致癌作用與皮膚 (Carcinogenesis and Skin) 精華筆記
流行病學與整體重要性
- 皮膚癌 (skin carcinomas) 年發生率至少為所有其他癌症總和的兩倍以上,是美國醫療系統的重大負擔。2017 年其他癌症估計 1,688,780 例;2012 年非黑色素瘤皮膚癌 (nonmelanoma skin cancers) 估計達 5,434,193 例(涉及 3,315,554 人)。
- 大多數皮膚癌源於陽光紫外線輻射 (ultraviolet radiation, UVR) 造成的突變;化學致癌物、致癌病毒等促成較小部分。
- 僅少數 BCC/SCC 病人會死於癌症,但因高頻率,美國每年估計約 2000 死(多為 SCC);黑色素瘤死亡率持續上升,估計每年 9730 例。
- 非黑色素瘤皮膚癌雖少致命,但常見於臉部等陽光暴露處,造成可觀外觀罹病負擔。
皮膚致癌作用一般原理 (General Principles)
- 多數惡性腫瘤經由逐步、多重遺傳與表觀遺傳 (epigenetic) 改變累積而成。此原則適用於 SCC(圖 19-1),但 BCC 為顯著例外:尚未鑑定前驅病變、轉移極罕見,單一致癌路徑不受控活化即可能足以致癌。
- 癌症的「特徵 (hallmarks)」(圖 19-2):降低生長刺激需求、對生長抑制與分化訊號反應受損、細胞凋亡 (apoptosis) 改變、衰老延遲、血管新生 (angiogenesis)、侵襲與轉移、代謝重新編程、免疫逃避。
- 驅動進展的關鍵是有缺陷的 DNA 修復,使致癌基因與腫瘤抑制基因突變得以累積。次世代定序 (next-generation sequencing) 已革新驅動突變的鑑定與「個人化腫瘤學」。
- 瘤內異質性 (intratumor heterogeneity):含分子、生化異質性,最顯著者為癌症幹細胞 (cancer stem cells)(又稱腫瘤起始細胞)。CSC 概念具臨床意涵——有效殺死 CSC 可治癒,僅針對非幹細胞則腫瘤消退後易復發(圖 19-3)。SCC 與 BCC 皆已描述 CSC 群。
- 瘤間異質性 (intertumor heterogeneity):如 BCC 表淺型 (superficial)、結節型 (nodular)、硬斑樣型 (morpheaform) 亞型,可由起源細胞 (cell of origin) 不同或內外在改變解釋。
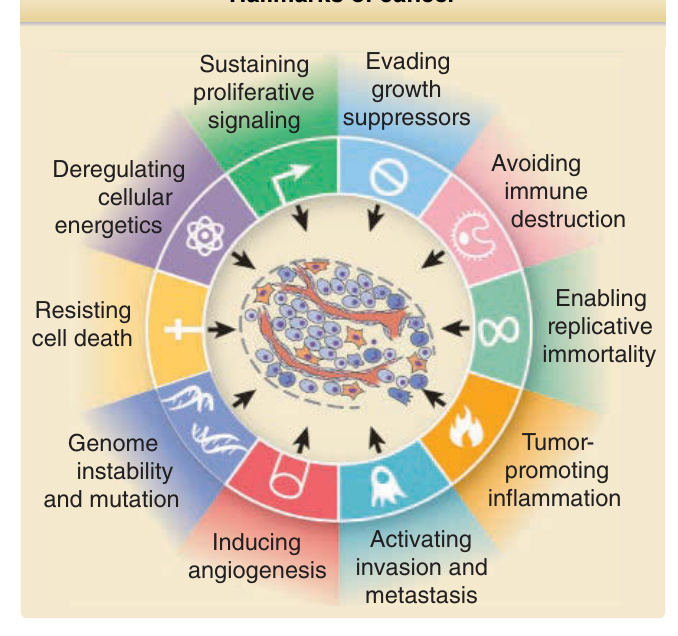
圖 19-2:癌症的特徵 (Hallmarks of cancer)。
皮膚癌的遺傳學 (Genetics)
- 致癌基因 (oncogenes):多衍生自原癌基因,僅需單一對偶基因經點突變、DNA 擴增或染色體易位即活化。
- 腫瘤抑制基因 (tumor suppressor genes):負調控增殖、誘發凋亡、修復 DNA;需兩個對偶基因皆去活化(常為點突變加雜合性喪失 loss of heterozygosity)。
- DNA 為致癌標的之重要性,由著色性乾皮症 (xeroderma pigmentosum) 等 DNA 修復缺陷病及 500 個轉移腫瘤分析(12.2% 帶生殖細胞系突變,其中 75% 影響 DNA 修復基因)所支持。
基底細胞癌的分子基礎 (BCC)
- 痣樣 BCC 症候群 (nevoid BCC syndrome, NBCCS / Gorlin):BCC 風險顯著增加、發病更早、數量更多;併髓母細胞瘤 (medulloblastoma)、分叉肋、鈣化大腦鐮、齒源性角化囊腫、額部隆凸、掌部凹陷。
- 病因為 PTCH1 生殖細胞系突變(Hedgehog 路徑關鍵組成)。失控的 Hedgehog 路徑活化幾乎可在所有 BCC 偵測到,且為 BCC 發展之充分條件、腫瘤維持所必需,故 Hedgehog 抑制劑可用於晚期/轉移性 BCC 治療。
- 其他潛在驅動突變:MYCN、PPP6C、STK19、LATS1、PIK3CA、RAS,以及 PTPN14、RB1、FBXW7 的功能喪失/錯義突變。
鱗狀細胞癌的突變與分子驅動 (SCC)
- 無專一性易罹皮膚 SCC 的遺傳症候群,也無類似黑色素瘤 BRAF 的高頻驅動因子;證據由高風險臨床情境、動物模型、基因體與蛋白質體學拼湊。
- 三種遺傳症候群凸顯重要性:著色性乾皮症(核苷酸切除修復缺陷,20 歲前皮膚癌風險增近 10,000 倍);隱性失養性表皮分解性水皰症 (recessive dystrophic epidermolysis bullosa,COL7A1 突變,慢性瘢痕致難治且常致命的 SCC);TGF-βR1 異合子去活化(Smith-Ferguson 症候群之角化棘皮瘤)。
- 經典發現:化學致癌作用中突變型 Hras 為主要驅動因子;1991 年已於人類 SCC 鑑定反覆 TP53 突變(DNA 結合區熱點,帶 UV 特徵 C→T 轉換)。
- 次世代定序:突變譜以 C→T 轉換與 UV 特徵突變為主,多為腫瘤抑制基因去活化。最常突變者:TP53、CDKN2A、NOTCH 家族、FAT 家族、KMT2C/KMT2D、KNSTRN;其他含 CASP8、CREBBP、CARD11;一系列中達 20% 帶突變型 HRAS;亦見 MYC、EGFR 擴增。
- 與其他部位 SCC 高度相似:TP53 突變在所有 SCC >70%;NOTCH 在皮膚 SCC >70%、HNSCC 20%、肺 SCC 13%、食道 SCC 10%;SOX2 擴增為譜系特異驅動因子。共享 TP53、TP63、NOTCH、SOX2 訊息傳遞改變。
- ERK 路徑為 SCC 重要路徑:兩個藥物誘發 SCC 情境佐證——黑色素瘤 BRAF 抑制(vemurafenib 約 22% 病人發生 SCC/類角化棘皮瘤,dabrafenib、encorafenib 較少;機轉為弔詭性 ERK 訊息傳遞與 JNK 抑制;合併 MEK 抑制劑幾可完全抑制);BCC 之 SMO 抑制劑 vismodegib 長期治療偶演變為抗藥性 SCC。MEK 抑制在 UV 臨床前模型有強效治療與化學預防效果。
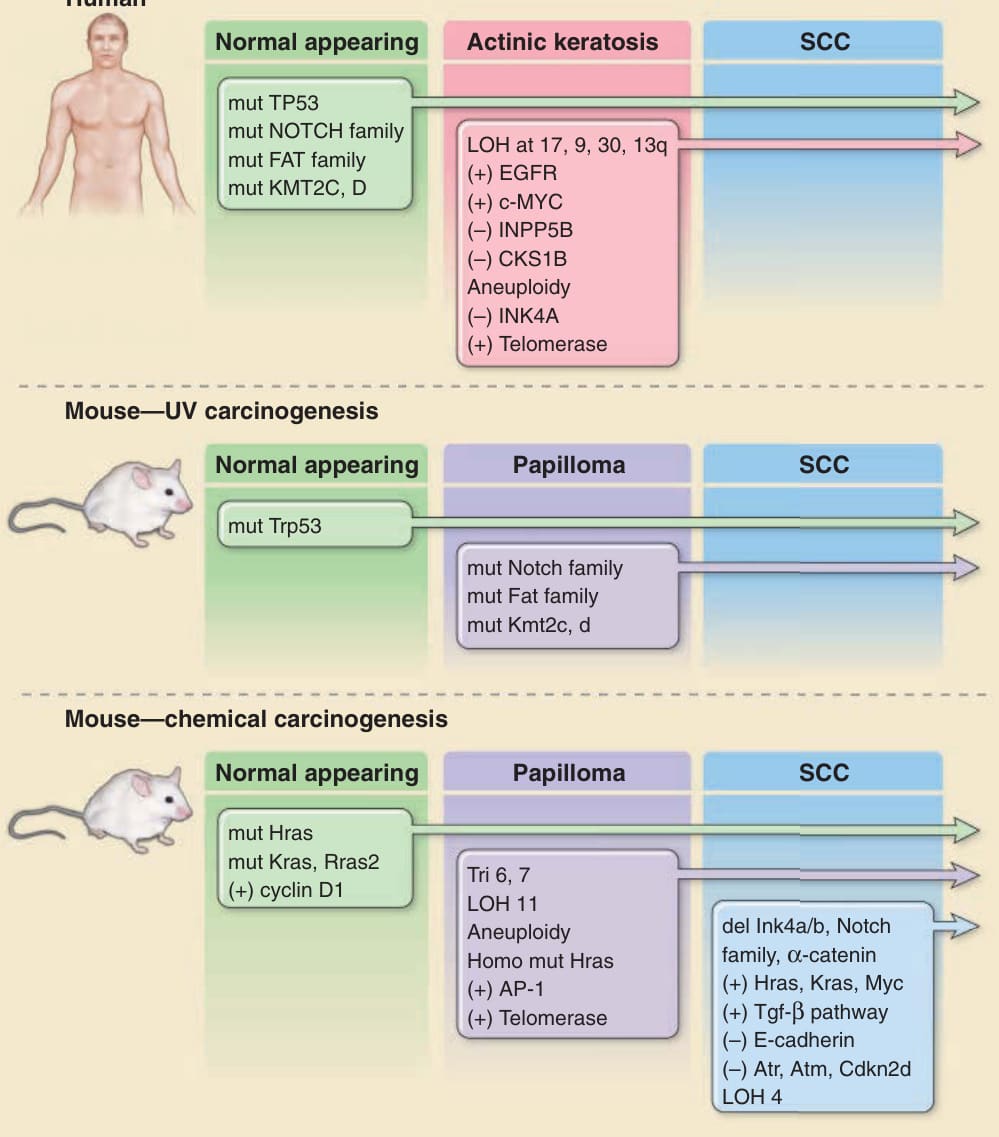
圖 19-1:與人類及小鼠皮膚 SCC 發展相關的分子與遺傳變化。
人類皮膚癌的病因學
光致癌作用 (Photocarcinogenesis)
- UVR 是所有皮膚癌的主要病因物質,也是人類環境主要致癌物。UV 光為完全致癌物 (complete carcinogen),單慢性暴露即足以致癌。
- 致癌活性來自:損傷 DNA 致突變、克隆擴增 (clonal expansion)、誘發活性氧物種 (reactive oxygen species, ROS)、免疫抑制。
- 光療風險:以 PUVA(補骨脂素與紫外線 A 光)最確切(Stern 前瞻 20 年追蹤)——SCC 風險高 100 倍以上、BCC 高 11 倍以上;窄波段 UVB 回溯分析未顯示風險升高(追蹤較短)。
UVR 誘發的 DNA 損傷與修復(圖 19-4)
- UVB/UVC 被嘧啶 (thymine、cytosine) 5–6 雙鍵吸收,相鄰嘧啶形成環丁烷嘧啶二聚體 (cyclobutane pyrimidine dimer, CPD)(最常 TT)或嘧啶 (6–4) 嘧啶酮光產物 [(6–4)PP](最常 TC)。兩者皆扭曲雙螺旋並被修復酶辨識。
- 陽光中 UVA 比 UVB 多 20 倍,但 DNA 損傷需高達 1000 倍劑量。膚質 II 型最小紅斑劑量:300 nm (UVB) 為 25 mJ/cm2,360 nm (UVA) 為 32,000 mJ/cm2。UVB 每 J/cm2 在人類皮膚每 100 萬個正常鹼基誘發 500 個光病變。
UVR 誘發的突變(圖 19-5)
- CPD 致突變兩途徑:DNA 複製時聚合酶將受損胞嘧啶讀為胸腺嘧啶(對面插腺嘌呤),或 CPD 加速胞嘧啶去胺基化 (deamination) 為尿嘧啶 → 皆致 C→T 取代。雙相鄰胞嘧啶突變則 CC→TT。
- UV 特徵突變 (UV signature mutation):C→T 且該 C 位於另一嘧啶旁(含 CC→TT),為 UVR 獨有,可推斷原始致癌物。約三分之二實驗性 UV 突變為特徵突變,其餘三分之一(G→T、T→C 等)多經 ROS 間接產生。
- 暗 CPD (dark CPDs):UV 誘發 ROS 過氧亞硝酸鹽與黑色素 (melanin) 互動,使電子化學激發,於 UV 暴露停止後數小時仍持續形成 CPD;占黑色素細胞中半數以上 CPD,提示超越「阻擋 UV」的新化學預防契機。
皮膚突變負荷與克隆動態(圖 19-6)
- 慢性 UV 暴露、外觀正常的表皮含約每百萬鹼基 5 個突變,超越許多人類癌症;皮膚癌具所有人類癌症最高突變負荷(僅錯配修復缺陷結腸癌可能例外)。
- TP53 突變克隆需持續 UV 暴露才以擴增克隆顯現選擇優勢;深度定序顯示廣泛存在高度易轉化的角質形成細胞斑塊。
光化性角化症演變為 SCC
- AK 進展為 SCC 速率:1 年 0.6% 至 4 年 2.6%。多數 AK 的分子譜與分化良好的侵襲性 SCC 大致無法區分,提示針對高度致突變化 UV 暴露皮膚的化學預防可能更有效。
病毒致癌作用 (Viral Carcinogenesis)
- 病毒相關癌症占人類惡性腫瘤多達 10%,皮膚者含卡波西肉瘤、SCC(疣狀表皮發育不良/免疫抑制者)、梅克爾細胞癌(表 19-1)。
- DNA 病毒早期蛋白劫持宿主細胞週期;病毒轉化常伴病毒 DNA 整合並持續表現早期病毒蛋白,靶向宿主訊息傳遞蛋白驅動轉化。
- 人類乳突瘤病毒 (HPV):>200 型分五屬。高風險 alpha HPV(HPV16、18)之 E6、E7 分別結合並降解 TP53 與 RB1 驅動腫瘤;beta HPV 與疣狀表皮發育不良及部分免疫抑制者皮膚 SCC 相關,但皮膚癌中無病毒轉錄本,提示非腫瘤維持所必需(可能「打帶跑 hit-and-run」機轉)。
- 人類多瘤病毒 (polyomavirus):感染無所不在。梅克爾細胞多瘤病毒 (MCPyV) 可在約 80% 梅克爾細胞癌偵測到,病毒 DNA 呈克隆性整合,提示早期因果角色。SV40 LTAg 破壞 TP53 與 RB1;MCPyV 則以 sTAg 為主要致癌驅動因子(與 SV40 相反)。
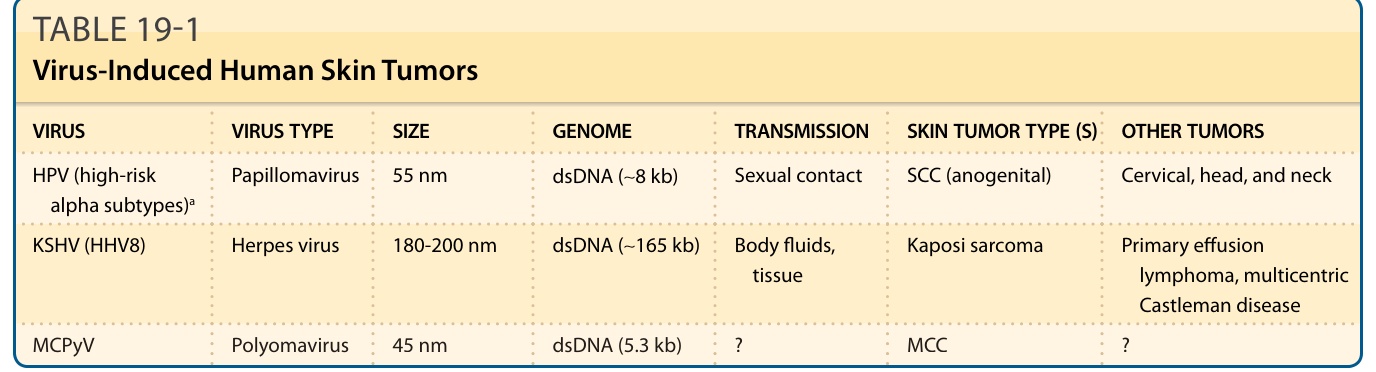
表 19-1:病毒誘發之人類皮膚腫瘤。
化學與其他致癌刺激
- 化學致癌史始於 1775 年 Pott 觀察煙囪清掃工陰囊癌與煤煙 (soot) 之關聯。國家毒理學計畫第 14 版致癌物報告(2016)列 248 種已知(62 種)或合理預期(186 種)人類致癌物。抗黴菌藥 voriconazole 連結至免疫抑制病人 SCC 增加。
- 游離輻射 (ionizing radiation, IR):過量風險幾乎僅限 BCC;兒童期暴露風險顯著增加(潛伏 >20 年,膚色白皙者尤甚)。飛行員暴露宇宙游離輻射,BCC 風險升 3 倍、黑色素瘤升 3.5 倍。
- 慢性傷口 / Marjolin 潰瘍:起源於慢性潰瘍或瘢痕(最常燒傷),>77% 與燒傷相關、近 90% 為 SCC、初始損傷至腫瘤平均 37 年;高度侵襲性(>30% 淋巴結侵犯、>11% 遠端轉移)。
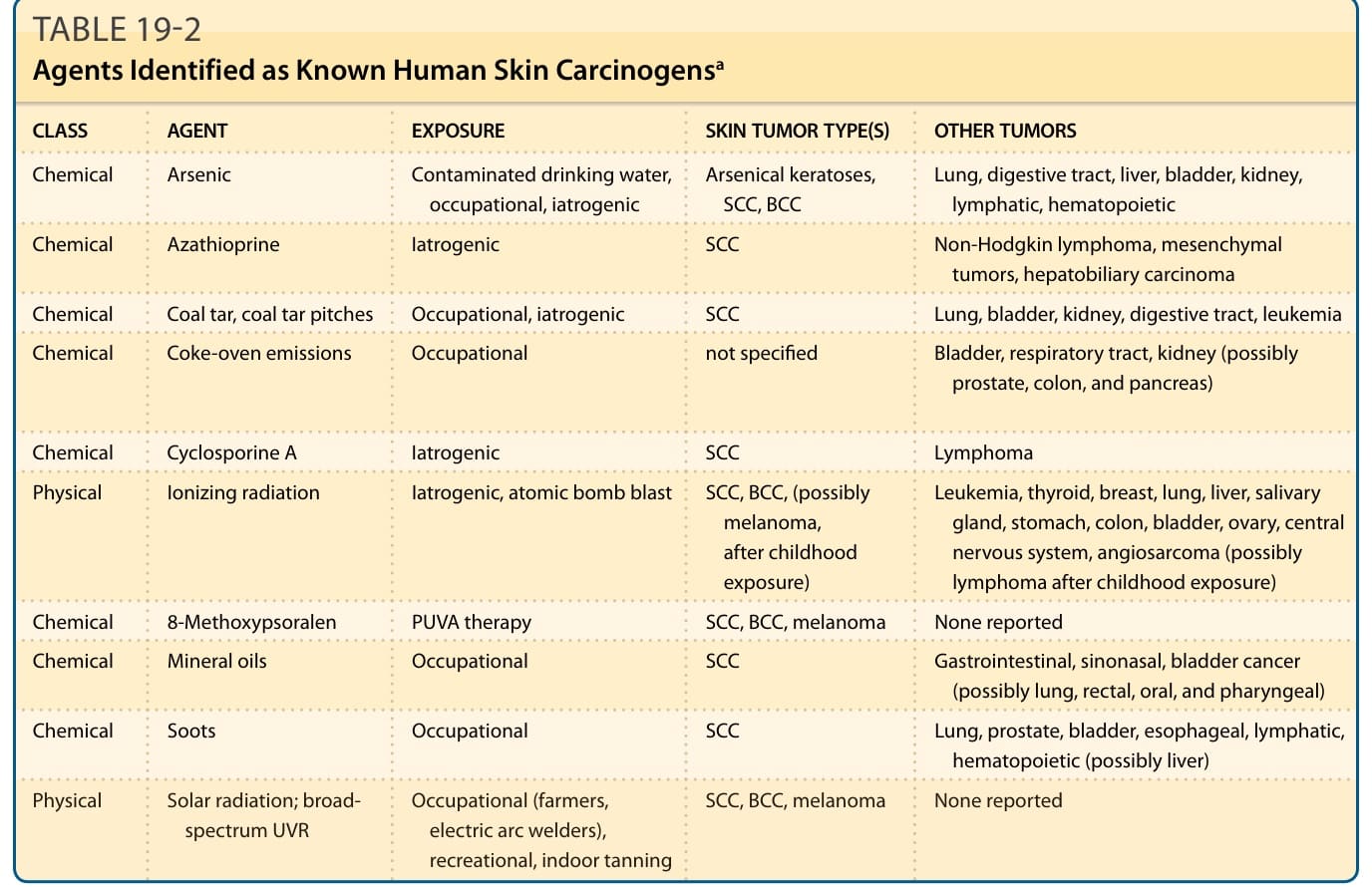
表 19-2:被鑑定為已知人類皮膚致癌物的物質(含砷、Azathioprine、煤焦油、環孢素 A、游離輻射、8-甲氧基補骨脂素、煤煙、太陽輻射等)。
人類皮膚癌的動物模型 (Animal Models)
- SCC 基因工程小鼠:致癌型 Hras 表現靶向毛囊間表皮 (interfollicular epidermis) 僅生良性乳突瘤(需傷口/腫瘤促進劑),靶向含幹細胞的毛囊上皮則自發 SCC。同時去活化 Rb1 與 Trp53 即足以誘發 SCC(與 HPV E6/E7 一致)。移除 Trp53 顯著加速化學與 UV 致癌;表皮特異移除 Notch1 增強致癌;間葉細胞缺乏 CSL(Notch 組成)小鼠無需起始物即自發多灶性 SCC。
- BCC 基因工程小鼠:表現 Hedgehog 活化因子(SHH、SMO、GLI1、GLI2)或刪除抑制因子(PTCH1、SUFU)可靠產生 BCC。
- 細胞起源:毛囊幹細胞對 Kras+Trp53 缺陷驅動之侵襲性 SCC 獨特敏感;毛基質細胞抗拒。表淺型 BCC 源自毛囊間表皮、結節型源自毛囊上皮;毛髮週期活化增強腫瘤;Hedgehog 訊息強弱決定表現型(低→基底樣錯構瘤,高→結節型)。
- 化學誘發(DMBA/TPA):研究逾 70 年。DMBA 致活化型 Hras 突變(最常 Q61,>90%),TPA 等促進。主要驅動為突變型 Hras(後可擴增);與人類對比為早期 ras 需求、Trp53 較晚(人類則 TP53 很早、RAS 較少)。
- UV 誘發(Hairless 小鼠):突變譜較化學模型更忠實於人類。Trp53 突變早發、見突變克隆;18 例 SCC 中位突變負荷達每百萬鹼基 155 個。繞過 Trp53、Survivin、Bcl2、E2f-1 調控之 UV 凋亡為完全易感性所必需;牽涉 COX-2、mTOR、AKT、ERK 及 WNT/β-catenin、miR-21/miR-31。
治療與預防 (Treatment and Prevention)
- 預防最有效:限制陽光與曬黑床暴露;限制未成年人使用曬黑床的立法預期顯著降低發生率。
- 化學預防:菸鹼醯胺 (nicotinamide) 第三期試驗於 3 個月時使 BCC 減 20%、SCC 減 30%、光化性角化症減 11%。
- 多數皮膚癌可手術有效治療。BCC:因幾乎皆由失控 Hedgehog 訊息傳遞所致,全身性 Hedgehog 抑制可治療局部晚期/轉移性 BCC(副作用常見、可發展抗藥性、停藥可復發),並能阻斷 Gorlin 症候群新發 BCC。SCC 因涉及多重不同程度的致癌改變,單一標靶治療不太可能同樣有效於預防。
結論
- 對 BCC/SCC 分子與細胞基礎的研究已促成 BCC 以機轉為基礎的標靶治療、揭示皮膚與其他器官 SCC 的分子相似性、提供強大致癌作用小鼠模型平台,並闡明腫瘤遺傳學與癌症生物學的基本原理。