Carcinogenesis and Skin
2
AT-A-GLANCE
■ The estimated annual incidence of skin carcinomas is at least twofold higher than that of all other cancers combined, making these cancers a major burden on the U.S. health care system and a source of significant morbidity and mortality.
■ Most skin cancers arise because of mutations caused by ultraviolet radiation in sunlight, with chemical carcinogens, oncogenic viruses, and other factors contributing to the development of a smaller proportion of tumors.
■ The convergence of evidence from epidemiology, inherited predisposition syndromes, cancer genetics, animal models, and most recently genomics have collectively revealed unprecedented insights into the key causative events that drive the development of basal cell carcinoma (BCC) and squamous cell carcinoma (SCC).
■ BCC and SCC display markedly different dependencies on specific pathways: whereas BCC is almost exclusively a Hedgehog pathway— dependent tumor, SCC appears to rely on a more varied set of gene mutations and oncogenic signaling.
■ The accessibility of skin and the ability to model cancer development in animals has been highly useful in identifying the components and functions of key oncogenic signaling pathways, facilitating development of targeted therapeutics, and uncovering general principles of cancer biology.
INTRODUCTION
It was estimated that in 2017, there would be 87,110 new cases of invasive melanoma and 74,680 new cases of melanoma in situ.1 Precise incidence data are not available for basal cell carcinoma (BCC) and cutaneous squamous cell carcinoma (SCC) because these tumors are not typically reported to cancer registries, but based on Medicare datasets, the total number of nonmelanoma skin cancers in 2006 was estimated at 4,013,890 cases (2,463,567 individuals) and in 2012 at 5,434,193 cases (affecting 3,315,554 individuals).2 Another study, using data from the Medical Expenditure Panel Survey, estimated the number of individuals treated annually for nonmelanoma skin cancers at 3,090,442 (based on data from 2002 to 2006) and 4,301,338 (based on 2007 to 2011 data), with annual treatment costs up from 4.8 billion, respectively.3 The combined incidence of BCC and SCC is thus likely to be at least twofold
higher than that of all other cancers combined, which is estimated at 1,688,780 for 2017.1 Only a small fraction of patients with BCC or SCC will die because of their cancer, but the high frequency of these malignancies nonetheless results in an estimated 2000 deaths per year, according to the American Cancer Society, and most cases are SCC. Although much less common than nonmelanoma skin cancer, melanoma has a continually rising death rate now estimated at 9730 per year.1
Although they are rarely lethal, nonmelanoma skin cancers cause considerable cosmetic morbidity because they frequently arise on sun-exposed sites such as the face. Understanding the etiology and pathogenesis of these malignancies is thus a significant public health goal, and development of mechanism-based, nondeforming therapies is urgently needed. The high prevalence of skin tumors, their external location, and well-defined preneoplastic lesions (for SCC and melanoma) all provide an excellent opportunities for studying the factors regulating cutaneous cancer induction in humans. Qualities that facilitate the study of human skin cancers have also been useful in establishing relevant animal models. Advances in molecular genetics, keratinocyte cell culture, and development of genetically altered mice and reconstructed human skin models have greatly facilitated the analysis of basic mechanisms of cutaneous carcinogenesis. The main focus in this chapter is on general aspects of cutaneous carcinogenesis using nonmelanoma skin cancers as illustrative examples, with more detailed discussion of specific cutaneous malignancies presented in other chapters.
CUTANEOUS CARCINOGENESIS
GENERAL PRINCIPLES
GENERAL PRINCIPLES
The majority of malignant tumors arise through a stepwise process marked by characteristic phenotypic changes that reflect the acquisition of multiple genetic and epigenetic alterations needed to drive tumorigenesis from its inception through to invasion and metastasis. This basic tenet of cancer biology holds true for SCC (Fig. 19-1), but the pathogenesis of BCC provides a notable exception to this rule: precursor lesions have not been identified, metastases are extremely rare, and uncontrolled activation of a single oncogenic pathway may be sufficient for BCC tumorigenesis.4 Established cancers exhibit fundamental alterations in behavior that distinguish them from the normal tissues in which they arise. These differences include a reduced requirement for growth stimuli, impaired responses to growth
2
Genetic changes associated with development of cutaneous squamous cell carcinomas in humans and mice
Human
Normal appearing Actinic keratosis SCC
mut TP53 mut NOTCH family mut FAT family mut KMT2C, D
Mouse—UV carcinogenesis
LOH at 17, 9, 30, 13q (+) EGFR (+) c-MYC (–) INPP5B (–) CKS1B Aneuploidy (–) INK4A (+) Telomerase
Normal appearing Papilloma SCC
mut Trp53
Mouse—chemical carcinogenesis
mut Notch family mut Fat family mut Kmt2c, d
Normal appearing Papilloma SCC
mut Hras mut Kras, Rras2 (+) cyclin D1 Tri 6, 7 LOH 11 Aneuploidy Homo mut Hras (+) AP-1 (+) Telomerase
del Ink4a/b, Notch family, α-catenin (+) Hras, Kras, Myc (+) Tgf-β pathway (–) E-cadherin (–) Atr, Atm, Cdkn2d LOH 4
inhibitory and differentiation signals, alterations in apoptosis, delayed or blocked senescence, angiogenesis, the capacity for invasion and metastasis, metabolic reprogramming, and the ability to evade elimination by the immune system5 (Fig. 19-2). Although one or more of these abnormalities can be detected at different stages of tumor progression and may thus be seen in premalignant lesions, all are typically present in advanced cancers.
A driving force underlying neoplastic progression is defective DNA repair,6 which enables the accumulation of mutations involving oncogenes and tumor suppressor genes that contribute to the observed aberrations in tumor cell function. In the past, uncovering the molecular basis of cancers relied on genetic linkage studies to identify the chromosomal locus which segregates with tumor phenotypes in cancer susceptibility syndromes or the use functional screens
311
2
Hallmarks of cancer
Evading growth suppressors
Sustaining proliferative signaling
Avoiding immune destruction
Deregulating cellular energetics
Enabling replicative immortality
Resisting cell death
Tumorpromoting inflammation
Genome instability and mutation
Activating invasion and metastasis
Inducing angiogenesis
to identify genes from tumor cells that can drive neoplastic transformation in culture. However, the recent development of rapid and affordable next-generation sequencing technology, which can be used to screen
for mutations in the coding portions of genes (exome sequencing) or the entire genome, has revolutionized the approach to identifying mutations that may drive cancer and serve as targets for therapeutic intervention through “personalized oncology.”7 Although some changes in cell function are cell-autonomous and can be studied in purified populations of tumor cells, others depend on various additional cell types in the tumor microenvironment that participate in the development and progression of cancer in intact organisms, including inflammatory cells, cancer-associated fibroblasts, nerves, blood vessels and lymphatics, and other components of tumor stroma.8
Many tumors exhibit intratumor heterogeneity,9
which may be apparent at multiple levels. Molecular heterogeneity provides a driving force for neoplastic progression, selecting for outgrowth of cells that have acquired mutations conferring a proliferative or survival advantage and the ability to invade and metastasize. Biochemical heterogeneity occurs when subsets of tumor cells exhibit distinct oncogenic signaling properties and behavior, driven by regional differences in secreted growth factors or other microenvironmental factors. Perhaps the most striking example of heterogeneity is the presence of a small subset of stem cell–like tumor cells, called cancer stem cells, in at least some tumor types. According to the cancer stem cell hypothesis, tumors contain a small number of self-renewing stem cells that produce transient-amplifying and differentiating progeny that constitute the overwhelming majority of cells in a tumor (reviewed by Nassar and coworkers10) (Fig. 19-3), reflecting the hierarchical organization of cell lineages in normal tissues. Cancer stem cells are also called tumor-initiating cells because they are functionally defined by their ability, when purified
Cancer stem cells as targets for treatment
Cancer stem cell (CSC) Transient-amplifying and differentiating tumor cells
Conventional cancer therapy
CSCtargeted cancer therapy
Recurrence
Cure
312
and tested in small numbers or even as single cells, to reform a tumor in cell transplantation experiments. Tumor cells that are not cancer stem cells, in contrast, fail to produce tumors even when tested in relatively large numbers.10 This functional characterization generally requires that cancer stem cells possess a unique marker profile that enables them to be isolated from a heterogeneous population of tumor cells. Although the cancer stem cell concept was initially established in work centered on acute myeloid leukemia,11 results of many subsequent studies support the existence of a hierarchical organization of tumor-initiating cells and progeny in many, but not all, solid tumors. Cancer stem cell populations have been described in SCC (reviewed by Nassar and coworkers10) and BCC.12
The cancer stem cell concept has important clinical implications because therapies that effectively kill cancer stem cells may lead to cures by eliminating the key cell population from which all remaining cells in a tumor arise. In contrast, treatments that target non– stem cells representing most of a tumor’s mass may lead to striking tumor regression, but this is eventually followed by tumor recurrence because of outgrowth from remaining cancer stem cells (see Fig. 19-3). Although there are technical limitations to some of the assays used to ascertain “stemness” of isolated cancer cells, the cancer stem cell concept has been validated in multiple mouse models, and selective targeting of cancer stem cells is being pursued in an attempt to treat cancer more effectively.13 The success of this approach will depend in part on whether transient-amplifying or differentiating tumor cells possess sufficient plasticity to revert back to a stem cell–like population after effective depletion of the original cancer stem cell pool. An additional general property of cancers is intertumor heterogeneity. This includes phenotypic differences between tumors of the same type that are not necessarily related to malignant progression but are sufficient to justify classification into distinct morphologic categories, for example, superficial, nodular, and morpheaform subtypes of BCC. One explanation for intertumor heterogeneity is based on the notion that transformation of different cell populations produces different tumor subtypes, so a tumor’s cell of origin is a key determinant of its ultimate phenotype. For example, mouse modeling suggests that oncogenic activation of the Hedgehog pathway in an epidermal basal cell may produce superficial BCC, but nodular BCCs may arise from responsive cell populations within hair follicle epithelium.14 Experimental evidence from other tumor types supports the concept that different cells of origin give rise to distinct tumor subtypes.15 Another example of intertumor heterogeneity is the disparate growth rates of histologically similar tumor types. This can be strikingly apparent in BCC patients, for example, in whom some nodular tumors are indolent but others grow at a much faster rate. Intertumor heterogeneity in this setting could be explained by intrinsic (genetic, epigenetic, or signaling) or extrinsic (microenvironmental) alterations that provide a growth or survival advantage to some tumors but not others.
2
GENETICS OF SKIN CANCER
GENETICS OF SKIN CANCER
GENERAL CONCEPTS
Studies on the molecular basis of cancer development have revealed two main classes of genes, oncogenes and tumor suppressor genes, that play a key role in the pathogenesis of cancer. An oncogene is any gene that can transform normal cells in culture and induce cancer in animals. Most oncogenes are derived from proto-oncogenes, which generally encode proteins that function as critical positive regulators of cell proliferation or inhibitors of apoptosis. Conversion to an oncogene can occur through point mutations resulting in a constitutively active protein, through DNA amplification, or through chromosomal translocations that link a highly active promoter with the proto-oncogene. The latter two mechanisms cause increased or inappropriate expression of a proto-oncogene and may alter growth regulation of a responsive normal cell. Less common mechanisms implicated in cancer entail fusion of the coding domains of two genes, producing a novel, chimeric molecule with oncogenic properties16 or mutations in non-coding DNA that control the expression of oncogenes.17
Tumor suppressor genes normally function to negatively regulate cell proliferation, cause apoptosis, repair damaged DNA, or induce cellular differentiation. In contrast to oncogenes, which typically require that only one allele undergo activation via mutation, both alleles of a tumor suppressor gene must be inactivated to promote tumor development. Frequently, inactivating point mutations occur in one copy of a tumor suppressor gene, and the remaining normal copy is lost through a process of chromosomal missegregation during mitosis that leads to loss of heterozygosity. However, mutations in noncoding DNA17
could also lead to functionally significant reductions in expression of tumor suppressor genes. In addition, altered expression of long noncoding RNAs could influence tumorigenesis by affecting signaling pathways at multiple levels.18
Which oncogenes and tumor suppressors contribute to development of cutaneous neoplasms? Considerable insight into the genetic basis of sporadic skin cancers has come from the identification of specific genes that underlie hereditary skin tumor syndromes.19
The importance of DNA as a target for carcinogenesis was strongly supported by the discovery of defects in DNA repair genes in skin cancer–prone patients with xeroderma pigmentosum (see later) and several other dermatoses characterized by photosensitivity.20 Moreover, a detailed genomic analysis of 500 metastatic tumors from various primary cancers has uncovered unanticipated germline mutations in 12.2% of cases, with 75% of them affecting DNA repair genes.21 Taken together, these findings underscore the importance of robust DNA repair mechanisms for preventing accumulation of mutations that drive cancer development and progression.
313
2
MOLECULAR BASIS OF BASAL CELL CARCINOMA
Patients with nevoid BCC syndrome (NBCCS) are at a markedly increased risk for developing BCCs, which arise at a younger age and appear in greater numbers than in the general population. Patients with NBCCS are also predisposed to the development of a pediatric brain tumor arising in the cerebellum, medulloblastoma, as well as a variety of other defects throughout the body, including bifid ribs, calcified falx cerebri, odontogenic keratocysts, frontal bossing, palmar pits, and bifid ribs.22 Some of these structural abnormalities must have taken place during fetal development, suggesting that the genetic alteration in NBCCS influences tissue or organ formation during embryogenesis, as well as cancer development after birth. The discovery that NBCCS is caused by germline mutations disrupting the PTCH1 gene,23,24 which encodes a key component of the Hedgehog signaling pathway, is in keeping with this idea. Physiologic Hedgehog signaling plays an important role in patterning and morphogenesis of various organs and tissues during development and contributes to tissue homeostasis and regeneration postnatally; in contrast, uncontrolled activation of the Hedgehog pathway (Fig. 114-1), caused by mutations affecting PTCH1 or other Hedgehog pathway components (Table 114-1), is tightly associated with BCC development both in NBCCS patients and the general population25-27 (see Chap. 111). Because deregulated activation of the Hedgehog pathway is detected in essentially all BCCs and pathway activation has been shown to be sufficient for BCC development and required for tumor maintenance, it was believed that pharmacologic inhibitors of this pathway may be useful in the medical management of BCC. This has proven to be the case in a significant proportion of patients with advanced or metastatic BCC treated with Hedgehog pathway inhibitors, as discussed later and in Chap. 111. Despite the pivotal role of deregulated Hedgehog signaling in BCC development, next-generation sequencing studies have uncovered additional potential driver mutations in genes encoding MYCN, PPP6C, STK19, LATS1, PIK3CA, RAS proteins, with loss-of-function mutations and missense mutations in PTPN14, RB1, and FBXW7.28 Additional studies will be needed to determine the functional significance of these genetic alterations and others27 on BCC biology and treatment response.
MUTATIONS AND MOLECULAR DRIVERS IN SQUAMOUS CELL CARCINOMA
In contrast to BCC, the identification of pivotal genetic events that drive the development of cutaneous SCC has been somewhat complicated by the fact that no inherited cancer syndromes exclusively predispose to cutaneous SCC, and there is no high-frequency
314
oncogenic driver analogous to mutant BRAF in melanoma. As a result, the key evidence implicating specific genetic alterations in SCC development has been pieced together through characterization of highrisk clinical scenarios, molecular analysis of animal models, genomics, and proteomics. Three rare inherited syndromes have highlighted the importance of environmental influences, specific clinical scenarios, and pathways that drive SCC development. Xeroderma pigmentosum, characterized by extreme photosensitivity and stark acceleration in the onset of skin cancer, is associated with a nearly 10,000- fold increased risk of skin cancer before the age of 20 years.29 The key unifying defect, caused by recessive inactivation of genes distributed across eight complementation groups, results in the lack of nucleotide excision repair, which is required for removing mutagenic DNA photoproducts caused by ultraviolet (UV) radiation. Recessive dystrophic epidermolysis bullosa is a skin fragility disorder caused by loss-of-function mutations in COL7A1, which results in subepidermal blistering, nonhealing wounds, and a high predisposition to SCC. The chronic scarring that results predisposes to SCC, which are clinically difficult to manage and frequently fatal.30 Finally, heterozygous germline inactivation of transforming growth factor (TGF)-βR1 results in a susceptibility to keratoacanthomas as part of Smith-Ferguson syndrome31 and indeed, inactivation of TGF-βR signaling more globally is associated with typical SCC development.32 Additional genodermatoses and genes linked to SCC development are listed in Table 112-1. By using the two-stage chemical carcinogenesis approach in mice (see later), Balmain and colleagues pioneered the molecular genetic analysis of tumor development in this classical skin cancer model and identified mutated Hras as the primary oncogenic driver of tumors in this model33 1 year after its isolation as the first human oncogene.34 By 1991, recurrent TP53 mutations had been identified in human SCC corresponding to hotspot sites in the DNA-binding domain and bearing characteristic C → T transitions attributed to UV exposure.35,36
Next-generation DNA sequencing has now provided extensive insight into the genetic lesions that may drive cutaneous SCC development. Although many of these reports are from primary tumors, some series of metastatic lesions has been reported now as well.37-41 The mutational spectrum across the exome is, as expected, strongly dominated by C → T transitions and UV signature mutations and is overwhelmingly represented by the inactivation of tumor suppressor genes, with very few highly recurrent mutations. Taken across published exome and targeted sequencing efforts, the most frequently mutated tumor suppressor genes are TP53, CDKN2A, NOTCH family members, atypical cadherin FAT family members, the histone methyltransferases KMT2C and KMT2D, and KNSTRN.37-42 The panoply of sample sources, sequencing methodologies and analysis pipelines makes direct comparisons difficult, but additional potential tumor suppressors include CASP8, CREBBP, and CARD11.43 Mutant HRAS is observed in
up to 20% of SCC in one series.38 Amplifications in MYC and EGFR as well as loss of CKS1B and INPP5A have also been reported in cutaneous SCC.44-47
Importantly, these data also show striking similarities to SCC arising in other sites. Because stratified squamous epithelia form interfaces with the environment, these tissues are frequent sites of interaction with carcinogens. TP53 mutations occur at more than 70% frequency across all SCCs; NOTCH family genes are mutated in more than 70% of cutaneous SCC (cuSCC), 20% of oral head and neck SCC (HNSCC), 13% of lung SCC, and 10% of esophageal SCC, and SOX2 amplification is a common lineage-specific driver of SCC.48
Coupled with previous results, this shows that SCCs from diverse sites share deep molecular commonalities, including alterations in global gene expression and in TP53, TP63, NOTCH family, and SOX2 signaling. There is less published transcriptomic and proteomic data, although the correspondence to carcinogeninduced SCC holds prominently for HNSCC and lung SCC. Transcriptional profiling implicates transcription factors downstream of wingless-related integration site (WNT), β-catenin, and ERK (extracellular signalregulated kinase) signaling.37,48 Emerging literature also implicates multiple microRNAs in the development of SCC (reviewed by Konicke and coworkers49). Several have been implicated in multiple contexts with tumor-promoting and tumor-suppressive functions in proliferation, apoptosis, and migration. Reverse-phase protein array-based interrogation of canonical cancer pathways showed upregulation of ERK and mTOR (mammalian target of rapamycin) pathway signaling across the progression sequence of SCC development.50 The importance of ERK pathway signaling has also been reflected in two clinical scenarios involving drug-induced SCC: BRAF (v-raf murine sarcoma viral oncogene homolog B) inhibition used for melanoma and smoothened (SMO) inhibition used for BCC. It was observed in the initial clinical trials with the first Food and Drug Administration–approved BRAF inhibitor, vemurafenib, that approximately 22% of treated melanoma patients developed SCC or keratoacanthoma-like lesions; this adverse effect is less frequently observed with the more potent BRAF inhibitors dabrafenib and encorafenib.51 Mechanistically, this has been attributed to both paradoxical ERK signaling resulting from the aberrant drug-induced hyperactivation of MEK (mitogen activated protein/ extracellular signal-related kinase kinase)/ERK signaling in the context of wild-type BRAF,52,53 often in the context of oncogenic mutant HRAS,54 and to the suppression of apoptosis that occurs as a result of offtarget suppression of c-Jun-N-terminal kinase (JNK) signaling.55 Accordingly, the concomitant use of MEK with BRAF inhibitors almost completely suppresses the emergence of these SCCs.51 Interestingly, the longterm treatment of BCCs with the SMO inhibitor vismodegib has occasionally been associated with the evolution of treated tumors to a SCC morphology that is associated with drug resistance and activation of ERK signaling.56,57 Collectively, these data strongly implicate ERK signaling as an important pathway
2
in SCC. This has also been validated in a UV-driven preclinical model in which MEK inhibition has potent therapeutic and chemopreventative effects.58
ETIOLOGY OF HUMAN SKIN CANCER
PHOTOCARCINOGENESIS
PHOTOCARCINOGENESIS
Ultraviolet radiation (UVR) in sunlight is the primary etiologic agent for all skin cancers, and thus UVR is the major carcinogen in the human environment. The powerful carcinogenic activity of UVR is attributable to its ability to damage DNA and cause mutations, its capacity to clonally expand incipient neoplastic cells whose altered signaling pathways provide a survival advantage in the face of UV-induced cytotoxicity, its ability to induce reactive oxygen species (ROS), and its activity as an immune suppressant. The association of UVR with skin cancer is so strongly supported by clinical, epidemiologic, and experimental data that it represents perhaps the most clear-cut etiologic factor in human malignancy. UV light is a complete carcinogen and chronic exposure alone is sufficient to induce skin cancers. The process begins with carcinogen exposure, DNA damage, and the progressive acquisition of mutations. Clonal expansion occurs at least partially because of selection for these mutations, increasing the target size for further damage. The combination of these changes with key microenvironmental and immunologic consequences of UV exposure collectively drive tumor development.59 In addition to environmental or occupational exposures, patients with various dermatoses are treated with UVR. The vast majority of experience in assessing the resulting risk for skin cancer has been in the context of psoriasis phototherapy. This risk was best established for psoralen and ultraviolet A light (PUVA) therapy in a prospective study with 20 years of follow-up initiated by Stern and colleagues, in which the risk of SCC was more than 100-fold higher and the risk of BCC was more than 11-fold higher.60 The data on narrowband UVB has been largely confined to retrospective analyses, which suggest that there is no elevated risk of skin cancer; however, these studies are additionally limited by shorter follow-up.
ULTRAVIOLET RADIATION-INDUCED DNA DAMAGE AND REPAIR DNA Photoproducts: The first molecular step in sunlight-induced carcinogenesis occurs when UVB photons induce DNA photoproducts (see Fig. 19-4). UVB and UVC tend to be absorbed at the 5–6 double bond of pyrimidines (thymine and cytosine). If two adjacent pyrimidines are activated, the resulting open bonds cross-react, creating a cyclobutane pyrimidine dimer (CPD). The most frequent is TT, but TC, CT, and CC cyclobutane dimers are also made. A single bond between the 6 position of one pyrimidine and the
315
2
Ultraviolet radiation (UVR) carcinogenesis
Natural terrestrial sunlight
5.4% of energy
Inflammation Immunosuppression
UV
UVA (95%)
UVB (5%)
Oxidative stress
DNA lesions Ratio
Cyclobutane dimer (half-life, 33 hr) 6
(6-4) Photoproduct (half-life, 2 hr) 1
Deamination Translesion synthesis
DNA damage responses Cell cycle arrest DNA repair (NER) -GGR (defective in XP) -TCR (defective in CS) Apoptosis
DNA mutations including C T “UV signature”
Dysfunctional tumor suppressors (p53, PTCH, etc.)
Enhance UV carcinogenesis
Mutator phenotype Cell survival advantage Aberrant proliferation
Clonal expansion
Additional mutations
Tumor formation
Malignant progression
316
exocyclic group of the other instead creates a pyrimidine (6–4) pyrimidone photoproduct [(6–4)PP]61 most frequently TC. Both photoproducts distort the DNA helix and are recognized by DNA repair enzymes. Although there is 20-fold more UVA than UVB in sunlight, UVA requires up to 1000-fold greater doses for some of its biological effects such as DNA damage and minimal erythemal doses at 300 nm (UVB) and at 360 nm (UVA) for skin type II are 25 mJ/cm2 and 32,000 mJ/cm2, respectively.62 UVB induces 500 photolesions per 106 normal bases per J/cm2 in human skin.63
Ultraviolet Radiation—Induced Mutations: A CPD can lead to a mutation in two ways (Fig. 19-5). When the lesion is copied during DNA replication, the DNA polymerase may read a damaged cytosine as a thymine and insert an adenine opposite it. At the next round of replication, the polymerase correctly inserts thymine across from adenine, resulting in a C → T substitution. Alternatively, CPDs accelerate spontaneous deamination of their cytosines to uracil, resulting in the same change. Although the TT CPD is the most common photoproduct, the addition of A across damaged T bases by DNA polymerase eta causes no mutations. If two adjacent cytosines mutate, the result is CC → TT. This distinctive pattern of mutation, C → T in which the C lies next to another pyrimidine, including CC → TT, is unique to UVR and is called the UV signature mutation.64
Signature mutations provide a tool for deducing the original carcinogen from mutations found in tumors.64,65 Nearly all experimentally created UVB or UVC mutations are located at adjacent pyrimidines, and about two thirds are signature mutations. The remaining third, typically G → T and T → C substitutions or small insertions or deletions, are caused by UV but probably arise indirectly by ROS. G → T transversion can be caused by incorporation of adenine opposite 8-hydroxy-2′-deoxyguanosine (see Fig. 19-5), a common oxidative DNA lesion. Because this oxidative class of damage can be caused by many carcinogens, these mutations do not reveal whether their source was UVB, UVA, tobacco smoke, or intracellular oxidative phosphorylation. However, tumors carrying classic UV signature mutations must also contain UV-induced oxidative mutations. UVA weakly induces UVB signature mutations by photosensitization but generates oxidation-like mutations and T → G changes, which have been proposed to be a UVA fingerprint.61
Recently, a chemically distinct pathway of UVinduced CPD generation was discovered.66 In this mechanism, UV-induced generation of the ROS peroxynitrite was found to interact with melanin, resulting in chemiexcitation of electrons in melanin to extremely high-energy states, analogous to reactions that culminate in bioluminescence. However, instead of generating light, these excited electrons can directly interact with DNA, resulting in continuous CPD formation hours after UVA or UVB exposure has ceased, so-called “dark CPDs.” Importantly, these dark CPDs account for more than half of CPDs in melanocytes after UV exposure. This discovery suggests that any
2
Ultraviolet radiation mutagenesis
Initial DNA Lesion Cellular responsees Net change
8-Hydroxy-2′- deoxyguanosine
UVA
Cyclobutane pyrimidine dimers
UVB
KEY
Misincorporation
Error-free TLS
Error-prone TLS
(6-4) Photoproduct
Deamination
Normal replication
G T
C G A A G T
^ ^
AA TT AA TT AA TT
No mutation
^ ^
AG TC AA TT AA TC
C T
^ ^ ^
AG TC AA TT AA TC AG TU
C T
^ ^
AA TT GA AA TT
CT
T C
source of ROS can potentially generate CPDs and that the kinetics of CPD generation by this mechanism may offer an opportunity for novel chemoprevention strategies beyond blocking UV exposure.66
MUTATION BURDEN AND CLONAL DYNAMICS IN SKIN
The repeated exposure of skin to UVR over a lifetime represents an enormous collective DNA damage burden. Indeed, initial reports dating back to 1996 showed that clones of keratinocytes aberrantly expressing stable (and therefore likely mutated) p53 could be detected in whole-mounted skin.67,68 These TP53-mutant clones have also been identified in UVexposed mouse skin. Recently, targeted and whole-exome sequencing have shown that chronically UV-exposed clinically and histologically normal-appearing epidermis harbors about five mutations per megabase of DNA37,69 (Fig. 19-6), a remarkably high mutational load that exceeds that of many human cancers. Indeed, skin cancers have the highest mutational loads of any human cancer reported with the possible exception of mismatch repair-deficient colon carcinomas. To be identified at all by existing DNA sequencing methodologies, there must be clones of cells bearing identical mutations of sufficient quantity to be reliably detectable at a given sequencing depth. Therefore, the extent of UV-induced mosaicism in epidermis is likely to be even greater than reported.
Although the presence of clones is interpreted as evidence of positive selection for specific mutations,69
the frequency distribution of clone sizes suggests that clones can also spontaneously arise without the need for such selection.70 In the case of TP53, it is clear that although TP53 mutations compromise apoptosis, continued UV exposure is necessary for a selective advantage for these mutations to manifest themselves in the form of expanding clones, presumably through the inhibition or killing of surrounding normal keratinocytes. Deep sequencing of TP53-mutant clones in archival samples of UV-exposed human skin showed that many subclonal mutations in genes associated with SCCs are present, suggesting the widespread existence of keratinocyte patches highly predisposed to transformation.71
EVOLUTION OF ACTINIC KERATOSES TO SQUAMOUS CELL CARCINOMA
Estimates of the rate of progression of AK to SCC range from 0.6% at 1 year to 2.6% in 4 years.72 At the genomic level, many of the key mutated genes observed in SCC and irradiated skin are also found in AK.37,71 There have been no consistently identified transcriptional differences between AK and SCC. Instead, the molecular profiles of most AKs are largely indistinguishable from those of well-differentiated invasive SCC, suggesting that chemoprevention targeted to heavily mutagenized UV-exposed skin may be more effective.
317
2
Burden of mutations in sun-exposed normal skin
KEY
NOTCH1-3
TP53
FAT1
RBM10
Other
1mm
FGFR3
VIRAL CARCINOGENESIS
VIRAL CARCINOGENESIS
GENERAL FEATURES
Virus-associated cancers comprise up to 10% of all human malignancies, including several that arise in skin: Kaposi sarcoma; SCC (arising in epidermodysplasia verruciformis patients and a small fraction of immunosuppressed individuals); and Merkel cell carcinoma73 (see Table 19-1). DNA viruses have evolved highly effective mechanisms, in permissive host cells, for replicating their genomes, synthesizing capsid proteins, and assembling infectious viral particles during their normal, vegetative life cycles. Because of their small size, viruses cannot produce the full complement of proteins needed for DNA replication; instead, viral “early proteins” needed during early
318
stages of the viral life cycle hijack the host cell cycle machinery to replicate the viral genome. After primary infection, most viruses are kept in check by the host immune system with minimal evidence of productive infection. Immunosuppression can lead to viral reactivation and disease, which may reflect lysis of host cells that accumulate large numbers of viral particles. Viral infection typically precedes cancer development by many years, and only a small subset of infected individuals develop cancer, frequently in the setting of systemic immunosuppression or impaired immune surveillance. Viral transformation is a dead end for viruses because it is usually associated with integration of viral DNA into the host cell genome in a manner that precludes viral genome replication and completion of the viral life cycle but allows for persistent expression of early viral proteins that drive cellular transformation by targeting key host cell signaling
2
VIRUS VIRUS TYPE SIZE GENOME TRANSMISSION SKIN TUMOR TYPE (S) OTHER TUMORS
HPV (high-risk alpha subtypes)a Papillomavirus 55 nm dsDNA (∼8 kb) Sexual contact SCC (anogenital) Cervical, head, and neck
KSHV (HHV8) Herpes virus 180-200 nm dsDNA (∼165 kb) Body fluids, tissue Kaposi sarcoma Primary effusion lymphoma, multicentric Castleman disease
MCPyV Polyomavirus 45 nm dsDNA (5.3 kb) ? MCC ?
MCPyV Polyomavirus 45 nm dsDNA (5.3 kb) ? MCC ?
aSee text for discussion of beta human papillomavirus (HPV) and cutaneous squamous cell carcinoma (SCC). dsDNA, double-stranded DNA; HHV8, human herpesvirus-8; KSHV, Kaposi sarcoma–associated herpesvirus; MCC, Merkel cell carcinoma; MCPyV, Merkel cell polyomavirus.
proteins.74 Thus, viral oncoproteins contribute to cancer by dysregulating many of the same proteins and pathways that are altered by exposure to UV light or other mutagens in sporadic cancers.
HUMAN PAPILLOMAVIRUSES
More than 200 types of human papillomaviruses (HPVs) have been identified and grouped into five genera, with the alpha and beta HPV types most closely linked to cancer.75 Some alpha HPVs infect skin and cause warts, but others infect mucosal surfaces, with low-risk HPVs producing benign lesions (condylomata) and high-risk HPVs, typically HPV16 or 18, associated with malignancy (cervical cancer, most anorectal cancers, and a sizeable fraction of genital and oropharyngeal cancers). Infection by high-risk alpha HPVs is relatively common in young, sexually active individuals, but the infection is cleared via the immune system in most individuals. Beta HPVs have been linked to development of cutaneous warts and SCC in individuals with epidermodysplasia verruciformis and may play a role in the pathogenesis of cutaneous SCC in some chronically immunosuppressed individuals.76 Nevertheless, unlike the alpha HPVs, there has been no evidence for viral transcripts in skin cancers,37,77 indicating that beta HPVs are not required for tumor maintenance. A hit-and-run mechanism that posits a temporally and functionally restricted requirement for HPV gene transcription is still compatible with a causal role but is difficult to demonstrate definitively. The early genes E6 and E7 from high-risk alpha HPVs drive tumorigenesis by binding and degrading the tumor suppressors TP53 and RB1, respectively. In contrast, beta HPVs influence other oncogenic effectors and processes to drive cancer, leading to deficient double-strand DNA repair, impaired apoptosis, disrupted epithelial differentiation, and altered NOTCH and TGF-β signaling.78
HUMAN POLYOMAVIRUSES
Human polyomavirus (PyV) infections are ubiquitous. The great majority of adults harbor serologic evidence of infection by multiple human PyVs occurring at an
early age followed by establishment of subclinical, persistent infection because of the presence of episomal viral DNA in the nucleus of the host cell.79 Viral reactivation and productive infection, which requires viral genome replication and packaging into infectious virions, can occur in immunosuppressed individuals and cause clinically evident disease.79 For example, BKPyV is associated with development of posttransplant nephropathy and hemorrhagic cystitis, and JCPyV causes progressive multifocal leukoencephalopathy. Several PyVs have been isolated from human skin, and some are associated with distinctive skin disorders, including pruritic and dyskeratotic dermatoses (HPyV6 and HPyV7) and the hair follicle disorder trichodysplasia spinulosa (TSPyV).80 In nearly all cases, these conditions arise in immunosuppressed hosts and are associated with productive infection, leading to accumulation of a large numbers of virions in affected cells. Merkel cell PyV (MCPyV) is also present in normal human skin but is not associated with any known skin disorders linked to productive viral infection. MCPyV is, however, detected in about 80% of Merkel cell carcinomas, which are rare but aggressive neuroendocrine malignancies that arise in skin (see Chap. 113). MCPyV viral DNA was found to be clonally integrated in MCCs,81 strongly suggesting that this virus plays an important role at early stages of MCC development in virus-positive tumors. Given the strong evidence pointing to a causal role for MCPyV in MCC, much effort has gone into investigating the transforming potential of the MCPyV early gene products large T and small T antigens (LTAg, sTAg). Historically, LTAg from a simian polyomavirus, SV40, has been used extensively to study fundamental aspects of cancer because of its potent ability to transform cells in cell culture and produce tumors in experimental animals.82 The transforming properties of SV40 LTAg have been attributed largely to its ability to disrupt the functions of both TP53 and RB1, identifying these two tumor suppressors as key targets for viral oncogenesis. In contrast to LTAg, SV40 sTAg alone is not a potent oncogene, although it may contribute to LTAg-driven transformation. Intriguingly, the contribution of MCPyV TAgs to transformation appears reversed relative to that of SV40 in that sTAg appears to be the main oncogenic driver both in vitro
319
2
and in vivo.83,84 Current studies in this area include work aimed at better defining the contributions of sTAg and LTAg to MCC development, identifying key TAg-driven changes in host cells and interacting proteins and pathways that contribute to tumorigenesis, and uncovering the MCC cell of origin.
CHEMICAL CARCINOGENESIS
CHEMICAL
CARCINOGENESIS
Also implicated in the development of a relatively small proportion of human skin cancers are various chemicals, as a result of environmental, occupational, or medicinal exposures (Table 19-2). In 1775, Sir Percivall Pott85 attributed the increased incidence of scrotal cancer in chimney sweeps to repeated exposure to soot. This report provided the first link between an occupational exposure and the development of cancer as well as the first example of chemical carcinogenesis. The National Toxicology Program’s 14th Report on Carcinogens (http://ntp.niehs.nih.gov/go/ roc14), released in 2016, lists 248 substances as either known (62 substances) or reasonably anticipated (186 substances) to be human carcinogens. Although most of these are chemicals, the list also includes physical agents (eg, ionizing and UVR) and infectious agents (eg, Kaposi sarcoma–associated herpesvirus and MCPyV), discussed earlier. Although not yet listed,
the antifungal voriconazole has been linked to an increased incidence of SCCs in immunosuppressed patients.86 Mechanisms by which chemicals cause cancer reveal striking similarities to those uncovered for UVR-induced cancer, including DNA damage, selective cytotoxicity, and immunosuppression.
OTHER CARCINOGENIC STIMULI
OTHER CARCINOGENIC
STIMULI
Ionizing radiation (IR) had been used to treat a variety of skin disorders, including acne and tinea capitus as well as malignant tumors. Interestingly the excess risk of skin cancer associated with IR exposure is almost exclusively confined to BCC and has been documented in atomic bomb survivors, radiologists, miners, and children treated for tinea capitis.87 Childhood exposure has been associated with a significantly increased risk of BCC with a latency of over 20 years, particularly in fair-complected individuals, suggesting a strong interactions between UV and IR exposures.88 Epidemiologically, pilots are highly exposed to cosmic IR, which is associated with a 3-fold elevated risk of BCC and 3.5- fold elevated risk of melanoma.89
Chronic wounds have long been recognized clinically as a risk factor for skin cancer, particularly SCC. Marjolin ulcers refer to skin cancers that arise in sites of chronic ulcers or scars, most often caused by burns90
CLASS AGENT EXPOSURE SKIN TUMOR TYPE(S) OTHER TUMORS
Chemical Arsenic Contaminated drinking water, occupational, iatrogenic Arsenical keratoses, SCC, BCC Lung, digestive tract, liver, bladder, kidney, lymphatic, hematopoietic
Chemical Azathioprine Iatrogenic SCC Non-Hodgkin lymphoma, mesenchymal tumors, hepatobiliary carcinoma
Chemical Coal tar, coal tar pitches Occupational, iatrogenic SCC Lung, bladder, kidney, digestive tract, leukemia
Chemical Coke-oven emissions Occupational not specified Bladder, respiratory tract, kidney (possibly prostate, colon, and pancreas)
Chemical Cyclosporine A Iatrogenic SCC Lymphoma
Physical Ionizing radiation Iatrogenic, atomic bomb blast SCC, BCC, (possibly melanoma, after childhood exposure)
Leukemia, thyroid, breast, lung, liver, salivary gland, stomach, colon, bladder, ovary, central nervous system, angiosarcoma (possibly lymphoma after childhood exposure)
Chemical 8-Methoxypsoralen PUVA therapy SCC, BCC, melanoma None reported
Chemical Mineral oils Occupational SCC Gastrointestinal, sinonasal, bladder cancer (possibly lung, rectal, oral, and pharyngeal)
Chemical Soots Occupational SCC Lung, prostate, bladder, esophageal, lymphatic, hematopoietic (possibly liver)
Physical Solar radiation; broad-
Occupational (farmers,
Physical Solar radiation; broadspectrum UVR Occupational (farmers, electric arc welders), recreational, indoor tanning
spectrum UVR
electric arc welders), recreational, indoor tanning
SCC, BCC, melanoma None reported
SCC, BCC, melanoma None reported
aInformation obtained primarily from the 14th Report on Carcinogens (2016), National Toxicology Program. Viral agents that cause human skin cancers are listed in Table 19-1. BCC, basal cell carcinoma; PUVA, psoralen and ultraviolet A light; SCC, squamous cell carcinoma; UVR, ultraviolet radiation.
320
and most often occurring on the lower extremities and scalp. Several series detailing long-term followup conclude that more than 77% of these cancers are associated with burns, almost 90% of tumors are SCC, and the average interval from initial injury to tumor development was 37 years.91,92 These SCCs are highly aggressive, resulting in nodal involvement in more than 30% of cases and distant metastases in more than 11%.93
ANIMAL MODELS OF HUMAN SKIN CANCER
GENETICALLY ENGINEERED MOUSE MODELS OF SKIN CANCER
GENETICALLY ENGINEERED
MOUSE MODELS OF
SKIN CANCER
SQUAMOUS CELL CARCINOMA MOUSE MODELS
Genetically engineered mouse models have enabled close focus on specific genetic events now known to be critical for SCC. Given the identification of activating Hras mutations in nearly all squamous papillomas and carcinomas arising during chemical carcinogenesis in mouse skin (see later), early transgenic mouse studies used oncogenic forms of Hras to establish the central role of sustained Hras-driven signaling in initiation and progression of squamous neoplasia. Expression of the Hras oncogene in skin led to development of either benign squamous papillomas or invasive SCC, depending on which cell populations were engineered to express the transgene. Only benign papillomas developed in mice with Hras expression targeted to interfollicular epidermis, and these required either wounding or treatment with a tumor promoter, but expression of the same oncogene in hair follicle epithelia that included stem cells led to spontaneous SCC development.94,95 These types of studies established the utility of genetically engineered mouse models for studying the genetic and cellular basis of skin tumors. With these initial models serving as a foundation, numerous subsequent studies explored the contribution of various other genes and pathways to squamous tumor development.96-99 Skin-targeted expression of TGF-α or other growth factors or receptors that activate the Ras pathway also lead to squamous tumors. Expression of components of the cell cycle machinery, including E2F1, cyclin D1, and Cdk4, could also contribute to SCC development, progression, or both. Transgenic overexpression of protein kinase C (PKC) isozymes, which are key regulators of epidermal differentiation and other cellular processes, leads to development of squamous papillomas and carcinomas (PKC-α) or less differentiated SCCs that rapidly metastasized to regional lymph nodes (PKC-ε). Spontaneous or carcinogen-induced tumor formation in genetically modified mice has revealed genes and pathways that
2
appear to be important in skin cancer induction but would not have been apparent from hereditary cancer syndromes or analysis of human skin cancers. Targeting of Myc to differentiating cells in transgenic mice permits proliferation in this normally postmitotic compartment and produces an actinic keratosis–like phenotype, and basal cell targeting of Myc using a Keratin 5 promoter yields spontaneous tumors. Mice carrying a deletion in c-fos developed v-rasHa–driven papillomas, but malignant progression to SCC was blocked. Multiple additional molecules and pathways have been implicated in SCC development through transgenic mouse modeling or recombinant human skin studies, including Smad2, Smad4, Stat3, PKC delta, PKC eta, Rac1, Pak1, Atf3, Sos, mTOR, TGF-β, Akt, Src, Fyn, and NF-κB (nuclear factor kappa B).96-100
The simultaneous inactivation of Rb1 and Trp53 in epidermis is sufficient to induce SCC in mice,101 consistent with the ability of high-risk HPV E6 and E7 expression to cause SCC in skin.102 Experiments using genetic ablation of the two most frequently mutated genes in SCC, Notch1 and Trp53, have implicated important pathways in tumor suppression, differentiation, proliferation, and stromal interactions. Removal of Trp53 in mice dramatically accelerates tumor induction in both chemical and UV carcinogenesis models.96 Epidermis-specific removal of Notch1 dramatically enhances carcinogenesis in the DMBA (7,12-dimethylbenz[a]-anthracene)/TPA (12-O-tetradecanoylphorbol-13-acetate) model resulting in both BCC and SCC induction.103 When removed in mosaic fashion, it was subsequently shown that Notch1 inactivation in skin resulted in non–cell-autonomous effects, resulting in an inflammatory woundlike phenotype, specifically acting in tumor promotion.104 The most dramatic example of this was seen in mice lacking the CSL gene, a vital component of Notch signaling, solely in mesenchymal cells, including dermal fibroblasts. Remarkably, these mice spontaneously developed multifocal SCC without the need for any initiating or promoting agent,105 demonstrating how simply disrupting Notch signaling between the mesenchyme and overlying epithelia is sufficient for tumorigenesis.
BASAL CELL CARCINOMA MOUSE MODELS
The discovery of PTCH1 mutations in NBCCS patients and either PTCH1 or SMO mutations in sporadic BCCs set the stage for studies directly testing the involvement of the Hedgehog pathway in BCC tumorigenesis. Skin-targeted mouse models developed to either express Hedgehog signaling activators (SHH, SMO, GLI1, GLI2), or delete Hedgehog signaling repressors (PTCH1, SUFU) reliably produce BCCs or BCC-like tumors in genetically engineered mice (reviewed in 4,106) (see Chap. 111). In addition to providing in vivo evidence strongly supporting a central role for deregulated Hedgehog signaling in BCC development, these models have yielded valuable insights into the requirement for sustained Hedgehog signaling in tumor maintenance; established the importance of tissue and cellular
321
2
context, as well as Hedgehog signaling levels, during BCC tumorigenesis; uncovered functionally significant interactions with other signaling pathways; and have provided powerful models for preclinical studies.
Cellular Origins of Skin Cancer: Historically, the cell of origin of human skin tumors was inferred from histopathology and the phenotypic characteristics of tumor cells. The undifferentiated appearance of BCC tumor cells pointed to a potential origin from the epidermal basal layer or hair follicle outer root sheath. In addition, the resemblance of early-stage BCCs to embryonic hair germs and consistent expression of the outer root sheath marker keratin 17, coupled with the absence of typical BCCs on palmar and plantar skin that is devoid of pilosebaceous units, were taken as evidence supporting a hair follicle origin for BCC. SCCs, on the other hand, harbor cells that undergo terminal differentiation to form squames resembling cells in the cornified layer of epidermis, leading to the assertion that SCCs are more likely to be derived from epidermis than hair follicles. Although these observations provide a useful starting point, they assume that the phenotype of a tumor cell faithfully reflects the cell type from which it originated. Experimental studies in mice have yielded insight into cell populations within skin that can give rise to nonmelanoma skin cancers. These studies are frequently performed by activating expression of a putative oncogene or deleting a tumor suppressor gene in specific cell populations and assessing tumor development over time. This approach typically requires breeding at least two different genetically engineered mouse models to generate bitransgenic mice carrying (1) a Cre recombinase–inducible oncogene or Cre-excisable tumor suppressor gene and (2) a hormone-activatable form of Cre recombinase expressed in specific cell types.107 Treating these bitransgenic mouse models with hormone activates Cre recombinase in defined cell types at a specific time, leading to activation of the dormant oncogene or excision of the tumor suppressor gene. For modeling SCC, inducible expression of oncogenic Hras or Kras is frequently combined with deletion of the Trp53 tumor suppressor gene; for modeling BCC, Hedgehog pathway oncogenes are activated or tumor suppressor genes are deleted. By combining different mouse models to target specific cell types, investigators have examined the tumorigenic potential of oncogenic drivers in hair follicle stem cells, transientamplifying hair follicle and other progenitor cells, or interfollicular epidermal basal cells.106-108
From these types of experiments, several general conclusions can be drawn regarding the potential cells of origin of SCC and BCC and the importance of cellular and tissue context in tumorigenesis. Hair follicle stem cells appear to be uniquely sensitive to invasive SCC development driven by oncogenic Kras combined with Trp53 deletion, with tumors progressing to advanced stages of development with a spindle cell morphology. Using the same set of oncogenic drivers, transiently amplifying hair matrix cells are completely resistant to tumor development, and interfollicular
322
epidermal cells form either benign squamous papillomas or SCC, depending on the targeting strategy.107,108
These studies also showed that although SCCs readily develop from follicle stem cells during the transition from the resting phase of the hair cycle (telogen) to active growth (anagen), they are largely resistant to SCC development when quiescent, in telogen. Genetic-based models have also provided insight into the potential cell of origin of BCC but have yielded some conflicting results. 106,107,109 Initial studies showed that BCCs preferentially arose from interfollicular epidermis or from hair follicle stem cells that migrated into epidermis after wounding, but subsequent studies using a different oncogenic driver showed that BCCs could arise from follicle stem cells as well as several progenitor populations, with superficial BCCs arising from interfollicular epidermis and nodular tumors from hair follicle epithelia. Similar to Kras+/Trp53-deficient SCC, development of nodular BCCs was potentiated by activation of the hair cycle, establishing the importance of tissue context in driving tumorigenesis. Moreover, the magnitude of Hedgehog signaling activity was a critical determinant of tumor phenotype, with low-level signaling producing basaloid hamartomas and high-level signaling yielding nodular tumors. Taken together, these studies underscore the importance of a tumor’s cell of origin in defining whether or not a tumor will develop, and if it does, its specific phenotype.
CHEMICALLY INDUCED SKIN CANCER IN RODENTS
CHEMICALLY INDUCED
SKIN CANCER IN RODENTS
The classical model of chemical carcinogenesis in skin has been studied for over 70 years and has proven to be a profoundly useful model for studying separable steps of tumor initiation, progression, and metastasis.97 In the most commonly used incarnation of the model, the carcinogen DMBA is applied initially, metabolized into a potent mutagen, and causes activating Hras mutations, most often at Q61 with over 90% frequency. Subsequent repeated application of tumor promoters, most commonly the phorbol ester TPA, results in highly reproducible tumor induction in terms of latency, multiplicity, incidence, and progression. Other tumor promoters can be used, including UVR, okadaic acid, and physical wounding.97 The dominant driver of these tumors is mutant Hras, which can undergo subsequent amplification. In this regard, one important contrast with human SCC is the early requirement for ras mutation as enforced by DMBA exposure, with the emergence of Trp53 mutations later.110 In humans, TP53 mutations occur very early and RAS mutations are detected less commonly.37-41 Nevertheless, the literature associated with this model mirrors the rise of the genetic paradigm of cancer because nearly every major cancer-related pathway has been investigated using this platform, particularly in conjunction with genetically engineered mouse models. These include the TP53, INK4A-RB1-E2F, TGF-β, PI3K/AKT, STAT3, mTOR,
and COX2 pathways, as well as multiple receptor tyrosine kinases, including EGFR (epidermal growth factor receptor) family members.96,97 In addition, the model has been critically important in elucidating important downstream effectors of RAS in cancer.96
ULTRAVIOLET LIGHT– INDUCED RODENT SKIN CANCER
ULTRAVIOLET LIGHT–
INDUCED RODENT
SKIN CANCER
The most frequently used rodent model for UVinduced skin cancers has been the immunocompetent Hairless mouse, an outbred strain that lacks a functional Hairless (Hr) gene. These mice undergo one round of anagen soon after birth, after which point follicles degenerate into cystic structures.111 The lack of hair obviates the need to shave the mice repeatedly and allows for consistent UV exposure over time. Although Hr gene function is implicated in NF-κB signaling112 and adipogenesis,113 the function of Hr in cancer is not completely understood. In terms of acquired mutations in the tumors, this model is significantly more faithful to the human condition than the DMBA/TPA model.114 UV irradiation has also been used in more common strains such as C57BL/6 with varying degrees of susceptibility to carcinoma, the SENCAR strain being among the most sensitive. Although highly informative, UV-driven mouse models have suffered somewhat from a lack of uniformity in the spectra, dose, and irradiances used across groups and strains. Some of this is unavoidable and caused by technical limitations in reproducing spectra across different light sources. Nevertheless, many important conclusions have been drawn from experiments with UV-driven models. Trp53 mutations occur early, and as in chronically irradiated human skin, Trp53 mutant clones are observed.115 Tumors exhibit very high mutational burdens with a median of 155 mutations per megabase in a series of 18 SCCs.114 Other mutations observed in tumors include Notch family members, Ink4a, and relatively rare mutations in ras and recurrent chromosomal alterations are seen that map to corresponding syntenic regions on human 3p, 11p, and 9q.114,116-121
As with the chemical carcinogenesis approach, many studies have focused on specific pathways, combining UV exposure with transgenic mouse models. Bypassing UV-induced apoptosis, as regulated by Trp53, Survivin, Bcl2, and E2f-1, has been shown to be necessary for full tumor susceptibility.122-126 Some of the same regulators implicated in chemical carcinogenesis are also known to be important in UV-driven models, including the COX-2, mTOR, AKT, and ERK pathways.127-131 Cross-species analysis with human SCC has also been performed using UV-driven models implicating WNT, β-catenin, and ERK pathways, as well as several microRNAs, including miR-21 and miR-31.37
2
TREATMENT AND PREVENTION OF SKIN CANCER
The most effective approach to reducing cancer-associated morbidity and mortality is prevention, and given the central role of UV-driven mutagenesis in skin cancer development, efforts aimed at effectively limiting exposure to sunlight and other UV sources, such as tanning beds, are likely to have a major impact on overall skin tumor development. Federal legislation restricting use of tanning beds by minors in nearly all US states is thus predicted to have a major beneficial effect at reducing skin cancer incidence in the future.132
Various approaches to chemoprevention may also play an important role in reducing the number of SCCs,133,134
with encouraging results presented for nicotinamide in a randomized phase 3 trial in high-risk patients, which showed a 20% and 30% reduction of BCC and SCC, respectively, at 3 months, and 11% fewer actinic keratoses.135
Although most skin cancers can be effectively treated with surgery, a deeper understanding of the molecular basis of these cancers may provide unique opportunities for targeting key oncogenic drivers for prevention or nonsurgical treatment. For example, because nearly all BCCs appear to be caused by unrestrained Hedgehog signaling, systemic Hedgehog pathway inhibition can be effective in treating patients with locally advanced or metastatic BCC, although side effects are common, drug resistance can develop, and tumors may recur after stopping therapy.136 In addition to causing regression of preexisting tumors, systemic Hedgehog pathway inhibitors effectively block development of new BCCs in patients with Gorlin syndrome.137 These findings suggest that regular use of a topical inhibitor, if effective at blocking oncogenic Hedgehog signaling, may be useful for BCC prevention in high-risk patients. Although this is an exciting possibility for patients with BCC, it seems unlikely that a single targeted therapeutic could be similarly useful for prevention of SCC tumors because multiple oncogenic alterations are engaged to variable degrees in SCC development.
CONCLUSIONS
Extensive clinical and experimental study of skin cancers has yielded important insights into the molecular and cellular basis of both BCC and SCC; led to the development of mechanism-based, targeted therapy for BCC; uncovered key molecular similarities between SCCs arising in skin and several other organs; provided powerful platforms for studying multistep cancer development using highly tractable mouse models of chemical and UV-driven carcinogenesis, as well as genetically engineered models; and shed light on fundamental aspects of tumor genetics and cancer biology that are relevant to understanding tumorigenesis in other organs. Continued progress in understanding
323
2
the pathogenesis of BCC and SCC is likely to lead to new approaches to preventing and treating these extremely common malignancies.
ACKNOWLEDGMENTS
We regret not being able to include citations to all relevant articles, given the space restrictions. We appreciate previous authors’ contributions to some of the material included in the current chapter, including Drs. Masaoki Kawasumi, Paul Nghiem, Timothy Heffernan, Douglas Brash, Adam Glick, and Stuart Yuspa.
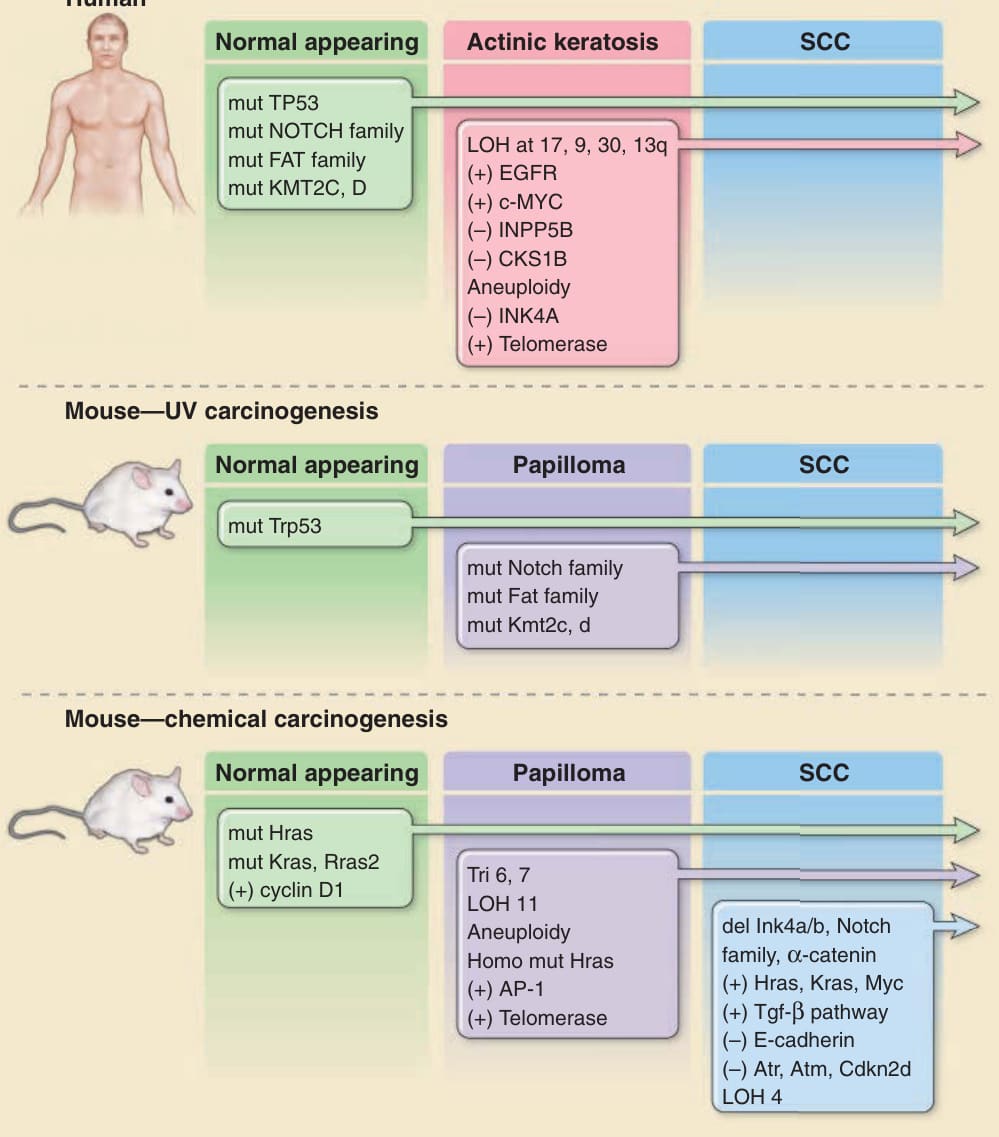
Figure 19-1 Molecular and genetic changes associated with development of cutaneous squamous cell carcinomas (SCCs) in humans and mice. The multistage evolution of cutaneous SCC in humans is depicted schematically with frequently associated genetic changes. Whereas single base mutations in early lesions frequently are characteristic of ultraviolet light-induced damage, later changes are associated with genomic instability. Increased activity of telomerase (deletion of inhibitor) or epidermal growth factor receptor (EGFR) tyrosine kinase (gene amplification) may also result from epigenetic changes. Similar changes are observed in ultraviolet (UV)-driven mouse models of cutaneous SCC. In chemically induced mouse cutaneous SCC, the multistage evolution to invasive tumors in this model is highly ordered both temporally and genetically. Operationally defined stages include initiation, promotion, and progression. Ras mutations are characteristic of chemical mutagens used to initiate tumor formation. Early upregulation of cyclin D1 and later upregulation of transforming growth factor (TGF)-β1 occur through epigenetic mechanisms and appear to be important components of carcinogenesis. Note that most events occur early in the progression sequence in UV-induced tumors (eg, TP53 mutations), but in chemical carcinogenesis, most events occur late and often do not bear the signatures of the original mutagen. It should be noted that the data are derived from many different sources and reflect a panoply of technologies used to survey these changes.
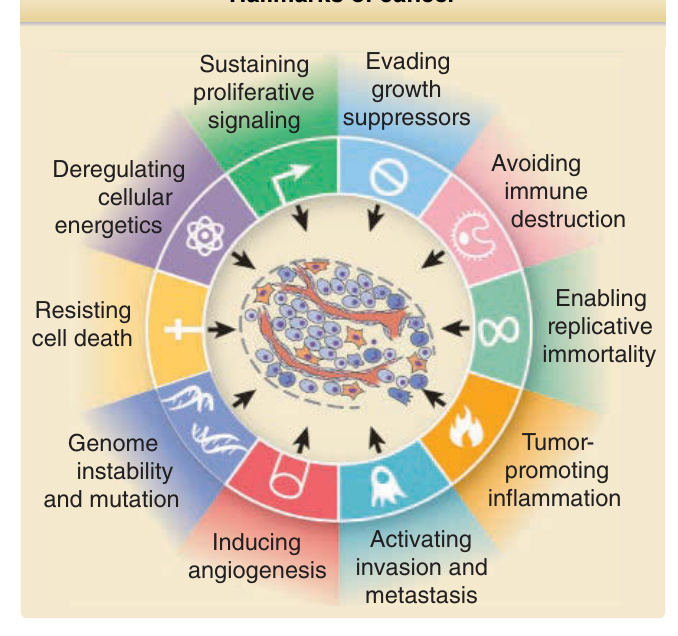
Figure 19-2 Hallmarks of cancer. Cancer cells acquire multiple properties distinguishing them from normal cells, including protection from cell death, sustained proliferation, evasion of growth inhibitory signals, the ability to invade and metastasize, immortalization, and induction of angiogenesis. More recently recognized properties important for tumorigenesis include deregulation of cellular energetics, immune evasion, genomic instability, and stimulation of inflammation. (Modified from Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.)
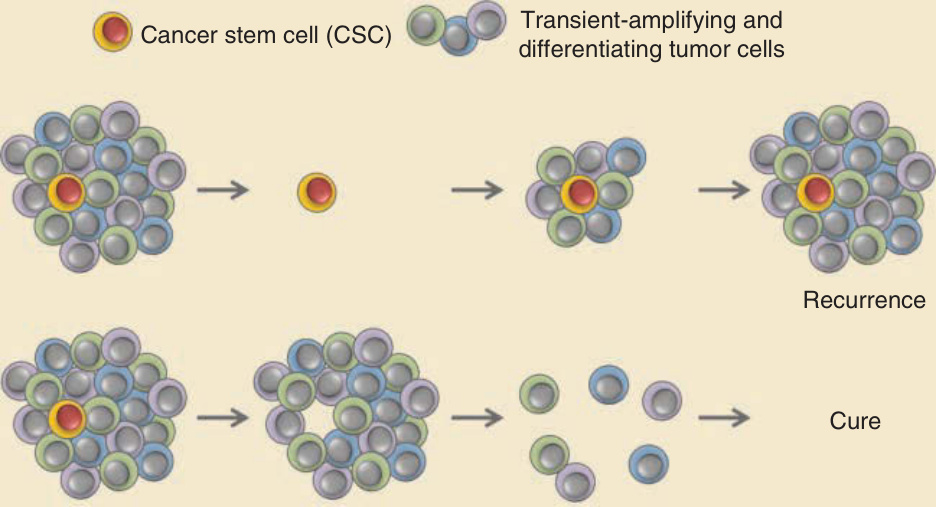
Figure 19-3 Cancer stem cells as targets for treatment. Representation of tumor mass containing a putative cancer stem cell (CSC) with its transient-amplifying and differentiating progeny that make up the bulk of the tumor. Treatment with conventional therapy may lead to rapid tumor regression, but if residual CSCs are present, tumors may recur after treatment is stopped. In contrast, CSC-targeted therapy may lead to a slower response, but after tumor cells regress, recurrence is unlikely if the CSC population has been eliminated. A hierarchical organization of CSCs and transiently amplifying progeny has been described in many, but not all, tumor types.
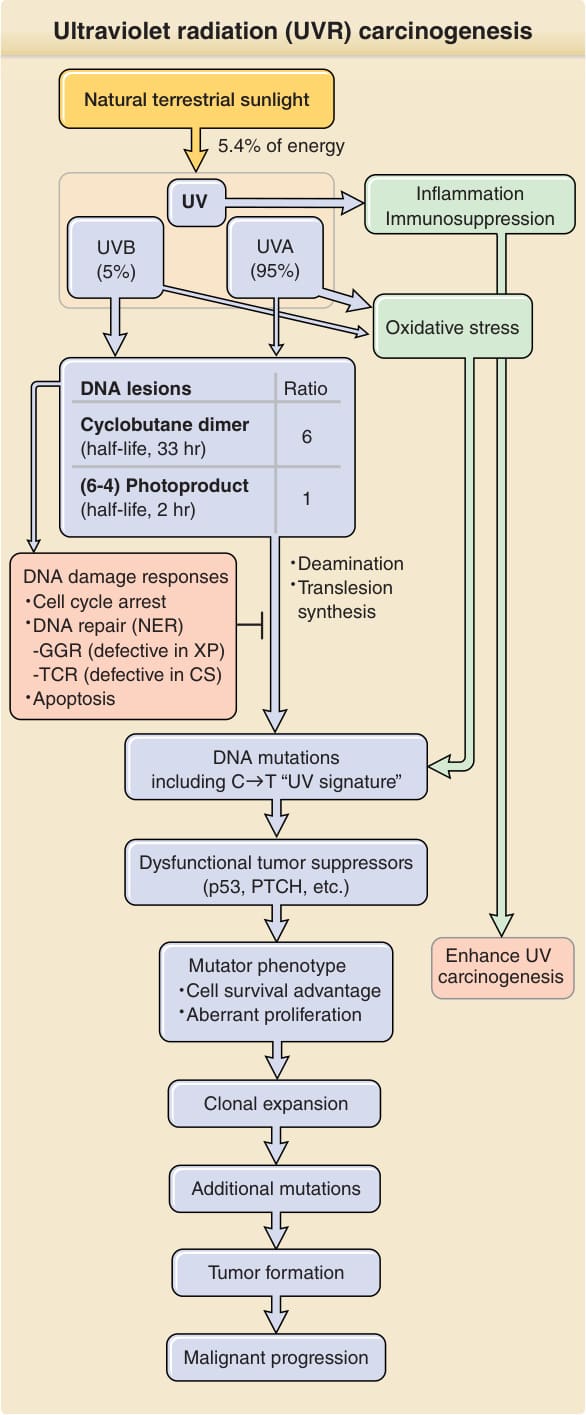
Figure 19-4 Ultraviolet radiation (UVR) carcinogenesis. UVR induces two major DNA lesions at dipyrimidine sites: cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidone photoproducts [(6–4)PPs]. Ratio given [6 CPDs per 1 (6–4)PP] is that induced by simulated sunlight. UVB accounts for only 5% of UVR (∼0.3% of all terrestrial sunlight energy) but produces the majority of UV-induced DNA lesions. Cells respond to UVR-induced DNA damage by activating DNA damage signaling pathways and inducing cell cycle arrest. Damaged DNA is repaired by nucleotide excision repair (NER). Unrepaired DNA lesions lead to genetic mutations through deamination or error-prone translesion synthesis. Unrepaired cytosine-containing CPDs contribute to UV signature mutation: C→T transition. Mutations in genes responsible for carcinogenesis confer a mutator phenotype including resistance to apoptosis. This expands the pool of clones susceptible to further damage and increases the potential for transformation. CS, Cockayne syndrome; GGR, global genome repair; PTCH, PATCHED1; TCR, transcription coupled repair; XP, xeroderma pigmentosum.
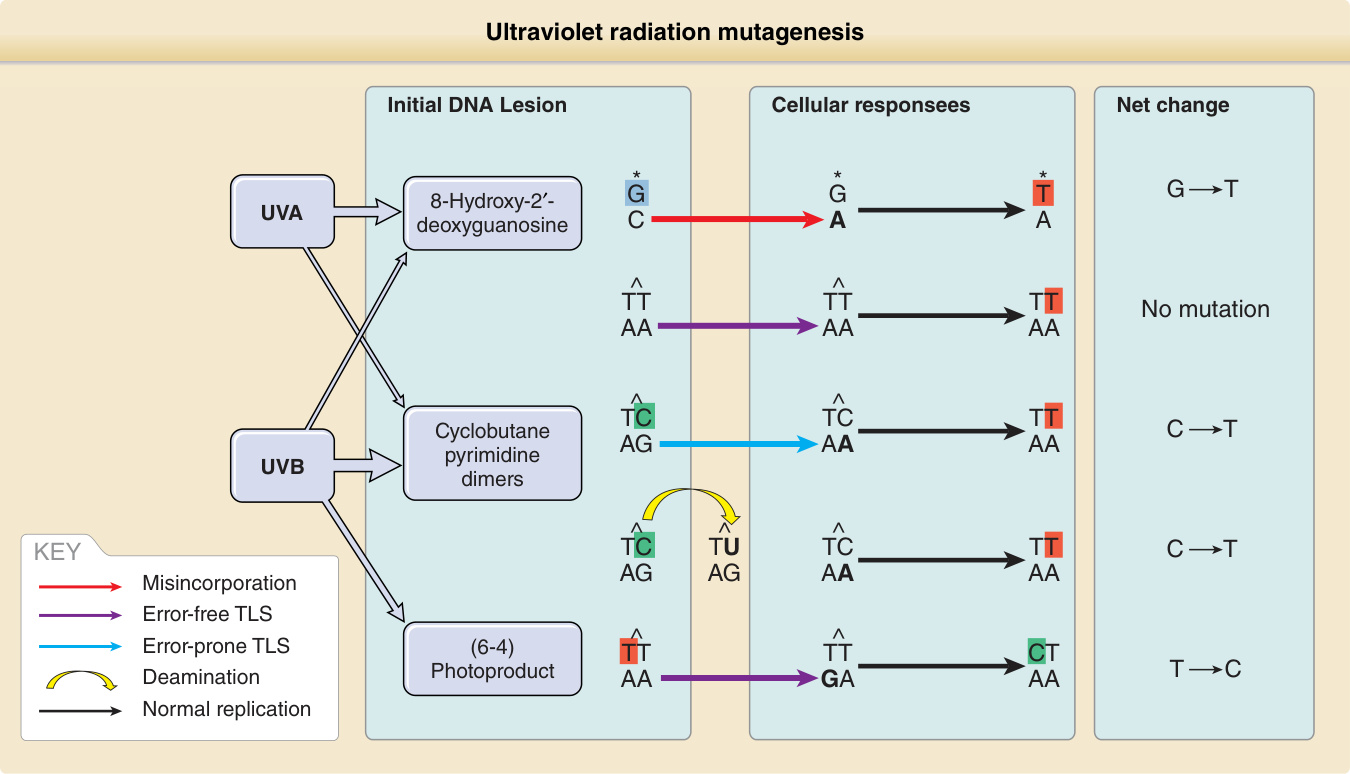
Figure 19-5 Ultraviolet radiation mutagenesis: how replication through unrepaired DNA lesions leads to DNA mutation. Cyclobutane pyrimidine dimers (CPDs) are the most relevant for ultraviolet (UV) mutagenesis and are primarily induced by UVB. Error-free translesion synthesis (TLS) adds adenines opposite CPDs, leaving thymine dimers unaffected but causing UV signature C→T transitions in cytosine dimers, which may also result from deamination. The (6–4) photoproduct induced by UVB and 8-hydroxy-2′-deoxyguanosine primarily induced by UVA are mutagenic but relatively rare.
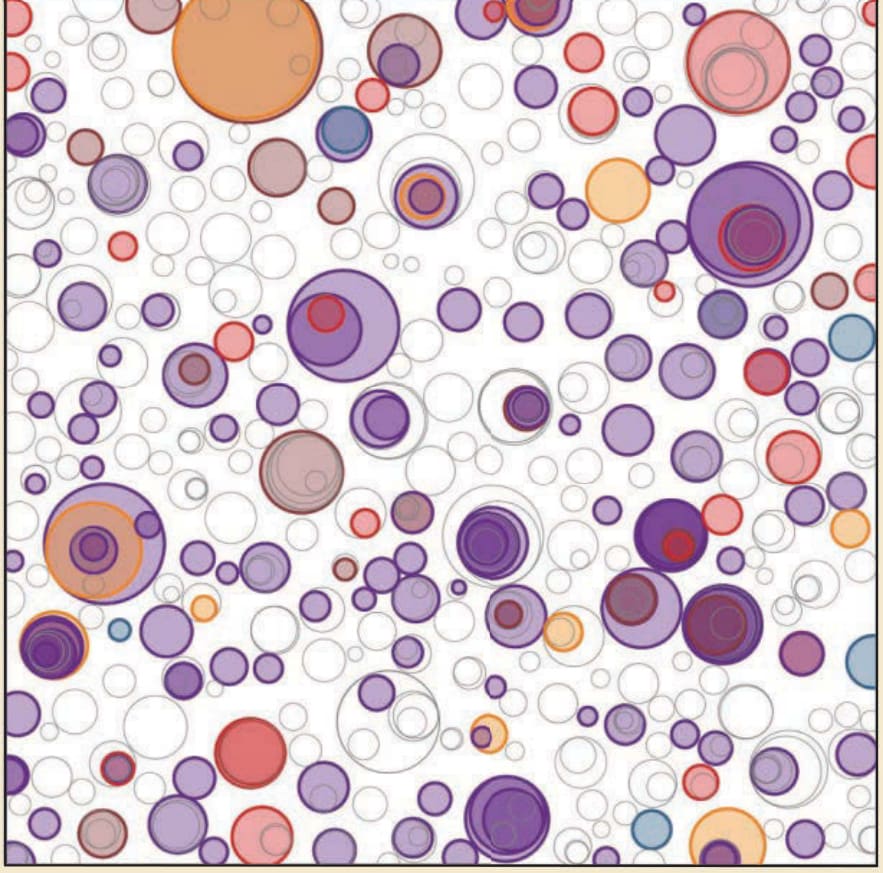
Figure 19-6 Graphical representation of mutant clone sizes in normal skin linked to specific genes implicated in SCC development. The plot represents estimated physical sizes of clones of epidermal cells based on the proportion of DNA sequences bearing a recurrent mutation and knowledge of skin biopsy sizes. Hundreds of clones bearing recurrently mutated cancer-relevant genes are found within a 1-cm2 patch of epidermis. Some mutations are inferred to represent subclones within larger clones (overlapping circles); however, bulk sequencing precludes definitive spatial mapping. (Adapted from Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880-886.69 Reprinted with permission from AAAS.)
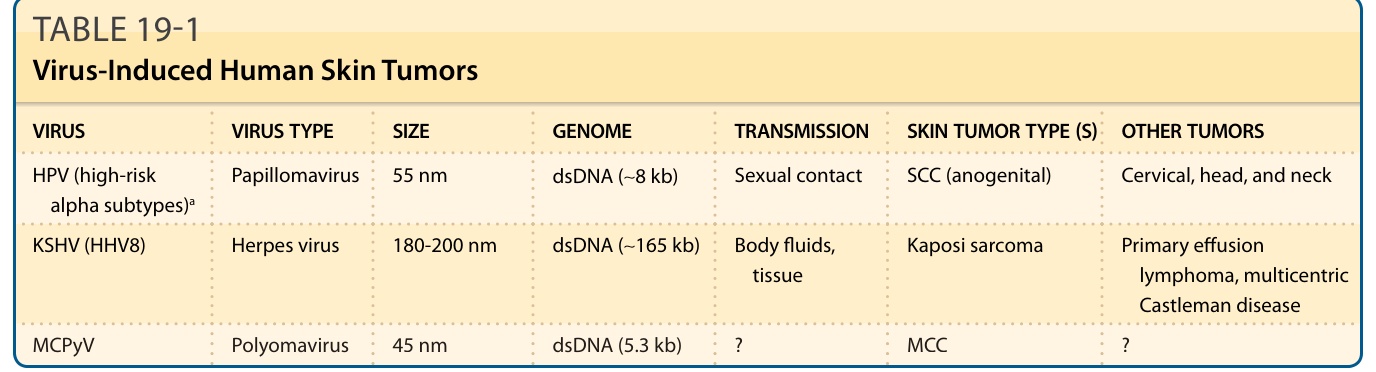
TABLE 19-1 Virus-Induced Human Skin Tumors
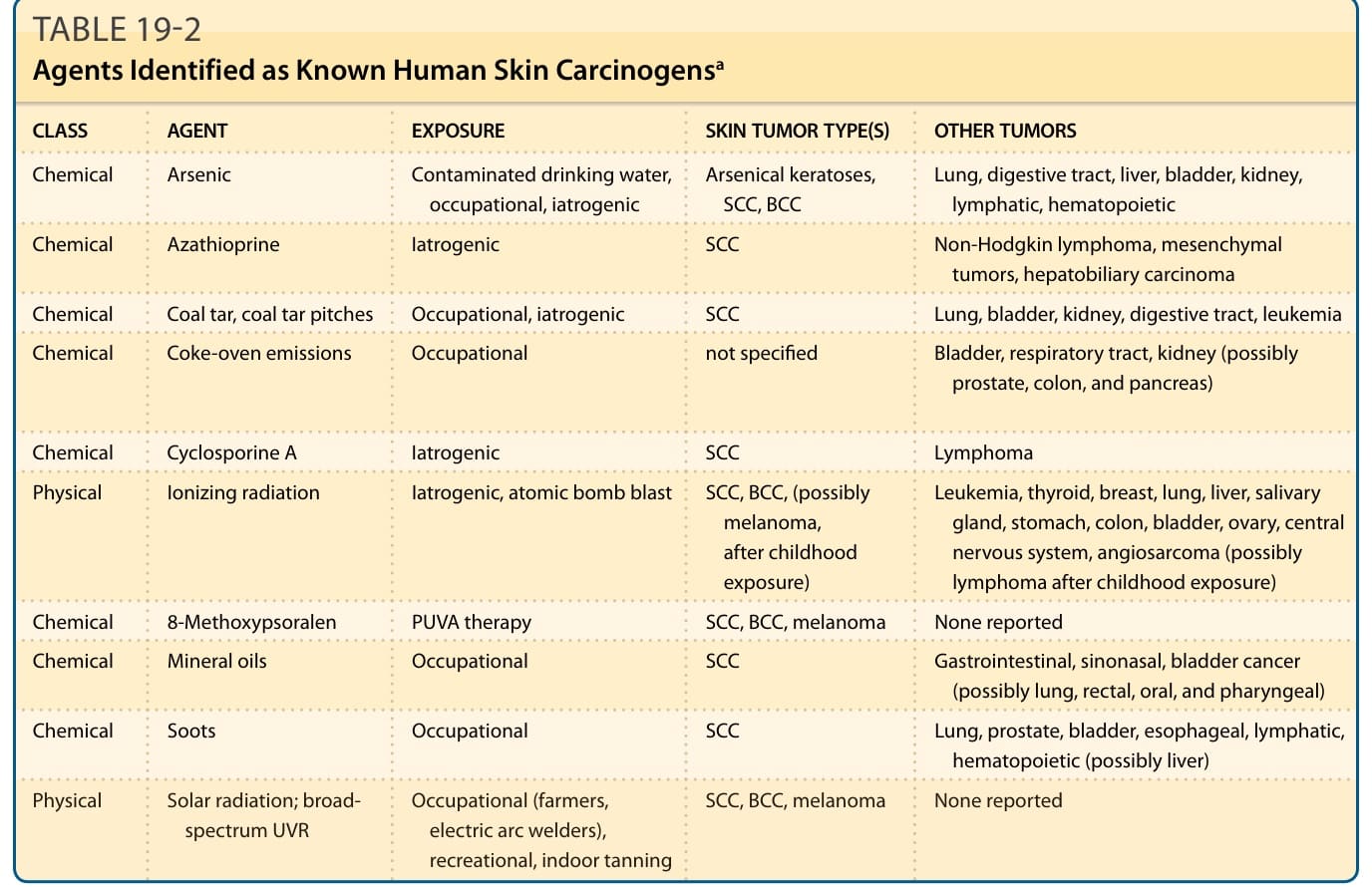
TABLE 19-2 Agents Identified as Known Human Skin Carcinogensa