皮膚光生物學 (Cutaneous Photobiology)
重點一覽
- 皮膚光生物學 (cutaneous photobiology) 研究非游離輻射(紫外線 UVR、可見光、紅外線)與皮膚的交互作用;游離輻射(X 光、伽瑪射線、α/β 粒子)能游離原子,非游離輻射僅能將電子提升至較高能量態。
- 輻射唯有先被皮膚中的發色團 (chromophore) 吸收才能引起光生物學反應(Grotthuß–Draper 定律 / 光化學第一定律)。每個發色團有特徵性吸收光譜與吸收極大值,反應具特定作用光譜 (action spectra)。
紫外線與可見光的來源與光物理
- 陽光:到達地表最短波長約 290 nm(大氣層臭氧過濾掉 UVC 100–290 nm)。波長越長能量越低、穿透越深。正午陽光中 UVA 約為 UVB 的 50 倍;UVA 可穿透窗戶玻璃,UVB 不能。
- UVR 細分(CIE):UVB 290–315 nm、UVA 315–400 nm(界線亦有用 320 nm 者)。
- 人工光源:螢光燈與鹵素燈可能發射少量 UVR;飛利浦 TL01 燈 (Phillips TL01) 為窄波段 UVB 重大進展,幾乎單色發射於 311–312 nm,兼具療效又避開主要造成曬傷的短波長。
- 皮膚發色團:DNA(吸收極大值 260 nm)、紫質 (porphyrins)(400–410 nm)、黑色素 (melanin)。穿透深度:UVA > UVB > UVC(UVC 僅達角質層與表皮上層)。
- 光物理/光化學:發色團吸收光子後電子躍升,不改變自旋為單重態 (singlet)、改變自旋為三重態 (triplet)。激發後可形成光產物 (photoproduct)(如前維生素 D3、環丁烷嘧啶二聚體),或將能量轉移給另一分子(光敏化反應 photosensitized reaction,如紫質激發氧產生單重態氧 singlet oxygen),或以熱、螢光 (fluorescence)、磷光 (phosphorescence) 釋放能量。螢光波長恆比激發波長長(Stokes 定律)。
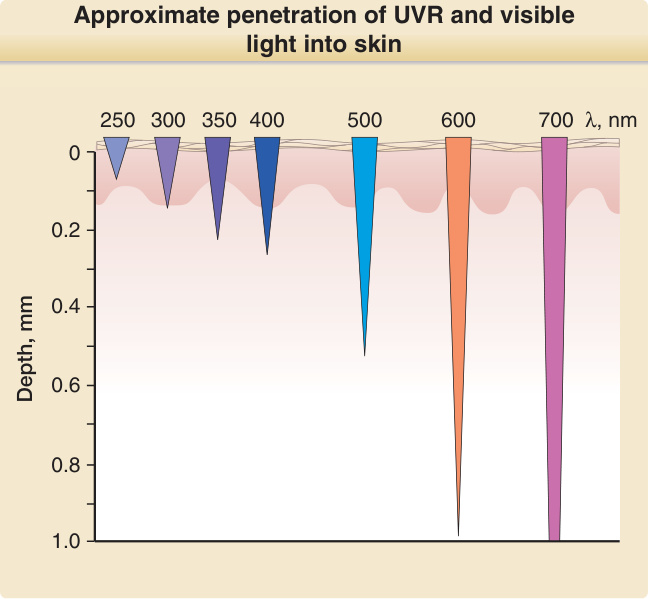
圖 17-3:紫外線輻射與可見光進入皮膚的概略穿透深度。
維生素 D 光生物學
- 維生素 D 調節鈣磷代謝,促進腸道鈣磷吸收與腎臟鈣再吸收,使骨骼正常礦化。缺乏導致兒童佝僂病 (rickets)、成人骨軟化症 (osteomalacia),並可能促成骨質疏鬆。高危族群:老年人、高緯度長冬居民、肥胖者、高緯度深膚色者。
- 毒性:過量表現為高鈣尿症 (hypercalciuria) 或高鈣血症 (hypercalcemia)(肌肉無力、意識混亂、噁心、骨痛,可致腎結石/腎衰竭)。成人安全上限 10,000 IU/天,慢性中毒劑量為每日超過 50,000 IU/day。
- 生化途徑:皮膚中前維生素 D3(7-去氫膽固醇 7-dehydrocholesterol)經 UVB 轉為前維生素 D3,自發異構化為維生素 D3 → 肝臟 25-羥化(需 NADPH、O₂、Mg²⁺)成 25-羥基維生素 D3(calcidiol)→ 腎近端小管經 25(OH)D-1α-羥化酶轉為 1,25-羥基維生素 D3(calcitriol,活性形式)。皮膚光合成最有效波長在 UVB 範圍、峰值 297 nm。
- 深膚色者需更長日照才能製造等量維生素 D;熱帶地區每週兩次、每次 10–15 分鐘日照臉手臂手部或背部即足夠。持續光防護可能導致缺乏,宜結合維生素 D 補充。
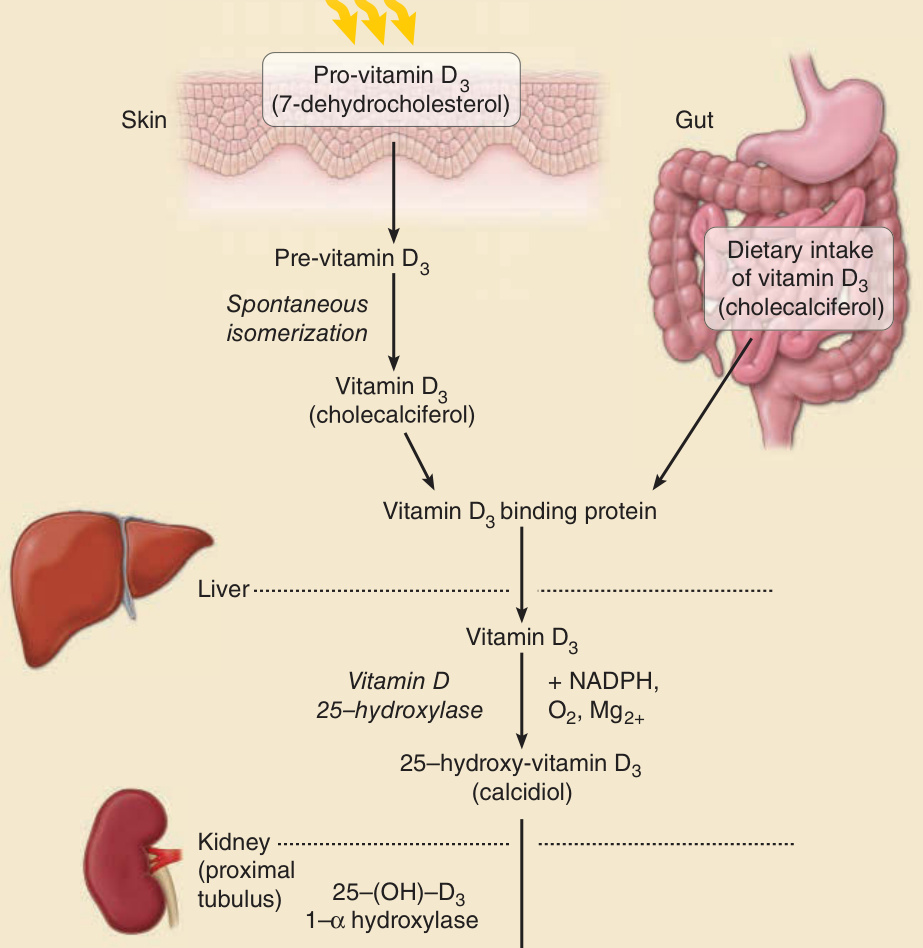
圖 17-6:活性維生素 D 的合成(發色團為 7-去氫膽固醇,吸收極大值 297 nm)。
UVR 對免疫系統的效應
- UVR 兼具促發炎與抗發炎特性。促發炎由先天免疫介導(血清素、前列腺素、IL-1、IL-6、IL-8、TNF-α,抗菌肽誘導,嗜中性球與淋巴細胞招募)。免疫抑制由蘭格罕細胞 (Langerhans cells) 遷移至淋巴結、抗發炎介質(IL-10、α-MSH)釋放、抗原特異性調節性 T 細胞 (regulatory T cells) 誘導介導。
- 二分法觀點過於簡化:多數乾癬 (psoriasis) 病人 UV 後改善(抗發炎),少數光加重型乾癬惡化(促發炎)。多形性日光疹 (polymorphous light eruption) 病人 UV 誘導免疫抑制較少,但對光致癌有較佳保護。
- 紅斑性狼瘡 (lupus erythematosus) 的光加重與光觸發可視為未能適當下調免疫反應。
- UVR 對皮膚癌是「三刃劍」:造成致突變 DNA 損傷、抑制腫瘤抗原辨識、誘導對腫瘤抗原的耐受。免疫效應發色團:DNA、RNA、尿刊酸 (urocanic acid)(反式→順式異構化)、紫質。多數免疫抑制由 UVB 造成,UVA 亦有類似效應。
曬傷 (Sunburning)
- 臨床:紅斑與疼痛灼熱(第一級)、可進展至水疱(第二級),曬太陽後 6 至 24 小時達高峰,隨後曬黑與脫屑。組織學:表皮水腫伴海綿狀水腫、曬傷細胞 (sunburn cells)、蘭格罕細胞耗竭、血管擴張、混合性發炎浸潤。
- 紅斑作用光譜在 300 nm 達高峰(UVB 範圍),向 UVA 呈指數下降。發色團為 DNA(紅斑作用光譜與嘧啶二聚體形成的作用光譜相似;著色性乾皮症 xeroderma pigmentosum 病人因 DNA 修復缺陷而有急性光敏感與降低的最小紅斑劑量 minimal erythema dose)。
曬黑 (Tanning)
- UVB 暴露後約 3 天達高峰,組織學見表皮基底與基底上層黑色素增加。發色團為 DNA;黑色素合成增加依賴 UV 誘導之 p53 誘導與活化。
- 皮膚光型 (skin phototypes) 由 Fitzpatrick 於 1975 年發展(依基礎色素、曬傷敏感性、曬黑能力分型),是 UVR 長期效應(特別是光致癌)的風險指標。
- 保護機轉:黑色素被轉移至角質細胞核上方形成「微型陽傘 (microparasol)」;曬傷後表皮過度增生使表皮與角質層增厚,減少 UVR 穿透至基底層;連同 DNA 修復酵素活性增加,造成紅斑閾值數倍增加。
- 立即性色素變深 (immediate pigment darkening)(20 分鐘內出現)由既存黑色素氧化重分布介導、主要由 UVA 誘導,幾乎不提供後續保護。
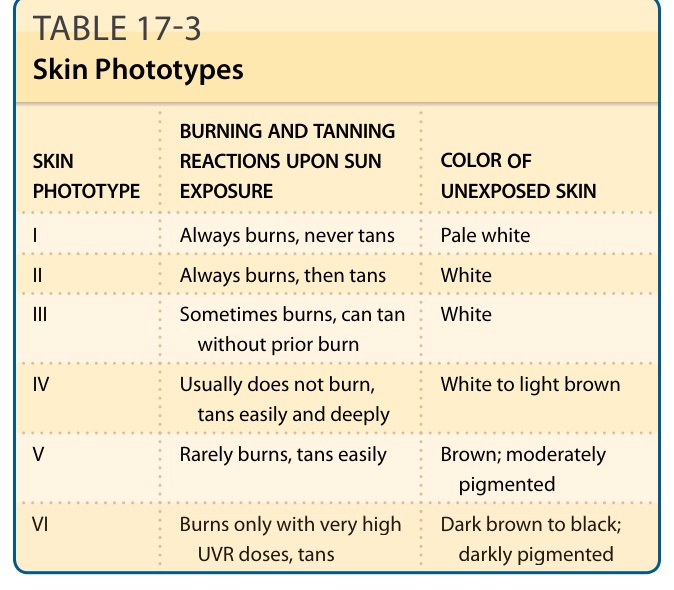
表 17-3:皮膚光型 (Skin Phototypes)。
光皮膚病的光生物學原理
- 血基質生合成途徑缺陷導致紫質症 (porphyrias):早期缺陷累積非芳香族中間產物(不作發色團,影響肝臟等其他器官,如急性間歇性紫質症);晚期缺陷累積芳香族中間產物(作發色團,UVR/可見光激發引起光毒性反應,如遲發性皮膚紫質症 porphyria cutanea tarda、紅血球生成性原紫質症 erythropoietic protoporphyria)。中間產物吸收極大值 400–410 nm(UVA 與可見藍光交界),故病人需同時防護 UVA 與可見光。
- 藥物誘發之光毒性/光過敏性皮膚炎:發色團為藥物或代謝物(芳香族、吸收於 UVA),故幾乎完全由 UVA 誘發。光植物性皮膚炎由呋喃香豆素 (furocoumarins) 介導;佛手柑皮膚炎發色團為佛手柑油(5-甲氧基補骨脂素)。多形性日光疹與日光性蕁麻疹 (solar urticaria) 誘發波長因人而異。
- 診斷工具:分別測 UVA-MED 與 UVB-MED、光貼布試驗 (photopatch testing)、尋找潛在發色團(血/尿/糞紫質)。
光照治療的光生物學原理
- 乾癬是最常見適應症。抗乾癬發色團為 DNA(治療作用光譜在約 300 nm 最大、與紅斑及嘧啶二聚體作用光譜重疊)。療效機轉現認為由免疫抑制介導(過度增生活化 T 細胞易受 UVB 促凋亡效應),而非早期認為的阻斷複製。
- 窄波段 UVB (nbUVB):燈泡發射 311–312 nm 幾乎單色 UVB,遠優於寬波段 UVB (bbUVB),因避開無療效的促紅斑短波長(280–290 nm)可用更高劑量。nbUVB 已取代 bbUVB 用於乾癬、異位性皮膚炎、白斑、皮膚 T 細胞淋巴瘤等。
- PUVA(光化學治療):補骨脂素 (psoralen,最常用 8-甲氧基補骨脂素 8-methoxy-psoralen / 8-MOP) 嵌入 DNA,UVA 照射後與嘧啶鹼基形成股間/股內交聯(只能由 DNA 重組修復,無法由核苷酸切除修復修復)。8-MOP 吸收極大值 248 與 301 nm,嵌入 DNA 後移至 313 nm、單加成物後移至 334 nm(UVA 範圍)。
- 高劑量 UVA1:不用光敏化劑,對急性異位性皮膚炎與硬化性病況(如硬斑病 morphea)有療效。
- 光動力療法 (PDT):美國常用 5-胺基乙醯丙酸 (5-ALA),規避血基質合成限速步驟使細胞內原紫質 IX (protoporphyrin IX) 增加;原紫質 IX 吸收極大值約 406 nm(Soret 帶),被藍光激發後與氧形成單重態氧。歐洲常用甲基胺基乙醯丙酸 (MAL) 合併紅光(親脂性較佳、穿透較深,適合較厚病灶)。
光致癌作用 (Photocarcinogenesis)
- 慢性 UV 暴露增加基底細胞癌、鱗狀細胞癌、惡性黑色素瘤與梅克爾細胞癌風險。
- DNA 損傷與突變:UVB 強烈激發 DNA(DNA 吸收極大值 260 nm)。直接光激發在相鄰嘧啶間形成共價鍵,主要光產物為環丁烷嘧啶二聚體 (CPDs) 與 6-4 嘧啶–嘧啶酮光產物 (6-4 PPs)。未修復則於 S 期複製時錯配,最常見為雙嘧啶位點的 C → T 鹼基置換(胞嘧啶對面錯誤嵌入腺嘌呤)。
- 紫外線特徵性突變 (UV signature mutations):雙嘧啶位點的 C → T 與 C:C → T:T 串聯突變,是 UVR 暴露的特徵性分子印記(非 UV 組織罕見)。「UVB 特徵性突變」一詞具誤導性(UVA 亦可形成),應避免。見於鱗狀細胞癌/光化性角化症的 p53、基底細胞癌的音蝟途徑基因 (ptch, shh, smo) 與 p53、黑色素瘤的 CDKN2A、PTEN、TERT 啟動子與 p53。
- 黑色素依賴性化學激發途徑 (melanin-dependent chemiexcitation pathway):Premi 等人描述 UV 暴露後長達 3 小時仍形成「暗 CPDs (dark CPDs)」,由黑色素(特別褐黑色素 pheomelanin)光激發、經過氧亞硝酸鹽 (peroxynitrite) 介導。黑色素因此不僅是光防護者,也可能促成癌症。
- 氧化性 DNA 損傷(氧化途徑):主要由 UVA 經單重態氧介導,最常見 8-oxoG(與腺嘌呤錯配產生 G → T 顛換)。然證據顯示氧化性損傷在致突變中僅次要角色;嘧啶二聚體對 UVB 與 UVA 都是最重要的前致突變損傷。UVA 形成嘧啶二聚體遠少於 UVB(約少 10,000 倍),但 UVA 不像 UVB 引發穩健的細胞週期停滯,純 UVA 暴露(窗玻璃後、曬黑裝置)可能特別致突變。
- 黑色素瘤突變負荷:黑色素瘤超過 70%(一系列中位數 77.7%)為 C → T 紫外線特徵性突變。慢性陽光損傷皮膚 (CSD) 黑色素瘤突變負荷約 60 突變/Mbase(常源自惡性小痣 lentigo maligna),非 CSD 約 30 突變/Mbase(好發女性小腿、男性背部,與間歇性高劑量曬傷相關)。
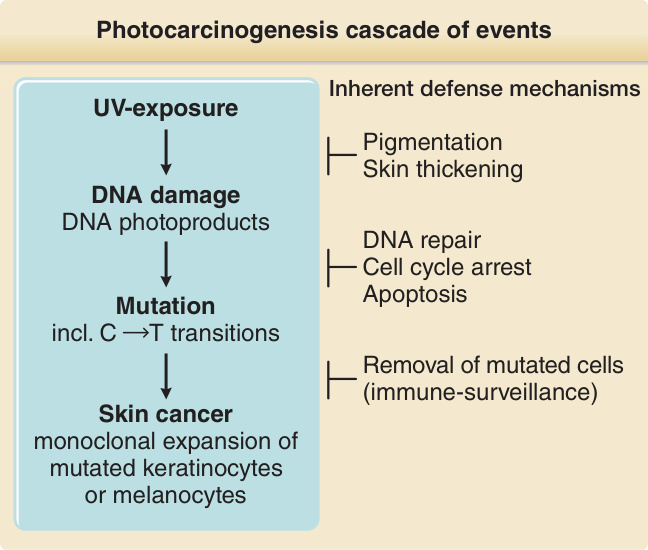
圖 17-9:光致癌作用的事件級聯。
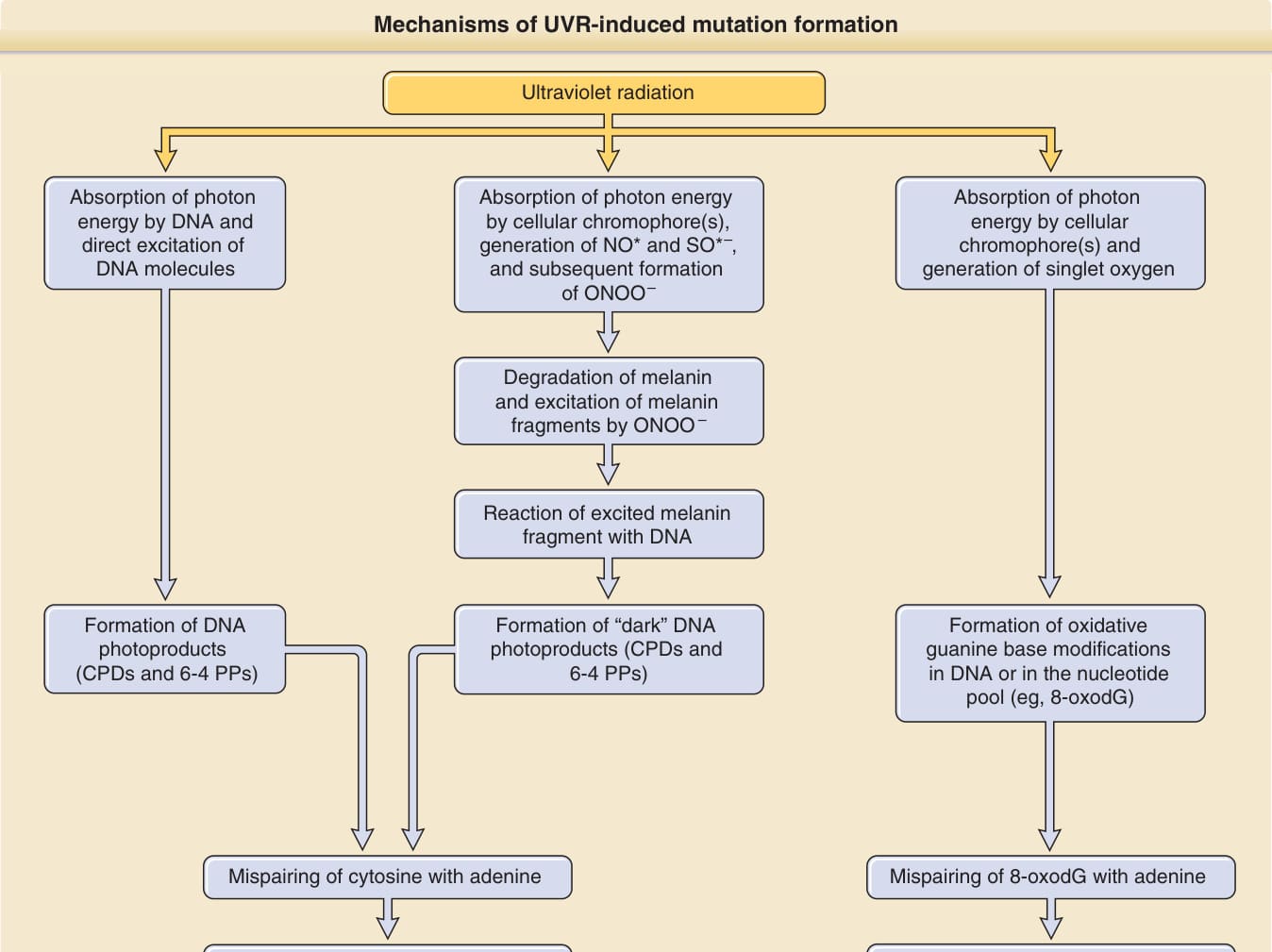
圖 17-11:UVR 誘導突變形成的三種機轉(古典途徑、黑色素依賴性化學激發途徑、氧化途徑)。
DNA 修復與其他防禦反應
- 防禦機轉(非僅 DNA 修復):DNA 修復、損傷誘導細胞週期停滯(給修復更多時間)、凋亡 (apoptosis)(移除嚴重受損細胞)、增加黑色素生成與皮膚增厚、免疫監視移除突變細胞。
- 核苷酸切除修復 (NER):修復 CPDs 與其他光產物。缺陷致著色性乾皮症 (XP)、Cockayne 症候群、毛髮硫營養不良症。XP 有 7 個互補群(XP-A 至 XP-G)。損傷辨識分全基因組 NER(XPE、XPC 結合)與轉錄偶聯 NER(停滯 RNA 聚合酶與 CSA/CSB)。XP 的 NER 缺陷賦予極高皮膚癌風險。
- 氧化性鹼基修飾由鹼基切除修復處理。p53(基因組守護者)協調細胞週期停滯、凋亡與 DNA 修復;上游為 ATM 與 ATR。重要檢查點效應子:p21(G1/S)。
- 跨損傷 DNA 合成由特化聚合酶(如 DNA 聚合酶 eta,在 XP 變異型中突變)執行,可能引入突變;失敗時用重組修復(范可尼貧血/BRCA 途徑)。
- 慢性陽光暴露皮膚帶有 p53 突變角質細胞的克隆性增生;p53 功能喪失造成「UV 突變子表型 (UV mutator phenotype)」,對鱗狀細胞癌發展至關重要。
哪些波長造成皮膚癌?
- UVB 致癌性久為人知;UVA 現已知在亞紅斑劑量下亦具致癌性,WHO 已將 UVA 歸為第一類致癌物 (class I carcinogen)。烏特勒支-費城作用光譜顯示致癌效力隨波長從 UVB 至 UVA 呈指數下降,但被陽光中 UVA 較高豐富度部分抵消。
- 黑色素瘤動物模型:DeFabo 等人發現 UVA 僅在有色素小鼠中誘導黑色素瘤(黑色素為 UVA 致黑色素瘤所必需),UVB 則不需。黑色素依賴性化學激發途徑為 UVA 致黑色素瘤的假定替代機轉。
- 曬黑床/日曬燈使用顯著增加黑色素瘤風險(瑞典與挪威 106,379 名女性世代研究相對風險 1.42 至 2.58)。
- 多數 UVA 誘導突變為 C → T 轉換,無獨立 UVA 特徵性突變訊號。綜合證據:UVB 與 UVA 都能誘導黑色素瘤(可能不同機轉),但相對貢獻仍不明確;無證據支持氧化性 DNA 鹼基損傷扮演主要角色。
光老化 (Photoaging)
- 光老化影響皮膚所有層次(表皮、色素系統、真皮結締組織、血管系統、皮下脂肪),常疊加於內源性老化 (intrinsic aging) 之上,特徵為克隆性增生事件與功能喪失事件並存。
- 增生性事件(雀斑等克隆性黑素細胞增生)屬腫瘤性事件、僅見於光損傷皮膚。功能喪失事件(表皮變薄、色素減退、真皮膠原喪失、微血管擴張 telangiectasias、皮下脂肪喪失)亦見於內源性老化,機轉很可能為加速增生耗竭。
- 光化性彈性纖維變性 (actinic elastosis):唯一不見於內源性老化的非增生性特徵。纖維狀嗜鹼性物質累積於真皮上中層,臨床呈黃色增厚、彈性喪失、深皺紋。主由彈性蛋白 (elastin) 與纖維蛋白 (fibrillin) 組成。彈性蛋白合成 UV 後上調;老化纖維母細胞無法因應 UVA 上調組織蛋白酶 K (cathepsin K) 以清除彈性蛋白片段,導致累積與交聯成不可降解巨分子,故大致不可逆。
- 早老素 (Progerin):核纖層蛋白 A (Lamin A) 的異常剪接變異體,由 UVA 經氧化壓力誘發,因失去法尼基化位點而不可降解,導致細胞衰老。
- UVA 為真皮細胞外基質變化的特殊禍首(穿透較深、誘導氧化壓力與衰老);UVB 雖不達真皮深層,亦可經旁分泌機轉(角質細胞訊號傳遞至真皮纖維母細胞)誘導基質金屬蛋白酶。
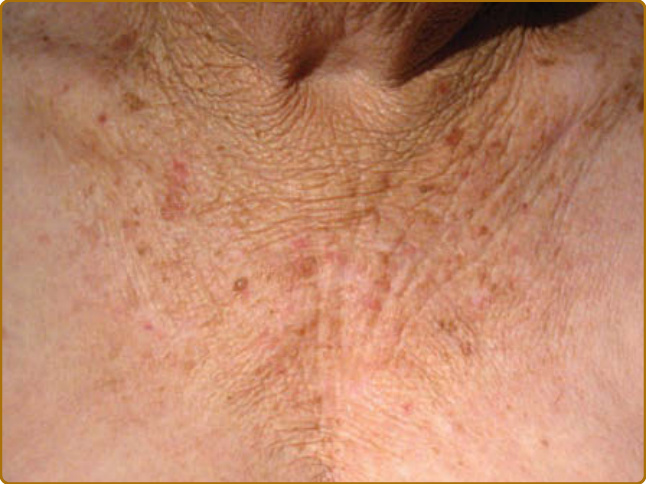
圖 17-15:慢性光老化皮膚中由光化性彈性纖維變性所致的深皺紋。
可見光與紅外線輻射的反應
- 可見光可誘導紅斑與色素沉著(立即與延遲性,特別深膚色者),並可誘發部分光皮膚病(日光性蕁麻疹、慢性光化性皮膚炎、皮膚紫質症)。潛在發色團:紫質、黑色素、β-胡蘿蔔素、核黃素、膽紅素、血紅素。已促成 IPL、雷射、PDT 等可見光裝置發展。
- 紅外線 (IR) 牽涉光老化與火激紅斑 (erythema ab igne);IRA 深入真皮、誘導基質金屬蛋白酶,可能促成膠原纖維喪失。IR 單獨似不致皮膚癌,但可能透過預防凋亡影響生長行為。
結論
- UVR 唯一正面效應(維生素 D 生成)被眾多有害效應抵消,光防護為預防所必需,且其對維生素 D 的負面效應可由口服補充解決。光防護策略須同時對抗 UVB 與 UVA。
- FDA 2011 年最終規則以臨界波長標準 (critical wavelength criterion) 定義廣譜 (broad-spectrum) 防曬(至少 370 nm 達 290–400 nm 吸光度曲線下面積 90%)。對 IR/可見光的光防護才剛起步。
- 抗氧化劑對皮膚癌的保護效應未確立,甚至可能因抑制 ROS 介導之衰老(如早老素形成)而促進光致癌作用。
- 尋求陽光/曬黑與愉悅的中樞神經效應有關(類似尼古丁成癮),增加公共衛生介入的難度。