Cutaneous Photobiology
2
AT-A-GLANCE
■ Radiation can only cause a photobiologic response, if it is first absorbed by a molecule (chromophore) in the skin.
■ Understanding the photophysical, photochemical, and photobiologic effects of radiation exposure is helpful to understand the wavelengthdependent consequences, including burning, tanning, photodermatoses, formation of skin cancer, photoaging, and effects of phototherapy.
■ Photoreactions have specific action spectra that depend on the absorption characteristics of various different intrinsic and extrinsic chromophores.
■ Cutaneous vitamin D production is mediated by wavelengths within the UVB range. Optimal vitamin D blood levels are essential for good bone health and are increasingly associated with a myriad of other potential health benefits.
■ Formation of DNA damage by ultraviolet radiation (UVR) mediates sunburning, tanning, and skin cancer formation.
■ Skin cells are equipped with a number of damage response pathways that limit the negative impact of radiation exposure, including several different DNA repair mechanisms.
■ Ultraviolet radiation–induced DNA damage can result in mutation formation. C to T and CC to TT mutations at dipyrimidine sites are highly characteristic for an induction by ultraviolet radiation and are called UV-signature mutations. Such UV-signature mutations are found in UV-induced skin cancers.
■ Ultraviolet radiation has both pro- and antiin- flammatory properties.
■ Photoaging affects all compartments of the skin and is characterized by both clonal proliferative events and loss-of-function events.
■ Photoaging is largely irreversible, possibly due in part to an UV-induced accumulation of undegradable abnormal proteins in the extra- and intracellular space.
■ Exposure to visible light and infrared radiation also has photobiological consequences, including erythema, tanning, and degradation of extracellular matrix proteins.
■ Rational phototherapy and photoprotection are based on these insights.
INTRODUCTION
As the outermost layer of the human body, skin is heavily exposed to damaging environmental agents, including different types of radiation (Table 17-1 and Fig. 17-1). Ionizing electromagnetic radiation, like, for example, x-rays or gamma rays, carries sufficient photon-energy to completely remove an electron from an atom or molecule (= ionization). Other types of ionizing radiation are alpha particles (2 protons and 2 neutrons) and beta particles (electrons). Alpha and beta particles are not part of the electromagnetic spectrum; they are energetic particles, as opposed to pure energy bundles (photons) of the electromagnetic radiation. Nonionizing radiation, which includes ultraviolet radiation (UVR), visible light, and infrared radiation, is able to move an electron to a higher energy state, but, in contrast to ionizing radiation, cannot remove an electron from atoms or molecules. Cutaneous photobiology is the science that studies the interaction of nonionizing radiation with skin. The term light is commonly reserved for wavelengths of the electromagnetic spectrum that are perceivable by the human eye: visible light. Both ionizing and nonionizing radiation have detrimental effects on skin. Because during the development of life on earth, electromagnetic radiation has always been present, cells and organisms could only evolve with development of mechanisms that protect against the damaging effects of radiation. This chapter covers the effects of UVR, visible light, and infrared radiation on skin. Insights into the photophysical, photochemical, and photobiological processes help to understand clinical manifestations of skin exposure to these types of radiation, including sunburning, tanning, skin cancer formation, photoaging, photodermatoses, and phototherapeutic effects on skin diseases.
SOURCES OF ULTRAVIOLET AND VISIBLE RADIATION
SUNLIGHT
SUNLIGHT
The shortest wavelength of solar electromagnetic radiation reaching the earth’s surface is approximately 290 nm, although slightly shorter wavelengths are detected at higher altitudes. The filtering of UVC (100 to 290 nm) by the earth’s atmosphere, in particular by ozone, is widely regarded a critical element in the development of life on earth, because the shorter wavelengths of UVR are highly damaging
2
NAME WAVELENGTH RANGE SUBDIVISION WAVELENGTH RANGE
Ionizing Radiation
Gamma-ray 10 fm-1 pm
X-ray 1 pm-10 nm
Vacuum ultraviolet 10 nm-200 nm
Nonionizing Radiation
Ultraviolet C 100 nm-290 nm
Ultraviolet B 290 nm-315 nm
Ultraviolet A 315 nm-400 nm UVA2 315 nm-340 nm
UVA1 340 nm-400 nm
Visible light 400 nm-760 nm Violet 400 nm-450 nm
Blue 450 nm-500 nm
Green 500 nm-570 nm
Yellow 570 nm-590 nm
Orange 590 nm-610 nm
Red 610 nm-760 nm
Infrared 760 nm-1 mm IR-A 760 nm-1.4 mm
IR-B 1.4 mm-3 mm
IR-C 3 mm-1 mm
Microwaves and radio waves >1 mm
Microwaves and radio waves >1 mm
to animals and plants. The subdivisions of UVR (Table 17-1 and Fig. 17-1) and of the whole spectrum of electromagnetic radiation are rather arbitrary, and it is important to recognize that photophysical properties of photons do not change abruptly between the defined ranges, but rather gradually with changing wavelength. In general, energy decreases with increasing wavelength. For example, a 300-nm UVB photon has twice the energy of a 600-nm orange photon. The most commonly used subdivision of
UVR is the one defined by the Commission Internationale de l’Eclairage, CIE, and sets 315 nm as the border between UVB and UVA. Other classifications use 320 nm. The defined threshold between ultraviolet and visible radiation is based on the properties of the human eye and its ability to sense radiation. Some species, for example, birds, insects, and fish, can also see some UVA radiation. The composition of solar radiation reaching the earth’s surface varies with the time of the day because
Spectrum of electromagnetic radiation
Gamma rays X-rays Ultraviolet
Energy
Visible Infrared Radio waves
Vacuum UV UVC UVB UVA
Visible
10 nm 100/200 nm 290 nm 760 nm
UVAII UVAI
315 nm 340 nm 400 nm
Wavelength, nanometers
266
of changing angles of sunlight and with differences in the length solar radiation has to travel through the atmosphere. Shorter wavelengths of visible light (violet and blue) are more filtered by a longer passage through the atmosphere than longer wavelength of visible light (orange and red), resulting in redder light at sunrise and sunset, as compared to midday sunlight. Likewise, the longer wavelengths of UVR (UVA, 315 to 400 nm) penetrate matter better than UVB (280 to 315 nm). Therefore, the fraction of UVA relative to UVB increases with lower angles of solar radiation. At midday, natural sunlight contains about 50 times more UVA than UVB. Although both components are less abundant early or late in the day, the ratio between UVA and UVB increases with declining solar angles. Similarly, UVA can penetrate through window glass, but not UVB.
ARTIFICIAL SOURCES
ARTIFICIAL SOURCES
There is a huge variety of different light sources that expose skin to visible, ultraviolet, and infrared radiation. These range from lamps used for conventional lighting purposes, for industrial applications, for phototherapy, and for tanning. Most of the light sources are designed to emit only the desired range of wavelength, for example, only visible light for lighting purposes. Nevertheless, some general lighting lamps sometimes emit small amounts of UVR, in particular fluorescent bulbs and halogen lamps. Unwanted wavelengths are often eliminated by filters. Fluorescent bulbs designed to emit UVR most often emit a broad range of wavelengths. For example, bulbs designed to emit UVA for photochemotherapy (PUVA) also emit some UVB, and those designed to emit UVB also emit some UVC. The Phillips TL01 fluorescent lamp has been a major advance in the design of UVB lamps for phototherapy, as it aligns its almost monochromatic emission at 311 to 312 nm, with therapeutic efficacy for inflammatory skin diseases, while avoiding shorter wavelengths that mostly cause sunburning, but are not effective for treatment.
PHOTOPHYSICS, PHOTOCHEMISTRY, AND PHOTOBIOLOGY: PRINCIPLES OF THE INTERACTION OF ELECTROMAGNETIC RADIATION WITH SKIN
For a photon to have biologically relevant consequences, a series of events must take place (Fig. 17-2). Optical factors of the skin and the wavelengths of the radiation determine how much and where a photon is reflected and scattered at the skin surface and within the skin. For ultraviolet and visible radiation, longer wavelengths
2
Cascade of photophysical, photochemical, and photobiological events that are involved when a photon exerts effects on exposed skin
UV and visible radiation
Tissue optics
Absorption by chromophores
Excited states
Photoproducts
Biochemical and cellular changes
Acute and chronic skin responses
penetrate matter better than shorter wavelengths. For example, UVA penetrates deeper into the dermis than UVB, of which only a small fraction penetrates deeper than the epidermis. UVC has the smallest penetration depth and in skin only reaches the stratum corneum and the upper layers of the epidermis (Fig. 17-3). The first law of photochemistry, also known as the Grotthuß–Draper law, named after the chemists Freiherr Christian Johann Dietrich Theodor von Grotthuß and John William Draper, states that that light must first be absorbed by a chemical substance for a photochemical reaction to take place. A compound that absorbs radiation is called a chromophore. Each chromophore has a characteristic absorption spectrum, typically with one wavelength that is most likely to excite it (= absorption maximum) and a distribution of wavelengths that are less likely to do so. Well characterized chromophores in the skin are DNA (absorption maximum at 260 nm), porphyrins (absorption maximum between 400 and 410 nm), and melanin (absorption maximum in the UVC range, but also very effectively excited by UVB and UVA) (Fig. 17-4). When a chromophore in the ground state absorbs the energy of a photon, electrons are raised to a higher orbit. If this involves no change in spin, the excited state is called singlet state. If it also undergoes a change in spin, the excited state is called a triplet state (Fig. 17-5). Upon photoexcitation, a photochemical reaction can occur to
267
2
Approximate penetration of UVR and visible light into skin
250 300 350 400 500 600 700 λ, nm
0
0.2
0.4
Depth, mm
0.6
0.8
1.0
Absorption spectra of cutaneous chromophores
1.2
1.0
Absorption (not to scale)
0.8
0.6
0.4
0.2
0 250
300 350 400
Wavelength, nm
KEY
NADH
Hemoglobin
7-DHC
Urocanic acid
DNA
Protein
Melanin
268
Excited states of chromophores, dissipation of the
absorbed energy, and formation of photoproducts after photon exposure
Singlet excited state
isc
Triplet excited state
ic
Photoproduct
hν Fl
isc
Ph
Photoproduct
Ground state
form a new, different molecule called photoproduct. For example, previtamin D3 is a photoproduct of photoexcited 7-dehydrocholesterol and the cyclobutane pyrimidine dimer the photoproduct of excited DNA. The process in which energy from an excited chromophore is transferred to another molecules is called a photosensitized reaction. A typical photobiologically relevant photosensitized reaction is energy transfer of an excited chromophore, eg, porphyrin, to oxygen with production of singlet oxygen, which in turn then reacts with other substrates, eg, guanine DNA bases. Instead of forming a photoproduct, the excited chromophore can also return to its ground state with the release of energy as heat or emission of a photon as fluorescence. The wavelengths of fluorescence is always longer (=less energetic) than the exciting wavelength (Stokes Law). For example, on excitation of protoporphyrin IX with blue light, fluorescence is red light, which has a longer wavelength than blue light and is less energetic. The excited singlet state is short-lived and the resulting fluorescence occurs in nanoseconds. Triplet states are longer-lived, as long as seconds. The photon emission from return of triplet excited states to ground states is called phosphorescence, which have longer wavelengths than fluorescence.
CUTANEOUS RESPONSES TO ULTRAVIOLET RADIATION
See Table 17-2.
VITAMIN D PHOTOBIOLOGY
VITAMIN D PHOTOBIOLOGY
FUNCTIONS OF VITAMIN D
Vitamin D regulates calcium and phosphorus metabolism. Its major role is to increase the flow of calcium into
UVA UVB
Immediate erythema 1000 × more erythemogenic than UVA; associated with sunburn
Immediate pigment darkening (within 20 minutes after UVR exposure) Delayed melanogenesis (peaks at 3 days)
Major role in drug induced photosensitivity; contributes to carcinogenesis
Major role in carcinogenesis
Central role in photoaging Role in photoaging
Penetrates down to deep dermis Penetrates no deeper than the upper dermis
Not associated with vitamin D production Role in vitamin D production
Penetrates window glass Does not penetrate window glass
Immunosuppression Immunosuppression
Immunosuppression Immunosuppression
the bloodstream, by promoting absorption of calcium and phosphorus from the intestines, and reabsorption of calcium in the kidneys, thus enabling normal mineralization of bone and muscle function. Vitamin D deficiency results in impaired bone mineralization that leads to bone softening diseases, rickets in children and osteomalacia in adults, and possibly contributes to osteoporosis.1-3 Deficiency can arise from inadequate intake, inadequate sunlight exposure, disorders that limit its absorption, and conditions that impair conversion of vitamin D into active metabolites, such as liver or kidney diseases. Most vulnerable to low vitamin D levels are elders, individuals living at high latitudes with long winters, obese persons, and all individuals with dark skin pigmentation living at high latitudes.4,5 Toxicity from excess vitamin D may manifest as hypercalciuria or hypercalcemia, the latter causing muscle weakness, apathy, headache, confusion, anorexia, irritability, nausea, vomiting, and bone pain, and may lead to complications such as kidney stones and kidney failure. Chronic toxicity includes the aforementioned symptoms along with constipation, anorexia, abdominal cramps, polydipsia, polyuria, backache, and hyperlipidemia. Findings may also include calcinosis, followed by hypertension and cardiac arrhythmias (due to shortened refractory period). Although information about the effects of high doses of vitamin D is limited, 10,000 IU daily is considered a safe upper limit for adult intake. A chronic toxic dose is more than 50,000 IU/day in adults. In addition to enhancing musculoskeletal health, vitamin D may also have an influence on the risk of a variety of other conditions, including cancer, cardiovascular disease, autoimmune disease, and infection. However, these effects remain a matter of significant controversy.6-16
There are 2 major sources of vitamin D, one is exogenous (diet) and the other is endogenous synthesis.
2
When obtained from food or supplements, vitamin D is absorbed in the small intestine. Natural food sources rich in vitamin D include certain oily fish such as salmon, mackerel, tuna, herring, catfish, cod, sardines and eel, butter, margarine, yogurt, liver, liver oil, and egg yolks, but, at least in the United States, most dietary vitamin D comes from the fortification of foods like cereal, milk, and orange juice. Fortified milk, for example, typically provides 100 IU per 250-mL glass, or only a quarter of the estimated adequate daily intake for adults.
BIOCHEMICAL PATHWAY OF VITAMIN D PRODUCTION
As a result of UV exposure of the skin, provitamin D3 (7-dehydrocholesterol, a precursor of cholesterol) is rapidly converted to previtamin D3, which spontaneously isomerizes to vitamin D3, entering the circulation on a binding protein and joining dietary D2 (or ergocalciferol) and D3 (cholecalciferol) absorbed from the gut (Fig. 17-6). Once reaching the liver, it is hydroxylated by the vitamin D 25-hydroxylase, a process that requires NADPH, O2, and Mg2+. The product 25-hydroxyvitamin D3 (calcidiol) is stored in the hepatocytes until it is needed. On release into the plasma, it makes its way to the proximal tubules of the kidneys, where it is acted upon by 25(OH)D-1α-hydroxylase, an enzyme whose activity is increased by parathyroid hormone and low PO4 2−. People with kidney disease may not be able to convert vitamin D to its active form. Following this conversion, 1,25-hydroxyvitamin D3 (calcitriol) is released into the circulation, and by binding to a carrier protein in the plasma, vitamin D–binding protein (VDBP), it is transported to various target organs. An action spectrum published by MacLaughlin et al. in 198217 shows that the most effective wavelengths to facilitate the photosynthesis of cutaneous vitamin D lie in the UVB range, with a peak at 297 nm, but the accuracy and validity of these data have been questioned more recently.18 In tropical areas, adequate amounts of vitamin D3 can be made in the skin with 10 to 15 minutes of biweekly sun exposure to the face, arms, and hands, or the back. Individuals with higher skin melanin content require more time in sunlight to produce the same amount of vitamin D as individuals with lower melanin content. It has been proposed that the evolutionary loss of pigment from human’s African origin to the white race living in more northern, less sunny latitudes provided advantages with regard to vitamin D production and bone health in areas where the photoprotective effects of melanin were not as critical as in lower latitudes.19 In today’s lifestyles with increased indoor activity and wearing of more extensive clothing, vitamin D deficiency is often a particular problem in people of color who need more UVB exposure to produce the same amount of vitamin in the skin, as compared to more fair-skinned individuals. It remains a matter of debate what are sufficient levels of vitamin D. These may be lower for the maintenance of bone health, and higher for the other, still controversial health benefits. Nevertheless, consistent
269
2
Synthesis of active vitamin D
UVB (max. with 297 nm)
Pro-vitamin D3 (7-dehydrocholesterol)
Skin
Pre-vitamin D3
Spontaneous isomerization
Vitamin D3 (cholecalciferol)
Gut
Dietary intake of vitamin D3 (cholecalciferol)
Vitamin D3 binding protein
Liver
Vitamin D 25–hydroxylase
Vitamin D3
- NADPH, O2, Mg2+
25–hydroxy-vitamin D3 (calcidiol)
Kidney (proximal tubulus)
25–(OH)–D3 1–α hydroxylase
1, 25-dihydroxy-vitamin D3 (calcitriol)
photoprotection for the prevention of the deleterious effects of UVR exposure, can undoubtedly result in vitamin D deficiency.20,21 Combining photoprotection with vitamin D supplementation therefore appears a wise choice, as dietary sources are often insufficient to maintain appropriate vitamin D levels.
EFFECTS ON THE IMMUNE SYSTEM
EFFECTS ON THE
IMMUNE SYSTEM
Several lines of observational evidence suggest that UV radiation has both proinflammatory and antiinflammatory properties. Proinflammatory properties
270
are supported by observations of sunburning, elucidation of inflammatory photodermatoses, induction and triggering of autoimmune connective tissue diseases, and photoaggravation of inflammatory skin diseases in some patients. Antiinflammatory/ immunosuppressive effects are supported by the observations of reactivation of labial herpes simplex with UVR exposure and efficacy of UV phototherapy for the treatment of inflammatory skin diseases. UVR has profound effects on the immune system, both locally at the sites of UV exposure and systemically. Currently, the immunostimulatory effects of UV radiation are thought to be mediated by innate immunity with release of proinflammatory mediators by skin-resident and nonresident cells (eg, serotonin, prostaglandins, IL-1, IL-6, IL-8, TNF-α), induction of
2
Mechanisms of photoimmunosuppression
Photophysics
Penetration of UVR into the epidermis and upper dermis
Photoexcitiation of DNA, RNA, trans-urocanic acid, and possibly of other chromophores
Photochemistry
Formation of DNA damage, RNA damage, cis-urocanic acid, and possibly other photoproducts
Photobiology
Suppression of pro-inflammatory cytokines (e.g. IFN-γ, IL-1β, -8, -12, -17, -18, -20, -22, -23)
Induction of anti-inflammatory cytokines (eg, IL-10, α-MSH)
Cellular effects:
• Apoptosis (T cells > keratinocytes, eg, via FasL secreted by KCs)
• Migration of Langerhans cells to regional lymph nodes with induction of antigen-specific Tregs/ tolerogenic state
• Inhibition of AG presentation by dermal dendritic cells
Alteration of autoantigen expression
antimicrobial peptides, and recruitment of neutrophils and lymphocytes.22 The immunosuppressive effects are thought to be mediated by migration of Langerhans cells from the epidermis to regional lymph nodes, release of antiinflammatory mediators of skin-resident and nonresident cells (eg, IL-10, α-MSH), and induction of antigen-specific regulatory T cells (Fig. 17-7). However, the view of such a pro- versus antiinflammatory dichotomy is probably overly simplistic. Instead, a variety of individual factors are likely influencing the pro- and antiinflammatory responses, resulting in different balances between these two poles. For example, for most patients with psoriasis, exposure to UV radiation results in an improvement of skin lesions, which showcases the antiinflammatory properties of UVR. A small subset of patients, however, experience worsening of their psoriasis. This demonstrates that for patients with such photoaggravated psoriasis, the net result of UVR is proinflammatory. Another example is the photodermatosis polymorphous light eruption, likely an inflammatory response to a UV-induced neoantigen. Patients with this condition have less UVinduced immunosuppression and are therefore more likely to develop this condition.23 On the other hand, this provides them with some protection against photocarcinogenesis, likely due to a more robust immuneresponse to UV-induced tumor antigens.24
The beneficial effect of UV-induced immunosuppression is probably the prevention of autoimmune reactions against self-antigens unmasked by UV-induced cell damage. An example of a failure to prevent such autoimmune reactions is lupus erythematosus with not only aggravation of skin lesions, but also triggering of new lesions and even systemic flares after UV exposure. From animal experimentation, is has been well established that the immunosuppressive effects of UV radiation are not only local, meaning limited to UV-exposed skin, but systemic as well, affecting also nonexposed skin. These effects have been shown to be mediated by IL-10 and subsequent induction of regulatory T cells. The photoaggravation and phototriggering of lupus erythematosus can be regarded a failure to appropriately downregulate immune reactions. The downside of photo-immunosuppression is that it prevents the recognition of UV-induced tumor antigens and with that promotes photocarcinogenesis. In addition to immunosuppression, UV radiation also induces long-lasting tolerance to antigens generated and/or unmasked following UV exposure. UV radiation can therefore be regarded a triple-edged sword for skin cancer development: it causes mutagenic DNA damage and not only inhibits the recognition of new tumor antigens but also induces tolerance against those. Chromophores of these effects on the immune
271
2
system are DNA with formation of DNA damage, RNA with formation of RNA damage, urocanic acid with UV-induced isomerization from the trans- to the cisisoform, and porphyrins that upon photoexcitation generate reactive oxygen species and subsequently change the redox potential of membrane lipids. Although most of the immunosuppressive effects are caused by UVB, UVA also has been shown to have similar effects.25
SUNBURNING
Sunburning (solar erythema) is clinically characterized by erythema and a burning sensation with pain (grade 1) and can progress to blister formation (grade 2), but does not progress to grade 3, at least not with solar available doses. It typically peaks 6 to 24 hours after sun exposure, followed by tanning and desquamation. Histologically, it is characterized by epidermal edema with spongiosis, few apoptotic keratinocytes (sunburn cells), depletion of Langerhans cells, vasodilation, and a mixed inflammatory infiltrate with lymphocytes and neutrophils. Action spectra for UV-induced erythema show that erythema formation peaks at 300 nm, which is within the UVB range, but then exponentially declines with increasing wavelength into the UVA range.26 Although very high doses of UVA can induce erythema, nonphotosensitive individuals do not develop erythema from solar available doses of UVA. UVC, albeit not present in solar radiation reaching the earth’s surface, can also induce sunburning. Because the erythema action spectrum is very similar to the action spectrum for the formation of directly UV-induced pyrimidine dimer type of DNA lesions, the chromophore for the sunburning reaction is thought to be DNA.27,28 This is further supported by the observation that many patients with defects in the repair of this type of lesions (xeroderma pigmentosum) have acute photosensitivity with a decreased UVB minimal erythema dose29,30 and similar observations from animal experimentation, where improvement of pyrimidine dimer repair reduces photosensitivity.31,32
Exposed skin cells with UV-induced DNA damage switch on a variety of damage response pathways, some of which result in the secretion of either preformed (eg, serotonin and TNFα) or newly synthesized inflammatory mediators (eg, prostaglandins, nitric oxide, and neuropeptides).33
TANNING
Darkening of the skin (tanning) is a characteristic consequence of sunburning, but also can be observed without prior burning (Fig. 17-8). It peaks approximately 3 days after UVB exposure, and is histologically characterized by an increase in basal and suprabasal melanin in the epidermis. Sensitivity for sunburning and ability to tan varies greatly among the human race and has been categorized in skin phototypes (Table 17-3), based on baseline pigmentation, sensitivity to sunburning, and ability to tan. This classification scheme was developed by Thomas B. Fitzpatrick for white skin
272
in 197534 and has since then been extended to darker skin types. In 2015, the computing industry standard Unicode has introduced Emoji modifiers to specify skin color based on Fitzpatrick’s skin phototype scale to represent diversity, which underlines the pervasiveness of skin color perception even in popular culture. An individual’s sensitivity to sunburning can be measured by determining the minimal erythema dose. This is the lowest dose that causes erythema in an individual, and it correlates, albeit poorly, with the clinically determined skin phototype.35 The skin phototypes, which are based on acute responses to UVR, are also indicators for an individual’s risk for long-term effects of UVR on skin, in particular photocarcinogenesis. The fact that the action spectrum for tanning is almost identical to the action spectrum for the formation of DNA photoproducts36,28 indicates that DNA is the chromophore for UVR-induced tanning, very similar to erythema formation (see above), and that cellular responses to DNA damage are critical to mediate the tanning response. The increased synthesis of melanin is dependent on the UV-induced induction
BURNING AND TANNING REACTIONS UPON SUN EXPOSURE COLOR OF UNEXPOSED SKIN
SKIN PHOTOTYPE
I Always burns, never tans Pale white
II Always burns, then tans White
III Sometimes burns, can tan without prior burn White
IV Usually does not burn, tans easily and deeply White to light brown
V Rarely burns, tans easily Brown; moderately pigmented
VI Burns only with very high UVR doses, tans Dark brown to black; darkly pigmented
VI Burns only with very high
Dark brown to black;
UVR doses, tans
darkly pigmented
and activation of p53, a central player in cellular DNA damage responses.37-39 Melanin produced by melanocytes is transferred through an active process involving specialized motor-proteins to keratinocytes, where it is preferentially placed over the nuclei, forming a microparasol.40 With this, the UV-absorbing properties of melanin shield the nuclei against the DNAdamaging effects of subsequent UV exposures. In response to the UV-induced inflammation (sunburning), the epidermal keratinocytes hyperproliferate, which results in thickening of the viable epidermis and of the stratum corneum. This thickening decreases penetration of UVR to the basal layer and with that also reduces formation of DNA damage and mutations in the skin’s stem cells. These two protective mechanisms, increased pigmentation and skin thickening, together with molecular adaptations, including, for example, increased activity of DNA repair enzymes, result in a several-fold increase of the erythema threshold.41 In addition to this delayed tanning, which is mostly induced by UVB, there is also an immediate pigment darkening, which can be observed within 20 minutes after UVR exposure. The latter is mediated by oxidation and redistribution of existing melanin, is mostly induced by UVA, and provides much less, if any, protection against subsequent exposures.
PHOTOBIOLOGIC PRINCIPLES OF PHOTODERMATOSES
The above-mentioned requirement for an excitation of a chromophore before a photobiologic reaction can occur also applies to photo-induced or photo-triggered dermatoses. For some, the chromophores are known, for others not. For example, defects in the heme biosynthesis pathways result in the accumulation of toxic pathway intermediates and cause porphyrias. Defects at an early stage of the heme biosynthesis pathway result in accumulation of nonaromatic intermediates that do not act as chromophores/photosensitizers. These porphyrias are not characterized by phototoxic reactions in the skin, but affect other organs, including, for example, the liver (eg, acute intermittent porphyria). Defects at later stages of the heme biosynthesis pathway result in the accumulation of larger, aromatic intermediates that act as chromophores. On exposure to UVR or visible light, excitation of these chromophores cause a phototoxic reaction (eg, subepidermal blisters) that is mediated by an energy transfer from the excited chromophore to oxygen (photosensitized reaction) and formation of singlet oxygen (eg, in porphyria cutanea tarda or erythropoietic protoporphyria). The absorption maximum of these intermediates, for example, uroporphyrinogen in porphyria cutanea tarda and protoporphyrin in erythropoietic protoporphyria, is between 400 and 410 nm, at the border between UVA and visible blue light. Affected patients, therefore, need to protect themselves not only against UVA but also against visible light to prevent phototoxic reactions. Different cutaneous porphyrias present with different symptoms. This is likely due to
2
differences in water solubility and subsequent difference in tissue distribution of the accumulated intermediates. The water-soluble uroporphyrinogen diffuses freely throughout the skin and induces most of its phototoxicity closest to the light source, at the epidermal junction, entailing blister formation. In erythropoietic protoporphyria, the much more lipophilic protoporphyrin remains in the endothelial lining and causes capillary damage with subsequent dermal necrosis and pain. In drug-induced phototoxic and photoallergic dermatitis, the chromophore is a medication or a metabolite of the medication. Such photosensitizing medications are aromatic molecules that absorb within the UVA range. Therefore, phototoxic and photoallergic drug reactions are almost exclusively induced by UVA. Photo-phytodermatitis is a phototoxic reaction mediated by plant-based chromophores, namely furocoumarins. These molecules are related to psoralens and have an absorption maximum in the UVA range. Berloque dermatitis is also a phototoxic dermatitis, in which the chromophore is the perfume ingredient bergamot oil (5-methoxy psoralen). Polymorphous light eruption and solar urticaria are likely heterogeneous conditions with regard to involved chromophores, as eliciting wavelength vary from patient to patient, ranging from UVB, to UVA, visible light, and possibly even infrared radiation.42-44
Diagnostic tools for the diagnosis of photodermatoses include determination of minimal erythema dose, separated for UVA and UVB (UVA-MED and UVB-MED), photopatch testing, and search for potential chromophores, for example, elevated porphyrins in blood, urine, and stool. Identification of the wavelength that elicit photodermatoses is important in tailoring appropriate photoprotection strategies for each individual patient.
PHOTOBIOLOGIC PRINCIPLES OF PHOTOTHERAPY
Therapeutic effects of electromagnetic radiation are used to treat a variety of skin diseases, either in combination with a photosensitizer, or without. For most phototherapy centers, psoriasis is the most common indication for phototherapy. In 1981, Parrish et al.45 published an action spectrum for the therapeutic effects of UVR for the treatment of this inflammatory skin disease. No therapeutic effect in the UVC range or with 280 or 290 nm, a maximum effect at around 300 nm, and an exponential decline of therapeutic efficacy with increasing wavelengths from 300 nm to the UVA range is similar to the erythema action spectrum described above and overlaps with the action spectrum for the formation of pyrimidine dimer type of DNA damage in the basal layer of the epidermis.27 This suggests that the chromophore that mediates the antipsoriasis therapeutic efficacy is DNA. At a time when psoriasis was mostly understood to be a condition that is driven by epidermal hyperproliferation, it was thought that UVinduced pyrimidine dimers would block replication of
273
2
hyperproliferating keratinocytes and with that improve psoriasis. However, slowing of epidermal hyperproliferation has since then been shown to be a later effect of UVB on psoriasis lesions, not a primary therapeutic effect. Today, the therapeutic effects of UVB are thought to be mediated by the above-mentioned immunosuppressive effects. Although the exact mechanisms remain to be clarified, hyperproliferating activated T cells may be particularly prone to the cytotoxic, proapoptotic effects of UVB. Induction of immune-deviation, eg, with transformation of TH1 and TH17 lymphocytes to Tregs, may be involved.46 Penetration of UVB is limited to the epidermis and the upper layers of the dermis, but it does not reach deeper levels of the dermis, which are often also infiltrated by immune cells in thick psoriatic plaques. It is also unclear whether skin-infiltrating lymphocytes or other immune cells are the primary target mediating efficacy of UVB phototherapy for psoriasis, or whether these are affected by signaling from UVexposed keratinocytes. The above-mentioned action spectrum for the therapeutic effects against psoriasis has led to the development of narrowband (nb) UVB phototherapy with bulbs that emit almost monochromatic UVB between 311 and 312 nm. This modality has been shown to be vastly more efficacious than the previously used broadband (bb) UVB phototherapy. This may be due to the avoidance of pro-erythemogenic short wavelengths of UVB (280-290 nm) that have no or only very little therapeutic efficacy and with that the ability to use significantly higher doses than with bbUVB. In addition, the higher proportion of UVB wavelengths penetrating deeper into the dermis may also facilitate a more effective reach of the target, the dermal inflammatory infiltrate. Today, nbUVB has replaced bbUVB not only for the treatment of psoriasis, but for all other UVB phototherapy indications as well, including, for example, atopic dermatitis, vitiligo, cutaneous T-cell lymphoma, etc. For these indications, the phototherapy mode of action is even less known than for psoriasis. The fact that nbUVB has been shown to be superior to bbUVB for all of these indications may suggest that the modes of action are similar to those in psoriasis. However, although the pathogenesis of these conditions is inflammatory in most of them, the exact inflammatory pathomechanisms are of course quite diverse. Much remains to learn how a single treatment modality, phototherapy with nbUVB, can improve such a diverse group of skin diseases. Crude coal tar is a potent photosensitizer, making exposed skin very sensitive to UVR-induced erythema. At a time when bbUVB was only partially effective, the use of tar in combination with bbUVB was a common practice, for example, with the treatment regimen named of the first describer, William H. Goeckerman. Tar is a mixture of many different compounds, many of which have photosensitizing properties. With nbUVB being highly effective as a monotherapy, the use of tar as a photosensitizer has become an uncommon practice. In contrast, photochemotherapy that combines either systemic or topical administration of psoralen
274
with subsequent irradiation with UVA remains a common phototherapy modality, although it is much less used today than at the time before the introduction of nbUVB. Psoralens (the most commonly used one is 8-methoxy-psoralen) intercalate with DNA and on photoexcitation react with pyrimidine bases to form mono- and divalent inter- and intrastrand crosslinks. For PUVA treatment, the chromophore is 8-MOP and psoralen adducts the UVA-induced photoproducts. The absorption maxima of 8-MOP are at 248 and 301 nm, which are in the UVC and UVB range. However, the absorption curve is quite broad and UVA still effectively excites this molecule and also produces the crosslinks quite effectively. The photochemistry of 8-MOP is quite complex. Intercalated with DNA, the absorption maximum is 313 nm, and after the formation of a monoadduct, the absorption maximum shifts to 334 nm, which is in the UVA range. These crosslinks are complex DNA lesions that can only be repaired by DNA recombination, not by nucleotide excision repair, as pyrimidine dimers. Similar to the above-described earlier belief that the replication-blocking properties of UVB-induced DNA damage mediates antiproliferative and with that antipsoriatic effects, the same was suggested for PUVA-induced crosslinks.47 As with UVB, this has since been refuted. The high efficacy of PUVA treatment may be explained by the deeper penetration of UVA into the dermis and its better reach to deeper inflammatory infiltrates. Because UVA is much less energetic and produces much less pyrimidine dimers than UVB, the addition of psoralen is likely required to bring UVA’s DNA-damaging effects into the therapeutic range. One way to circumvent the limited biologic effects of UVA is to just increase its dosing. This principle has been used for the development of high-dose UVA1 phototherapy without the use of a photosensitizer. This treatment has been shown to have some efficacy for the treatment of acute atopic dermatitis and for sclerosing conditions, for example, morphea. Chromophores that mediate this effect have not been identified, and the exact mode of action of this type of phototherapy also remains to be worked out. Another mode of phototherapy that employs the use of a photosensitizer is photodynamic therapy (PDT). In the United States, the most commonly used agent to photosensitize skin for PDT is 5-amino-levulinic acid (5-ALA). However, 5-ALA is not the chromophore that mediates the therapeutic effects of PDT. Application of 5-ALA circumvents the rate-limiting step of 5-ALA synthesis of the heme biosynthesis pathway and results in an increase in the cellular content of protoporphyrin IX. This aromatic molecule has an absorption maximum at around 406 nm, the so-called Soret band. It is readily excited by such blue light, upon which it reacts with oxygen to form singlet oxygen. PDT with 5-ALA therefore uses blue light, and its effects are thought to be mediated by the cytotoxic effects of singlet oxygen. The destruction of premalignant and malignant skin neoplasms by PDT may be due to the complete destruction of premalignant or malignant cells, or, more likely, to the destruction of some of these cells
with unmasking of tumor-antigens and subsequent immune-mediated destruction of others. At least partial tumor selectivity may be mediated by the more effective uptake of 5-ALA by premalignant and malignant cells. There are a number of different treatment modalities for PDT, varying by the route of application, the nature of the photosensitizer, and the wavelengths used. In either case, the wavelengths used must be carefully chosen to not only match the absorption characteristics of the photosensitizer, but also to facilitate sufficient penetration. PDT with 5-ALA and blue light is limited by the depth of penetration of blue light. In Europe, the most commonly used skin-directed PDT modality uses methyl aminolevulinic acid (MAL) in combination with red light. MAL has the advantage of better uptake into cells because of its more lipophilic properties, as compared to 5-ALA. The resulting chromophore, however, is the same as with 5-ALA. Although protoporphyrin IX is most effectively excited by blue light, its absorption curve shows a few small peaks in the green, orange, yellow, and red light range (Q-bands), which still provide sufficient photoexcitation with red light. The combination of deeper penetration of MAL, as compared to 5-ALA with the deeper penetration of red light, as compared to blue light, may make this PDT modality more effective for thicker skin lesions, for example, infiltrating skin cancers. PDT is not limited to regular, broad-spectrum sources of visible light but can also use monochromatic radiation from a variety of different laser sources, and research on the use of lasers for PDT is currently ongoing. Other modifications of current PDT practices involve the development of other photosensitizers with different photoabsorbing properties. The elegance of the current PDT, however, is that it uses a small molecule that is easily taken up into cells and then results in an increased production of the photosensitizer within the cell. This circumvents that problem that photoabsorbing molecules are usually larger and aromatic and are therefore less likely to be internalized by cells.
PHOTOCARCINOGENESIS
PHOTOCARCINOGENESIS
Chronic exposure of the skin to ultraviolet light increases the risk for a number of different cutaneous malignancies, including basal cell carcinoma, squamous cell carcinoma, malignant melanoma, and Merkel cell carcinoma. Photocarcinogenesis is the result of a chain of photophysical, photochemical, and photobiological events in UVR-exposed skin cells (Fig. 17-9).
FORMATION OF DNA DAMAGE AND MUTATIONS BY UVR
DNA has an absorption maximum at 260 nm, which is within the UVC range. UVB is still able to strongly excite DNA and penetrates deeper into the epidermis
2
Photocarcinogenesis cascade of events
UV-exposure Inherent defense mechanisms
Pigmentation Skin thickening
DNA damage DNA photoproducts
DNA repair Cell cycle arrest Apoptosis
Mutation incl. C T transitions
Removal of mutated cells (immune-surveillance)
Skin cancer monoclonal expansion of mutated keratinocytes or melanocytes
than UVC and therefore induces more photochemical reactions in the basal keratinocytes and in melanocytes. A critical consequence of directly UVR-induced photoexcitation of DNA is the formation of covalent chemical bindings between 2 adjacent pyrimidine bases, thymine and cytosine. There are 2 main types of such DNA photoproducts formed by UVR, cis-syn cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone photoproducts (6-4 PPs). Both types of dimers can form between any combination of thymine and cytosine— T:T, T:C, C:T, or C:C (Fig. 17-10). Unrepaired CPDs or 6,4-pyrimidine–pyrimidone dimers impair DNA replication and can result in mispairing if not removed prior to cells entering the S-phase of the cell cycle for DNA replication. A mispairing results in the change of the DNA sequence, a mutation. Several types of such mutations are observed after UV exposure, but by far the most common ones are C → T base exchange mutations at sites of 2 adjacent pyrimidines (dipyrimidine sites) (Fig. 17-10). This mutation is the result of misincorporation of adenine opposite cytosine during replication of the photoproduct. Without UV exposure, for example, in noncutaneous tissues or tumors, such mutations are observed only rarely, and the C → T and the C:C → T:T tandem mutations at dipyrimidine sites have therefore been named UV signature mutations.48 Once these mutations are formed, they remain for the life span of the affected cells and are also propagated during cell division. They can be considered the memory of a cell’s lifetime exposure to UVR. Some authors use the term UVB signature mutations. However, this term is
275
2
Generation of UVR-induced DNA damage and subsequent mutation formation
Formation of DNA damage (photoproduct)
Repair
UV
P
P P
Two adjacent pyrimidine bases
P P P
Cyclobutane pyrimidine dimer (CPD)
No repair
T G C T T T T C A G T
C
Normal sequence
T G C T T T T C A G T
T
C T mutation
misleading, as they are also formed by UVA,48,49 and should be avoided. In squamous cell carcinomas and actinic keratoses, such UV signature mutations are found in the p53 gene,48,50 in basal cell carcinomas in the genes of the sonic hedgehog signaling pathway (ptch, shh, smo) and in p53,51 and in melanoma in the CDKN2A, PTEN, TERT promoter, and p53 genes,52-55 demonstrating a molecular imprint of UVR as the transforming agent in tumor-specific gene mutations. The best characterized mechanism of how UVR induces formation of pyrimidine dimers is via direct absorption of photons by DNA bases, with DNA itself acting as the chromophore (classic pathway; Fig. 17-11; left panel). Recently, however, Premi et al.56 (see also Brash57) described an alternative, melanin-dependent mechanism that likely contributes to pyrimidine dimer formation in melanocytes and in melanin-containing keratinocytes. They had observed formation of DNA photoproducts up to 3 hours after UV exposure, and then showed that the formation of these so-called “dark CPDs” is mediated by photoexcitation of melanin, in particular of pheomelanin, through formation of peroxynitrite from UV-induced nitric oxide and superoxide (melanin-dependent chemiexcitation pathway; Fig. 17-11; middle panel). With this, it appears
276
that melanin is not only a photoprotector but may also contribute to cancer formation by increasing formation of DNA damage. This observation also provides an attractive explanation why UVA induces melanoma only in pigmented mice but not albino mice.58
The bystander effect describes the phenomenon that non-exposed cells in the vicinity of irradiated cells also demonstrate stress responses similar to exposed cells. UVA in particular has been shown to exert such a bystander effect in melanocytes, and long-lived UVRinduced radicals have been proposed to mediate it.59,60
It is tempting to speculate that the long-lived peroxynitrite described by Premi et al.56 could possibly not only generate “dark CPDs” in irradiated cells, but also in unirradiated bystander cells. In addition to pyrimidine dimers, UVR can also induce oxidative DNA damage (oxidative pathway, Fig. 17-11; right panel), for example, through absorption of photons by other cellular chromophores and subsequent energy transfer to DNA (type I photosensitized reaction) or to molecular oxygen with formation of reactive oxygen species (ROS), which in turn react with DNA to form oxidative DNA base modifications (type II photosensitized reaction). Although UVB can generate some ROS, most of the oxidative DNA damage after exposure to natural sunlight is caused by UVA.61,62 UVR-induced ROS are mostly singlet oxygen,
2
Mechanisms of UVR-induced mutation formation
Ultraviolet radiation
Absorption of photon energy by cellular chromophore(s), generation of NO* and SO*–, and subsequent formation of ONOO–
Absorption of photon energy by DNA and direct excitation of DNA molecules
Absorption of photon energy by cellular chromophore(s) and generation of singlet oxygen
Degradation of melanin and excitation of melanin fragments by ONOO –
Reaction of excited melanin fragment with DNA
Formation of “dark” DNA photoproducts (CPDs and 6-4 PPs)
Formation of DNA photoproducts (CPDs and 6-4 PPs)
Formation of oxidative guanine base modifications in DNA or in the nucleotide pool (eg, 8-oxodG)
Mispairing of cytosine with adenine Mispairing of 8-oxodG with adenine
Formation of UV-signature mutations (C T and CC TT transitions) Formation of mutations typical for oxidative base damage (G T and A C transversions)
Classic pathway UVB >>> UVA Melanin-dependent chemiexcitation pathway UVA and UVB
Oxidative pathway UVA (?)
but others, for example, hydrogen peroxide, the superoxide radical, and the hydroxyl radical also may be formed, albeit in much lower numbers. ROS-induced DNA damage involves predominantly guanine bases. Several such oxidative guanine lesions have been described after UVR exposure, of which 8-dihydro- 8-oxoguanosine (8-oxoG) is the best studied.
During DNA replication, 8-oxoG mispairs with adenine, giving rise to G → T transversion mutations. Similarly, oxidation of a guanosine nucleotide can result in misincorporation opposite an adenine during replication, giving rise to A → C transversions. There is no notable increase of such mutations in UVR-induced cutaneous malignancies, and it remains a matter of
277
2
debate how much oxidative DNA damage contributes to photocarcinogenesis. An action spectrum for the formation of pyrimidine dimers and oxidative guanine base modifications in mammalian cells shows gradually changing DNA-damaging capacity with increasing wavelengths from UVC, to UVB, UVA, and visible light.62 Few pyrimidine dimers are formed by UVA, approximately 10,000-fold less than with UVB when comparing, for example, 360 nm with 300 nm. Even when considering the much higher abundance of UVA in natural sun, UVB is most likely generating the majority of solar radiation–induced pyrimidine dimers. Nevertheless, there is mounting evidence from both in vitro and in vivo mutation spectra that pyrimidine dimers are the most important premutagenic DNA lesions not only with UVB but also with UVA, and that oxidative DNA damage plays only a minor role in mutagenesis.49,63
One hypothesis we have brought forward to explain a higher rate of mutations at UVA-induced pyrimidine dimers, as compared to the mutation rate of the UVBinduced ones, is that UVA does not induce as robust a cellular DNA damage response as UVB, in particular with cell cycle arrest.64 This may not be relevant for mutagenesis following exposure to broad-spectrum solar radiation, because with that the damage response can be evoked by the UVB component, which would also protect against mutagenesis at the few UVAinduced pyrimidine dimers. However, with exposures to pure UVA sources, as, for example, found with exposure to solar radiation through window glass or with use of tanning devices, UVA may be particularly mutagenic and carcinogenic. Although oxygen radicals have been described following UVR exposure, we found no evidence that DNA double-strand breaks, a common type of DNA damage generated by such ROS, are formed by either UVB or UVA.65,66 This is consistent with the fact that deletions or insertions, types of mutations that are commonly generated by DNA double-strand breaks, are not a common observation in UV-induced mutation spectra. Upon accumulation of a sufficient number of mutations in specific genes (driver mutations), cells undergo malignant transformation. In this process, mutation formation is a random event. Usually, many mutations occur all over the genome of exposed cells to ultimately also accumulate the critical number of specific driver mutations. For example, the high mutagenic burden in melanoma was demonstrated by Pleasance et al.,67 who were the first to report the full genomic sequence of a melanoma as compared to the normal sequence of a lymphoblast cell line from the same patient. Of 33,345 base substitution mutations observed in the melanoma, more than 70% were C → T UV-signature mutations. The predominance of this type of mutation that is otherwise rare in non-UV-exposed tumors is an impressive molecular account of a lifetime of sun exposure of a single melanocyte that has ultimately given rise to this melanoma. These data have since been confirmed in a large series of more than 300 melanomas, showing that a median of 77.7% of all mutations in melanoma
278
are UV-signature mutations.68 These data also show that the mean mutation rate in melanomas from sunexposed sites is one of the highest of all cancers investigated in this way. This high burden of mutations with mostly C → T transitions again emphasizes the role of UVR’s high mutagenic properties and its role in the pathogenesis of cutaneous melanoma. A genetically distinct melanoma subtype presents most commonly on the face and in the elderly, but can also occur in other areas with heavily photodamaged skin. These melanomas on chronically sun-damaged skin (CSD melanomas) show an extremely high mutation burden (approx. 60 mutations/Mbase)68,69 and often arise from an in situ melanoma termed lentigo maligna. Desmoplastic melanomas have an even higher mutation burden70 and have a similar age and anatomic site distribution as CSD melanomas. Melanomas on sun-exposed skin without signs of chronic sun-induced damage (non-CSD melanomas) show a lower mutation burden of approximately 30 mutations/Mbase. The somatic mutations found in all of the above-mentioned melanoma subtypes show a clear UV signature, whereas mucosal and acral melanomas, which are not thought to be UVR-induced, do not. Non-CSD melanomas are most common on the lower legs in women and on the back in men.71 At least in Western cultures, these areas tend to be photoprotected for long periods of time, for example, during winter, and are then exposed to the sun in the spring and summer, often suddenly and with subsequent sunburning. These types of melanoma therefore appear to be associated with sudden, intermittent high-dose UVR exposures of not sun-adapted skin.72 Chronically UVR-exposed skin is characterized by an upregulation of anti-mutagenic mechanisms. The absence of such protective states in photoprotected skin, in combination with a relative inability of melanocytes to undergo apoptosis on high-dose UVR exposures has been suggested as an explanation for the induction of melanoma by intermittent UVR exposures73 and may indicate the importance of cellular mechanisms that prevent formation of DNA damage and of mutations, and malignant transformation in chronically UVRexposed skin (Fig. 17-9).
DNA REPAIR AND OTHER RESPONSES TO UVR-INDUCED DNA DAMAGE
DNA repair is an important cellular defense mechanism that prevents mutation formation at sites of DNA damage after UV exposure. However, it is not the only defense mechanism (Fig. 17-9). Most mutations are generated during replication of damaged DNA. Therefore, a damage-induced arrest in cell cycling, which allows more time for repair, is another important cellular damage response that prevents mutation formation.64,74 Furthermore, programmed cell death (apoptosis) prevents the survival of cells with overwhelming DNA damage, and through that mechanism
the frequency of cells with UV-induced mutations is also reduced. Other inherent defense mechanisms against the carcinogenic properties of UVR include increased melanogenesis and thickening of the epidermis and stratum corneum, which protect from future DNA damage, as well as removal of mutated cells through host immune responses. More than 100 DNA repair genes have been identified (http://sciencepark. mdanderson.org/labs/wood/DNA_Repair_Genes .html). The nucleotide excision repair (NER) pathway acts on DNA damaged by UVR, repairing CPDs and other photoproducts, as well as on DNA damaged by certain other carcinogens (such as benzo-[a]-pyrene). In NER, the damaged nucleotide is removed and replaced with undamaged DNA. A simplified schema of the NER system describing some of the many proteins that act in concert to repair UV-induced DNA damage is shown in Fig. 17-12. Defects in these repair
2
genes can cause human diseases including xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy (TTD) (Chap. 130). For instance, XP can be caused by a defect in any one of several genes involved in NER. Based on cell fusion experiments, cells/patients with defects in the same gene are considered to be in the same complementation group, and in different complementation groups if different genes are affected. If DNA repair in the fused cell is normalized, with the wildtype gene from each cell giving rise to a functional protein that is absent in the other cell, the cells “complement” each other and are in different complementation groups. Seven such complementation groups have been identified (XP-A to XP-G), which correspond with mutations in 7 distinct genes that can cause XP. Transcribed genes are repaired faster than the rest of the genome. In the NER pathway, the first steps
DNA nucleotide excision repair
A DNA damage recognition
Global genome repair Transcription-coupled repair
XPE
XPC
5’ 3’
B Unwinding of DNA helix
TFIIH
XPB XPD
5’ 3’
RNA-polymerase
5’ 3’
CSA CSB
XPA
C Incision and release of 24-34 residue oligonucleotide
XPF complexed with ERCC1 nuclease
XPB XPD
5’ 3’
D Gap filling and ligation
5’ 3’
XPG
PCNA RPA DNA polymerase ε/δ
279
2
involving DNA damage recognition are different in nontranscribed (global genome NER) and transcribed genes (transcription-coupled NER). In nontranscribed genes and noncoding areas, which represent most of the genome, the XPE and XPC gene products bind to UV-damaged DNA, marking it for further processing. In contrast, DNA damage in transcribed genes appears to be sensed by a stalled RNA polymerase acting in conjunction with the CSA and CSB (CS complementation groups A and B) gene products. After the DNA damage recognition steps, global genome NER and transcription-coupled NER follow the same pathway (Fig. 17-12). In XP, pronounced NER deficiencies confer a very high risk to develop nonmelanoma skin cancer and melanoma. More subtle variations in DNA repair capacity, for example, as a consequence of polymorphisms in NER genes or of an aging-related decline, also have been linked to an increased skin cancer risk.75-78
Oxidative base modifications are a simpler type of DNA damage than pyrimidine dimers, as they only involve chemical changes in a single DNA base. These lesions are processed by a different DNA repair pathway, called base excision repair. Several cellular responses to DNA damage contribute to the maintenance of genome integrity by preventing mutation formation. These include cell cycle arrest, apoptosis (programmed cell death), and DNA repair. Because these responses need to be carefully orchestrated, there are many proteins involved in the signaling of DNA damage and in the regulation of cellular DNA damage responses. Different types of DNAdamaging agents and different types of DNA damage require different DNA damage responses. A simplified version of this complex pathway is presented in Fig. 17-13. As with defects in DNA repair genes, defects in many of these DNA damage–signaling genes (boxed in Fig. 17-13) are also implicated in
Cellular DNA damage responses—pathways and effects
DNA damage
ATM ATR
Chk1 Chk2
p53
Cdc25A, B, C degradation
p95/Nbs1
p21
A, B, C A
Cell cycle checkpoints:
intra S G2/M G1/S
Caspase 2
Bax
Caspase 9
Caspase 3
FA/BRCA pathway
Apoptosis
Recombination repair
XPC
GG-NER
XPE/DDB2
GADD45
Error-prone translesional DNA synthesis
280
hereditary disorders of genome instability, including, for example, ataxia telangiectasia, Fanconi anemia, familial breast cancer, Li-Fraumeni syndrome, and others (Chap. 130). The tumor suppressor gene p53, often called the guardian of the genome, plays a pivotal role in regulating and orchestrating these responses and is mutated in many cancers, including cutaneous squamous cell carcinomas. Upstream regulators of p53 in the cellular DNA damage response pathway are ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia- and Rad3-related) genes. One of p53’s several functions is the regulation of the cell cycle in response to DNA damage. After cell division (mitosis), cells have 23 pairs of chromosomes and are in the G1 phase of the cell cycle. The chromosomes then replicate during DNA synthesis, or S phase, and as a result have twice the number of chromosomes (G2 phase) just before mitosis (M phase). In response to damage, the cell may stop cycling (arrest) in specific cell cycle phases called cell cycle checkpoints. An important downstream effector in preventing cells from entering S phase (G1/S checkpoint) is p21. p53 also induces NER by transcriptionally inducing XPC, XPE/p48, and GADD45. If cells enter S phase with unrepaired DNA damage, or if cells are UV exposed during S phase, regular DNA polymerases stall at DNA photoproducts and detach from the DNA strand.79 For these instances, cells are equipped with several specialized DNA polymerases for translesional DNA synthesis. DNA polymerase eta is one of these; it is specialized to bypass DNA photoproducts but may introduce mutations while doing so. It is mutated in XP variant patients, who are clinically indistinguishable from other XP patients with defects in NER (Chap. 130).80,81 This demonstrates the importance of this second line of defense against the mutagenic and carcinogenic consequences of DNA photoproducts. p53 and p21 also downregulate the activity of this translesional DNA synthesis to maintain a low mutagenic activity at the price of reduced damage bypass. If this translesional DNA synthesis fails, cells can use recombination repair to resolve stalled replication forks.82 When invoked in response to damage from UV light, this third line of defense is mediated by activation of the Fanconi anemia/BRCA DNA damage response pathway.65
Apoptosis is a regulated physiologic process leading to cell death characterized by cell shrinkage, membrane blebbing, and DNA fragmentation. A group of cysteine proteases called caspases are central regulators of apoptosis. Triggers may be extrinsic or intrinsic to the cell (eg, DNA damage) and involve separate initiator caspases (eg, caspase 2 in response to DNA damage) but share the same downstream effector caspases. Mutations in the p53 tumor suppressor gene are very commonly found in cutaneous squamous cell carcinomas and their precursors, actinic keratoses. In addition, chronically sun-exposed skin harbors many clonal proliferations of keratinocytes with p53 mutations, which are not detectable by macroscopic or microscopic abnormalities.83 Loss of p53 function as
2
a central regulator of antimutagenic responses entails a higher rate of mutation formation with subsequent UVR exposures. This so-called UV mutator phenotype is critical in the development of cutaneous squamous cell cancers, as it makes it much more likely that all the mutations necessary for complete malignant transformation of a single keratinocyte are accumulated with repeated UV exposures.84
WHICH WAVELENGTHS OF UVR CAUSE SKIN CANCER?
Although the carcinogenic properties of UVB have been well known for decades, UVA has long been considered harmless because of its inability to induce sunburning with solar available doses. However, it is now recognized that UVR is also damaging and carcinogenic in suberythemogenic doses and that UVA can induce cutaneous squamous cell carcinomas by itself in mice. Consequently, the WHO has classified UVA as an independent class I carcinogen.85 The Utrecht- Philadelphia action spectrum for photocarcinogenesis in mice shows an exponential decline in carcinogenic efficacy in mice with increasing wavelengths from UVB to UVA.86 Some of this decline is offset by the higher abundance of UVA in natural solar radiation. In addition, exposures to pure UVA by window glass–filtered solar radiation and in tanning parlors may significantly increase the contribution of UVA to photocarcinogenesis, as detailed above. Therefore, although it appears that UVB contributes more to photocarcinogenesis than UVA, the relative contributions of either one to a lifetime risk to develop nonmelanoma skin cancer remains unclear. Several animal models have been used to answer the wavelengths question for melanoma. Ley87 observed the induction of melanoma precursor lesions in the opossum with UVA, more than with UVB. However, no infiltrative melanomas could be induced. Setlow et al88 used swordfish and observed that UVA induces melanomas more effectively than UVB. However, this observation could not be reproduced and the opposite result was reported by Mitchell et al.89 DeFabo et al90 used HGF/SF transgenic mice and observed melanoma induction only with UVB, but not with UVA. Later, the same group58 observed that UVA does induce melanoma, but only in pigmented mice, not in the previously studied nonpigmented mice. With that, it appears that, at least in this mouse model, the presence of melanin is required for the induction of melanoma with UVA, but not with UVB, and that both wavelengths, UVA and UVB, can induce melanoma, but possibly via a different mechanism. It is very plausible that the recently described melanin-dependent chemiexcitation pathway for the generation of pyrimidine dimers and UV signature mutations (Fig. 17-11; middle panel) is that postulated alternative mechanism for melanomagenesis with UVA. In addition, increases of melanoma risk with tanning parlor use provide further support for UVA being a particular risk factor for melanoma. Many studies
281
2
have shown a significantly increased risk for melanoma subsequent to sunbed/sunlamp use,91,92 including a large prospective cohort study in 106,379 women in Sweden and Norway, showing a relative melanoma risk of 1.42 to 2.58 with use of tanning devices.93 A melanoma epidemic in Iceland with increasing rates of melanoma between 1990 and 2006, mainly in young women, also has been associated with increased tanning bed use.94 Melanomas at usually covered sites, for example, skin in the sacral and pubic areas, also may be attributable to UVR exposure from sunbed use.95
When a few decades ago it was increasingly recognized that UVB (280 to 315 nm) had skin-damaging effect, but UVA (315 to 400 nm) was still considered relatively safe, the tanning industry reduced the UVB output in their devices and claimed that a UVA-induced tan would be safe. This is why the increased melanoma risk with tanning bed use may support the role of UVA as a particular risk factor for melanoma. However, tanning machines differ widely in their spectral output.96,97 Therefore, the increased melanoma risk with sunbed use also may be due to the UVB that is still emitted by tanning devices. The ability to generate oxidative DNA lesions increases with increasing wavelengths from UVB to UVA and it has been hypothesized that such damage, in particular when induced by UVA, could play a role in melanomagenesis.63 When contemplating this hypothesis, it is important to recognize that at least in fibroblasts and keratinocytes, UVA has been shown to produce more pyrimidine dimers than 8-oxodG.98
Unlike an early report of a separate UVA signature mutation observed in transformed rodent cells,99 the majority of UVA-induced mutations has later been shown to be C → T transitions, without a particular signal for a separate UVA signature mutation or a high rate of mutations typical for oxidative base modifications, both in vitro and in vivo.100-103
An attractive candidate for generation of oxidative stress on exposure to UVR in melanocytes is melanin. Melanin has been described to have some photosensitizing properties, in particular, the more reddishcolored pheomelanin.104,105 Wang et al.106,107 reported that, on irradiation with UVA, melanocytes generate
more oxidative DNA damage, have less efficient repair of oxidative DNA damage, and produce more mutations. They proposed that oxidative DNA damage is a major driver in melanomagenesis. However, they did not sequence the UVA-induced mutations and with that did not provide ultimate proof that the UVAinduced mutations are indeed the G → T or A → C transversions that are typical for oxidative DNA damage. In addition, the above-mentioned mutation data from melanomas show only a small percentage of such mutations typical for oxidative DNA damage.67,68
Taken together, there is no proof that oxidative DNA base lesions play a major role in UVR-induced mutation formation that drives melanoma. In summary, the evidence supports that both UVB and UVA can induce melanoma, possibly through different mechanisms. However, the relative contribution to the overall melanoma risk of UVB and UVA remain unclear.
PHOTOAGING
Photoaging describes changes observed in chronically sun-exposed skin, whereas intrinsic aging of the skin describes changes that occur with advancing age without extrinsic damage from chronic sun exposure. Skin alterations with photoaging are observed in all compartments of the skin, including the epidermis, the pigmentary system, the dermal connective tissue, the vasculature, and the subcutaneous fat (Figs. 17-14 and 17-15).108,109 Although subtle changes of photoaging can sometimes be observed in young individuals already, manifestations of photoaging are more common in older patients, indicating that skin changes of photoaging are typically superimposed on changes of intrinsic skin aging. Many manifestations of photoaging are also observed in intrinsically aged skin. This raises the question whether photoaging is solely an acceleration of the normal aging process by chronic photodamage. Table 17-4 lists typical manifestation of cutaneous photoaging. Symptoms that involve benign, premalignant, and malignant clonal proliferations of keratinocytes and melanocytes are associated with
282
aging, as cancer in general is associated with aging. They are better classified as neoplastic events, rather than aging events. They are not observed in intrinsically aged, only in photoprotected, skin. The mechanisms that drive these events are described under section “Photocarcinogenesis”. In contrast, thinning of the epidermis, hypopigmentations, loss of dermal collagen, telangiectasias (as a consequence of a diminished extracellular matrix scaffold), and loss of subcutaneous fat are loss-of-function events (Table 17-4), which are also observed with intrinsic aging. Accelerated proliferative exhaustion by chronic photodamage is a likely mechanism for these features, which is consistent with the view that photoaging represents damage-accelerated aging.
LOSS OF FUNCTION CLONAL PROLIFERATION
Epidermis Epidermal atrophy Expansion of mutant clones Malignant transformation
✓ ✓ ✓
Pigmentary System Freckles/lentigines Hypopigmentations Melanocytic nevi Malignant melanoma
✓ ✓
✓ ✓
Dermal Connective Tissue Loss of collagen Actinic elastosis ✓ ✓
Vasculature Telangiectasias (loss of ECM scaffolding) ✓
Subcutaneous Fat Loss of fat ✓
Subcutaneous Fat Loss of fat ✓
2
The only nonproliferative feature of photoaged skin that is not observed in intrinsically aged skin is actinic elastosis, which is characterized by accumulation of fibrillary basophilic material in the upper and middermis. Clinically, it presents with yellowish thickening of the skin, sometimes nodular, loss of elasticity, and formation of deep wrinkles (Fig. 17-15). It is composed mainly of elastin and fibrillin, stains strongly with elastin stains, and can be degraded by elastase, but not collagenase. Elastin synthesis is upregulated following UV exposure,110 but the UVR-induced influx of neutrophils, as seen, for example, in sunburned skin, also provides a mechanism for increased degradation via activity of elastase secreted by neutrophils. Following degradation of elastin in the extracellular space, dermal fibroblast internalize elastin fragments through receptor-mediated endocytosis to lysosomes for further degradation by the lysosomal protease cathepsin K.111
Cathepsin K is upregulated by UVA to promote this intracellular clearing.112 Aged fibroblasts do not upregulate cathepsin K in response to UVA, and it is hypothesized that this entails a failure to properly clear elastin fragments from the extracellular space and ultimately results in the accumulation of elastin with crosslinking to a large, abnormal macromolecule.112 With this, actinic elastosis can be classified as a loss-of-function event. Actinic elastosis is largely irreversible, even with consistent photoprotection, which is most likely due to the fact that the large abnormal macromolecule has become undegradable. This is reminiscent of the intracellular accumulation of abnormal macromolecules and subsequent impairment of cellular function as a result of a failure to clear them from the cytoplasm via a mechanism called macroautophagy, which is an important mechanism how aged cells in general loose functions.113
Progerin is an abnormal splice variant of the nuclear membrane protein Lamin A. It is found in high concentrations in cells from patients with the premature aging syndrome Hutchinson Gilford progeria, but also accumulates in normally aging cells.114,115 It interferes with many nuclear functions and ultimately results in cellular senescence and an aged cellular phenotype. This aging protein is induced by UVA via oxidative stress and induction of abnormal splicing.116 This is yet another example how UVA in particular promotes an aging phenotype. As a result of a loss of a farnesylation site that allows degradation of lamin A, progerin is largely undegradable. This is a second example how exposure to UVA results in the accumulation of an undegradable protein that interferes with cell function. Although some of the effects of acute UVR-induced inflammation induce metabolic changes that may contribute to photoaging, for example, increased elastin synthesis, increased expression of matrix metalloproteinase (ie, collagenases), and decreased collagen synthesis, these should be reversible on cessation of UV exposure. The observation, however, is that changes of the extracellular matrix, which manifest as wrinkling and increased laxity of skin, are not reversible, may be explained by the accumulation of abnormal and undegradable proteins in the extra- and intracellular
283
2
space. Although the contribution of oxidative DNA damage to photocarcinogenesis is unproven, UVAinduced oxidative stress is likely a central mediator in these loss-of-function events in photoaging. As ROS mediate senescence, for example, via the formation of progerin,116 they probably entail an anticancer effect that counteracts the pro-carcinogenic effects of UVR. With this, antioxidants may prevent UVR-induced loss of function events of photoaging, but may in turn also promote photocarcinogenesis. In addition to its specific roles in inducing a cellular aging phenotype with loss of cell functions, UVA also may be a particular culprit for the changes in the dermal extracellular matrix because of its deeper penetration into the dermis, as compared to UVB. However, UVB does affect metabolism in the deep dermis even without reaching into it. For example, the induction of matrix metalloproteinases in the dermis after irradiation with UVB has been shown to be mediated by a paracrine mechanism via excretion of inflammatory mediators from keratinocytes and signaling to dermal fibroblasts.117-119 Further signaling into the subcutaneous fat may even mediate loss of subcutaneous fat as a feature of photoaging.120
CUTANEOUS RESPONSES TO VISIBLE LIGHT AND INFRARED RADIATION
In comparison to UVR, the effects of other types of nonionizing radiation of the electromagnetic spectrum, namely, visible light and infrared radiation (IR), have been studied much less. However, there is increasing evidence that these also have photobiological effects on the skin. Visible light can induce erythema and pigmentation (immediate and delayed pigment darkening, in particular in darker skin types)121,122 and some photodermatoses can sometimes also be elicited by visible light, including, for example, solar urticaria, chronic actinic dermatitis, and cutaneous porphyrias. Potential chromophores that absorb visible light and could be mediating these effects are porphyrins, melanin, β-carotene, riboflavin, bilirubin, and hemoglobin. In addition, the action spectra for the formation of pyrimidine dimer types of DNA damage and oxidative DNA base modification extend well into the range of visible light.62
Although the mode of action of these effects is still only very incompletely understood, these insights have led to the development of visible light–based treatment devices, including, for example, intense pulsed light (IPL), lasers, and photodynamic therapy. IR has been implicated in photoaging and in erythema ab igne.123,124 IRA penetrates deep into the dermis and has profound effects on the gene expression profile of dermal fibroblasts, including, for example, the induction of matrix metalloproteinase. This may contribute to the loss of collagen fiber in chronically sunexposed skin. Although infrared radiation alone does
284
not appear to cause skin cancer, growth behavior may be affected, for example, via prevention of apoptosis.125
CONCLUSIONS
The only positive effect of UVR on skin, vitamin D production, is offset by a number of serious and potentially life-threatening detrimental effects that mandate effective photoprotection for prevention. Fortunately, the negative effect of photoprotection on vitamin D metabolism can easily be addressed with oral vitamin D supplementation. From the described effects of UVB and UVA, is it clear that photoprotection strategies must include protection against both qualities. Fortunately, the Food and Drug Administration (FDA) of the US Department of Health and Human Services issued a final role on labeling and effectiveness testing of sunscreen drug products for nonprescription human use in 2011 that addresses such broad-spectrum protection. This new standard adds an in vitro measurement for broadspectrum protection to include UVA. It uses the critical wavelength criterion originally suggested by Diffey et al, defining broad spectrum by a wavelength of at least 370 nm at which 90% of the area under the 290- to 400-nm absorbance curve is met.126
The recognition that IR and visible light can damage skin has given rise in efforts to extend photoprotection to these types of radiation.127,128 Attempts to filter visible light will probably be hampered by poor acceptability, as such filters will be visible. There may be an opportunity to filter IR, but the development of such filters has only just begun. Systemic and topical antioxidants have been advocated to reduce UVR-, visible light-, and IR-induced oxidative damage, in particular, oxidative DNA damage.129 However, their protective effects against any of the above-mentioned endpoints have not been well established, or are marginal at best. In addition, the evidence that oxidative DNA damage might not contribute significantly to UVA-induced mutagenesis (as was previously thought) raises the question whether antioxidants provide any protection against skin cancer. Reactive oxygen species are not only damaging but also induce cellular responses that can protect against photocarcinogenesis, including, for example, induction of senescence through generation of the aging-associated protein called progerin.116 This suggests that antioxidants may actually be promoting photocarcinogenesis, as already suggested for other types of cancer. In 2014, the Surgeon General and the U.S. Department of Health and Human Services published a Call to Action to Prevent Skin Cancer. Stated goals include (1) increase opportunities for sun protection in outdoor settings; (2) provide individuals with the information they need to make informed, healthy choices about UV exposure; (3) promote policies that advance the national goal of preventing skin cancer; (4) reduce harms from indoor tanning; and (5) strengthen research, surveillance, monitoring, and evaluation related to skin cancer prevention.
When thinking about public awareness campaigns, it is important to recognize that sun-seeking and tanning device–seeking behavior is, at least in some individuals, associated with a pleasurable central nervous system effect.130-132 Such features of addiction to UVR exposure are reminiscent of the addictive nature of nicotine that has made smoking cessation programs for lung cancer prevention difficult. The fact that antismoking campaigns have been able to overcome nicotine addiction and have resulted in a decline in smoking and consequently in lung cancer deaths are encouraging for efforts to reduce rates of nonmelanoma skin cancer and melanoma by addressing addictive tanning and sun seeking. The dermatologic community has the opportunity to make a huge impact on public health by supporting the Surgeon General’s Call to Action.
ACKNOWLEDGMENTS
Parts of this chapter used significant portions of Chap. 90 of the previous edition of this book, “Fundamentals of Cutaneous Photobiology and Photoimmunology,” written by Irene E. Kochevar, Charles R. Taylor, and Jean Krutmann. Other parts used elements of Chap. 110, “Genome Instability, DNA Repair, and Cancer,” written by Thomas M. Rünger and Kenneth H. Kraemer.

Figure 17-1 Spectrum of electromagnetic radiation divided into major wavelength regions and further subdivisions.
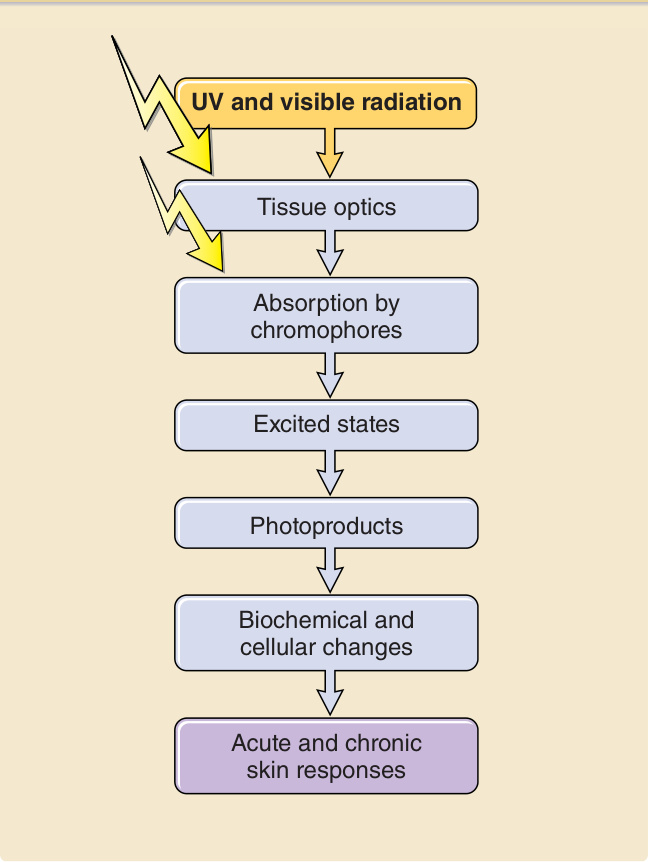
Figure 17-2 Series of photophysical, photochemical, and photobiological events that are required for a photon to have an effect on exposed skin.
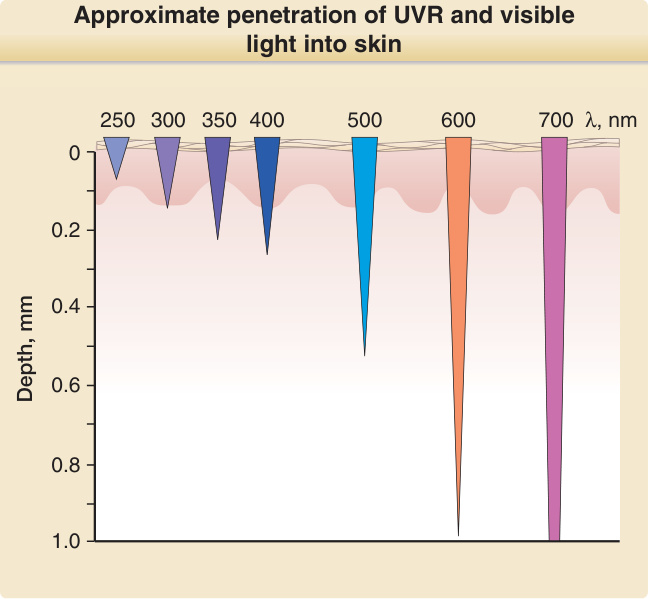
Figure 17-3 Approximate penetration of ultraviolet radiation and visible light into skin. λ = wavelength.
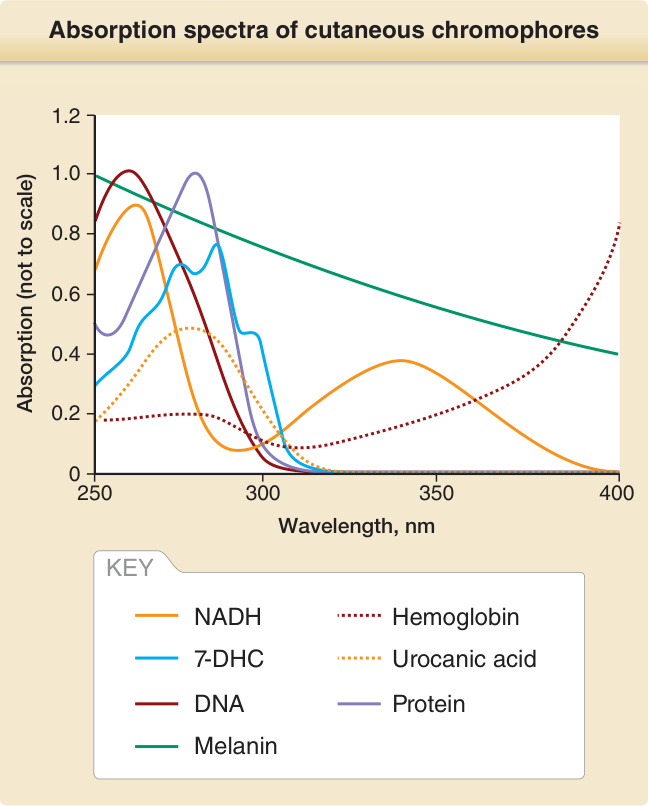
Figure 17-4 Absorption spectra of cutaneous chromophores absorbing ultraviolet radiation. Note that the relative amounts of UVR absorbed by these chromophores in skin depend on the heights of the absorption peak, the amount of each chromophore in skin, and the penetration of each wavelength into skin. For example, although the absorption maximum of naked DNA is at 260 nm, the most effective wavelengths to excite DNA in basal keratinocytes is 300 nm. This is because shorter wavelengths are absorbed and scattered in the stratum corneum and the upper layers of the epidermis and do not reach the basal keratinocytes as effectively. 7-DHC, 7-dihydrocholesterol; NADH, reduced nicotinamide adenine dinucleotide.
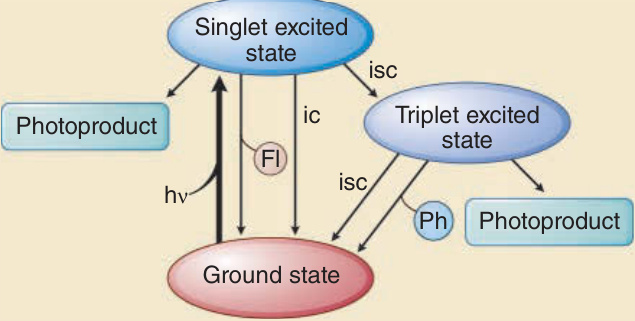
Figure 17-5 Excited states of chromophores, dissipation of the absorbed energy, and formation of photoproducts after exposure to photons of ultraviolet radiation and visible light. FI, fluorescence; ic, internal conversion; isc, intersystem crossing; Ph, phosphorescence.
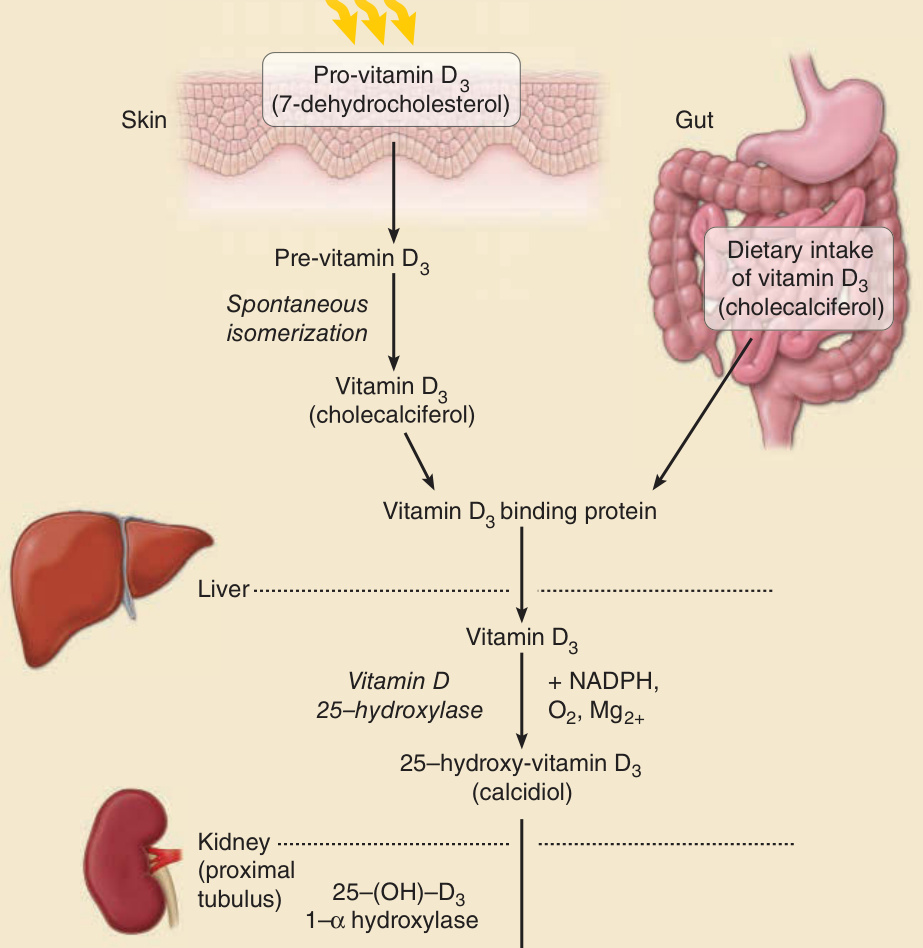
Figure 17-6 Synthesis of active vitamin D. The chromophore that absorbs UVB and initiates this pathway is 7-dehydrocholesterol. Its absorption maximum is 297 nm. The photoproduct of photoexcited 7-dehydrocholesterol is pre-vitamin D3, which quickly isomerizes to vitamin D3. See text for more details.

Figure 17-7 Mechanisms of photoimmunosuppression. Photoimmunosuppression is mediated by photoexcitation of chromophores, including DNA and trans-urocanic acid, formation of photoproduct (eg, cyclobutane pyrimidine dimers, cis-urocanic acid), and subsequently a range off immunologic endpoints. This cascade demonstrates a typical cascade of events following exposure of the skin to UVR, starting with photophysical responses (penetration of UVR into the skin and photoexcitation of chromophores), followed by photochemical reactions (production of photoproducts), and ultimately photobiologic consequences.
Figure 17-8 Tanning in a patient with nbUVB phototherapy. Note the lack of tanning in skin folds not exposed to UVR. This patient with skin phototype V did not have a sunburn prior to tanning.
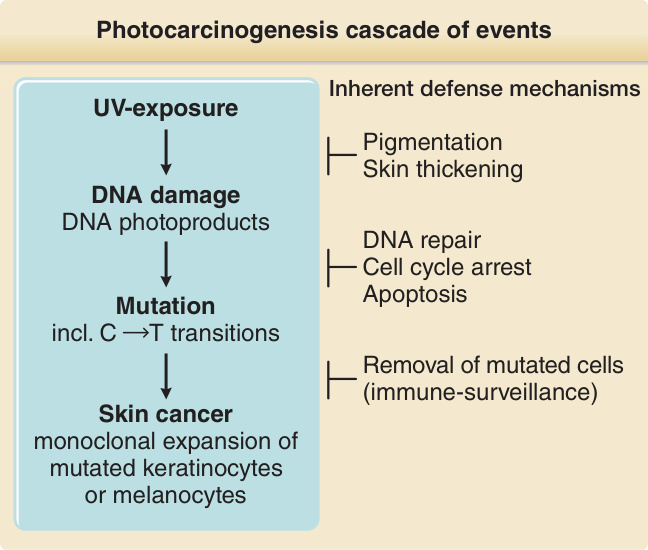
Figure 17-9 Photocarcinogenesis cascade of events. Exposure to UVR induces typical types of DNA damage, namely, cyclobutane pyrimidine dimers and 6,4-pyrimidine–pyrimidone photo-products. These often generate single and tandem base substitution mutations (C→T and CC→TT) at di-pyrimidine sites that are typical for UVR exposure and are therefore termed UV-signature mutations. With sufficient numbers of inactivating mutations in crucial genes (tumor suppressor genes), individual cells may undergo malignant transformation, clonally expand, and form skin cancers. Several inherent defense mechanisms counteract this chain of events.
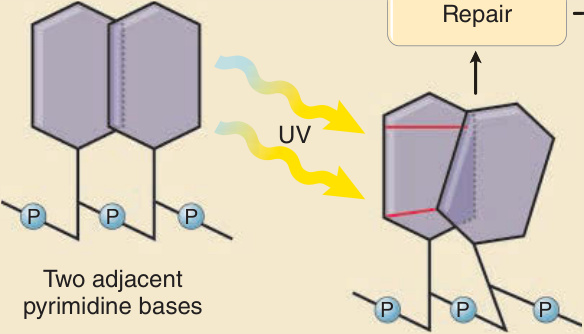
Figure 17-10 Example of UVR-induced generation of DNA damage and subsequent mutation. Upon direct excitation of the DNA molecule by UVR, adjacent pyrimidine bases (cytosine or thymine) may form covalent bonds between them, which leads to creation of pyrimidine dimers. The illustration shows an example in which 2 covalent bindings generate a tricyclic cyclobutane ring between 2 pyrimidines. Hence, this type of UV light–induced DNA damage is called a cyclobutane pyrimidine dimer, a common type of DNA photoproduct. The nucleotide excision repair DNA repair system functions to remove this damage, which results in the normal DNA sequence (upper box). If the damage is not repaired, this type of DNA photoproduct can lead to formation of a typical C→T single base substitution mutation (lower box). This is most likely to occur on replication of damaged DNA and misincorporation of adenine opposite the cytosine-containing photoproduct. An example of such a UV-signature mutation is shown on the right. Note that the mutation is located within a run of 7 pyrimidines, a common location for UV-signature mutations.
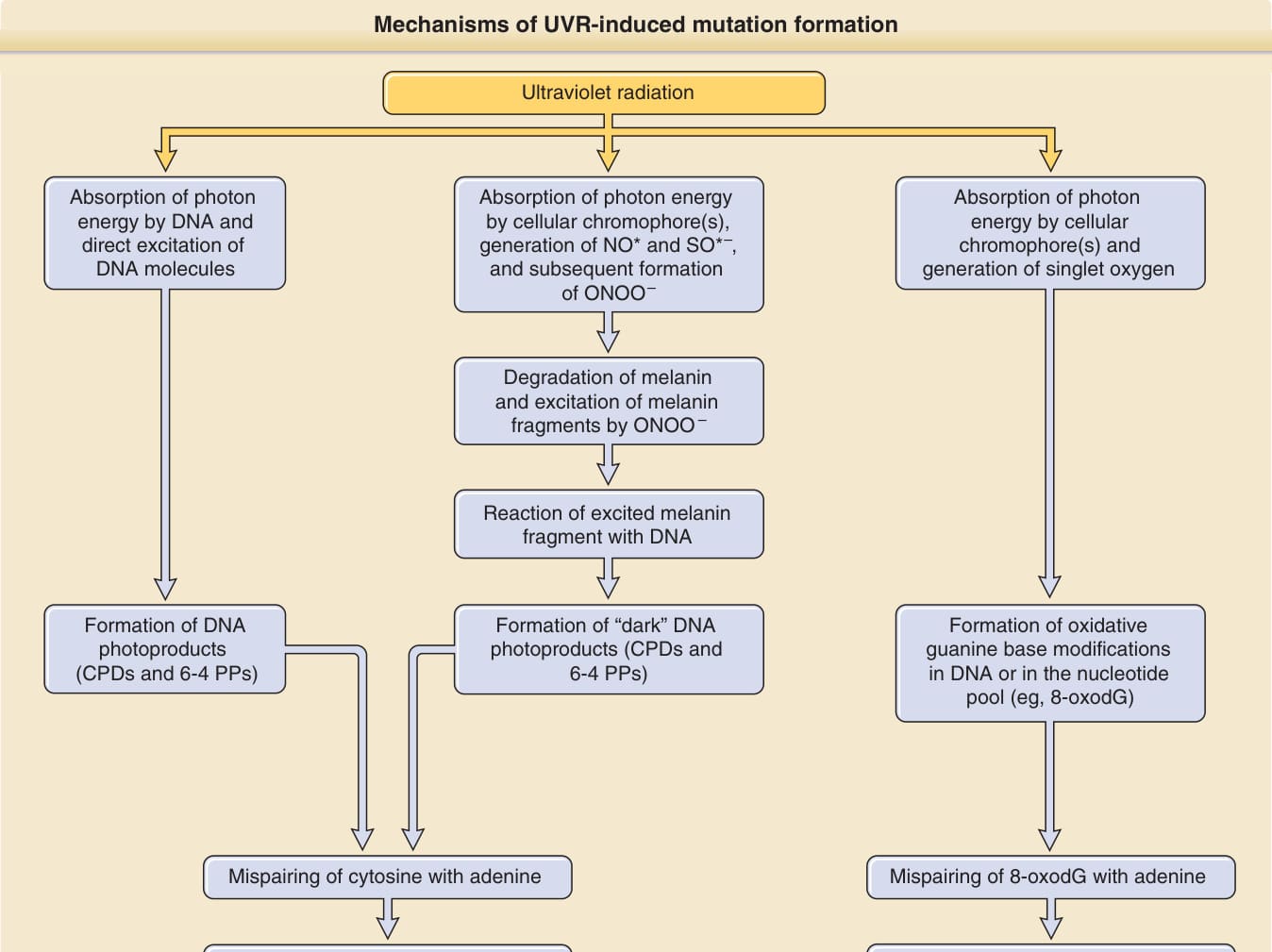
Figure 17-11 Mechanisms of UVR-induced mutation formation. In the classic pathway (left panel), adjacent pyrimidines react with each other and dimerize to form DNA photoproducts after direct excitation of DNA molecules by photons. The C → T and CC → TT UV-signature mutations may form at sites of DNA photoproducts by mispairing of cytosine(s) with adenine. Although 260 nm is the absorption maximum of DNA, UVB still strongly excites DNA and generates mutations through this pathway. UVA’s ability to excite DNA is much weaker, but may still generate mutations via this mechanism. CPDs, cis-syn cyclobutane pyrimidine dimers; 6-4 PPs, pyrimidine (6-4) pyrimidone photoproducts. The melanin-dependent chemiexcitation pathway (middle panel) was recently described by Premi et al.56 It describes the formation of DNA photoproducts not by direct excitation of DNA, as in the classic pathway, but by energy transfer from excited melanin fragments to DNA, which involves nitric oxide (NO∗), superoxide (SO∗–), and peroxynitrite (ONOO–). This mechanism generates UV-signature mutations both after irradiation with UVA and with UVB, and contributes to mutation formation in melanocytes and melanin-containing keratinocytes. The oxidative pathway (right panel) involves a photosensitized reaction with formation of singlet oxygen after excitation of a cellular chromophore other than DNA. Singlet oxygen then reacts with guanine either in DNA or in the nucleotide pool. A common oxidative base lesion is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG). By mispairing with adenine, G → T and A → C transversions can form. UVA has long been thought to contribute to UVR-induced mutation load through this mechanism. However, molecular evidence, in particular the low frequency of such transversions in melanomas, does not support a major role in the formation of skin cancer.
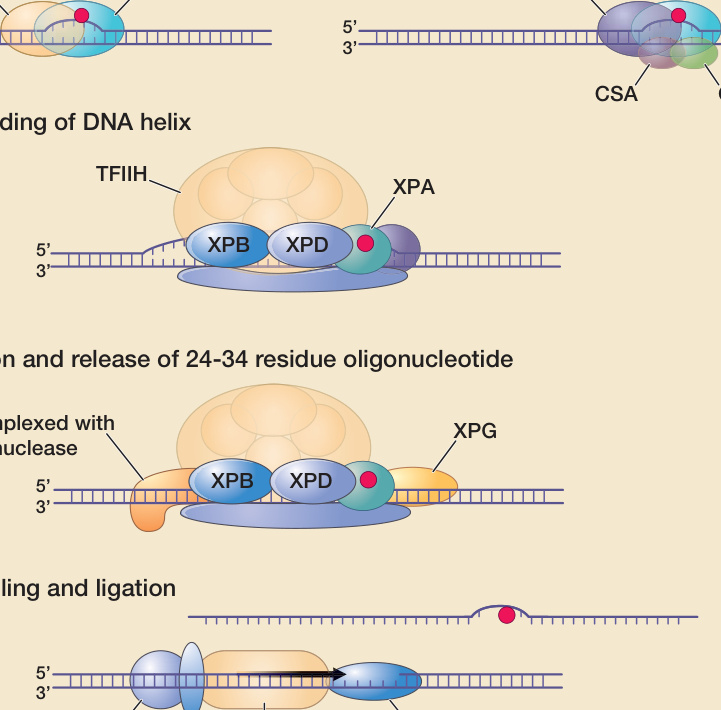
Figure 17-12 DNA nucleotide excision repair scheme. (A) Right: Damaged DNA in actively transcribed genes results in stalling of the RNA polymerase in a process that involves the CSA and CSB proteins. This serves as a signal to initiate transcription-coupled DNA repair. Left: Damaged DNA in the remainder of the genome is bound by the XPE and XPC gene products. This serves as a signal to initiate global genome repair. (B) A portion of the DNA, including the damage, is unwound by a complex of proteins including the XPB and XPD gene products. These proteins are also part of the 10-subunit basal transcription factor IIH (TFIIH). The XPA protein may stabilize the unwound DNA. (C) The XPF and XPG nucleases make single-strand cuts on either side of the damage, releasing a 24- to 34-residue segment of DNA. (D) The resulting gap is filled by DNA polymerase in a process that includes the proteins proliferating cell nuclear antigen (PCNA) and replication protein A (RPA). CSA, CSB, Cockayne syndrome complementation groups A and B; ERCC1, excision repair cross-complementing gene 1; LIG1, ligase 1; XPA, XPC, etc, xeroderma pigmentosum complementation groups A, C, etc.
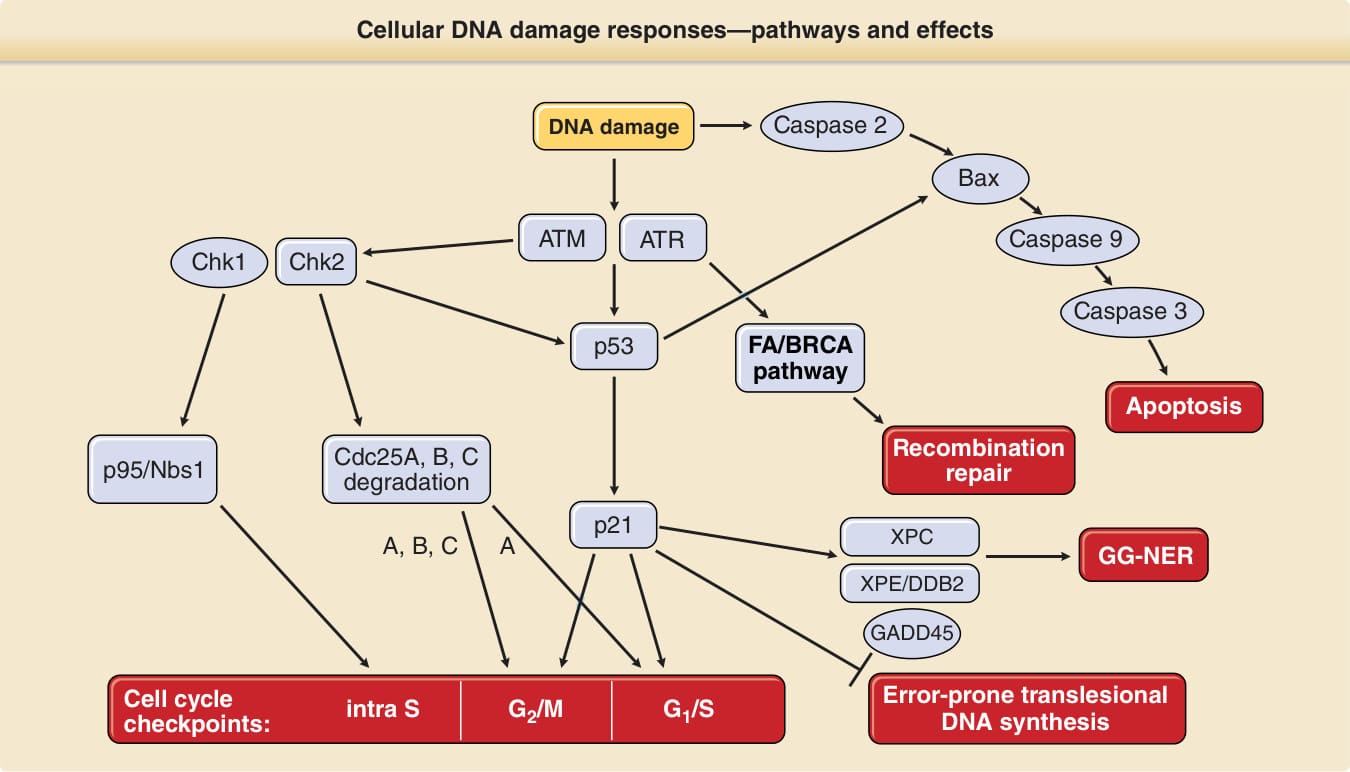
Figure 17-13 Signaling cascades that regulate apoptosis, DNA repair, translesional DNA synthesis, and activation of cell cycle checkpoints in response to UVR-induced DNA damage. This is a highly simplified diagram that depicts only the most important players in the intricate and interwoven DNA damage–signaling networks. The traditional thinking was that ATM (ataxia telangiectasia mutated gene) is activated (phosphorylated) in response to ionizing radiation (IR) and ATR (ataxia telangiectasia- and Rad3-related gene) in response to ultraviolet (UV) radiation, but newer data indicate that both are activated by IR and UV. ATM/ATR can activate (ie, phosphorylate) p53 either directly or indirectly through activation (phosphorylation) of Chk2 (an ATM target) or Chk1 (an ATR target). Through transcriptional activation of p21 and subsequent inhibition of cyclin-dependent kinases, which usually drive cells from the G1 into the S phase of the cell cycle, activated p53 activates the G1/S checkpoint (ie, arrests cells in G1). The G1/S checkpoint is also activated by Chk1/Chk2- induced phosphorylation and then degradation of Cdc25A and subsequent failure to activate cyclin-dependent kinases. Phosphorylation and subsequent degradation of Cdc25A, Cdc25B, and Cdc25C also mediate the G2/M arrest, as does p21. Intra–S phase arrest is mediated through activation (phosphorylation) of p95/Nbs1. p53 also induces global genome nucleotide excision repair (GG-NER) through transcriptional activation of XPC, XPE/p48, and GADD45. Translesional DNA synthesis has been shown to be downregulated by p53 via p21. Recombination repair is mediated through the Fanconi anemia (FA)/BRCA pathway, which in turn is dependent on ATR activation. The mitochondrial pathway of apoptosis is activated through activation of Bax by caspase 2 and p53, the initiator caspase 9, and the effector caspases 3, 6, and 7. Gene products that are defective in hereditary human diseases are boxed. DDB2, DNA damage binding protein 2; XPC, XPE, xeroderma pigmentosum complementation groups C and E.
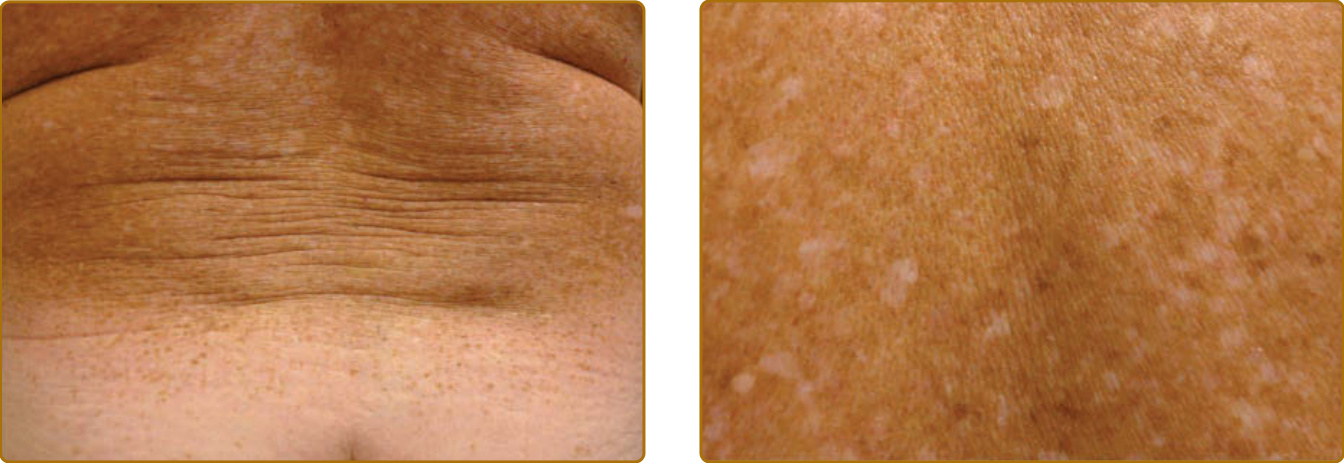
Figure 17-14 Pigmentary changes of photoaged skin include freckles, which are the result of clonal melanocyte proliferation events, and hypopigmentations, which are melanocyte loss-of-function events.
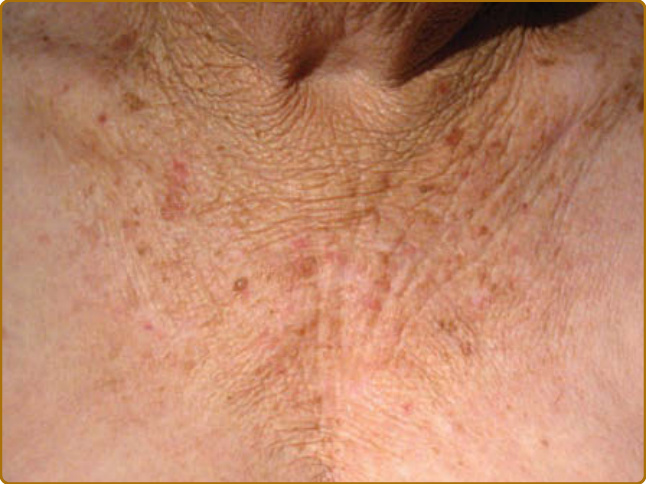
Figure 17-15 Deep wrinkling, in chronically photoaged skin due to actinic elastosis. Note the absence of such wrinkling in the more photoprotected areas on the lateral aspects of the chest.
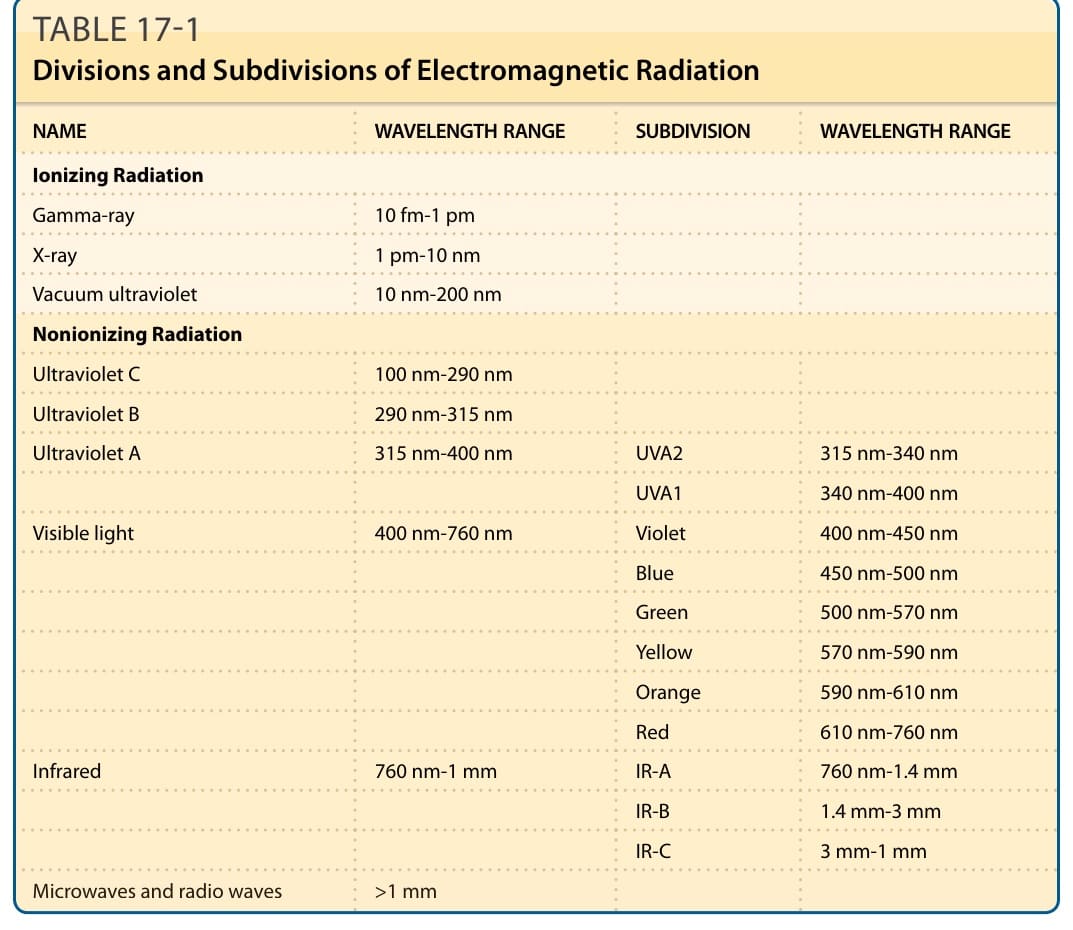
TABLE 17-1 Divisions and Subdivisions of Electromagnetic Radiation
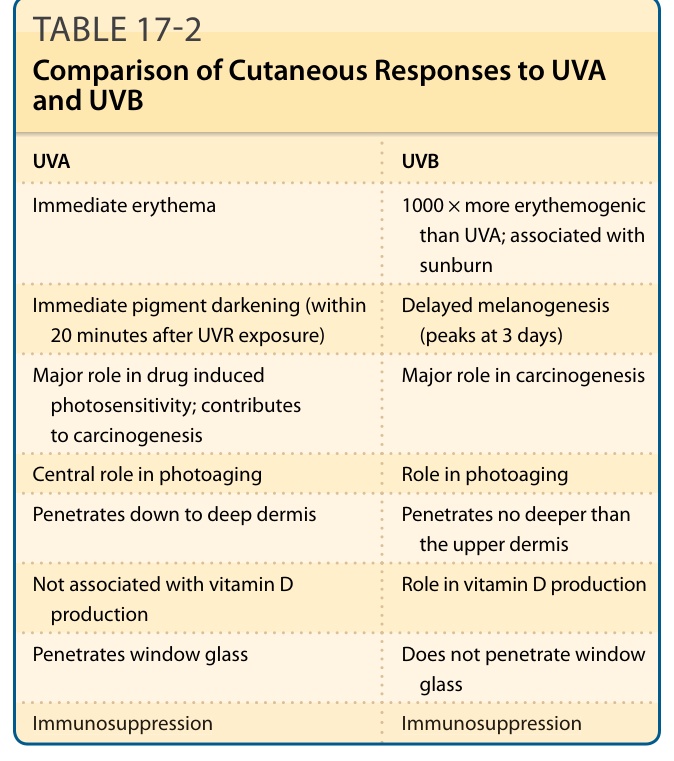
TABLE 17-2 Comparison of Cutaneous Responses to UVA and UVB
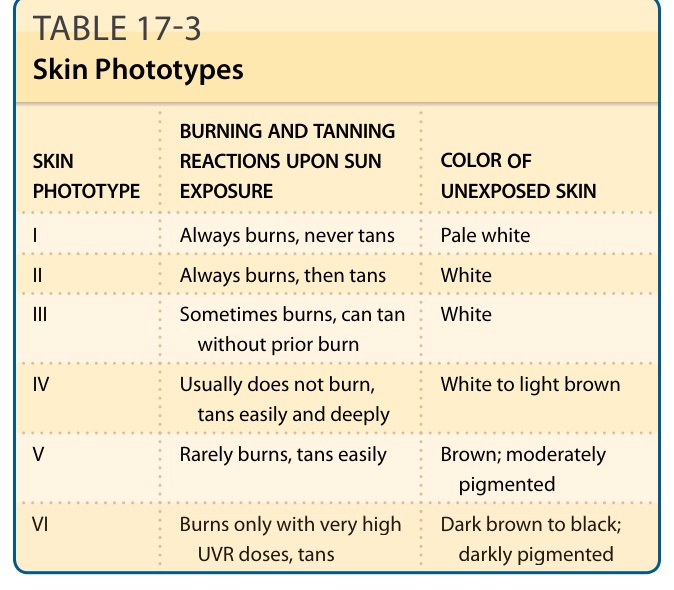
TABLE 17-3 Skin Phototypes
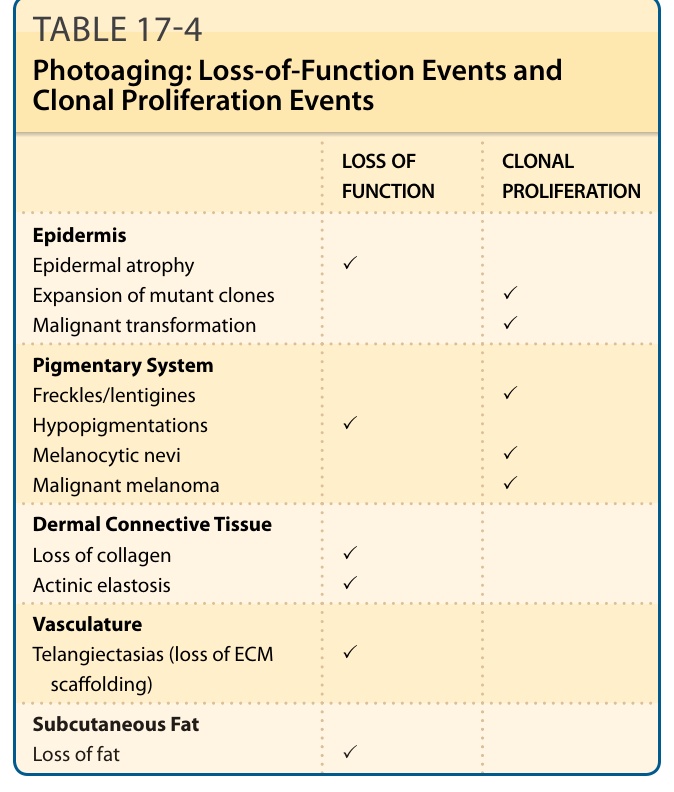
TABLE 17-4 Photoaging: Loss-of-Function Events and Clonal Proliferation Events