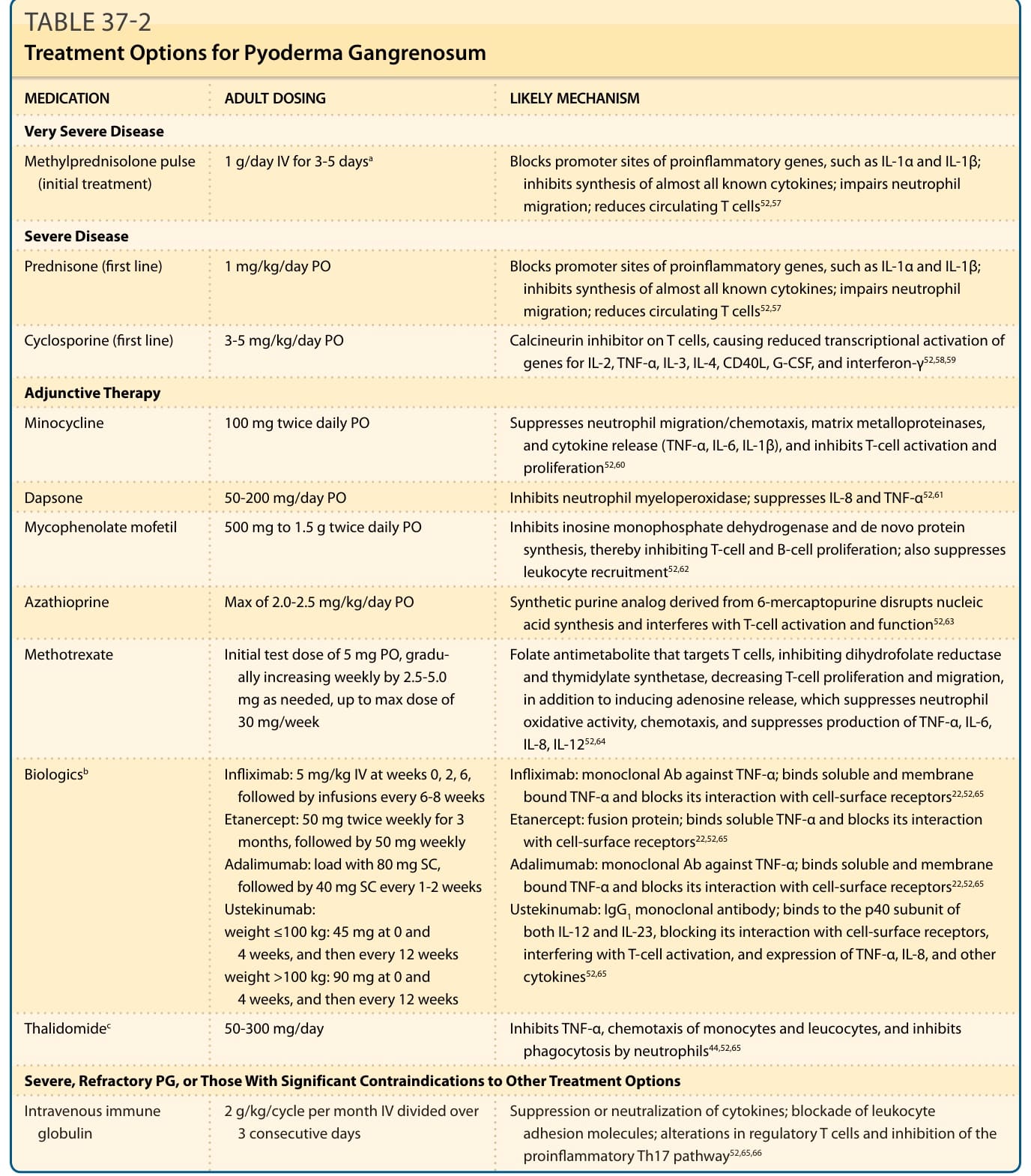
表 37-2:壞疽性膿皮症的治療選項。