皮膚光生物學 (Cutaneous Photobiology)
重點一覽 (AT-A-GLANCE)
■ 輻射唯有先被皮膚中的某個分子(發色團 chromophore)吸收,才能引起光生物學反應。
■ 了解輻射暴露的光物理 (photophysical)、光化學 (photochemical) 與光生物學 (photobiologic) 效應,有助於理解其隨波長而異的後果,包括曬傷 (burning)、曬黑 (tanning)、光皮膚病 (photodermatoses)、皮膚癌的形成、光老化 (photoaging),以及光照治療 (phototherapy) 的效應。
■ 光反應 (photoreactions) 具有特定的作用光譜 (action spectra),取決於各種不同的內源性與外源性發色團的吸收特性。
■ 皮膚維生素 D 的生成由 UVB 範圍內的波長所介導。最佳的維生素 D 血中濃度對良好的骨骼健康至關重要,並日益被認為與其他眾多潛在的健康益處有關。
■ 紫外線輻射 (ultraviolet radiation, UVR) 所造成的 DNA 損傷介導了曬傷、曬黑與皮膚癌的形成。
■ 皮膚細胞配備有許多損傷反應途徑 (damage response pathways),以限制輻射暴露的負面衝擊,包括數種不同的 DNA 修復機轉。
■ 紫外線輻射所誘導的 DNA 損傷可導致突變的形成。在雙嘧啶 (dipyrimidine) 位點上的 C 變 T 與 CC 變 TT 突變極具紫外線輻射誘導的特徵性,被稱為紫外線特徵性突變 (UV-signature mutations)。此類紫外線特徵性突變可見於 UV 誘導的皮膚癌中。
■ 紫外線輻射兼具促發炎與抗發炎特性。
■ 光老化影響皮膚的所有層次,其特徵為克隆性增生事件 (clonal proliferative events) 與功能喪失事件 (loss-of-function events) 兩者並存。
■ 光老化大致上是不可逆的,部分原因可能是 UV 誘導之不可降解異常蛋白質在細胞外與細胞內空間中的累積。
■ 暴露於可見光 (visible light) 與紅外線輻射 (infrared radiation) 也具有光生物學後果,包括紅斑 (erythema)、曬黑,以及細胞外基質蛋白 (extracellular matrix proteins) 的降解。
■ 合理的光照治療與光防護 (photoprotection) 即建立在這些見解之上。
前言 (INTRODUCTION)
作為人體的最外層,皮膚大量暴露於具破壞性的環境因子,包括不同類型的輻射(表 17-1 與圖 17-1)。游離性電磁輻射 (ionizing electromagnetic radiation),例如 X 光 (x-rays) 或伽瑪射線 (gamma rays),攜帶足夠的光子能量 (photon-energy),能將電子從原子或分子中完全移除(即游離 ionization)。其他類型的游離輻射為 α 粒子 (alpha particles)(2 個質子與 2 個中子)與 β 粒子 (beta particles)(電子)。α 與 β 粒子並非電磁波譜的一部分;它們是高能粒子,相對於電磁輻射的純能量束(光子 photons)。非游離輻射 (Nonionizing radiation),包括紫外線輻射 (ultraviolet radiation, UVR)、可見光 (visible light) 與紅外線輻射 (infrared radiation),能將電子移動到較高的能量狀態,但與游離輻射相反,無法將電子從原子或分子中移除。皮膚光生物學 (Cutaneous photobiology) 是研究非游離輻射與皮膚交互作用的科學。「光 (light)」一詞通常專指人眼可感知的電磁波譜波長:可見光。游離與非游離輻射對皮膚都有有害效應。由於在地球上生命的發展過程中,電磁輻射始終存在,因此細胞與生物體唯有發展出對抗輻射破壞性效應的保護機轉才得以演化。本章涵蓋 UVR、可見光與紅外線輻射對皮膚的效應。對光物理、光化學與光生物學過程的見解,有助於了解皮膚暴露於這些類型輻射的臨床表現,包括曬傷、曬黑、皮膚癌的形成、光老化、光皮膚病,以及對皮膚疾病的光照治療效應。
紫外線與可見光輻射的來源 (SOURCES OF ULTRAVIOLET AND VISIBLE RADIATION)
陽光 (SUNLIGHT)
到達地球表面的太陽電磁輻射其最短波長約為 290 nm,儘管在較高海拔處可偵測到略短的波長。地球大氣層(尤其是臭氧 ozone)對 UVC(100 至 290 nm)的過濾作用,被廣泛認為是地球生命發展的關鍵要素,因為較短波長的 UVR 對動物與植物具有高度破壞性。UVR 的細分(表 17-1 與圖 17-1)以及整個電磁輻射波譜的細分相當任意,重要的是要認識到光子的光物理特性在所定義的範圍之間並非突然改變,而是隨波長變化逐漸改變。一般而言,能量隨波長增加而減少。例如,一個 300-nm 的 UVB 光子具有 600-nm 橙光光子兩倍的能量。最常用的 UVR 細分是由國際照明委員會(Commission Internationale de l’Eclairage, CIE)所定義者,其設定 315 nm 為 UVB 與 UVA 之間的界線。其他分類則使用 320 nm。紫外線與可見光輻射之間所定義的閾值,是基於人眼的特性及其感知輻射的能力。某些物種,例如鳥類、昆蟲與魚類,也能看到部分 UVA 輻射。到達地球表面的太陽輻射組成會隨一天中的時間而變化,因為陽光的角度改變,以及太陽輻射穿越大氣層所需行進的長度有所差異。較短波長的可見光(紫光與藍光)比較長波長的可見光(橙光與紅光)更會被穿越大氣層較長路徑所過濾,因此相較於正午陽光,日出與日落時的光線較為偏紅。同樣地,較長波長的 UVR(UVA,315 至 400 nm)比 UVB(280 至 315 nm)更能穿透物質。因此,UVA 相對於 UVB 的比例會隨太陽輻射角度的降低而增加。在正午時,自然陽光中所含的 UVA 約為 UVB 的 50 倍。雖然這兩種成分在一天的早晨或傍晚都較不豐富,但 UVA 與 UVB 之間的比例會隨太陽角度的下降而增加。同樣地,UVA 可穿透窗戶玻璃,但 UVB 不能。
人工光源 (ARTIFICIAL SOURCES)
有種類繁多的不同光源會使皮膚暴露於可見光、紫外線與紅外線輻射。這些光源涵蓋用於傳統照明用途、工業應用、光照治療與曬黑的燈具。大多數光源被設計為僅發射所需範圍的波長,例如僅供照明用途的可見光。儘管如此,某些一般照明燈具有時會發射少量 UVR,特別是螢光燈泡 (fluorescent bulbs) 與鹵素燈 (halogen lamps)。不需要的波長常以濾光片 (filters) 消除。被設計用以發射 UVR 的螢光燈泡最常發射廣範圍的波長。例如,被設計用以發射 UVA 供光化學治療(photochemotherapy, PUVA)的燈泡也會發射一些 UVB,而被設計用以發射 UVB 的燈泡也會發射一些 UVC。飛利浦 TL01 螢光燈(Phillips TL01 fluorescent lamp)是光照治療用 UVB 燈設計上的一大進展,因為它將其幾乎單色 (monochromatic) 的發射對準於 311 至 312 nm,兼具對發炎性皮膚疾病的治療效力,同時避開了主要造成曬傷、但對治療無效的較短波長。
光物理、光化學與光生物學:電磁輻射與皮膚交互作用的原理 (PHOTOPHYSICS, PHOTOCHEMISTRY, AND PHOTOBIOLOGY: PRINCIPLES OF THE INTERACTION OF ELECTROMAGNETIC RADIATION WITH SKIN)
要使一個光子產生具生物學意義的後果,必須發生一連串事件(圖 17-2)。皮膚的光學因子與輻射的波長決定了一個光子在皮膚表面及皮膚內部被反射與散射的程度與位置。對於紫外線與可見光輻射,較長的波長比較短的波長更能穿透物質。例如,UVA 比 UVB 更深入真皮,而 UVB 中只有一小部分穿透至比表皮更深處。UVC 的穿透深度最小,在皮膚中只到達角質層 (stratum corneum) 與表皮的上層(圖 17-3)。光化學第一定律,又稱為 Grotthuß–Draper 定律(以化學家 Freiherr Christian Johann Dietrich Theodor von Grotthuß 與 John William Draper 命名),陳述光必須先被某一化學物質吸收,光化學反應才能發生。吸收輻射的化合物稱為發色團 (chromophore)。每一個發色團都有其特徵性的吸收光譜,通常具有一個最可能將其激發的波長(即吸收極大值 absorption maximum),以及一個較不可能將其激發的波長分布。皮膚中特徵明確的發色團包括 DNA(吸收極大值在 260 nm)、紫質 (porphyrins)(吸收極大值在 400 至 410 nm 之間),以及黑色素 (melanin)(吸收極大值在 UVC 範圍,但也能被 UVB 與 UVA 非常有效地激發)(圖 17-4)。當處於基態 (ground state) 的發色團吸收一個光子的能量時,電子會被提升到較高的軌道。如果這不涉及自旋 (spin) 的改變,則該激發態稱為單重態 (singlet state)。如果它同時經歷自旋的改變,則該激發態稱為三重態 (triplet state)(圖 17-5)。光激發 (photoexcitation) 後,可能發生光化學反應而形成一個新的、不同的分子,稱為光產物 (photoproduct)。例如,前維生素 D3 (previtamin D3) 是光激發 7-去氫膽固醇 (7-dehydrocholesterol) 的光產物,而環丁烷嘧啶二聚體 (cyclobutane pyrimidine dimer) 則是激發態 DNA 的光產物。能量從激發態發色團轉移到另一分子的過程稱為光敏化反應 (photosensitized reaction)。一個典型且具光生物學意義的光敏化反應,是激發態發色團(例如紫質 porphyrin)將能量轉移給氧並產生單重態氧 (singlet oxygen),而單重態氧接著再與其他受質(例如鳥嘌呤 guanine DNA 鹼基)反應。激發態發色團除了形成光產物外,也可以以熱的形式釋放能量,或以螢光 (fluorescence) 的形式發射一個光子,而回到其基態。螢光的波長總是比激發波長更長(即能量較低)(Stokes 定律)。例如,以藍光激發原紫質 IX (protoporphyrin IX) 時,螢光為紅光,其波長比藍光更長且能量較低。激發單重態是短暫的,所產生的螢光在數奈秒 (nanoseconds) 內發生。三重態存在的時間較長,可長達數秒。從三重激發態回到基態時的光子發射稱為磷光 (phosphorescence),其波長比螢光更長。
皮膚對紫外線輻射的反應 (CUTANEOUS RESPONSES TO ULTRAVIOLET RADIATION)
見表 17-2。
維生素 D 光生物學 (VITAMIN D PHOTOBIOLOGY)
維生素 D 的功能 (FUNCTIONS OF VITAMIN D)
維生素 D 調節鈣與磷的代謝。其主要角色是藉由促進腸道對鈣與磷的吸收,以及腎臟對鈣的再吸收,來增加鈣進入血流的流量,從而使骨骼的正常礦化與肌肉功能得以進行。維生素 D 缺乏會導致骨骼礦化受損,進而引起骨骼軟化疾病——兒童的佝僂病 (rickets) 與成人的骨軟化症 (osteomalacia),並可能促成骨質疏鬆症 (osteoporosis)。¹⁻³ 缺乏可能源自攝取不足、日照不足、限制其吸收的疾病,以及損害維生素 D 轉換為活性代謝物的狀況,例如肝臟或腎臟疾病。最容易出現低維生素 D 濃度者為老年人、居住於冬季漫長之高緯度地區的個體、肥胖者,以及所有居住於高緯度且皮膚色素深的個體。⁴,⁵ 過量維生素 D 的毒性可能表現為高鈣尿症 (hypercalciuria) 或高鈣血症 (hypercalcemia),後者會引起肌肉無力、淡漠、頭痛、意識混亂、厭食、易怒、噁心、嘔吐與骨痛,並可能導致腎結石與腎衰竭等併發症。慢性毒性除上述症狀外,還包括便秘、厭食、腹部絞痛、多飲 (polydipsia)、多尿 (polyuria)、背痛與高血脂 (hyperlipidemia)。表現也可能包括鈣質沉著 (calcinosis),隨後出現高血壓與心律不整(因不反應期縮短所致)。雖然有關高劑量維生素 D 效應的資訊有限,但 10,000 IU/天被認為是成人攝取的安全上限。成人的慢性中毒劑量為每日超過 50,000 IU/day。除增進肌肉骨骼健康外,維生素 D 也可能影響多種其他病況的風險,包括癌症、心血管疾病、自體免疫疾病與感染。然而,這些效應仍存在重大爭議。⁶⁻¹⁶
維生素 D 有兩大來源,一是外源性(飲食),另一是內源性合成。
當從食物或補充劑中獲得時,維生素 D 在小腸被吸收。富含維生素 D 的天然食物來源包括某些油性魚類,如鮭魚 (salmon)、鯖魚 (mackerel)、鮪魚 (tuna)、鯡魚 (herring)、鯰魚 (catfish)、鱈魚 (cod)、沙丁魚 (sardines) 與鰻魚 (eel)、奶油、人造奶油、優格、肝臟、魚肝油與蛋黃,但至少在美國,大多數飲食中的維生素 D 來自於穀片、牛奶與柳橙汁等食物的強化 (fortification)。例如,強化牛奶通常每 250-mL 一杯提供 100 IU,僅為成人估計足夠每日攝取量的四分之一。
維生素 D 生成的生化途徑 (BIOCHEMICAL PATHWAY OF VITAMIN D PRODUCTION)
皮膚暴露於 UV 的結果,前維生素 D3(provitamin D3,7-去氫膽固醇 7-dehydrocholesterol,為膽固醇的前驅物)迅速轉換為前維生素 D3 (previtamin D3),後者自發性異構化 (spontaneously isomerizes) 為維生素 D3,藉由結合蛋白進入循環,並與從腸道吸收的飲食性 D2(或麥角鈣化醇 ergocalciferol)與 D3(膽鈣化醇 cholecalciferol)會合(圖 17-6)。一旦到達肝臟,它便被維生素 D 25-羥化酶(vitamin D 25-hydroxylase)羥化,此過程需要 NADPH、O2 與 Mg2+。產物 25-羥基維生素 D3(25-hydroxyvitamin D3,鈣二醇 calcidiol)儲存於肝細胞中,直到有需要時。釋放至血漿後,它前往腎臟的近端小管 (proximal tubules),在此被 25(OH)D-1α-羥化酶(25(OH)D-1α-hydroxylase)作用,該酵素的活性因副甲狀腺素 (parathyroid hormone) 與低 PO4²⁻ 而增加。腎臟疾病患者可能無法將維生素 D 轉換為其活性形式。在此轉換之後,1,25-羥基維生素 D3(1,25-hydroxyvitamin D3,鈣三醇 calcitriol)被釋放至循環中,並藉由與血漿中的載體蛋白——維生素 D 結合蛋白(vitamin D–binding protein, VDBP)結合,被運送至各個標的器官。MacLaughlin 等人於 1982 年發表的一份作用光譜¹⁷顯示,最有效促進皮膚維生素 D 光合成的波長位於 UVB 範圍,峰值在 297 nm,但這些資料的準確性與有效性近期已受到質疑。¹⁸ 在熱帶地區,皮膚每週兩次、每次 10 至 15 分鐘的日照於臉、手臂與手部,或背部,即可製造足夠量的維生素 D3。皮膚黑色素含量較高的個體需要在陽光下較久的時間,才能製造出與黑色素含量較低個體相同量的維生素 D。有人提出,人類從非洲起源到居住於較北方、較不晴朗緯度的白種人之色素在演化上的喪失,在黑色素的光防護效應不如低緯度地區那麼關鍵的地區,於維生素 D 生成與骨骼健康方面提供了優勢。¹⁹ 在當今室內活動增加、穿著更為遮蔽衣物的生活型態下,維生素 D 缺乏對於有色人種往往是特別的問題,相較於膚色較白的個體,他們需要更多的 UVB 暴露才能在皮膚中製造出相同量的維生素。何謂足夠的維生素 D 濃度仍是個爭論的議題。維持骨骼健康所需的濃度可能較低,而對於其他仍有爭議的健康益處所需的濃度可能較高。儘管如此,為了預防 UVR 暴露之有害效應而持續進行的光防護,無疑可能導致維生素 D 缺乏。²⁰,²¹ 因此,將光防護與維生素 D 補充相結合似乎是明智的選擇,因為飲食來源往往不足以維持適當的維生素 D 濃度。
對免疫系統的效應 (EFFECTS ON THE IMMUNE SYSTEM)
數條觀察性證據線索顯示,UV 輻射兼具促發炎與抗發炎特性。促發炎特性受到下列觀察的支持:曬傷、發炎性光皮膚病的闡明、自體免疫結締組織疾病的誘發與觸發,以及某些病人發炎性皮膚疾病的光加重 (photoaggravation)。抗發炎/免疫抑制效應則受到下列觀察的支持:UVR 暴露使唇部單純疱疹 (labial herpes simplex) 再活化,以及 UV 光照治療對發炎性皮膚疾病的療效。UVR 對免疫系統有深遠的效應,既存在於 UV 暴露的局部部位,也存在於全身性。目前認為,UV 輻射的免疫刺激效應是由先天免疫 (innate immunity) 所介導,伴隨皮膚常駐與非常駐細胞釋放促發炎介質(例如血清素 serotonin、前列腺素 prostaglandins、IL-1、IL-6、IL-8、TNF-α)、抗菌肽 (antimicrobial peptides) 的誘導,以及嗜中性球與淋巴細胞的招募。²² 免疫抑制效應則被認為是由蘭格罕細胞 (Langerhans cells) 從表皮遷移至局部淋巴結、皮膚常駐與非常駐細胞釋放抗發炎介質(例如 IL-10、α-MSH),以及抗原特異性調節性 T 細胞 (antigen-specific regulatory T cells) 的誘導所介導(圖 17-7)。然而,這種促發炎相對於抗發炎的二分法觀點,可能過於簡化。相反地,各種個別因子很可能影響著促發炎與抗發炎反應,導致這兩極之間達成不同的平衡。例如,對大多數乾癬 (psoriasis) 病人而言,暴露於 UV 輻射會使皮膚病灶改善,這展現了 UVR 的抗發炎特性。然而,一小部分病人的乾癬會惡化。這說明對於這類光加重型乾癬 (photoaggravated psoriasis) 的病人而言,UVR 的淨結果是促發炎的。另一個例子是光皮膚病多形性日光疹 (polymorphous light eruption),這很可能是對 UV 誘導之新抗原 (neoantigen) 的一種發炎反應。罹患此病況的病人有較少的 UV 誘導免疫抑制,因此更可能發展出此病況。²³ 另一方面,這也為他們提供了一些對抗光致癌作用 (photocarcinogenesis) 的保護,這很可能是由於對 UV 誘導之腫瘤抗原有更穩健的免疫反應所致。²⁴
UV 誘導免疫抑制的有益效應,很可能是預防對抗因 UV 誘導細胞損傷而暴露之自體抗原 (self-antigens) 的自體免疫反應。未能預防此類自體免疫反應的一個例子是紅斑性狼瘡 (lupus erythematosus),UV 暴露後不僅使皮膚病灶加重,還會觸發新病灶,甚至引起全身性惡化。從動物實驗中已充分確立,UV 輻射的免疫抑制效應不僅是局部的(即限於 UV 暴露的皮膚),也是全身性的,亦會影響未暴露的皮膚。這些效應已被證明是由 IL-10 及其隨後對調節性 T 細胞的誘導所介導。紅斑性狼瘡的光加重與光觸發 (phototriggering) 可被視為未能適當下調免疫反應的結果。光免疫抑制的負面之處在於,它阻止了對 UV 誘導腫瘤抗原的辨識,並因此促進光致癌作用。除了免疫抑制外,UV 輻射也誘導對 UV 暴露後所生成及/或暴露出之抗原的長久耐受性 (tolerance)。因此,UV 輻射對於皮膚癌的發展可被視為一把三刃劍:它造成致突變的 DNA 損傷,不僅抑制對新腫瘤抗原的辨識,還誘導對這些抗原的耐受。這些對免疫系統效應的發色團包括:形成 DNA 損傷的 DNA、形成 RNA 損傷的 RNA、發生 UV 誘導從反式 (trans-) 到順式 (cis-) 異構化的尿刊酸 (urocanic acid),以及光激發後產生活性氧物種 (reactive oxygen species) 並隨後改變膜脂質氧化還原電位 (redox potential) 的紫質。雖然大多數免疫抑制效應由 UVB 所造成,但 UVA 也被證明具有類似的效應。²⁵
曬傷 (SUNBURNING)
曬傷(日光性紅斑 solar erythema)的臨床特徵為紅斑與伴隨疼痛的灼熱感(第一級 grade 1),並可進展為水疱形成(第二級 grade 2),但不會進展至第三級,至少在太陽可提供的劑量下不會。它通常在曬太陽後 6 至 24 小時達到高峰,隨後出現曬黑與脫屑。組織學上,其特徵為伴隨海綿狀水腫 (spongiosis) 的表皮水腫、少數凋亡的角質細胞(曬傷細胞 sunburn cells)、蘭格罕細胞的耗竭、血管擴張,以及伴有淋巴細胞與嗜中性球的混合性發炎浸潤。UV 誘導紅斑的作用光譜顯示,紅斑形成在 300 nm 達到高峰(位於 UVB 範圍內),然後隨波長增加進入 UVA 範圍而呈指數下降。²⁶ 雖然非常高劑量的 UVA 可誘導紅斑,但非光敏感的個體不會因太陽可提供劑量的 UVA 而產生紅斑。UVC 雖然不存在於到達地球表面的太陽輻射中,但也能誘導曬傷。由於紅斑作用光譜與直接 UV 誘導之嘧啶二聚體型 DNA 損傷形成的作用光譜非常相似,曬傷反應的發色團被認為是 DNA。²⁷,²⁸ 這進一步得到下列觀察的支持:許多在此類損傷修復方面有缺陷的病人(著色性乾皮症 xeroderma pigmentosum)具有急性光敏感性,並有降低的 UVB 最小紅斑劑量 (minimal erythema dose)²⁹,³⁰,以及來自動物實驗的類似觀察,其中嘧啶二聚體修復的改善可降低光敏感性。³¹,³²
具有 UV 誘導 DNA 損傷的暴露皮膚細胞會啟動各種損傷反應途徑,其中某些會導致預先形成的(例如血清素與 TNFα)或新合成的發炎介質(例如前列腺素、一氧化氮與神經肽 neuropeptides)的分泌。³³
曬黑 (TANNING)
皮膚的變黑(曬黑 tanning)是曬傷的一個特徵性後果,但也可在沒有先前曬傷的情況下被觀察到(圖 17-8)。它在 UVB 暴露後約 3 天達到高峰,組織學上的特徵為表皮中基底層與基底上層黑色素的增加。對曬傷的敏感性與曬黑的能力在人類之間差異很大,並已根據基礎色素沉著、對曬傷的敏感性與曬黑能力,被歸類為皮膚光型 (skin phototypes)(表 17-3)。此分類方案由 Thomas B. Fitzpatrick 於 1975 年針對白皮膚所發展³⁴,此後已延伸至較深的皮膚類型。2015 年,計算機產業標準 Unicode 引入了 Emoji 修飾符,根據 Fitzpatrick 的皮膚光型量表來指定膚色以代表多樣性,這凸顯了膚色認知即使在大眾文化中也無所不在。個體對曬傷的敏感性可藉由測定最小紅斑劑量 (minimal erythema dose) 來衡量。這是在個體身上引起紅斑的最低劑量,它與臨床判定的皮膚光型相關,儘管相關性不佳。³⁵ 皮膚光型雖以對 UVR 的急性反應為基礎,但也是個體對 UVR 長期效應(特別是光致癌作用)風險的指標。曬黑的作用光譜幾乎與 DNA 光產物形成的作用光譜相同³⁶,²⁸,這一事實表明 DNA 是 UVR 誘導曬黑的發色團,與紅斑形成非常相似(見上文),且細胞對 DNA 損傷的反應對於介導曬黑反應至關重要。黑色素合成的增加取決於 UV 誘導之 p53 的誘導與活化,p53 是細胞 DNA 損傷反應的核心參與者。³⁷⁻³⁹ 黑素細胞 (melanocytes) 所製造的黑色素,透過涉及特化馬達蛋白 (motor-proteins) 的主動過程被轉移到角質細胞,並在角質細胞中優先被置於細胞核上方,形成微型陽傘 (microparasol)。⁴⁰ 藉此,黑色素的吸收 UV 特性使細胞核免於後續 UV 暴露之 DNA 損傷效應。為了回應 UV 誘導的發炎(曬傷),表皮角質細胞過度增生,這導致活表皮 (viable epidermis) 與角質層的增厚。這種增厚減少了 UVR 對基底層的穿透,並因此也減少了皮膚幹細胞中 DNA 損傷與突變的形成。這兩種保護機轉——色素沉著增加與皮膚增厚——連同分子適應(例如 DNA 修復酵素活性的增加),共同造成紅斑閾值數倍的增加。⁴¹ 除了這種主要由 UVB 誘導的延遲性曬黑外,還有一種立即性色素變深 (immediate pigment darkening),可在 UVR 暴露後 20 分鐘內被觀察到。後者由既存黑色素的氧化與重新分布所介導,主要由 UVA 誘導,對後續暴露提供的保護少得多(如果有的話)。
光皮膚病的光生物學原理 (PHOTOBIOLOGIC PRINCIPLES OF PHOTODERMATOSES)
上述發色團在光生物學反應發生之前必須先被激發的要求,也適用於光誘導 (photo-induced) 或光觸發 (photo-triggered) 皮膚病。對某些光皮膚病而言,發色團是已知的,對其他則否。例如,血基質生合成途徑 (heme biosynthesis pathways) 的缺陷會導致毒性途徑中間產物的累積,並引起紫質症 (porphyrias)。血基質生合成途徑早期階段的缺陷會導致非芳香族中間產物的累積,這些中間產物不作為發色團/光敏化劑。這些紫質症的特徵並非皮膚的光毒性反應,而是影響其他器官,例如肝臟(如急性間歇性紫質症 acute intermittent porphyria)。血基質生合成途徑較晚階段的缺陷則導致較大的芳香族中間產物累積,這些中間產物作為發色團。暴露於 UVR 或可見光時,這些發色團的激發會引起光毒性反應(例如表皮下水疱),此反應是由激發態發色團向氧的能量轉移(光敏化反應)與單重態氧的形成所介導(例如在遲發性皮膚紫質症 porphyria cutanea tarda 或紅血球生成性原紫質症 erythropoietic protoporphyria 中)。這些中間產物(例如遲發性皮膚紫質症中的尿紫質原 uroporphyrinogen 與紅血球生成性原紫質症中的原紫質 protoporphyrin)的吸收極大值在 400 至 410 nm 之間,位於 UVA 與可見藍光之間的交界處。因此,受影響的病人不僅需要保護自己對抗 UVA,也需保護對抗可見光,以預防光毒性反應。不同的皮膚紫質症呈現不同的症狀。這很可能是由於所累積中間產物水溶性的差異以及隨之而來的組織分布差異。水溶性的尿紫質原可自由瀰漫至整個皮膚,並在最接近光源處(表皮交界處 epidermal junction)誘發大部分的光毒性,導致水疱形成。在紅血球生成性原紫質症中,親脂性高得多的原紫質滯留於內皮襯裡 (endothelial lining),並造成微血管損傷,隨後引起真皮壞死與疼痛。在藥物誘發的光毒性 (phototoxic) 與光過敏性 (photoallergic) 皮膚炎中,發色團為藥物或藥物的代謝物。此類光敏化藥物是吸收於 UVA 範圍內的芳香族分子。因此,光毒性與光過敏性藥物反應幾乎完全由 UVA 所誘發。光植物性皮膚炎 (Photo-phytodermatitis) 是由植物性發色團(即呋喃香豆素 furocoumarins)所介導的光毒性反應。這些分子與補骨脂素 (psoralens) 相關,其吸收極大值在 UVA 範圍。佛手柑皮膚炎 (Berloque dermatitis) 也是一種光毒性皮膚炎,其中發色團是香水成分佛手柑油(bergamot oil,5-甲氧基補骨脂素 5-methoxy psoralen)。多形性日光疹與日光性蕁麻疹 (solar urticaria) 在所涉及的發色團方面很可能是異質性病況,因為誘發波長因病人而異,範圍從 UVB 到 UVA、可見光,甚至可能到紅外線輻射。⁴²⁻⁴⁴
光皮膚病的診斷工具包括:分別測定 UVA 與 UVB 的最小紅斑劑量(UVA-MED 與 UVB-MED)、光貼布試驗 (photopatch testing),以及尋找潛在的發色團(例如血液、尿液與糞便中升高的紫質)。辨識誘發光皮膚病的波長,對於為每位個別病人量身打造適當的光防護策略而言很重要。
光照治療的光生物學原理 (PHOTOBIOLOGIC PRINCIPLES OF PHOTOTHERAPY)
電磁輻射的治療效應被用以治療各種皮膚疾病,可與光敏化劑 (photosensitizer) 合併使用,亦可不使用。對大多數光照治療中心而言,乾癬是光照治療最常見的適應症。1981 年,Parrish 等人⁴⁵發表了一份關於 UVR 治療此一發炎性皮膚疾病之治療效應的作用光譜。在 UVC 範圍或以 280 或 290 nm 時無治療效應,在約 300 nm 處有最大效應,且治療效力隨波長從 300 nm 增加至 UVA 範圍而呈指數下降——這與上述的紅斑作用光譜相似,並與表皮基底層中嘧啶二聚體型 DNA 損傷形成的作用光譜重疊。²⁷ 這顯示介導抗乾癬治療效力的發色團是 DNA。在當時,乾癬主要被理解為一種由表皮過度增生所驅動的病況,因此認為 UV 誘導的嘧啶二聚體會阻斷過度增生角質細胞的複製,並藉此改善乾癬。然而,此後已證明表皮過度增生的減緩是 UVB 對乾癬病灶的較晚期效應,而非主要的治療效應。如今,UVB 的治療效應被認為是由上述的免疫抑制效應所介導。雖然確切機轉仍待釐清,但過度增生的活化 T 細胞可能特別容易受到 UVB 的細胞毒性、促凋亡效應的影響。免疫偏移 (immune-deviation) 的誘導(例如將 TH1 與 TH17 淋巴細胞轉變為 Tregs)可能參與其中。⁴⁶ UVB 的穿透僅限於表皮與真皮上層,但無法到達真皮較深的層次,而這些層次在厚的乾癬斑塊中也常被免疫細胞浸潤。亦不清楚浸潤皮膚的淋巴細胞或其他免疫細胞是否為介導 UVB 光照治療療效的主要標的,或這些細胞是否受到來自 UV 暴露角質細胞之訊號傳遞的影響。上述針對乾癬治療效應的作用光譜,已促成窄波段 (narrowband, nb) UVB 光照治療的發展,使用發射 311 至 312 nm 之間幾乎單色 UVB 的燈泡。此種模式已被證明遠比先前使用的寬波段 (broadband, bb) UVB 光照治療更有效。這可能是由於避開了無治療效力或僅有極少治療效力的促紅斑性 (pro-erythemogenic) 短波長 UVB(280-290 nm),並因此能使用比 bbUVB 顯著更高的劑量。此外,較高比例的 UVB 波長穿透至真皮較深處,也可能促成更有效地觸及標的——即真皮的發炎浸潤。如今,nbUVB 不僅在乾癬的治療上取代了 bbUVB,也在所有其他 UVB 光照治療的適應症上取而代之,例如異位性皮膚炎 (atopic dermatitis)、白斑 (vitiligo)、皮膚 T 細胞淋巴瘤 (cutaneous T-cell lymphoma) 等。對於這些適應症,其光照治療的作用機制比乾癬更不為人所知。nbUVB 在所有這些適應症上都被證明優於 bbUVB,這一事實可能暗示其作用機制與乾癬中的相似。然而,雖然這些病況中大多數的致病機轉是發炎性的,但確切的發炎病理機轉當然相當多樣。對於單一治療模式(nbUVB 光照治療)如何能改善如此多樣的一組皮膚疾病,仍有許多有待學習之處。粗煤焦油 (Crude coal tar) 是一種強效光敏化劑,使暴露的皮膚對 UVR 誘導的紅斑非常敏感。在 bbUVB 只有部分效果的時代,使用焦油合併 bbUVB 是一種常見的做法,例如以首位描述者 William H. Goeckerman 命名的治療方案。焦油是許多不同化合物的混合物,其中許多具有光敏化特性。隨著 nbUVB 作為單一療法即高度有效,使用焦油作為光敏化劑已成為一種不常見的做法。相對地,結合補骨脂素 (psoralen) 全身性或局部投予並隨後以 UVA 照射的光化學治療 (photochemotherapy),仍是一種常見的光照治療模式,儘管如今的使用遠少於 nbUVB 引入之前的時代。補骨脂素(最常使用者為 8-甲氧基補骨脂素 8-methoxy-psoralen)嵌入 (intercalate) DNA,並在光激發後與嘧啶鹼基反應,形成單價與雙價的股間 (inter-) 與股內 (intrastrand) 交聯。對 PUVA 治療而言,發色團是 8-MOP,而補骨脂素加成物 (adducts) 是 UVA 誘導的光產物。8-MOP 的吸收極大值在 248 與 301 nm,位於 UVC 與 UVB 範圍。然而,其吸收曲線相當寬廣,UVA 仍能有效激發此分子,並也相當有效地產生交聯。8-MOP 的光化學相當複雜。嵌入 DNA 後,吸收極大值為 313 nm,而在形成單加成物 (monoadduct) 後,吸收極大值移至 334 nm,位於 UVA 範圍。這些交聯是複雜的 DNA 損傷,只能藉由 DNA 重組 (DNA recombination) 來修復,而無法像嘧啶二聚體那樣由核苷酸切除修復 (nucleotide excision repair) 修復。類似於上述較早期認為 UVB 誘導 DNA 損傷的阻斷複製特性介導抗增生並藉此抗乾癬效應的觀念,同樣的觀念也曾被提出用於 PUVA 誘導的交聯。⁴⁷ 如同 UVB 的情況,這一觀念此後也已被推翻。PUVA 治療的高療效可能可由 UVA 較深穿透至真皮及其更能觸及較深發炎浸潤來解釋。由於 UVA 的能量遠較低,產生的嘧啶二聚體遠少於 UVB,因此很可能需要添加補骨脂素來將 UVA 的 DNA 損傷效應提升至治療範圍。規避 UVA 有限生物效應的一種方法就是單純增加其劑量。此一原理已被用於發展不使用光敏化劑的高劑量 UVA1 光照治療。此治療已被證明對急性異位性皮膚炎及硬化性病況(例如硬斑病 morphea)的治療具有一些療效。介導此效應的發色團尚未被辨識,此類光照治療的確切作用機制也仍待釐清。另一種採用光敏化劑的光照治療模式是光動力療法(photodynamic therapy, PDT)。在美國,最常用以使皮膚對 PDT 光敏化的製劑是 5-胺基乙醯丙酸(5-amino-levulinic acid, 5-ALA)。然而,5-ALA 並非介導 PDT 治療效應的發色團。5-ALA 的施用規避了血基質生合成途徑中 5-ALA 合成的限速步驟,並導致細胞內原紫質 IX (protoporphyrin IX) 含量的增加。此芳香族分子的吸收極大值約在 406 nm,即所謂的 Soret 帶 (Soret band)。它易被此種藍光激發,激發後與氧反應形成單重態氧。因此,以 5-ALA 進行的 PDT 使用藍光,其效應被認為是由單重態氧的細胞毒性效應所介導。PDT 對癌前與惡性皮膚腫瘤的破壞,可能是由於癌前或惡性細胞的完全破壞,或更可能是由於這些細胞中部分的破壞並伴隨腫瘤抗原的暴露,隨後對其餘細胞進行免疫介導的破壞。至少部分的腫瘤選擇性可能是由癌前與惡性細胞對 5-ALA 更有效的攝取所介導。PDT 有許多不同的治療模式,因施用途徑、光敏化劑的性質與所使用的波長而異。無論在哪種情況下,所使用的波長都必須謹慎選擇,不僅要匹配光敏化劑的吸收特性,也要促進足夠的穿透。以 5-ALA 與藍光進行的 PDT 受限於藍光的穿透深度。在歐洲,最常用的皮膚導向 PDT 模式使用甲基胺基乙醯丙酸(methyl aminolevulinic acid, MAL)合併紅光。MAL 具有較佳被細胞攝取的優勢,因為相較於 5-ALA,它具有更高的親脂性。然而,所產生的發色團與 5-ALA 相同。雖然原紫質 IX 最有效地被藍光激發,但其吸收曲線在綠光、橙光、黃光與紅光範圍顯示幾個小峰(Q 帶 Q-bands),這些仍能以紅光提供足夠的光激發。MAL 相較於 5-ALA 的較深穿透,與紅光相較於藍光的較深穿透相結合,可能使此 PDT 模式對較厚的皮膚病灶(例如浸潤性皮膚癌)更為有效。PDT 不限於常規的廣譜可見光光源,也可使用來自各種不同雷射光源的單色輻射,而使用雷射進行 PDT 的研究目前正在進行中。當前 PDT 實務的其他改良涉及發展具有不同光吸收特性的其他光敏化劑。然而,當前 PDT 的精妙之處在於,它使用一種容易被細胞攝取的小分子,並隨後導致細胞內光敏化劑生成的增加。這規避了光吸收分子通常較大且為芳香族、因此較不易被細胞內化的問題。
光致癌作用 (PHOTOCARCINOGENESIS)
皮膚慢性暴露於紫外線會增加多種不同皮膚惡性腫瘤的風險,包括基底細胞癌 (basal cell carcinoma)、鱗狀細胞癌 (squamous cell carcinoma)、惡性黑色素瘤 (malignant melanoma) 與梅克爾細胞癌 (Merkel cell carcinoma)。光致癌作用是 UVR 暴露皮膚細胞中一連串光物理、光化學與光生物學事件的結果(圖 17-9)。
UVR 造成的 DNA 損傷與突變的形成 (FORMATION OF DNA DAMAGE AND MUTATIONS BY UVR)
DNA 的吸收極大值在 260 nm,位於 UVC 範圍內。UVB 仍能強烈激發 DNA,且穿透表皮的深度比 UVC 更深,因此在基底角質細胞與黑素細胞中誘導更多的光化學反應。直接 UVR 誘導之 DNA 光激發的一個關鍵後果,是在兩個相鄰嘧啶鹼基——胸腺嘧啶 (thymine) 與胞嘧啶 (cytosine)——之間形成共價化學鍵。UVR 所形成的此類 DNA 光產物有兩種主要類型:順式-順式環丁烷嘧啶二聚體(cis-syn cyclobutane pyrimidine dimers, CPDs)與嘧啶-嘧啶酮光產物(pyrimidine-pyrimidone photoproducts, 6-4 PPs)。這兩種類型的二聚體都可在胸腺嘧啶與胞嘧啶的任何組合之間形成——T:T、T:C、C:T 或 C:C(圖 17-10)。未修復的 CPDs 或 6,4-嘧啶–嘧啶酮二聚體會損害 DNA 複製,若在細胞進入細胞週期的 S 期 (S-phase) 進行 DNA 複製之前未被移除,則可能導致錯配 (mispairing)。錯配導致 DNA 序列的改變,即突變。UV 暴露後可觀察到數種此類突變,但目前為止最常見者是在兩個相鄰嘧啶(雙嘧啶 dipyrimidine 位點)處的 C → T 鹼基置換突變(圖 17-10)。此突變是複製光產物時於胞嘧啶對面錯誤嵌入腺嘌呤 (adenine) 的結果。在沒有 UV 暴露的情況下(例如在非皮膚的組織或腫瘤中),此類突變僅罕見地被觀察到,因此雙嘧啶位點處的 C → T 與 C:C → T:T 串聯突變 (tandem mutations) 被命名為紫外線特徵性突變 (UV signature mutations)。⁴⁸ 一旦這些突變形成,它們便在受影響細胞的整個壽命中持續存在,並在細胞分裂時被傳遞。它們可被視為細胞一生中 UVR 暴露的記憶。有些作者使用「UVB 特徵性突變」一詞。然而,此一名詞具誤導性,因為它們也由 UVA 所形成⁴⁸,⁴⁹,應予避免。在鱗狀細胞癌與光化性角化症 (actinic keratoses) 中,此類紫外線特徵性突變見於 p53 基因⁴⁸,⁵⁰;在基底細胞癌中,見於音蝟訊號途徑(sonic hedgehog signaling pathway)的基因(ptch、shh、smo)與 p53⁵¹;在黑色素瘤中,見於 CDKN2A、PTEN、TERT 啟動子 (promoter) 與 p53 基因⁵²⁻⁵⁵,這證明了 UVR 作為腫瘤特異性基因突變之轉化因子的分子印記。UVR 如何誘導嘧啶二聚體形成的最佳特徵化機轉,是透過 DNA 鹼基直接吸收光子,由 DNA 本身作為發色團(古典途徑 classic pathway;圖 17-11;左圖)。然而,近期 Premi 等人⁵⁶(亦見 Brash⁵⁷)描述了一種替代的、黑色素依賴性的機轉,這很可能促成黑素細胞及含黑色素角質細胞中的嘧啶二聚體形成。他們曾觀察到 DNA 光產物在 UV 暴露後長達 3 小時仍持續形成,並隨後證明這些所謂的「暗 CPDs(dark CPDs)」的形成,是由黑色素(特別是褐黑色素 pheomelanin)的光激發、透過 UV 誘導之一氧化氮與超氧化物形成過氧亞硝酸鹽 (peroxynitrite) 所介導(黑色素依賴性化學激發途徑 melanin-dependent chemiexcitation pathway;圖 17-11;中圖)。藉此,黑色素似乎不僅是光防護者,也可能藉由增加 DNA 損傷的形成而促成癌症的形成。此一觀察也提供了一個有吸引力的解釋,說明為何 UVA 只在有色素的小鼠中誘導黑色素瘤,而不在白化 (albino) 小鼠中誘導。⁵⁸
旁觀者效應 (bystander effect) 描述的是這樣一種現象:受照射細胞鄰近的未暴露細胞也展現出與暴露細胞相似的壓力反應。UVA 尤其已被證明在黑素細胞中發揮此種旁觀者效應,且有人提出長壽命的 UVR 誘導自由基介導此效應。⁵⁹,⁶⁰
人們不禁推測,Premi 等人⁵⁶所描述的長壽命過氧亞硝酸鹽,不僅可能在受照射細胞中產生「暗 CPDs」,也可能在未受照射的旁觀者細胞中產生。除了嘧啶二聚體外,UVR 也能誘導氧化性 DNA 損傷(氧化途徑 oxidative pathway,圖 17-11;右圖),例如透過其他細胞發色團吸收光子並隨後將能量轉移給 DNA(第 I 型光敏化反應 type I photosensitized reaction)或轉移給分子氧並形成活性氧物種(reactive oxygen species, ROS),而後者再與 DNA 反應形成氧化性 DNA 鹼基修飾。雖然 UVB 能產生一些 ROS,但暴露於自然陽光後大部分的氧化性 DNA 損傷由 UVA 所造成。⁶¹,⁶² UVR 誘導的 ROS 大多為單重態氧,但其他自由基,例如過氧化氫 (hydrogen peroxide)、超氧化物自由基 (superoxide radical) 與氫氧自由基 (hydroxyl radical) 也可能形成,儘管數量低得多。ROS 誘導的 DNA 損傷主要涉及鳥嘌呤鹼基。UVR 暴露後已描述數種此類氧化性鳥嘌呤損傷,其中以 8-二氫-8-氧鳥苷(8-dihydro-8-oxoguanosine, 8-oxoG)研究最為透徹。
在 DNA 複製期間,8-oxoG 與腺嘌呤錯配,產生 G → T 顛換突變 (transversion mutations)。同樣地,鳥苷核苷酸的氧化可導致複製時於腺嘌呤對面錯誤嵌入,產生 A → C 顛換。在 UVR 誘導的皮膚惡性腫瘤中,此類突變並無顯著增加,且氧化性 DNA 損傷對光致癌作用的貢獻有多少仍是一個爭論的議題。哺乳動物細胞中嘧啶二聚體與氧化性鳥嘌呤鹼基修飾形成的作用光譜顯示,隨波長從 UVC 增加至 UVB、UVA 與可見光,DNA 損傷能力逐漸改變。⁶² UVA 形成的嘧啶二聚體很少,例如比較 360 nm 與 300 nm 時,約比 UVB 少 10,000 倍。即使考慮到自然陽光中 UVA 豐富得多,UVB 仍最有可能產生大部分太陽輻射誘導的嘧啶二聚體。儘管如此,來自體外與體內突變光譜的證據日益增多,顯示嘧啶二聚體不僅對 UVB、也對 UVA 而言都是最重要的前致突變 (premutagenic) DNA 損傷,而氧化性 DNA 損傷在致突變作用中只扮演次要角色。⁴⁹,⁶³
我們曾提出一個假說來解釋 UVA 誘導之嘧啶二聚體相較於 UVB 誘導者有較高的突變率,那就是 UVA 不像 UVB 那樣誘導出穩健的細胞 DNA 損傷反應,特別是細胞週期停滯 (cell cycle arrest) 方面。⁶⁴ 這對於暴露於廣譜太陽輻射後的致突變作用可能並不相關,因為在這種情況下,損傷反應可由 UVB 成分所引發,這也會保護對抗在少數 UVA 誘導之嘧啶二聚體處的致突變作用。然而,在暴露於純 UVA 光源時(例如,透過窗戶玻璃的太陽輻射,或使用曬黑裝置時),UVA 可能特別具致突變性與致癌性。雖然 UVR 暴露後已描述有氧自由基,但我們並未發現 DNA 雙股斷裂(DNA double-strand breaks,此類 ROS 所產生的一種常見 DNA 損傷類型)由 UVB 或 UVA 所形成的證據。⁶⁵,⁶⁶ 這與下列事實一致:缺失 (deletions) 或插入 (insertions)(DNA 雙股斷裂常產生的突變類型)在 UV 誘導的突變光譜中並非常見的觀察。當特定基因中累積足夠數量的突變(驅動突變 driver mutations)時,細胞便發生惡性轉化。在此過程中,突變的形成是隨機事件。通常,許多突變遍布於暴露細胞的整個基因組中,最終才也累積到關鍵數量的特定驅動突變。例如,黑色素瘤中高度的突變負荷由 Pleasance 等人⁶⁷所證明,他們是首批報告黑色素瘤完整基因組序列、並與同一病人淋巴母細胞 (lymphoblast) 細胞株正常序列相比較的人。在黑色素瘤中觀察到的 33,345 個鹼基置換突變中,超過 70% 為 C → T 紫外線特徵性突變。這種在非 UV 暴露腫瘤中原本罕見之突變類型的優勢,是單一黑素細胞終其一生陽光暴露的一個令人印象深刻的分子記錄,而此黑素細胞最終產生了這個黑色素瘤。這些資料此後已在一個超過 300 個黑色素瘤的大型系列中得到證實,顯示黑色素瘤中所有突變的中位數有 77.7% 為紫外線特徵性突變。⁶⁸ 這些資料也顯示,來自陽光暴露部位的黑色素瘤其平均突變率是所有以此方式研究之癌症中最高者之一。這種以 C → T 轉換為主的高突變負荷,再次強調了 UVR 高度致突變特性的角色及其在皮膚黑色素瘤致病機轉中的角色。一種遺傳上獨特的黑色素瘤亞型最常出現於臉部與老年人,但也可發生於其他有嚴重光損傷皮膚的部位。這些位於慢性陽光損傷皮膚上的黑色素瘤(chronically sun-damaged skin, CSD 黑色素瘤)顯示極高的突變負荷(約 60 個突變/Mbase)⁶⁸,⁶⁹,且常源自一種稱為惡性小痣 (lentigo maligna) 的原位黑色素瘤 (in situ melanoma)。促結締組織增生性黑色素瘤 (Desmoplastic melanomas) 具有更高的突變負荷⁷⁰,且與 CSD 黑色素瘤有相似的年齡與解剖部位分布。位於陽光暴露皮膚但無慢性陽光誘導損傷徵象的黑色素瘤(非 CSD 黑色素瘤)顯示較低的突變負荷,約為 30 個突變/Mbase。上述所有黑色素瘤亞型中所發現的體細胞突變 (somatic mutations) 都顯示明確的紫外線特徵,而被認為非 UVR 誘導的黏膜型 (mucosal) 與肢端型 (acral) 黑色素瘤則否。非 CSD 黑色素瘤最常見於女性的小腿與男性的背部。⁷¹ 至少在西方文化中,這些部位往往長期受光防護(例如在冬季期間),然後在春夏季暴露於陽光,往往是突然暴露並隨後曬傷。因此,這些類型的黑色素瘤似乎與未經陽光適應之皮膚的突然、間歇性高劑量 UVR 暴露有關。⁷² 慢性 UVR 暴露的皮膚以抗突變機轉 (anti-mutagenic mechanisms) 的上調為特徵。光防護皮膚中此類保護性狀態的缺乏,結合黑素細胞於高劑量 UVR 暴露時相對無法進行凋亡,已被提出作為間歇性 UVR 暴露誘導黑色素瘤的一種解釋⁷³,並可能顯示細胞機轉在預防 DNA 損傷與突變形成以及慢性 UVR 暴露皮膚之惡性轉化方面的重要性(圖 17-9)。
DNA 修復與對 UVR 誘導 DNA 損傷的其他反應 (DNA REPAIR AND OTHER RESPONSES TO UVR-INDUCED DNA DAMAGE)
DNA 修復是一種重要的細胞防禦機轉,可預防 UV 暴露後在 DNA 損傷位點的突變形成。然而,它並非唯一的防禦機轉(圖 17-9)。大多數突變是在受損 DNA 複製期間產生的。因此,損傷誘導的細胞週期停滯(讓修復有更多時間)是另一種預防突變形成的重要細胞損傷反應。⁶⁴,⁷⁴ 此外,程序性細胞死亡(凋亡 apoptosis)可阻止具有壓倒性 DNA 損傷之細胞的存活,並透過此機轉減少帶有 UV 誘導突變之細胞的頻率。對抗 UVR 致癌特性的其他固有防禦機轉包括:增加黑色素生成與表皮和角質層的增厚(保護免於未來的 DNA 損傷),以及透過宿主免疫反應移除突變細胞。已辨識出超過 100 個 DNA 修復基因(http://sciencepark.mdanderson.org/labs/wood/DNA_Repair_Genes.html)。核苷酸切除修復(nucleotide excision repair, NER)途徑作用於被 UVR 損傷的 DNA,修復 CPDs 與其他光產物,也作用於被某些其他致癌物(例如苯并[a]芘 benzo-[a]-pyrene)損傷的 DNA。在 NER 中,受損的核苷酸被移除並以未受損的 DNA 取代。圖 17-12 顯示了 NER 系統的簡化示意圖,描述了協同作用以修復 UV 誘導 DNA 損傷的眾多蛋白質中的一部分。這些修復基因的缺陷可造成人類疾病,包括著色性乾皮症(xeroderma pigmentosum, XP)、Cockayne 症候群(Cockayne syndrome, CS)與毛髮硫營養不良症(trichothiodystrophy, TTD)(見第 130 章)。例如,XP 可由參與 NER 之數個基因中任一個的缺陷所造成。根據細胞融合 (cell fusion) 實驗,在同一基因有缺陷的細胞/病人被認為屬於同一互補群 (complementation group),而若不同基因受影響則屬於不同互補群。如果融合細胞中的 DNA 修復恢復正常——即來自每個細胞的野生型 (wildtype) 基因產生另一細胞所缺乏的功能性蛋白質——則這些細胞彼此「互補」,並屬於不同的互補群。已辨識出 7 個此類互補群(XP-A 至 XP-G),對應於可造成 XP 的 7 個不同基因的突變。被轉錄的基因比基因組的其餘部分修復得更快。在 NER 途徑中,涉及 DNA 損傷辨識的第一步驟,在未轉錄基因(全基因組 NER global genome NER)與被轉錄基因(轉錄偶聯 NER transcription-coupled NER)中有所不同。在代表大部分基因組的未轉錄基因與非編碼區域中,XPE 與 XPC 基因產物結合至 UV 損傷的 DNA,標記它以供進一步處理。相對地,被轉錄基因中的 DNA 損傷似乎由停滯的 RNA 聚合酶 (RNA polymerase) 與 CSA 和 CSB(CS 互補群 A 與 B)基因產物協同作用所感知。在 DNA 損傷辨識步驟之後,全基因組 NER 與轉錄偶聯 NER 遵循相同的途徑(圖 17-12)。在 XP 中,顯著的 NER 缺陷賦予發展非黑色素瘤皮膚癌與黑色素瘤的極高風險。DNA 修復能力較細微的變異(例如,作為 NER 基因多型性 polymorphisms 或老化相關衰退的結果)也與皮膚癌風險增加有關。⁷⁵⁻⁷⁸
氧化性鹼基修飾是比嘧啶二聚體更簡單的一種 DNA 損傷類型,因為它們僅涉及單一 DNA 鹼基的化學改變。這些損傷由一個不同的 DNA 修復途徑——稱為鹼基切除修復 (base excision repair)——所處理。數種對 DNA 損傷的細胞反應藉由預防突變形成來促成基因組完整性的維持。這些反應包括細胞週期停滯、凋亡(程序性細胞死亡)與 DNA 修復。由於這些反應需要被謹慎協調,因此有許多蛋白質參與 DNA 損傷的訊號傳遞與細胞 DNA 損傷反應的調節。不同類型的 DNA 損傷劑與不同類型的 DNA 損傷需要不同的 DNA 損傷反應。此複雜途徑的簡化版本呈現於圖 17-13。如同 DNA 修復基因的缺陷一樣,這些 DNA 損傷訊號傳遞基因(在圖 17-13 中以方框標示)中許多基因的缺陷也牽涉到基因組不穩定性的遺傳性疾患,包括例如運動失調性微血管擴張症 (ataxia telangiectasia)、范可尼貧血 (Fanconi anemia)、家族性乳癌、Li-Fraumeni 症候群等(見第 130 章)。腫瘤抑制基因 p53(常被稱為基因組的守護者)在調節與協調這些反應方面扮演關鍵角色,並在許多癌症(包括皮膚鱗狀細胞癌)中發生突變。在細胞 DNA 損傷反應途徑中,p53 的上游調節因子是 ATM(ataxia telangiectasia mutated,運動失調性微血管擴張症突變)與 ATR(ataxia telangiectasia- and Rad3-related,運動失調性微血管擴張症與 Rad3 相關)基因。p53 的數種功能之一是因應 DNA 損傷調節細胞週期。細胞分裂(有絲分裂 mitosis)後,細胞有 23 對染色體並處於細胞週期的 G1 期。染色體接著在 DNA 合成(S 期)期間複製,因而在有絲分裂(M 期)前具有兩倍數量的染色體(G2 期)。因應損傷,細胞可能在稱為細胞週期檢查點 (cell cycle checkpoints) 的特定細胞週期階段停止週期(停滯)。預防細胞進入 S 期(G1/S 檢查點)的一個重要下游效應子是 p21。p53 也藉由轉錄誘導 XPC、XPE/p48 與 GADD45 來誘導 NER。如果細胞帶著未修復的 DNA 損傷進入 S 期,或細胞在 S 期被 UV 暴露,則常規 DNA 聚合酶會在 DNA 光產物處停滯並從 DNA 股上脫離。⁷⁹ 對於這些情況,細胞配備有數種特化的 DNA 聚合酶以進行跨損傷 DNA 合成 (translesional DNA synthesis)。DNA 聚合酶 eta (DNA polymerase eta) 是其中之一;它特化於跨越 DNA 光產物,但在此過程中可能引入突變。它在 XP 變異型 (XP variant) 病人中發生突變,這些病人在臨床上與其他具 NER 缺陷的 XP 病人無法區分(見第 130 章)。⁸⁰,⁸¹ 這證明了此第二道防線對抗 DNA 光產物致突變與致癌後果的重要性。p53 與 p21 也下調此種跨損傷 DNA 合成的活性,以維持低致突變活性,代價是降低損傷的跨越。如果此種跨損傷 DNA 合成失敗,細胞可使用重組修復 (recombination repair) 來解決停滯的複製叉 (replication forks)。⁸² 當因應 UV 光損傷而啟動時,此第三道防線是由范可尼貧血/BRCA DNA 損傷反應途徑的活化所介導。⁶⁵
凋亡是一個受調節的生理過程,導致以細胞皺縮、膜泡形成 (membrane blebbing) 與 DNA 片段化為特徵的細胞死亡。一群稱為半胱天冬酶 (caspases) 的半胱胺酸蛋白酶 (cysteine proteases) 是凋亡的核心調節因子。觸發因子可能是細胞外源性或內源性的(例如 DNA 損傷),並涉及各別的啟動子半胱天冬酶(例如因應 DNA 損傷的 caspase 2),但共用相同的下游效應子半胱天冬酶。p53 腫瘤抑制基因的突變很常見於皮膚鱗狀細胞癌及其前驅病灶——光化性角化症。此外,慢性陽光暴露的皮膚帶有許多具 p53 突變之角質細胞的克隆性增生,這些增生無法以肉眼或顯微鏡的異常偵測到。⁸³ p53 作為抗突變反應核心調節因子之功能的喪失,會在後續 UVR 暴露時導致較高的突變形成率。此種所謂的 UV 突變子表型 (UV mutator phenotype) 在皮膚鱗狀細胞癌的發展中至關重要,因為它使得單一角質細胞透過反覆 UV 暴露累積完全惡性轉化所需之所有突變的可能性大為提高。⁸⁴
哪些波長的 UVR 造成皮膚癌?(WHICH WAVELENGTHS OF UVR CAUSE SKIN CANCER?)
雖然 UVB 的致癌特性數十年來已廣為人知,但 UVA 長期以來因其在太陽可提供劑量下無法誘導曬傷而被認為無害。然而,現在已認識到 UVR 在亞紅斑劑量 (suberythemogenic doses) 下也具破壞性與致癌性,且 UVA 本身就能在小鼠中誘導皮膚鱗狀細胞癌。因此,世界衛生組織 (WHO) 已將 UVA 歸類為獨立的第一類致癌物 (class I carcinogen)。⁸⁵ 烏特勒支-費城作用光譜(Utrecht-Philadelphia action spectrum)顯示,小鼠光致癌作用的致癌效力隨波長從 UVB 增加至 UVA 而呈指數下降。⁸⁶ 這種下降的一部分被自然太陽輻射中 UVA 較高的豐富度所抵消。此外,如上文所述,透過窗戶玻璃過濾的太陽輻射及曬黑沙龍中對純 UVA 的暴露,可能顯著增加 UVA 對光致癌作用的貢獻。因此,雖然看來 UVB 對光致癌作用的貢獻比 UVA 更大,但兩者對發展非黑色素瘤皮膚癌之終生風險的相對貢獻仍不明確。已使用數種動物模型來回答黑色素瘤的波長問題。Ley⁸⁷觀察到以 UVA 在負鼠 (opossum) 中誘導黑色素瘤前驅病灶,比 UVB 更多。然而,無法誘導出浸潤性黑色素瘤。Setlow 等人⁸⁸使用劍魚 (swordfish),觀察到 UVA 比 UVB 更有效地誘導黑色素瘤。然而,此觀察無法被重現,Mitchell 等人⁸⁹報告了相反的結果。DeFabo 等人⁹⁰使用 HGF/SF 轉殖基因小鼠,觀察到僅以 UVB 誘導黑色素瘤,而非 UVA。後來,同一團隊⁵⁸觀察到 UVA 確實誘導黑色素瘤,但僅在有色素的小鼠中,而非先前研究的無色素小鼠中。藉此,看來至少在此小鼠模型中,黑色素的存在對於以 UVA 誘導黑色素瘤是必需的,但對 UVB 則否,且兩種波長 UVA 與 UVB 都能誘導黑色素瘤,但可能透過不同的機轉。非常合理的是,近期所描述的黑色素依賴性化學激發途徑(用以產生嘧啶二聚體與紫外線特徵性突變;圖 17-11;中圖)就是 UVA 致黑色素瘤所假定的替代機轉。此外,曬黑沙龍使用所伴隨的黑色素瘤風險增加,為 UVA 作為黑色素瘤特殊危險因子提供了進一步的支持。許多研究已顯示,使用日曬床/日曬燈 (sunbed/sunlamp) 後罹患黑色素瘤的風險顯著增加⁹¹,⁹²,包括一項在瑞典與挪威 106,379 名女性中進行的大型前瞻性世代研究,顯示使用曬黑裝置的黑色素瘤相對風險為 1.42 至 2.58。⁹³ 冰島 1990 至 2006 年間黑色素瘤發生率上升的黑色素瘤流行(主要在年輕女性中),也與曬黑床使用的增加有關。⁹⁴ 在通常被遮蓋部位的黑色素瘤(例如薦部與恥骨區的皮膚),也可能歸因於日曬床使用的 UVR 暴露。⁹⁵
幾十年前,當人們日益認識到 UVB(280 至 315 nm)具有皮膚破壞性效應、但 UVA(315 至 400 nm)仍被認為相對安全時,曬黑產業減少了其裝置中的 UVB 輸出,並聲稱 UVA 誘導的曬黑是安全的。這就是為什麼曬黑床使用所伴隨的黑色素瘤風險增加可能支持 UVA 作為黑色素瘤特殊危險因子的角色。然而,曬黑機在其光譜輸出方面差異甚大。⁹⁶,⁹⁷ 因此,日曬床使用所伴隨的黑色素瘤風險增加,也可能是由於曬黑裝置仍發射的 UVB 所致。產生氧化性 DNA 損傷的能力隨波長從 UVB 增加至 UVA 而增加,且有人假設此類損傷(特別是當由 UVA 誘導時)可能在致黑色素瘤作用中扮演角色。⁶³ 在思考此假說時,重要的是要認識到,至少在纖維母細胞 (fibroblasts) 與角質細胞中,UVA 已被證明產生的嘧啶二聚體多於 8-oxodG。⁹⁸
不同於一份關於在轉化的囓齒類細胞中觀察到獨立 UVA 特徵性突變的早期報告⁹⁹,後來已證明大多數 UVA 誘導的突變是 C → T 轉換,無論在體外或體內,都沒有獨立 UVA 特徵性突變的特殊訊號,也沒有氧化性鹼基修飾所典型之突變的高發生率。¹⁰⁰⁻¹⁰³
黑素細胞中暴露於 UVR 時產生氧化壓力的一個有吸引力的候選者是黑色素。黑色素已被描述具有一些光敏化特性,特別是顏色較偏紅的褐黑色素。¹⁰⁴,¹⁰⁵ Wang 等人¹⁰⁶,¹⁰⁷報告,黑素細胞在以 UVA 照射時,會產生更多的氧化性 DNA 損傷、對氧化性 DNA 損傷的修復效率較低,並產生更多的突變。他們提出氧化性 DNA 損傷是致黑色素瘤作用的主要驅動因子。然而,他們並未對 UVA 誘導的突變進行定序,因此並未提供 UVA 誘導之突變確實是氧化性 DNA 損傷所典型之 G → T 或 A → C 顛換的最終證明。此外,上述來自黑色素瘤的突變資料顯示,此類氧化性 DNA 損傷所典型的突變僅佔一小百分比。⁶⁷,⁶⁸
綜合而言,沒有證據證明氧化性 DNA 鹼基損傷在驅動黑色素瘤的 UVR 誘導突變形成中扮演主要角色。總之,證據支持 UVB 與 UVA 都能誘導黑色素瘤,可能透過不同的機轉。然而,UVB 與 UVA 對整體黑色素瘤風險的相對貢獻仍不明確。
光老化 (PHOTOAGING)
光老化描述的是慢性陽光暴露皮膚中所觀察到的變化,而皮膚的內源性老化 (intrinsic aging) 描述的則是隨年齡增長、不伴隨慢性陽光暴露之外源性損傷而發生的變化。光老化的皮膚改變可見於皮膚的所有層次,包括表皮、色素系統、真皮結締組織、血管系統與皮下脂肪(圖 17-14 與 17-15)。¹⁰⁸,¹⁰⁹ 雖然有時在年輕個體中已可觀察到光老化的細微變化,但光老化的表現在年長病人中更為常見,這顯示光老化的皮膚變化通常疊加於內源性皮膚老化的變化之上。許多光老化的表現也可見於內源性老化的皮膚。這引發了一個問題:光老化是否僅是慢性光損傷對正常老化過程的加速。表 17-4 列出皮膚光老化的典型表現。涉及角質細胞與黑素細胞之良性、癌前與惡性克隆性增生的症狀與老化相關,因為癌症一般而言與老化相關。它們最好被歸類為腫瘤性事件 (neoplastic events),而非老化事件。它們不見於內源性老化的皮膚,只見於光防護的皮膚。驅動這些事件的機轉已於「光致癌作用」一節中描述。相對地,表皮的變薄、色素減退、真皮膠原蛋白的喪失、微血管擴張 (telangiectasias)(作為細胞外基質支架減少的後果),以及皮下脂肪的喪失,都是功能喪失事件(表 17-4),也可見於內源性老化。慢性光損傷所致的加速增生耗竭 (proliferative exhaustion) 很可能是這些特徵的機轉,這與光老化代表損傷加速老化 (damage-accelerated aging) 的觀點一致。
光老化皮膚中唯一不見於內源性老化皮膚的非增生性特徵是光化性彈性纖維變性 (actinic elastosis),其特徵為纖維狀嗜鹼性物質 (fibrillary basophilic material) 在真皮上層與中層的累積。臨床上,它呈現為皮膚的黃色增厚(有時呈結節狀)、彈性喪失,以及深皺紋的形成(圖 17-15)。它主要由彈性蛋白 (elastin) 與纖維蛋白 (fibrillin) 組成,以彈性蛋白染色強烈著色,可被彈性蛋白酶 (elastase) 降解,但不能被膠原蛋白酶 (collagenase) 降解。彈性蛋白合成在 UV 暴露後上調¹¹⁰,但 UVR 誘導的嗜中性球流入(例如曬傷皮膚中所見者)也提供了一個透過嗜中性球分泌之彈性蛋白酶活性增加降解的機轉。在細胞外空間的彈性蛋白降解之後,真皮纖維母細胞透過受體介導的胞吞作用 (receptor-mediated endocytosis) 將彈性蛋白片段內化至溶體 (lysosomes),以供溶體蛋白酶組織蛋白酶 K (cathepsin K) 進一步降解。¹¹¹
組織蛋白酶 K 由 UVA 上調以促進此細胞內清除。¹¹² 老化的纖維母細胞不會因應 UVA 上調組織蛋白酶 K,而有人假設這導致無法適當地從細胞外空間清除彈性蛋白片段,並最終導致彈性蛋白的累積,並交聯成為一個大型、異常的巨分子。¹¹² 藉此,光化性彈性纖維變性可被歸類為功能喪失事件。光化性彈性纖維變性大致上是不可逆的,即使持續光防護也是如此,這很可能是由於該大型異常巨分子已變得不可降解的事實。這令人聯想到異常巨分子在細胞內的累積,以及由於無法透過一種稱為巨自噬 (macroautophagy) 的機轉將它們從細胞質清除而隨後造成的細胞功能損害,巨自噬是老化細胞一般而言喪失功能的一個重要機轉。¹¹³
早老素 (Progerin) 是核膜蛋白核纖層蛋白 A (Lamin A) 的一種異常剪接變異體 (splice variant)。它在早老症候群 (premature aging syndrome) Hutchinson Gilford 早老症 (Hutchinson Gilford progeria) 病人的細胞中以高濃度被發現,但也累積於正常老化的細胞中。¹¹⁴,¹¹⁵ 它干擾許多核功能,並最終導致細胞衰老 (cellular senescence) 與老化的細胞表型。此種老化蛋白由 UVA 透過氧化壓力與異常剪接的誘導所誘發。¹¹⁶ 這是 UVA 特別促進老化表型的又一個例子。由於失去一個允許核纖層蛋白 A 降解的法尼基化 (farnesylation) 位點,早老素大致上不可降解。這是暴露於 UVA 導致干擾細胞功能之不可降解蛋白質累積的第二個例子。雖然急性 UVR 誘導發炎的某些效應誘導了可能促成光老化的代謝變化(例如彈性蛋白合成增加、基質金屬蛋白酶 matrix metalloproteinase(即膠原蛋白酶 collagenases)表現增加,以及膠原蛋白合成減少),但這些變化在停止 UV 暴露後應可逆轉。然而,觀察到的是,表現為皺紋與皮膚鬆弛增加的細胞外基質變化是不可逆的,這可由異常且不可降解蛋白質在細胞外與細胞內空間的累積來解釋。雖然氧化性 DNA 損傷對光致癌作用的貢獻尚未被證實,但 UVA 誘導的氧化壓力很可能是光老化中這些功能喪失事件的核心介導者。由於 ROS 介導衰老(例如,透過早老素的形成)¹¹⁶,它們很可能蘊含一種對抗 UVR 促致癌效應的抗癌效應。藉此,抗氧化劑可能預防 UVR 誘導的光老化功能喪失事件,但反過來也可能促進光致癌作用。除了在誘導伴隨細胞功能喪失之細胞老化表型方面的特定角色外,UVA 也可能是真皮細胞外基質變化的一個特殊禍首,因為相較於 UVB,它較深入真皮。然而,UVB 即使不到達真皮深層,也會影響其代謝。例如,UVB 照射後真皮中基質金屬蛋白酶的誘導,已被證明是由一種旁分泌機轉 (paracrine mechanism) 所介導,即角質細胞分泌發炎介質並向真皮纖維母細胞傳遞訊號。¹¹⁷⁻¹¹⁹ 進一步傳遞訊號至皮下脂肪甚至可能介導皮下脂肪的喪失,作為光老化的一個特徵。¹²⁰
皮膚對可見光與紅外線輻射的反應 (CUTANEOUS RESPONSES TO VISIBLE LIGHT AND INFRARED RADIATION)
相較於 UVR,電磁波譜中其他類型非游離輻射(即可見光與紅外線輻射 infrared radiation, IR)的效應研究得少得多。然而,越來越多的證據顯示,這些輻射對皮膚也有光生物學效應。可見光可誘導紅斑與色素沉著(立即與延遲性色素變深,特別是在較深的皮膚類型中)¹²¹,¹²²,且某些光皮膚病有時也可由可見光誘發,包括例如日光性蕁麻疹、慢性光化性皮膚炎 (chronic actinic dermatitis) 與皮膚紫質症。能吸收可見光並可能介導這些效應的潛在發色團為紫質、黑色素、β-胡蘿蔔素 (β-carotene)、核黃素 (riboflavin)、膽紅素 (bilirubin) 與血紅素 (hemoglobin)。此外,嘧啶二聚體型 DNA 損傷與氧化性 DNA 鹼基修飾形成的作用光譜延伸至可見光範圍內。⁶²
雖然這些效應的作用機制仍只有非常不完整的理解,但這些見解已促成以可見光為基礎之治療裝置的發展,包括例如脈衝光(intense pulsed light, IPL)、雷射與光動力療法。IR 已被牽涉於光老化與火激紅斑 (erythema ab igne)。¹²³,¹²⁴ IRA 深入真皮,並對真皮纖維母細胞的基因表現譜有深遠的效應,包括例如基質金屬蛋白酶的誘導。這可能促成慢性陽光暴露皮膚中膠原蛋白纖維的喪失。雖然紅外線輻射單獨似乎不會造成皮膚癌,但生長行為可能受到影響,例如透過預防凋亡。¹²⁵
結論 (CONCLUSIONS)
UVR 對皮膚唯一的正面效應——維生素 D 的生成——被許多嚴重且可能危及生命的有害效應所抵消,這些有害效應使有效的光防護成為預防所必需。所幸,光防護對維生素 D 代謝的負面效應,可透過口服維生素 D 補充輕易解決。從所描述的 UVB 與 UVA 效應可以清楚看出,光防護策略必須包括對抗這兩種性質的保護。所幸,美國衛生及公共服務部的食品藥物管理局 (FDA) 於 2011 年針對非處方人用防曬藥品的標示與效力測試發布了一項最終規則,處理了此種廣譜 (broad-spectrum) 保護。此新標準增加了一項針對廣譜保護的體外測量,以納入 UVA。它使用 Diffey 等人最初建議的臨界波長標準 (critical wavelength criterion),以至少 370 nm 的波長定義廣譜,在此波長下,達到 290 至 400 nm 吸光度曲線下面積的 90%。¹²⁶
對 IR 與可見光可損傷皮膚的認識,已催生將光防護延伸至這些類型輻射的努力。¹²⁷,¹²⁸ 過濾可見光的嘗試很可能會因可接受度差而受阻,因為此類濾片將會是可見的。過濾 IR 可能有機會,但此類濾片的發展才剛起步。全身性與局部抗氧化劑已被倡導用以減少 UVR、可見光與 IR 誘導的氧化損傷,特別是氧化性 DNA 損傷。¹²⁹ 然而,它們對抗上述任何終點的保護效應尚未被充分確立,或頂多是微不足道的。此外,氧化性 DNA 損傷可能對 UVA 誘導的致突變作用沒有顯著貢獻(與先前的想法相反)這一證據,引發了抗氧化劑是否提供任何對抗皮膚癌之保護的問題。活性氧物種不僅具破壞性,也誘導能保護對抗光致癌作用的細胞反應,包括例如透過產生稱為早老素的老化相關蛋白來誘導衰老。¹¹⁶ 這顯示抗氧化劑實際上可能正在促進光致癌作用,如同已對其他類型癌症所提出的那樣。2014 年,美國醫務總監 (Surgeon General) 與美國衛生及公共服務部發布了一份《預防皮膚癌行動呼籲》(Call to Action to Prevent Skin Cancer)。所陳述的目標包括:(1) 增加戶外環境中防曬的機會;(2) 為個人提供做出明智、健康之 UV 暴露選擇所需的資訊;(3) 推動促進預防皮膚癌之國家目標的政策;(4) 減少室內曬黑的危害;以及 (5) 加強與皮膚癌預防相關的研究、監測、追蹤與評估。
在思考公眾意識宣傳活動時,重要的是要認識到,尋求陽光與尋求曬黑裝置的行為,至少在某些個體中,與一種令人愉悅的中樞神經系統效應有關。¹³⁰⁻¹³² 此類對 UVR 暴露成癮的特徵,令人聯想到尼古丁的成癮性,後者使肺癌預防的戒菸計畫變得困難。反菸宣傳活動已能克服尼古丁成癮,並導致吸菸率以及隨之而來的肺癌死亡率下降,這一事實對於透過處理成癮性曬黑與尋求陽光來降低非黑色素瘤皮膚癌與黑色素瘤發生率的努力是令人鼓舞的。皮膚科學界有機會透過支持醫務總監的行動呼籲,對公共衛生產生巨大的影響。
致謝 (ACKNOWLEDGMENTS)
本章部分內容大量使用了本書前一版第 90 章「皮膚光生物學與光免疫學基礎」(Fundamentals of Cutaneous Photobiology and Photoimmunology) 的部分內容,該章由 Irene E. Kochevar、Charles R. Taylor 與 Jean Krutmann 撰寫。其他部分使用了第 110 章「基因組不穩定性、DNA 修復與癌症」(Genome Instability, DNA Repair, and Cancer) 的元素,該章由 Thomas M. Rünger 與 Kenneth H. Kraemer 撰寫。
圖表 (FIGURES AND TABLES)

圖 17-1:電磁輻射的波譜,劃分為主要波長區域與進一步的細分。
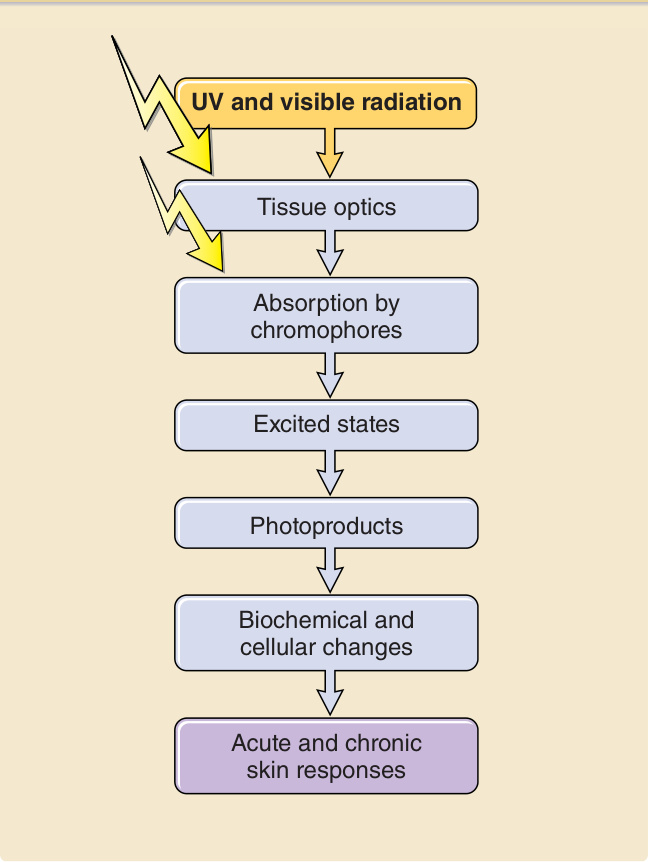
圖 17-2:一個光子要對暴露的皮膚產生效應所必需的一系列光物理、光化學與光生物學事件。
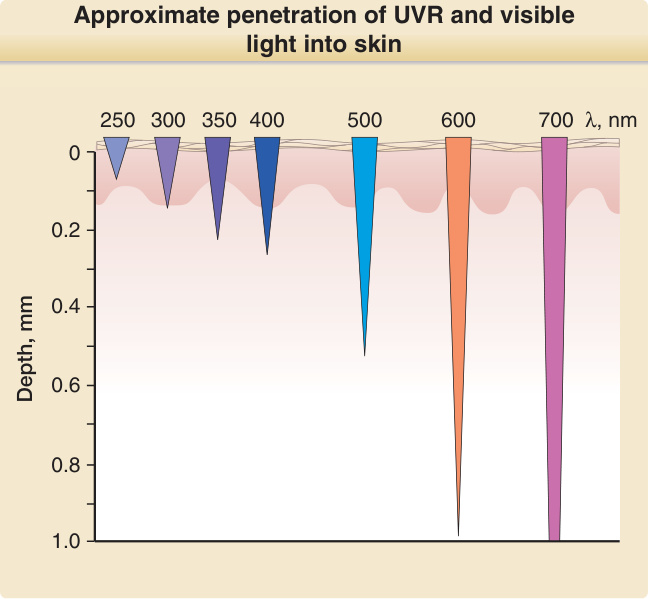
圖 17-3:紫外線輻射與可見光進入皮膚的概略穿透深度。λ = 波長 (wavelength)。
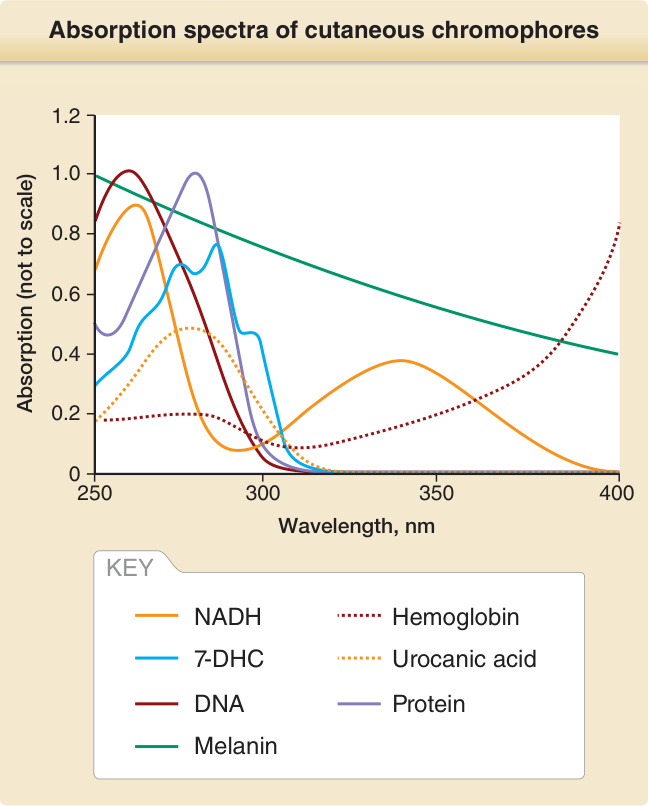
圖 17-4:吸收紫外線輻射之皮膚發色團 (cutaneous chromophores) 的吸收光譜。請注意,這些發色團在皮膚中所吸收 UVR 的相對量,取決於吸收峰的高度、皮膚中每種發色團的量,以及每個波長進入皮膚的穿透。例如,雖然裸露 DNA (naked DNA) 的吸收極大值在 260 nm,但激發基底角質細胞中 DNA 最有效的波長是 300 nm。這是因為較短的波長在角質層與表皮上層被吸收與散射,無法那麼有效地到達基底角質細胞。7-DHC, 7-去氫膽固醇 (7-dihydrocholesterol);NADH, 還原型菸鹼醯胺腺嘌呤二核苷酸 (reduced nicotinamide adenine dinucleotide)。
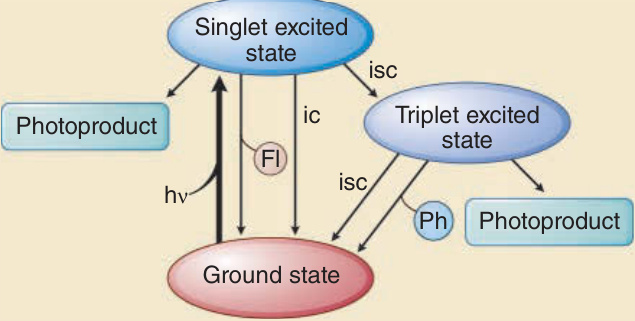
圖 17-5:發色團的激發態、所吸收能量的耗散,以及暴露於紫外線輻射與可見光的光子後光產物的形成。Fl, 螢光 (fluorescence);ic, 內轉換 (internal conversion);isc, 系統間跨越 (intersystem crossing);Ph, 磷光 (phosphorescence)。
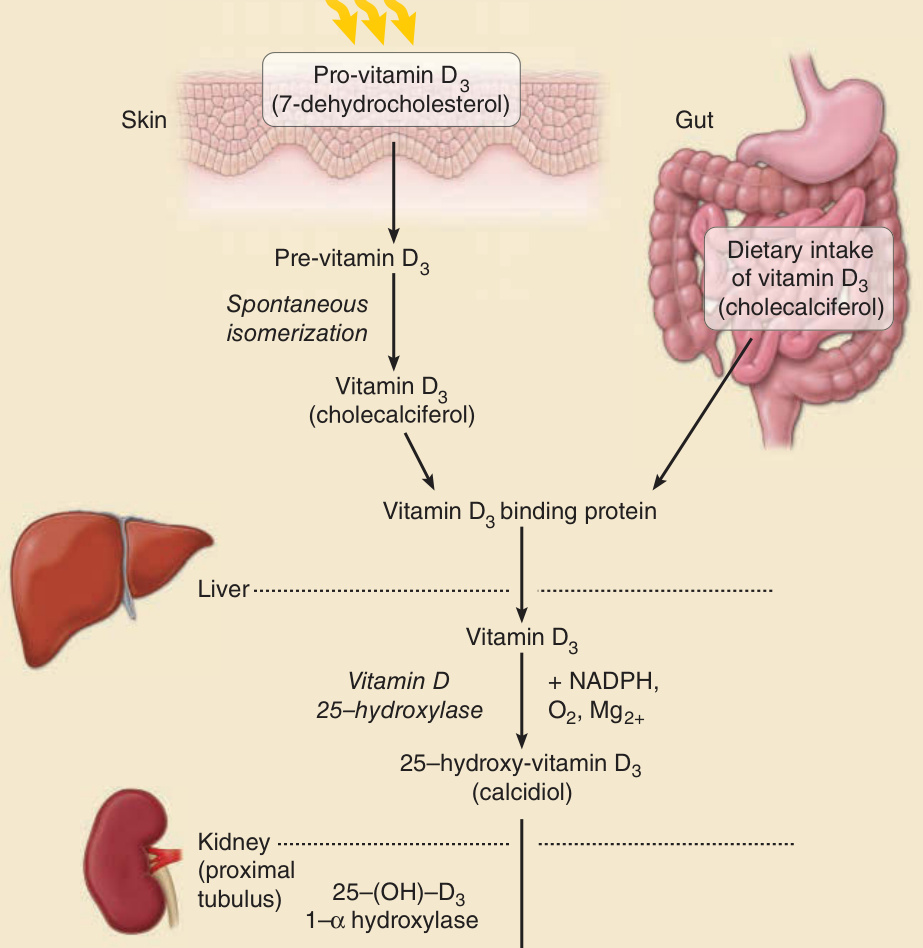
圖 17-6:活性維生素 D 的合成。吸收 UVB 並啟動此途徑的發色團是 7-去氫膽固醇 (7-dehydrocholesterol)。其吸收極大值為 297 nm。光激發 7-去氫膽固醇的光產物是前維生素 D3 (pre-vitamin D3),它迅速異構化為維生素 D3。詳見內文。

圖 17-7:光免疫抑制的機轉。光免疫抑制由發色團(包括 DNA 與反式尿刊酸 trans-urocanic acid)的光激發、光產物(例如環丁烷嘧啶二聚體、順式尿刊酸 cis-urocanic acid)的形成,以及隨後一系列免疫學終點所介導。此級聯展示了皮膚暴露於 UVR 後的一個典型事件級聯,始於光物理反應(UVR 進入皮膚與發色團的光激發),接著是光化學反應(光產物的產生),並最終達到光生物學後果。
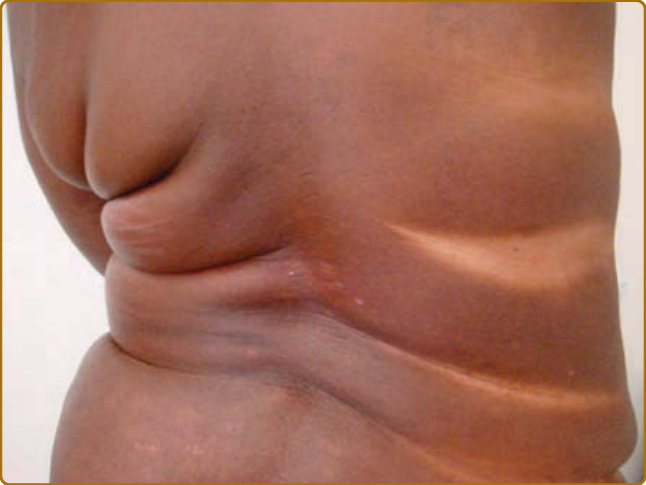
圖 17-8:一位接受 nbUVB 光照治療之病人的曬黑。請注意未暴露於 UVR 的皮膚皺褶中缺乏曬黑。這位皮膚光型 V (skin phototype V) 的病人在曬黑前並未發生曬傷。
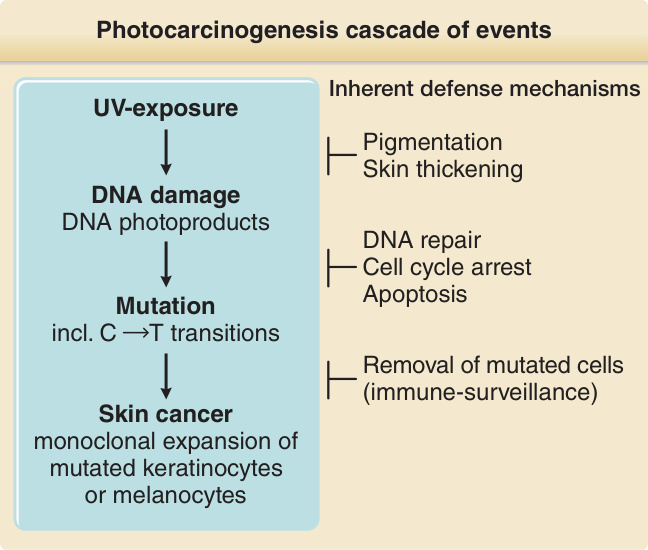
圖 17-9:光致癌作用的事件級聯。暴露於 UVR 誘導典型類型的 DNA 損傷,即環丁烷嘧啶二聚體與 6,4-嘧啶–嘧啶酮光產物。這些常在雙嘧啶位點產生單一與串聯鹼基置換突變(C→T 與 CC→TT),這些是 UVR 暴露所典型的突變,因此被稱為紫外線特徵性突變。當關鍵基因(腫瘤抑制基因)中有足夠數量的失活突變時,個別細胞可能發生惡性轉化、克隆性擴增並形成皮膚癌。數種固有的防禦機轉對抗此一事件連鎖。
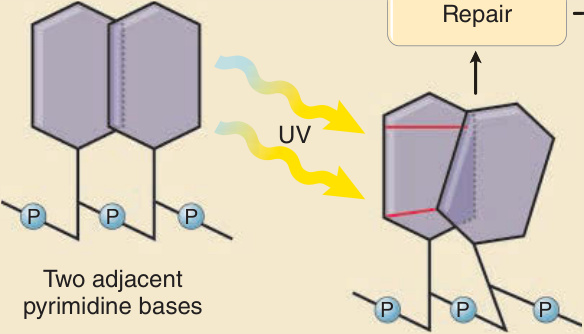
圖 17-10:UVR 誘導 DNA 損傷及隨後突變產生的範例。在 UVR 直接激發 DNA 分子後,相鄰的嘧啶鹼基(胞嘧啶或胸腺嘧啶)可能在彼此之間形成共價鍵,導致嘧啶二聚體的產生。此圖示顯示一個範例,其中兩個共價鍵在兩個嘧啶之間產生一個三環環丁烷環 (tricyclic cyclobutane ring)。因此,此類紫外線誘導的 DNA 損傷稱為環丁烷嘧啶二聚體,是一種常見的 DNA 光產物類型。核苷酸切除修復 DNA 修復系統的功能是移除此損傷,這會恢復正常的 DNA 序列(上方方框)。如果損傷未被修復,此類 DNA 光產物可導致典型 C→T 單一鹼基置換突變的形成(下方方框)。這最有可能在受損 DNA 的複製過程中、於含胞嘧啶之光產物對面錯誤嵌入腺嘌呤時發生。右側顯示一個此類紫外線特徵性突變的範例。請注意,該突變位於連續 7 個嘧啶的序列中,這是紫外線特徵性突變的常見位置。
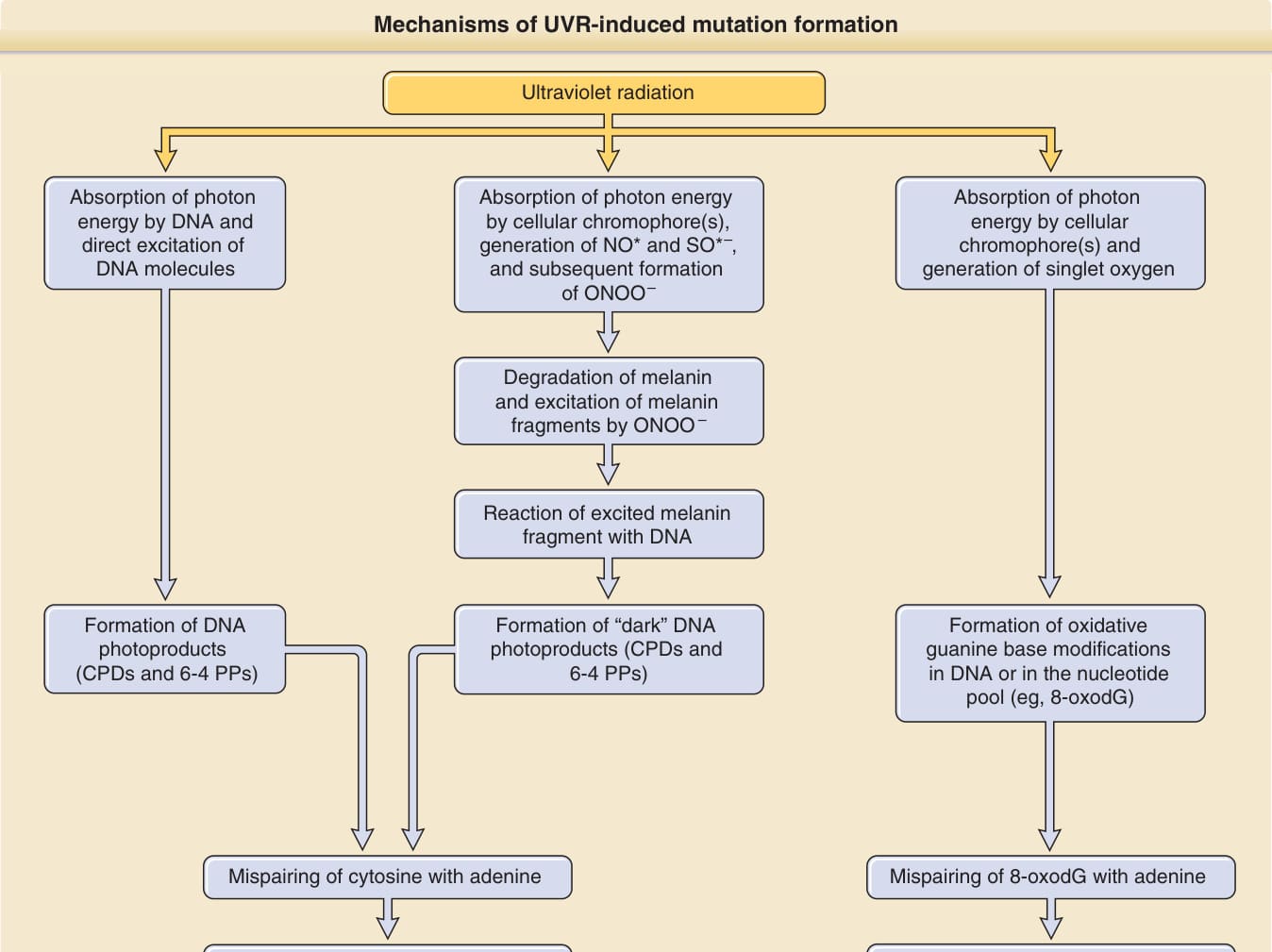
圖 17-11:UVR 誘導突變形成的機轉。在古典途徑 (classic pathway,左圖) 中,相鄰的嘧啶在 DNA 分子被光子直接激發後彼此反應並二聚化形成 DNA 光產物。C → T 與 CC → TT 紫外線特徵性突變可能在 DNA 光產物的位點上,透過胞嘧啶與腺嘌呤的錯配而形成。雖然 260 nm 是 DNA 的吸收極大值,但 UVB 仍強烈激發 DNA 並透過此途徑產生突變。UVA 激發 DNA 的能力弱得多,但仍可能透過此機轉產生突變。CPDs, 順式-順式環丁烷嘧啶二聚體 (cis-syn cyclobutane pyrimidine dimers);6-4 PPs, 嘧啶 (6-4) 嘧啶酮光產物 (pyrimidine (6-4) pyrimidone photoproducts)。黑色素依賴性化學激發途徑 (melanin-dependent chemiexcitation pathway,中圖) 近期由 Premi 等人⁵⁶所描述。它描述 DNA 光產物的形成並非透過 DNA 的直接激發(如古典途徑中),而是透過從激發態黑色素片段向 DNA 的能量轉移,此過程涉及一氧化氮 (NO∗)、超氧化物 (SO∗–) 與過氧亞硝酸鹽 (ONOO–)。此機轉在以 UVA 與 UVB 照射後都會產生紫外線特徵性突變,並促成黑素細胞與含黑色素角質細胞中的突變形成。氧化途徑 (oxidative pathway,右圖) 涉及一個光敏化反應,在 DNA 以外的細胞發色團被激發後形成單重態氧。單重態氧接著與 DNA 中或核苷酸池中的鳥嘌呤反應。一種常見的氧化性鹼基損傷是 8-氧-7,8-二氫-2′-去氧鳥苷 (8-oxo-7,8-dihydro-2′-deoxyguanosine, 8-oxodG)。透過與腺嘌呤錯配,可形成 G → T 與 A → C 顛換。長期以來認為 UVA 透過此機轉促成 UVR 誘導的突變負荷。然而,分子證據(特別是黑色素瘤中此類顛換的低頻率)並不支持其在皮膚癌形成中扮演主要角色。
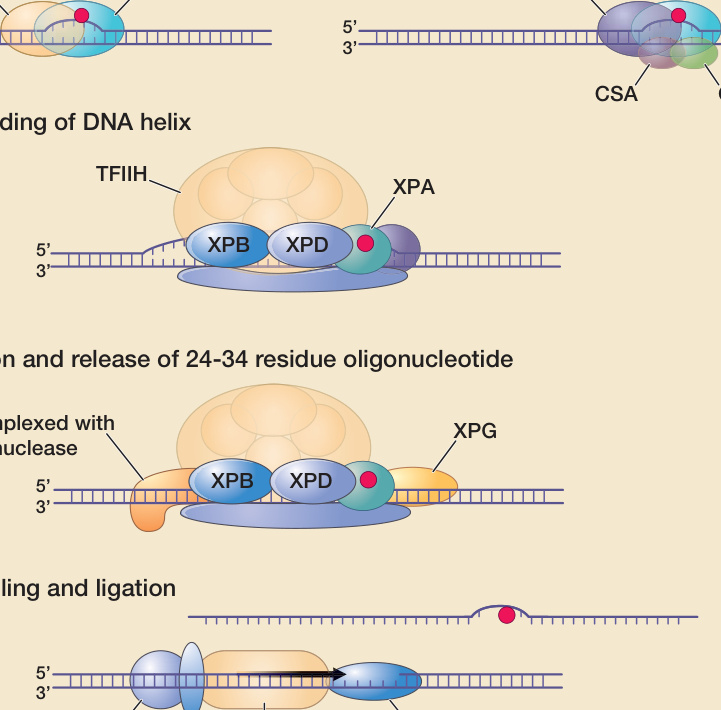
圖 17-12:DNA 核苷酸切除修復示意圖。(A) 右:活躍轉錄基因中的受損 DNA 導致 RNA 聚合酶停滯,此過程涉及 CSA 與 CSB 蛋白。這作為啟動轉錄偶聯 DNA 修復的訊號。左:基因組其餘部分的受損 DNA 由 XPE 與 XPC 基因產物所結合。這作為啟動全基因組修復的訊號。(B) 包含損傷在內的一部分 DNA,由包括 XPB 與 XPD 基因產物在內的一個蛋白質複合體所解旋。這些蛋白質也是 10 次單元基礎轉錄因子 IIH (basal transcription factor IIH, TFIIH) 的一部分。XPA 蛋白可能穩定解旋的 DNA。(C) XPF 與 XPG 核酸酶 (nucleases) 在損傷兩側各做一個單股切口,釋放一段 24 至 34 個殘基的 DNA。(D) 所產生的缺口由 DNA 聚合酶填補,此過程包括增殖細胞核抗原 (proliferating cell nuclear antigen, PCNA) 與複製蛋白 A (replication protein A, RPA) 等蛋白質。CSA, CSB, Cockayne 症候群互補群 A 與 B;ERCC1, 切除修復交叉互補基因 1 (excision repair cross-complementing gene 1);LIG1, 連接酶 1 (ligase 1);XPA, XPC 等, 著色性乾皮症互補群 A、C 等 (xeroderma pigmentosum complementation groups A, C, etc)。
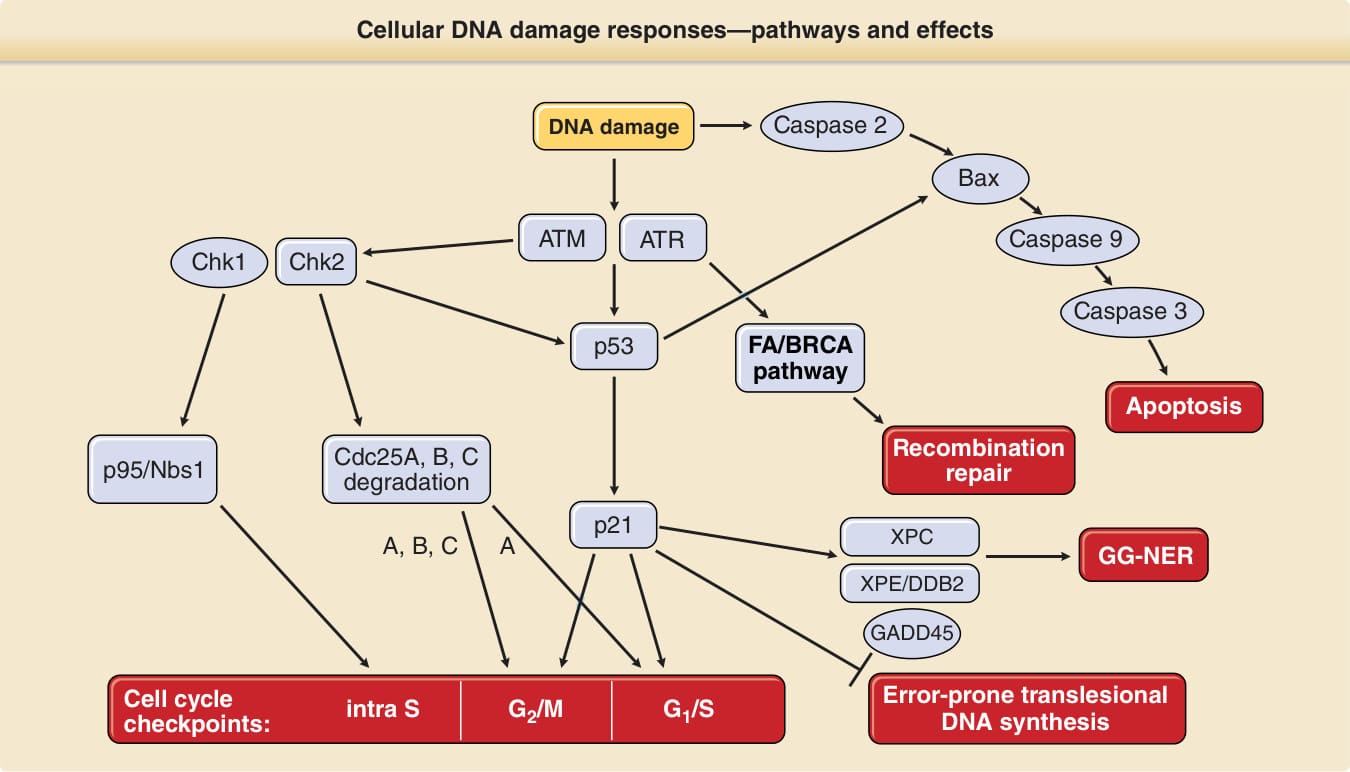
圖 17-13:因應 UVR 誘導 DNA 損傷而調節凋亡、DNA 修復、跨損傷 DNA 合成與細胞週期檢查點活化的訊號傳遞級聯。這是一個高度簡化的圖示,僅描繪複雜交織之 DNA 損傷訊號傳遞網絡中最重要的參與者。傳統的想法是 ATM(運動失調性微血管擴張症突變基因)因應游離輻射 (IR) 而被活化(磷酸化),而 ATR(運動失調性微血管擴張症與 Rad3 相關基因)則因應紫外線 (UV) 輻射而被活化,但較新的資料顯示兩者都被 IR 與 UV 所活化。ATM/ATR 可直接活化(即磷酸化)p53,或透過活化(磷酸化)Chk2(ATM 的標的)或 Chk1(ATR 的標的)間接活化 p53。透過 p21 的轉錄活化及隨後對細胞週期蛋白依賴性激酶(通常驅動細胞從 G1 進入細胞週期 S 期)的抑制,被活化的 p53 活化 G1/S 檢查點(即將細胞停滯於 G1)。G1/S 檢查點也由 Chk1/Chk2 誘導之 Cdc25A 的磷酸化及隨後降解、以及隨後無法活化細胞週期蛋白依賴性激酶所活化。Cdc25A、Cdc25B 與 Cdc25C 的磷酸化及隨後降解也介導 G2/M 停滯,p21 亦然。S 期內停滯由 p95/Nbs1 的活化(磷酸化)所介導。p53 也透過 XPC、XPE/p48 與 GADD45 的轉錄活化來誘導全基因組核苷酸切除修復 (GG-NER)。跨損傷 DNA 合成已被證明由 p53 透過 p21 下調。重組修復由范可尼貧血 (FA)/BRCA 途徑所介導,而後者又依賴於 ATR 的活化。凋亡的粒線體途徑透過 caspase 2 與 p53 對 Bax 的活化、啟動子 caspase 9,以及效應子 caspase 3、6 與 7 而被活化。在遺傳性人類疾病中有缺陷的基因產物以方框標示。DDB2, DNA 損傷結合蛋白 2 (DNA damage binding protein 2);XPC, XPE, 著色性乾皮症互補群 C 與 E。
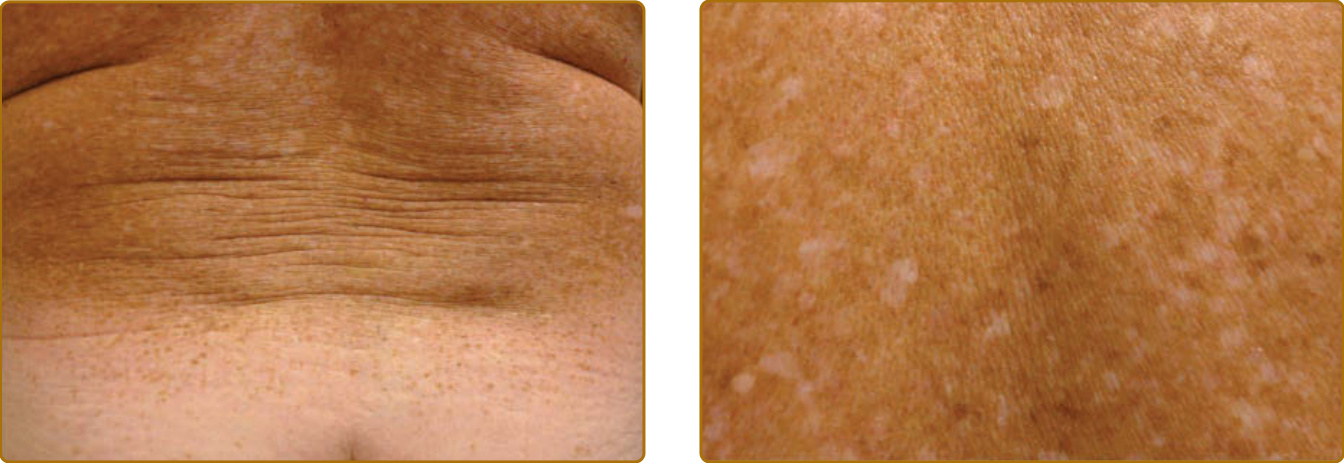
圖 17-14:光老化皮膚的色素變化包括雀斑(為克隆性黑素細胞增生事件的結果)與色素減退(為黑素細胞功能喪失事件)。
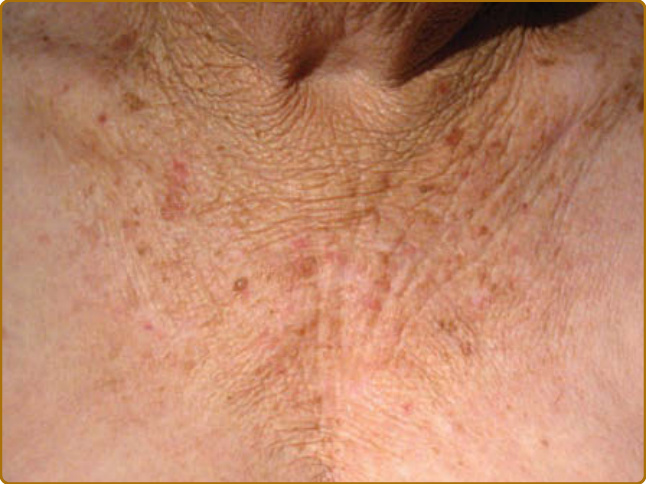
圖 17-15:慢性光老化皮膚中由光化性彈性纖維變性所致的深皺紋。請注意在胸部側面較受光防護的區域缺乏此類皺紋。
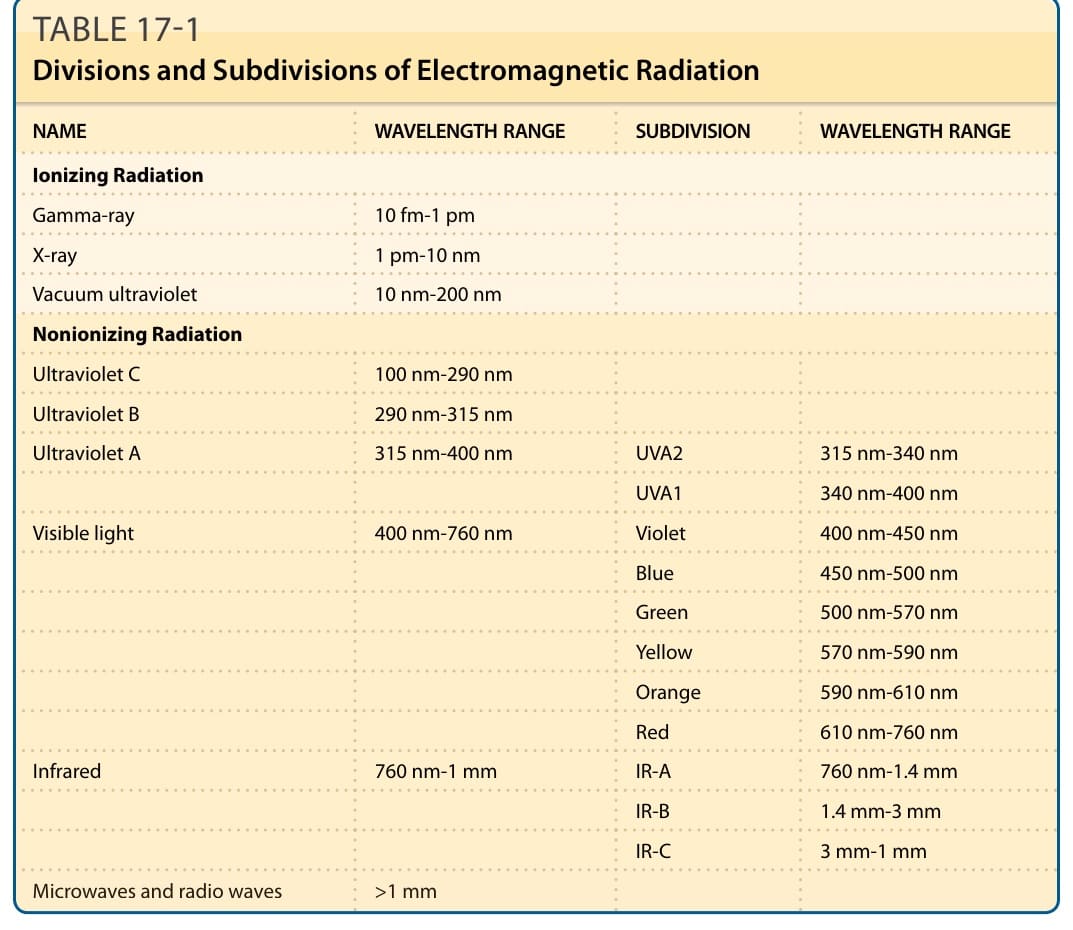
表 17-1:電磁輻射的劃分與細分 (Divisions and Subdivisions of Electromagnetic Radiation)
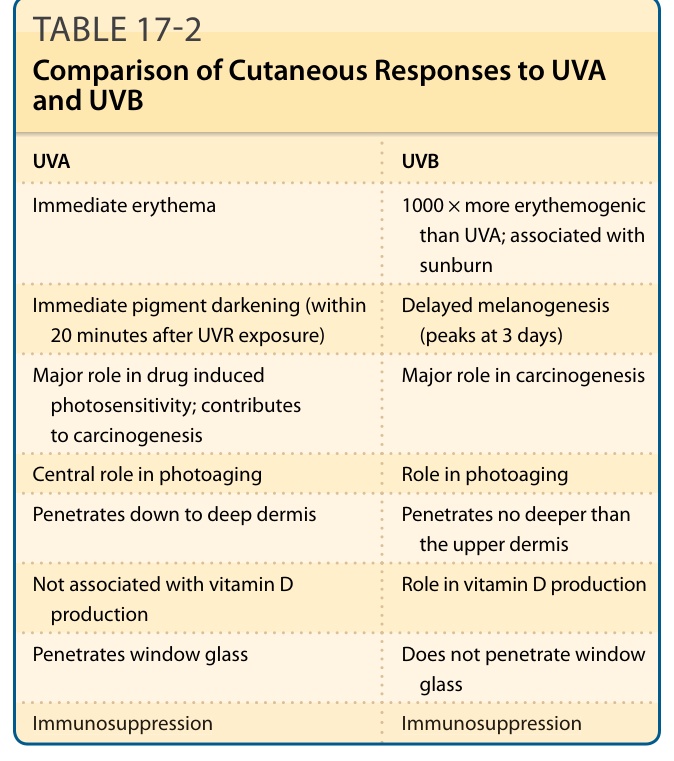
表 17-2:皮膚對 UVA 與 UVB 反應的比較 (Comparison of Cutaneous Responses to UVA and UVB)
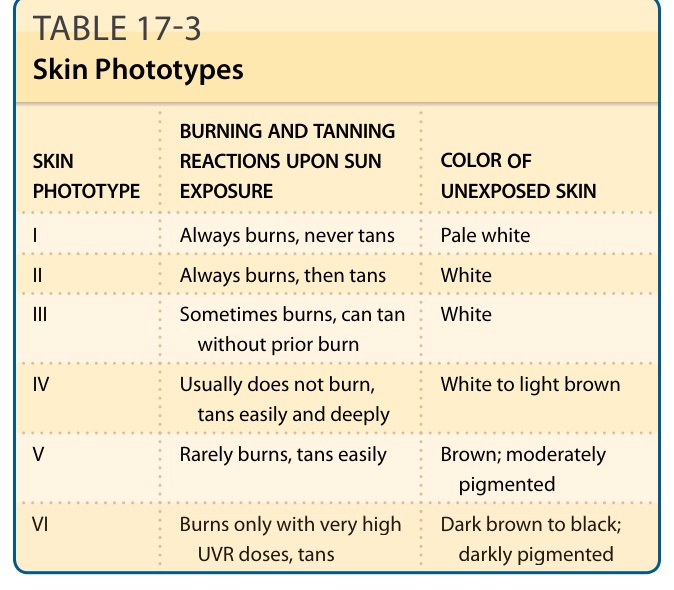
表 17-3:皮膚光型 (Skin Phototypes)
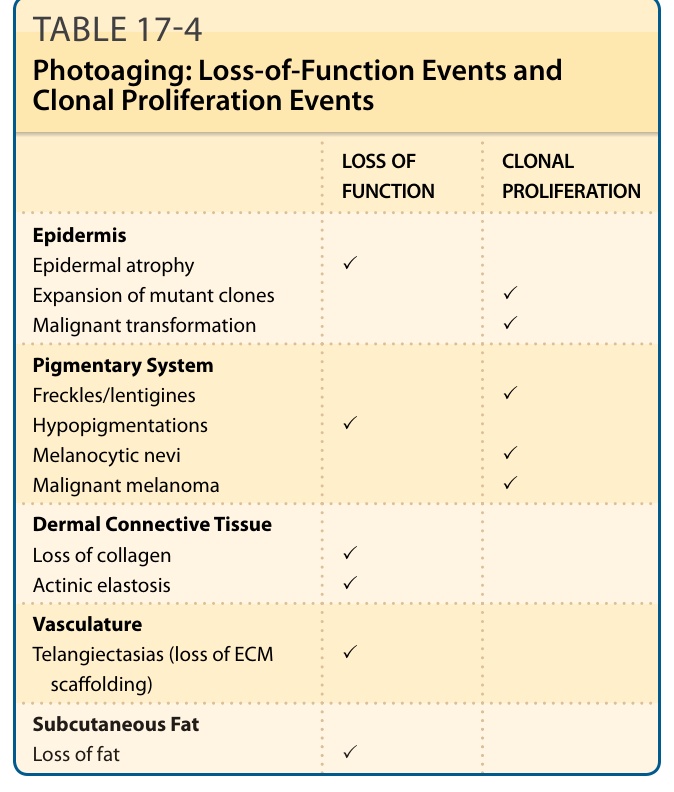
表 17-4:光老化:功能喪失事件與克隆性增生事件 (Photoaging: Loss-of-Function Events and Clonal Proliferation Events)