血管畸形 (Vascular Malformations) 精華筆記
總論 (Overview)
- 定義:血管畸形 (vascular malformations) 起因於子宮內第 4 至第 10 週血管發育的錯誤;隨病人成比例生長、通常不消退、界線清楚且局部性。大多為散發性 (sporadic),少數家族為遺傳性。
- 分類(Mulliken/Glowacki 生物學分類,後由 ISSVA 採用):血管異常分兩大類——(1) 血管腫瘤 (vascular tumors,有細胞增殖,如血管瘤);(2) 血管畸形 (結構性異常),依血管類型分為微血管 (capillary)、靜脈 (venous)、淋巴管 (lymphatic)、動靜脈 (arteriovenous) 及混合型。
- 依血流動力學分為慢流型 (slow flow:微血管、淋巴管、靜脈、混合) 與快流型 (fast flow:動脈、動靜脈、混合)。
- 流行病學:影響約 0.3% 人口,多數為微血管畸形 (CM);大多先天性,但可能晚期才診斷。
- 致病機轉:由各種基因的遺傳性或體細胞 (somatic) 突變引起(見表 147-3)。
- 診斷:90% 淺表畸形靠臨床診斷。病理可見增大、迂曲、內皮靜止的血管,無實質腫塊、無細胞增殖。影像首選都卜勒超音波 (Doppler) 與 MRI。基因檢測:體細胞突變需切片受累組織;遺傳性突變抽血即可。
- 處置:治療困難、常無法完全治癒且會復發;廣泛/複雜病灶須多專科團隊處置。
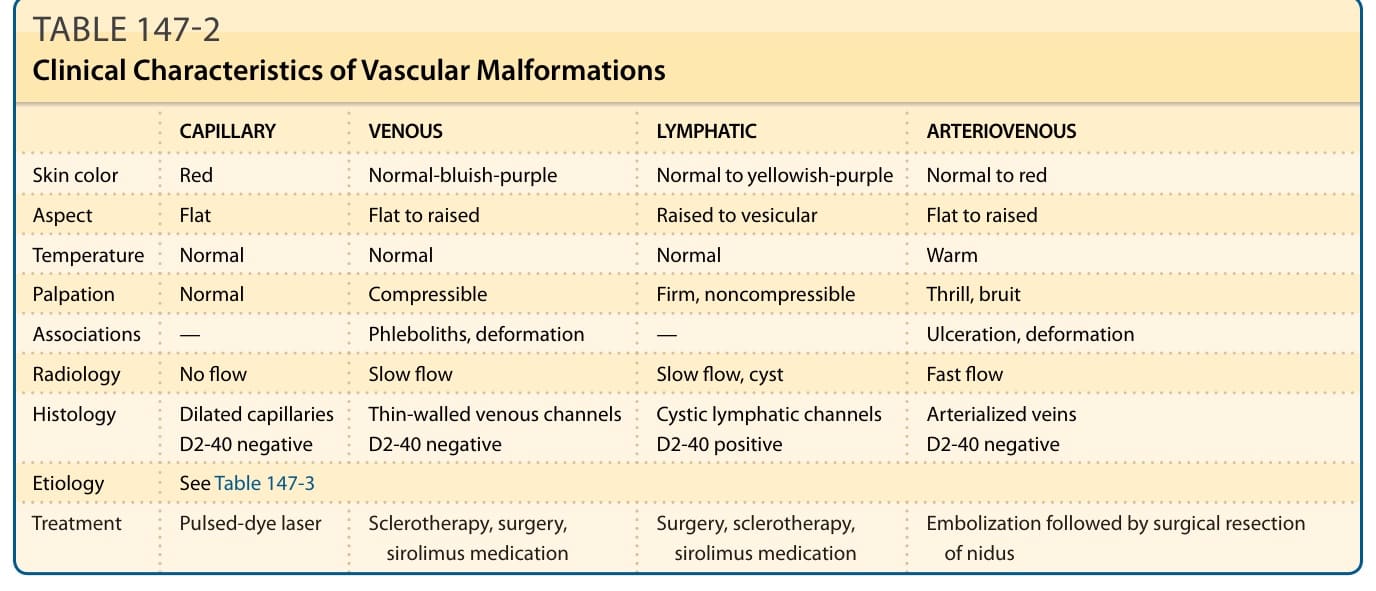
表 147-2:血管畸形的臨床特徵(依血管類型比較膚色、觸診、放射學、組織學與治療)
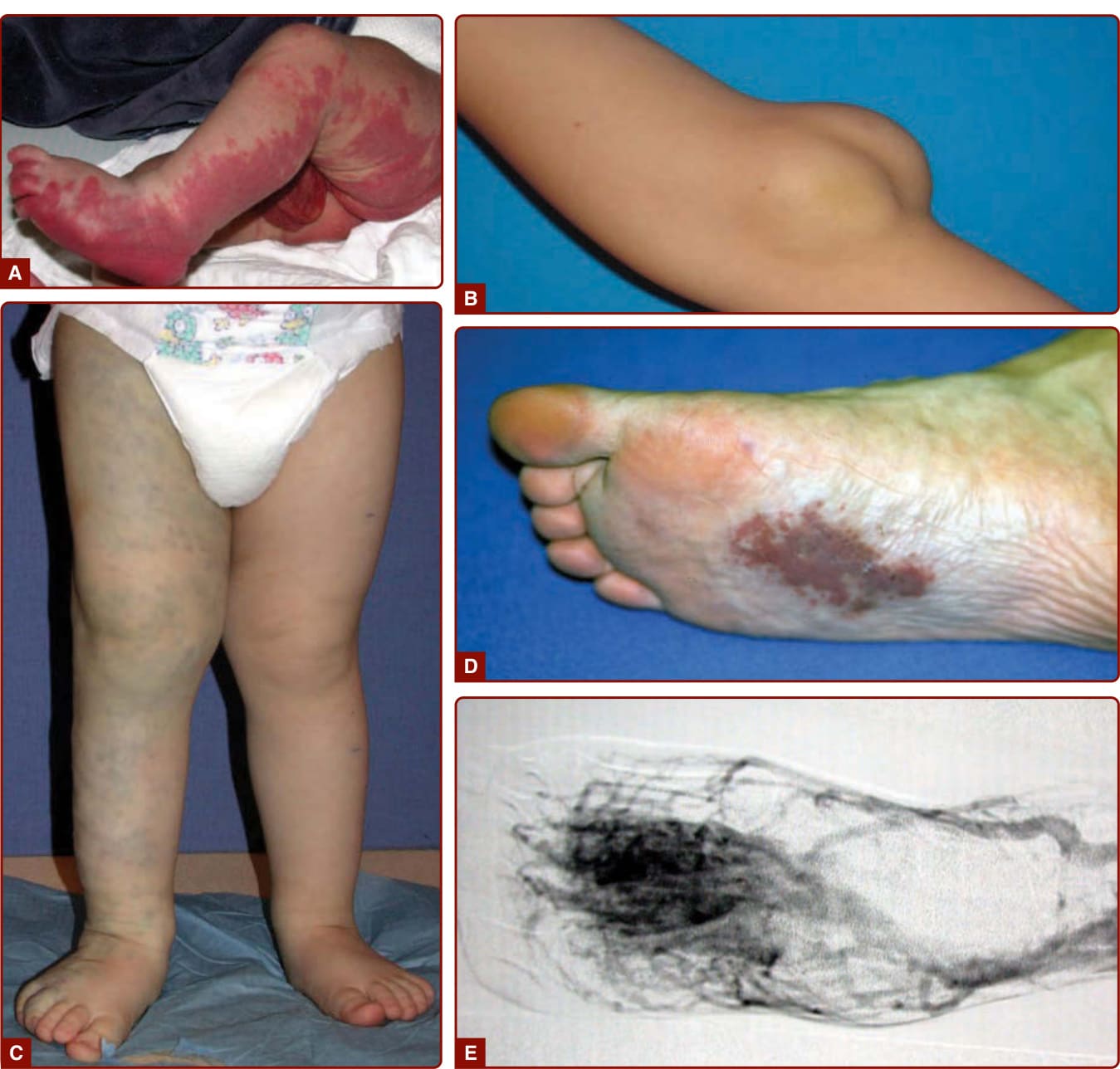
圖 147-1:各類血管類型的血管畸形(A 微血管、B 淋巴管、C 靜脈、D-E 動靜脈含病灶核心 nidus)
微血管畸形 (Capillary Malformations, CM)
- 定義/流行病學:俗稱葡萄酒色斑 (port-wine stain),微血管床的先天性慢流型畸形,盛行率 0.3%,無性別偏向。多為散發性;遺傳性時通常多發,屬 CM-AVM 表型。
- 臨床表現:紅色、均質、先天性、常單側、非正中線;顏色由粉紅紅至深紫,呈地圖狀或皮節分布;扁平、無痛、不自發出血、觸診不溫熱(與 AVM 相反)。半數位於顏面,沿三叉神經分布(V1 前額上眼瞼、V2 下眼瞼臉頰上唇、V3 下唇下巴下頜)。遺傳性 CM (CM-AVM1/2) 通常小、多灶、圓至橢圓、常有蒼白暈 (pale halo)。
- 須與「鸛咬痕/天使之吻/鮭魚斑/單純母斑/Unna 母斑」鑑別:後者位於頸後 (81%)、眼瞼 (45%)、眉間 (33%),約 1–4 歲自發消失(Unna 母斑除外);真正 CM 終生持續。
- 症候群(皆非遺傳性):
- 斑痣性血管錯構瘤病 (PPV):體節分布 CM 合併色素病灶(蒙古斑、斑點母斑等),60% 合併全身/內臟/神經徵象。
- Sturge-Weber 症候群 (SWS):額眼瞼區 (V1) CM 合併同側軟腦膜及脈絡膜微血管-靜脈畸形 (CVM);高癲癇、智能不足風險,合併青光眼、牛眼、視網膜剝離。
- 伴過度生長的瀰漫性微血管畸形 (DCMO):半身肥大、網狀界線不清 CM、不依循 Blaschko 線。
- 巨頭症-微血管畸形 (M-CM):唇紅緣/鼻尖深色 CM 合併巨頭症(特異徵象),有巨腦症及智能不足風險。
- CM-AVM 表型:CM-AVM1 由 RASA1 失活突變、CM-AVM2 由 EPHB4 失活突變引起;20%–30% 風險合併皮膚、腦或脊椎 AVM。CM-AVM1:97% 有 CM、24% 有 AVM/AVF、8% Parkes Weber;CM-AVM2 各為 93%、19%、7%。
- 病程/併發症:出生即存在、從不自發消退;新生兒初期略褪色後紅色穩定。青春期及晚期緩慢增厚變深、變隆起結節狀,可發生化膿性肉芽腫 (pyogenic granuloma) 與軟組織/骨性肥大。V2/V3 或四肢的 CM 易致軟硬組織肥大、骨骼不對稱、開咬畸形。Meyerson 現象(CM 區內異位性皮膚炎/乾癬/痤瘡更嚴重)。
- 致病機轉:顏面 CM 由神經嵴異常細胞克隆性擴增引起;GNAQ 體細胞活化突變為顏面 CM 與 SWS 之因。
- 診斷與追蹤:病理見真皮乳頭層及上網狀層擴張微血管、S100 顯示異常神經支配。SWS 之 CM 需眼科與神經學檢查(出生最初數月開始、每年至青春期)加腦部 MRI;下肢廣泛 CM 約 4–6 歲起做肢體長度測量 (scaniometry);腰薦部 CM 做脊髓 MRI;CM-AVM 篩 RASA1/EPHB4,合併巨頭症者篩 PTEN。
- 處置:
- 雷射為大多數 CM 的黃金標準:脈衝染料雷射 (pulsed-dye laser),波長 585 或 595 nm、短脈衝持續 400–1500 ms;需多次療程(6–12 次)、可能需全身麻醉;頸顏面與軀幹比四肢有效;併用動態冷卻系統 (dynamic cooling)。雷射對肥大無效,停止後可能復發。
- 化膿性肉芽腫或唇肥大可考慮輪廓切除 (contour resection)。
- 腿長差異 >1.5 cm 需鞋墊;約 11–13 歲時有時需骨骺固定術 (epiphysiodesis)。
- 遮瑕(如 Covermark)、遺傳與心理諮詢。
- 預防:SWS 出生後立即眼科追蹤;預防性抗癲癇藥物有人倡議但缺乏隨機研究支持。
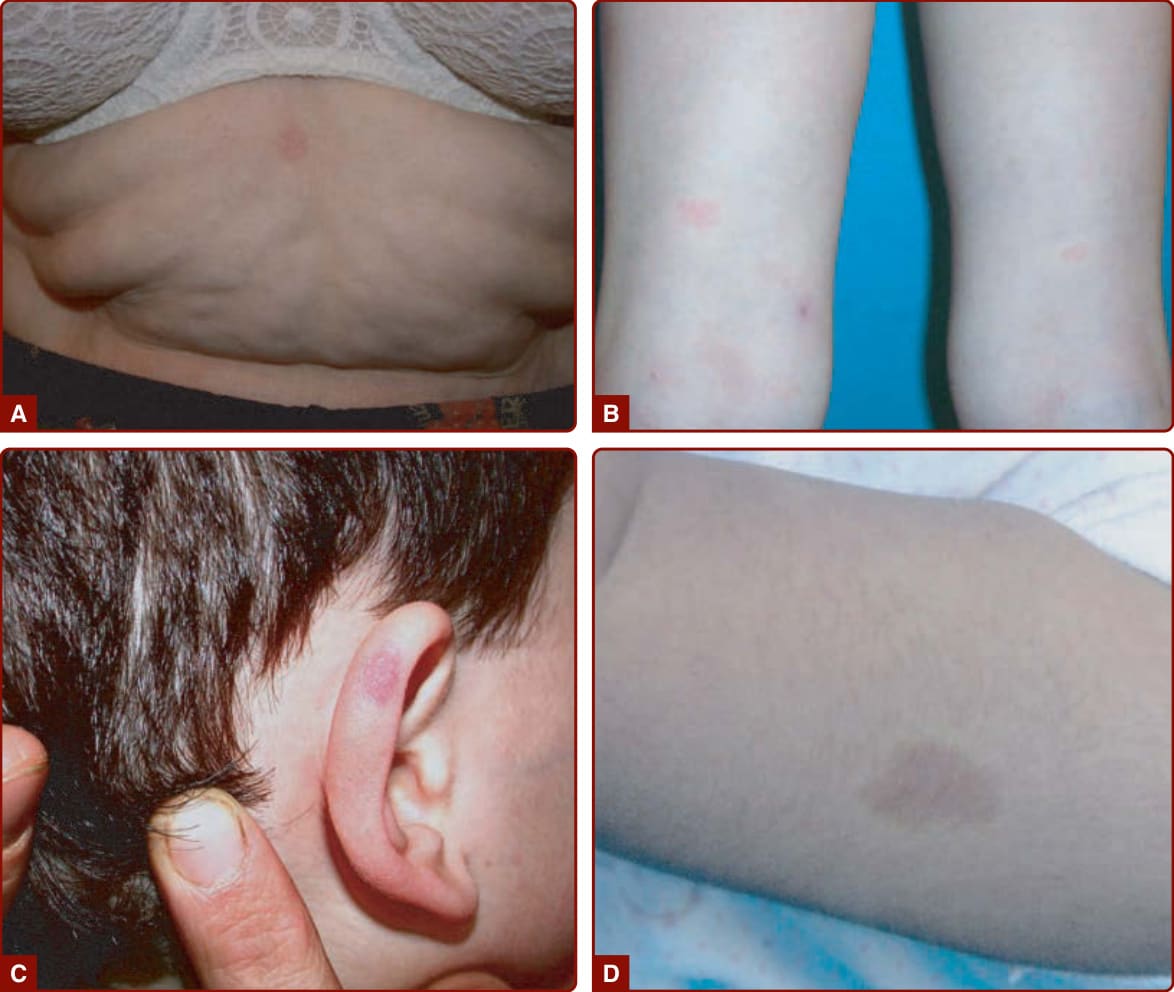
圖 147-6:屬於 CM-AVM 的小型非典型微血管畸形,D 圖可見病灶周圍蒼白暈 (pale halo)
靜脈異常 (Venous Anomalies, VM)
- 定義/流行病學:靜脈床的先天性慢流型畸形;轉介專科中心最常見,整體發生率約 1/10,000(遠低於 CM)。半數位於頸顏面、37% 在四肢。1% 為遺傳性皮膚黏膜靜脈畸形 (VMCM)、5% 為遺傳性球體靜脈畸形 (GVM);VMCM 與 GVM 外顯率隨年齡上升,20 歲達最大(VMCM 87%、GVM 92.7%)。
- 臨床表現:偏藍、可壓陷 (compressible)、有靜脈石 (phleboliths)、皮溫正常、無震顫雜音;下垂位較大、加壓可排空;除非血栓否則不痛。深部 VM 表皮色正常,常青春期才因疼痛診斷。
- 遺傳/症候群亞型:
- VMCM:小、多灶 (>80% 病人有 >2 病灶,多 <5 cm)、侵犯唇舌頰黏膜。
- 多灶性靜脈畸形 (MVM):散發但多發,類似 VMCM 但無家族史。
- GVM(球體靜脈畸形):藍紫色、隆起、鵝卵石面、角化過度、多灶、常在四肢;觸診常疼痛、不可壓陷、無法完全排空(與 VM 相反)、罕見於黏膜、無腸道出血。
- 疣狀靜脈畸形 (VVM,舊稱疣狀血管瘤):WT1 與 GLUT-1 陽性但出生即有、不消退;主在下肢,常潰瘍,不侵犯肌肉/深部。
- 藍色橡皮泡母斑症候群 (BRBN):一大型「主導性」VM 加多發小型深藍乳頭狀病灶(掌蹠),合併多發胃腸道 VM 致慢性貧血、可急性出血致死。
- Maffucci 症候群:多發內生軟骨瘤 (enchondromas) 合併遠端四肢皮下 VM,IDH1/IDH2 突變,40% 惡性化(主為軟骨肉瘤)。
- 併發症:不對稱與進行性變形;疼痛(晨起)、出血、阻塞重要結構;顳肌 VM 致偏頭痛、咽喉 VM 致睡眠呼吸中止;四肢 VM 侵犯肌肉關節致無力、腿長差異、膝關節內出血致早發關節病。慢性局部血管內凝血障礙 (LIC):約 40% 病人 D-二聚體升高 (>500 ng/mL);併低纖維蛋白原為嚴重 LIC,手術/硬化療法/荷爾蒙可觸發轉為 DIC。
- 致病機轉:VMCM 為體染色體顯性,TEK 基因(編碼 TIE2)生殖細胞突變,造成受體過度磷酸化、活化 PI3K/AKT。GVM 為 glomulin 顯性功能喪失突變加體細胞二次打擊。散發 VM:60% 由 TIE2 體細胞活化突變、另 20% 由 PIK3CA 突變引起(皆活化 PI3K/AKT)。BRBN 亦由 TIE2 突變(體細胞、血中測不到)。
- 診斷:純 VM 或含靜脈成分者應查凝血(血小板、纖維蛋白原、D-二聚體;首診時做、青春期前後及疼痛時複查);GVM 則否。病理:VM 為薄壁擴張靜脈通道、不連續管壁平滑肌細胞;GVM 見圓/多角形「球體細胞 (glomus cells)」(SMA、vimentin 陽性,desmin、vWF、S100、glomulin 陰性)。影像:平片可見靜脈石鈣化(LIC 特異徵象);都卜勒確認慢流、靜脈通道可被探頭壓陷(與 LM 相反);MRI (T1/T2 + 脂肪抑制) 定位;BRBN 須內視鏡/結腸鏡查胃腸道病灶。
- 病程/預後:成比例生長、不自發消退、無惡性轉化(VVM/BRBN 等);外傷或荷爾蒙(青春期、懷孕)可致疼痛或擴大;主要併發症為危及生命的胃腸道出血與呼吸道壓迫;Maffucci 惡性化高 (40%)。
- 處置(多專科):
- 醫療:四肢 VM 用壓迫衣物(GVM 反而加痛、不適用);有 LIC 徵象者圍手術期給低分子量肝素 (LMWH,100 IU anti-Xa/kg/day),術前 24 小時起、術後續 5 天;局部血栓疼痛則治療約 2 週。
- 標靶藥:雷帕黴素 (rapamycin / sirolimus) 對難治廣泛 VM 與 BRBN 有效(疼痛、凝血障礙、體積改善;出血 24 小時內止),副作用輕微(黏膜炎、頭痛、疲倦、腹瀉);不適用於有反應於標準照護的小型無症狀慢流畸形。
- 程序:經皮病灶內硬化療法為 VM 主要治療(GVM 例外);無水乙醇 (absolute ethanol) 最有效;替代為 sodium tetradecyl-sulphate 泡沫或 lauromacrogol。手術切除常於硬化療法後;GVM/VVM 切除常需植皮;BRBN 用 Nd:YAG 雷射、氬離子電漿凝固、band-ligation。
- 諮詢:VMCM/GVM 告知 50% 遺傳風險。
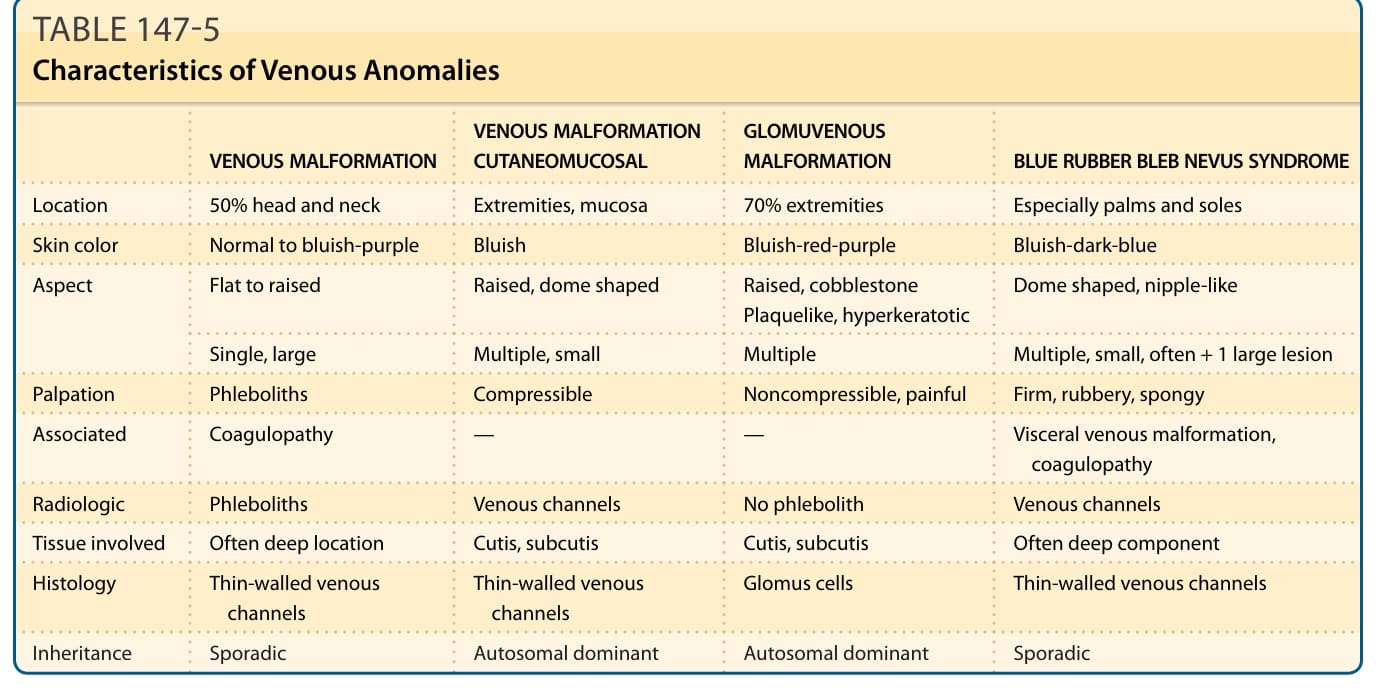
表 147-5:靜脈異常的特徵(VM、VMCM、GVM、BRBN 之位置、膚色、觸診、組織學與遺傳比較)
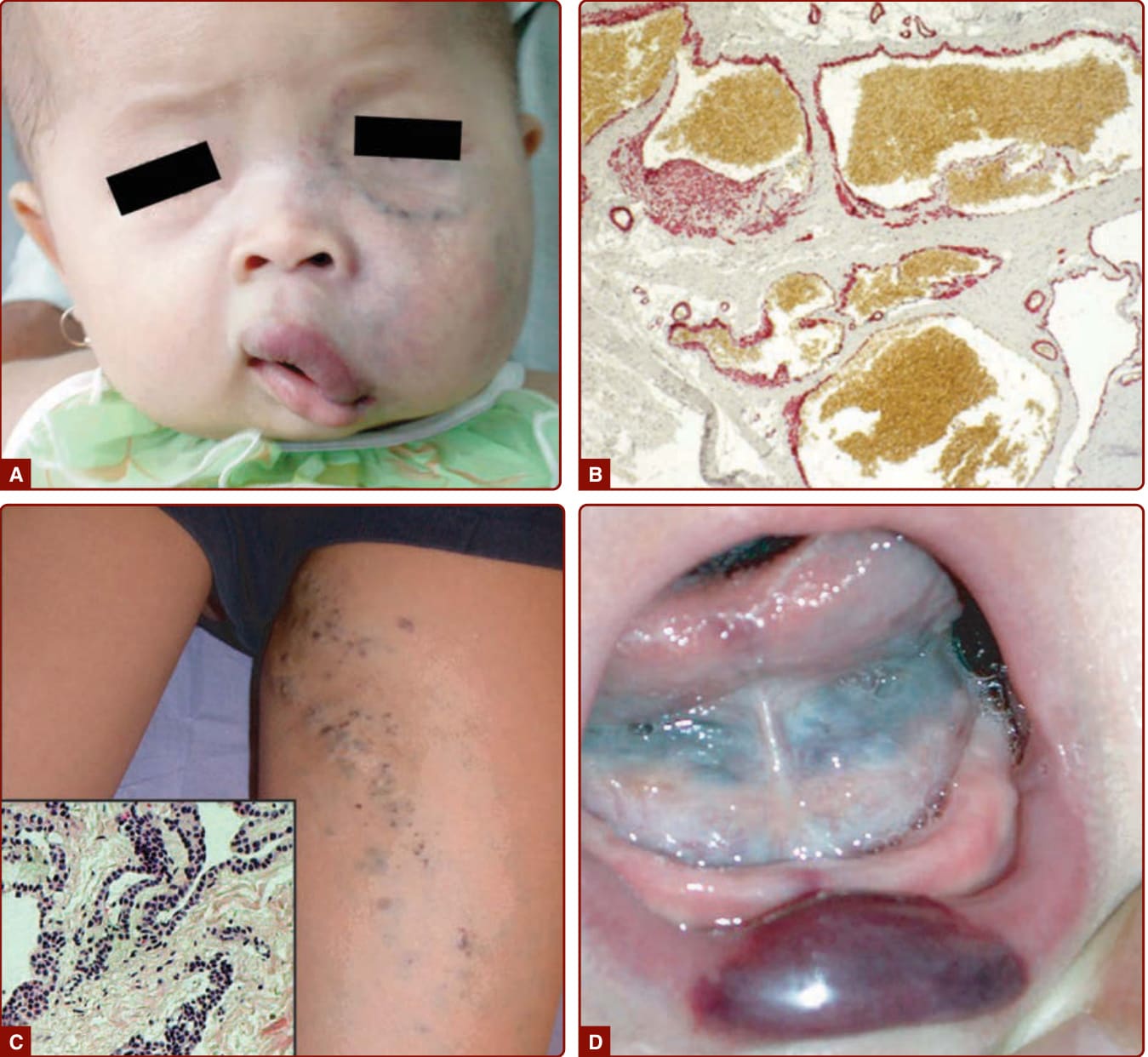
圖 147-13:靜脈畸形。C 為大腿內側淺表球體靜脈畸形 (GVM),組織學見管壁球體細胞 (glomus cells)
淋巴管異常 (Lymphatic Anomalies, LM)
- 定義/流行病學:淋巴管的先天性、散發性、慢流型畸形,由充滿淋巴液的微囊 (microcysts) 或巨囊 (macrocysts, >1 cm) 構成;發生率未知,較 VM 少見。巨囊型可第一孕期子宮內診斷,多數 2 歲前診斷。原發性淋巴水腫 (primary lymphedema) 為較全身性病況,可遺傳(多體染色體顯性),高達 20% 為遺傳性,分先天型(Milroy 病)與晚發型(Meige 病)。
- 症候群:
- Klippel-Trenaunay 症候群 (KTS):微血管-淋巴管-靜脈混合畸形合併肢體肥大,70% 侵犯下肢;大腿外側持續性胚胎靜脈為特異徵象。
- CLOVES 症候群:進行性不對稱肥大、軀幹脂肪瘤樣腫塊、血管畸形、表皮母斑、骨骼異常;須與 Proteus 症候群鑑別。
- 全身性淋巴異常 (GLA):LM 侵犯縱膈、肺、胸膜、胃腸道、骨骼等。
- Gorham-Stout 症候群(骨消失病):骨進行性脫鈣破壞、被淋巴/微血管取代,預後不佳(16% 致死)。
- 臨床表現:微囊(舊稱局限性淋巴管瘤)或巨囊(舊稱囊狀水瘤);充清澈或血清血性液;巨囊型柔軟多分葉界線清楚,微囊型界線不清且侵犯鄰近結構。局限性血管角化瘤 (CLM):混合性、粉紅至藍紅、角化過度。淋巴水腫表現為下肢腫脹;Milroy 病出生即雙側、膝以下,可合併陰囊水腫 (37%)、明顯靜脈、上斜趾甲、乳頭瘤病等。
- 併發症:可因咳嗽、發炎、感染或囊內出血突然增大;復發性蜂窩性組織炎為主要併發症(尤其 KTS,可演為敗血症);舌根/頸顏面受累致呼吸道阻塞;廣泛肢體 LM 致象皮病;可致乳糜胸、蛋白質流失性腸病變;KTS/CLOVES 高肺栓塞風險。
- 致病機轉:LM 及 KTS、CLOVES 由 PIK3CA 鑲嵌/體細胞突變(活化 PI3K/AKT/mTor)引起。原發性淋巴水腫已知 >20 基因突變:Milroy 病為 VEGFR3 遺傳性功能喪失;Lymphedema-distichiasis 為 FOXC2;Emberger 症候群(GATA2,合併血液惡性腫瘤)等。約 35%–40% 家族性病例可找到生殖細胞突變。
- 診斷:病理見擴張、內皮扁平、表現淋巴標記(podoplanin、D2-40、VEGFR-3)的通道。影像:都卜勒見微/巨囊由薄隔膜分隔、不可被探頭壓陷(與 VM 相反);MRI 見囊性與液-液平面;CT 顯示骨侵犯最佳;KTS/CLOVES 須查深部靜脈系統,CLOVES 每 6 個月追蹤至青春期結束(含腹部超音波查威爾姆斯腫瘤)。
- 病程/預後:隨兒童生長,感染/出血時增大;偶有發炎後自體硬化致自發消退。
- 處置:
- 醫療:全身性抗生素治感染與早期蜂窩性組織炎;CLOVES/KTS 圍手術期給預防劑量 LMWH 100 anti-Xa/kg/day,術後續 10–20 天降低肺栓塞風險;雷帕黴素對難治廣泛 LM 及 KTS、GLA 有效。淋巴水腫用彈性襪、按摩、氣壓壓迫裝置。
- 程序:Nd:YAG 或二氧化碳雷射治真皮 LM 水疱;巨囊型抽吸後病灶內注射硬化劑(sodium tetradecyl sulfate、純乙醇、OK432/picibanil、doxycycline、博來黴素 bleomycin)。手術切除為替代方案,復發頻繁(微囊型難與正常組織區分)。
- 諮詢:原發性淋巴水腫須衛教成因/治療與監測計畫。
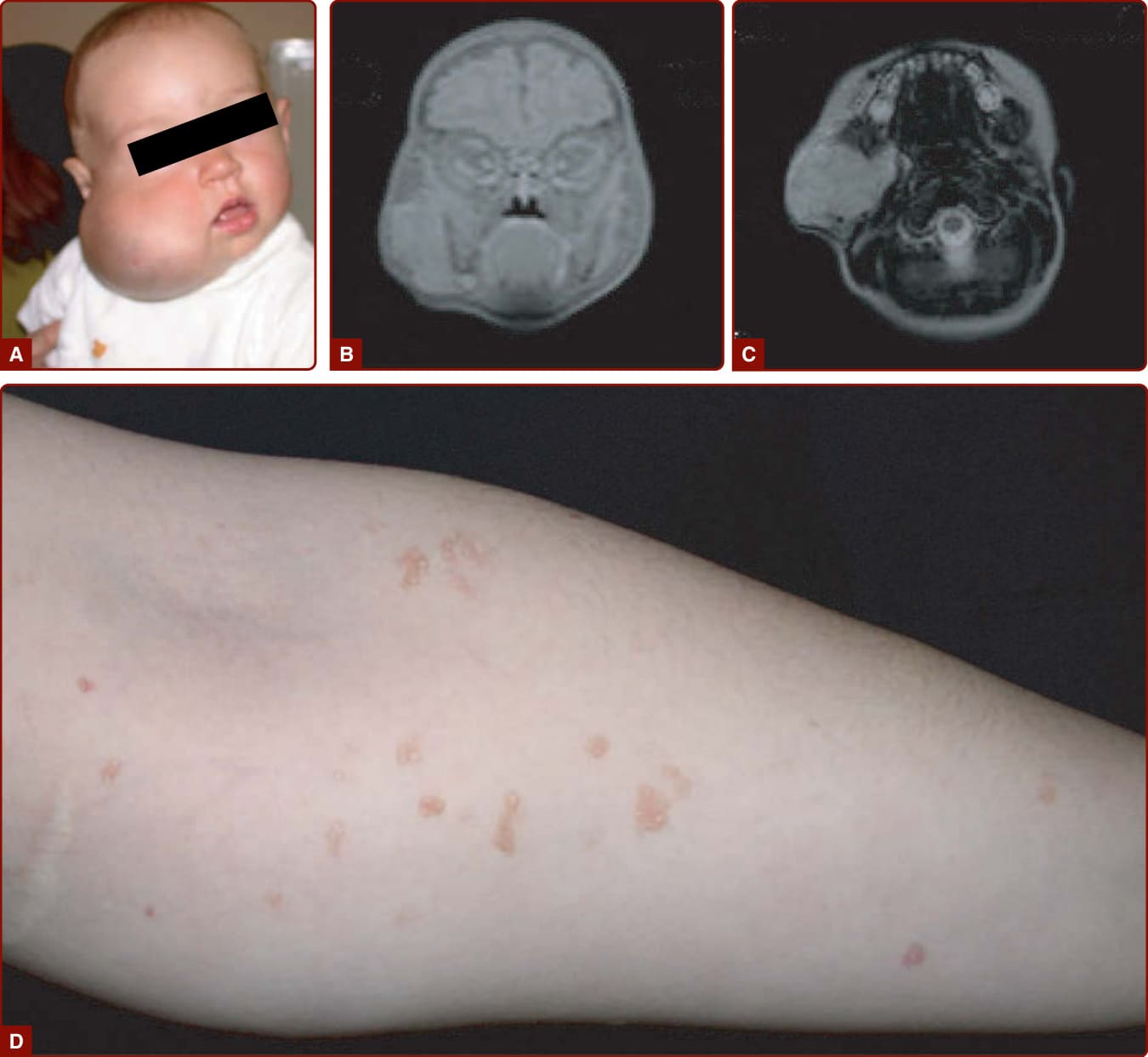
圖 147-16:淋巴管畸形 (LM) 各種樣貌(臉頰廣泛 LM 之藍色變色與 CT、真皮 LM 小型清澈水疱)
動靜脈畸形 (Arteriovenous Malformations, AVM)
- 定義/流行病學:先天性快流型畸形,可隱匿至青春期;發生率未知、罕見、通常散發。特徵為「病灶核心 (nidus)」——多條供應動脈與引流靜脈間直接交通、無正常微血管床。AVF 多為外傷結果。遺傳傾向見 CM-AVM、HHT(盛行率 1/5000)、PHTS。
- 臨床表現:皮膚性、淡色、紅至紫色、界線不清的腫塊,伴震顫 (thrill)、雜音 (bruit)、振幅增加搏動。約 1/3 出生即有、1/3 兒童/青春期出現、其餘成年表現(荷爾蒙與外傷觸發生長)。70% 在頭頸部,不自發消退、隨時間惡化。
- Schobinger 分期:I 期(紅色染斑+雜音搏動)→ II 期(靜脈明顯迂曲)→ III 期(變深、疼痛、潰瘍、出血)→ IV 期(心衰竭)。
- 症候群:
- Bonnet-Dechaume-Blanc / Wyburn-Mason 症候群:顏面+眼眶+腦 AVM,致鼻出血、眼球突出、偏盲。
- Cobb 症候群:同體節皮膚與脊髓 AVM,背/下肢疼痛合併感覺障礙、神經性膀胱。
- 遺傳性出血性微血管擴張 (HHT):三聯徵(多發皮膚黏膜微血管擴張、鼻出血、陽性家族史,Curaçao 標準)。頻率:自發復發鼻出血 90%、皮膚微血管擴張 75%、肝/肺 AVM 30%、胃腸道出血 15%;肺 AVM 致中風/腦膿瘍風險升高。
- Parkes Weber 症候群:四肢大片紅色血管染斑+軟組織骨骼肥大+多發小動靜脈微瘻管;可為 CM-AVM 一部分,可致充血性心衰竭。
- PHTS(PTEN 錯構瘤腫瘤症候群):含 Bannayan-Riley-Ruvalcaba 與 Cowden;巨頭症、陰莖雀斑、快流型 VM (54%)、惡性化風險增加。
- 致病機轉:HHT 由 TGF-β 訊號改變引起(ENG=HHT1、ACVRL1/ALK1=HHT2,較少 SMAD4、GDF2)。CM-AVM1 為 RASA1、CM-AVM2 為 EPHB4 功能喪失突變(活化 RAS-MAPK)。PHTS 之 AVM 由 PTEN 功能喪失。散發顱外 AVM 由 MAP2K1 (MEK) 體細胞突變、腦 AVM 由 KRAS 突變引起。
- 診斷:病理見管壁肌肉增厚的扭曲動靜脈、動靜脈分流。影像:彩色都卜勒見低阻高速動脈血流與搏動性靜脈血流聚集(無腫塊);MRI 優於 CT,血流空隙 (flow voids) 為特異徵象;治療前須動脈攝影 (arteriography) 找供應動脈與病灶核心。基因檢測:合併多灶 CM (CM-AVM) 或微血管擴張 (HHT) 者有指徵;合併巨頭症者篩 PTEN。
- 鑑別:兒童期常被誤認為 CM 或血管瘤,但不成比例的溫熱、搏動、震顫、雜音支持 AVM;都卜勒可區分。
- 病程/預後:隨時間惡化、局部破壞或危及生命出血;不當處置(結紮供應動脈、部分切除病灶核心)會戲劇性擴大 AVM,甚至需截肢。
- 處置(最難治、多專科):
- 治癒目標為栓塞阻塞並完全移除病灶核心;忌部分切除與近端結紮動脈(會招募新供應動脈、阻礙進入病灶核心栓塞)。
- 藥物:肢體 AVM/多發 AVF 用彈性襪穩定保護皮膚;Thalidomide 減少 HHT 鼻出血、亦用於無法切除之第 3 期 AVM。
- 程序:超選擇性栓塞 (superselective embolization)——顆粒須達病灶震央,注射部位盡量靠近病灶核心;除 AVF 外栓塞少能治癒,常為緩解或為手術前置。根除性切除通常於栓塞後進行以減少術中出血,盡量採腫瘤外科切緣;覆蓋可用組織擴張或皮瓣。任何治療後須每年都卜勒/MRI 追蹤至少 5 年。Parkes Weber 應盡量保守。
- 諮詢:CM-AVM、HHT、PHTS 須遺傳諮詢。
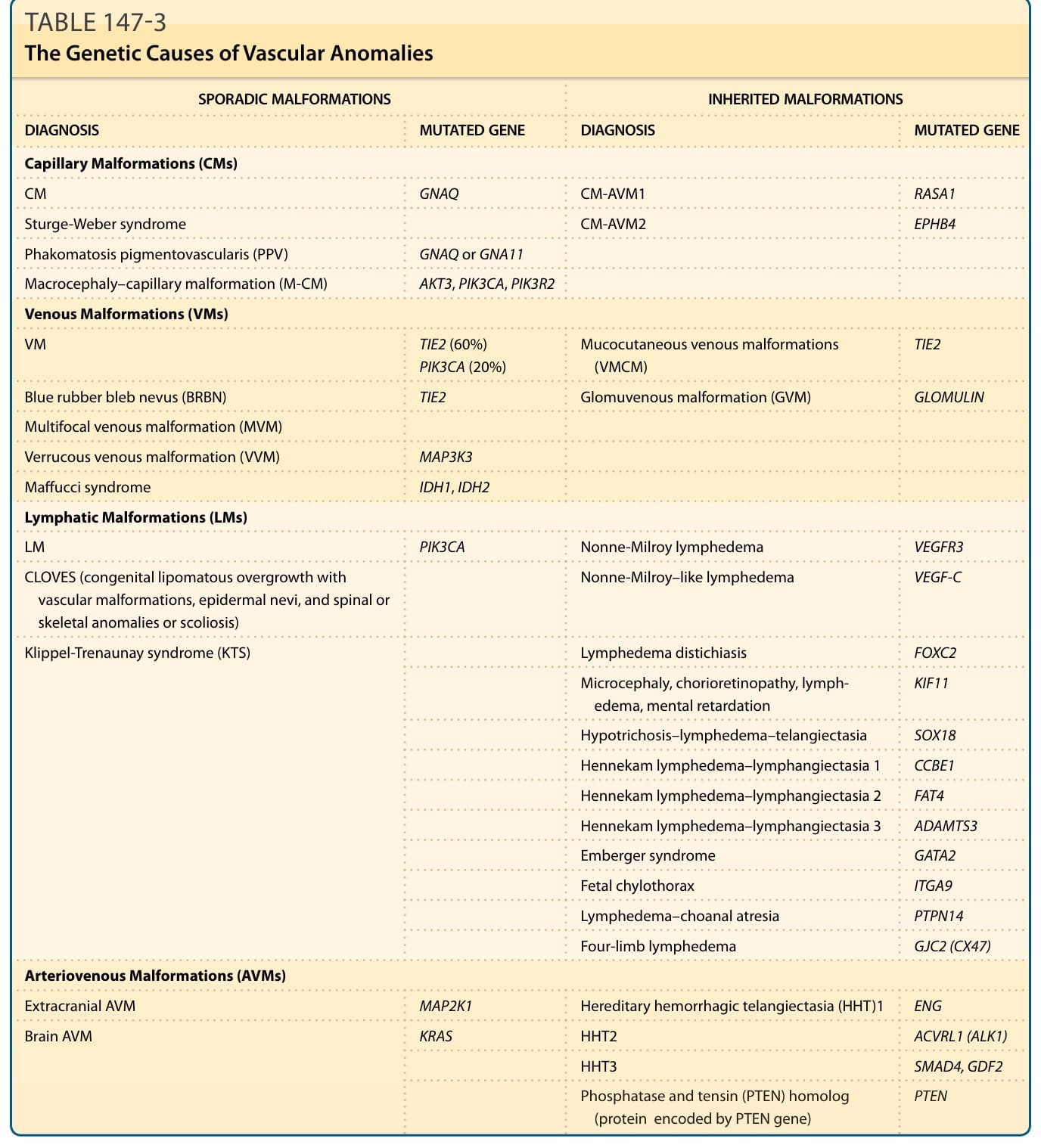
表 147-3:血管異常的遺傳成因(散發性與遺傳性畸形之突變基因對照)
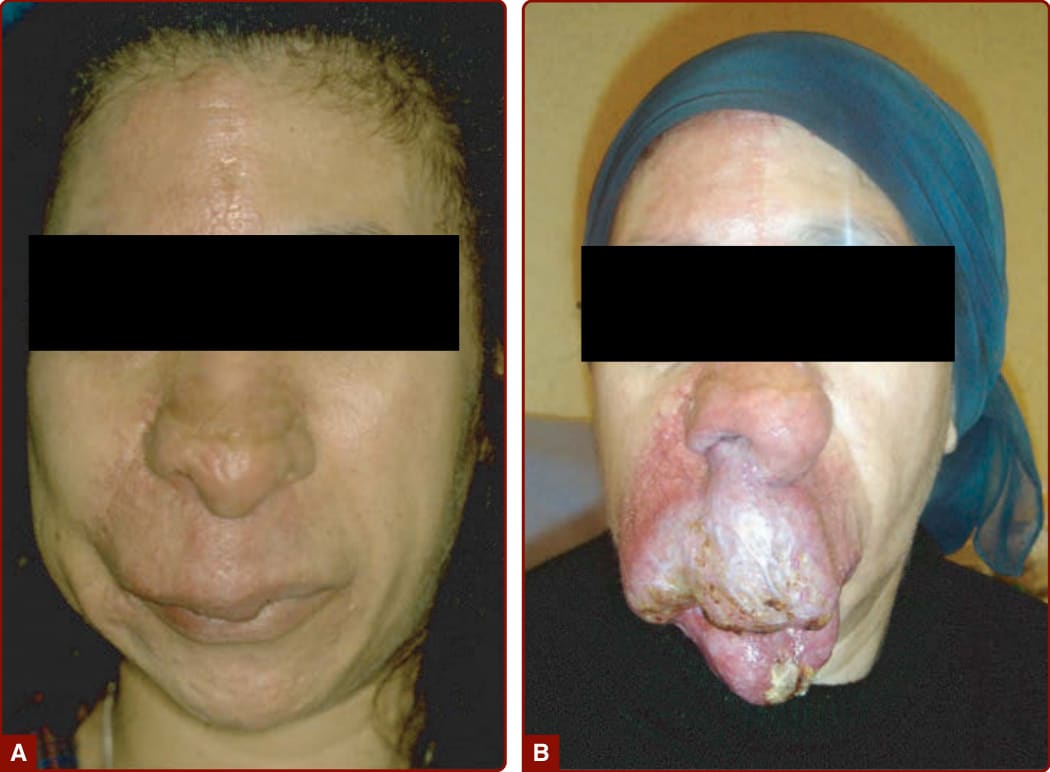
圖 147-23:第 2 期動靜脈畸形 (AVM) 演變為第 4 期 AVM