致癌作用與皮膚 (Carcinogenesis and Skin)
重點一覽 (AT-A-GLANCE)
■ 估計皮膚癌 (skin carcinomas) 的年發生率至少是所有其他癌症總和的兩倍以上,使這些癌症成為美國醫療照護系統的重大負擔,並為顯著罹病率與死亡率的來源。
■ 大多數皮膚癌起因於陽光中紫外線輻射 (ultraviolet radiation) 所造成的突變,化學致癌物 (chemical carcinogens)、致癌病毒 (oncogenic viruses) 及其他因素則促成較少部分腫瘤的發生。
■ 來自流行病學、遺傳性易感症候群 (inherited predisposition syndromes)、癌症遺傳學、動物模型,以及最近的基因體學等多方面證據的匯聚,共同揭示了驅動基底細胞癌 (basal cell carcinoma, BCC) 與鱗狀細胞癌 (squamous cell carcinoma, SCC) 發生之關鍵致病事件的前所未有的洞見。
■ BCC 與 SCC 對特定路徑展現出明顯不同的依賴性:BCC 幾乎完全是一種依賴 Hedgehog 路徑 (Hedgehog pathway-dependent) 的腫瘤,而 SCC 似乎依賴更為多樣化的基因突變組合與致癌訊息傳遞。
■ 皮膚的可及性,以及在動物身上模擬癌症發展的能力,對於鑑定關鍵致癌訊息傳遞路徑的組成與功能、促進標靶治療藥物的開發,以及揭示癌症生物學的一般性原理,皆極具助益。
前言 (INTRODUCTION)
據估計,在 2017 年將有 87,110 例新的侵襲性黑色素瘤 (invasive melanoma) 與 74,680 例原位黑色素瘤 (melanoma in situ)。基底細胞癌 (basal cell carcinoma, BCC) 與皮膚鱗狀細胞癌 (cutaneous squamous cell carcinoma, SCC) 並無精確的發生率資料,因為這些腫瘤通常不會被通報至癌症登記系統,但根據 Medicare 資料集,2006 年非黑色素瘤皮膚癌 (nonmelanoma skin cancers) 的總數估計為 4,013,890 例(涉及 2,463,567 人),2012 年則為 5,434,193 例(涉及 3,315,554 人)。另一項使用 Medical Expenditure Panel Survey 資料的研究估計,每年接受非黑色素瘤皮膚癌治療的人數分別為 3,090,442 人(依據 2002 至 2006 年資料)與 4,301,338 人(依據 2007 至 2011 年資料),而每年治療費用分別由 4.8 billion。因此,BCC 與 SCC 合計的發生率很可能至少是所有其他癌症總和的兩倍以上,後者 2017 年估計為 1,688,780 例。
只有一小部分 BCC 或 SCC 病人會死於其癌症,但這些惡性腫瘤的高頻率,依據美國癌症協會 (American Cancer Society) 的資料,仍導致每年估計約 2000 人死亡,且大多數病例為 SCC。儘管黑色素瘤遠較非黑色素瘤皮膚癌少見,其死亡率卻持續上升,目前估計為每年 9730 例。
雖然非黑色素瘤皮膚癌極少致命,但因為它們常出現於臉部等陽光暴露部位,會造成可觀的外觀上之罹病負擔。因此,理解這些惡性腫瘤的病因學與發病機轉,是一項重要的公共衛生目標,而開發以機轉為基礎、不造成毀容的治療方法則迫切需要。皮膚腫瘤的高盛行率、其外在的位置,以及明確定義的癌前病變(就 SCC 與黑色素瘤而言),皆為研究調控人類皮膚癌誘發之因素提供了絕佳機會。促進人類皮膚癌研究的這些特質,也有助於建立相關的動物模型。分子遺傳學的進展、角質形成細胞 (keratinocyte) 的細胞培養,以及基因改造小鼠與重組人類皮膚模型 (reconstructed human skin models) 的發展,大幅促進了對皮膚致癌作用基本機轉的分析。本章的主要重點在於皮膚致癌作用的一般性面向,並以非黑色素瘤皮膚癌作為說明範例,至於特定皮膚惡性腫瘤的更詳盡討論則呈現於其他章節。
皮膚致癌作用 (CUTANEOUS CARCINOGENESIS)
一般原理 (GENERAL PRINCIPLES)
大多數惡性腫瘤是經由一個逐步進行的過程而產生,此過程以特徵性的表現型變化為標誌,這些變化反映了驅動腫瘤發生(從起始、經侵襲、到轉移)所需的多重遺傳與表觀遺傳 (epigenetic) 改變的累積。這項癌症生物學的基本原則對 SCC 成立(圖 19-1),但 BCC 的發病機轉則為此規則的一個顯著例外:尚未鑑定出前驅病變、轉移極為罕見,且單一致癌路徑的不受控活化可能即足以導致 BCC 腫瘤發生。已形成的癌症展現出根本性的行為改變,使其有別於其所源自的正常組織。這些差異包括:對生長刺激的需求降低、對生長抑制與分化訊號的反應受損、細胞凋亡 (apoptosis) 的改變、衰老 (senescence) 延遲或受阻、血管新生 (angiogenesis)、侵襲與轉移的能力、代謝重新編程 (metabolic reprogramming),以及逃避被免疫系統清除的能力(圖 19-2)。雖然這些異常中的一或多項可在腫瘤進展的不同階段被偵測到,因而可能出現於惡性前病變中,但所有這些異常通常都會出現於晚期癌症中。
腫瘤新生進展背後的一個驅動力是有缺陷的 DNA 修復 (defective DNA repair),它使涉及致癌基因 (oncogenes) 與腫瘤抑制基因 (tumor suppressor genes) 的突變得以累積,從而促成所觀察到的腫瘤細胞功能異常。過去,揭示癌症的分子基礎仰賴遺傳連鎖分析 (genetic linkage studies),以鑑定在癌症易感症候群中與腫瘤表現型共分離的染色體位點,或運用功能性篩選 (functional screens) 以從腫瘤細胞中鑑定能在培養中驅動腫瘤新生轉化的基因。然而,近期快速且負擔得起的次世代定序 (next-generation sequencing) 技術之發展——可用於篩選基因編碼區的突變(外顯子體定序,exome sequencing)或整個基因體——已徹底革新了鑑定可能驅動癌症並可作為「個人化腫瘤學」(personalized oncology) 治療性介入標的之突變的方法。雖然某些細胞功能的變化是細胞自主性 (cell-autonomous) 的,可在純化的腫瘤細胞群中研究,其他變化則取決於腫瘤微環境 (tumor microenvironment) 中各種額外的細胞類型,這些細胞在完整生物體中參與癌症的發展與進展,包括發炎細胞、癌症相關纖維母細胞 (cancer-associated fibroblasts)、神經、血管與淋巴管,以及腫瘤基質 (tumor stroma) 的其他組成成分。
許多腫瘤展現出瘤內異質性 (intratumor heterogeneity),此異質性可在多個層次上顯現。分子異質性 (molecular heterogeneity) 為腫瘤新生進展提供了驅動力,篩選出已獲得賦予增殖或存活優勢以及侵襲、轉移能力之突變的細胞,使其得以增生。生化異質性 (biochemical heterogeneity) 發生於腫瘤細胞的某些亞群展現出不同的致癌訊息傳遞特性與行為時,此乃由分泌的生長因子或其他微環境因素的區域性差異所驅動。或許最顯著的異質性範例,是在至少某些腫瘤類型中存在一小群類似幹細胞的腫瘤細胞,稱為癌症幹細胞 (cancer stem cells)。根據癌症幹細胞假說,腫瘤含有少數能自我更新的幹細胞,這些幹細胞產生過渡性放大 (transient-amplifying) 與分化中的後代,構成腫瘤中絕大多數的細胞(由 Nassar 及其同事回顧)(圖 19-3),反映了正常組織中細胞譜系的階層式組織。癌症幹細胞也被稱為腫瘤起始細胞 (tumor-initiating cells),因為它們在功能上是由其能力所定義的:當以少量甚至單一細胞純化並測試時,能在細胞移植實驗中重新形成腫瘤。相對地,非癌症幹細胞的腫瘤細胞即使以相對大量測試,仍無法產生腫瘤。此功能性特徵描述通常要求癌症幹細胞具備獨特的標記譜 (marker profile),使其得以從異質性的腫瘤細胞群中被分離出來。雖然癌症幹細胞概念最初是在以急性骨髓性白血病 (acute myeloid leukemia) 為中心的研究中建立的,許多後續研究的結果支持腫瘤起始細胞及其後代之階層式組織存在於許多(但非所有)固態腫瘤中。SCC(由 Nassar 及其同事回顧)與 BCC 中皆已描述過癌症幹細胞群。
癌症幹細胞概念具有重要的臨床意涵,因為能有效殺死癌症幹細胞的治療,可藉由消除腫瘤中所有其餘細胞所源自的關鍵細胞群而達到治癒。相對地,針對代表腫瘤大部分質量之非幹細胞的治療,可能導致明顯的腫瘤消退,但最終會因殘存癌症幹細胞的增生而出現腫瘤復發(見圖 19-3)。雖然用以確認分離出的癌細胞之「幹性」(stemness) 的某些檢測法存在技術上的限制,癌症幹細胞概念已在多個小鼠模型中獲得驗證,且為更有效治療癌症,正在追求選擇性靶向癌症幹細胞。此方法的成功,部分將取決於過渡性放大或分化中的腫瘤細胞,在原始癌症幹細胞庫被有效耗竭後,是否具備足夠的可塑性以回復為類似幹細胞的細胞群。
癌症的另一項一般性特性是瘤間異質性 (intertumor heterogeneity)。這包括同一類型腫瘤之間的表現型差異,這些差異不必然與惡性進展相關,但已足以正當化將其分類為不同的形態學類別,例如 BCC 的表淺型 (superficial)、結節型 (nodular) 與硬斑樣型 (morpheaform) 亞型。瘤間異質性的一種解釋是基於以下概念:不同細胞群的轉化產生不同的腫瘤亞型,因此腫瘤的起源細胞 (cell of origin) 是其最終表現型的關鍵決定因素。例如,小鼠模型研究顯示,在表皮基底細胞中致癌性活化 Hedgehog 路徑可能產生表淺型 BCC,但結節型 BCC 可能源自毛囊上皮 (hair follicle epithelium) 內有反應性的細胞群。來自其他腫瘤類型的實驗證據支持不同起源細胞產生不同腫瘤亞型的概念。瘤間異質性的另一範例是組織學上相似之腫瘤類型具有迥異的生長速率。這在 BCC 病人身上可能極為明顯,例如某些結節型腫瘤呈惰性生長,但其他則以快得多的速率生長。此情境下的瘤間異質性可由內在(遺傳、表觀遺傳或訊息傳遞)或外在(微環境)的改變來解釋,這些改變賦予某些腫瘤而非其他腫瘤生長或存活的優勢。
皮膚癌的遺傳學 (GENETICS OF SKIN CANCER)
一般概念 (GENERAL CONCEPTS)
針對癌症發展之分子基礎的研究已揭示兩大類基因——致癌基因 (oncogenes) 與腫瘤抑制基因 (tumor suppressor genes)——它們在癌症的發病機轉中扮演關鍵角色。致癌基因是任何能在培養中轉化正常細胞並在動物中誘發癌症的基因。大多數致癌基因衍生自原癌基因 (proto-oncogenes),後者通常編碼作為細胞增殖之關鍵正向調節因子或細胞凋亡抑制因子的蛋白質。轉化為致癌基因可經由以下途徑發生:導致組成型活化蛋白質的點突變、DNA 擴增 (DNA amplification),或將高度活化的啟動子與原癌基因相連的染色體易位 (chromosomal translocations)。後兩種機轉導致原癌基因的表現增加或不適當,並可能改變有反應之正常細胞的生長調控。較不常見而與癌症有關的機轉,包括兩個基因編碼區域的融合,產生具有致癌特性的新型嵌合分子 (chimeric molecule),或控制致癌基因表現之非編碼 DNA (non-coding DNA) 的突變。
腫瘤抑制基因正常的功能是負向調節細胞增殖、誘發細胞凋亡、修復受損的 DNA,或誘導細胞分化。相對於致癌基因——通常只需一個對偶基因 (allele) 經由突變活化——腫瘤抑制基因的兩個對偶基因都必須被去活化才能促進腫瘤發展。常見的情況是,去活化的點突變發生於腫瘤抑制基因的一個複本中,而剩餘的正常複本則經由有絲分裂期間染色體錯誤分離 (chromosomal missegregation) 的過程而喪失,導致雜合性喪失 (loss of heterozygosity)。然而,非編碼 DNA 的突變也可能導致腫瘤抑制基因表現出現具功能意義的減少。此外,長鏈非編碼 RNA (long noncoding RNAs) 的表現改變,可能藉由在多個層次影響訊息傳遞路徑而影響腫瘤發生。
哪些致癌基因與腫瘤抑制基因促成皮膚腫瘤的發展?對於偶發性皮膚癌之遺傳基礎的相當多洞見,來自於鑑定出構成遺傳性皮膚腫瘤症候群基礎的特定基因。
DNA 作為致癌作用標的之重要性,因下列發現而獲得有力支持:在易罹患皮膚癌的著色性乾皮症 (xeroderma pigmentosum)(見後文)病人,以及數種以光敏感性 (photosensitivity) 為特徵的其他皮膚病中,發現 DNA 修復基因的缺陷。此外,一項針對來自各種原發性癌症之 500 個轉移性腫瘤的詳盡基因體分析,揭示了 12.2% 的病例存在未預期的生殖細胞系突變 (germline mutations),其中 75% 影響 DNA 修復基因。綜合來看,這些發現強調了健全的 DNA 修復機轉對於防止驅動癌症發展與進展之突變累積的重要性。
基底細胞癌的分子基礎 (MOLECULAR BASIS OF BASAL CELL CARCINOMA)
痣樣 BCC 症候群 (nevoid BCC syndrome, NBCCS) 病人發生 BCC 的風險顯著增加,這些 BCC 較一般族群於更年輕時出現且數量更多。NBCCS 病人也易發展出一種起源於小腦的小兒腦瘤——髓母細胞瘤 (medulloblastoma),以及全身各處多種其他缺陷,包括分叉肋 (bifid ribs)、鈣化大腦鐮 (calcified falx cerebri)、齒源性角化囊腫 (odontogenic keratocysts)、額部隆凸 (frontal bossing)、掌部凹陷 (palmar pits) 與分叉肋。這些結構異常中有些必然發生於胎兒發育期間,顯示 NBCCS 中的遺傳改變不僅影響出生後的癌症發展,也影響胚胎發生期間的組織或器官形成。NBCCS 是由破壞 PTCH1 基因(編碼 Hedgehog 訊息傳遞路徑的關鍵組成成分)的生殖細胞系突變所致,此一發現與上述概念相符。生理性 Hedgehog 訊息傳遞在發育期間各種器官與組織的型態形成 (patterning and morphogenesis) 中扮演重要角色,並於出生後促成組織恆定與再生;相對地,由影響 PTCH1 或其他 Hedgehog 路徑組成成分(表 114-1)之突變所引起的 Hedgehog 路徑不受控活化(圖 114-1),與 NBCCS 病人及一般族群的 BCC 發展皆有緊密關聯(見第 111 章)。由於失控的 Hedgehog 路徑活化幾乎可在所有 BCC 中偵測到,且路徑活化已被證明對 BCC 發展為充分條件並為腫瘤維持所必需,因此曾認為此路徑的藥理性抑制劑可能有助於 BCC 的內科治療。在相當比例接受 Hedgehog 路徑抑制劑治療的晚期或轉移性 BCC 病人中,事實證明確實如此,此將於後文及第 111 章討論。儘管失控的 Hedgehog 訊息傳遞在 BCC 發展中扮演樞紐角色,次世代定序研究已揭示其他潛在的驅動突變,涉及編碼 MYCN、PPP6C、STK19、LATS1、PIK3CA、RAS 蛋白的基因,以及 PTPN14、RB1 與 FBXW7 的功能喪失突變與錯義突變 (missense mutations)。仍需進一步研究以確定這些及其他遺傳改變對 BCC 生物學與治療反應的功能意義。
鱗狀細胞癌中的突變與分子驅動因子 (MUTATIONS AND MOLECULAR DRIVERS IN SQUAMOUS CELL CARCINOMA)
與 BCC 相對,鑑定驅動皮膚 SCC 發展之樞紐遺傳事件,因下列事實而頗為複雜:沒有任何遺傳性癌症症候群是專一性地易罹患皮膚 SCC,且不存在類似黑色素瘤中突變型 BRAF 那樣的高頻率致癌驅動因子。因此,將特定遺傳改變與 SCC 發展連結起來的關鍵證據,是經由對高風險臨床情境的特徵描述、動物模型的分子分析、基因體學與蛋白質體學 (proteomics) 拼湊而成。三種罕見的遺傳性症候群凸顯了環境影響、特定臨床情境,以及驅動 SCC 發展之路徑的重要性。以極度光敏感性及皮膚癌發生顯著加速為特徵的著色性乾皮症 (xeroderma pigmentosum),與 20 歲前皮膚癌風險增加近 10,000 倍有關。其關鍵的統一性缺陷,是由分布於八個互補群 (complementation groups) 的基因發生隱性去活化所致,導致缺乏核苷酸切除修復 (nucleotide excision repair),而此修復對於移除紫外線 (ultraviolet, UV) 輻射所造成的致突變性 DNA 光產物 (photoproducts) 為必需。隱性失養性表皮分解性水皰症 (recessive dystrophic epidermolysis bullosa) 是一種皮膚脆弱性疾病,由 COL7A1 的功能喪失突變所致,造成表皮下水皰、不癒合傷口,以及對 SCC 的高度易感性。其所導致的慢性瘢痕化使病人易罹患 SCC,這些 SCC 在臨床上難以處置且常為致命。最後,轉化生長因子 (transforming growth factor, TGF)-βR1 的異合子生殖細胞系去活化,導致對角化棘皮瘤 (keratoacanthomas) 的易感性,此為 Smith-Ferguson 症候群的一部分;而事實上,更廣泛地去活化 TGF-βR 訊息傳遞與典型 SCC 的發展有關。其他與 SCC 發展相關的遺傳性皮膚病 (genodermatoses) 與基因列於表 112-1。藉由在小鼠中使用兩階段化學致癌作用 (two-stage chemical carcinogenesis) 方法(見後文),Balmain 及其同事開創了在此經典皮膚癌模型中對腫瘤發展的分子遺傳學分析,並在突變型 Hras 被分離為第一個人類致癌基因後一年,鑑定出突變型 Hras 為此模型中腫瘤的主要致癌驅動因子。到了 1991 年,已在人類 SCC 中鑑定出反覆出現的 TP53 突變,對應到 DNA 結合區 (DNA-binding domain) 中的熱點位置,並帶有歸因於 UV 暴露之特徵性的 C → T 轉換 (transitions)。
次世代 DNA 定序如今已提供對可能驅動皮膚 SCC 發展之遺傳病變的廣泛洞見。雖然其中許多報告來自原發性腫瘤,現在也已有部分轉移性病灶的系列報告。如預期所料,跨外顯子體的突變譜強烈地以 C → T 轉換與 UV 特徵突變為主導,且絕大部分表現為腫瘤抑制基因的去活化,而高度反覆出現的突變極少。綜合已發表的外顯子體與標靶定序工作,最常發生突變的腫瘤抑制基因為 TP53、CDKN2A、NOTCH 家族成員、非典型鈣黏蛋白 FAT 家族成員 (atypical cadherin FAT family members)、組蛋白甲基轉移酶 (histone methyltransferases) KMT2C 與 KMT2D,以及 KNSTRN。樣本來源、定序方法與分析流程的繁多使得直接比較有困難,但其他潛在的腫瘤抑制基因包括 CASP8、CREBBP 與 CARD11。在一個系列中,多達 20% 的 SCC 觀察到突變型 HRAS。皮膚 SCC 中也曾報告 MYC 與 EGFR 的擴增,以及 CKS1B 與 INPP5A 的喪失。
重要的是,這些資料也顯示出與起源於其他部位之 SCC 的顯著相似性。因為複層鱗狀上皮 (stratified squamous epithelia) 與環境形成界面,這些組織是與致癌物互動的常見部位。TP53 突變在所有 SCC 中以超過 70% 的頻率發生;NOTCH 家族基因在超過 70% 的皮膚 SCC (cuSCC)、20% 的口腔頭頸 SCC (HNSCC)、13% 的肺 SCC 與 10% 的食道 SCC 中發生突變,而 SOX2 擴增是 SCC 一個常見的譜系特異性驅動因子。
結合先前的結果,這顯示來自不同部位的 SCC 共享深層的分子共通性,包括整體基因表現以及 TP53、TP63、NOTCH 家族與 SOX2 訊息傳遞的改變。雖然已發表的轉錄體與蛋白質體資料較少,但與致癌物誘發之 SCC 的對應性在 HNSCC 與肺 SCC 中尤為顯著。轉錄譜分析牽涉到位於 wingless 相關整合位點 (wingless-related integration site, WNT)、β-catenin 與 ERK(細胞外訊號調節激酶,extracellular signal-regulated kinase)訊息傳遞下游的轉錄因子。新興文獻也牽涉到多種 microRNA 參與 SCC 的發展(由 Konicke 及其同事回顧)。數種 microRNA 已在多種情境中被認為在增殖、細胞凋亡與遷移方面具有促腫瘤與抑腫瘤功能。以反相蛋白質陣列 (reverse-phase protein array) 為基礎對典範癌症路徑的探查,顯示在 SCC 發展的進展序列中 ERK 與 mTOR(雷帕黴素哺乳動物標的,mammalian target of rapamycin)路徑訊息傳遞上調。ERK 路徑訊息傳遞的重要性也反映在兩個涉及藥物誘發 SCC 的臨床情境中:用於黑色素瘤的 BRAF(v-raf 鼠肉瘤病毒致癌基因同源物 B)抑制,以及用於 BCC 的 smoothened (SMO) 抑制。在首個美國食品藥物管理局 (Food and Drug Administration) 核准的 BRAF 抑制劑 vemurafenib 的初期臨床試驗中觀察到,約 22% 接受治療的黑色素瘤病人發展出 SCC 或類角化棘皮瘤 (keratoacanthoma-like) 病灶;此一不良反應在效力更強的 BRAF 抑制劑 dabrafenib 與 encorafenib 較少觀察到。在機轉上,此現象被歸因於兩方面:其一是在野生型 BRAF 情境下(常合併致癌性突變型 HRAS),藥物誘發 MEK(絲裂原活化蛋白/細胞外訊號相關激酶激酶,mitogen activated protein/extracellular signal-related kinase kinase)/ERK 訊息傳遞之異常過度活化所導致的弔詭性 ERK 訊息傳遞;其二是因脫靶抑制 c-Jun-N 端激酶 (c-Jun-N-terminal kinase, JNK) 訊息傳遞所造成的細胞凋亡抑制。據此,合併使用 MEK 與 BRAF 抑制劑幾乎完全抑制了這些 SCC 的出現。有趣的是,以 SMO 抑制劑 vismodegib 長期治療 BCC 偶爾與下列情形有關:接受治療的腫瘤演變為一種與藥物抗性及 ERK 訊息傳遞活化相關的 SCC 形態。綜合而言,這些資料強烈牽涉 ERK 訊息傳遞為 SCC 的一個重要路徑。此點也在一個 UV 驅動的臨床前模型中獲得驗證,於該模型中 MEK 抑制具有強效的治療與化學預防效果。
人類皮膚癌的病因學 (ETIOLOGY OF HUMAN SKIN CANCER)
光致癌作用 (PHOTOCARCINOGENESIS)
陽光中的紫外線輻射 (ultraviolet radiation, UVR) 是所有皮膚癌的主要病因物質,因此 UVR 是人類環境中的主要致癌物。UVR 強大的致癌活性可歸因於:其損害 DNA 並造成突變的能力、其讓那些訊息傳遞路徑改變而在面對 UV 誘發之細胞毒性時具有存活優勢的初期腫瘤新生細胞進行克隆擴增 (clonally expand) 的能力、其誘發活性氧物種 (reactive oxygen species, ROS) 的能力,以及其作為免疫抑制劑 (immune suppressant) 的活性。UVR 與皮膚癌的關聯獲得臨床、流行病學與實驗資料如此強力的支持,使其或許代表人類惡性腫瘤中最明確的病因因素。UV 光是一種完全致癌物 (complete carcinogen),單是慢性暴露即足以誘發皮膚癌。此過程始於致癌物暴露、DNA 損傷,以及突變的逐步累積。克隆擴增至少部分是因為對這些突變的篩選而發生,從而增大了進一步損傷的標靶範圍。這些變化與 UV 暴露之關鍵微環境及免疫學後果相結合,共同驅動腫瘤發展。除了環境或職業暴露外,患有各種皮膚病的病人也接受 UVR 治療。評估由此產生之皮膚癌風險的絕大部分經驗,來自於乾癬 (psoriasis) 光療的情境。此風險最確切地建立於補骨脂素與紫外線 A 光 (psoralen and ultraviolet A light, PUVA) 療法,係由 Stern 及其同事所發起、具有 20 年追蹤的前瞻性研究,其中 SCC 風險高出 100 倍以上,BCC 風險高出 11 倍以上。關於窄波段 UVB (narrowband UVB) 的資料則大致侷限於回溯性分析,這些分析顯示皮膚癌風險未升高;然而,這些研究另受限於追蹤時間較短。
紫外線輻射誘發的 DNA 損傷與修復 (ULTRAVIOLET RADIATION-INDUCED DNA DAMAGE AND REPAIR)
DNA 光產物 (DNA Photoproducts): 陽光誘發之致癌作用的第一個分子步驟,發生於 UVB 光子誘發 DNA 光產物時(見圖 19-4)。UVB 與 UVC 傾向被吸收於嘧啶 (pyrimidines)(胸腺嘧啶 thymine 與胞嘧啶 cytosine)的 5–6 雙鍵處。若兩個相鄰嘧啶被活化,所產生的開放鍵會交叉反應,形成環丁烷嘧啶二聚體 (cyclobutane pyrimidine dimer, CPD)。最常見者為 TT,但 TC、CT 與 CC 環丁烷二聚體也會生成。一個嘧啶的 6 號位置與另一個嘧啶的環外基團 (exocyclic group) 之間若形成單鍵,則改為產生嘧啶 (6–4) 嘧啶酮光產物 [pyrimidine (6–4) pyrimidone photoproduct, (6–4)PP],最常見者為 TC。兩種光產物皆會扭曲 DNA 雙螺旋,並被 DNA 修復酶辨識。雖然陽光中 UVA 比 UVB 多 20 倍,但對於其某些生物學效應(如 DNA 損傷)而言,UVA 需要高達 1000 倍的劑量;而對於膚質 II 型,在 300 nm(UVB)與 360 nm(UVA)的最小紅斑劑量 (minimal erythemal doses) 分別為 25 mJ/cm2 與 32,000 mJ/cm2。UVB 在人類皮膚中每 J/cm2 誘發每 106 個正常鹼基 500 個光病變 (photolesions)。
紫外線輻射誘發的突變 (Ultraviolet Radiation—Induced Mutations): 一個 CPD 可經由兩種方式導致突變(圖 19-5)。當該病變在 DNA 複製期間被複製時,DNA 聚合酶 (polymerase) 可能將受損的胞嘧啶讀為胸腺嘧啶,並在其對面插入一個腺嘌呤 (adenine)。在下一輪複製中,聚合酶正確地在腺嘌呤對面插入胸腺嘧啶,導致 C → T 取代。或者,CPD 會加速其胞嘧啶自發地去胺基化 (deamination) 為尿嘧啶 (uracil),導致相同的變化。雖然 TT CPD 是最常見的光產物,但由 DNA 聚合酶 eta 在受損 T 鹼基對面添加 A 不會造成突變。若兩個相鄰胞嘧啶突變,結果為 CC → TT。此種獨特的突變模式——即 C → T 且該 C 位於另一個嘧啶旁邊(包括 CC → TT)——為 UVR 所獨有,稱為 UV 特徵突變 (UV signature mutation)。
特徵突變提供了一項工具,可從腫瘤中發現的突變推斷出原始致癌物。幾乎所有實驗性產生的 UVB 或 UVC 突變都位於相鄰嘧啶處,且約三分之二為特徵突變。其餘三分之一,通常為 G → T 與 T → C 取代或小的插入或缺失,是由 UV 所造成,但很可能經由 ROS 間接產生。G → T 顛換 (transversion) 可由在 8-羥基-2′-去氧鳥苷 (8-hydroxy-2′-deoxyguanosine)(一種常見的氧化性 DNA 病變)對面摻入腺嘌呤所造成(見圖 19-5)。由於此氧化類別的損傷可由許多致癌物造成,這些突變無法揭示其來源是 UVB、UVA、菸草煙霧,抑或細胞內的氧化磷酸化。然而,帶有典型 UV 特徵突變的腫瘤必然也含有 UV 誘發的氧化性突變。UVA 經由光敏化 (photosensitization) 弱誘發 UVB 特徵突變,但會產生類氧化突變與 T → G 變化,後者已被提議為 UVA 的指紋特徵。
最近,發現了一種化學上不同的 UV 誘發 CPD 生成途徑。在此機轉中,發現 UV 誘發生成的 ROS 過氧亞硝酸鹽 (peroxynitrite) 會與黑色素 (melanin) 互動,導致黑色素中的電子發生化學激發 (chemiexcitation) 至極高能量狀態,類似於最終產生生物發光 (bioluminescence) 的反應。然而,這些被激發的電子並非產生光,而是能直接與 DNA 互動,導致在 UVA 或 UVB 暴露停止後數小時仍持續形成 CPD,即所謂的「暗 CPD」(dark CPDs)。重要的是,在 UV 暴露後,這些暗 CPD 占了黑色素細胞 (melanocytes) 中半數以上的 CPD。此一發現提示任何 ROS 來源皆有可能產生 CPD,且此機轉之 CPD 生成的動力學特性,可能為超越阻擋 UV 暴露的新型化學預防策略提供契機。
皮膚中的突變負荷與克隆動態 (MUTATION BURDEN AND CLONAL DYNAMICS IN SKIN)
皮膚在一生中反覆暴露於 UVR,代表了龐大的集體 DNA 損傷負荷。事實上,早自 1996 年的初期報告即顯示,可在全裝片 (whole-mounted) 皮膚中偵測到異常表現穩定(因而很可能突變)p53 的角質形成細胞克隆。這些 TP53 突變克隆也已在 UV 暴露的小鼠皮膚中被鑑定出。最近,標靶與全外顯子體定序已顯示,慢性 UV 暴露、臨床與組織學上外觀正常的表皮,含有每百萬鹼基 (megabase) 約五個突變的 DNA(圖 19-6),此一驚人的高突變負荷超越了許多人類癌症。事實上,皮膚癌具有所有已報告之人類癌症中最高的突變負荷,唯一可能的例外是錯配修復缺陷 (mismatch repair-deficient) 的結腸癌。要能被既有的 DNA 定序方法偵測到,必須存在帶有相同突變、數量足以在特定定序深度下可靠偵測的細胞克隆。因此,表皮中 UV 誘發之鑲嵌現象 (mosaicism) 的程度,很可能比報告所述更為廣泛。
雖然克隆的存在被詮釋為對特定突變正向篩選的證據,克隆大小的頻率分布卻顯示克隆也可在無需此種篩選的情況下自發出現。就 TP53 而言,很清楚的是,雖然 TP53 突變損害細胞凋亡,但要使這些突變的選擇性優勢以擴增克隆的形式顯現,仍需持續的 UV 暴露,推測是經由抑制或殺死周圍的正常角質形成細胞。對 UV 暴露之人類皮膚的存檔樣本中 TP53 突變克隆進行深度定序,顯示存在許多與 SCC 相關之基因的次克隆 (subclonal) 突變,提示高度易於轉化的角質形成細胞斑塊普遍存在。
光化性角化症演變為鱗狀細胞癌 (EVOLUTION OF ACTINIC KERATOSES TO SQUAMOUS CELL CARCINOMA)
光化性角化症 (actinic keratosis, AK) 進展為 SCC 的速率估計,範圍從 1 年 0.6% 至 4 年 2.6%。在基因體層次,許多在 SCC 與受照射皮膚中觀察到的關鍵突變基因,也可在 AK 中發現。AK 與 SCC 之間尚未一致地鑑定出轉錄上的差異。相反地,大多數 AK 的分子譜大致無法與分化良好的侵襲性 SCC 區分,提示針對被高度致突變化之 UV 暴露皮膚的化學預防可能更為有效。
病毒致癌作用 (VIRAL CARCINOGENESIS)
一般特徵 (GENERAL FEATURES)
病毒相關癌症占所有人類惡性腫瘤的多達 10%,包括數種起源於皮膚者:卡波西肉瘤 (Kaposi sarcoma);SCC(起源於疣狀表皮發育不良 (epidermodysplasia verruciformis) 病人及一小部分免疫抑制個體);以及梅克爾細胞癌 (Merkel cell carcinoma)(見表 19-1)。DNA 病毒已演化出高度有效的機轉,在容許性宿主細胞 (permissive host cells) 中,於其正常的營養性 (vegetative) 生命週期期間複製其基因體、合成衣殼蛋白 (capsid proteins) 並組裝具感染力的病毒顆粒。由於體積小,病毒無法產生 DNA 複製所需的全部蛋白質;取而代之的是,病毒生命週期早期所需的病毒「早期蛋白」(early proteins) 劫持宿主細胞週期機制以複製病毒基因體。在初次感染後,大多數病毒被宿主免疫系統所控制,僅有極少的活躍性感染 (productive infection) 跡象。免疫抑制可導致病毒再活化與疾病,這可能反映了累積大量病毒顆粒之宿主細胞的溶解。病毒感染通常在癌症發展前數年即已發生,且只有一小部分受感染的個體會發展出癌症,常見於全身性免疫抑制或免疫監視 (immune surveillance) 受損的情境。對病毒而言,病毒性轉化是一條死路,因為它通常與病毒 DNA 以一種阻止病毒基因體複製及病毒生命週期完成、但容許早期病毒蛋白持續表現的方式整合進宿主細胞基因體有關,而這些早期病毒蛋白藉由靶向關鍵宿主細胞訊息傳遞蛋白來驅動細胞轉化。因此,病毒致癌蛋白 (viral oncoproteins) 藉由失調許多在偶發性癌症中因 UV 光或其他致突變物暴露而改變的相同蛋白質與路徑,從而促成癌症。
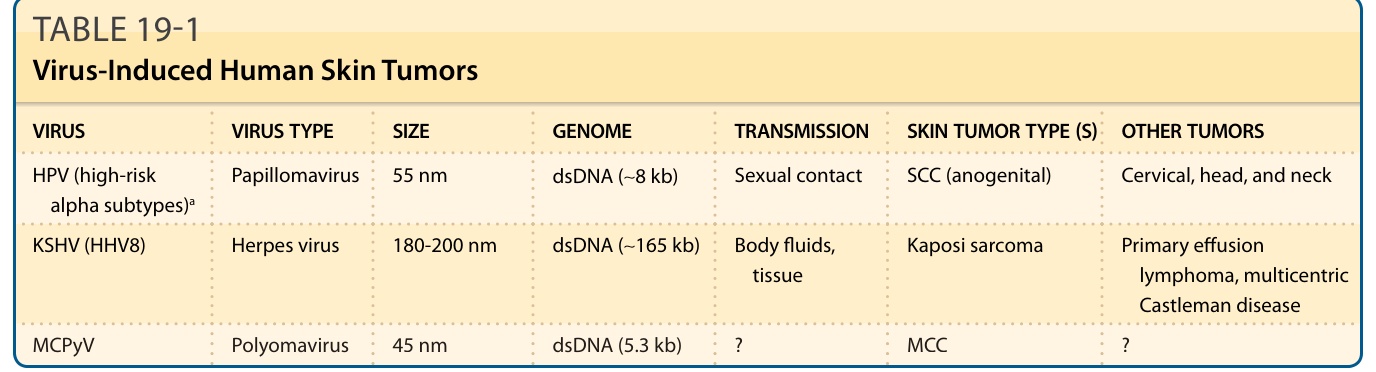
表 19-1:病毒誘發之人類皮膚腫瘤 (Virus-Induced Human Skin Tumors)
| 病毒 (VIRUS) | 病毒類型 (VIRUS TYPE) | 大小 (SIZE) | 基因體 (GENOME) | 傳播 (TRANSMISSION) | 皮膚腫瘤類型 (SKIN TUMOR TYPE(S)) | 其他腫瘤 (OTHER TUMORS) |
|---|---|---|---|---|---|---|
| HPV(高風險 alpha 亞型)a | 乳突瘤病毒 (Papillomavirus) | 55 nm | dsDNA(∼8 kb) | 性接觸 | SCC(肛門生殖器) | 子宮頸、頭頸 |
| KSHV (HHV8) | 皰疹病毒 (Herpes virus) | 180-200 nm | dsDNA(∼165 kb) | 體液、組織 | 卡波西肉瘤 (Kaposi sarcoma) | 原發性滲出液淋巴瘤、多中心型 Castleman 病 |
| MCPyV | 多瘤病毒 (Polyomavirus) | 45 nm | dsDNA(5.3 kb) | ? | MCC | ? |
a 關於 beta 人類乳突瘤病毒 (human papillomavirus, HPV) 與皮膚鱗狀細胞癌 (SCC) 的討論見內文。dsDNA,雙股 DNA (double-stranded DNA);HHV8,人類皰疹病毒-8 (human herpesvirus-8);KSHV,卡波西肉瘤相關皰疹病毒 (Kaposi sarcoma–associated herpesvirus);MCC,梅克爾細胞癌 (Merkel cell carcinoma);MCPyV,梅克爾細胞多瘤病毒 (Merkel cell polyomavirus)。
人類乳突瘤病毒 (HUMAN PAPILLOMAVIRUSES)
已鑑定出超過 200 種人類乳突瘤病毒 (human papillomaviruses, HPVs),並分為五個屬 (genera),其中 alpha 與 beta HPV 類型與癌症關聯最為密切。某些 alpha HPV 感染皮膚並造成疣 (warts),但其他則感染黏膜表面,其中低風險 HPV 產生良性病變(尖形濕疣 condylomata),而高風險 HPV(通常為 HPV16 或 18)則與惡性腫瘤(子宮頸癌、大多數肛門直腸癌,以及相當部分的生殖器與口咽癌)有關。高風險 alpha HPV 感染在年輕、性活躍的個體中相對常見,但在大多數個體中此感染會經由免疫系統被清除。Beta HPV 已與疣狀表皮發育不良個體之皮膚疣與 SCC 的發展連結在一起,並可能在某些慢性免疫抑制個體的皮膚 SCC 發病機轉中扮演角色。然而,與 alpha HPV 不同,皮膚癌中尚無病毒轉錄本 (viral transcripts) 的證據,顯示 beta HPV 並非腫瘤維持所必需。一種「打帶跑」(hit-and-run) 機轉——假設 HPV 基因轉錄具有時間上與功能上受限的需求——仍與因果角色相容,但難以確切證明。來自高風險 alpha HPV 的早期基因 E6 與 E7 藉由分別結合並降解腫瘤抑制因子 TP53 與 RB1 來驅動腫瘤發生。相對地,beta HPV 則影響其他致癌效應因子與過程以驅動癌症,導致雙股 DNA 修復缺陷、細胞凋亡受損、上皮分化受擾,以及 NOTCH 與 TGF-β 訊息傳遞改變。
人類多瘤病毒 (HUMAN POLYOMAVIRUSES)
人類多瘤病毒 (polyomavirus, PyV) 感染無所不在。絕大多數成人血清學上帶有早年由多種人類 PyV 感染的證據,隨後因宿主細胞核內存在游離型 (episomal) 病毒 DNA 而建立亞臨床、持續性的感染。病毒再活化與活躍性感染(需要病毒基因體複製及包裝為具感染力的病毒體 virions)可發生於免疫抑制個體並造成臨床上明顯的疾病。例如,BKPyV 與移植後腎病變及出血性膀胱炎的發展有關,而 JCPyV 則造成進行性多灶性白質腦病 (progressive multifocal leukoencephalopathy)。數種 PyV 已從人類皮膚分離出來,其中某些與特殊的皮膚疾患有關,包括搔癢性與異常角化性皮膚病 (HPyV6 與 HPyV7),以及毛囊疾患棘狀毛髮發育不良 (trichodysplasia spinulosa, TSPyV)。在幾乎所有病例中,這些情況皆發生於免疫抑制宿主並與活躍性感染有關,導致受影響細胞中大量病毒體的累積。梅克爾細胞 PyV (Merkel cell PyV, MCPyV) 也存在於正常人類皮膚中,但與任何已知連結至活躍性病毒感染的皮膚疾患無關。然而,MCPyV 可在約 80% 的梅克爾細胞癌中偵測到,後者為起源於皮膚之罕見但具侵襲性的神經內分泌惡性腫瘤(見第 113 章)。發現 MCPyV 病毒 DNA 在 MCC 中呈克隆性整合,強烈提示此病毒在病毒陽性腫瘤之 MCC 發展的早期階段扮演重要角色。鑑於強力證據指向 MCPyV 在 MCC 中的因果角色,已投入大量心力研究 MCPyV 早期基因產物大 T 抗原與小 T 抗原 (large T and small T antigens, LTAg, sTAg) 的轉化潛能。歷史上,來自一種猴多瘤病毒 SV40 的 LTAg 因其在細胞培養中轉化細胞並在實驗動物中產生腫瘤的強大能力,而被廣泛用於研究癌症的基本面向。SV40 LTAg 的轉化特性大致歸因於其破壞 TP53 與 RB1 兩者功能的能力,從而將這兩種腫瘤抑制因子鑑定為病毒致癌作用的關鍵標靶。與 LTAg 相對,SV40 sTAg 單獨並非強效致癌基因,雖然它可能促成 LTAg 驅動的轉化。耐人尋味的是,MCPyV TAg 對轉化的貢獻相對於 SV40 似乎是相反的,因為 sTAg 在體外與體內似乎皆為主要的致癌驅動因子。此領域目前的研究包括:旨在更妥善界定 sTAg 與 LTAg 對 MCC 發展的貢獻、鑑定 TAg 驅動之宿主細胞變化以及促成腫瘤發生之互動蛋白與路徑,並揭示 MCC 的起源細胞。
化學致癌作用 (CHEMICAL CARCINOGENESIS)
由於環境、職業或醫療暴露,各種化學物質也被認為與相對小部分人類皮膚癌的發展有關(表 19-2)。1775 年,Sir Percivall Pott 將煙囪清掃工人陰囊癌 (scrotal cancer) 發生率增加歸因於反覆暴露於煤煙 (soot)。此報告提供了職業暴露與癌症發展之間的第一個關聯,也是化學致癌作用的第一個範例。美國國家毒理學計畫 (National Toxicology Program) 於 2016 年發布的第 14 版致癌物報告 (14th Report on Carcinogens)(http://ntp.niehs.nih.gov/go/roc14),列出 248 種物質為已知(62 種物質)或合理預期(186 種物質)為人類致癌物。雖然其中大多數為化學物質,該清單也包括物理性物質(如游離輻射與 UVR)與感染性物質(如卡波西肉瘤相關皰疹病毒與 MCPyV),前文已討論。雖然尚未列入,抗黴菌藥 voriconazole 已被連結至免疫抑制病人中 SCC 發生率的增加。化學物質致癌的機轉揭示出與 UVR 誘發之癌症所發現者的顯著相似性,包括 DNA 損傷、選擇性細胞毒性與免疫抑制。
其他致癌刺激 (OTHER CARCINOGENIC STIMULI)
游離輻射 (ionizing radiation, IR) 曾被用於治療各種皮膚疾患,包括痤瘡 (acne) 與頭癬 (tinea capitis),以及惡性腫瘤。有趣的是,與 IR 暴露相關的皮膚癌過量風險幾乎完全侷限於 BCC,且已在原子彈倖存者、放射科醫師、礦工,以及因頭癬接受治療的兒童中獲得記載。兒童期暴露已與 BCC 風險顯著增加有關,潛伏期超過 20 年,尤其在膚色白皙的個體,提示 UV 與 IR 暴露之間存在強烈互動。在流行病學上,飛行員高度暴露於宇宙游離輻射,此與 BCC 風險升高 3 倍及黑色素瘤風險升高 3.5 倍有關。
慢性傷口 (chronic wounds) 長久以來在臨床上被認定為皮膚癌(尤其是 SCC)的危險因子。Marjolin 潰瘍 (Marjolin ulcers) 指起源於慢性潰瘍或瘢痕部位的皮膚癌,最常由燒傷所致,且最常發生於下肢與頭皮。數個詳述長期追蹤的系列研究結論為:超過 77% 的這些癌症與燒傷有關,將近 90% 的腫瘤為 SCC,且從初始損傷至腫瘤發展的平均間隔為 37 年。這些 SCC 具高度侵襲性,在超過 30% 的病例造成淋巴結侵犯,並在超過 11% 的病例造成遠端轉移。
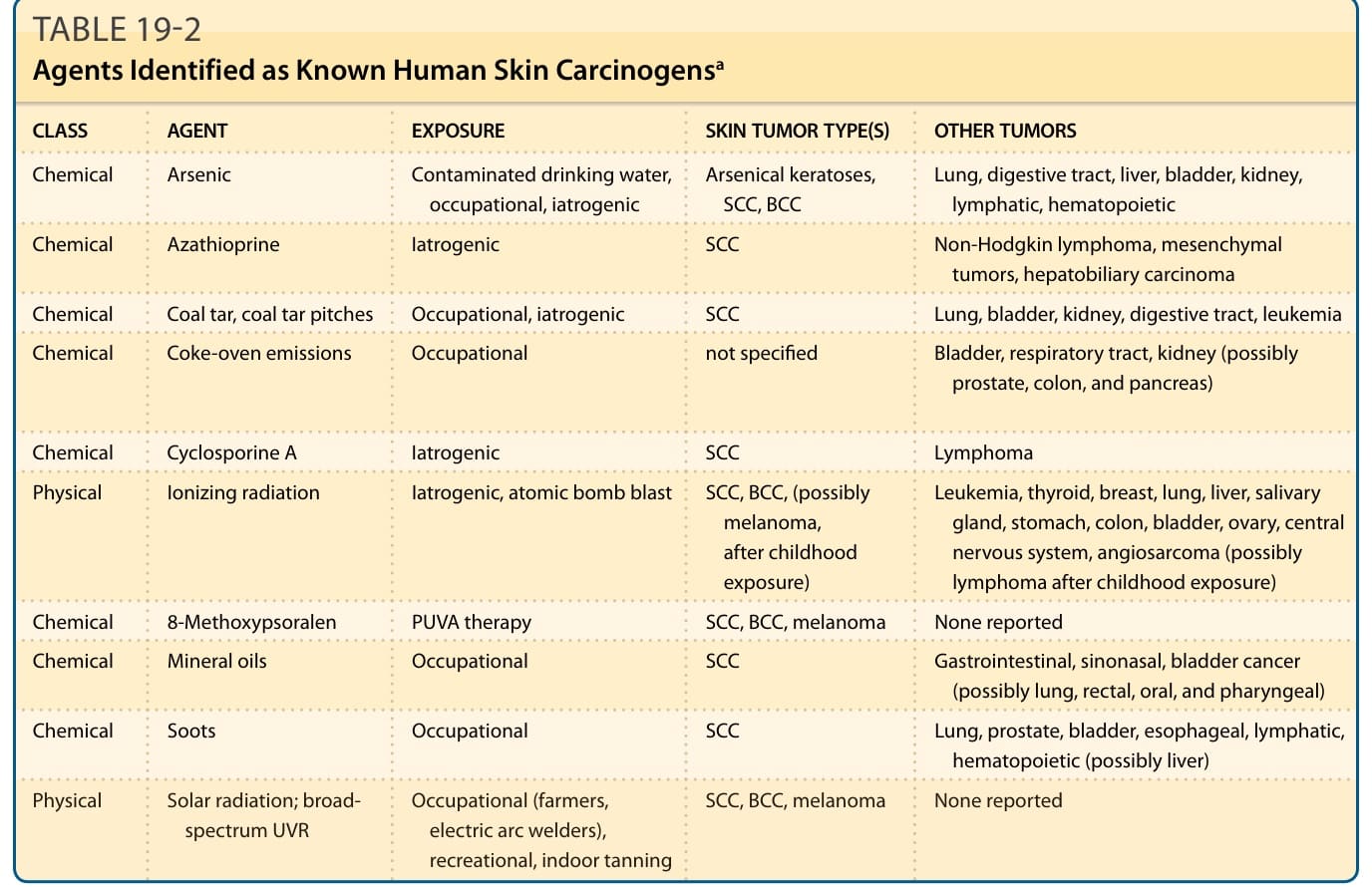
表 19-2:被鑑定為已知人類皮膚致癌物的物質a (Agents Identified as Known Human Skin Carcinogens)
| 類別 (CLASS) | 物質 (AGENT) | 暴露 (EXPOSURE) | 皮膚腫瘤類型 (SKIN TUMOR TYPE(S)) | 其他腫瘤 (OTHER TUMORS) |
|---|---|---|---|---|
| 化學性 | 砷 (Arsenic) | 受污染飲用水、職業性、醫源性 | 砷性角化症 (Arsenical keratoses)、SCC、BCC | 肺、消化道、肝、膀胱、腎、淋巴、造血系統 |
| 化學性 | Azathioprine | 醫源性 | SCC | 非何杰金氏淋巴瘤、間葉組織腫瘤、肝膽癌 |
| 化學性 | 煤焦油、煤焦油瀝青 (Coal tar, coal tar pitches) | 職業性、醫源性 | SCC | 肺、膀胱、腎、消化道、白血病 |
| 化學性 | 焦爐排放物 (Coke-oven emissions) | 職業性 | 未特別指明 | 膀胱、呼吸道、腎(可能包括前列腺、結腸與胰臟) |
| 化學性 | 環孢素 A (Cyclosporine A) | 醫源性 | SCC | 淋巴瘤 |
| 物理性 | 游離輻射 (Ionizing radiation) | 醫源性、原子彈爆炸 | SCC、BCC(兒童期暴露後可能包括黑色素瘤) | 白血病、甲狀腺、乳房、肺、肝、唾液腺、胃、結腸、膀胱、卵巢、中樞神經系統、血管肉瘤(兒童期暴露後可能包括淋巴瘤) |
| 化學性 | 8-甲氧基補骨脂素 (8-Methoxypsoralen) | PUVA 療法 | SCC、BCC、黑色素瘤 | 未有報告 |
| 化學性 | 礦物油 (Mineral oils) | 職業性 | SCC | 胃腸道、鼻竇鼻腔、膀胱癌(可能包括肺、直腸、口腔與咽部) |
| 化學性 | 煤煙 (Soots) | 職業性 | SCC | 肺、前列腺、膀胱、食道、淋巴、造血系統(可能包括肝) |
| 物理性 | 太陽輻射;廣譜 UVR (Solar radiation; broad-spectrum UVR) | 職業性(農夫、電弧焊接工)、休閒性、室內曬黑 | SCC、BCC、黑色素瘤 | 未有報告 |
a 資訊主要取自美國國家毒理學計畫 (National Toxicology Program) 第 14 版致癌物報告 (14th Report on Carcinogens, 2016)。造成人類皮膚癌的病毒性物質列於表 19-1。BCC,基底細胞癌 (basal cell carcinoma);PUVA,補骨脂素與紫外線 A 光 (psoralen and ultraviolet A light);SCC,鱗狀細胞癌 (squamous cell carcinoma);UVR,紫外線輻射 (ultraviolet radiation)。
人類皮膚癌的動物模型 (ANIMAL MODELS OF HUMAN SKIN CANCER)
皮膚癌的基因工程小鼠模型 (GENETICALLY ENGINEERED MOUSE MODELS OF SKIN CANCER)
鱗狀細胞癌小鼠模型 (SQUAMOUS CELL CARCINOMA MOUSE MODELS)
基因工程小鼠模型已能讓研究密切聚焦於現今已知對 SCC 至關重要的特定遺傳事件。鑑於在小鼠皮膚化學致癌作用期間產生的幾乎所有鱗狀乳突瘤 (squamous papillomas) 與癌症中皆鑑定出活化型 Hras 突變(見後文),早期的轉殖基因小鼠研究使用致癌型的 Hras,以確立持續性 Hras 驅動之訊息傳遞在鱗狀腫瘤新生之起始與進展中的核心角色。在皮膚中表現 Hras 致癌基因會導致發展出良性鱗狀乳突瘤或侵襲性 SCC,取決於哪些細胞群被改造以表現該轉殖基因。在 Hras 表現靶向毛囊間表皮 (interfollicular epidermis) 的小鼠中只發展出良性乳突瘤,且這些乳突瘤需要傷口或以腫瘤促進劑 (tumor promoter) 處理;但在包含幹細胞的毛囊上皮中表現相同致癌基因,則導致自發性 SCC 發展。這類研究確立了基因工程小鼠模型對於研究皮膚腫瘤之遺傳與細胞基礎的效用。以這些初始模型為基礎,眾多後續研究探討了各種其他基因與路徑對鱗狀腫瘤發展的貢獻。皮膚靶向表現 TGF-α 或其他活化 Ras 路徑的生長因子或受體,也會導致鱗狀腫瘤。表現細胞週期機制的組成成分,包括 E2F1、cyclin D1 與 Cdk4,也可能促成 SCC 的發展、進展或兩者。轉殖基因過度表現蛋白激酶 C (protein kinase C, PKC) 同工酶——表皮分化與其他細胞過程的關鍵調節因子——會導致鱗狀乳突瘤與癌症(PKC-α)的發展,或分化較差、快速轉移至局部淋巴結的 SCC(PKC-ε)。在基因改造小鼠中自發性或致癌物誘發的腫瘤形成,揭示了一些似乎在皮膚癌誘發中重要的基因與路徑,而這些從遺傳性癌症症候群或人類皮膚癌的分析中並不會顯現。在轉殖基因小鼠中將 Myc 靶向分化中的細胞,容許此一通常為有絲分裂後 (postmitotic) 之區室進行增殖,並產生類光化性角化症的表現型;而使用 Keratin 5 啟動子將 Myc 靶向基底細胞則產生自發性腫瘤。帶有 c-fos 缺失的小鼠發展出 v-rasHa 驅動的乳突瘤,但向 SCC 的惡性進展被阻斷。經由轉殖基因小鼠模型或重組人類皮膚研究,多種其他分子與路徑已被牽涉於 SCC 發展,包括 Smad2、Smad4、Stat3、PKC delta、PKC eta、Rac1、Pak1、Atf3、Sos、mTOR、TGF-β、Akt、Src、Fyn 與 NF-κB(核因子 kappa B,nuclear factor kappa B)。
同時去活化表皮中的 Rb1 與 Trp53,即足以在小鼠中誘發 SCC,這與高風險 HPV E6 與 E7 表現能在皮膚中造成 SCC 的能力一致。使用基因剔除 SCC 中最常突變的兩個基因 Notch1 與 Trp53 的實驗,已牽涉到腫瘤抑制、分化、增殖與基質互動的重要路徑。在小鼠中移除 Trp53 在化學與 UV 致癌作用模型中皆顯著加速腫瘤誘發。表皮特異性移除 Notch1 顯著增強 DMBA(7,12-二甲基苯并[a]蒽,7,12-dimethylbenz[a]-anthracene)/TPA(12-O-十四醯佛波-13-乙酸酯,12-O-tetradecanoylphorbol-13-acetate)模型中的致癌作用,導致 BCC 與 SCC 兩者的誘發。當以鑲嵌方式移除時,隨後顯示皮膚中 Notch1 去活化導致非細胞自主性 (non-cell-autonomous) 效應,造成一種類似傷口的發炎表現型,特別作用於腫瘤促進。此現象最戲劇性的範例見於僅在間葉細胞(包括真皮纖維母細胞)中缺乏 CSL 基因(Notch 訊息傳遞的關鍵組成成分)的小鼠。值得注意的是,這些小鼠在無需任何起始或促進物質的情況下自發發展出多灶性 SCC,顯示僅僅破壞間葉與其上覆蓋上皮之間的 Notch 訊息傳遞,即足以導致腫瘤發生。
基底細胞癌小鼠模型 (BASAL CELL CARCINOMA MOUSE MODELS)
在 NBCCS 病人中發現 PTCH1 突變,以及在偶發性 BCC 中發現 PTCH1 或 SMO 突變,為直接測試 Hedgehog 路徑參與 BCC 腫瘤發生的研究奠定了基礎。為了表現 Hedgehog 訊息傳遞活化因子(SHH、SMO、GLI1、GLI2)或刪除 Hedgehog 訊息傳遞抑制因子(PTCH1、SUFU)而開發的皮膚靶向小鼠模型,可靠地在基因工程小鼠中產生 BCC 或類 BCC 腫瘤(見第 111 章)。除了提供強力支持失調之 Hedgehog 訊息傳遞在 BCC 發展中扮演核心角色的體內證據外,這些模型還產生了寶貴的洞見:揭示腫瘤維持對持續 Hedgehog 訊息傳遞的需求;確立組織與細胞情境以及 Hedgehog 訊息傳遞水準在 BCC 腫瘤發生期間的重要性;揭示與其他訊息傳遞路徑具功能意義的互動;並提供了用於臨床前研究的強大模型。
皮膚癌的細胞起源 (Cellular Origins of Skin Cancer): 歷史上,人類皮膚腫瘤的起源細胞是由組織病理學與腫瘤細胞的表現型特徵推斷而來。BCC 腫瘤細胞未分化的外觀指向其可能起源於表皮基底層或毛囊外根鞘 (outer root sheath)。此外,早期 BCC 與胚胎毛芽 (embryonic hair germs) 的相似性、外根鞘標記 keratin 17 的一致表現,加上缺乏毛皮脂腺單位 (pilosebaceous units) 之掌蹠皮膚上典型 BCC 的缺如,皆被視為支持 BCC 毛囊起源的證據。另一方面,SCC 帶有經歷終末分化以形成類似表皮角質層 (cornified layer) 細胞之鱗屑 (squames) 的細胞,導致 SCC 較可能源自表皮而非毛囊的論斷。雖然這些觀察提供了有用的起點,但它們假設腫瘤細胞的表現型忠實地反映其源自的細胞類型。小鼠的實驗研究已對皮膚內可產生非黑色素瘤皮膚癌的細胞群提供了洞見。這些研究常經由在特定細胞群中活化某一假定致癌基因或刪除某一腫瘤抑制基因,並隨時間評估腫瘤發展來進行。此方法通常需要交配至少兩種不同的基因工程小鼠模型,以產生帶有以下兩者的雙轉殖基因 (bitransgenic) 小鼠:(1) 一個可由 Cre 重組酶 (Cre recombinase) 誘導的致癌基因或可被 Cre 切除的腫瘤抑制基因,以及 (2) 一種在特定細胞類型中表現之可由激素活化的 Cre 重組酶形式。以激素處理這些雙轉殖基因小鼠模型,可在特定時間於界定的細胞類型中活化 Cre 重組酶,導致休眠致癌基因的活化或腫瘤抑制基因的切除。對於 SCC 的建模,誘導表現致癌型 Hras 或 Kras 常與刪除 Trp53 腫瘤抑制基因結合;對於 BCC 的建模,則活化 Hedgehog 路徑致癌基因或刪除腫瘤抑制基因。藉由結合不同小鼠模型以靶向特定細胞類型,研究者已檢驗致癌驅動因子在毛囊幹細胞、過渡性放大的毛囊與其他前驅細胞,或毛囊間表皮基底細胞中的致瘤潛能。
從這類實驗中,可對 SCC 與 BCC 之潛在起源細胞,以及細胞與組織情境在腫瘤發生中的重要性,得出數項一般性結論。毛囊幹細胞似乎對由致癌型 Kras 結合 Trp53 刪除所驅動的侵襲性 SCC 發展具獨特敏感性,腫瘤進展至具梭形細胞 (spindle cell) 形態的晚期階段。使用相同組致癌驅動因子時,過渡性放大的毛基質 (hair matrix) 細胞則完全抗拒腫瘤發展,而毛囊間表皮細胞則形成良性鱗狀乳突瘤或 SCC,取決於靶向策略。
這些研究也顯示,雖然 SCC 在毛髮週期由靜止期 (telogen) 過渡至活躍生長 (anagen) 期間易於從毛囊幹細胞發展出來,但這些幹細胞在靜止狀態(telogen)時則大致抗拒 SCC 發展。基於遺傳的模型也對 BCC 的潛在起源細胞提供了洞見,但產生了一些相互矛盾的結果。初期研究顯示 BCC 優先源自毛囊間表皮,或源自傷口後遷移進入表皮的毛囊幹細胞,但後續使用不同致癌驅動因子的研究顯示,BCC 可源自毛囊幹細胞及數種前驅細胞群,其中表淺型 BCC 源自毛囊間表皮,結節型腫瘤則源自毛囊上皮。類似於 Kras+/Trp53 缺陷的 SCC,結節型 BCC 的發展因毛髮週期的活化而增強,確立了組織情境在驅動腫瘤發生中的重要性。此外,Hedgehog 訊息傳遞活性的強弱是腫瘤表現型的關鍵決定因素,低水準訊息傳遞產生基底樣錯構瘤 (basaloid hamartomas),高水準訊息傳遞則產生結節型腫瘤。綜合來看,這些研究強調了腫瘤起源細胞在決定腫瘤是否會發展、以及若發展則其特定表現型方面的重要性。
囓齒動物的化學誘發皮膚癌 (CHEMICALLY INDUCED SKIN CANCER IN RODENTS)
皮膚化學致癌作用的經典模型已被研究超過 70 年,並已證明是研究腫瘤起始、進展與轉移之可分離步驟的極為有用模型。在此模型最常用的形式中,先施用致癌物 DMBA,使其被代謝為強效致突變物,並造成活化型 Hras 突變,最常見於 Q61,頻率超過 90%。隨後反覆施用腫瘤促進劑(最常為佛波酯 phorbol ester TPA),就潛伏期、多發性、發生率與進展而言,導致高度可重現的腫瘤誘發。也可使用其他腫瘤促進劑,包括 UVR、岡田酸 (okadaic acid) 與物理性傷口。這些腫瘤的主要驅動因子是突變型 Hras,其後可發生擴增。在此方面,與人類 SCC 的一個重要對比是因 DMBA 暴露而強制的早期 ras 突變需求,而 Trp53 突變則於較晚出現。在人類中,TP53 突變發生得非常早,而 RAS 突變則較不常被偵測到。儘管如此,與此模型相關的文獻反映了癌症遺傳典範的興起,因為幾乎每個主要的癌症相關路徑都已使用此平台被研究過,尤其是與基因工程小鼠模型結合時。這些包括 TP53、INK4A-RB1-E2F、TGF-β、PI3K/AKT、STAT3、mTOR 與 COX2 路徑,以及多種受體酪胺酸激酶 (receptor tyrosine kinases),包括 EGFR(表皮生長因子受體,epidermal growth factor receptor)家族成員。此外,此模型在闡明 RAS 在癌症中的重要下游效應因子方面至關重要。
紫外線誘發的囓齒動物皮膚癌 (ULTRAVIOLET LIGHT–INDUCED RODENT SKIN CANCER)
UV 誘發皮膚癌最常使用的囓齒動物模型是免疫健全的無毛 (Hairless) 小鼠,這是一個缺乏功能性 Hairless (Hr) 基因的遠交品系。這些小鼠在出生後不久經歷一輪 anagen,此後毛囊退化為囊狀結構。缺乏毛髮免去了反覆剃毛的需求,並容許隨時間進行一致的 UV 暴露。雖然 Hr 基因功能被牽涉於 NF-κB 訊息傳遞與脂肪生成 (adipogenesis),但 Hr 在癌症中的功能尚未完全明瞭。就腫瘤中獲得的突變而言,此模型遠較 DMBA/TPA 模型更忠實於人類的情況。UV 照射也被用於 C57BL/6 等較常見的品系,這些品系對癌症的易感性程度不一,其中 SENCAR 品系是最敏感者之一。雖然極具資訊價值,UV 驅動的小鼠模型在一定程度上受困於各組與品系所用之光譜、劑量與輻照度缺乏一致性。其中部分是無可避免的,是由於跨不同光源重現光譜的技術限制所致。儘管如此,已從 UV 驅動模型的實驗中得出許多重要結論。Trp53 突變發生得早,且如同慢性受照射的人類皮膚,可觀察到 Trp53 突變克隆。在一個 18 例 SCC 的系列中,腫瘤展現出極高的突變負荷,中位數為每百萬鹼基 155 個突變。腫瘤中觀察到的其他突變包括 Notch 家族成員、Ink4a,以及 ras 中相對罕見的突變;並可見到反覆出現的染色體改變,這些改變對應到人類 3p、11p 與 9q 上的相應同源 (syntenic) 區域。
如同化學致癌作用方法,許多研究聚焦於特定路徑,將 UV 暴露與轉殖基因小鼠模型結合。已證明繞過由 Trp53、Survivin、Bcl2 與 E2f-1 所調控的 UV 誘發細胞凋亡,是完全腫瘤易感性所必需。一些在化學致癌作用中被牽涉的相同調節因子,也已知在 UV 驅動模型中重要,包括 COX-2、mTOR、AKT 與 ERK 路徑。也已使用 UV 驅動模型與人類 SCC 進行跨物種分析,牽涉到 WNT、β-catenin 與 ERK 路徑,以及數種 microRNA,包括 miR-21 與 miR-31。
皮膚癌的治療與預防 (TREATMENT AND PREVENTION OF SKIN CANCER)
降低癌症相關罹病率與死亡率最有效的方法是預防;鑑於 UV 驅動之致突變作用在皮膚癌發展中的核心角色,旨在有效限制暴露於陽光及曬黑床 (tanning beds) 等其他 UV 來源的努力,很可能對整體皮膚腫瘤發展產生重大影響。因此,幾乎所有美國各州限制未成年人使用曬黑床的聯邦立法,預計未來在降低皮膚癌發生率方面將產生重大有益效果。
各種化學預防 (chemoprevention) 方法也可能在減少 SCC 數量方面扮演重要角色,菸鹼醯胺 (nicotinamide) 在高風險病人的一項隨機第三期試驗中呈現出令人鼓舞的結果,顯示在 3 個月時 BCC 與 SCC 分別減少 20% 與 30%,光化性角化症則減少 11%。
雖然大多數皮膚癌可經由手術有效治療,但對這些癌症分子基礎的更深入理解,可能為靶向關鍵致癌驅動因子以進行預防或非手術治療提供獨特機會。例如,因為幾乎所有 BCC 似乎皆由不受約束的 Hedgehog 訊息傳遞所造成,全身性 Hedgehog 路徑抑制可有效治療局部晚期或轉移性 BCC 病人,雖然副作用常見、可能發展出藥物抗性,且停止治療後腫瘤可能復發。除了造成既存腫瘤消退外,全身性 Hedgehog 路徑抑制劑也能有效阻斷 Gorlin 症候群病人新發 BCC 的發展。這些發現提示,若局部抑制劑能有效阻斷致癌性 Hedgehog 訊息傳遞,則規律使用此類抑制劑可能有助於高風險病人的 BCC 預防。雖然這對 BCC 病人是令人振奮的可能性,但似乎不太可能有單一標靶治療藥物能同樣有效用於 SCC 腫瘤的預防,因為 SCC 發展涉及不同程度的多重致癌改變。
結論 (CONCLUSIONS)
對皮膚癌的廣泛臨床與實驗研究,已對 BCC 與 SCC 兩者的分子與細胞基礎產生重要洞見;促成了針對 BCC 之以機轉為基礎的標靶治療之開發;揭示了起源於皮膚與數種其他器官之 SCC 之間的關鍵分子相似性;提供了使用高度可操作之化學與 UV 驅動致癌作用小鼠模型以及基因工程模型來研究多步驟癌症發展的強大平台;並闡明了與理解其他器官腫瘤發生相關之腫瘤遺傳學與癌症生物學的基本面向。在理解 BCC 與 SCC 發病機轉方面的持續進展,很可能將帶來預防與治療這些極為常見之惡性腫瘤的新方法。
致謝 (ACKNOWLEDGMENTS)
由於篇幅限制,我們很遺憾無法納入所有相關文章的引用。我們感謝先前作者對本章部分內容的貢獻,包括 Drs. Masaoki Kawasumi、Paul Nghiem、Timothy Heffernan、Douglas Brash、Adam Glick 與 Stuart Yuspa。
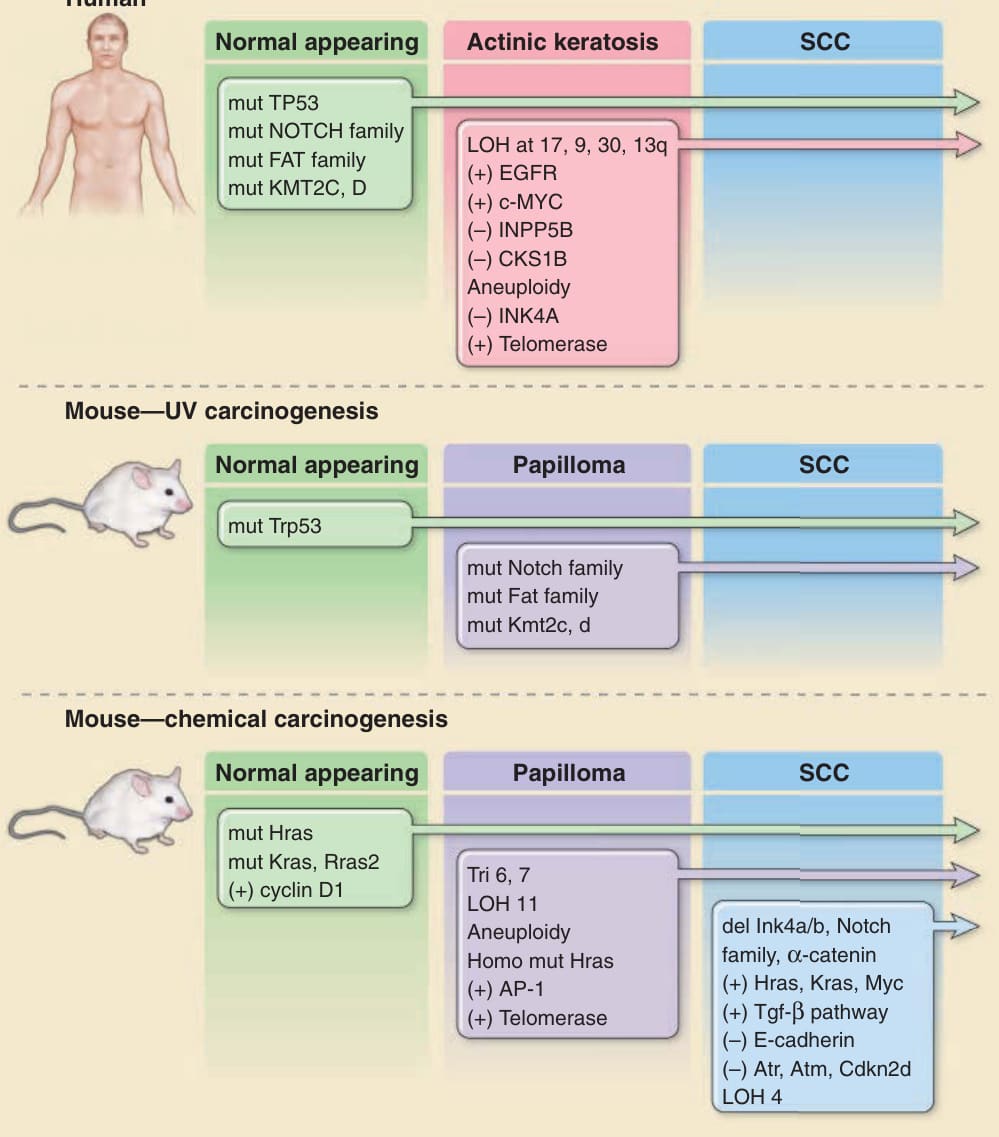
圖 19-1:與人類及小鼠皮膚鱗狀細胞癌 (SCCs) 發展相關的分子與遺傳變化。圖中以示意方式描繪人類皮膚 SCC 的多階段演變及其常合併的遺傳變化。早期病變中的單一鹼基突變常具有紫外線誘發損傷的特徵,而較晚期的變化則與基因體不穩定 (genomic instability) 有關。端粒酶 (telomerase)(抑制因子的缺失)或表皮生長因子受體 (EGFR) 酪胺酸激酶(基因擴增)活性的增加,也可能源自表觀遺傳變化。在 UV 驅動的皮膚 SCC 小鼠模型中可觀察到類似的變化。在化學誘發的小鼠皮膚 SCC 中,此模型向侵襲性腫瘤的多階段演變在時間上與遺傳上皆高度有序。操作上定義的階段包括起始 (initiation)、促進 (promotion) 與進展 (progression)。Ras 突變是用以起始腫瘤形成之化學致突變物的特徵。cyclin D1 的早期上調與 TGF-β1 的較晚上調經由表觀遺傳機轉發生,且似乎是致癌作用的重要組成成分。請注意,在 UV 誘發的腫瘤中大多數事件發生於進展序列的早期(如 TP53 突變),但在化學致癌作用中,大多數事件發生於晚期且常不帶有原始致突變物的特徵。應注意這些資料源自許多不同來源,反映了用以調查這些變化的多種技術。
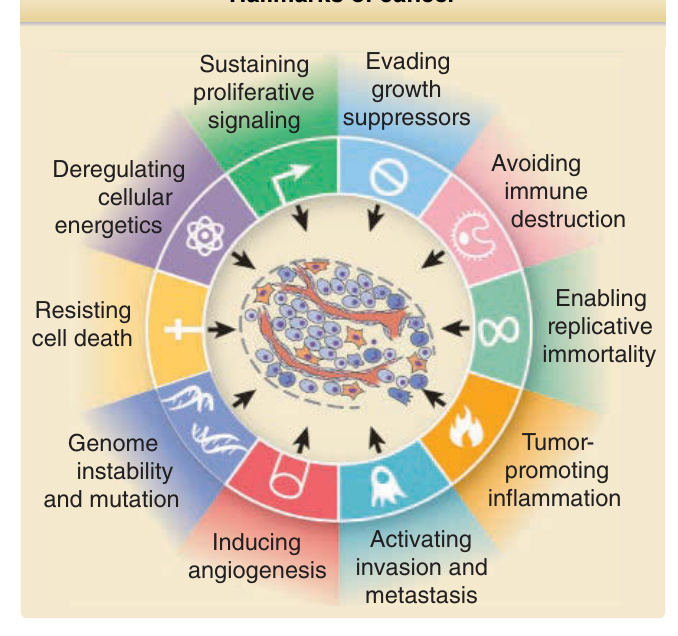
圖 19-2:癌症的特徵 (Hallmarks of cancer)。癌細胞獲得多種使其有別於正常細胞的特性,包括免於細胞死亡的保護、持續增殖、逃避生長抑制訊號、侵襲與轉移的能力、永生化 (immortalization) 與血管新生 (angiogenesis) 的誘導。較近期被認定對腫瘤發生重要的特性包括細胞能量代謝 (cellular energetics) 的失調、免疫逃避、基因體不穩定,以及發炎的刺激。(修改自 Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.)
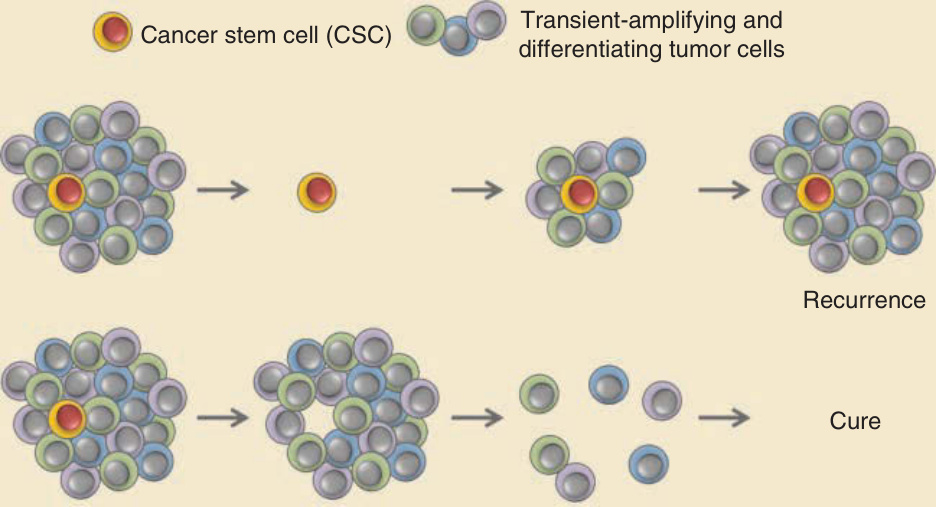
圖 19-3:作為治療標靶的癌症幹細胞 (Cancer stem cells as targets for treatment)。圖示一個腫瘤質塊,含有一個假定的癌症幹細胞 (cancer stem cell, CSC) 及其構成腫瘤主體的過渡性放大與分化中的後代。以常規治療處理可能導致腫瘤迅速消退,但若殘存 CSC 存在,停止治療後腫瘤可能復發。相對地,CSC 標靶治療可能導致較慢的反應,但在腫瘤細胞消退後,若 CSC 細胞群已被消除,則不太可能復發。許多(但非所有)腫瘤類型已描述過 CSC 與過渡性放大後代的階層式組織。
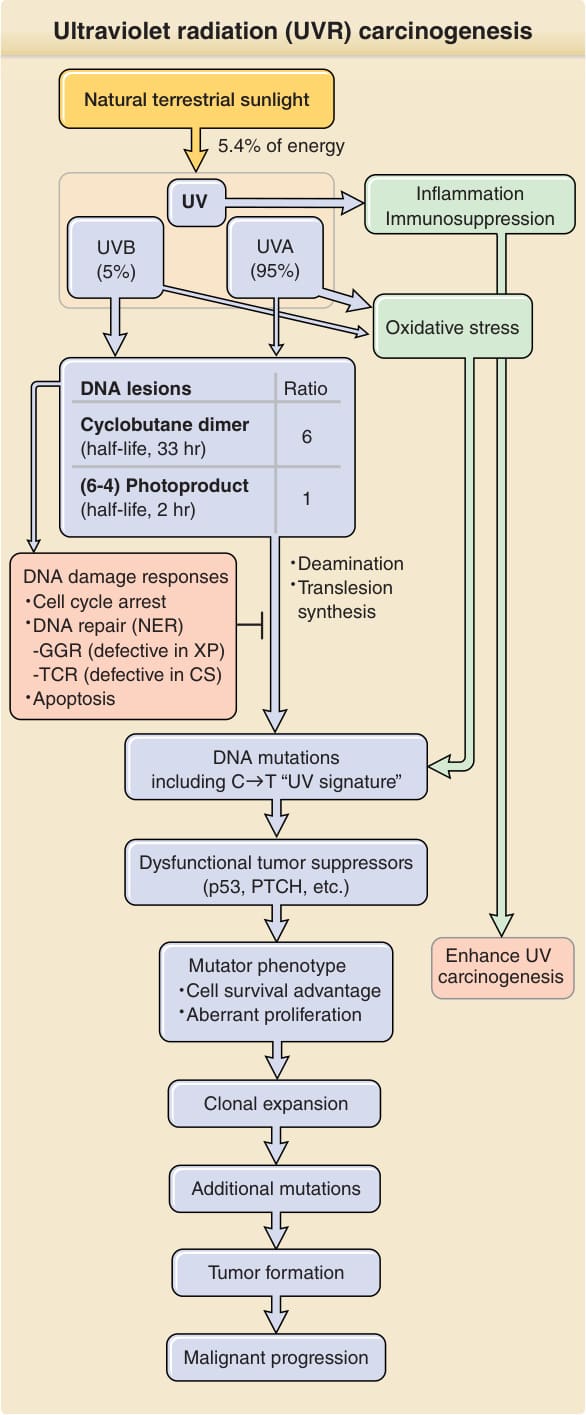
圖 19-4:紫外線輻射 (UVR) 致癌作用。UVR 在雙嘧啶 (dipyrimidine) 位點誘發兩種主要 DNA 病變:環丁烷嘧啶二聚體 (cyclobutane pyrimidine dimers, CPDs) 與嘧啶 (6–4) 嘧啶酮光產物 [(6–4)PPs]。所示比例 [每 1 個 (6–4)PP 對應 6 個 CPD] 為模擬陽光所誘發者。UVB 僅占 UVR 的 5%(約占所有陸地陽光能量的 0.3%),但卻產生大多數 UV 誘發的 DNA 病變。細胞藉由活化 DNA 損傷訊息傳遞路徑與誘導細胞週期停滯 (cell cycle arrest) 來回應 UVR 誘發的 DNA 損傷。受損的 DNA 由核苷酸切除修復 (nucleotide excision repair, NER) 修復。未修復的 DNA 病變經由去胺基化或易出錯的跨損傷合成 (translesion synthesis) 導致基因突變。未修復且含胞嘧啶的 CPD 促成 UV 特徵突變:C→T 轉換。負責致癌作用之基因的突變賦予一種致突變表現型 (mutator phenotype),包括對細胞凋亡的抗性。這擴大了易受進一步損傷之克隆的細胞庫,並增加了轉化的潛能。CS,Cockayne 症候群;GGR,全基因體修復 (global genome repair);PTCH,PATCHED1;TCR,轉錄偶聯修復 (transcription coupled repair);XP,著色性乾皮症 (xeroderma pigmentosum)。
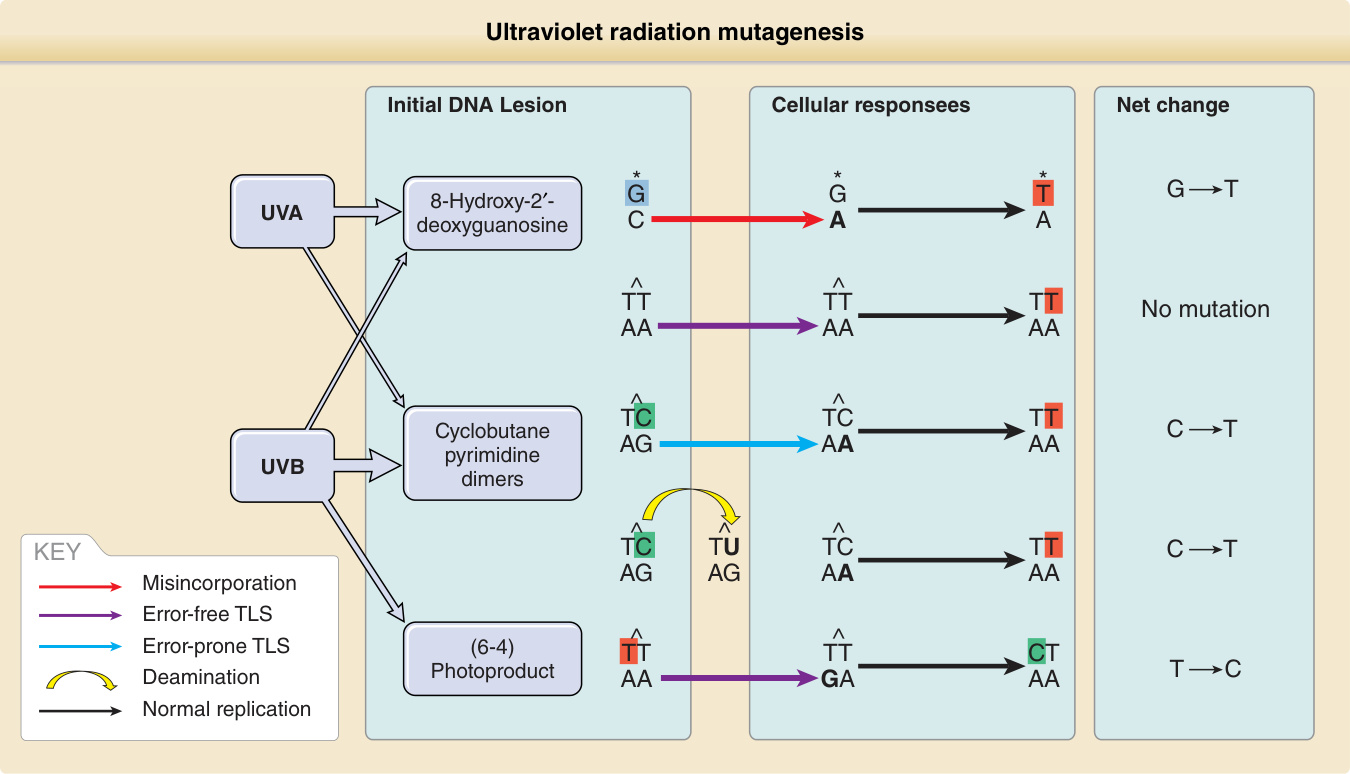
圖 19-5:紫外線輻射致突變作用:透過未修復 DNA 病變的複製如何導致 DNA 突變。環丁烷嘧啶二聚體 (CPDs) 對紫外線 (UV) 致突變作用最為相關,主要由 UVB 誘發。無錯誤的跨損傷合成 (error-free translesion synthesis, TLS) 在 CPD 對面添加腺嘌呤,使胸腺嘧啶二聚體不受影響,但在胞嘧啶二聚體中造成 UV 特徵性的 C→T 轉換,後者也可能源自去胺基化。由 UVB 誘發的 (6–4) 光產物與主要由 UVA 誘發的 8-羥基-2′-去氧鳥苷雖具致突變性,但相對罕見。
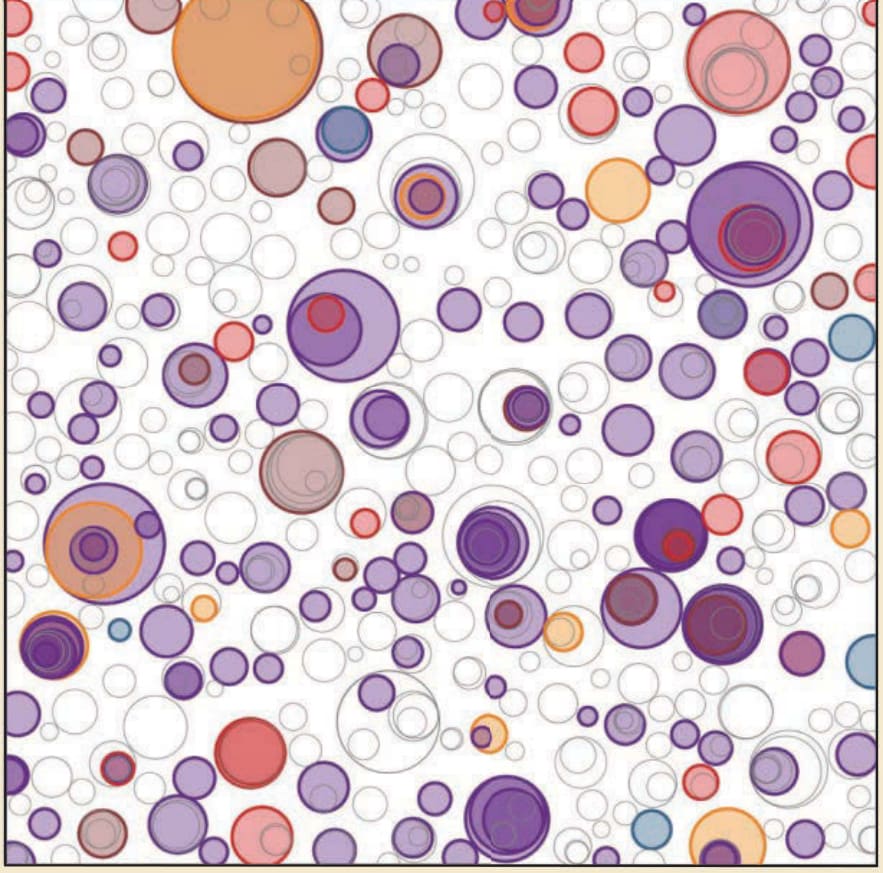
圖 19-6:與 SCC 發展相關之特定基因所連結之正常皮膚中突變克隆大小的圖示。此圖根據帶有反覆突變之 DNA 序列的比例以及皮膚切片大小的知識,呈現表皮細胞克隆的估計物理大小。在 1-cm2 的表皮斑塊內可發現數百個帶有反覆突變之癌症相關基因的克隆。某些突變被推斷為較大克隆內的次克隆(重疊的圓圈);然而,整體定序 (bulk sequencing) 無法進行確切的空間定位。(改編自 Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880-886. 經 AAAS 許可轉載。)