壞疽性膿皮症 (Pyoderma Gangrenosum)
PART 6
嗜中性球、嗜酸性球與肥大細胞疾病 (Neutrophilic, Eosinophilic, and Mast Cell Disorders)
重點一覽 (AT-A-GLANCE)
■ 壞疽性膿皮症(pyoderma gangrenosum, PG)是一種不常見的嗜中性球發炎性皮膚病況,典型表現為一個疼痛的結節、斑塊或膿疱,逐漸擴大並崩解,形成一個進行性擴大的潰瘍,邊緣隆起、有潛掘 (undermined)、呈紫紅色 (violaceous),並圍繞著一圈紅斑帶。癒合中的 PG 病灶會發展出篩狀 (cribriform) 外觀。
■ PG 因疼痛激惹現象(pathergic phenomenon,即外傷或刺激可誘發 PG 惡化),傾向於發生與復發在外傷部位。
■ PG 最常與全身性發炎相關,多數通報的病例都有相關的潛在疾病(如發炎性腸道疾病 inflammatory bowel disease、單株免疫球蛋白病 monoclonal gammopathy、血液疾病、發炎性關節炎、惡性腫瘤、化膿性汗腺炎 hidradenitis suppurativa 等);然而,PG 也可能先於這些疾病出現。PG 亦曾被報告與基因突變及症候群相關,例如化膿性關節炎、壞疽性膿皮症與痤瘡(pyogenic arthritis, pyoderma gangrenosum, and acne, PAPA)症候群,或在無可辨識之潛在疾病的情況下發生。
■ PG 最常以典型的潰瘍型 (ulcerative variant) 表現,但也可能以大疱型 (bullous)、膿疱型 (pustular) 或增殖型 (vegetative) 出現。不同型別的臨床特徵有時在個別病人身上重疊,但通常會有一種型別主導臨床表現。
■ 沒有任何實驗室檢查或檢查項目能確切地建立 PG 的診斷。組織病理學發現並非診斷性的,但在適當的臨床情境下能支持 PG 的診斷,且對於排除其他診斷至關重要。
■ 特定的標準(見「診斷演算法 (Diagnostic Algorithm)」一節)可提示 PG 的診斷,但其他病況(尤其是感染、血管疾病與惡性腫瘤)必須被排除。
■ 治療的主軸是全身性免疫抑制劑,加上適當的局部 (local) 與外用 (topical) 治療。
■ 典型潰瘍型 PG 是一種慢性病況。完全癒合通常需要數個月的治療;許多病人需要維持治療,且復發很常見。潰瘍型與大疱型 PG 的病人會經歷顯著的病態 (morbidity) 與死亡率。
定義 (DEFINITIONS)
壞疽性膿皮症(pyoderma gangrenosum, PG)是一種不常見、慢性的嗜中性球發炎性皮膚病況,其典型的潰瘍型通常初期表現為一個疼痛的結節、斑塊或膿疱,逐漸擴大並崩解,形成一個進行性擴大的潰瘍,邊緣隆起、有潛掘 (undermined)、呈紫紅色,並圍繞著一圈紅斑帶。¹,² 這些病灶通常非常疼痛,癒合時發展出篩狀 (cribriform) 外觀,並在高達 50% 的病例中傾向於發生與復發在外傷部位。¹,² PG 被認為與全身性發炎相關,多數通報的病例都伴隨自體發炎 (autoinflammatory) 或血液性病況,且通常對免疫抑制有反應。¹,³⁻⁵ PG 最常以典型的潰瘍型表現,但也可能以大疱型、膿疱型或增殖型出現。除非另有說明,本文中提及 PG 時,所指的均為典型的潰瘍型。
流行病學 (EPIDEMIOLOGY)
PG 估計每年每百萬人中影響高達 10 例,並占慢性腿部潰瘍病例的高達 3%,但可發生於身體任何部位。PG 曾在所有年齡層的人身上被通報過;然而,多數病例出現於生命中的第二至第六個十年,可能有女性偏多的現象。²,⁶
疾病關聯 (DISEASE ASSOCIATIONS)
高達 70% 通報的 PG 病例與已知的潛在全身性疾病相關,例如發炎性腸道疾病(inflammatory bowel disease, IBD)、單株免疫球蛋白病 (monoclonal gammopathy)、血液疾病、發炎性關節炎、惡性腫瘤、化膿性汗腺炎 (hidradenitis suppurativa) 及其他,即使 PG 可能先於這些疾病出現。PG 亦曾與基因突變相關聯,作為某個遺傳症候群的一部分,例如化膿性關節炎、PG 與痤瘡(pyogenic arthritis, PG, and acne, PAPA)症候群,或在無可辨識之潛在疾病的情況下發生。⁷
將近三分之一的 PG 病例與 IBD 相關,克隆氏症 (Crohn disease) 與潰瘍性結腸炎(ulcerative colitis, UC)的頻率相近,雖然多數克隆氏症病例為結腸侵犯。³,⁸ 副蛋白血症(paraproteinemia,單株免疫球蛋白病)見於 10% 的 PG 病例⁹,最常見為免疫球蛋白 A 單株免疫球蛋白病。¹⁰ 血液惡性腫瘤在高達 7% 的 PG 病例中被見到,最常通報的亞型為急性骨髓性白血病 (acute myeloid leukemia),且多數白血病病例先有血液異常(如骨髓增生不良症候群 myelodysplastic syndrome)為前驅。¹,¹¹ 骨髓增生性疾病(myeloproliferative diseases),例如真性紅血球增多症 (polycythemia vera)¹²、原發性血小板增多症 (essential thrombocythemia)¹³,以及骨髓纖維化(myelofibrosis,特發性骨髓化生 agnogenic myeloid metaplasia)¹⁴,¹⁵,還有類白血病反應 (leukemoid reaction)¹⁶,也曾與 PG 相關。亦有較少的報告指出 PG 與何杰金氏 (Hodgkin) 及非何杰金氏淋巴瘤 (non-Hodgkin lymphomas)、皮膚 T 細胞淋巴瘤 (cutaneous T-cell lymphoma),以及源自乳房、卵巢、膀胱、攝護腺、結腸、類癌 (carcinoid) 與支氣管的實體腫瘤相關⁹,¹⁷,雖然這些較不常見之關聯的意義較不確定。亦有藥物誘發 PG 的報告,包括數例與顆粒球群落刺激因子(granulocyte colony-stimulating factor, G-CSF)相關的 PG,在停用 G-CSF 後消退。¹⁸
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
PG 在型態學上可分類為 (a) 潰瘍型、(b) 大疱型、(c) 膿疱型或 (d) 增殖型。雖然有些病人可能表現出超過一種型別(例如孤立的膿疱性病灶常發生於潰瘍型 PG 的病人),但通常會有一種型別主導臨床表現,病人應據此分類。潰瘍型 PG 是 PG 最常見且最早被描述的型別。潰瘍型 PG 病人最常見的初始臨床病灶是一個劇痛的紅斑性丘疹、膿疱或結節性癤腫 (nodular furuncle)(這些病灶通常為單一,但也可能為多發)。它們在外觀看似正常的皮膚上爆發,最常見的部位為腿部。逐漸擴大的初始病灶發展出一圈周圍的紅斑帶,向周圍皮膚延伸。隨著它擴大,中央退化、壞死並糜爛,轉變為一個糜爛性潰瘍(圖 37-1),其發展伴隨疼痛令人警覺地增加。潰瘍常有一個藍色/紫紅色的潛掘 (undermined) 邊緣,基底覆蓋著膿性壞死物質。潰瘍型 PG 在某些病例中可能深層糜爛,露出肌肉或肌腱。¹,²
PG 也可發生於外傷或手術部位(圖 37-2),此即所謂的疼痛激惹現象 (pathergic phenomenon),外傷或刺激可誘發 PG 誇大的惡化。¹,²
大疱型 PG(有時稱為非典型 PG, atypical PG)表現為一個疼痛、快速擴大的淺表發炎性水疱,迅速糜爛。在早期急性階段,病灶的大疱本質很明顯,但因水疱頂部迅速壞死,仔細檢查已形成病灶的邊緣對於揭示其大疱本質是必要的(圖 37-3)。大疱型 PG 常與血液疾病相關,且最常出現於上肢。¹⁹
此型 PG 可能在臨床與組織學上與 Sweet 症候群(嗜中性球皮膚病之一,其本身常與血液疾病相關)重疊。膿疱型 PG(亦稱為 IBD 的膿疱性疹)是一種泛發性疹,幾乎只發生於急性 IBD(通常為 UC)惡化的情境中。其發作戲劇化,軀幹(以及較少程度的顏面與四肢)上迅速發展出多發、大型、圓形至卵圓形的疼痛膿疱。若不控制腸道疾病,此疹難以控制,在某些病例中可能需要切除病變的腸道。¹,²
增殖型 PG(或淺表肉芽腫性膿皮症, superficial granulomatous pyoderma)通常表現為單一的癤腫樣 (furunculoid) 結節、膿瘍、斑塊或淺表潰瘍,典型位於軀幹(圖 37-4)。與其他型別相反,此不常見的型別發作緩慢,所造成的不適輕微,且通常不伴隨全身性疾病。此型 PG 通常比其他型別對局部或溫和形式的全身性治療更有反應。²⁰
術後 (postoperative) 與造口旁 (parastomal) PG 被認為是潰瘍型 PG 展現疼痛激惹現象的例子。¹,²
非皮膚表現 (NONCUTANEOUS FINDINGS)
由於多數 PG 病例與並存的潛在全身性疾病相關或先於其出現,因此必須進行詳盡的病史詢問與身體檢查,特別要尋找 IBD、發炎性關節炎、血液疾病(單株免疫球蛋白病與其他血液惡質病 dyscrasias)、血液惡性腫瘤與內臟惡性腫瘤的臨床與生物標記。此外,臨床醫師應意識到內臟器官(肺、骨關節、中樞神經系統 CNS、心血管系統、腹腔內臟、眼)的無菌性嗜中性球浸潤 (sterile neutrophilic infiltration) 可與皮膚 PG 相關發生,甚至先於其發作。²¹
併發症 (COMPLICATIONS)
活躍或控制不良的皮膚 PG 造成顯著的病態,包括嚴重疼痛、行動能力喪失、慢性病貧血 (anemia of chronic disease) 以及治療介入的併發症。許多 PG 的治療必須投予許多個月,且可能有顯著的副作用。例如,皮質類固醇 (corticosteroids) 可誘發或惡化糖尿病,環孢素 (cyclosporine) 可誘發或惡化腎功能不全,而免疫療法可導致免疫功能低下 (immunocompromise),增加常見與非典型微生物感染的風險。因此,對病人進行密切監測與追蹤是必要的。必須小心避免對皮膚造成外傷,且選擇性手術應謹慎進行,因為有透過疼痛激惹現象誘發新 PG 病灶的可能性。此外,未能辨識 PG 中內臟器官的嗜中性球浸潤,可能導致不必要的外科手術。¹,²²
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
PG 的病因不明,其致病機轉所知甚少,而其病理生理機制很可能依相關的潛在促成疾病而異。促成、誘發與調控這些異常的因子很可能是多因子的,包括基因傾向、副免疫 (paraimmune) 及/或副腫瘤 (paraneoplastic) 現象⁷,這些可能造成對免疫反應與發炎的敏感性增加,或其活性增加。有趣的是,PG 與廣泛範圍的疾病相關發生,然而它們全都導致或關聯於 PG 這個共同的表型,使人提出一個假說,即這些疾病以某種方式匯聚於導致異常嗜中性球活性與功能的發炎途徑上。²³ 儘管有此相似性,關於 PG 的疾病活性與潛在疾病狀態活性之間的關聯,各報告仍有差異。在 IBD 的情境中,PG 的活性傳統上與腸道的發炎過程相關。根據 Agarwal 等人的系統性回顧⁸(納入 60 名病人,其中 35 名(58%)在 PG 診斷時有活躍的 IBD,僅 9 名(15%)有不活躍的 IBD,16 名(27%)的 IBD 活性未明)。雖然有些人報告腸道疾病的惡化與皮膚病灶的惡化之間無明顯關聯²⁴,²⁵,但有多份報告證明 PG 在針對病變腸道進行治療後癒合。Powell 等人¹² 報告了一個病例系列,其中 9 名 UC 病人在接受全大腸直腸切除術 (total proctocolectomy) 後,PG 病灶全部改善;Talansky 等人²⁶ 報告了在切除病變腸道後 2 個月內 PG 皮膚迅速癒合的病例,包括 3 名嚴重迴腸結腸炎 (ileocolitis)、1 名嚴重 UC 與 1 名中度 UC 的病人。值得注意的是,亦有一例 PG 報告發生於一名 UC 病人在大腸直腸切除術後 10 年⁹,這引發了是否仍有殘留病變腸道的疑問。大疱型 PG 的改善曾在急性骨髓性白血病經治療並緩解後被報告²⁷,這或許可部分由以下發現解釋:在一名急性骨髓性白血病病人的 PG 皮膚樣本中發現了由非典型骨髓芽細胞 (atypical myeloblasts) 組成的白血病細胞浸潤。²⁸ PG 的完全康復曾在以克拉屈濱 (cladribine) 成功治療潛在的毛細胞白血病 (hairy cell leukemia) 後被報告。²⁹ 與腎細胞癌 (renal cell carcinoma) 相關之難治性 PG 的成功治療曾以烏特克單抗 (ustekinumab) 被報告,但僅在切除癌瘤後才見效。³⁰
發炎介質與異常嗜中性球活性 (INFLAMMATORY MEDIATORS AND ABERRANT NEUTROPHIL ACTIVITY)
T 細胞可能透過各種機制(包括細胞激素訊號傳遞與抗原刺激)在 PG 的發展中扮演重要角色³¹,並很可能參與了部分所觀察到的異常嗜中性球活性。Marzano 等人³² 檢視了 21 名 PG 病人的病灶皮膚切片,發現傷口邊緣的浸潤主要為 CD3+ T 細胞與 CD163+ 巨噬細胞,並伴隨傷口床中腫瘤壞死因子-α(tumor necrosis factor-α, TNF-α)、介白素(interleukin, IL)-8(強效嗜中性球趨化劑)、IL-17(促發炎細胞激素;刺激 IL-8 與 G-CSF 的表現)、基質金屬蛋白酶(matrix metalloproteinase, MMP)-2/MMP-9(介導組織損傷的膠原酶)的增加。基於 PG 病灶活躍的進展邊緣存在淋巴球浸潤,有人推測淋巴球抗原活化伴隨細胞激素釋放與嗜中性球招募的發生。這可能不僅發生於皮膚,也發生於其他組織,如肺、腸與關節。³³ 基於 PG 病理標本中顯著的富含嗜中性球的真皮浸潤,以及 PG 對抗嗜中性球藥物(如達普頌 dapsone)的反應,嗜中性球被認為在 PG 中扮演重要角色。多項研究報告了 PG 病人在嗜中性球趨化性 (chemotaxis)、訊號傳遞與遷移 (trafficking) 方面的異常。⁴,³⁴,³⁵ 這些嗜中性球異常,以及組織學上嗜中性球的優勢,也使 PG 在更廣泛的嗜中性球皮膚病譜系中占有一席之地,這些疾病包括 Sweet 症候群(急性發熱性嗜中性球皮膚病 acute febrile neutrophilic dermatosis)、貝賽特氏病 (Behçet disease)、疱疹樣皮膚炎 (dermatitis herpetiformis) 與嗜中性球小汗腺汗腺炎 (neutrophilic eccrine hidradenitis)。除了真皮嗜中性球增多外,這些病況共有的其他常見特徵包括相關的潛在疾病、治療上的相似性,以及疼痛激惹 (pathergy) 的傾向,某些疾病比其他疾病更明顯(PG、Sweet 氏、貝賽特氏、疱疹樣皮膚炎)。⁷
TNF-α 在促發炎細胞激素(包括 IL-1、IL-6、IL-8 與干擾素-γ)的產生中扮演重要角色,並已被證明能刺激並增強嗜中性球的脫顆粒 (degranulation) 與超氧化物 (superoxide) 的產生。³⁶⁻³⁸ TNF-α、IL-6 與可溶性 IL-2 受體的升高曾在一名 PG 病人身上被報告,其血清濃度與潰瘍活性相關。³⁹ 除了在 PG 中增加外,IL-8 也已被證明能在轉染重組人類 IL-8 的人類皮膚異種移植 (xenografts) 中造成類似的潰瘍。⁴⁰ Guenova 等人⁴¹ 以聚合酶連鎖反應 (polymerase chain reaction) 發現 PG 病灶相較於正常皮膚有升高的 IL-23 表現,這具有意義,因為 IL-23 在驅動與 IL-17 產生及嗜中性球招募相關的發炎中扮演重要角色。此外,PAPA 症候群與某些型別的病人已被發現有 IL-1β 與 IL-18 產生的增加。⁴²,⁴³
遺傳學 (GENETICS)
揭示 PG 的基因關聯很重要,因為它能提供對 PG 病理生理更佳的理解,並協助闡明治療的新靶點。PG 曾被報告與全身性疾病、基因突變與症候群相關,或單獨發生。DeFilippis 等人⁴⁴ 關於與 PG 相關之症候群與基因突變的系統性回顧納入了 823 例 PG,揭示了 31 例(3.8%)PAPA 症候群及其變異型、2 名(0.2%)有 Janus 激酶(Janus kinase, JAK)2 突變的病人、3 名(0.4%)有亞甲基四氫葉酸還原酶(methylenetetrahydrofolate reductase, MTHFR)突變並發展出 PG 樣潰瘍的病人,以及 14 名(1.7%)無特定通報突變的家族性病例,此外還有與 IBD、多發性關節炎與血液疾病相關的病例。PAPA 症候群是一種體染色體顯性疾病,已被連結到位於第 15q 號染色體上的脯胺酸-絲胺酸-蘇胺酸磷酸酶交互作用蛋白 1(proline-serine-threonine phosphatase interacting protein 1, PSTPIP1;亦稱為 CD2 抗原結合蛋白 1 [CD2BP1])基因的突變,該基因編碼 PSTPIP1,與 pyrin 結合並調控發炎體 (inflammasome)。PSTPIP1 的突變導致發炎體的抑制減少,以及 IL-1β 與 IL-18 的產生增加,這可能造成 PAPA 所見的發炎。⁴²,⁴³
文獻中已描述 PAPA 症候群的多種變異型,包括 PAPASH 症候群(化膿性無菌性關節炎、PG、囊腫性痤瘡與化膿性汗腺炎 pyogenic sterile arthritis, PG, cystic acne, and hidradenitis suppurativa),其由 PSTPIP1 基因的 E277D 錯義突變 (missense mutation) 所致⁴⁵;PASH 症候群(PG、痤瘡與化膿性汗腺炎 PG, acne, and suppurative hidradenitis),其由 PSTPIP1 啟動子區域中 CCTG 微衛星重複序列 (microsatellite repeat) 的增加所致⁴⁶;以及其他變異型。JAKs 是細胞分化、增生與凋亡所需的細胞內酪胺酸激酶。JAKs 的突變可造成發炎性與骨髓增生性疾病,也曾與 PG 病例相關。有趣的是,此途徑由 G-CSF 與 TNF-α,以及 ILs 與干擾素所活化。⁴⁴
已發現某些與 IBD 易感性連結的特定基因座與 PG 的發展相關。⁴⁷ Weizman 等人⁴⁷ 使用全基因組關聯資料 (genome-wide association data) 回顧了 5756 名 IBD 病人,並描述了數個與 IBD 易感性相關且與 PG 顯著相關的基因,包括 IL8RA(嗜中性球遷移的介導者)、MUC17、MMP24、WNK2、DOCK9、PRDM1 與 NDIFIP1²²,此外還有其他與 IBD 及 PG 相關的基因座,例如 GPBAR1、TIMP3 與 TRAF31P2。⁴⁴
荷爾蒙影響 (HORMONAL INFLUENCES)
有報告指出,數種皮膚病況(如化膿性汗腺炎與尋常性痤瘡 acne vulgaris)有經前惡化 (premenstrual flares) 的情形;醫學文獻描述雌激素 (estrogen) 在皮膚傷口癒合中扮演正面角色,而雄激素 (androgens),或雄激素受體活化,扮演負面角色。Jourabchi 等人⁴⁸ 報告了一例有經前惡化的 PG,以複方口服避孕藥 (combined oral contraceptive) 與抗雄激素藥(乙炔雌二醇/屈螺酮 ethinyl estradiol/drospirenone)控制。⁴⁸
有趣的是,化膿性汗腺炎與痤瘡已知在某些病人身上會與 PG 同時發生,提示有相關的病理生理機制。此外,PG、化膿性汗腺炎與痤瘡有一些其他重疊的特徵,包括病灶皮膚中升高的 IL-17 與 TNF-α,以及對 TNF-α 抑制劑的正面反應。²²,⁴⁹,⁵⁰
診斷 (DIAGNOSIS)
PG 目前的診斷是臨床性的,而且非常重要的是,要在排除皮膚表現的其他潛在病因之後(尤其是感染性病因,如非典型分枝桿菌 atypical mycobacteria 與深部黴菌感染 deep fungal infections),因為迄今尚未發現特異性的血清學、組織學或基因標記。
診斷演算法 (DIAGNOSTIC ALGORITHM)
由於沒有針對 PG 的特異性診斷檢查,因此提出以下標準以協助做出 PG 的診斷,其需要同時符合兩項主要標準與至少 2 項次要標準:¹,³,⁷,²²
- 主要標準 (Major criteria):
a. 在一名沒有發燒、顯著毒血症 (toxemia) 或相關藥物攝入的病人身上,突然發作一個疼痛的病灶,具有「皮膚表現 (Cutaneous Findings)」一節所描述的特徵性型態。
b. 以特殊組織學檢查/染色及陰性的組織培養,在組織病理學上排除顯著的血管炎 (vasculitis)、惡性腫瘤與感染性微生物,並以適當的檢查排除顯著的血管鬱滯/阻塞 (vascular stasis/occlusion)。
- 支持診斷的次要標準 (Minor criteria) 如下:
a. 發生於患有全身性疾病的個體,如「疾病關聯 (Disease Associations)」一節所述。
b. 典型的組織學 PG 發現。(註:在曾接受全身性皮質類固醇的病人中,嗜中性球的存在可能被鈍化,而可能改以單核細胞為主。)
c. 在開始高劑量全身性皮質類固醇治療時疼痛與發炎迅速減輕,以及對高劑量全身性皮質類固醇治療有迅速的潰瘍癒合反應,潰瘍大小在 1 個月內減少 50%。
d. 提示疼痛激惹 (pathergy) 的病史,或篩狀瘢痕 (cribriform scarring) 的臨床發現。
提供照護者必須意識到,許多病人可能在皮膚表現初次發展後數個月或數年才就診,期間正在服用或曾嘗試過各種藥物(包括全身性皮質類固醇或其他免疫抑制劑),因此可能不會以典型的臨床發現或診斷標準表現。因此,取得詳細的病史對於做出適當的診斷至關重要。
輔助檢查 (SUPPORTIVE STUDIES)
組織病理學檢查與感染性染色及培養 (HISTOPATHOLOGIC STUDIES AND INFECTIOUS STAINS AND CULTURES)
皮膚的組織病理學變化並非診斷性的,但能高度提示 PG。病理變化必須結合整體臨床表現來考量。所見的顯微鏡變化取決於 (a) PG 的臨床型別(潰瘍型、大疱型、膿疱型或增殖型)、(b) 切片的時機,以及 (c) 切片相對於發炎過程的部位。應從 PG 病灶的邊緣取一個切開楔形皮膚切片 (incisional wedge skin biopsy),小心取樣一部分由邊緣穿過進入活躍發炎區域的正常皮膚,以便辨別各種組織學型態。切除的組織接著應分割,一部分(新鮮組織)送去做細菌培養、含抹片的分枝桿菌培養,以及含顯微鏡檢的黴菌培養,另一部分則以福馬林送去做組織學評估,要求進行蘇木精與伊紅 (hematoxylin and eosin) 製備,並加上過碘酸希夫染色 (periodic acid–Schiff)、Giemsa、Fite、Gram 以及其他認為相關的染色。若無法進行切開式切片,可改做兩個 4-mm 環鑽切片 (punch biopsies)。免疫螢光檢查 (Immunofluorescence studies) 在病灶皮膚中可能顯示陽性的血管周圍染色,這可能繼發於深度發炎;它並非診斷所必需,除非鑑別診斷中懷疑血管炎,否則可省略。由於 PG 可能因上述微生物的繼發感染而複雜化,不癒合或復發的 PG 應重新切片以排除合併感染。切片在病灶內的位置特別重要,因為從已形成病灶的中央取的切片,可能與從周邊皮膚取的切片有不同的發現。在從圍繞潰瘍型 PG 活躍病灶的「帶 (zone)」或紅斑區域所取的切片中,可能見到明顯的血管周圍淋巴球浸潤。可見淋巴球浸潤血管壁,伴隨壁內 (intramural) 與血管內 (intravascular) 纖維蛋白沉積,顯示血管損傷(有時稱為淋巴球性血管炎 lymphocytic vasculitis)。膿瘍形成伴隨強烈的真皮嗜中性球浸潤可能延伸至脂肪層 (panniculus) 與組織壞死區域,主導從潰瘍型 PG 病灶中央區域所取切片的組織學發現。白血球破裂 (Leukocytoclasis) 並非顯著的發現,雖然偶爾可在壞死中心附近見到白血球破裂性血管炎 (leukocytoclastic vasculitis) 的證據,但這是次要特徵,被認為是強烈發炎變化的繼發結果,而非原發事件。對一名大疱型 PG 病人的病灶皮膚進行組織學檢查,將顯示一個表皮下或表皮內水疱,伴隨其上覆的表皮壞死與明顯的上真皮水腫,且嗜中性球突出。膿疱型 PG 的切片將顯示緻密的真皮嗜中性球浸潤(常以毛囊為中心),伴隨表皮下水腫與嗜中性球浸潤至表皮,並有角質層下聚集 (subcorneal aggregations)。增殖型 PG 在組織學上的特徵為假上皮瘤性增生 (pseudoepitheliomatous hyperplasia)、竇道形成 (sinus tract formation),以及在局灶性真皮嗜中性球膿瘍的情境中出現柵欄狀肉芽腫 (palisading granulomas)。³,⁷,³⁸
實驗室檢查 (LABORATORY TESTING)
所有 PG 病人都應進行這些檢查:含白血球分類計數的全血球計數 (full blood cell count with differential white cell count);完整代謝套組 (complete metabolic panel);紅血球沉降速率 (erythrocyte sedimentation rate);自體抗體篩檢(包括抗核抗體 antinuclear antibodies、抗 Ro/La 抗體 anti-Ro/La antibodies);類風濕因子 (rheumatoid factor);抗磷脂質抗體篩檢 (antiphospholipid antibody screen);抗嗜中性球細胞質抗體 (antineutrophilic cytoplasmic antibodies);血清蛋白電泳 (serum protein electrophoresis) 與免疫固定法檢查 (immunofixation studies);以及胸部 X 光攝影 (chest radiography)。
血管檢查 (VASCULAR STUDIES)
所有腿部潰瘍的病人都應進行靜脈逆流檢查 (venous reflux studies),以排除顯著的靜脈功能不全 (venous insufficiency) 與深部靜脈血栓 (deep venous thrombosis),此外還要進行踝/趾肱指數檢查 (ankle/toe brachial index studies),以排除缺血 (ischemia) 作為潰瘍的潛在病因。
其他檢查 (OTHER INVESTIGATIONS)
在多數病人中,以下額外的檢查可能有其必要:內視鏡檢查 (endoscopy)(上及/或下消化道)以排除常相關的 IBD;在有指徵時仔細檢查周邊血液型態,若發現任何異常,應進行骨髓抽吸檢查 (bone marrow aspirate examination);腹部超音波(包括肝/脾/主動脈);以及電腦斷層攝影 (computed tomography) 以視覺化胸腔、腹部與骨盆。圖 37-5 概述了其他導向性檢查。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
PG 病人需要考量的鑑別診斷相當廣泛。⁵¹ PG 的不同型別(潰瘍型、大疱型、增殖型、膿疱型)提示了不同的替代診斷,而 PG 發生於某些皮膚部位則為臨床醫師帶來進一步的診斷問題,如表 37-1 所示。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
預後取決於 PG 的型別;病人的年齡與性別;潛在全身性疾病與其他共病的存在;以及將疾病控制所需治療的類型、劑量與持續時間。典型潰瘍型 PG 是一種慢性復發性疾病,具有顯著的病態與死亡率。此型別中年齡大於 65 歲的病人與男性病人似乎有較差的預後。增殖型 PG 的病人一般有良好的預後,且與典型 PG 相比,增殖型 PG 病灶更可能在不使用口服皮質類固醇的情況下癒合。²⁰ 造口旁 PG 有良好的預後,常對外用或病灶內類固醇治療有反應。膿疱型 PG 的病人若能控制通常伴隨此型別的嚴重 IBD,常能使其皮膚病灶完全緩解。患有大疱型 PG 並伴隨相關血液疾病的病人預後不佳。大疱型 PG 在一名穩定的真性紅血球增多症 (polycythemia rubra vera) 病人身上發作,曾被報告為白血病性轉變 (leukemic change) 發作的前兆。³⁰,³⁸,⁵²
治療 (MANAGEMENT)
治療途徑 (APPROACH TO MANAGEMENT)
了解 PG 發生的背景對於決定適當的治療途徑至關重要。PG 不可預測的本質及其在個別病人身上多變的侵襲性,使得治療需要彈性的途徑。治療藥物必須適應病人的生理狀態(懷孕、老年、腎臟疾病、糖尿病、免疫抑制狀態等),也要配合其潛在的相關疾病狀態。雖然潛在疾病的病程並不總是與 PG 的惡化相關,但治療它可導致 PG 的改善。無論潛在疾病狀態為何,全身性類固醇及/或環孢素 (cyclosporine) 都可能有效控制 PG,但 PG 常需要合併治療與多次藥物試驗才能達到癒合,在這些病例中,應利用潛在疾病狀態來協助指引藥物選擇與治療途徑。充分休息、有效的疼痛緩解,以及對任何影響癒合之共病(靜脈鬱滯疾病、糖尿病、伴隨體液過多的鬱血性心衰竭等)的適當治療,也是 PG 病人整體治療策略中的關鍵。若存在其他全身性疾病,需要與內科專科醫師及相關次專科醫師密切合作。
藥物 (MEDICATIONS)
PG 的確切治療指引並不存在,部分原因是與不同潛在疾病相關的 PG 似乎表現不同,也因為文獻中的數據匱乏。治療途徑是基於導向性的治療性免疫調節 (immunomodulation),以減少發炎並促進癒合。治療的選擇取決於 PG 的嚴重度,且在可能時也應嘗試治療病人的潛在病況(例如,IBD 病人用英利昔單抗 infliximab,類風濕性關節炎病人用甲胺喋呤 methotrexate)。具有小而淺潰瘍的輕度 PG 或增殖型 PG 的斑塊,可能對外用第 I 級皮質類固醇(clobetasol propionate 0.05%)、病灶內皮質類固醇,或鈣調神經磷酸酶抑制劑(如 tacrolimus 0.1%)每日兩次塗抹有反應。較嚴重或廣泛 PG 的第一線治療是全身性糖皮質素 (systemic glucocorticoids)。我們通常以每日 1 至 2 mg/kg 的口服 prednisone 開始治療,最大劑量為 150 mg/day。對於非常侵襲性的疾病,靜脈注射脈衝式 methylprednisone 1 g/day 連續 3 至 5 天作為初始治療可能有幫助,接續每日口服糖皮質素。¹,⁵³,⁵⁴ 對全身性糖皮質素的反應通常迅速,典型在 1 至 2 週內停止進展。由於長期治療伴隨顯著的不良反應,一旦 PG 的進展停止,應開始緩慢的糖皮質素減量。同時開始一種類固醇節省輔助劑(如 cyclosporine、dapsone、infliximab)可在減量時協助預防惡化。¹
口服環孢素 (cyclosporine) 是無法耐受全身性糖皮質素之病人的另一種第一線治療,也用作輔助治療。一項多中心、平行組、觀察者盲性、隨機對照試驗比較了口服 prednisolone 0.75 mg/kg/day 與 cyclosporine 4 mg/kg/day 治療 PG,發現兩組間療效相當。⁵⁵ 不良反應包括腎毒性與高血壓,必須加以監測,建議的治療時程為少於 1 年。⁵⁵,⁵⁶
全身性糖皮質素與環孢素作用快速,使其成為初始治療的理想選擇,而其他某些療法可能需要數週才能產生治療效果,較適合作為輔助或維持治療,尤其是對於嚴重病例。嚴重的 PG 病例需要合併治療以控制病況同時將治療副作用降至最低,這並不罕見。表 37-2 概述了額外的療法。請注意,這些藥物多數需要特定的基線實驗室檢查結果、對不良反應的密切監測、預防藥物交互作用的謹慎,以及對免疫抑制的小心。維持治療應持續到傷口完全癒合,然後緩慢減量停藥。此外,潰瘍型 PG 的病人有顯著的復發風險,因此需要長期追蹤,且部分病人需要長期維持治療。
無反應的壞疽性膿皮症與治療考量 (UNRESPONSIVE PYODERMA GANGRENOSUM AND TREATMENT CONSIDERATIONS)
當 PG 對治療無反應,或若癒合停滯時,提供照護者必須重新評估此病例:
- 是否有未受控制的共病(如靜脈鬱滯、血管缺血、糖尿病)正在抑制傷口癒合且需要處理?
- 是否有新的合併感染需要檢查與治療?若有,是否應再進行一次切片以做全面培養 (pan-culture)?
- 是否有更好、更具導向性的藥物應該嘗試?
在難治性或難以治療的 PG 病人中,可考慮進行基因分析,這可能有助於辨識更具導向性的治療。DeFilippis 等人⁴⁴ 回顧了已發表的關於與 PG 相關之症候群與基因突變的文獻,發現 PG 對不同治療的反應取決於潛在的疾病過程。例如,PAPA 症候群與伴隨 IL-1β 增加的變異型病例,在抗 IL-1β 單株抗體 canakinumab、IL-1 受體拮抗劑 anakinra 與 TNF-α 抑制劑下改善;而一例伴隨 JAK2 突變的真性紅血球增多症對 JAK1/JAK2 抑制劑 ruxolitinib 有反應;伴隨 MTHFR 突變的 PG 樣病例(其途徑依賴維生素 B6、B9 與 B12)則在 B 群維生素治療下改善。⁴⁴
傷口照護 (WOUND CARE)
每個病灶的位置、型態、大小與深度應在就診時及後續複診時記錄(並攝影),以協助監測病程。PG 的皮膚病灶通常極度觸痛,因此清潔應每日以微溫的無菌生理食鹽水或溫和的消毒溶液進行。1% 磺胺嘧啶銀 (silver sulfadiazine) 乳膏塗抹於 PG 潰瘍病灶上通常有舒緩作用,並可能促進肉芽組織 (granulation tissue) 形成,同時抑制細菌生長。應將非黏性敷料覆蓋於病灶上,並以縐布彈性繃帶 (crêpe elasticized bandage) 牢固但不緊繃地纏繞固定。某些病人,特別是淺表病灶者,使用水膠體敷料 (hydrocolloid dressings) 可獲得顯著緩解,這類敷料可留置 2 至 3 天並「融入」病灶。對病人與護理人員的仔細指導很重要,以確保配合度,並避免使用刺激物,例如化學去腐劑 (chemical desloughing agents)、腐蝕劑(如硝酸銀 silver nitrate),或可能黏附於潰瘍基底的敷料(如浸有軟石蠟及/或抗菌劑的紗布)。傷口表面可能培養出各種細菌,但這些通常代表污染物,除非傷口周圍有早期蜂窩性組織炎 (incipient cellulitis) 的臨床徵象,否則不需要導向性的抗生素治療。
處置 (PROCEDURES)
清創 (Debridement) 不可積極進行,且若可能應避免植皮 (skin grafting),因為有疼痛激惹現象與在供皮區 (donor sites) 誘發新 PG 病灶的風險。在疾病已受控制但再上皮化 (re-epithelialization) 不完全的病人中,曾報告培養的組織同種異體移植/自體移植 (cultured tissue allografts/autografts) 與使用牛膠原蛋白基質 (bovine collagen matrix) 是有用的。⁶⁷
諮詢 (COUNSELING)
應給予病人對於此疾病可能的恢復速度的實際期望。因此,雖然病灶在數天內發展與演變,但癒合過程可能需要數週至數個月。
預防/篩檢 (PREVENTION/SCREENING)
曾有 PG 病史的病人應被建議避免對皮膚造成外傷,因為有透過疼痛激惹現象誘發新病灶的可能性。若這類病人必須接受手術,他們應由皮膚科醫師密切監督其術後病程。有侵襲性 PG 病史的病人,可能值得在手術期間及術後一段時間(2 週或更久)給予一個療程的全身性類固醇,以預防新 PG 病灶的發展,且在可能處應使用皮內縫合 (subcuticular sutures)。有 PG 與克隆氏症病史且即將接受迴腸造口術 (ileostomy) 的病人,應被警告造口旁 PG 病灶可能發展,並應盡量避免對該區域的刺激,以協助預防疼痛激惹。
致謝 (ACKNOWLEDGMENTS)
我們特別感謝本章前一版的作者:Frank C. Powell、Bridget C. Hackett 與 Daniel Wallach。
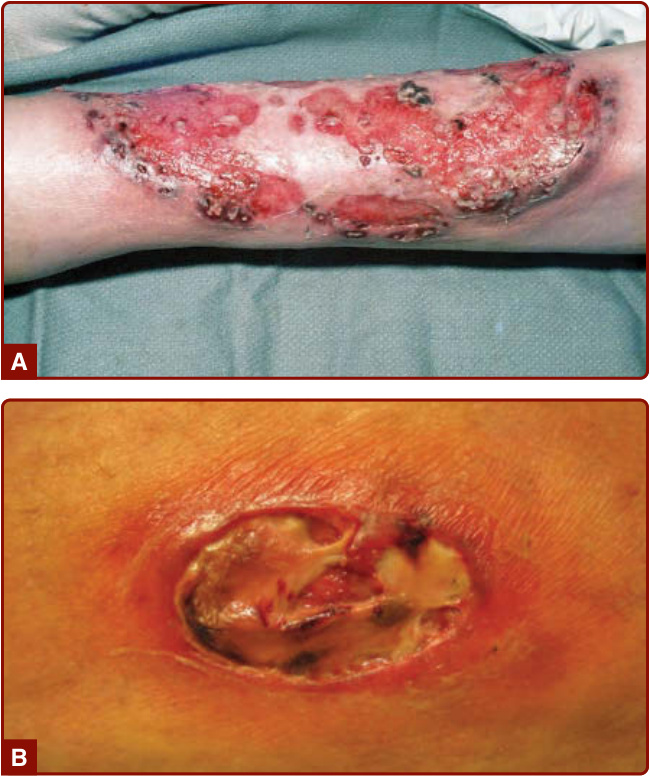
圖 37-1 A、B,典型潰瘍型壞疽性膿皮症 (classic ulcerative pyoderma gangrenosum) 的已形成病灶,顯示界線分明的潰瘍、潛掘 (undermining) 與周圍的紅斑帶。
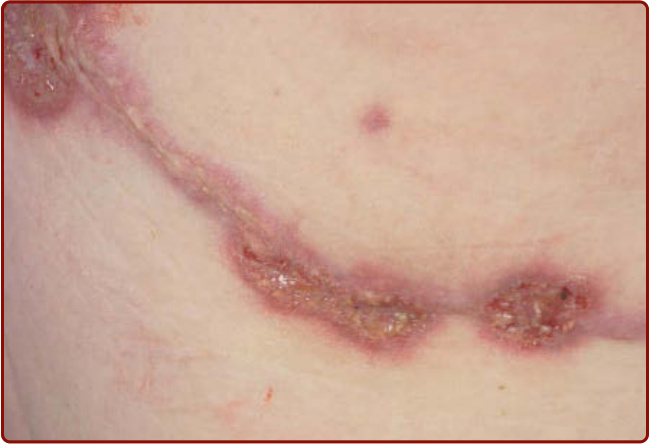
圖 37-2 沿著開胸術瘢痕 (thoracotomy scar) 部位發生的疼痛激惹性壞疽性膿皮症病灶。注意中央潰瘍、紫紅色邊緣與周邊的紅斑環。
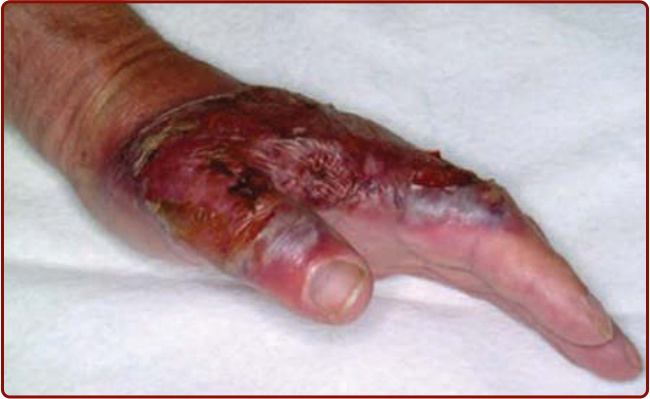
圖 37-3 大疱型壞疽性膿皮症 (bullous pyoderma gangrenosum) 病灶,顯示塌陷的水疱頂部與隨後潰瘍的淺表糜爛性特質。
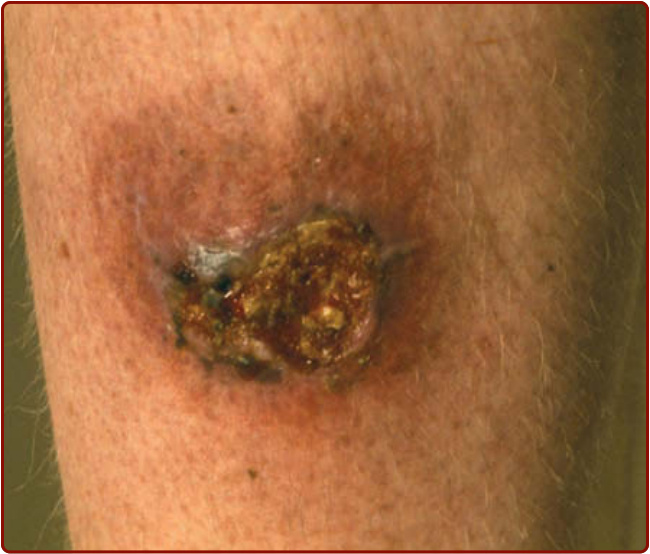
圖 37-4 增殖型壞疽性膿皮症 (vegetative pyoderma gangrenosum)——一個持續數月的慢性發炎與潰瘍的緩慢性區域。
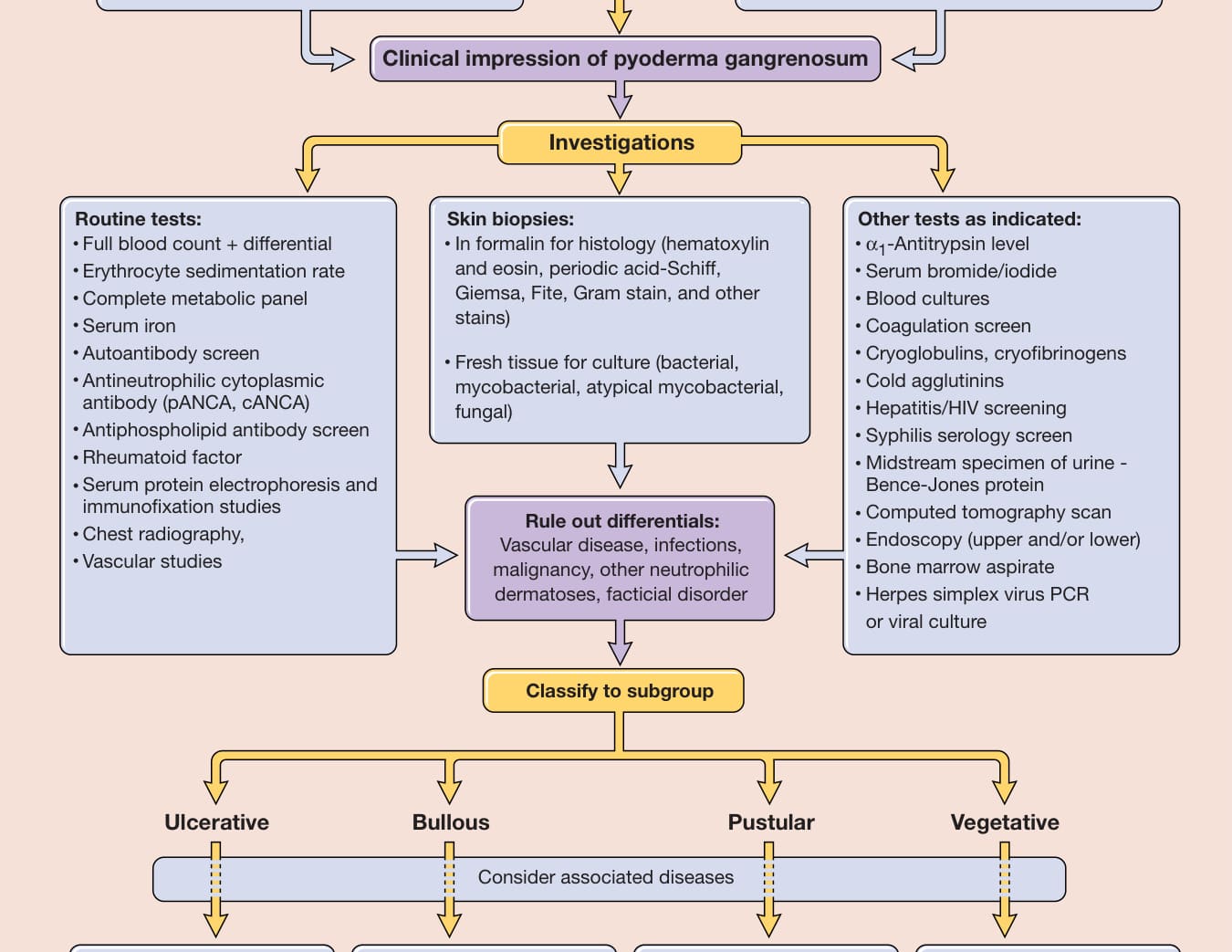
圖 37-5 壞疽性膿皮症病人的處置途徑。cANCA,細胞質型抗嗜中性球細胞質抗體 (cytoplasmic antineutrophil cytoplasmic antibody);pANCA,核周型抗嗜中性球細胞質抗體 (perinuclear antineutrophil cytoplasmic antibody);PCR,聚合酶連鎖反應 (polymerase chain reaction)。
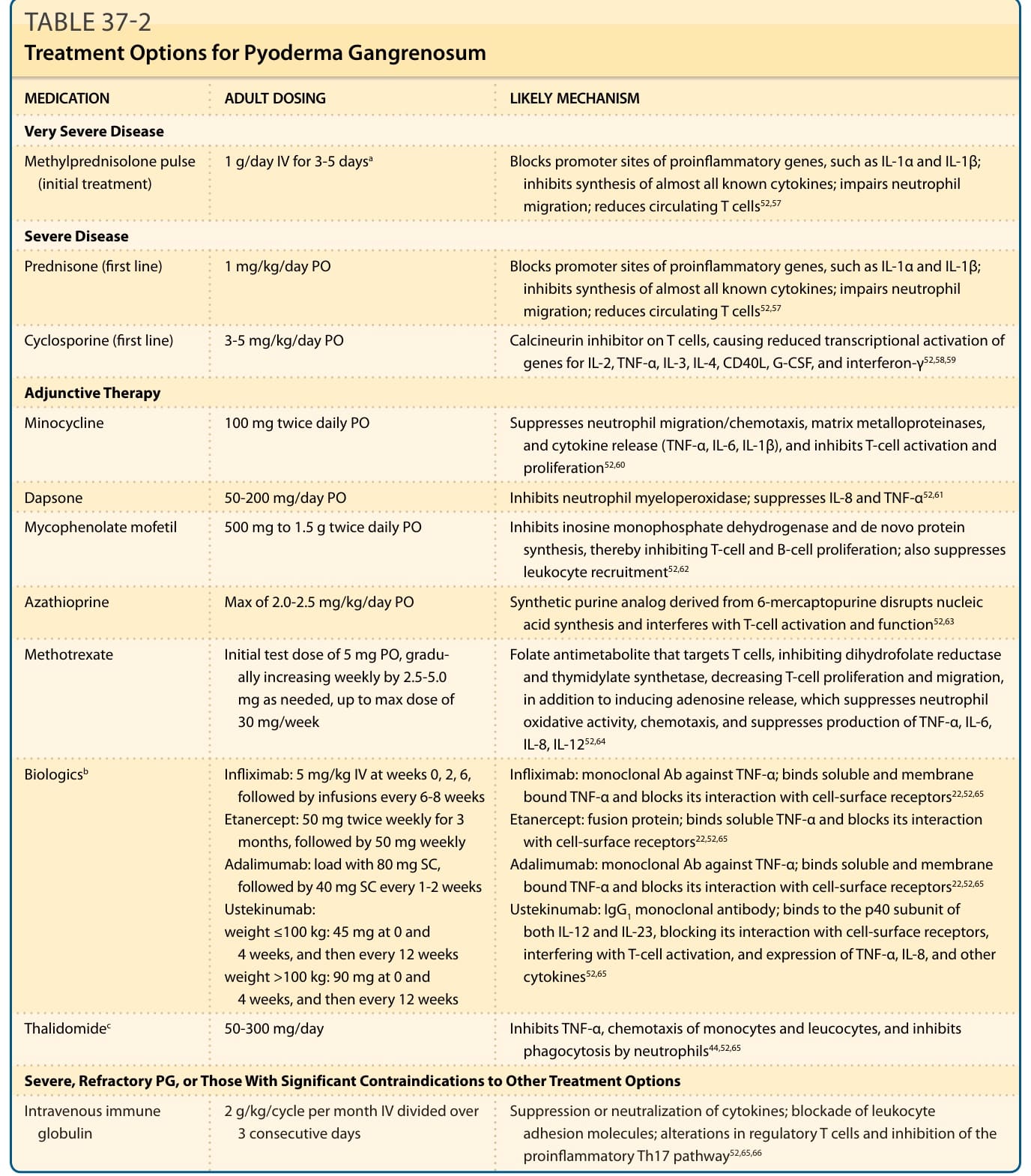
表 37-2 壞疽性膿皮症的治療選項 (Treatment Options for Pyoderma Gangrenosum)