Pyoderma Gangrenosum
6
AT-A-GLANCE
■ Pyoderma gangrenosum (PG) is an uncommon, neutrophilic inflammatory skin condition, which classically presents as a painful nodule, plaque, or pustule that enlarges and breaks down to form a progressively enlarging ulcer with raised, undermined, violaceous borders and a surrounding zone of erythema. Healing PG lesions develop a cribriform appearance.
■ PG tends to occur and recur in areas of trauma because of the pathergic phenomenon, where trauma or irritation can induce flaring of PG.
■ PG most often occurs in association with systemic inflammation, and most reported cases have an associated underlying disease (such as inflammatory bowel disease, monoclonal gammopathy, hematologic disease, inflammatory arthritis, malignancy, hidradenitis suppurativa, etc); however, PG may precede these disorders. PG has also been reported in association with genetic mutations and syndromes, such as the pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome, or without an identifiable underlying disease.
■ PG most commonly presents as the classic ulcerative variant, but may also arise as bullous, pustular, or vegetative variants. Clinical features of different variants sometimes overlap in individual patients but usually one variant dominates the clinical picture.
■ There is no laboratory test or investigation that establishes the diagnosis of PG with certainty. The histopathologic findings are not diagnostic but can be supportive of the diagnosis of PG in the appropriate clinical setting and are essential to rule out alternative diagnoses.
■ Specified criteria (see “Diagnostic Algorithm” section) suggest the diagnosis of PG, but other conditions (particularly infection, vascular disease, and malignancy) must be excluded.
■ The mainstays of management are systemic immunosuppressive agents together with appropriate local and topical therapy.
■ Classic ulcerative PG is a chronic condition. Complete healing usually requires months of treatment; maintenance therapy is necessary in many and relapses are common. Significant morbidity and mortality are experienced by patients with ulcerative and bullous PG.
DEFINITIONS
Pyoderma gangrenosum (PG) is an uncommon, chronic, neutrophilic inflammatory skin condition, with the classic ulcerative variant typically presenting initially as a painful nodule, plaque, or pustule that enlarges and breaks down to form a progressively enlarging ulcer with raised, undermined, violaceous borders and a surrounding zone of erythema.1,2 The lesions are commonly very painful, develop a cribriform appearance with healing, and tend to occur and recur in areas of trauma in up to 50% of cases.1,2 PG is believed to occur in association with systemic inflammation, and most reported cases are associated with autoinflammatory or hematologic conditions, and typically respond to immune suppression.1,3-5 PG most commonly presents as the classic ulcerative variant, but may also present as bullous, pustular, or vegetative variants. Unless otherwise specified, when we refer to PG in this text, we are referring to the classic ulcerative variant.
EPIDEMIOLOGY
PG is estimated to affect up to 10 cases per million people per year, and represents up to 3% of chronic leg ulcer cases, but may occur anywhere on the body. PG has been reported in people of all ages; most cases, however, present in the second to sixth decades of life, with a possible female predominance.2,6
DISEASE ASSOCIATIONS
DISEASE ASSOCIATIONS
Up to 70% of reported PG cases are associated with a known underlying systemic disease, such as inflammatory bowel disease (IBD), monoclonal gammopathy, hematologic disease, inflammatory arthritis, malignancy, hidradenitis suppurativa, and others, even though PG may precede these disorders. PG also has been associated with genetic mutations, as part of a genetic syndrome such as the pyogenic arthritis, PG, and acne (PAPA) syndrome, or without an identifiable underlying disorder.7
Nearly one-third of PG cases are associated with IBD, with similar frequency of both Crohn disease and ulcerative colitis (UC), although most of the Crohn disease cases have colonic involvement.3,8 Paraproteinemia (monoclonal gammopathy) is seen in 10% of PG cases,9 most commonly immunoglobulin A monoclonal gammopathy.10 Hematologic malignancy has been seen in up to 7% of PG cases, with the most common subtype reported being acute myeloid leukemia,
6
and most cases of leukemia being preceded by an hematologic abnormality such as myelodysplastic syndrome.1,11 Myeloproliferative diseases, such as polycythemia vera,12 essential thrombocythemia,13
and myelofibrosis (agnogenic myeloid metaplasia),14,15
as well as leukemoid reaction,16 also have been associated with PG. There also have been fewer reports of PG associated with Hodgkin and non-Hodgkin lymphomas, cutaneous T-cell lymphoma, and solid neoplasms originating in the breast, ovaries, bladder, prostate, colon, carcinoid, and bronchus,9,17 although the significance of these less-frequent associations is less certain. There also are reports of medications inducing PG, including several cases of PG associated with granulocyte colony-stimulating factor (G-CSF), which resolved after G-CSF was discontinued.18
CLINICAL FEATURES
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
PG can be classified morphologically as being (a) ulcerative, (b) bullous, (c) pustular, or (d) vegetative. Although some patients may show more than 1 variant (eg, isolated pustular lesions frequently occur in patients with ulcerative PG), usually 1 variant of PG dominates the clinical picture and the patient should be classified accordingly. Ulcerative PG is the most common and originally described variant of PG. The most common initial clinical lesion in a patient with ulcerative PG is an intensely painful erythematous papule, pustule, or nodular furuncle (these lesions are usually single but may be multiple). They erupt on apparently normal-looking skin, with the most common site being the leg. The enlarging initial lesion develops a surrounding zone of erythema that extends into the surrounding skin. As it enlarges, the center degenerates, necroses, and erodes, converting it into an eroding ulcer (Fig. 37-1), the development of which is accompanied by an alarming increase in pain. The ulcer often has a bluish/violaceous undermined edge and the base is covered with purulent necrotic material. Ulcerative PG may erode deeply with exposure of muscle or tendon in some cases.1,2
PG can also occur at the site of trauma or surgery (Fig. 37-2), which is known as the pathergic phenomenon, where trauma or irritation can induce exaggerated flaring of PG.1,2
Bullous PG (sometimes called atypical PG) presents as a painful, rapidly expanding superficial inflammatory blister that quickly erodes. In the early acute stage, the bullous nature of the lesion is evident, but because the roof of the blister necroses rapidly, close inspection of the border of established lesions is necessary to reveal its bullous nature (Fig. 37-3). Bullous PG is commonly associated with hematologic disease and most often appears on the upper limbs.19
606
This variant of PG may show clinical and histologic
A
B
overlap with Sweet syndrome (one of the neutrophilic dermatoses which is itself often associated with hematologic disease). Pustular PG (also called the pustular eruption of IBD) is a generalized eruption that occurs almost exclusively in the setting of an exacerbation of acute IBD (usually UC). Its onset is dramatic, with the rapid development of multiple, large, circular-to-oval, painful pustules on the trunk and, to a lesser extent, the face and limbs. Control of this eruption is difficult without controlling the bowel disease, which may require resection of diseased bowel in some cases.1,2
Vegetative PG (or superficial granulomatous pyoderma) usually presents as a single furunculoid nodule, abscess, plaque, or superficial ulcer, typically on the trunk (Fig. 37-4). In contrast to the other variants, this uncommon variant is gradual in its onset, mild in the discomfort it generates, and not usually associated with the presence of systemic disease. This form of PG is usually more responsive to localized or mild forms of systemic therapy than the other variants.20
Postoperative and parastomal PG are considered to be examples of ulcerative PG demonstrating the pathergic phenomenon.1,2
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
As the majority of PG cases are associated with, or precede, a concurrent underlying systemic disease, a thorough history and physical examination is mandatory with a particular search for clinical and biologic markers of IBD, inflammatory arthritis, hematologic disease (monoclonal gammopathy and other dyscrasias), hematologic malignancy, and internal malignancy. Additionally, The clinician should be aware that sterile neutrophilic infiltration of internal organs (lung,
6
bone joints, CNS, cardiovascular system, intraabdominal viscera, eye) can occur in association with or even precede the onset of cutaneous PG.21
COMPLICATIONS
COMPLICATIONS
Active or poorly controlled cutaneous PG causes significant morbidity, including severe pain, loss of mobility, anemia of chronic disease, and complications from therapeutic interventions. Many of the treatments for PG must be administered for many months and may have significant side effects. For example, corticosteroids can induce or worsen diabetes, cyclosporine can induce or worsen renal dysfunction, and immunotherapy can lead to immunocompromise, which increases risk for infection by both common and atypical microorganisms. Consequently, close monitoring and followup of patients is necessary. Care must be taken to avoid trauma to the skin and elective surgery should be undertaken with caution because of the possibility of inducing new PG lesions through the pathergic phenomenon. Additionally, lack of recognition of the neutrophilic infiltration of internal organs in PG can lead to unnecessary surgical procedures.1,22
ETIOLOGY AND PATHOGENESIS
The etiology of PG is unknown, its pathogenesis is poorly understood, and the mechanisms of its pathophysiology likely differ depending on the associated underlying contributory disease. The factors predisposing to, inciting, and regulating these abnormalities are likely multifactorial, including genetic predisposition, paraimmune and/or paraneoplastic phenomena,7
which may cause increased sensitivity to, or increased activity of, the immune response and inflammation. Interestingly, PG occurs in association with a broad spectrum of diseases, and yet they all lead to or associate with the common phenotype of PG, lending to the hypothesis that these diseases somehow converge on inflammatory pathways leading to aberrant neutrophil activity and function.23 Despite this similarity, there is variation in reports of the association between the disease activity of PG and that of the underlying disease state. In the setting of IBD, PG activity classically has been associated with the inflammatory process in the bowel. According to the systematic review of Agarwal et al,8 which included 60 patients, of whom 35 (58%) had active IBD and only 9 (15%) had inactive IBD at PG diagnosis (16 (27%) with IBD activity unspecified). While some have reported no apparent association between exacerbations of bowel disease with worsening of the skin lesions,24,25 there are multiple reports demonstrating healing of PG following therapy for the diseased bowel. Powell et al12 reported a case series where PG lesions improved in all of 9 patients with UC following total proctocolectomy, and Talansky et al26
607
6
reported prompt PG skin healing within 2 months after resection of diseased bowel in 3 patients with severe ileocolitis, 1 with severe UC and 1 with moderate UC. Notably, there also has been a case of PG reported in a patient with UC 10 years after proctocolectomy,9 which raises the question of whether there was any remaining diseased bowel. Improvement of bullous PG has been reported after treatment and remission of acute myeloid leukemia,27 which may be partially explained by the presence of a leukemic cell infiltrate consisting of atypical myeloblasts discovered within a PG skin sample from a patient with acute myeloid leukemia.28 Complete recovery of PG has been reported after successful treatment of underlying hairy cell leukemia with cladribine.29 Successful treatment of refractory PG associated with renal cell carcinoma has been reported with ustekinumab, but only after excision of the carcinoma.30
INFLAMMATORY MEDIATORS AND ABERRANT NEUTROPHIL ACTIVITY
INFLAMMATORY MEDIATORS
AND ABERRANT NEUTROPHIL
ACTIVITY
T cells may be important in the development of PG through various mechanisms, including cytokine signaling and antigenic stimulus,31 and are likely involved in some of the of the observed aberrant neutrophil activity. Marzano et al32 looked at lesional skin biopsies from 21 patients with PG and found wound edge infiltrates of predominately CD3+ T cells and CD163+ macrophages, along with increased tumor necrosis factor-α (TNF-α), interleukin (IL)-8 (potent neutrophil chemotactic agent), IL-17 (proinflammatory cytokine; stimulates the expression of IL-8 and G-CSF), matrix metalloproteinase (MMP)-2/MMP-9 (collagenase mediating tissue damage) in the wound bed. Based on the presence of a lymphocytic infiltrate at the active advancing border of PG lesions, it has been postulated that lymphocytic antigen activation occurs with cytokine release and neutrophil recruitment. This may take place not only in the skin but also in other tissues, such as the lung, intestine, and joints.33 Neutrophils are thought to play a significant role in PG based on the prominent neutrophil-rich dermal infiltrate in PG pathology specimens, and PG response to antineutrophil medications like dapsone. Multiple studies have reported abnormalities in neutrophil chemotaxis, signaling, and trafficking in patients with PG.4,34,35 These neutrophil abnormalities, and the predominance of neutrophils on histology, also give PG a place among the broader spectrum of neutrophilic dermatoses, which include Sweet syndrome (acute febrile neutrophilic dermatosis), Behçet disease, dermatitis herpetiformis, and neutrophilic eccrine hidradenitis. Besides dermal neutrophilia, other common features these conditions share include an associated underlying disorder, similarities in treatment, and a tendency
608
for pathergy, in some more than others (PG, Sweets, Behçet, dermatitis herpetiformis).7
TNF-α plays an important role in production of proinflammatory cytokines, including IL-1, IL-6, IL-8, and interferon-γ, and has been shown to stimulate and enhance neutrophil degranulation and superoxide production.36-38 TNF-α, IL-6, and soluble IL-2 receptor elevation has been reported in a patient with PG, with serum levels correlating with ulcer activity.39 In addition to being increased in PG, IL-8 also has been shown to cause similar ulceration in human skin xenografts transfected with recombinant human IL-8.40 Guenova et al41 found elevated expression of IL-23 by polymerase chain reaction in a PG lesion compared with normal skin, which is significant because IL-23 plays an important role in driving inflammation associated with IL-17 production and neutrophil recruitment. Additionally, patients with the PAPA syndrome and some variants have been found to have increased production of IL-1β and IL-18.42,43
GENETICS
GENETICS
Uncovering genetic associations with PG is important as it can provide a better understanding of the pathophysiology of PG and help elucidate new targets for treatment. PG has been reported in association with systemic diseases, genetic mutations and syndromes, or in isolation. The systematic review of syndromes and genetic mutations associated with PG by DeFilippis et al44 included 823 cases of PG, and revealed 31 (3.8%) cases of PAPA syndrome and its variants, 2 (0.2%) patients with mutations in Janus kinase (JAK) 2, 3 (0.4%) patients with mutations in methylenetetrahydrofolate reductase (MTHFR) who developed PGlike ulcerations, and 14 (1.7%) familial cases with no specific reported mutations, in addition to cases associated with IBD, polyarthritis, and hematologic disease. The PAPA syndrome, which is an autosomal dominant disorder, has been linked to mutations in the proline-serine-threonine phosphatase interacting protein 1(PSTPIP1; also known as CD2 antigen-binding protein 1 [CD2BP1]) gene on chromosome 15q, encoding PST- PIP1, which binds to pyrin and regulates the inflammasome. Mutations in PSTPIP1 lead to decreased inhibition of the inflammasome and increased production of IL-1β and IL-18, which can result in the inflammation seen with PAPA.42,43
Variations of the PAPA syndrome have been described in the literature, including PAPASH syndrome (pyogenic sterile arthritis, PG, cystic acne, and hidradenitis suppurativa), which results from an E277D missense mutation in the PSTPIP1 gene,45 PASH syndrome (PG, acne, and suppurative hidradenitis), which results from an increased CCTG microsatellite repeat in the promoter region of PSTPIP1,46 as well as other variations. JAKs are intracellular tyrosine kinases that are needed for cell differentiation, proliferation, and apoptosis. Mutations in JAKs can cause inflammatory and
myeloproliferative disorders, and also have been associated with cases of PG. Interestingly, the pathway is activated by G-CSF and TNF-α, as well as ILs and interferons.44
Some specific genetic loci linked to IBD susceptibility have been found to correlate with the development of PG.47 Weizman et al47 reviewed 5756 patients with IBD using genome-wide association data and described several genes associated with susceptibility to IBD that were significantly associated with PG, including IL8RA (a mediator of neutrophil migration), MUC17, MMP24, WNK2, DOCK9, PRDM1, and NDIFIP1,22 in addition to other genetic loci associated with IBD and PG, such as GPBAR1, TIMP3, and TRAF31P2.44
HORMONAL INFLUENCES
HORMONAL INFLUENCES
There are reports of premenstrual flares associated with several dermatologic conditions such as hidradenitis suppurativa and acne vulgaris; the medical literature describes a positive role of estrogen and a negative role of androgens, or androgen receptor activation, in cutaneous wound healing. Jourabchi et al48
reported a case of PG with premenstrual flares that was controlled with use of a combined oral contraceptive and antiandrogen (ethinyl estradiol/drospirenone).48
Interestingly, hidradenitis suppurativa and acne are known to occur with PG in some patients, suggesting a related pathophysiologic mechanism. Additionally, PG, hidradenitis suppurativa, and acne have some other overlapping features, including elevated IL-17 and TNF-α in lesional skin, and positive response to TNF-α inhibitors.22,49,50
DIAGNOSIS
PG is currently diagnosed clinically and, very importantly, after excluding other potential causes for the skin manifestations (especially infectious causes such as atypical mycobacteria and deep fungal infections) as no specific serologic, histologic, or genetic markers have yet been found.
DIAGNOSTIC ALGORITHM
DIAGNOSTIC ALGORITHM
Because there are no specific diagnostic tests for PG, the following criteria are proposed to help make the diagnosis of PG, which requires the fulfillment of both major criteria and at least 2 minor criteria:1,3,7,22
- Major criteria:
a. Sudden onset of a painful lesion with the characteristic morphology described in “Cutaneous Findings” section in a patient who does not have fever, significant toxemia, or relevant drug intake. b. Histopathologic exclusion of significant vasculitis, malignancy, and infective organisms by special histologic studies/stains and negative tissue
6
cultures, as well as exclusion of significant vascular stasis/occlusion by appropriate studies.
2. Minor criteria that are supportive of the diagnosis are as follows:
a. Occurrence in an individual with systemic disease, as described in “Disease Associations” section. b. Classic histologic PG findings. (Note: In patients who have received systemic corticosteroids, the presence of neutrophils may be blunted and mononuclear cells may instead predominate.) c. Rapid reduction of pain and inflammation on initiation of high-dose systemic corticosteroid therapy and rapid ulcer healing response to high-dose systemic corticosteroid therapy with a 50% decrease in ulcer size within 1 month. d. History suggestive of pathergy or a clinical finding of cribriform scarring.
Providers must be aware that many patients may present months or years after initial development of skin manifestations, while taking or having tried various medications, including systemic corticosteroids or other immunosuppressants, and consequently may not present with the classic clinical findings or diagnostic criteria. Consequently, obtaining a detailed history is crucial to making the appropriate diagnosis.
SUPPORTIVE STUDIES
SUPPORTIVE STUDIES
HISTOPATHOLOGIC STUDIES AND INFECTIOUS STAINS AND CULTURES
The histopathologic changes in the skin are not diagnostic but can be highly suggestive of PG. Pathologic changes must be considered in view of the total clinical picture. The microscopic changes that are seen depend on (a) the clinical variant of PG (ulcerative, bullous, pustular, or vegetative), (b) the timing of the biopsy, and (c) the site of the biopsy relative to the inflammatory process. An incisional wedge skin biopsy should be taken from the edge of the PG lesion, taking care to sample a portion of normal skin progressing through the border into the area of active inflammation, to allow the various histologic patterns to be discerned. The excised tissue should then be divided with one section (fresh tissue) sent for bacterial culture, mycobacterial culture with smear, and fungal culture with microscopy, and the other portion sent in formalin for histologic evaluation requesting hematoxylin and eosin preparation with periodic acid–Schiff, Giemsa, Fite, Gram, and other stains considered relevant. If an incisional biopsy is not possible, two 4-mm punch biopsies may be performed instead. Immunofluorescence studies may show positive perivascular staining in lesional skin, which may be secondary to profound inflammation; it is not required for diagnostic purposes and can be omitted unless vasculitis is suspected in the differential diagnosis. Because PG can be complicated by secondary infection with the organisms mentioned above,
609
6
nonhealing PG or recurring PG should be rebiopsied to exclude superimposed infection. The location of the biopsy within the lesion is particularly important as biopsies taken from the center of established lesions may have different findings from biopsies taken from peripheral skin. A marked perivascular lymphocytic infiltration may be seen in biopsies taken from the “zone” or area of erythema that surrounds active lesions of ulcerative PG. Lymphocytes may be seen to infiltrate vessel walls with intramural and intravascular fibrin deposition indicative of vascular damage (sometimes called lymphocytic vasculitis). Abscess formation with intense dermal neutrophilic infiltration may extend to the panniculus and areas of tissue necrosis, dominating the histologic findings in biopsies taken from central areas of ulcerative PG lesions. Leukocytoclasis is not a prominent finding and although occasionally evidence of leukocytoclastic vasculitis is seen close to the necrotic center, this is a minor feature and considered secondary to the intense inflammatory changes rather than the primary event. Histologic examination of lesional skin from a patient with bullous PG will show a subepidermal or intraepidermal bulla with overlying epidermal necrosis and marked upper dermal edema with prominence of neutrophils. Biopsy of pustular PG will show a dense dermal neutrophilic infiltration (often centered about a follicle) with subepidermal edema and infiltration of neutrophils into the epidermis with subcorneal aggregations. Vegetative PG is characterized histologically by the presence of pseudoepitheliomatous hyperplasia, sinus tract formation, and the presence of palisading granulomas in the setting of focal dermal neutrophilic abscesses.3,7,38
LABORATORY TESTING
All patients with PG should have these tests performed: full blood cell count with differential white cell count; complete metabolic panel; erythrocyte sedimentation rate; autoantibody screen (including antinuclear antibodies, anti-Ro/La antibodies); rheumatoid factor; antiphospholipid antibody screen; antineutrophilic cytoplasmic antibodies; serum protein electrophoresis and immunofixation studies; and chest radiography.
VASCULAR STUDIES
All patients with leg ulcers should have venous reflux studies performed to rule out significant venous insufficiency and deep venous thrombosis, in addition to ankle/toe brachial index studies to rule out ischemia as an underlying cause for the ulceration.
OTHER INVESTIGATIONS
In most patients, the following additional tests may are warranted: endoscopy (upper and/or lower gastrointestinal) to rule out often-associated IBD; careful examination of peripheral blood morphology when
610
indicated, and if any abnormality is found, bone marrow aspirate examination should be performed; ultrasound of abdomen (including liver/spleen/aorta); and computed tomography for visualization of the thorax, abdomen, and pelvis. Figure 37-5 outlines other directed investigations.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis to be considered in a patient with PG is extensive.51 Different variants of PG (ulcerative, bullous, vegetative, pustular) suggest alternative diagnoses, and the occurrence of PG at certain cutaneous sites raises further diagnostic issues for the clinician, as shown in Table 37-1.
CLINICAL COURSE AND PROGNOSIS
The prognosis depends on the PG variant; the age and sex of the patient; presence of underlying systemic disease and other comorbid conditions; and the type, dosage, and duration of therapy required to bring the disease under control. Classic ulcerative PG is a chronic recurrent disease with a significant morbidity and mortality. Patients with this variant who are older than 65 years of age and male patients seem to have a worse prognosis. Patients with vegetative PG generally have a good prognosis, and compared to classic PG, vegetative PG lesions are more likely to heal without the use of oral corticosteroids.20 Parastomal PG has a good prognosis, often responding to topical or intralesional steroid therapy. Patients with pustular PG often have complete remission of their cutaneous lesions if the severe IBD that usually accompanies this variant is controlled. Patients with bullous PG who have an associated hematologic disorder have a poor prognosis. The onset of bullous PG in a patient with stable polycythemia rubra vera has been reported to herald the onset of leukemic change.30,38,52
MANAGEMENT
APPROACH TO MANAGEMENT
APPROACH TO
MANAGEMENT
Understanding the context in which PG is occurring is crucial in deciding on proper treatment approach. The unpredictable nature of PG and its variable aggressiveness in individual patients necessitate a flexible approach to treatment. Therapeutic agents must be adapted to the patient’s physiologic state (pregnancy, old age, renal disease, diabetes, immunosuppressed status, etc) and also with their underlying associated disease state. Although the course of the underlying disease does not always correlate with PG flares, its treatment can result in improvement of PG. Systemic
6
Approach to the patient with pyoderma gangrenosum
General examination
Detailed history (includes drugs, trauma, systems review) Detailed physical exam: location, type, size, outline, depth
Clinical impression of pyoderma gangrenosum
Investigations
Routine tests: Full blood count + differential Erythrocyte sedimentation rate Complete metabolic panel Serum iron Autoantibody screen Antineutrophilic cytoplasmic antibody (pANCA, cANCA) Antiphospholipid antibody screen Rheumatoid factor Serum protein electrophoresis and immunofixation studies Chest radiography, Vascular studies
Skin biopsies: In formalin for histology (hematoxylin and eosin, periodic acid-Schiff, Giemsa, Fite, Gram stain, and other stains)
Other tests as indicated: α1-Antitrypsin level Serum bromide/iodide Blood cultures Coagulation screen Cryoglobulins, cryofibrinogens Cold agglutinins Hepatitis/HIV screening Syphilis serology screen Midstream specimen of urine - Bence-Jones protein Computed tomography scan Endoscopy (upper and/or lower) Bone marrow aspirate Herpes simplex virus PCR or viral culture
Fresh tissue for culture (bacterial, mycobacterial, atypical mycobacterial, fungal)
Rule out differentials:
Vascular disease, infections, malignancy, other neutrophilic dermatoses, facticial disorder
Classify to subgroup
Ulcerative Bullous Pustular Vegetative
Consider associated diseases
Chronic renal impairment – Frequent – Frequent – Frequent – Uncommon Arthritis, inflammatory bowel disease, monoclonal gammopathy, malignancy
Hematologic dyscrasias/malignancy Inflammatory bowel disease
steroids and/or cyclosporine may be effective in controlling PG regardless of the underlying disease state, but PG often requires combination therapy and multiple medication trials to achieve healing, and in these cases the underlying disease state should be used to help guide medication selection and treatment approach. Adequate rest, efficient pain relief, and appropriate therapy for any comorbidities (venous stasis disease, diabetes, congestive heart failure with fluid overload, etc) that affect healing are also pivotal in the overall management strategy of a patient with PG. If other systemic illnesses are present, close collaboration with
an internal medicine specialist and pertinent subspecialty physicians is needed.
MEDICATIONS
Definitive treatment guidelines for PG do not exist, partly because PG associated with different underlying diseases seems to behave differently, and also because there is a paucity of data in the literature. Approach to treatment is based on directed therapeutic immunomodulation to decrease inflammation and promote healing. Choice of treatment depends on the severity of the PG, and when possible should also attempt to
611
6
Differential Diagnosis of Pyoderma Gangrenosum TABLE 37-1
VARIANT SPECIFIC
Ulcerative pyoderma gangrenosum (PG) Most Likely
■Vascular
■Venous stasis ulceration
■Vasculopathy
■Arterial occlusive disease
■Vasculitis
■Antiphospholipid–antibody syndrome
■Infection
■Bacterial (ecthyma gangrenosum)
■Mycobacterial/atypical mycobacterial (eg, Mycobacterium chelonae, Mycobacterium marinum)
■Deep fungal infection (eg, North American blastomycosis, sporotrichosis, Aspergillus, Cryptococcus) Consider
■Infection
■Tertiary syphilitic ulcer
■Amebiasis
■Viral (herpes simplex)
■Drugs (eg, hydroxyurea, halogenoderma)
■Other
■Dermatitis artefacta
■Calciphylaxis
■Necrotizing insect bite (eg, brown recluse spider)
■Malignant rheumatoid disease (with high-titer rheumatoid factor and depressed complement levels)
■Cutaneous involvement of malignancy process (eg, lymphoma, leukemia cutis)
Bullous PG
Most Likely
■Infection
■Bacterial (cellulitis/impetigo)
■Viral (especially in immunocompromised)
■Fungal (eg, mucormycosis in persons with diabetes)
■Other
■Sweet syndrome/Behçet disease
Consider
■Bullous dermatoses
■Erythema multiforme
■Other
■Insect/arthropod bite
Pustular PG
Most Likely
■Infection
■Bacterial/viral/fungal
■Vasculitis
■Pustular vasculitis
Consider
■Other
■Pustular psoriasis
■Sneddon-Wilkinson disease
■Pustular drug eruption
■Bowel bypass syndrome
SITE SPECIFICa
Parastomal
Most Likely
■Dermatoses
■Irritant/allergic contact dermatitis
■Other (eg, psoriasis, eczema)
■Infection
■Bacterial (Staphylococcus/Streptococcus)/cellulitis
■Fungal (Candida)
■Other
■Extraintestinal inflammatory
■Bowel disease
■Malignancy
Postsurgical Wounds
Most Likely
■Infection
■Bacterial/cellulitis
■Mycobacterial/atypical mycobacterial
■Fungal (eg, mucormycosis)
■Breakdown
■Suture allergy
■Mechanical
Consider
■Necrotizing fasciitis
■Malignancy
Perineum
Most Likely
■Infection
■Bacterial/viral infection (herpes simplex virus, Epstein-Barr virus, cytomegalovirus)
■Fournier gangrene
■Other
■Hidradenitis suppurativa
■Extraintestinal Crohn disease
■Squamous cell/extramammary Paget disease
Consider
■Infection
■Syphilis
■Lymphogranuloma venereum/granuloma inguinale
■Leishmaniasis
■Other
■Dermatitis artefacta
■Behçet disease
Vegetative PG
Most Likely
■Infection
■Bacterial/viral/fungal
■Mycobacterial/atypical mycobacterial
■Leishmaniasis
Consider
■Blastomycosis-like pyoderma
■Dermatitis artefacta/malignancy
■Pyoderma vegetans
■Other
■Dermatitis artefacta
aThe differential diagnosis of lower-limb PG is essentially that delineated for variant-specific ulcerative PG.
612
treat the patient’s underlying condition (eg, infliximab for patients with IBD and methotrexate for patients with rheumatoid arthritis). Mild PG with small shallow ulcers or a plaque of vegetative PG may respond to topical class I corticosteroids (clobetasol propionate 0.05%), intralesional corticosteroids, or a calcineurin inhibitor such as tacrolimus 0.1%, with twice-a-day application. First-line therapy for more severe or extensive PG is systemic glucocorticoids. We typically begin treatment at 1 to 2 mg/kg of oral prednisone daily, with a maximum dose of 150 mg/day. For very aggressive disease, intravenous pulse methylprednisone 1 g/day for 3 to 5 days can be helpful as initial therapy, followed by daily oral glucocorticoids.1,53,54 Response to systemic glucocorticoids is typically rapid, typically halting progression within 1 to 2 weeks. Because long-term therapy is associated with significant adverse effects, slow glucocorticoid taper should begin once progression of PG has stopped. Simultaneously beginning a steroidsparing adjunctive agent (eg, cyclosporine, dapsone, infliximab) can help prevent flares while tapering.1
Oral cyclosporine is an alternative first-line treatment for patients who cannot tolerate systemic glucocorticoids and is also used as adjunctive therapy. A multicenter, parallel group, observer blind, randomized, controlled trial comparing oral prednisolone 0.75 mg/kg/day with cyclosporine 4 mg/kg/day for PG found comparable effectiveness between the 2 groups.55 Adverse effects include renal toxicity and hypertension, which must be monitored, and recommended time course for treatment is less than 1 year.55,56
Systemic glucocorticoids and cyclosporine are rapid acting, which makes them ideal for initial therapy, whereas some of the other therapies can take weeks for therapeutic effect and are better suited as adjunctive or maintenance therapy, especially for severe cases. It is not uncommon for severe cases of PG to require combination therapy to control the condition while minimizing side effects from therapy. Table 37-2 outlines additional therapies. Note that most of these medications require specific baseline laboratory test results, close monitoring for adverse effects, care to prevent drug interactions, and caution for immunosuppression. Maintenance therapy should be continued until there is complete wound healing and then slowly tapered off. In addition, patients with ulcerative PG have a significant risk of relapse, so long-term followup is required, and some patients require longterm maintenance therapy.
UNRESPONSIVE PYODERMA GANGRENOSUM AND TREATMENT CONSIDERATIONS
When PG is not responding to treatment, or if healing stalls, the provider must reassess the case:
- Is there an uncontrolled comorbidity (eg, venous stasis, vascular ischemia, diabetes) that is inhibiting wound healing and needs to be addressed?
6
- Is there a new superimposed infection that needs workup and treatment? And if so, should another biopsy be performed for pan-culture?
- Is there a better, more directed medication that should be tried?
In patients with refractory or difficult-to-treat PG, genetic analysis can be considered and may help identify better directed therapy. DeFilippis et al44 reviewed the published literature regarding syndromes and genetic mutations associated with PG, and found that PG responded to different treatments depending on the underlying disease process. For example, cases with the PAPA syndrome and variants with increased IL-1β improved with the anti–IL-1β monoclonal antibody canakinumab, the IL-1 receptor antagonist anakinra, and TNF-α inhibitors, whereas a case of polycythemia vera with JAK2 mutation responded to the JAK1/JAK2 inhibitor ruxolitinib, and PG-like cases with MTHFR mutations (whose pathway relies on vitamins B6, B9, and B12) improved with B-vitamin therapy.44
WOUND CARE
The location, morphology, size, and depth of each lesion should be recorded (with photography) on presentation and subsequent review to help monitor course. The cutaneous lesions of PG are usually extremely tender so cleansing should be carried out daily with tepid sterile saline or a mild antiseptic solution. Silver sulfadiazine 1% cream is usually soothing when applied to the ulcerated lesions of PG and may facilitate granulation tissue formation while also inhibiting bacterial growth. A nonadhesive dressing should be applied over the lesion and held in place with a crêpe elasticized bandage wrapped firmly, but not tightly, over it. Some patients, particularly those with superficial lesions, obtain significant relief with the use of hydrocolloid dressings, which can be left on for 2 to 3 days and “melt” into the lesion. Careful instruction to the patient and nurse is important to ensure compliance and to avoid the use of irritants such as chemical desloughing agents, caustics (such as silver nitrate), or dressings such as gauze impregnated with soft paraffin and/or antibacterial agents that may adhere to the ulcer base. A variety of bacteria may be cultured from the wound surface, but these usually represent contaminants and directed antibiotic therapy is not required unless there are clinical signs of incipient cellulitis around the wound.
PROCEDURES
Debridement must not be performed aggressively and skin grafting should be avoided if possible because of pathergy and the risk of inducing new PG lesions at the donor sites. Cultured tissue allografts/autografts and the use of bovine collagen matrix have been reported to be useful in patients in whom the disease is controlled but re-epithelialization incomplete.67
613
6
MEDICATION ADULT DOSING LIKELY MECHANISM
Very Severe Disease
Methylprednisolone pulse (initial treatment) 1 g/day IV for 3-5 daysa Blocks promoter sites of proinflammatory genes, such as IL-1α and IL-1β; inhibits synthesis of almost all known cytokines; impairs neutrophil migration; reduces circulating T cells52,57
Severe Disease
Prednisone (first line) 1 mg/kg/day PO Blocks promoter sites of proinflammatory genes, such as IL-1α and IL-1β; inhibits synthesis of almost all known cytokines; impairs neutrophil migration; reduces circulating T cells52,57
Cyclosporine (first line) 3-5 mg/kg/day PO Calcineurin inhibitor on T cells, causing reduced transcriptional activation of genes for IL-2, TNF-α, IL-3, IL-4, CD40L, G-CSF, and interferon-γ52,58,59
Adjunctive Therapy
Minocycline 100 mg twice daily PO Suppresses neutrophil migration/chemotaxis, matrix metalloproteinases, and cytokine release (TNF-α, IL-6, IL-1β), and inhibits T-cell activation and proliferation52,60
Dapsone 50-200 mg/day PO Inhibits neutrophil myeloperoxidase; suppresses IL-8 and TNF-α52,61
Mycophenolate mofetil 500 mg to 1.5 g twice daily PO Inhibits inosine monophosphate dehydrogenase and de novo protein synthesis, thereby inhibiting T-cell and B-cell proliferation; also suppresses leukocyte recruitment52,62
Azathioprine Max of 2.0-2.5 mg/kg/day PO Synthetic purine analog derived from 6-mercaptopurine disrupts nucleic acid synthesis and interferes with T-cell activation and function52,63
Methotrexate Initial test dose of 5 mg PO, gradually increasing weekly by 2.5-5.0 mg as needed, up to max dose of 30 mg/week
Biologicsb Infliximab: 5 mg/kg IV at weeks 0, 2, 6, followed by infusions every 6-8 weeks Etanercept: 50 mg twice weekly for 3 months, followed by 50 mg weekly Adalimumab: load with 80 mg SC, followed by 40 mg SC every 1-2 weeks Ustekinumab: weight ≤100 kg: 45 mg at 0 and 4 weeks, and then every 12 weeks weight >100 kg: 90 mg at 0 and 4 weeks, and then every 12 weeks
Folate antimetabolite that targets T cells, inhibiting dihydrofolate reductase and thymidylate synthetase, decreasing T-cell proliferation and migration, in addition to inducing adenosine release, which suppresses neutrophil oxidative activity, chemotaxis, and suppresses production of TNF-α, IL-6, IL-8, IL-1252,64
Infliximab: monoclonal Ab against TNF-α; binds soluble and membrane bound TNF-α and blocks its interaction with cell-surface receptors22,52,65
Etanercept: fusion protein; binds soluble TNF-α and blocks its interaction with cell-surface receptors22,52,65
Adalimumab: monoclonal Ab against TNF-α; binds soluble and membrane bound TNF-α and blocks its interaction with cell-surface receptors22,52,65
Ustekinumab: IgG1 monoclonal antibody; binds to the p40 subunit of both IL-12 and IL-23, blocking its interaction with cell-surface receptors, interfering with T-cell activation, and expression of TNF-α, IL-8, and other cytokines52,65
Thalidomidec 50-300 mg/day Inhibits TNF-α, chemotaxis of monocytes and leucocytes, and inhibits phagocytosis by neutrophils44,52,65
Severe, Refractory PG, or Those With Significant Contraindications to Other Treatment Options
Intravenous immune
2 g/kg/cycle per month IV divided over
Suppression or neutralization of cytokines; blockade of leukocyte
Intravenous immune globulin 2 g/kg/cycle per month IV divided over 3 consecutive days Suppression or neutralization of cytokines; blockade of leukocyte adhesion molecules; alterations in regulatory T cells and inhibition of the proinflammatory Th17 pathway52,65,66
globulin
3 consecutive days
aFollowed by daily oral prednisone.
adhesion molecules; alterations in regulatory T cells and inhibition of the proinflammatory Th17 pathway52,65,66
bConsider for severe PG in IBD (except etanercept, which is not helpful for IBD), rheumatoid arthritis, or hidradenitis suppurativa.
cConsider for severe PG in myelodysplastic syndrome. Ab, antibody; G-CSF, granulocyte colony-stimulating factor; Ig, immunoglobulin; IL, interleukin; PO, per os (by mouth); SC, subcutaneously; Th, T-helper cell; TNF, tumor necrosis factor.
COUNSELING
The patient should be given realistic expectations of the speed of recovery likely in this disease. Thus, although lesions develop and evolve within days, the healing process can take weeks to months.
614
PREVENTION/SCREENING
PREVENTION/SCREENING
A patient who has had a history of PG should be advised to avoid trauma to the skin as there is the possibility of precipitating a new lesion through
the pathergic phenomenon. If such patients have to undergo surgery, they should have close supervision by a dermatologist of their postoperative course. Patients with a history of aggressive PG may warrant a course of systemic steroids during and for a period (2 weeks or longer) postoperatively to prevent the development of new PG lesions, and subcuticular sutures should be used where possible. Patients with a history of PG and Crohn disease who are to have an ileostomy should be warned about the possible development of parastomal PG lesions, and to try to avoid irritation to the area to help prevent pathergy.
ACKNOWLEDGMENTS
We give special thanks to the authors of the prior version of this chapter, Frank C. Powell, Bridget C. Hackett, and Daniel Wallach.
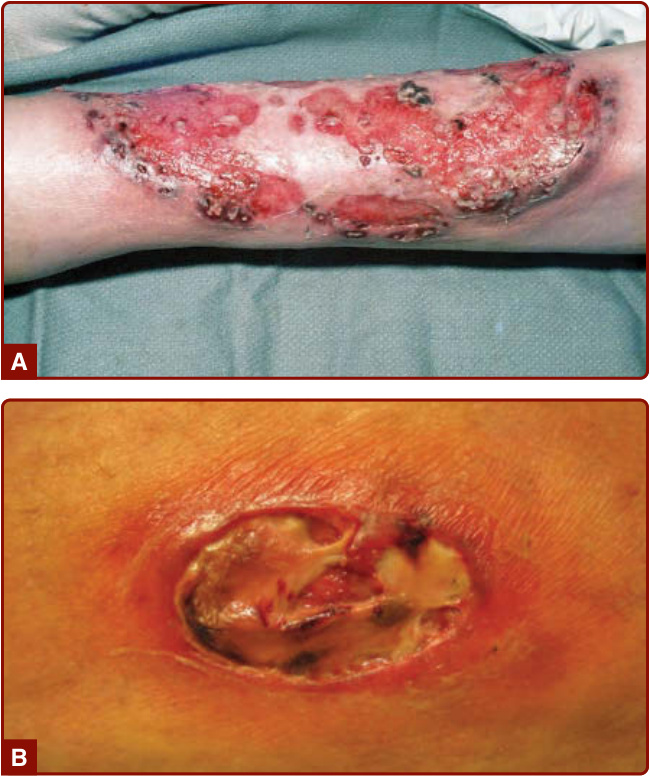
Figure 37-1 A, B, Established lesion of classic ulcerative pyoderma gangrenosum showing well-defined ulceration, undermining, and surrounding zone of erythema.
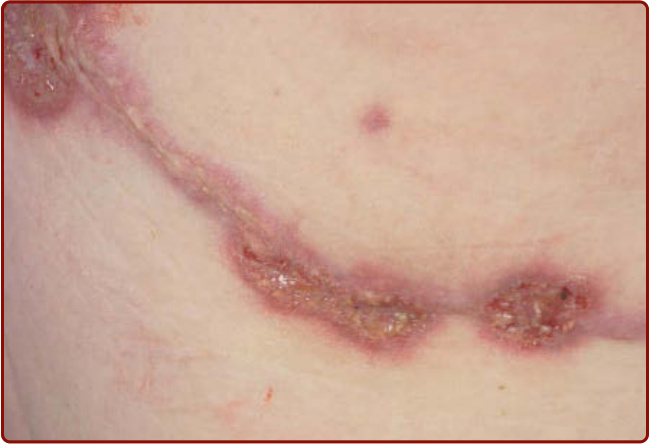
Figure 37-2 Pathergic pyoderma gangrenosum lesions occurring along a thoracotomy scar site. Note central ulceration, violaceous borders, and peripheral rim of erythema.
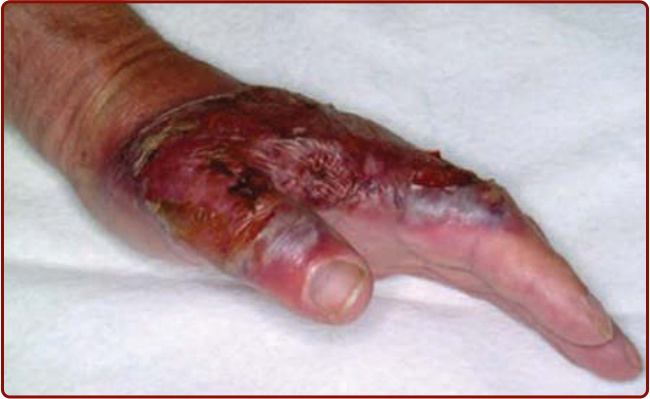
Figure 37-3 Bullous pyoderma gangrenosum lesion showing collapsed roof of blister and superficial erosive quality of the subsequent ulceration.
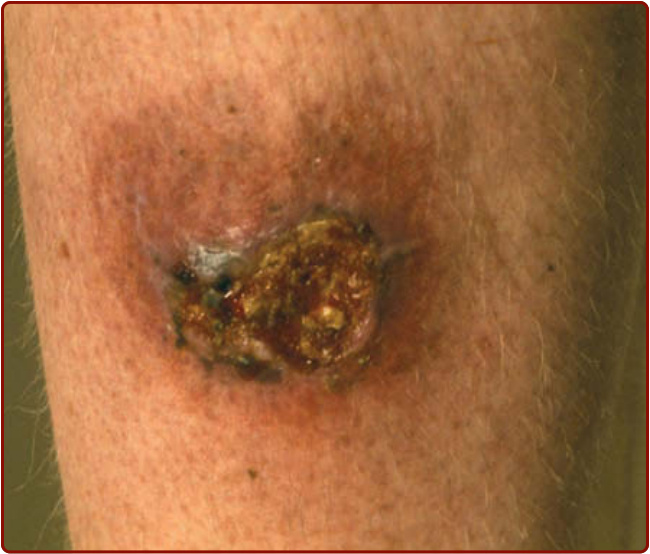
Figure 37-4 Vegetative pyoderma gangrenosum—an indolent area of chronic inflammation and ulceration that was present for months.
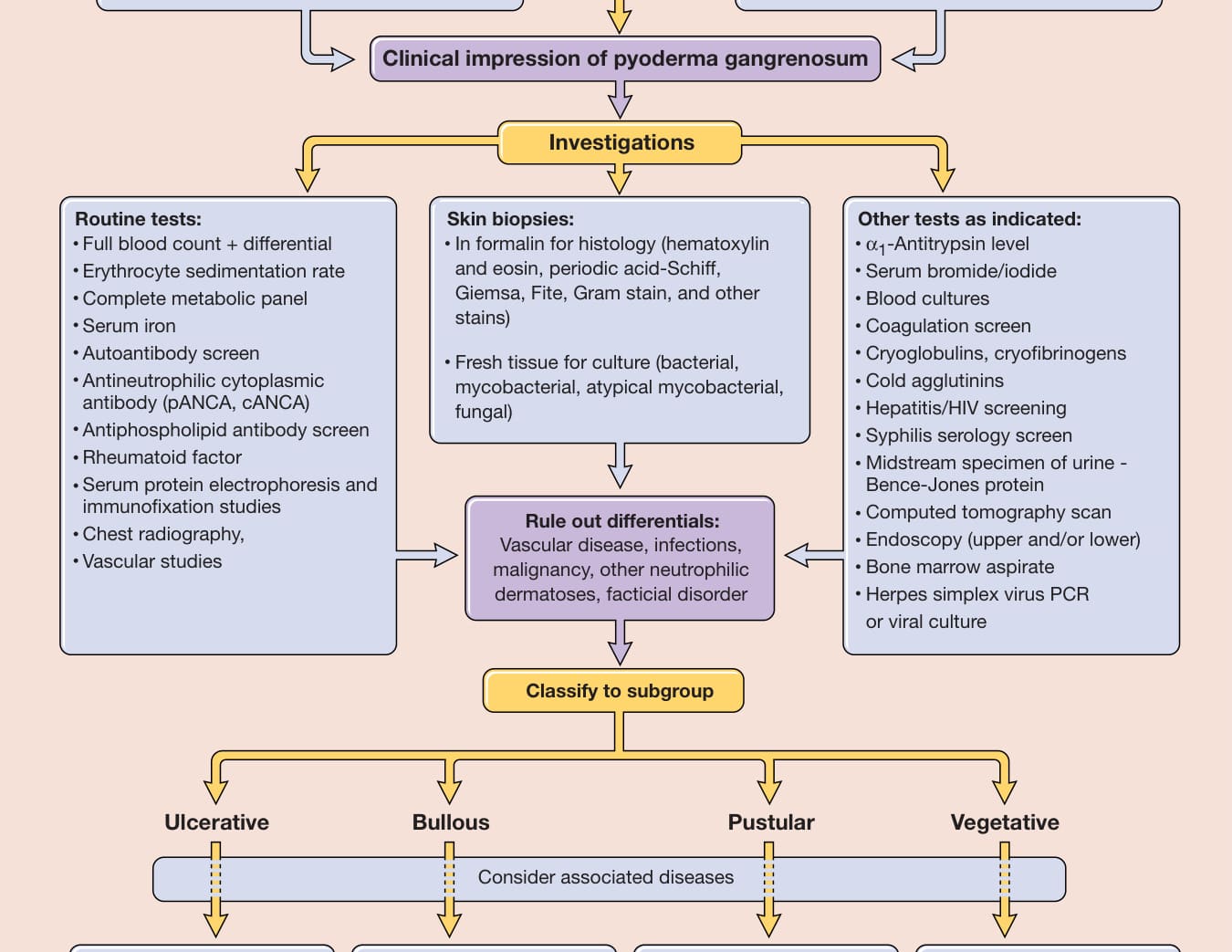
Figure 37-5 Approach to the patient with pyoderma gangrenosum. cANCA, cytoplasmic antineutrophil cytoplasmic antibody; pANCA, perinuclear antineutrophil cytoplasmic antibody; PCR, polymerase chain reaction.
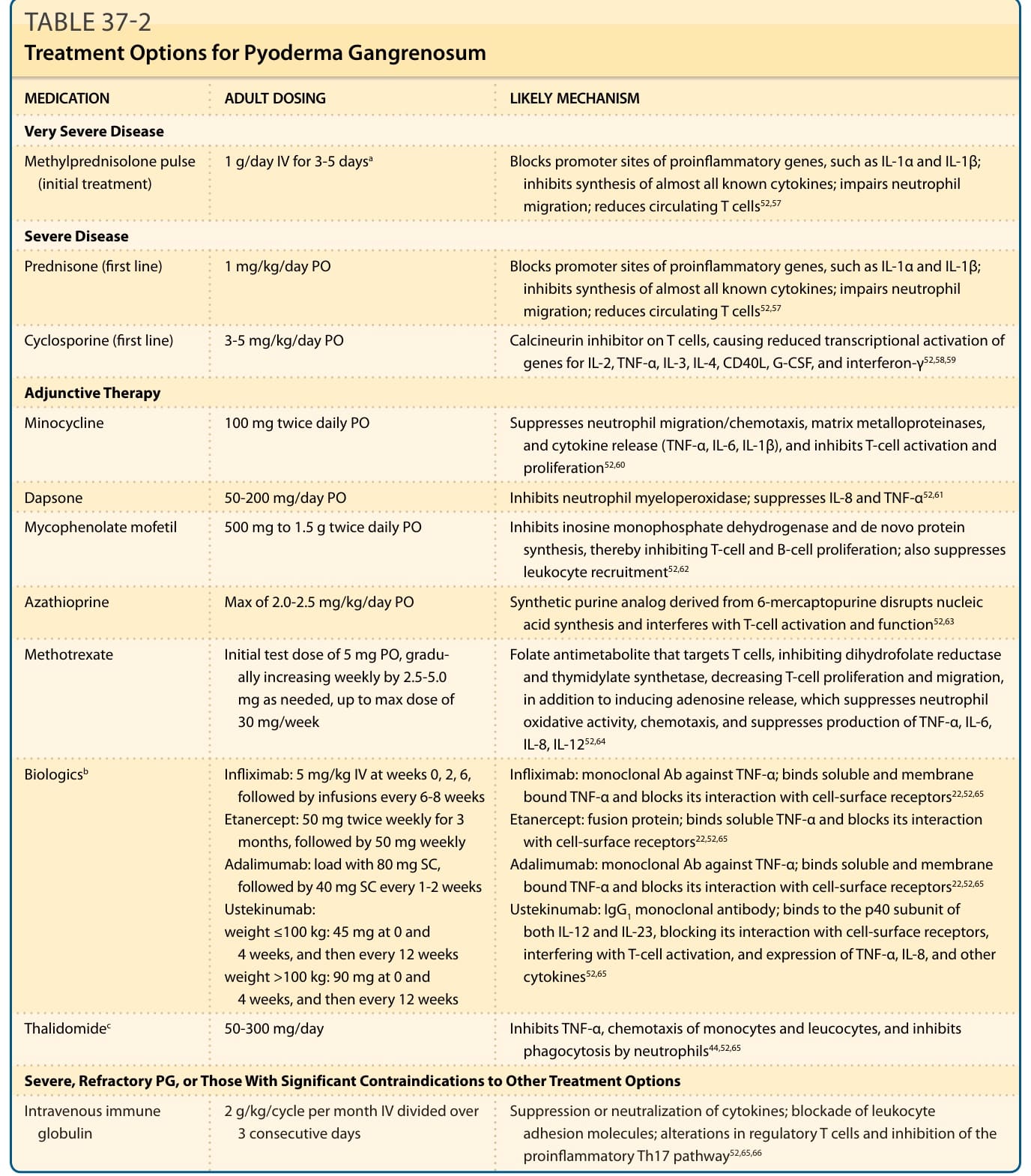
TABLE 37-2 Treatment Options for Pyoderma Gangrenosum