血管畸形 (Vascular Malformations)
PART 22
血管疾病 (Vascular Diseases)
重點一覽 (AT-A-GLANCE)
■ 全球盛行率約為 0.3%,其中大多數為微血管畸形 (capillary malformations)。
■ 為先天性、局部性(雖然有時範圍廣泛或多發)、界線清楚的各類畸形血管病灶:微血管 (capillary)、靜脈 (venous)、淋巴管 (lymphatic)、動靜脈 (arteriovenous) 及混合型。
■ 組織學上由各類型增大、迂曲的血管所構成。
■ 由各種基因的遺傳性或體細胞 (somatic) 突變所引起。
■ 可為孤立性、混合性,或為某症候群的一部分。
■ 處置:多專科團隊合作 (multidisciplinary approach)。
前言 (INTRODUCTION)
定義 (DEFINITION)
一般認為血管畸形 (vascular malformations) 起因於子宮內生命第 4 至第 10 週期間血管發育過程中的錯誤。大多數血管畸形為散發性 (sporadic),但也已辨識出數個具遺傳形式的家族。它們的異質性很高。¹
分類 (CLASSIFICATION)
多年來,血管異常 (vascular anomalies) 被統稱為血管瘤 (angioma),這妨礙了精確的分類,並導致錯誤的診斷與不當的處置。例如,hemangioma(血管瘤)一詞既被用於血管畸形(常為靜脈性,即海綿狀血管瘤 cavernous hemangioma),也被用於血管腫瘤(草莓狀血管瘤 strawberry hemangioma)。1982 年,Mulliken 與 Glowacki 發展出一套生物學分類,改變了這套命名法。²,³
這套分類系統根據臨床、血流動力學、放射學與組織學特徵來組織血管異常。它將血管異常分為兩大類:(1) 血管腫瘤(具有細胞增殖,其中以血管瘤最為常見;見第 118 章),以及 (2) 血管畸形(血管的結構性異常),後者再依受累的血管類型細分為動脈性 (arterial)、微血管性 (capillary)、淋巴管性 (lymphatic) 或靜脈性畸形 (venous malformations, VMs)。1996 年,此分類被國際血管異常研究學會(International Society for the Study of Vascular Anomalies, ISSVA;表 147-1)採用並進一步發展。⁴
血管畸形大多僅影響單一血管類型(圖 147-1),但也存在混合型畸形。它們依受累的血管類型命名,例如微血管-靜脈畸形 (capillary-venous) 或靜脈-淋巴管畸形 (venolymphatic malformation)。除了孤立型外,血管畸形也出現於某些症候群中,例如 Klippel-Trenaunay 症候群(KTS,微血管-淋巴管-靜脈畸形合併肢體肥大)、Maffucci 症候群(多發性內生軟骨瘤合併多發性靜脈異常及高惡性化發生率)、CLOVES 症候群(先天性脂肪瘤樣過度生長合併血管畸形、表皮母斑及脊椎或骨骼異常或脊柱側彎),或 Parkes Weber 症候群(肢體高流量血管畸形合併軟組織肥大)。⁴
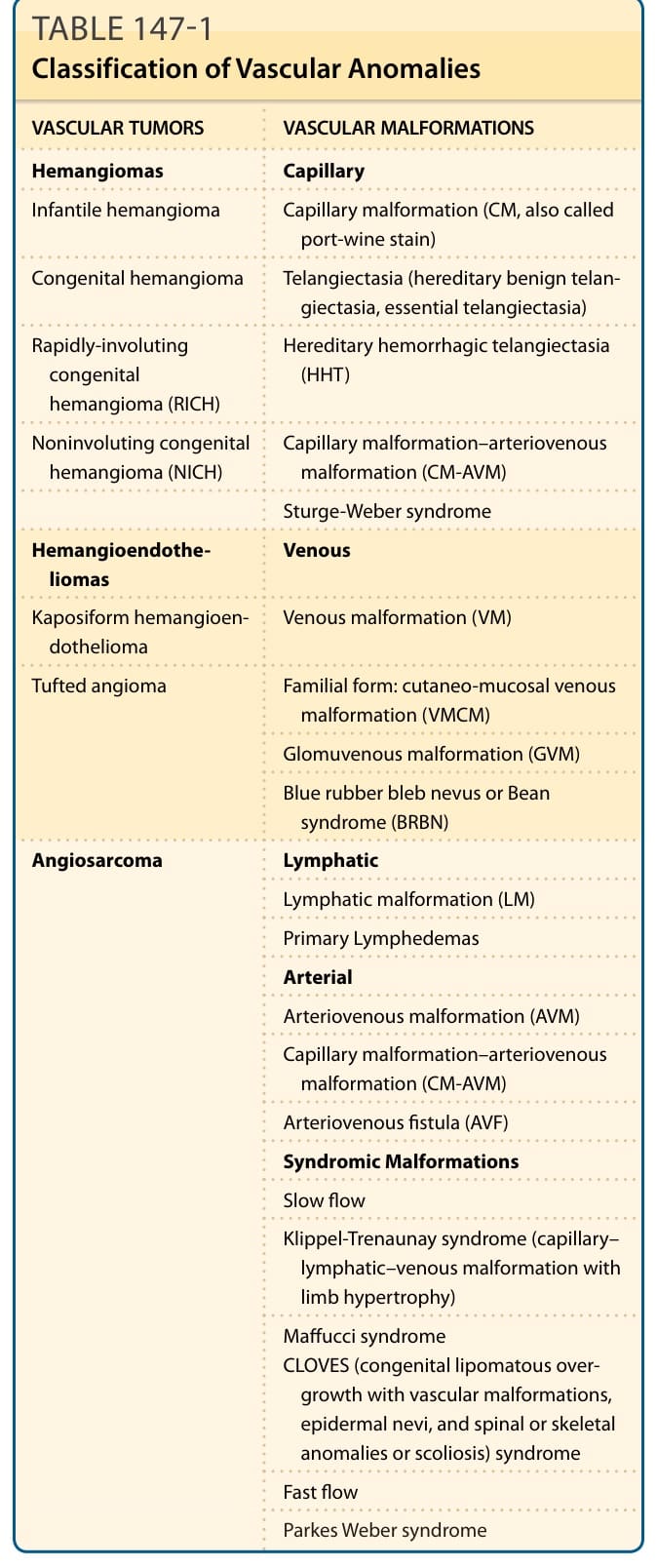
表 147-1:血管異常的分類 (Classification of Vascular Anomalies)
| 血管腫瘤 (Vascular Tumors) | 血管畸形 (Vascular Malformations) |
|---|---|
| 血管瘤 (Hemangiomas) | 微血管性 (Capillary) |
| 嬰兒血管瘤 (Infantile hemangioma) | 微血管畸形(CM,又稱葡萄酒色斑 port-wine stain) |
| 先天性血管瘤 (Congenital hemangioma) | 微血管擴張 (Telangiectasia)(遺傳性良性微血管擴張、本態性微血管擴張) |
| 快速消退型先天性血管瘤 (RICH) | 遺傳性出血性微血管擴張 (HHT) |
| 非消退型先天性血管瘤 (NICH) | 微血管畸形-動靜脈畸形 (CM-AVM) |
| Sturge-Weber 症候群 | |
| 血管內皮瘤 (Hemangioendotheliomas) | 靜脈性 (Venous) |
| 卡波西樣血管內皮瘤 (Kaposiform hemangioendothelioma) | 靜脈畸形 (VM) |
| 叢狀血管瘤 (Tufted angioma) | 家族型:皮膚黏膜靜脈畸形 (VMCM) |
| 球體靜脈畸形 (GVM) | |
| 藍色橡皮泡母斑或 Bean 症候群 (BRBN) | |
| 血管肉瘤 (Angiosarcoma) | 淋巴管性 (Lymphatic) |
| 淋巴管畸形 (LM) | |
| 原發性淋巴水腫 (Primary Lymphedemas) | |
| 動脈性 (Arterial) | |
| 動靜脈畸形 (AVM) | |
| 微血管畸形-動靜脈畸形 (CM-AVM) | |
| 動靜脈瘻管 (AVF) | |
| 症候群性畸形 (Syndromic Malformations) | |
| 慢流型 (Slow flow):Klippel-Trenaunay 症候群(微血管-淋巴管-靜脈畸形合併肢體肥大)、Maffucci 症候群、CLOVES 症候群(先天性脂肪瘤樣過度生長合併血管畸形、表皮母斑及脊椎或骨骼異常或脊柱側彎) | |
| 快流型 (Fast flow):Parkes Weber 症候群 |
流行病學 (EPIDEMIOLOGY)
血管畸形影響約 0.3% 的人口,其中大多數為微血管畸形 (capillary malformations, CMs)。血管畸形大多為先天性,儘管可能在生命較晚期才被診斷出來。⁵
臨床特徵 (CLINICAL FEATURES)
血管畸形隨病人成比例地生長。它們通常不會消退。最常見的情況是界線清楚且局部性。它們可影響身體任何部位,包括內臟。在罕見情況下,它們可能是深部病灶的標記,或是某症候群的首發徵象。血管畸形依血流動力學分為慢流型(微血管性、淋巴管性、靜脈性與混合型)與快流型(動脈性、動靜脈性與混合型)(表 147-2)。⁴ 它們可導致美觀或功能上的損害,罕見情況下甚至危及生命。⁶
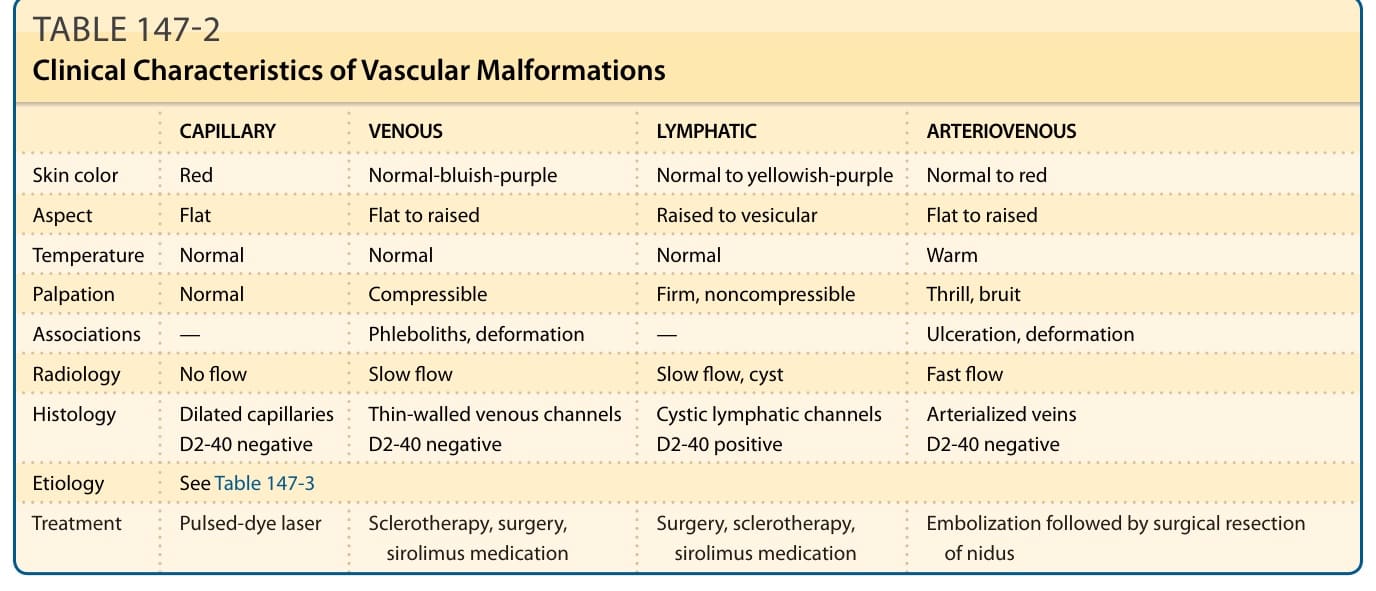
表 147-2:血管畸形的臨床特徵 (Clinical Characteristics of Vascular Malformations)
| 微血管性 (Capillary) | 靜脈性 (Venous) | 淋巴管性 (Lymphatic) | 動靜脈性 (Arteriovenous) | |
|---|---|---|---|---|
| 膚色 | 紅色 | 正常至藍紫色 | 正常至黃紫色 | 正常至紅色 |
| 外觀 | 扁平 | 扁平至隆起 | 隆起至水疱狀 | 扁平至隆起 |
| 溫度 | 正常 | 正常 | 正常 | 溫熱 |
| 觸診 | 正常 | 可壓陷 (compressible) | 堅實、不可壓陷 | 震顫 (thrill)、雜音 (bruit) |
| 相關特徵 | — | 靜脈石 (phleboliths)、變形 | — | 潰瘍、變形 |
| 放射學 | 無血流 | 慢流 | 慢流、囊腫 | 快流 |
| 組織學 | 擴張的微血管,D2-40 陰性 | 薄壁靜脈通道,D2-40 陰性 | 囊性淋巴管通道,D2-40 陽性 | 動脈化靜脈,D2-40 陰性 |
| 病因 | 見表 147-3 | |||
| 治療 | 脈衝染料雷射 (pulsed-dye laser) | 硬化療法、手術、sirolimus 藥物 | 手術、硬化療法、sirolimus 藥物 | 栓塞後手術切除病灶核心 (nidus) |
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
已辨識出數個基因在遺傳形式的血管畸形中發生突變(表 147-3)。體細胞遺傳突變 (somatic genetic mutations) 也已被闡明為許多常見散發性病灶的成因。⁷⁻¹⁵
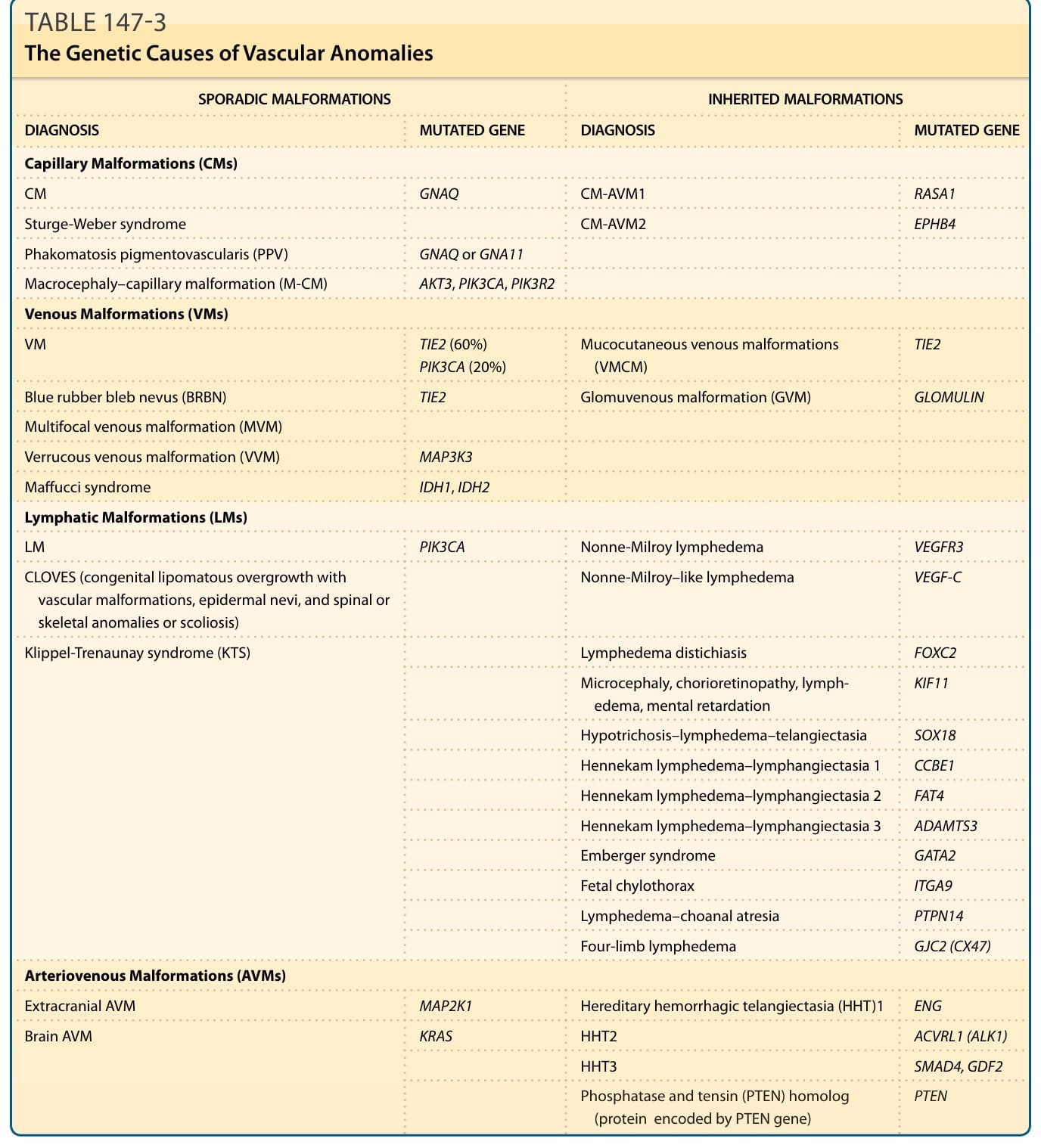
表 147-3:血管異常的遺傳成因 (The Genetic Causes of Vascular Anomalies)
| 診斷 | 散發性畸形之突變基因 | 遺傳性畸形之診斷 | 遺傳性畸形之突變基因 |
|---|---|---|---|
| 微血管畸形 (CMs) | |||
| CM | GNAQ | CM-AVM1 | RASA1 |
| Sturge-Weber 症候群 | GNAQ | CM-AVM2 | EPHB4 |
| 斑痣性血管錯構瘤病 (PPV) | GNAQ 或 GNA11 | ||
| 巨頭症-微血管畸形 (M-CM) | AKT3、PIK3CA、PIK3R2 | ||
| 靜脈畸形 (VMs) | |||
| VM | TIE2 (60%)、PIK3CA (20%) | 皮膚黏膜靜脈畸形 (VMCM) | TIE2 |
| 藍色橡皮泡母斑 (BRBN) | TIE2 | 球體靜脈畸形 (GVM) | GLOMULIN |
| 多發性靜脈畸形 (MVM) | |||
| 疣狀靜脈畸形 (VVM) | MAP3K3 | Maffucci 症候群 | IDH1、IDH2 |
| 淋巴管畸形 (LMs) | |||
| LM | PIK3CA | Nonne-Milroy 淋巴水腫 | VEGFR3 |
| CLOVES 症候群 | PIK3CA | Nonne-Milroy 樣淋巴水腫 | VEGF-C |
| Klippel-Trenaunay 症候群 (KTS) | PIK3CA | 淋巴水腫-雙行睫 (distichiasis) | FOXC2 |
| 小頭症、脈絡視網膜病變、淋巴水腫、智能不足 | KIF11 | ||
| 少毛症-淋巴水腫-微血管擴張 | SOX18 | ||
| Hennekam 淋巴水腫-淋巴管擴張 1 | CCBE1 | ||
| Hennekam 淋巴水腫-淋巴管擴張 2 | FAT4 | ||
| Hennekam 淋巴水腫-淋巴管擴張 3 | ADAMTS3 | ||
| Emberger 症候群 | GATA2 | ||
| 胎兒乳糜胸 | ITGA9 | ||
| 淋巴水腫-後鼻孔閉鎖 | PTPN14 | ||
| 四肢淋巴水腫 | GJC2 (CX47) | ||
| 動靜脈畸形 (AVMs) | |||
| 顱外 AVM | MAP2K1 | 遺傳性出血性微血管擴張 (HHT) 1 | ENG |
| 腦部 AVM | KRAS | HHT2 | ACVRL1 (ALK1) |
| HHT3 | SMAD4、GDF2 | ||
| PTEN(磷酸酶與張力蛋白同源物,由 PTEN 基因編碼之蛋白) | PTEN |
診斷 (DIAGNOSIS)
90% 的淺表性畸形之診斷通常基於臨床特徵。
輔助檢查 (SUPPORTIVE STUDIES)
病理 (Pathology):組織學上,血管畸形由增大、迂曲且內皮處於靜止狀態的血管所構成。與血管瘤相反,既沒有實質性腫塊,也沒有明顯的細胞增殖。
影像 (Imaging):放射學檢查用於勾勒畸形的範圍,但除非畸形位置深在,否則很少用於診斷。都卜勒超音波 (Doppler ultrasonography) 是一種非常有用的非侵入性放射學檢查,能提供區分各種類型的線索。磁振造影(magnetic resonance imaging, MRI)能詳細顯示病灶的延伸範圍與精確位置。¹⁶
基因檢測 (Genetic Testing):由於大量血管異常已知具有生殖細胞 (germline) 或體細胞遺傳成因,基因檢測可用於確認或協助做出精確診斷。對於體細胞突變,需要對受累組織進行切片。對於遺傳性突變,血液檢測即足以做出診斷。
處置 (MANAGEMENT)
血管畸形的治療取決於受累的血管類型、病灶位置、病人年齡與症狀。由於許多病灶範圍廣泛,病人應了解通常無法完全治癒,且會復發。治療可能困難並伴隨嚴重併發症。範圍廣泛或複雜的病灶應始終由多專科團隊處置。
微血管畸形 (CAPILLARY MALFORMATIONS)
重點一覽 (AT-A-GLANCE)
■ 全球皆有發生。盛行率約為 0.3%。
■ 為微血管床的先天性、慢流型畸形。
■ 顏色呈粉紅紅色至紫色;隨時間傾向變深與增厚。
■ 可為某散發性症候群的一部分,例如 Sturge-Weber 症候群或 KTS。
■ 可為遺傳性微血管畸形-動靜脈畸形表型的一部分。
■ 病理:微血管數量與大小增加,並有異常的神經支配。
微血管畸形主要為散發性發生,但也有記載完整的家系顯示體染色體顯性遺傳 (autosomal dominant inheritance)。¹⁷ 當為遺傳性時,它們通常為多發性,並為微血管畸形-動靜脈畸形 (CM-AVM) 表型的一部分,該表型將非典型 CM 與動靜脈畸形 (AVM,見後文「動靜脈畸形」) 相關聯。¹⁸⁻²³
流行病學 (EPIDEMIOLOGY)
CM 俗稱葡萄酒色斑 (port-wine stain),是一種慢流型血管畸形,盛行率為 0.3%。⁵ 無性別偏向。
雖然 CM 最常為孤立性發現,但在罕見情況下,CM 可為隱性脊柱裂閉合不全 (occult spinal dysraphism) 的皮膚標記,尤其當位於腰薦部 (lumbosacral area) 時。²⁴ 其他可受壓退色的粉紅色斑塊,稱為鸛咬痕 (stork bite)、天使之吻 (angel’s kiss)、鮭魚斑 (salmon patch)、單純母斑 (nevus simplex) 或新生兒火焰母斑 (nevus flammeus neonatorum),常與 CM 混淆。它們位於頸後 (nape of the neck, 81%)、眼瞼 (eyelids, 45%) 或眉間 (glabella, 33%)。²⁵ 當位於枕部 (occiput) 時,可使用 Unna 母斑 (Unna nevus) 這一別稱(圖 147-2)。這些斑塊在白人嬰兒中的發生率(42%)遠高於黑人嬰兒(31%)。²⁶ 它們也出現於各種症候群中,例如 Beckwith-Wiedemann 與 Rubinstein-Taybi 症候群。這些病灶(Unna 母斑除外)約在 1 至 4 歲時自發消失。相反地,真正的 CM 則終生持續存在。
症候群性疾患 (SYNDROMIC DISORDERS)
CM 可為某症候群的一部分,例如斑痣性血管錯構瘤病(phakomatosis pigmentovascularis, PPV)、Sturge-Weber 症候群(SWS)、KTS、Parkes Weber 症候群、CLOVES 症候群、PTEN(磷酸酶與張力蛋白同源物,phosphatase and tensin homolog)錯構瘤腫瘤症候群、伴過度生長的瀰漫性微血管畸形(diffuse capillary malformation with overgrowth, DCMO),以及巨頭症-微血管畸形(macrocephaly–capillary malformation, M-CM,又稱巨腦症-微血管畸形-多小腦回症候群)。這些症候群皆非遺傳性。
斑痣性血管錯構瘤病 (PHAKOMATOSIS PIGMENTOVASCULARIS)
一般認為 PPV 是一種影響血管運動神經 (vasomotor nerves) 與黑色素細胞 (melanocytes) 的胚胎發生異常,兩者皆源自神經嵴 (neural crest)。它表現為一大片、按體節 (metameric) 分布的 CM,通常位於軀幹或四肢,並合併色素性皮膚病灶,例如色素母斑 (pigmented nevus)、斑點狀母斑 (nevus spilus)、咖啡牛奶斑 (café-au-lait patch),或位於非薦部的非典型蒙古斑 (atypical Mongolian spot)(見第 77 章)(圖 147-3)。²⁷
貧血母斑 (Nevus anemicus) 也可在鄰近處以雙生斑 (twin spot) 的形式出現。²⁸
Sturge-Weber 症候群 (STURGE-WEBER SYNDROME)
當位於額眼瞼區 (frontopalpebral area) 時,CM 可為 SWS 的一部分(圖 147-4A)。這種神經-眼-皮膚症候群將三叉神經眼支 (V1) 的皮膚 CM 與同側軟腦膜微血管-靜脈畸形(leptomeningeal capillary-venous malformation, CVM)及脈絡膜 CVM 相關聯。常合併青光眼(圖 147-4B)。SWS 因腦部靜脈引流異常而合併高癲癇與智能不足風險,並合併青光眼、牛眼 (buphthalmos),有時還有視網膜剝離 (retinal detachment)。²⁹
伴過度生長的瀰漫性微血管畸形 (DIFFUSE CAPILLARY MALFORMATION WITH OVERGROWTH)
DCMO 病人有半身肥大 (hemihypertrophy),可為全身性、區域性或對側性。CM 可瀰漫遍及全身。此疾病以網狀、界線不清的 CM 為特徵。³⁰ 病灶不依循 Blaschko 線(圖 147-5A)。
巨頭症-微血管畸形 (MACROCEPHALY–CAPILLARY MALFORMATION)
唇紅緣 (vermillion border)、鼻尖或兩者出現界線清楚的深色 CM 並合併巨頭症,常為 M-CM 的特異徵象 (pathognomonic)。這些病人有巨腦症 (megalencephaly) 並有智能不足風險。³¹ 文獻中有兩例 M-CM 合併威爾姆斯腫瘤 (Wilms tumor) 的報告。³²
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
CM 是一種紅色、均質、先天性病灶,常為單側性,有時為雙側性,但通常非正中線分布。CM 侵犯皮膚與皮下,有時侵犯黏膜(見圖 147-5)。其顏色從粉紅紅色到深紫色不等,呈地圖狀輪廓 (geographic contour) 或皮節 (dermatomal) 分布。病灶扁平且無痛,不會自發出血,觸診時也從不溫熱,這與 AVM(見後文)相反。後天性 CM 可見於外傷後。³³
半數 (Fifty percent) 的 CM 位於顏面,在該處依循三叉神經 (trigeminal nerve) 的分布:眼支 V1(前額與上眼瞼)、上頜支 V2(下眼瞼、臉頰與上唇)(見圖 147-4A 及 147-5B),或下頜支 V3(下唇、下巴與下頜)。CM 可瀰漫遍及全身,常合併半身肥大(見圖 147-5A)。此疾病稱為伴過度生長的瀰漫性微血管畸形(見前文討論)。當為遺傳性時,CM 通常較小且多灶性,例如 CM-AVM1 與 2。這些小 CM 大小從數毫米到數公分不等。它們常呈圓形至橢圓形,顏色為粉紅、紅或棕色,且常被一圈蒼白暈 (pale halo) 環繞(圖 147-6)。¹⁸,³⁴ 病灶界線清楚,並在以玻片壓診 (diascopy) 加壓時退色。其中一些於出生時即存在,但其他則較晚出現。它們隨兒童生長,且無症狀。Bier 斑(白色皮膚斑點,周圍環繞一圈蒼白的紅暈)在兩種疾病中皆可見。與 CM-AVM1 相反,CM-AVM2 中的 CM 外觀可更具微血管擴張性 (telangiectatic),尤其在口周與上胸部。²³ CM-AVM1 的特徵為:97% 的病人有 CM,24% 有 AVM 或動靜脈络管(arteriovenous fistulas, AVFs)(10% 在中樞神經系統 [CNS] 內,13% 在 CNS 外),8% 有 Parkes Weber 表型。²¹,³⁵ 在 CM-AVM2 中,快流型病灶的風險似乎較不常見,因為其各別外顯率分別為 93%(CM)、19%(AVM)與 7%(Parkes Weber)。
非皮膚表現 (NONCUTANEOUS FINDINGS)
在罕見情況下,CM 可為某潛在異常的標記,例如薦部 CM 下方的腰部脊柱裂閉合不全 (lumbar dysraphism)。PPV 的皮膚病灶在 60% 的病例中可合併全身性、內臟(喉部發育不全、腸道息肉症)、肌肉(脊柱側彎)或神經(智能不足、癲癇、顱內)的徵象與症狀。³⁶
SWS 因腦部靜脈引流異常而合併高癲癇與智能不足風險,並合併青光眼、牛眼,有時還有視網膜剝離。²⁹
DCMO 病人有半身肥大,可為全身性、區域性或對側性。他們也合併腳趾異常(佔 30%)與明顯的靜脈。³⁰ 在 CM-AVM1 或 2 中,有 20% 至 30% 的風險合併位於皮膚、腦部或脊椎的 AVM。²¹,³⁵ 唇紅緣或鼻尖界線清楚的深色 CM 合併巨頭症常為 M-CM 的特異徵象,這些病人有巨腦症並有智能不足風險。³¹
併發症 (COMPLICATIONS)
CM 病人的主要顧慮為美觀,因可見的變色所致。隨時間可能發生軟組織(常為唇與牙齦)與下方硬組織的肥大,尤其當 CM 影響 V2 與 V3 皮節或四肢時(圖 147-7D 及 147-7)。上頜或下頜的過度生長會導致骨骼不對稱、咬合傾斜 (occlusal tilt) 與開咬畸形 (open-bite deformity)。當兒童有異位性皮膚炎、乾癬或痤瘡時,CM 區域的病灶會更嚴重,此一現象稱為 Meyerson 現象。³⁷
到了青春期可能發展出均勻增厚的皮膚、紫色結節與化膿性肉芽腫 (pyogenic granulomas)(圖 147-7B 及 147-7C)。³⁸,³⁹
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
一般認為顏面 CM 是由源自神經嵴的異常細胞之克隆性擴增 (clonal expansion) 所引起。⁴⁰
GNAQ 的體細胞活化性突變已被辨識為合併肥大的顏面 CM 以及 SWS 的成因。¹³ GNAQ 是一類 q 型 G 蛋白 α 次單元,介導 G 蛋白偶聯受體與其下游效應器之間的訊號。GNAQ 與 GNA11 的突變也已在色素性病灶中被辨識,例如藍母斑 (blue nevi)、太田母斑 (nevus of Ota)、葡萄膜黑色素瘤 (uveal melanoma) 與 PPV。⁴¹,⁴²
遺傳性 CM-AVM1 是由 RASA1 的失活性突變所引起。²¹ 此基因編碼一種 GTP 酶活化蛋白 (GTPase-activating protein),負向調節 Ras 活性。CM-AVM2 是由 EPHB4 的失活性突變所引起。²³ EPHB4 通常表現於靜脈內皮細胞,其配體 EPHRINB2 則表現於動脈內皮細胞。它們調節動靜脈識別 (arteriovenous identity)。EPHB4 與 RASA1 交互作用以調節 RAS-MAPK 訊號,而 CM-AVM 中 EPHB4 或 RASA1 的功能喪失很可能活化此訊號途徑。M-CM 是由 AKT3、PIK3CA 與 PIK3R2 的活化性突變所引起。⁴³
診斷 (DIAGNOSIS)
輔助檢查 (SUPPORTIVE STUDIES)
病理 (Pathology):組織學上,CM 的特徵為真皮乳頭層與上網狀層的擴張微血管,並合併正常外觀微血管數量增加的區域(圖 147-8)。⁴⁴ 內皮細胞扁平。第 VIII 因子 (Factor VIII)、纖維連接蛋白 (fibronectin) 與基底膜蛋白 (basement membrane protein) 正常,但 S100 染色顯示異常的神經支配。⁴⁵,⁴⁶
影像 (Imaging):除罕見情況外,CM 不需強制進行影像檢查。若所謂的「CM」會疼痛、溫熱或自發出血,則有指徵進行都卜勒超音波,以排除快流型畸形的診斷,例如 AVM、Parkes Weber 症候群或增殖性血管瘤 (proliferating hemangioma)。
位於額眼瞼區的 CM,尤其若侵犯上眼瞼內側部分,可為 SWS 的一部分。因此,對於這些 CM,眼科與神經學檢查是強制性的。它們需要在生命的最初數個月內進行,並每年重複一次直到青春期,即使在較年幼時正常亦然。⁴⁷ 也應進行腦部 MRI 以評估相關軟腦膜 CVM 的發生。在 PPV 病人中,由於 CM 常合併全身性病灶,眼科、神經學與骨科的追蹤是強制性的。在下肢有廣泛 CM 的病人(如 DCMO)中,需在約 4 至 6 歲時進行肢體測量研究(scaniometry,下肢長度 X 光片)以評估可能的進行性生長差異,並需重複至生長結束,以讓骨科醫師在適當時機矯正差異。也有指徵進行連續腎臟超音波檢查,以排除威爾姆斯腫瘤的出現。在出現腰薦部 CM 時,有指徵進行脊髓 MRI。在 CM-AVM 病人中,應考慮進行腦部與脊椎 MRI。
基因檢測 (Genetic Testing):基因檢測有指徵,尤其在有多灶性病灶的病人(CM-AVM1 與 2)中,因其顱內快流型病灶風險增加。這些病人應接受 RASA1 與 EPHB4 的篩檢。蒼白、不具特徵且不顯眼的 CM 可為 PHTS 的徵象,尤其若這些血管病灶合併巨頭症(見後文 AVM)。由於癌症風險增加,這類病人應接受 PTEN 突變篩檢。辨識出生殖細胞突變將能進行精確的諮詢與監測。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 147-4。

表 147-4:微血管畸形的鑑別診斷 (Differential Diagnosis of Capillary Malformation)
| 依型態 (Based on Morphology) | 鑑別診斷 |
|---|---|
| 最可能 (Most Likely) | |
| 淡粉紅色正中線斑塊 | ■ Unna 母斑 ■ 新生兒火焰母斑(單純母斑) |
| 多發、遺傳性 | ■ CM-AVM 之 CM |
| 微血管擴張性斑塊 | ■ CMTC(先天性大理石樣微血管擴張症) ■ HHT ■ CM-AVM2 ■ 本態性微血管擴張 ■ 單側母斑樣微血管擴張 |
| 觸診溫熱 | ■ 增殖性血管瘤 ■ AVM ■ AVF |
| 潰瘍或疼痛 | ■ AVM ■ AVF |
| 須排除 (Rule Out) | |
| 小、多發 | ■ 肥大細胞增生症 (Mastocytosis) ■ CM-AVM 之 CM |
| 依部位 (Based on Location) | |
| 最可能 (Most Likely) | |
| 枕部 | ■ Unna 母斑 |
| 眉間、上眼瞼、頸後 | ■ Unna 母斑 ■ 鸛咬痕、天使之吻、鮭魚斑 |
| 考慮 (Consider) | |
| 四肢 | ■ CMTC ■ Klippel-Trenaunay 症候群 ■ Parkes Weber 症候群 |
| 軀幹上淡色 | ■ M-CM |
| 須排除 (Rule Out) | |
| 頭皮 | ■ Adams-Oliver 症候群 |
| 眼眥附近多發性微血管擴張 | ■ 共濟失調性微血管擴張症 (Ataxia telangiectasia) |
縮寫:AVF,動靜脈络管;AVM,動靜脈畸形;CM,微血管畸形;CM-AVM,微血管畸形-動靜脈畸形;CMTC,先天性大理石樣微血管擴張症 (cutis marmorata telangiectatica congenita);HHT,遺傳性出血性微血管擴張;M-CM,巨頭症-微血管畸形。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
CM 於出生時即存在,且從不自發消退。在生命的最初數週,隨著新生兒血紅素濃度下降,它們可能略微褪色。隨後,紅色色調穩定下來,病灶與身體其餘部分成比例地生長。約在青春期及生命較晚期時,CM 隨時間緩慢增厚與變深。它常變得隆起且呈結節狀(見圖 147-7)。³⁸ 隨時間可能發生化膿性肉芽腫,以及軟組織或骨性肥大。³⁹
處置 (MANAGEMENT)
處置的目標是監測相關併發症,並將 CM 對自尊與生活品質的影響降至最低。使用 Covermark 等粉底進行遮瑕仍是一種有效的方法(圖 147-9)。
介入 (INTERVENTIONS)
骨科考量 (Orthopedic Considerations):在出現腿長差異時,若腿長差異超過 1.5 cm,需及早配置適合的鞋墊以防止骨盆的代償性傾斜。當兒童約 11 至 13 歲時,有時需要進行骨骺固定術 (epiphysiodesis)。
治療程序 (Procedures):雷射治療是大多數 CM 的黃金標準療法。具有特定波長(585 或 595 nm)與短脈衝持續時間(400–1500 ms)的脈衝染料雷射,目前在嬰兒與兒童中藉由淡化 CM 的紅色而給出最佳結果。併發症少。需要多次療程(6–12 次),且由於該程序疼痛,可能需要全身麻醉。雷射治療在頸顏面與軀幹區域比在四肢更有效。兒童期早期治療並不會減少雷射療程的次數。⁴⁸ 使用動態冷卻系統 (dynamic cooling system) 以避免加熱表皮,可允許更高的雷射能量密度,從而達到病灶的最佳淡化。療法停止後可能復發。⁴⁹ 雷射對相關肥大無影響。⁶,⁵⁰
考慮進行輪廓切除 (contour resection) 以治療併發症,例如化膿性肉芽腫或唇肥大。⁶
諮詢 (Counseling):針對病人及其家屬的遺傳與心理諮詢是多專科處置的一部分。專門針對血管異常的病友組織在支持病人及其家屬方面也扮演重要角色。
預防 (PREVENTION)
在 SWS 病人中,出生後立即開始的眼科追蹤至關重要,因為減少視力損害將取決於治療的及時性。預防性抗癲癇藥物以預防神經細胞死亡也已被倡議,儘管目前缺乏支持其療效的隨機前瞻性研究。⁵¹,⁵²
靜脈異常 (VENOUS ANOMALIES)
重點一覽 (AT-A-GLANCE)
■ 為轉介至血管異常專科中心最常見者;發生率未知但大幅低於 CM,估計約為 1/10,000。
■ 為靜脈床的先天性慢流型畸形。
■ 顏色偏藍,局部性或廣泛性,孤立性或多灶性。
■ 觸診時可壓陷;有靜脈石 (phleboliths)。
■ 約 40% 的病例合併消耗性凝血異常 (consumptive coagulation abnormalities)。
■ 組織學上由擴張的靜脈樣通道構成,並有管壁細胞 (mural cells) 異常。
■ 主要為散發性,但可作為體染色體顯性性狀遺傳。
■ 遺傳性靜脈異常:皮膚黏膜靜脈畸形 (1%)、球體靜脈畸形 (>5%)。
■ 症候群性靜脈畸形:KTS、藍色橡皮泡母斑、Maffucci 症候群。
前言 (INTRODUCTION)
VM 是一種由皮膚或黏膜的靜脈型血管所構成的先天性病灶,但可侵犯任何結構(皮下、肌肉、骨骼與神經)與任何器官(CNS、胃腸道 [GI])。半數 (Fifty percent) 的 VM 位於頸顏面區,37% 位於四肢。VM 主要為孤立性,但可為複雜血管疾患的一部分,例如 KTS、Maffucci 與藍色橡皮泡母斑 (BRBN) 症候群。⁵³,⁵⁴
流行病學 (EPIDEMIOLOGY)
靜脈異常是轉介至專科中心最常見的 VM,在人群中的整體發生率為 1/10,000。⁵⁴ 它們主要為散發性發生,但 1% 為遺傳性皮膚黏膜靜脈畸形(VMCMs),5% 為遺傳性球體靜脈畸形(GVMs)。無性別偏向報告。⁵⁵ VMCM 與 GVM 的外顯率皆有年齡依賴性變異,在 20 歲時達到最大值(VMCM 為 87%,GVM 為 92.7%)。⁵⁶ 大型 VMCM 與 GVM 於出生時即存在。然而,17% 的受影響個體會隨時間發展出新的小病灶。⁵⁵ BRBN 症候群罕見且為散發性發生,文獻中約報告 200 例。
症候群性疾患 (SYNDROMIC DISORDERS)
具有靜脈異常的症候群包括 BRBN 症候群、KTS(見後文「淋巴管畸形」)與 Maffucci 症候群。
藍色橡皮泡母斑症候群 (BLUE RUBBER BLEB NEVUS SYNDROME)
BRBN 的特徵為侵犯皮膚與內臟器官的多發性靜脈系統畸形。它通常以一個大型「主導性 (dominant)」VM 病灶合併多發性小型、深藍色、乳頭狀病灶為特徵,後者典型位於手掌與足底(圖 147-10)。¹⁵ 這些皮膚病灶合併多發性小型胃腸道 VM,常為慢性貧血的成因。¹⁵,⁵⁷
Maffucci 症候群 (MAFFUCCI SYNDROME)
Maffucci 症候群是一種罕見疾患,特徵為多發性內生軟骨瘤 (enchondromas) 合併遠端四肢的皮下 VM(圖 147-11)。此病始於兒童期,伴隨手足骨骼與長骨的內生軟骨瘤發展。常發生四肢的變形與縮短。皮下血管結節較晚出現,約在青春期,位於手指與腳趾。可能出現靜脈石。⁵⁸
臨床特徵 (CLINICAL FEATURES)
靜脈異常的特徵總結於表 147-5。
皮膚表現 (CUTANEOUS FINDINGS)
VM 通常為孤立性,但可為多灶性,後者在大多數情況下提示遺傳性疾患。⁵⁴ 它們在某解剖區域內可相對局部性或廣泛性。最常見的位置是頭頸部區域(圖 147-12A 及 147-12D–F)。VM 可影響任何組織或器官(圖 147-12 至 147-14)。VM 為先天性、淺至深藍色的病灶。深部 VM 的覆蓋皮膚顏色正常,常僅在青春期或生命較晚期隨疼痛發作而被診斷。大小從小的海綿狀泡 (spongy blebs) 到直徑數公分的大型病灶不等。皮膚溫度正常。無震顫或雜音。VM 在下垂位置時較大,並可藉由加壓或促進靜脈引流而輕易排空。除非發生血栓,否則觸診不會疼痛。⁵⁴,⁵⁹
VMCM 中的 VM 病灶為小型且多灶性。它們更近似於散發性發生的小型 VM,而非 BRBN。它們可隨時間增大且常無症狀。這些 VM 一般較小(<5 cm),且超過 80% 的病人有超過兩個病灶。一些於出生時即存在;大多數病灶在青春期出現。黏膜病灶侵犯唇、舌與頰黏膜。⁶⁰,⁶¹
在罕見情況下,VM 可為散發性但多發。這些多灶性靜脈畸形(multifocal venous malformations, MVMs)與 VMCM 病灶非常相似,但無相同表現的家族史。¹⁵
GVM 是一種藍至紫色、隆起的靜脈異常,特徵為多灶性、角化過度 (hyperkeratosis) 與具有鵝卵石表面 (cobblestone surface) 的結節性(見圖 147-13C)。在罕見情況下(尤其在新生兒),病灶可能扁平且呈紫色,類似 CM⁶²;這種斑塊樣 GVM 通常隨時間變深。GVM 通常於出生時即存在,並在兒童期緩慢擴大。
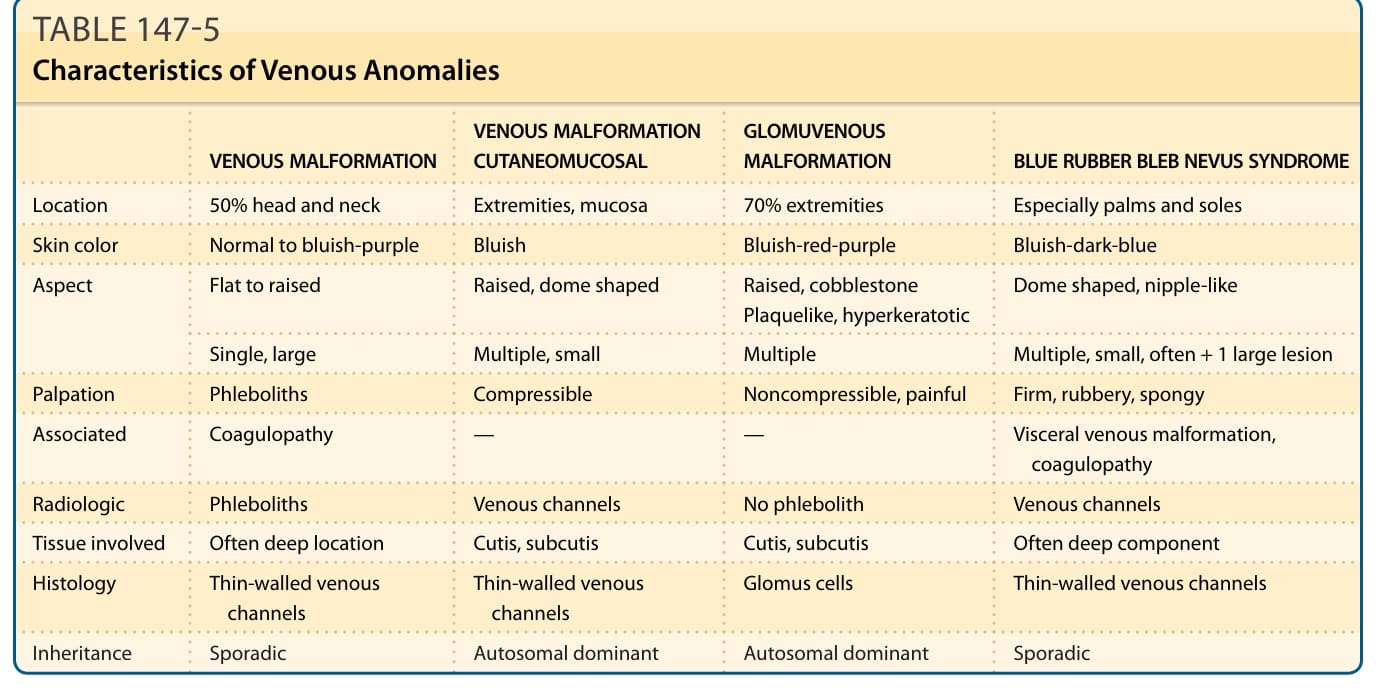
表 147-5:靜脈異常的特徵 (Characteristics of Venous Anomalies)
| 靜脈畸形 (VM) | 皮膚黏膜靜脈畸形 (VMCM) | 球體靜脈畸形 (GVM) | 藍色橡皮泡母斑症候群 (BRBN) | |
|---|---|---|---|---|
| 位置 | 50% 頭頸部 | 四肢、黏膜 | 70% 四肢 | 尤其手掌與足底 |
| 膚色 | 正常至藍紫色 | 偏藍 | 藍紅紫色 | 藍至深藍色 |
| 外觀 | 扁平至隆起;單一、大型 | 隆起、圓頂狀;多發、小型 | 隆起、鵝卵石狀、斑塊樣、角化過度;多發 | 圓頂狀、乳頭狀;多發、小型,常 + 1 個大病灶 |
| 觸診 | 靜脈石、可壓陷 | 可壓陷 | 不可壓陷、疼痛 | 堅實、橡膠樣、海綿狀 |
| 相關特徵 | 凝血障礙 (coagulopathy) | — | — | 內臟靜脈畸形、凝血障礙 |
| 放射學 | 靜脈石、靜脈通道 | 靜脈通道 | 無靜脈石 | 靜脈通道 |
| 受累組織 | 常為深部位置 | 皮膚、皮下 | 皮膚、皮下 | 常有深部成分 |
| 組織學 | 薄壁靜脈通道 | 薄壁靜脈通道 | 球體細胞 (glomus cells) | 薄壁靜脈通道 |
| 遺傳 | 散發性 | 體染色體顯性 | 體染色體顯性 | 散發性 |
新的小病灶隨時間出現。與 VM 相反,GVM 觸診時常疼痛,且無法藉由加壓完全排空。GVM 通常為多灶性並位於四肢,侵犯皮膚與皮下。它們很少出現於黏膜,且無腸道出血。GVM 病人的智能與身體發展正常。GVM 的診斷基於皮膚病灶的臨床與組織學評估。基因檢測可區分小型多灶性 GVM 與 VMCM。⁵⁵
疣狀靜脈畸形 (Verrucous Venous Malformation):疣狀靜脈畸形(verrucous venous malformation, VVM),先前稱為疣狀血管瘤 (verrucous hemangioma),是一種罕見的先天性血管異常。雖然 VVM 表現出與血管腫瘤相似的免疫組織化學特徵(威爾姆斯腫瘤 1 [Wilms tumor 1] 與 GLUT-1 陽性),但它與嬰兒血管瘤不同,因為它通常於出生時即被注意到,且從不自發消退。VVM 主要位於下肢,但也可見於頭部與軀幹。它最初表現為藍色斑點,之後變為紅斑-紫紅色,並在外傷與繼發感染後常演變為疣狀結節(見圖 147-14)。它從不侵犯肌肉或深部組織。⁶³
非皮膚表現 (NONCUTANEOUS FINDINGS)
VMCM 病人,如同散發性發生的 MVM 病人,很少有位於內部器官(如肺、腎、腦或胃腸道)的 VM。¹⁵ BRBN 的皮膚外表現包括胃腸道侵犯。小腸是主要區域;然而,VM 可發生於從口腔到肛門黏膜的任何部位。在罕見情況下,VM 也可見於腦、肺與腎,以及其他器官。⁵⁷
併發症 (COMPLICATIONS)
VM 通常為單側性,造成不對稱與進行性、緩慢惡化的變形(見圖 147-12 及 147-13)。依其大小與位置而定,VM 可造成疼痛(尤其在早晨醒來時)與解剖變形,甚至可因出血、擴大或阻塞重要結構而危及生命。當 VM 位於顳肌 (temporal muscle) 時,偏頭痛 (migraine) 是常見特徵。口咽部 VM 可損害言語並造成吞嚥困難,而咽部或喉部病灶則可危及呼吸道並造成打鼾與睡眠呼吸中止 (sleeping apnea)。⁵⁴
在四肢,VM 常侵犯肌肉與關節。它們造成肌肉無力、發育不全 (hypotrophy),有時造成肥大,導致腿長差異(較 KTS 為輕)。若畸形位於膝關節,關節內出血會導致早發性關節病 (arthrosis)。⁶⁴ 生殖器 VM 常與肢體 VM 同時發生,可造成性交疼痛 (dyspareunia)。胃腸道 VM 可導致慢性貧血。局部血栓通常是持續數天並隨靜脈石形成而消退的急性疼痛的原因,靜脈石可藉由觸診或放射攝影辨識。VM 很少造成肺栓塞 (pulmonary embolism),儘管當存在大型引流靜脈時可能發生此併發症。⁶⁵,⁶⁶
慢性局部血管內凝血障礙(localized intravascular coagulopathy, LIC)合併於孤立性或症候群性海綿狀 VM。它在 40% 的病人中以 D-二聚體濃度升高(>500 ng/mL)為特徵。伴有低纖維蛋白原 (fibrinogen) 濃度的嚴重 LIC 常見於四肢的大型 VM,而在頸顏面區罕見。許多事件,例如手術、硬化療法與荷爾蒙影響,可觸發 LIC 轉變為瀰漫性血管內凝血障礙(disseminated intravascular coagulopathy, DIC)。⁶⁷
與皮膚病灶相反,如 BRBN 病人所見的胃腸道病灶有出血傾向。它們可能自發破裂,造成急性出血甚至死亡。然而,大多數出血傾向緩慢進展,導致慢性與隱性失血,可導致缺鐵性貧血。其他併發症包括腸套疊 (intussusception)、腸扭轉 (volvulus) 與腸梗塞 (bowel infarction),這些都應在有 BRBN 與腹痛的病人中加以考慮。罹病率取決於胃腸道侵犯的範圍。⁵⁷ VVM 常潰瘍。⁶³
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
VMCM 以體染色體顯性疾患遺傳,由 TEK 基因的生殖細胞突變所引起,該基因編碼血管生成素受體 (angiopoietin receptor) TIE2。⁶⁸,⁶⁹ 遺傳性突變即使在無配體存在的情況下也造成受體的過度磷酸化 (hyperphosphorylation)。在兩名病人的 VMCM 組織中發現了體細胞二次打擊 (somatic second hits):一例造成第二個野生型等位基因的膜表現喪失;另兩例則發生在帶有遺傳性突變的等位基因上,造成過度磷酸化增加。¹⁵,⁷⁰ 這些突變活化內皮細胞中的 PI3K/AKT 訊號途徑。⁷¹,⁷²
GVM 是由 glomulin 基因的顯性、功能喪失 (loss-of-function) 突變所引起。⁷³ 此外,病灶形成需要一次體細胞二次打擊,導致 glomulin 完全功能喪失。最常見的二次打擊是後天性單親同源二倍體 (uniparental isodisomy),這導致遺傳性突變的同型合子化 (homozygosity)。⁷³,⁷⁴ 這解釋了家族成員間在外顯率、病灶範圍與病灶數量方面的可變表現度 (variable expressivity)。⁷⁵ 迄今,在幾乎所有接受檢測的 GVM 家族中皆發現 glomulin 突變,顯示其基因座同質性 (locus homogeneity)。⁷⁵⁻⁷⁷ Glomulin 似乎表現於血管平滑肌細胞,但其確切功能仍屬未知。散發性 VM 也由遺傳突變所引起。六十 (Sixty percent) 是由 TIE2 的活化性體細胞突變所引起。⁷⁰ 這些突變與 VMCM 所見的遺傳性突變不同。VM 中最常見的突變(L914F)尚未在 VMCM 中被辨識,而 VMCM 中最常見的遺傳性突變(R849W)也尚未在單一體細胞 VM 中被辨識。這顯示,反覆出現的體細胞突變所造成的效應過於有害以致無法在生殖細胞中被支持並可能造成致死,而遺傳性突變則具有較弱的效應,需要額外的變化(體細胞二次打擊)。⁷⁰⁻⁷²
另有 20% 的散發性發生 VM 是由 PIK3CA 基因的體細胞突變所引起。¹⁰
癌症中可見相同的突變。它們活化 PI3K/AKT 訊號途徑,正如 TIE2 中的突變一樣,這強調此訊號活性的擾動是 VM 致病機轉的基礎。¹⁰,¹¹
MVM 也由 TIE2 突變所引起。與 VMCM 相似,這些病人常有兩個 TIE2 突變。最常見的是以鑲嵌性 (mosaic) 變化存在的 R915C 突變,而體細胞 Y897C 突變則發生在同一等位基因上 R915C 變化之上。這解釋了缺乏家族史(第一個突變以鑲嵌方式在病人身上重新發生 [de novo])、多灶性(鑲嵌突變是一種傾向性變化,如同 VMCM 中的遺傳性突變),以及與 VMCM 病灶的臨床相似性(兩者皆有相似但不相同的雙重順式 [cis] 突變)。¹⁵ VVM 是由體細胞 MAP3K3 突變所引起。⁷⁸ BRBN 症候群,如同 VMCM、MVM 與 60% 的 VM,也是由 TIE2 突變所引起。BRBN 突變在血液中無法偵測;它們為體細胞性。¹⁵ 然而,在同一 BRBN 病人不同的、遠端分布的病灶中,可以相同的等位基因頻率辨識出相同的雙重突變。最常見的是 T1105N–T1106P 雙重突變。這顯示分開且甚至新出現的病灶是由源自單一共同部位(例如主導性病灶或骨髓)的內皮細胞所形成。引起 BRBN 的 TIE2 突變造成受體強烈的過度磷酸化,且內皮細胞獲得增加的克隆形成能力、存活率與更快的遷移。¹⁵
Maffucci 症候群是由 IDH1 與 2 的體細胞突變所引起。⁷⁹⁻⁸¹
診斷 (DIAGNOSIS)
輔助檢查 (SUPPORTIVE STUDIES)
實驗室檢查 (Laboratory Testing):任何純 VM 或具靜脈成分的複雜畸形病人,皆應進行包含血小板計數、纖維蛋白原與 D-二聚體濃度的凝血功能檢查,因為約 40% 的 VM 其 D-二聚體濃度升高。此血液檢測應在首次諮詢時進行,並在青春期前後及每當畸形變得疼痛時重複。GVM 或其他單純血管異常的病人則從不如此。伴有低纖維蛋白原是嚴重 LIC 的特異徵象。嚴重 LIC 可在介入或外科手術期間輕易代償失調為 DIC 並伴隨嚴重出血。⁶⁷,⁸²⁻⁸⁴
病理 (Pathology):組織學上,VM 由靜脈型的擴張血管通道構成,內皮扁平。血管壁薄,並由不連續的一層管壁平滑肌細胞 (mural smooth muscle cells) 內襯,該細胞對平滑肌-α-肌動蛋白 (smooth muscle-α-actin) 呈陽性。⁸⁵ 基底膜也薄。相反地,GVM 在組織學上的特徵為擴張的靜脈通道被管壁「球體細胞 (glomus cells)」環繞,這些球體細胞是異常分化的平滑肌細胞。球體細胞為圓形或多角形,而非如正常血管平滑肌細胞般延長。⁸⁶ 在免疫組織化學上,球體細胞對平滑肌-α-肌動蛋白與波形蛋白 (vimentin) 呈陽性染色,但對結蛋白 (desmin)、馮維勒布蘭德因子 (von Willebrand factor) 與 S100 呈陰性。⁸⁷
以原位雜交 (in situ hybridization),它們對 glomulin 也呈陰性。⁷⁶
VVM 的特徵為角化過度的表皮,下方為位於真皮與皮下、具多層膜 (multilaminated membrane) 的小血管。可見 GLUT-1 的局部陽性染色。⁶³ Maffucci 症候群病人的組織病理學檢查顯示梭形細胞血管內皮瘤 (spindle cell hemangioendothelioma) 的特徵。
影像 (Imaging):平片放射攝影 (Plain radiography) 可辨識靜脈石的鈣化,這是 VM 中 LIC 的特異徵象。它也可顯示 Maffucci 症候群病人多發性內生軟骨瘤的存在(見圖 147-11)。都卜勒超音波是非侵入性的,可確認病灶的慢流性質。與淋巴管畸形(lymphatic malformations, LMs)相反,靜脈通道可被探頭壓陷。⁸⁸
具有自旋迴波 (spin-echo) T1 與 T2 加權及脂肪抑制 (fat-saturation) 序列的 MRI 影像可描繪病灶的精確解剖位置(見圖 147-12)。⁸⁹ 這將凸顯 GVM 相較於 VM 更具細胞性的成分以及更淺表的位置。非典型的 MRI 表現需要切片以排除肉瘤 (sarcoma) 或神經纖維瘤 (neurofibroma)。在出現 BRBN 典型的掌蹠多灶性病灶時(見圖 147-10),應進行內視鏡、結腸鏡或無線膠囊內視鏡以偵測胃腸道病灶。這些放射學檢查對於任何靜脈異常的治療前評估都是強制性的。
基因檢測 (Genetic Testing):對於有多灶性病灶的病人,應考慮進行 TIE2 與 glomulin 突變的基因檢測。這可區分這些病況並使遺傳諮詢成為可能。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 147-6。
表 147-6:靜脈畸形的鑑別診斷 (Differential Diagnosis of Venous Malformation)
| 依型態 (Based on Morphology) | 鑑別診斷 |
|---|---|
| 最可能 (Most Likely) | |
| 藍色皮下腫塊、不可壓陷 | ■ 嬰兒血管瘤(深部) ■ 淋巴管畸形 |
| 伴多發性內生軟骨瘤 | ■ Maffucci 症候群 |
| 考慮 (Consider) | |
| 角化過度 | ■ GVM(若位於四肢) ■ CCM 之皮膚病灶 ■ VVM |
| 須排除 (Rule Out) | |
| 淺表、側支正常靜脈 | ■ 深部靜脈功能不全、醫源性狹窄或先天性發育不全 ■ 藍色非血管性病灶 |
| 依部位 (Based on Location) | |
| 考慮 (Consider) | |
| 鼻基部 | ■ 正常明顯靜脈 |
| 頭皮或前額 | ■ 正常明顯靜脈 ■ 顱骨竇 (sinus pericranii) ■ 下方 Galen 畸形 |
| 甲下 | ■ 孤立性球體瘤 (solitary glomus tumor) |
| 臉頰 | ■ 藍母斑 |
| 腦部 | ■ 發育性靜脈異常 (developmental venous anomaly) ■ CCM |
| 須排除 (Rule Out) | |
| 頸部;深部腫塊 | ■ 甲狀舌管 (thyroglossal duct) ■ 支氣管源性囊腫 (bronchogenic cyst) |
縮寫:CCM,腦海綿狀畸形 (cerebral cavernous malformation);GVM,球體靜脈畸形;VVM,疣狀靜脈畸形。
一種不尋常的腦部引流途徑,即腦發育性靜脈異常 (cerebral developmental venous anomaly),由匯入較大引流靜脈的擴張髓內靜脈所構成,可見於 0.5% 的人口。相反地,它存在於 20% 有廣泛頭頸部 VM 的病人中。雖然通常無症狀,此異常可造成頭痛。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
VM 隨病人成比例地生長。它們從不自發消退。最初無症狀的病灶常因外傷或荷爾蒙狀態改變(青春期或懷孕)而變得疼痛。它們也可能在血栓後擴大。尚未有此病況惡性轉化 (oncogenic transformation) 的描述。主要併發症為危及生命的胃腸道出血與呼吸道壓迫。有遺傳性靜脈異常的病人,以及 BRBN 與 MVM 病人,傾向隨時間發展出額外的小病灶。¹⁵ Maffucci 症候群病人有高惡性化發生率(40%),主要為軟骨肉瘤 (chondrosarcoma),但也包括神經膠質瘤 (glioma)、纖維肉瘤 (fibrosarcoma) 與血管肉瘤 (angiosarcoma)。⁵⁸
處置 (MANAGEMENT)
當病灶有症狀或基於美觀目的時,靜脈異常的治療有其必要。最常見的治療指徵是疼痛與功能損害。⁵⁴ 其處置通常為多專科性,涉及血液科醫師、外科醫師與介入放射科醫師。
介入 (INTERVENTIONS)
醫療處置 (Medical Management):壓迫衣物 (Compression garments) 可藉由降低靜脈壓而有助於減少四肢 VM 的腫脹與疼痛。相反地,加壓會增加 GVM 病人的疼痛,在此情況下不適用。低分子量肝素(low-molecular-weight heparin, LMWH;LMWH,100 IU anti-Xa/kg/day)通常給予有 LIC 徵象的病人,在外科手術前 24 小時及術後另 5 天使用,以將出血風險降至最低。同樣的治療給予有局部血栓疼痛發作的病人。在此情況下,治療持續時間通常為 2 週。⁵⁴,⁶⁷,⁸⁴
來自胃腸道 BRBN 病灶的出血可藉由鐵劑補充與輸血進行保守處置。隨著對潛在病理生理機轉(PI3K-AKT 訊號的活化)的理解,標靶分子療法正變得可能。雷帕黴素(rapamycin,sirolimus)在臨床前 VM 小鼠模型試驗中很有前景。⁹⁰ 一項針對難以治療的廣泛 VM 或無法以傳統處置處理之複雜慢流型血管異常的第二期臨床試驗也給出了令人鼓舞的結果。⁹⁰,⁹¹
病人經歷了疼痛與症狀幾乎完全緩解、凝血障礙(若存在)減少、功能改善,以及自我感知生活品質提升。散發性 VM 與 BRBN 的出血在 24 小時內停止。副作用輕微,包括黏膜炎 (mucositis)、輕微頭痛、疲倦與腹瀉。此外,在大多數達到 1 年追蹤的病人中,MRI 觀察到統計上顯著的體積減少。一項多中心歐洲研究(VASE;NCT 02638389)正在進行,以確定哪些 VM 亞型最適合雷帕黴素治療以及治療應持續多久。雷帕黴素不應被視為對標準照護有反應的小型、局部性且無症狀慢流型血管畸形的治療方法。⁹²
治療程序 (Procedures):經皮病灶內硬化療法 (Percutaneous intralesional sclerotherapy) 是 VM 的主要治療(與 GVM 相反)。無水乙醇 (Absolute ethanol) 是最有效的硬化劑。⁹³ 使用局部加壓或病灶內線圈 (intralesional coils) 以防止乙醇擴散進入全身循環。局部併發症包括發炎、水腫、起水疱、壞死、慢性滲出與暫時或永久性神經缺損。已有全身性併發症的報告,例如腎或肺毒性、心肌抑制與心搏停止。由於 VM 傾向再通 (recanalize) 與復發,常需多次硬化療法療程。乙醇硬化療法的替代方案為四癸基硫酸鈉泡沫 (sodium tetradecyl-sulphate foam) 或聚多卡醇 (lauromacrogol),它們對小型 VM 有效並造成較少的局部不良反應。在一項涉及 86 名病人(91 個 VM)的研究中,可見 49.5%(疼痛)與 52.7%(腫塊縮小)的陽性反應。⁹⁴ 清潔劑類硬化劑 (Detergent sclerosants) 已被用作微泡沫 (microfoams),使用空氣泡或二氧化碳以增加體積與表面與內皮的接觸。然而,已有 2% 病例描述神經學併發症。因此發展出一種改良的不透射線乙醇硬化劑。它將乙醇捕陷於乙基纖維素 (ethylcellulose) 的網狀結構中以增加黏度。⁹⁵,⁹⁶
手術切除常在硬化療法後進行。⁵³ 在 BRBN 病人中,以 Nd:YAG(摻釹釔鋁石榴石,neodymium-doped yttrium aluminum garnet)雷射進行內視鏡凝固、雙極或氬離子電漿凝固 (argon plasma coagulation),以及結紮 (band-ligation) 都很有用。允許手術切除所有胃腸道病灶的開放手術可消除輸血需求,以及由腸套疊引起的常見間歇性腹痛。⁹⁷,⁹⁸ 在 GVM 病人中,若進行切除,常需植皮 (skin graft)。VVM 也最好以手術切除治療。閉合常需植皮。
諮詢 (Counseling):在 VMCM 或 GVM 病人中,應為受影響家庭提供遺傳諮詢,告知病人有 50% 的風險遺傳致病突變,以及臨床表現的可變性。針對血管異常的社會心理諮詢與病友組織對病人及其家屬也是一項重要資產。
淋巴管異常 (LYMPHATIC ANOMALIES)
重點一覽 (AT-A-GLANCE)
■ LM 的全球發生率未知,但較 VM 為少見。
■ 為淋巴管的先天性、散發性、慢流型畸形。
■ LM 由充滿淋巴液的微囊 (microcysts) 或巨囊 (macrocysts) 構成。
■ 組織學上由內皮扁平、表現 D2-40 的擴張淋巴管通道構成。
■ 原發性淋巴水腫 (Primary lymphedema) 可作為體染色體性狀遺傳。
■ 感染是最常見的併發症,可導致敗血症。
前言 (INTRODUCTION)
LM 是淋巴管的局部形態發生錯誤 (morphogenic errors)。它們由充滿淋巴液的小水疱或大囊腫構成。巨囊型 LM 早在懷孕第一孕期即可於子宮內診斷。⁹⁹ 然而,大多數 LM 在嬰兒期、2 歲前被診斷。有些可能僅在青春期或成年期才表現。它們可為孤立性、與其他血管異常混合,或為某症候群的一部分。原發性淋巴水腫並非局部病灶,而是一種較全身性的病況,其中淋巴液累積於間質組織 (interstitial tissue)。下肢最常受影響。它可為單側或雙側。原發性淋巴水腫可為孤立性病況或某症候群的一部分,並可散發性發生或作為遺傳性疾患。
流行病學 (EPIDEMIOLOGY)
LM 是一種發生率未知的先天性疾患。它為散發性發生,這與原發性淋巴水腫相反,後者可遺傳(通常為體染色體顯性性狀),高達 20% 的病例為遺傳性。原發性淋巴水腫分為先天型(Milroy 病,線上人類孟德爾遺傳資料庫 [OMIM] #153100)與晚發型(青春期表現,Meige 病,OMIM #153200)。它可為孤立性或某症候群的一部分,例如特納症候群 (Turner) 與努南症候群 (Noonan syndromes)。¹⁰⁰
症候群性疾患 (SYNDROMIC DISORDERS)
Klippel-Trenaunay 症候群 (KLIPPEL-TRENAUNAY SYNDROME)
Klippel-Trenaunay 症候群是一種微血管-淋巴管-靜脈混合畸形,合併受累肢體的肥大。70% 的病例侵犯下肢。它的特徵為一片地圖狀、廣泛的 CM 合併淋巴管小水疱。位於大腿外側的持續性胚胎靜脈 (persistent embryonic vein) 之存在為特異徵象。¹⁰¹,¹⁰²
CLOVES 症候群 (CLOVES SYNDROME)
CLOVES 症候群是「先天性脂肪瘤樣過度生長合併血管畸形、表皮母斑及骨骼異常 (congenital lipomatous overgrowth with vascular malformations, epidermal nevi, and skeletal anomalies)」的縮寫。¹⁰³ 這種非遺傳性疾患的特徵為進行性不對稱肥大、多發性軀幹脂肪瘤樣腫塊伴脊椎旁快流或慢流型血管異常(或兩者)、表皮母斑、肢端病灶,以及骨骼或脊椎異常(圖 147-15)。
最重要的鑑別診斷是其他過度生長症候群,例如 Proteus 症候群與 Proteus 樣症候群。¹⁰⁴
全身性淋巴異常 (GENERALIZED LYMPHATIC ANOMALY)
全身性淋巴異常(generalized lymphatic anomaly, GLA)是一種罕見病況,其中 LM 可侵犯數個器官,例如縱膈、肺、胸膜、胃腸道、骨骼與軟組織。¹⁰⁵
Gorham-Stout 症候群 (GORHAM-STOUT SYNDROME)
Gorham-Stout 症候群,又稱「骨消失病 (vanishing bone disease)」,是一種侵襲性的罕見淋巴疾患,特徵為骨骼進行性脫鈣與破壞,並被淋巴管與微血管所取代。¹⁰⁶
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
LM 由微囊(圖 147-16 及 147-17,先前稱為局限性淋巴管瘤 lymphangioma circumscriptum)或巨囊(直徑 >1 cm,先前稱為囊狀水瘤 cystic hygroma)構成,並與兒童成比例地生長(見圖 147-1B、147-16 及 147-17)。它們充滿清澈或血清血性 (serosanguineous) 液體(見圖 147-17B)。巨囊型 LM 表現為柔軟、多分葉、界線清楚的腫塊,而微囊型 LM 則界線不清且常侵犯鄰近結構。真皮 LM 可表現為小型、毫米大小的水疱,清澈(見圖 147-16D 及 147-17A)或深紅色(當有囊內出血時),可出血並變為紫色與結節狀。
局限性血管角化瘤 (Angiokeratoma Circumscriptum)
局限性血管角化瘤 (Angiokeratoma circumscriptum),或微血管-淋巴管畸形(capillary-lymphatic malformation, CLM),是一種混合性、界線清楚的病灶,常位於四肢。病灶顏色為粉紅色至藍紅色,略微隆起,且通常角化過度。臨床上,可辨識出局限性血管角化瘤、Mibelli 血管角化瘤(遠端四肢界線清楚、深紅色、角化過度的斑塊)與 Fordyce 血管角化瘤(年長男性陰囊上非常常見的角化過度、藍黑色丘疹)。它們須與法布瑞氏病 (Fabry disease) 的血管角化瘤(見第 127 章)以及 VVM(先前稱為疣狀血管瘤;見前文 VM 章節)相鑑別。
淋巴水腫 (Lymphedema)
淋巴水腫的特徵為受累身體部位(通常為下肢)的腫脹,由內在淋巴功能障礙導致淋巴液累積於細胞外腔所引起(圖 147-18)。依發病年齡、位置與相關異常而存在各種表型。¹⁰⁷,¹⁰⁸
先天性淋巴水腫 (Congenital Lymphedema)
當足背腫脹合併淋巴水腫家族史時,懷疑為 Milroy 病。淋巴水腫於出生時即存在,常為雙側,並影響膝以下的下肢。¹⁰⁹ 其他特徵可合併於先天性淋巴水腫,例如陰囊水腫 (hydrocele, 男性中佔 37%)、明顯靜脈 (23%)、上斜趾甲 (upslanting toenails, 14%)、乳頭瘤病 (papillomatosis, 10%),或男性尿道異常 (4%)。
非皮膚表現 (NONCUTANEOUS FINDINGS)
KTS 或 CLOVES 症候群病人可發生肺栓塞。¹¹⁰ 依受累器官而定,GLA 病人可發生胸膜積液 (pleural effusion)、腹水 (ascites) 與吸收不良 (malabsorption) 等症狀。¹⁰⁵ Gorham-Stout 症候群病人常見疼痛性病理性骨折 (pathological fractures)。¹⁰⁶
併發症 (COMPLICATIONS)
LM 可因咳嗽、發炎、發燒、病毒或細菌感染或病灶內出血而突然增大(圖 147-19)。復發性蜂窩性組織炎 (Recurrent cellulitis) 是主要併發症,尤其在 KTS 病人中,若不及時治療可演變為敗血症。出現局部發紅與溫熱,病灶變得疼痛。顏面不對稱(尤其下頜)常合併微囊型 LM(見圖 147-16)。眼眶內 LM 是眼球異位 (ocular dystopia)、眼球突出 (exophthalmia) 以及眼眶增大的原因。舌頭上的 LM 損害言語並產生滲液與口臭 (halitosis)。當舌根或頸顏面區受累時,呼吸道阻塞常見。¹¹¹ 廣泛的肢體 LM 可造成象皮病 (elephantiasis)。可延伸至胸膜並造成乳糜胸 (chylothorax)。內臟 LM 可造成蛋白質流失性腸病變 (protein-losing enteropathy) 與低白蛋白血症 (hypoalbuminemia)。KTS 病人與 CLOVES 病人分別因持續性胚胎靜脈與擴張胸腔靜脈的存在,而有高肺栓塞風險。¹¹⁰
淋巴水腫在 20% 的病例中併發蜂窩性組織炎。更罕見地,可觀察到胸膜積液(甚至於子宮內)、胎兒水腫 (hydrops fetalis) 與乳糜性腹水 (chylous ascites)。¹⁰⁸,¹¹²,¹¹³
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
LM 以及 KTS 與 CLOVES 症候群,是由 PIK3CA 的鑲嵌性或體細胞突變所引起,這些突變活化 PI3K/AKT/mTor 訊號途徑。¹¹,¹²,¹¹⁴,¹¹⁵
已辨識出超過 20 個基因在各種形式的原發性淋巴水腫中發生突變。¹⁰⁸
Milroy 病是由血管內皮生長因子受體 3(vascular endothelial growth factor receptor 3, VEGFR3)的遺傳性功能喪失突變所引起。¹¹⁶⁻¹¹⁸ 同一基因的突變也是散發性胎兒水腫與全身性皮下水腫的原因。¹¹⁹ 淋巴水腫-雙行睫 (Lymphedema-distichiasis) 是由 FOXC2 轉錄因子的功能喪失突變所引起。¹²⁰ 合併小頭症(伴或不伴脈絡視網膜病變或發展遲緩)的淋巴水腫,是由 KIF11 的顯性遺傳突變所引起。¹²¹,¹²² 少毛症-淋巴水腫-微血管擴張 (Hypotrichosis–lymphedema–telangiectasia),具有體染色體顯性或隱性遺傳模式,由 SOX18 的突變所引起。¹¹⁸ Hennekam 症候群(OMIM #235510)是一種體染色體隱性的全身性淋巴發育不良,特徵為腸道淋巴管擴張 (intestinal lymphangiectasia) 合併四肢、生殖器與顏面的嚴重且進行性淋巴水腫,以及嚴重智能不足。¹²³ 已在 CCBE1(膠原蛋白與鈣結合 EGF 結構域 1,collagen and calcium-binding EGF domains 1)中辨識出突變。¹⁰⁷,¹²⁴ 在由 GATA2 突變引起的 Emberger 症候群中,淋巴水腫合併血液惡性腫瘤。¹²⁵ 在約 35% 至 40% 的家族性原發性淋巴水腫病人中,可辨識出生殖細胞突變。¹⁰⁸,¹²⁶
診斷 (DIAGNOSIS)
輔助檢查 (SUPPORTIVE STUDIES)
病理 (Pathology):LM 的特徵為擴張、內皮扁平內襯、管壁厚度不一的通道(見圖 147-8C)。除非囊內出血後或存在混合淋巴-靜脈畸形,否則這些腔隙中看不到血球。巨囊型 LM 由單一或多發、被厚纖維膜環繞的淋巴囊腫構成。這些囊腫彼此不相通。內皮細胞表現特定的淋巴標記,例如足泡蛋白 (podoplanin)、D2-40 與 VEGFR-3。⁴⁴,¹²⁷ 淋巴水腫的特徵為原發性周邊淋巴微血管、收集淋巴管,或淋巴瓣膜與淋巴靜脈瓣膜的異常。這在受累肢體中明顯,但有時也見於對側。常存在廣泛的真皮纖維化。⁴⁴
影像 (Imaging):都卜勒超音波顯示由薄隔膜 (thin septa) 分隔的微囊、巨囊或兩者。與 VM 相反,它們無法被探頭壓陷。MRI 也顯示病灶的囊性性質,常可辨別出液-液平面 (fluid-fluid levels)。¹²⁸,¹²⁹ 電腦斷層 (Computed tomography, CT) 是顯示骨性侵犯的最佳檢查。在 KTS 與 CLOVES 病人中,需要對受累下肢與骨盆區的深部靜脈系統進行都卜勒超音波。此外,由於 CLOVES 症候群的特徵為嬰兒期進行性過度生長,建議每 6 個月仔細追蹤直到青春期結束。追蹤包括臨床檢查、肢體測量(scaniometry,下肢長度 X 光片)與腹部超音波,因為隱睪症 (cryptorchidism)、陰囊水腫、腎萎縮與威爾姆斯腫瘤的風險增加。¹³⁰ 應依症狀進行額外檢查。
基因檢測 (Genetic Testing):在通過認證的診斷實驗室中可進行以基因組套為基礎的檢測。此類篩檢有助於辨識生殖細胞突變並可協助處置。例如,辨識出生殖細胞 GATA2 突變表示需要特定的癌症監測計畫,而 KIF11 突變則意味著需要詳細的眼科與發展檢查。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 147-7。

表 147-7:淋巴管畸形的鑑別診斷 (Differential Diagnosis of Lymphatic Malformation)
| 鑑別診斷 | |
|---|---|
| 最可能 (Most Likely) | ■ 嬰兒血管瘤 |
| 考慮 (Consider) | ■ 靜脈畸形 ■ 畸胎瘤 (Teratoma) ■ 纖維肉瘤或橫紋肌肉瘤 (Fibrosarcoma or rhabdomyosarcoma) |
| 須排除(外陰部 Vulvar)(Rule Out) | ■ 放射治療引起的後天性淋巴管擴張 ■ 克隆氏病 (Crohn disease) 引起的後天性淋巴管擴張 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
LM 通常隨兒童生長。它可造成伴骨性過度生長的不對稱。在感染或病灶內出血期間可見大小增加。有偶發性自發消退的報告。由於局部感染後常見消退,這可能是由發炎後自體硬化 (postinflammatory autosclerosis) 所引起。Gorham-Stout 症候群病人預後不佳;16% 的病例為致死性。
處置 (MANAGEMENT)
介入 (INTERVENTIONS)
醫療處置 (Medical Management):在孤立性與症候群性 LM 中,皆應使用全身性抗生素治療病灶內細菌感染以及早期蜂窩性組織炎。可能需要抗發炎藥物以緩解疼痛。CLOVES 或 KTS 症候群病人需在任何外科手術前接受預防劑量 100 anti-Xa/kg/day 的 LMWH;這應在術後持續另 10 至 20 天,以避免其凝血異常於圍手術期代償失調,更重要的是降低危及生命的術後肺栓塞風險。¹¹⁰ 雷帕黴素 (Rapamycin) 已有效用於對標準治療有抗藥性的廣泛 LM,以及 KTS 與 GLA 病人。雖然無人達到完全消退,但數名病人經歷了淋巴滲漏與感染的停止以及畸形體積的減少。副作用輕微。¹³¹,¹³²
淋巴水腫最好以彈性襪 (elastic stockings)、按摩與氣壓壓迫裝置 (pneumatic compression devices) 治療。Sildenafil 曾被建議對 LM 的治療有效¹³³,但進一步的研究並未證實其療效,並凸顯了潛在副作用,例如感染與出血。¹³⁴,¹³⁵
治療程序 (Procedures):Nd:YAG 雷射或二氧化碳雷射光凝固術 (photocoagulation) 已被用於治療造成滲液的真皮 LM 水疱(圖 147-20)。巨囊型 LM 以抽吸液體後,由介入放射科醫師進行經皮、病灶內注射硬化劑來治療。硬化劑包括四癸基硫酸鈉 (sodium tetradecyl sulfate)、純乙醇、OK432(由滅活的 A 群化膿性鏈球菌 [group A Streptococcus pyogenes] 菌株萃取物:picibanil)、doxycycline 與博來黴素 (bleomycin)。副作用從發燒與紅斑到水腫不等。¹³⁶,¹³⁷
手術切除是 LM、CLOVES 與 KTS 症候群的另一替代方案。¹³⁸,¹³⁹ 結果取決於病灶的解剖部位與延伸範圍。復發頻繁,因為難以區分微囊型 LM 與鄰近正常組織。這導致切除不完全或不必要地犧牲正常結構。
諮詢 (Counseling):對於原發性淋巴水腫,必須對家庭與病人進行關於成因與治療的衛教,因為它可遺傳並合併其他重要的醫療問題,需要納入特殊監測計畫。針對血管異常的社會心理諮詢與病友組織對病人及其家屬也是一項重要幫助。
動靜脈畸形 (ARTERIOVENOUS MALFORMATIONS)
重點一覽 (AT-A-GLANCE)
■ 全球皆有發生;罕見但確切頻率未知。
■ 為先天性、快流型畸形,可隱匿至青春期。
■ 組織學上由動脈與靜脈之間的直接交通構成。
■ 通常為散發性,但可有遺傳傾向,如 CM-AVM、遺傳性出血性微血管擴張或 PHTS。
■ 為最難治療的血管畸形;需要多專科團隊合作。
前言 (INTRODUCTION)
快流型血管畸形,即 AVM 與 AVF,是最嚴重且最具破壞性的畸形。AVF 通常為外傷的結果。AVM 的特徵為存在一個「病灶核心 (nidus)」,這是病灶的震央,由多條供應動脈與引流靜脈之間的直接交通所構成,其間沒有正常的微血管床。AVM 於出生時即存在,儘管它們不一定可見。它們可為局部性或廣泛性,並隨年齡演進。
流行病學 (EPIDEMIOLOGY)
AVM 的發生率未知。尚未辨識出性別偏向。它是一種罕見、通常為散發性的快流型血管畸形。遺傳性出血性微血管擴張(hereditary hemorrhagic telangiectasia, HHT)是一種體染色體遺傳疾患,盛行率為 1/5000。¹⁴⁰
CM-AVM 是另一種體染色體顯性遺傳的家族形式,盛行率估計值相似。¹⁹⁻²³
症候群性疾患 (SYNDROMIC DISORDERS)
Bonnet-Dechaume-Blanc 或 Wyburn-Mason 症候群 (BONNET-DECHAUME-BLANC OR WYBURN-MASON SYNDROME)
Bonnet-Dechaume-Blanc 或 Wyburn-Mason 症候群是一種散發性、症候群性 AVM,位於顏面中央或半側顏面區(或兩者),合併眼-眼眶 (oculo-orbital) 與腦部侵犯。¹⁴¹ 它很少像 SWS 那樣依循三叉神經分布。腦內 AVM 在 Bonnet-Dechaume 症候群中常見,可造成鼻出血 (epistaxis)、眼球突出與偏盲 (hemianopia)。也可發生智能不足。
Cobb 症候群 (COBB SYNDROME)
Cobb 症候群是另一種散發性、症候群性 AVM,將同一體節 (metamere) 的皮膚與脊髓 AVM 相關聯。¹⁴² 皮膚病灶偽裝為 CM,儘管它觸診溫熱。Cobb 症候群在兒童期表現為突然發作的背部或下肢疼痛合併感覺障礙。依 AVM 的位置與延伸範圍,可發生其他神經學併發症(疼痛、感覺與運動障礙,以及神經性膀胱 neurogenic bladder)。
遺傳性出血性微血管擴張 (HEREDITARY HEMORRHAGIC TELANGIECTASIA)
HHT 個體表現出以下三聯徵 (triad) 的組合:多發性皮膚與黏膜微血管擴張(常位於黏膜唇)、鼻出血,以及陽性家族史。¹⁴³ 診斷為臨床診斷,依照 Curaçao 標準。HHT 病人各表現的估計頻率為:自發性、復發性鼻出血,90%;皮膚微血管擴張,75%;肝或肺 AVM,30%;胃腸道出血,15%。¹⁴⁴ 30% 的 HHT 病人可見肝、肺或腦 AVM(或這些 AVM 的任何組合)。¹⁴⁴ 相反地,23% 的 CM-AVM1 病人與 13% 的 CM-AVM2 病人存在腦內或脊髓內 AVM¹⁸,¹⁹,²²,²³,³⁵,但不像 HHT 那樣有內臟 AVM 風險。Galen 靜脈瘤樣畸形 (Vein of Galen aneurysmal malformation) 也是表型的一部分。復發性鼻出血通常是 HHT 的初始表現。流鼻血可嚴重到需要輸血。肺 AVM 影響約 30% 的 HHT 病人,在 HHT1 中尤其常見。由於肺的正常過濾功能喪失,這些病人發生中風與腦膿瘍 (brain abscess) 的風險高於健康人群。偏頭痛發生於 13% 至 50% 的病例。復發性無痛胃腸道出血發生於 10% 至 40% 的病人。症狀可能包括腹痛、黃疸、高輸出性心衰竭 (high-output cardiac failure) 症狀,以及食道靜脈曲張 (esophageal varices) 出血。¹⁴⁵
Parkes Weber 症候群 (PARKES WEBER SYNDROME)
Parkes Weber 症候群的特徵為四肢上一大片先天性、皮膚性、紅色血管染斑,合併受累肢體的軟組織與骨骼肥大,以及下方多發性小動脈-小靜脈微瘻管 (arteriolar-venular microfistulas)。¹⁴⁶ 受累肢體(常為下肢)較對側更長更大。雖然常為散發性,但它可為 CM-AVM 的一部分。在這些情況下,身體其他部位有小型、多灶性 CM。徵象與症狀隨年齡惡化。受影響病人可發展出充血性心衰竭 (congestive heart failure)。
磷酸酶與張力蛋白同源物錯構瘤腫瘤症候群 (PHOSPHATASE AND TENSIN HOMOLOG HAMARTOMA TUMOR SYNDROME)
PHTS 是一種體染色體顯性疾患,包括 Bannayan-Riley-Ruvalcaba 症候群與 Cowden 症候群病人,因為他們之中分別有 60% 與 81% 帶有 PTEN 突變。¹⁴⁷ 這些病人典型有巨頭症、陰莖雀斑 (penile freckling)、腦中多發性發育性靜脈異常、快流型 VM (54%),以及惡性化風險增加。這些 VM 常為多灶性 (57%) 且為肌肉骨骼性,並合併異位脂肪沉積 (ectopic fat deposition) 與正常組織結構的破壞。¹⁴⁷,¹⁴⁸
臨床特徵 (CLINICAL FEATURES)
皮膚表現 (CUTANEOUS FINDINGS)
AVM 通常表現為皮膚性、淡色、紅至紫色、界線不清的腫塊,伴有震顫、雜音或振幅增加的搏動(圖 147-21 至 147-23;另見圖 147-1D 及 147-1E)。約三分之一的 AVM 於出生時即存在,另三分之一在兒童期或青春期出現,其餘則在成年期表現,因為荷爾蒙變化與外傷通常會觸發 AVM 的生長。AVM 可影響任何組織與器官。70% 的 AVM 位於頭頸部。它們從不自發消退並隨時間惡化。¹⁴⁹
1985 年,Schobinger 依嚴重度將 AVM 分期。Schobinger 第 I 期為伴有雜音與振幅增加搏動的紅色染斑。隨年齡、青春期或外傷,AVM 可惡化,靜脈變得明顯且迂曲(Schobinger 第 II 期;圖 147-22),隨後變深且疼痛,並潰瘍與出血(Schobinger 第 III 期;見圖 147-23)。大型 AVM 的最終階段為心衰竭(Schobinger 第 IV 期;見圖 147-23)。¹⁵⁰ 病人常表示能聽到自己的心臟搏動。
CM-AVM(見前文 CM-AVM 討論)表現為隨機分布的多發性非典型 CM(見圖 147-6),並在 30%(CM-AVM1)或 20%(CM-AVM2)的病人中合併快流型病灶——AVM、AVF 或 Parkes Weber 症候群。家族內的表現度高度可變,從小型、無症狀的 CM 到危及生命的 AVM。快流型病灶(AVM 或 AVF)為皮膚性或皮下性,伴或不伴肌肉內與骨內侵犯。約 10% 至 15% 的 CM-AVM 病人有侵犯下肢(此類病例的三分之二)或上肢的 Parkes Weber 表型。這些 AVM 可無症狀,但依位置與大小會發生併發症。¹⁹⁻²¹
非皮膚表現 (NONCUTANEOUS FINDINGS)
30% 的 HHT 病人有肝、肺或腦 AVM(或這些 AVM 的任何組合)。¹⁴⁴ 23% 的 CM-AVM1 病人與 13% 的 CM-AVM2 病人存在腦內或脊髓內 AVM(或兩者)。¹⁸,¹⁹,²²,²³,³⁵
併發症 (COMPLICATIONS)
侵犯顏面的 AVM 也可破壞下方的骨結構,並可造成牙齦出血或鼻出血以及肥大性不對稱。骨性肥大是顏面 AVM 的常見特徵,造成不對稱。四肢 AVM 常因血流竊取現象 (blood flow steal phenomenon) 而造成周邊缺血。心衰竭罕見(Schobinger 第 IV 期),尤其在兒童期。青春期、外傷以及荷爾蒙影響可觸發 AVM 的出現。腦內 AVM,如 Bonnet-Dechaume 症候群所見,可造成鼻出血、眼球突出與偏盲。也可發生智能不足。Cobb 症候群在兒童期表現為突然發作的背部或下肢疼痛合併感覺障礙。依 AVM 的位置與延伸範圍,可發生其他神經學併發症(疼痛、感覺與運動障礙,以及神經性膀胱)。復發性鼻出血通常是 HHT 的初始表現。流鼻血可嚴重到需要輸血。HHT1 病人的腦 AVM 盛行率比一般人群(1/10,000)高 1000 倍,HHT2 病人則高 100 倍。CM-AVM1 與 2 也可見類似的風險增加。有肺 AVM 與胃腸道微血管擴張的 HHT 病人有危及生命的出血風險。其他出血部位可能包括腎、脾、膀胱、肝、腦膜與腦的部位。中風可為出血性或缺血性。由於肺的正常過濾功能喪失,這些病人發生中風與腦膿瘍的風險高於健康人群。偏頭痛發生於 13% 至 50% 的病例。復發性無痛胃腸道出血發生於 10% 至 40% 的病人。症狀可能包括腹痛、黃疸、高輸出性心衰竭症狀,以及食道靜脈曲張出血。¹⁴⁵
CM-AVM 病人可因 Galen 靜脈瘤樣畸形而出現子宮內危及生命的腦內出血。²²,¹⁵¹ 腦與延髓 AVM 也可在出生後出血。²²,³⁵,¹⁵¹
病因與致病機轉 (ETIOLOGY AND PATHOGENESIS)
CM-AVM(OMIM #608354)、HHT(OMIM #187300)與 PHTS(OMIM #153480 及 158350)的快流型血管畸形之潛在遺傳缺陷已被辨識。這些疾患以體染色體顯性模式遺傳,皮膚 CM 或微血管擴張的外顯率約為 95%。¹⁸,¹⁹,²²,¹⁵¹
HHT 是由轉化生長因子-β(transforming growth factor-β)訊號途徑的改變所引起。涉及 HHT 的基因為 ENG(編碼 endoglin,HHT 第 1 型)與 ACVRL1(先前稱為 ALK1,HHT 第 2 型);SMAD4 與 GDF2 則較不常涉及。¹⁵²⁻¹⁵⁵
Endoglin 與 ACVRL1 分別為第 III 型與第 I 型轉化生長因子-β 受體,兩者皆在血管內皮細胞上良好表現。此訊號的功能喪失會同時增加 PI3K-AKT 訊號。¹⁵⁶
CM-AVM1 是由 RASA1 的功能喪失突變所引起。¹⁸,²⁰ RASA1 編碼 P120RASGAP,一種調節 RAS 活性的 GTP 酶。它將活化的 GTP 結合型 RAS 轉化為其失活的 GDP 結合型。⁶¹,⁶²,¹⁵⁷ 高度的家族內表型可變性可由體細胞二次打擊突變發生的必要性來解釋。²¹,¹⁵⁸ 30% 的 CM-AVM1 病人,其突變為重新發生 (de novo)。CM-AVM2 是由 EPHB4 的功能喪失突變所引起,該基因編碼靜脈血管中的內皮細胞受體。與 EPHB4 相反,配體 EPHRINB2 表現於動脈內皮細胞。此雙向配體-受體系統對動靜脈識別與分離很重要。EPHB4 使用 p120RASGAP 傳訊,任一基因的功能喪失皆造成 RAS-MAPK 訊號增加。¹⁸,¹⁵⁹⁻¹⁶²
AVM 也可由腫瘤抑制基因 PTEN 的功能喪失突變所引起,如 PHTS 病人所見。¹⁶³ PTEN 調節 PI3K-AKT 活性,而這似乎至少部分由 HHT 受體複合體所調節(見前文)。此複合體的某個夥伴或 PTEN 本身的功能喪失會導致 PI3K 的活化。散發性顱外 AVM 是由體細胞活化性 MAP2K1 (MEK) 突變所引起。¹⁴ 腦 AVM 是由 KRAS 的另一個活化性突變所引起,KRAS 也涉及 RAS-MAPK 訊號途徑。¹⁶⁴
診斷 (DIAGNOSIS)
輔助檢查 (SUPPORTIVE STUDIES)
病理 (Pathology):組織學上,AVM 界線不清,由因動靜脈分流 (arteriovenous shunting) 與纖維化而管壁肌肉增厚的扭曲動脈與靜脈構成(見圖 147-22C)。
影像 (Imaging):超音波與彩色都卜勒不顯示腫塊,而顯示低阻力的高速動脈血流與搏動性靜脈血流的聚集。血管迂曲。在四肢,藉由比較受累肢體與未受累肢體的動脈流出進行非侵入性追蹤。MRI 優於 CT,可勾勒 AVM 的範圍並區分 AVM 與血管瘤及其他 VM。對應於快流型血管的血流空隙 (flow voids) 是 AVM 的特異徵象。任何治療前皆需進行動脈攝影 (arteriography) 以確定供應動脈與病灶核心(見圖 147-1E 及 147-22B)。
基因檢測 (Genetic Testing):基因檢測有指徵於合併多灶性皮膚 CM(CM-AVM)或微血管擴張(HHT)的 AVM 病人。分子診斷有助於遺傳諮詢並指引進一步影像檢查以發現可能未被偵測的畸形。合併巨頭症的 AVM 病人應接受 PTEN 突變篩檢。辨識出生殖細胞突變將能進行精確諮詢並納入癌症監測計畫。
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
見表 147-8。在兒童期,AVM 因其表現為淡色、界線不清的斑狀紅色染斑而常被誤認為 CM 或血管瘤。然而,觸診時不成比例的溫熱是其快流成分的線索。搏動、震顫與雜音是支持 AVM 的其他徵象。都卜勒超音波可協助區分慢流型 CM 與快流型 AVM 或血管瘤。血管瘤有赤道供應動脈、周邊靜脈、可變的回音性 (echogenicity) 與快流,但無真正的動靜脈分流。其他皮膚病灶可模擬 AVM。上皮樣血管內皮瘤 (Epithelioid hemangioendothelioma) 發生於四肢,為紫色且局部侵襲性的病灶。腫脹性紅斑性狼瘡 (Tumid lupus erythematosus)、顏面類肉瘤病 (sarcoidosis) 與唇部的 Melkersson-Rosenthal 症候群可模擬 AVM。Dabska 腫瘤位於兒童耳部時可模擬 AVM。
表 147-8:動靜脈畸形的鑑別診斷 (Differential Diagnosis of Arteriovenous Malformation)
| 鑑別診斷 | |
|---|---|
| 最可能(出生時 At birth)(Most Likely) | ■ 先天性血管瘤 ■ 嬰兒纖維肉瘤 (Infantile fibrosarcoma) ■ 其他肉瘤 |
| 出生後快速生長 (Rapid growth after birth) | ■ 嬰兒血管瘤 |
| 須排除(耳、鼻 Ear, nose)(Rule Out) | ■ 腫脹性紅斑性狼瘡 (Lupus erythematosus tumidus) ■ 類肉瘤病 (Sarcoidosis) ■ Dabska 腫瘤 |
| 唇 (Lip) | ■ Melkersson-Rosenthal 症候群 |
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
AVM 傾向隨時間惡化,造成局部破壞或危及生命的出血。青春期與外傷觸發病灶的生長。不當的處置(常因誤診),例如結紮供應動脈,或部分切除病灶核心,可造成戲劇性後果與 AVM 的擴大(見圖 147-23)。CM-AVM 中的 CM 無害,通常僅具美觀重要性,儘管血流特性常異常。
處置 (MANAGEMENT)
這種快流型 VM 是最複雜且最難治療的血管異常。其處置為多專科性。AVM 應藉由定期評估追蹤。治癒 AVM 的目標基於阻塞(藉由栓塞 embolization)並完全移除病灶核心。部分切除與近端結紮供應動脈會導致嚴重併發症、復發,並隨時間再擴大。此外,結紮近端動脈會阻礙進入病灶核心進行栓塞、刺激新供應動脈的招募,因此使畸形擴大。AVM 處置不當可導致截肢。¹⁹
介入 (INTERVENTIONS)
藥物 (Medications):在肢體 AVM 或多發性 AVF 中,彈性襪可穩定病灶並保護皮膚,尤其在早期階段。由於 AVM 難以根除,處置的重點常為控制畸形的演進,而非治癒病人。⁴⁰,⁴⁵ 一種以 Marimastat(一種合成基質金屬蛋白酶抑制劑 matrix metalloproteinase inhibitor)的藥理學方法已被用於治療廣泛的 AVM,效果良好。它減少了畸形的延伸並降低了疼痛與雜音。⁴⁶ 不幸的是,該藥物已不再供應。Thalidomide 已成功用於減少 HHT 病人流鼻血的頻率與持續時間。¹⁶⁵ 它也被用於標準治療未成功的無法切除的第 3 期 AVM。對某些病人,它與栓塞合併使用以提高療效。
治療程序 (Procedures):超選擇性栓塞 (Superselective embolization):與 AVF 相反,單獨的超選擇性動脈栓塞僅為緩解性,且僅用於無法切除、複雜的 AVM。被栓塞的顆粒需到達病灶的震央,以避免病灶核心透過新側支再充填。¹⁶⁶ 對於先前曾接受動脈結紮或栓塞的病人,直接穿刺病灶核心是另一種可能性。¹⁶⁷ 最常見的材料為液體(乙醇、異丁基氰基丙烯酸酯 isobutylcyanoacrylate)、顆粒(Ivalon)、泡沫,以及可植入裝置(線圈、微球體 microspheres)。除非為動靜脈络管而進行,否則栓塞很少能治癒。栓塞可單獨使用(緩解性方法)或隨後完全手術切除(治癒性方法)。注射部位必須盡可能靠近病灶核心,以允許超選擇性方法。近端阻塞是病灶核心透過側支動脈繼發性再充填的原因。²⁴
根除性切除 (Radical excision):手術切除通常僅在栓塞後進行,以將圍手術期出血降至最低。¹⁶⁸ 切除應完全且根除性,盡可能採用如腫瘤外科的切緣 (carcinologic margins)。切除前先進行超選擇性栓塞以阻塞病灶核心並減少術中出血。¹⁹,²⁶ 若覆蓋皮膚正常則可保留;否則應廣泛切除。覆蓋選項包括組織擴張 (tissue expansion) 或局部或游離皮瓣 (free flap)。²⁹ 皮膚擴張可在栓塞或手術前進行,但可能作為刺激畸形生長的觸發因素。對「靜止性 AVM」(Schobinger 第 I 期)的早期介入有爭議,僅在可能完全切除時才應考慮。¹⁶⁹ 任何治療後,皆強制每年以都卜勒超音波或 MRI(或兩者)追蹤至少 5 年。對於 Parkes Weber 症候群病人,治療應盡可能保守。可能需要骨骺固定術以控制腿長差異。此程序有時可能加重 AVM。¹⁴⁶
對於 HHT 病人,已嘗試多種治療以減少出血,包括局部塗抹抗發炎藥物、雷射或手術。由於病灶範圍廣泛,它們皆只能給出有限的無症狀間隔。
諮詢 (Counseling):遺傳諮詢在 CM-AVM、HHT 與 PHTS 中為強制性。
圖片 (FIGURES)
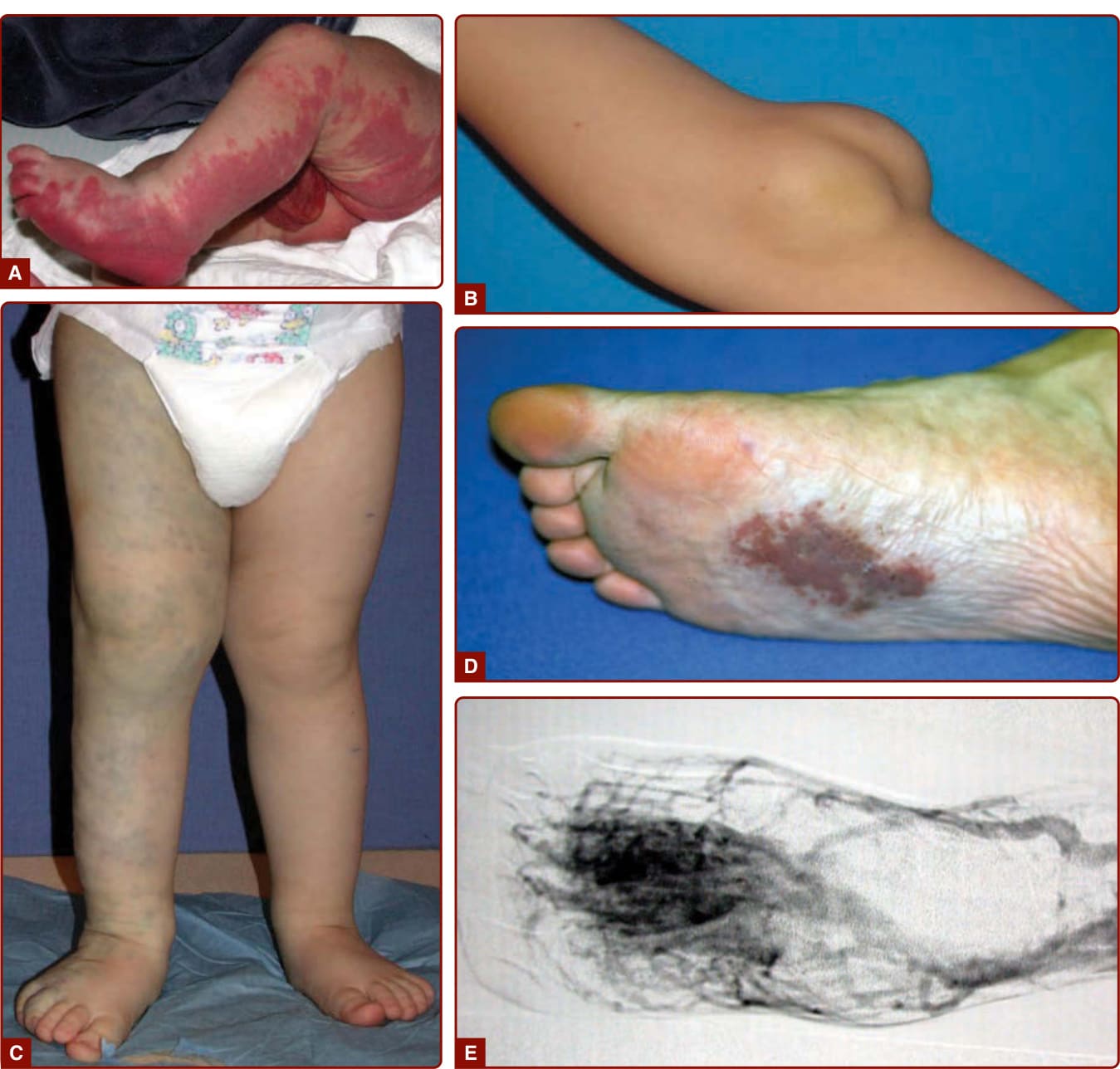
圖 147-1:各類血管類型的血管畸形。A,左下肢與生殖器的微血管畸形 (capillary malformation)。B,侵犯肘部神經 (cubital nerve) 的皮下淋巴管畸形。C,右下肢的廣泛靜脈畸形,造成疼痛、腫脹與凝血障礙。D,足底的動靜脈畸形。E,動脈攝影上病灶的病灶核心 (nidus)。
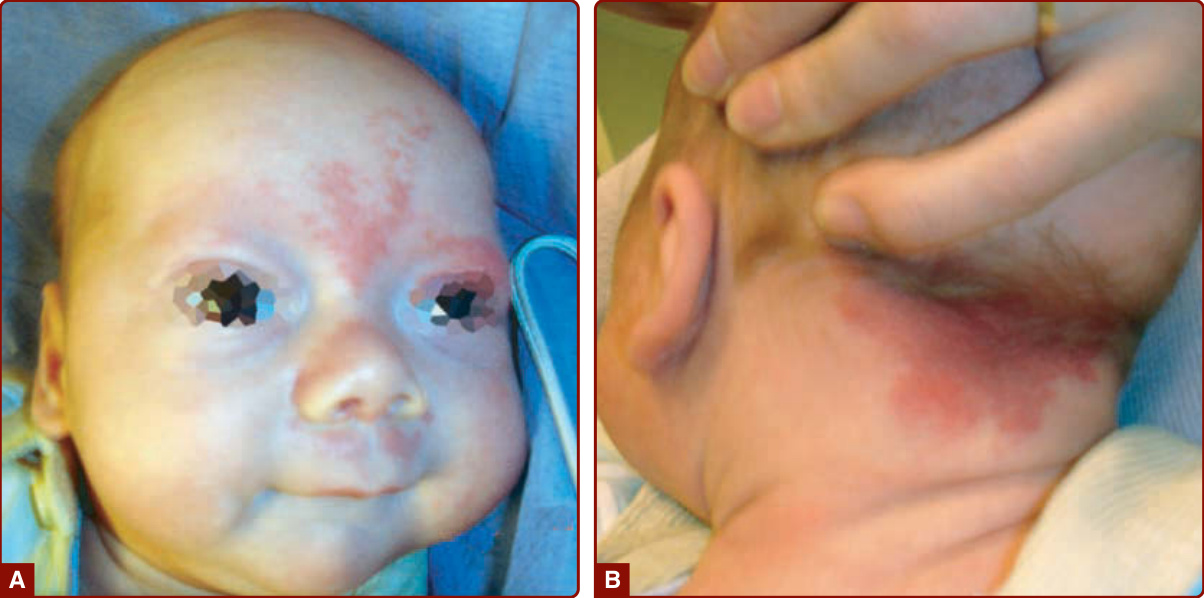
圖 147-2:一名 6 週大新生兒眉間、上眼瞼與上唇 (A) 的新生兒火焰母斑 (nevus flammeus neonatorum,又稱單純母斑 nevus simplex),該嬰兒枕部 (B) 也有 Unna 母斑。
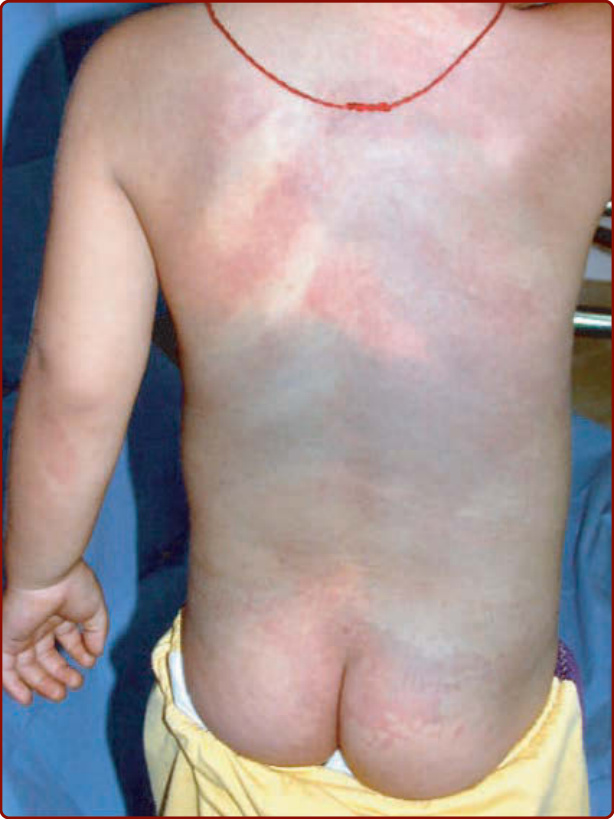
圖 147-3:斑痣性血管錯構瘤病 (phakomatosis pigmentovascularis) 的臨床特徵。位於背部的一大片按體節分布的微血管畸形合併非典型廣泛性真皮黑色素增多 (dermal melanocytosis)。
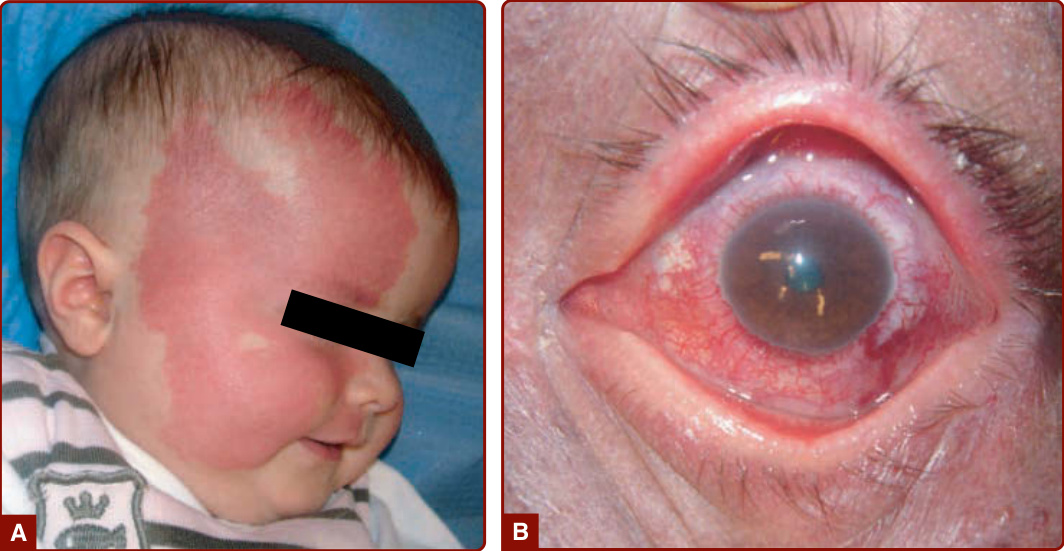
圖 147-4:廣泛的右側顏面微血管畸形。A,一名 6 個月大、患有 Sturge-Weber 症候群的女嬰,侵犯三叉神經的眼支與上頜支。B,一名 50 歲、患有 Sturge-Weber 症候群的男性,脈絡膜微血管-靜脈畸形 (choroid capillary–venous malformation) 造成視力喪失。
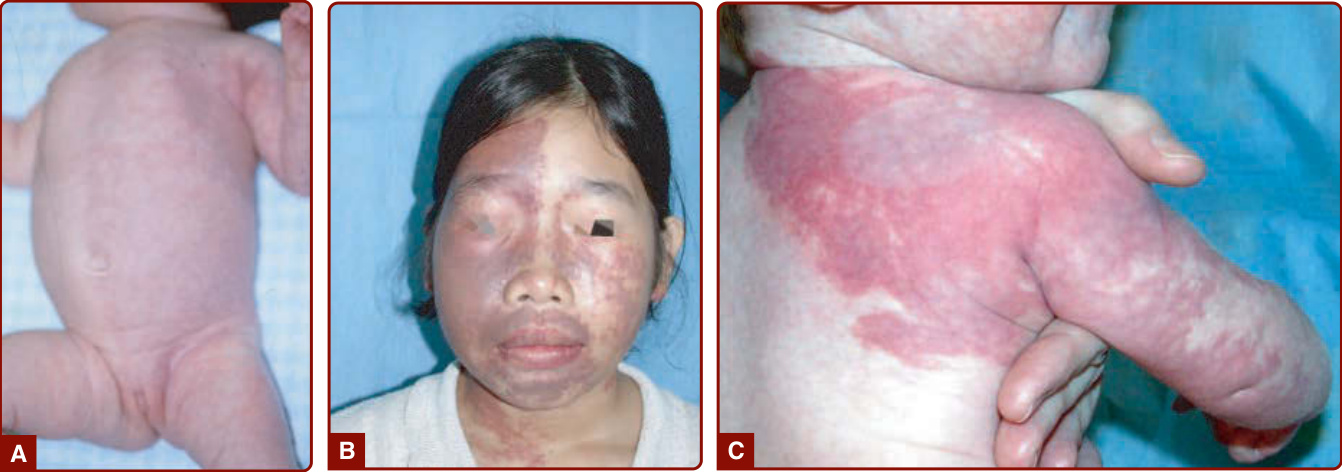
圖 147-5:微血管畸形的數種樣貌。A,侵犯半側身體的淺紅色病灶。B,顏面深紅色病灶合併黏膜侵犯與軟組織肥大。C,較局限於肩部與手臂。
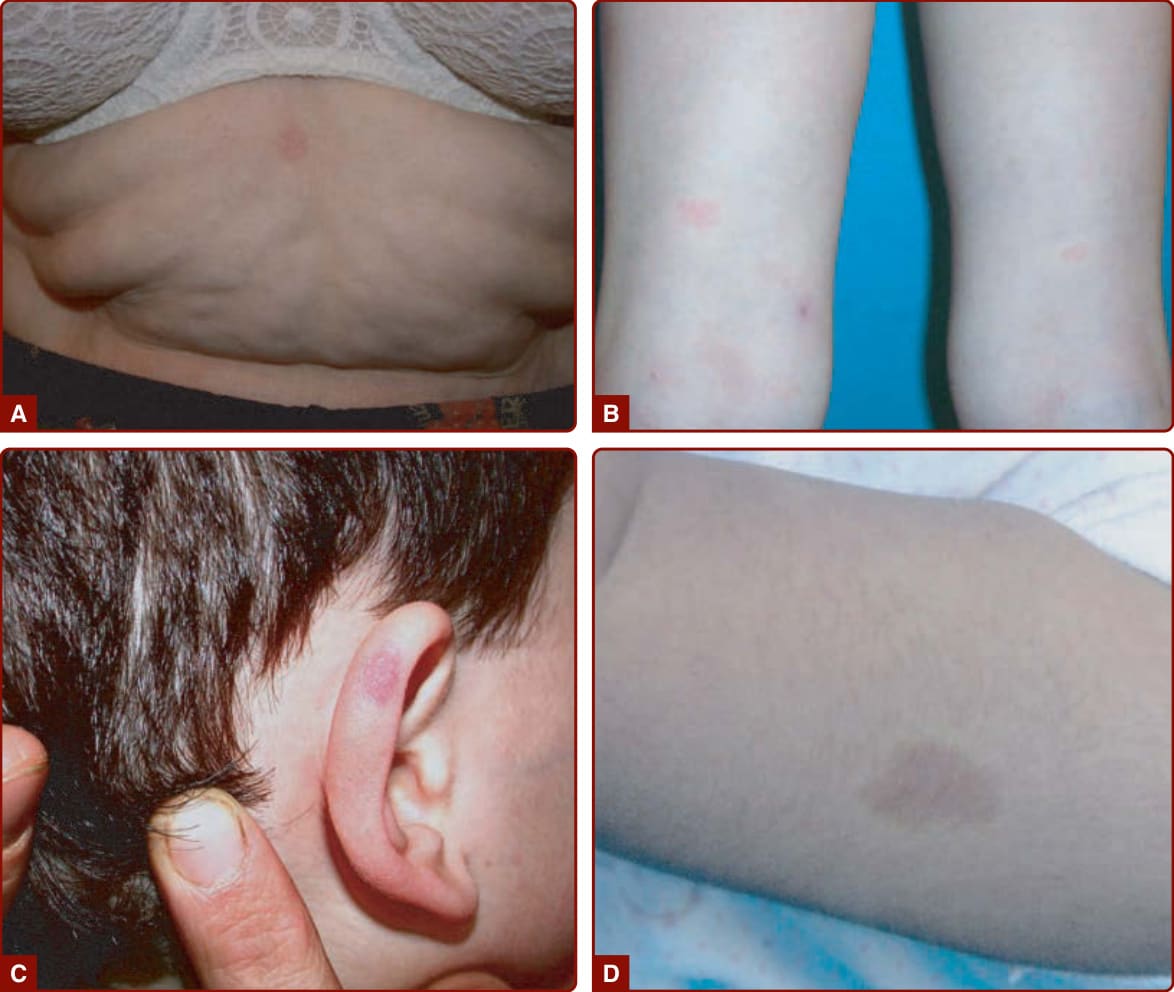
圖 147-6:A–D,屬於 CM-AVM(微血管畸形-動靜脈畸形)之小型非典型微血管畸形範例。D,注意病灶周圍環繞的蒼白暈 (pale halo)。
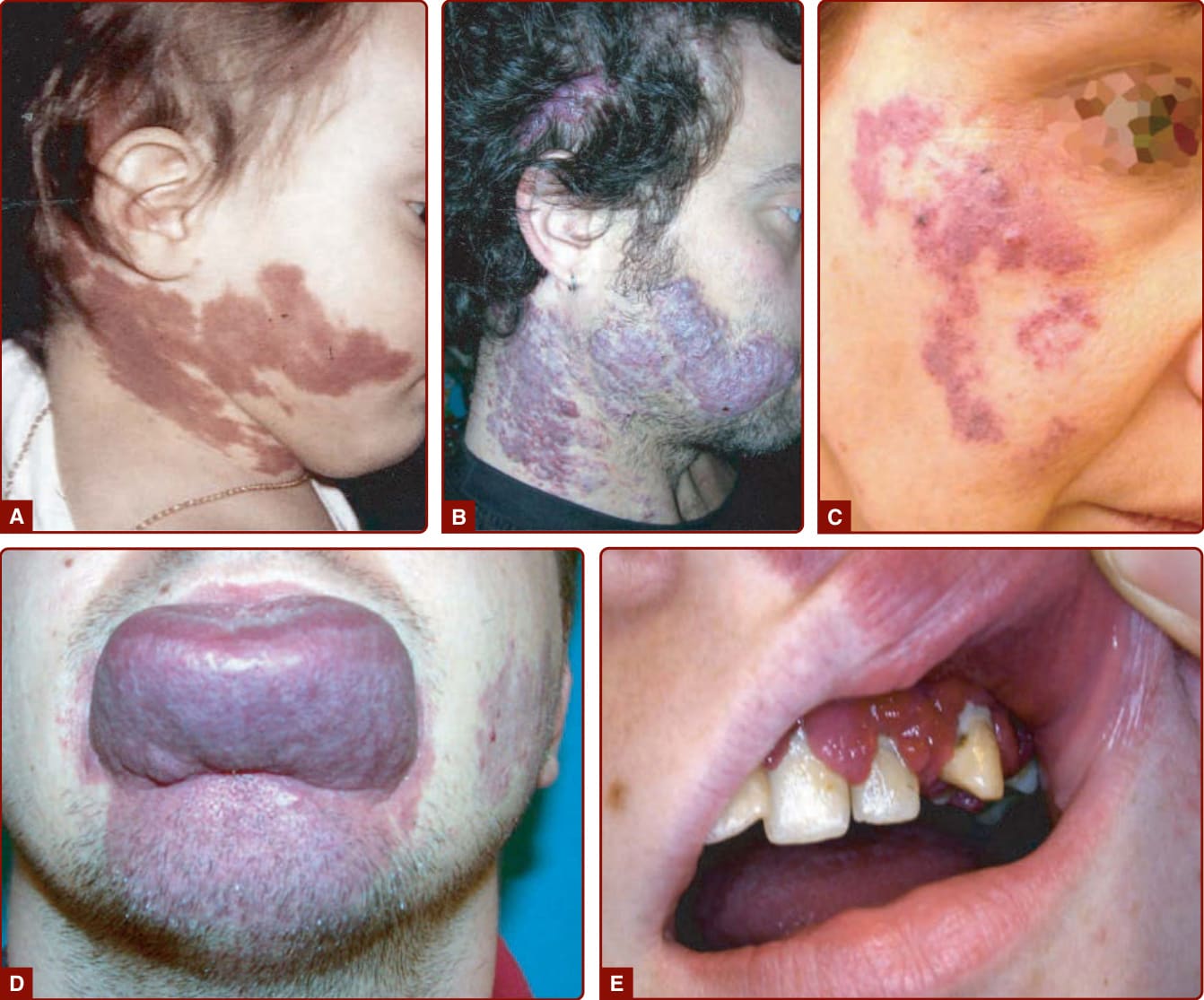
圖 147-7:微血管畸形的臨床演變:隨時間變深與增厚。A,6 個月大時。B,33 歲時。C,化膿性肉芽腫 (pyogenic granuloma) 的發展。D,軟組織肥大。E,黏膜肥大。
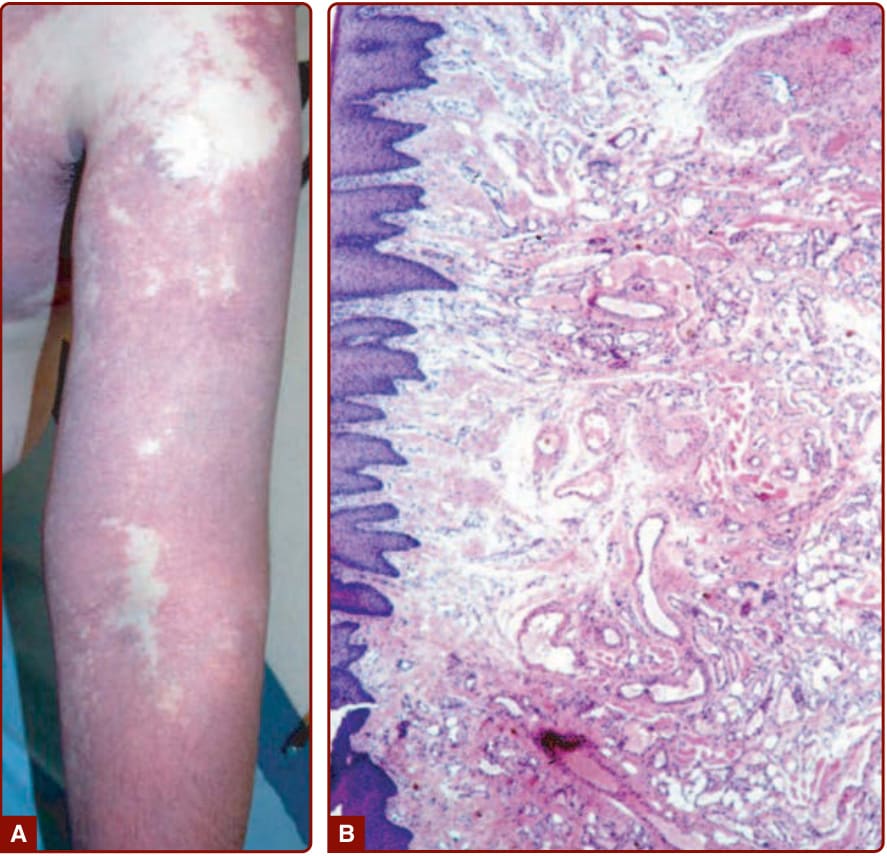
圖 147-8:A,上肢的廣泛微血管畸形。B,組織學上,微血管畸形的特徵為真皮乳頭層與上網狀層中正常數量的擴張微血管,並合併正常外觀微血管數量增加的區域(蘇木精與伊紅染色 hematoxylin and eosin staining)。
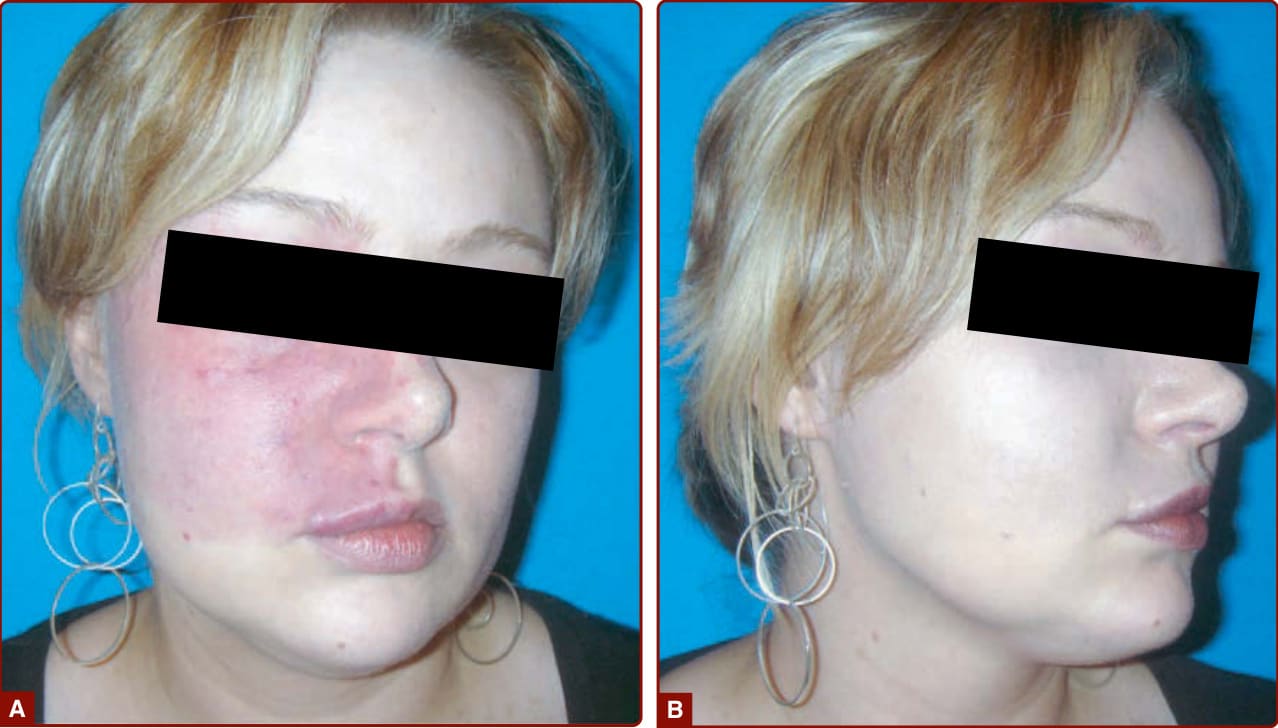
圖 147-9:使用化妝品有效地非侵入性處置顏面微血管畸形。
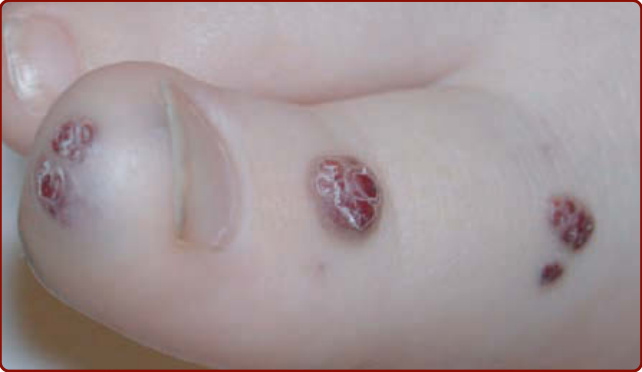
圖 147-10:藍色橡皮泡母斑症候群 (Blue rubber bleb nevus syndrome)。腳趾上特異徵象性的多發性小型、深藍色、乳頭狀病灶。
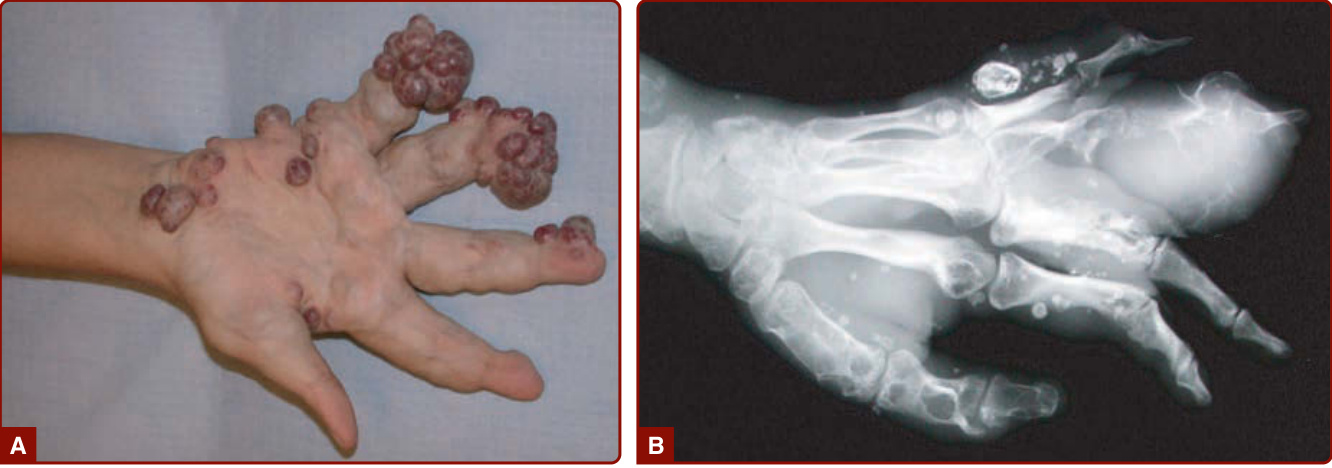
圖 147-11:Maffucci 症候群的臨床 (A) 與放射學 (B) 樣貌。多發性靜脈畸形與內生軟骨瘤使手部與手指變形。
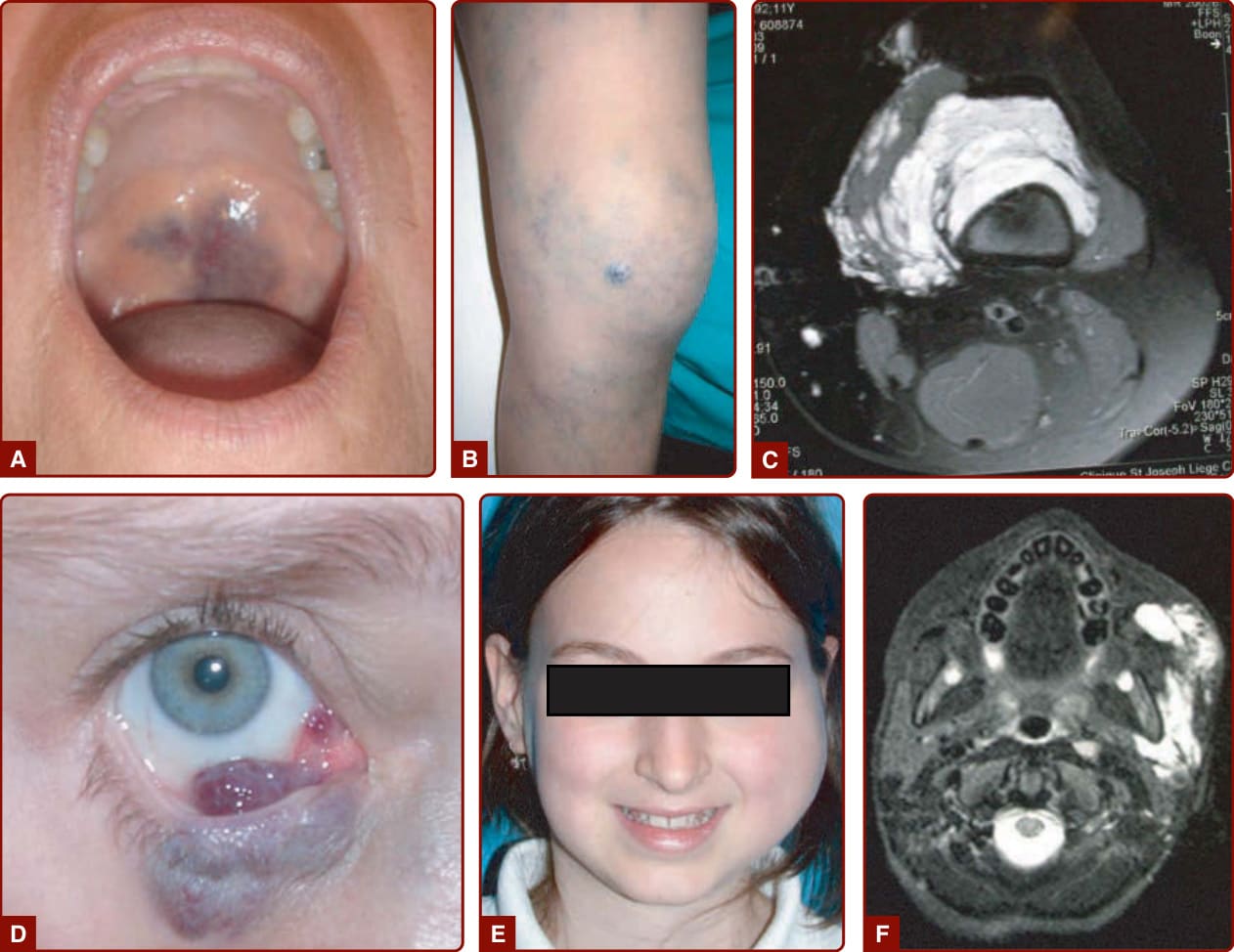
圖 147-12:各種靜脈畸形。A,軟顎處,造成睡眠呼吸中止。B 與 C,膝部處,造成關節積血 (hemarthroses);以 T2 加權影像確認。D,黏膜眼瞼處。E 與 F,臉頰處,造成顏面不對稱與疼痛。
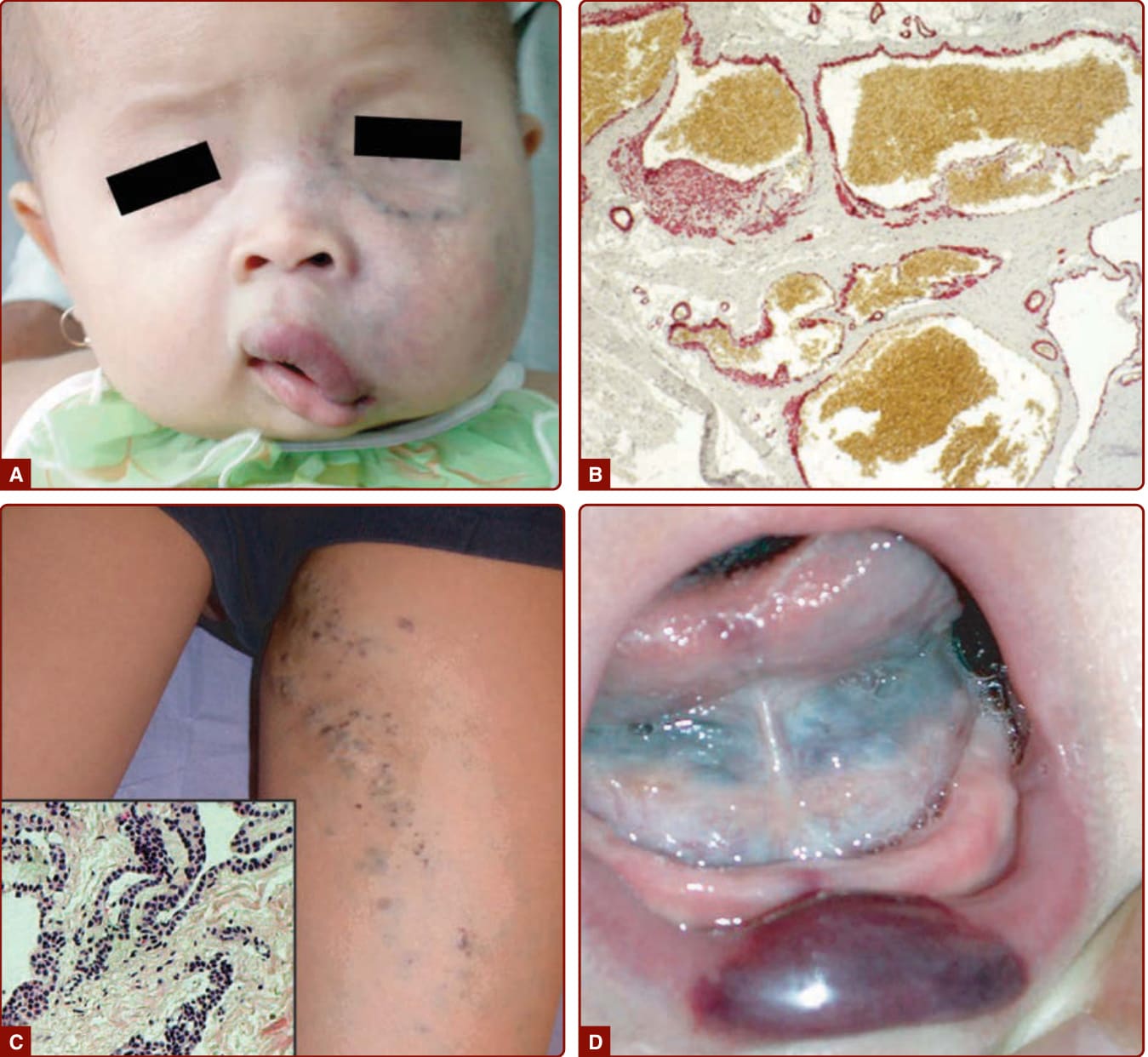
圖 147-13:靜脈畸形 (VMs)。A,一名 6 個月大男嬰廣泛的半側顏面 VM,造成軟組織肥大與唇部變形。B,組織學上,VM 由具管壁細胞 (mural cells) 異常的擴張靜脈樣通道構成(蘇木精與伊紅)。C,大腿內側的淺表球體靜脈畸形 (glomuvenous malformation)。注意組織學上管壁球體細胞 (glomus cells) 的存在(蘇木精與伊紅)。D,侵犯下唇與口底的黏膜 VM。
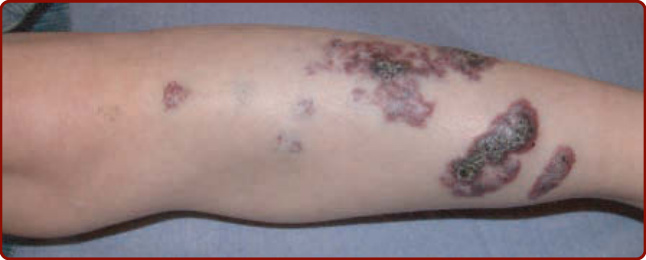
圖 147-14:下肢的疣狀靜脈畸形 (verrucous venous malformation,先前稱為疣狀血管瘤),造成滲液。它呈紫紅色且略微隆起,並有角化過度。
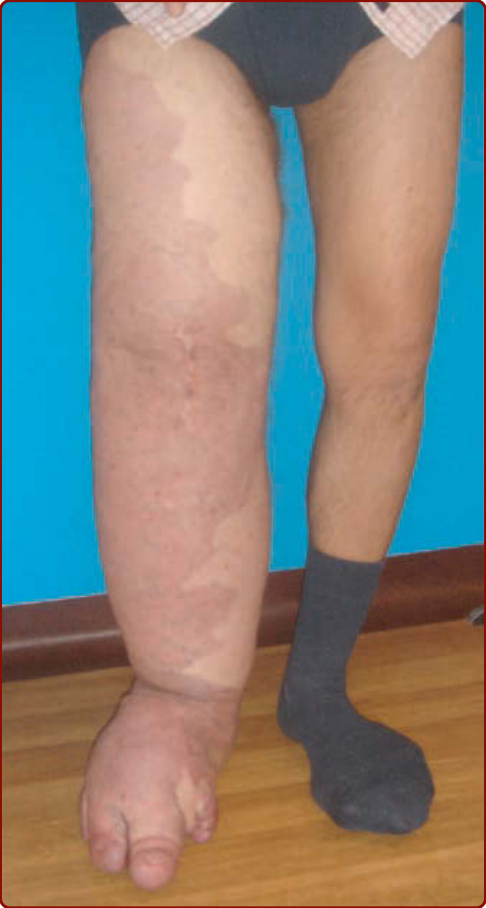
圖 147-15:右下肢的微血管-淋巴管-靜脈畸形合併軟組織肥大(CLOVES 症候群)。
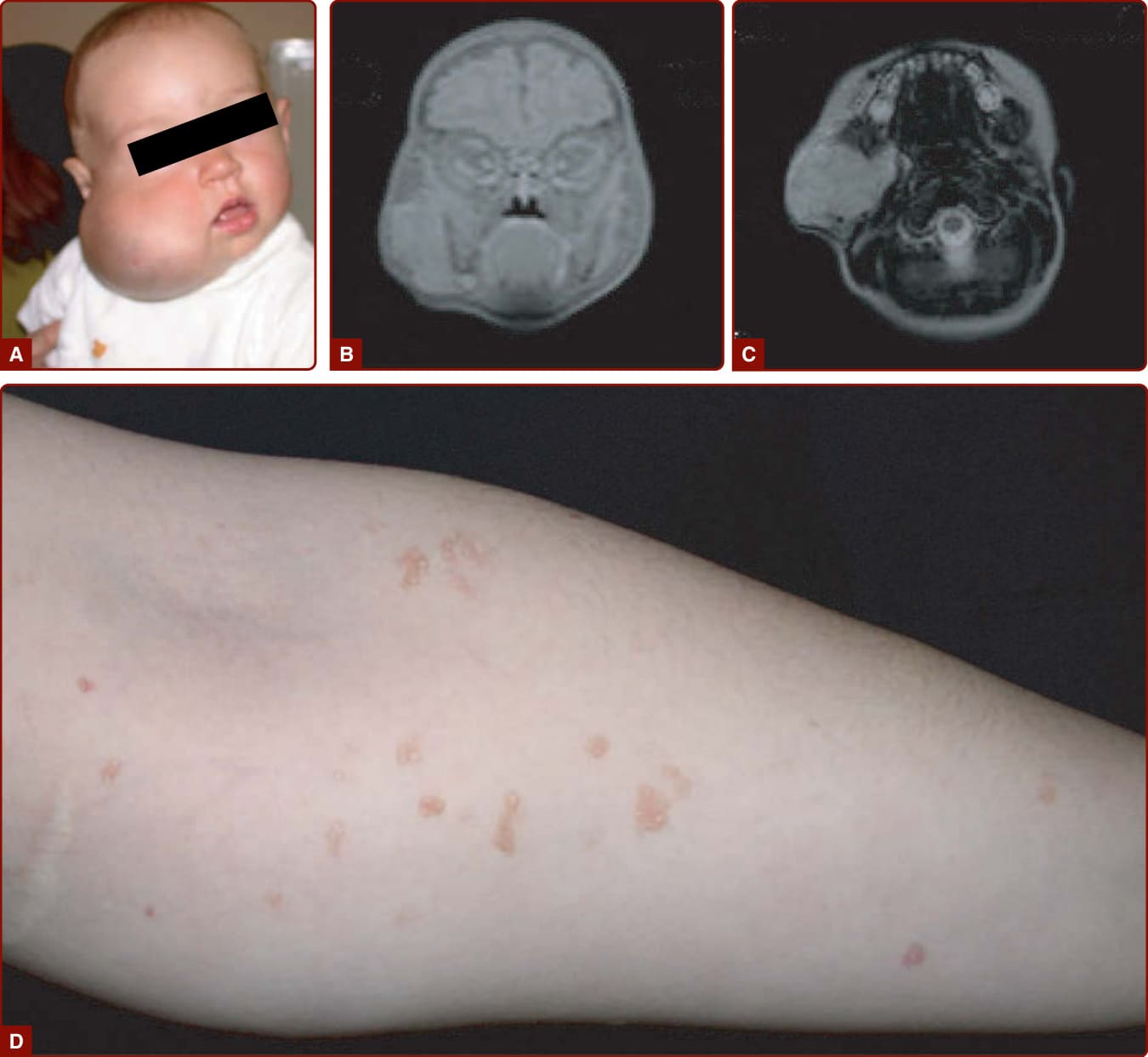
圖 147-16:淋巴管畸形 (LM) 的各種樣貌。臉頰廣泛 LM 的藍色變色 (A),如電腦斷層掃描所見 (B 與 C)。真皮 LM 的小型清澈水疱 (D)。
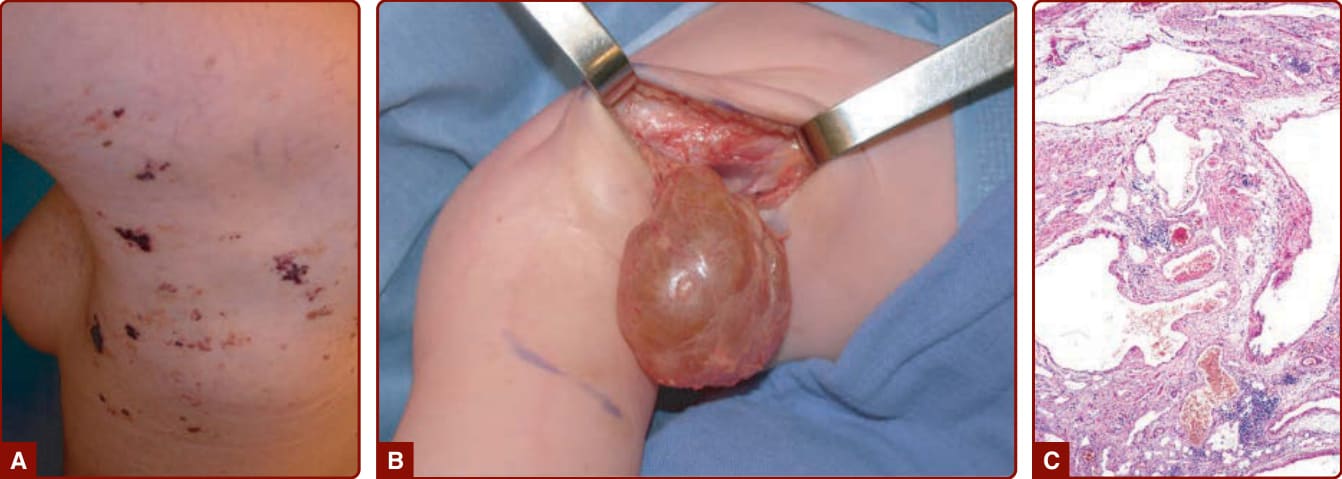
圖 147-17:A,由清澈與深紅色水疱構成的微囊型、真皮淋巴管畸形 (LM)。B,腋窩巨囊型 LM 的術中視野,顯示一個充滿清澈淋巴液、界線清楚的囊腫。C,組織學上,LM 由內皮扁平的擴張淋巴管通道構成(蘇木精與伊紅)。
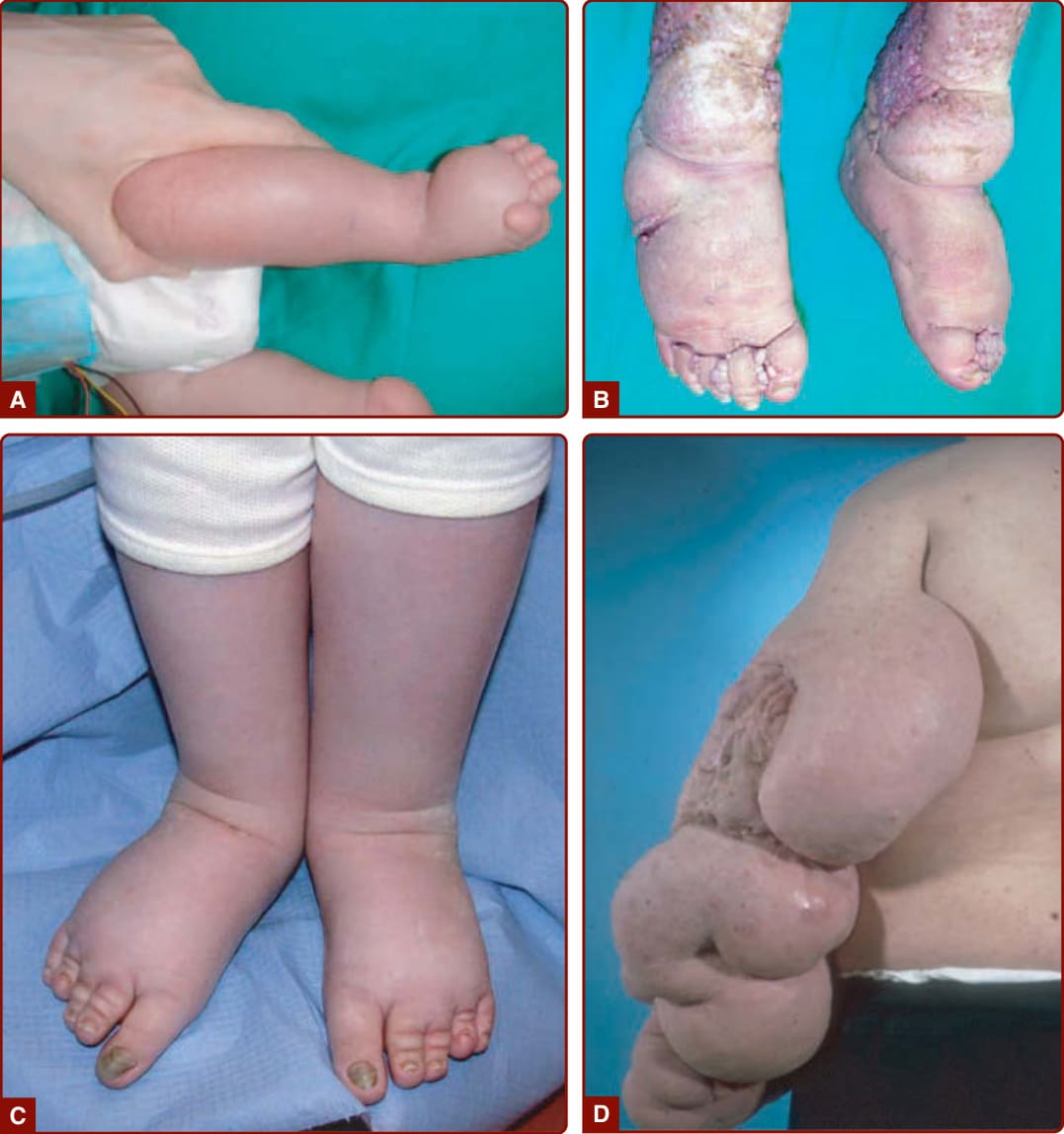
圖 147-18:各類型先天性原發性淋巴水腫,特徵為膝以下的下肢淋巴水腫 (A);伴乳頭瘤病 (B);伴上斜趾甲 (C);可演變為象皮病 (D)。
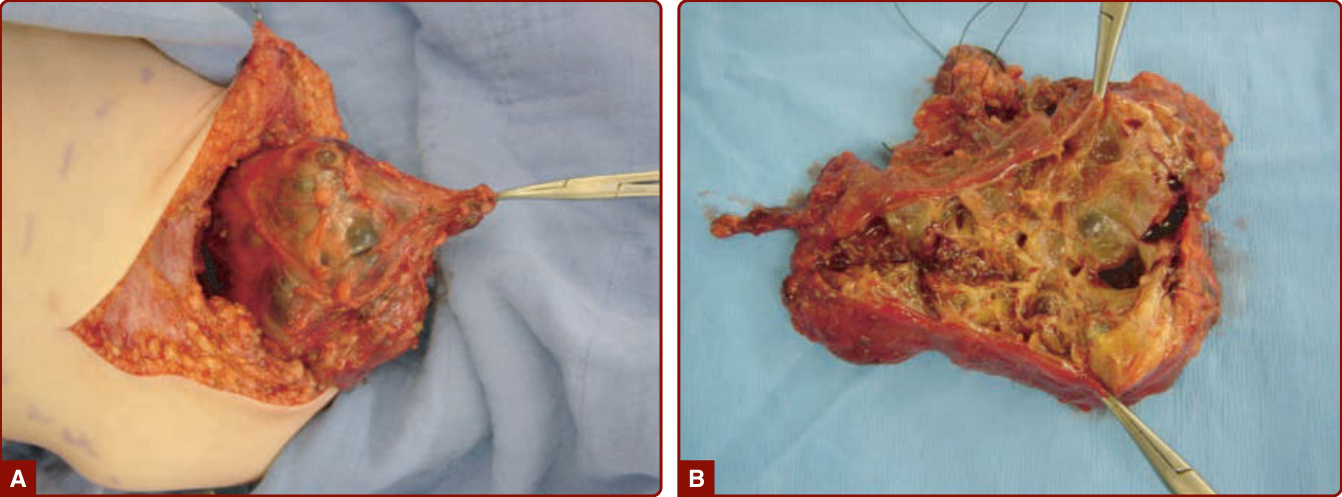
圖 147-19:一個因囊內出血而突然增大的腋窩淋巴管畸形之術中視野。
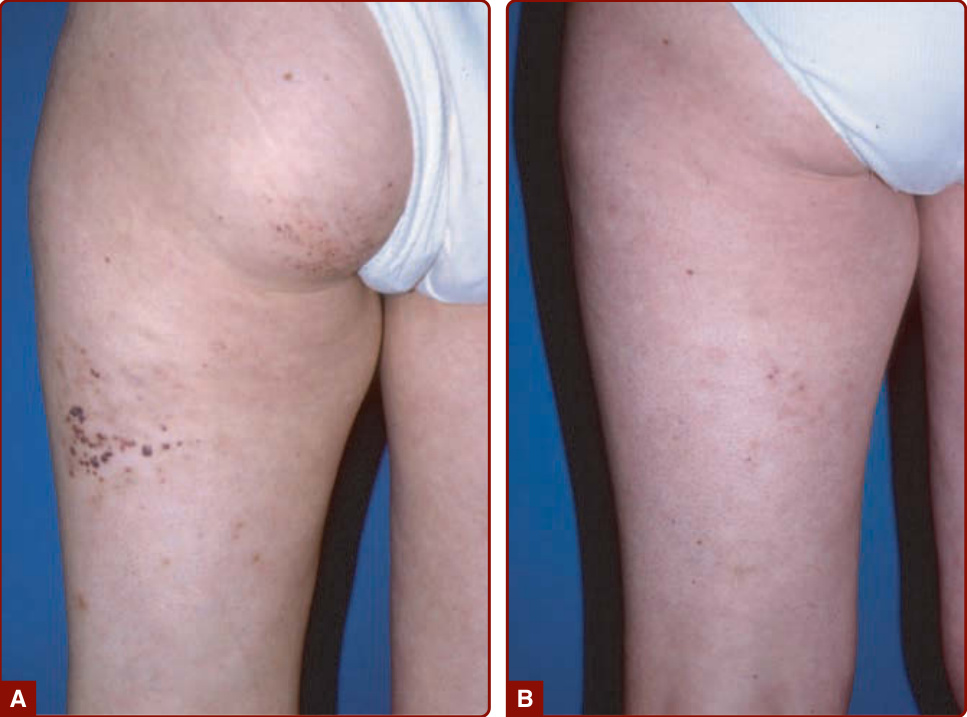
圖 147-20:真皮淋巴管畸形 (A) 以二氧化碳雷射有效治療 (B)。
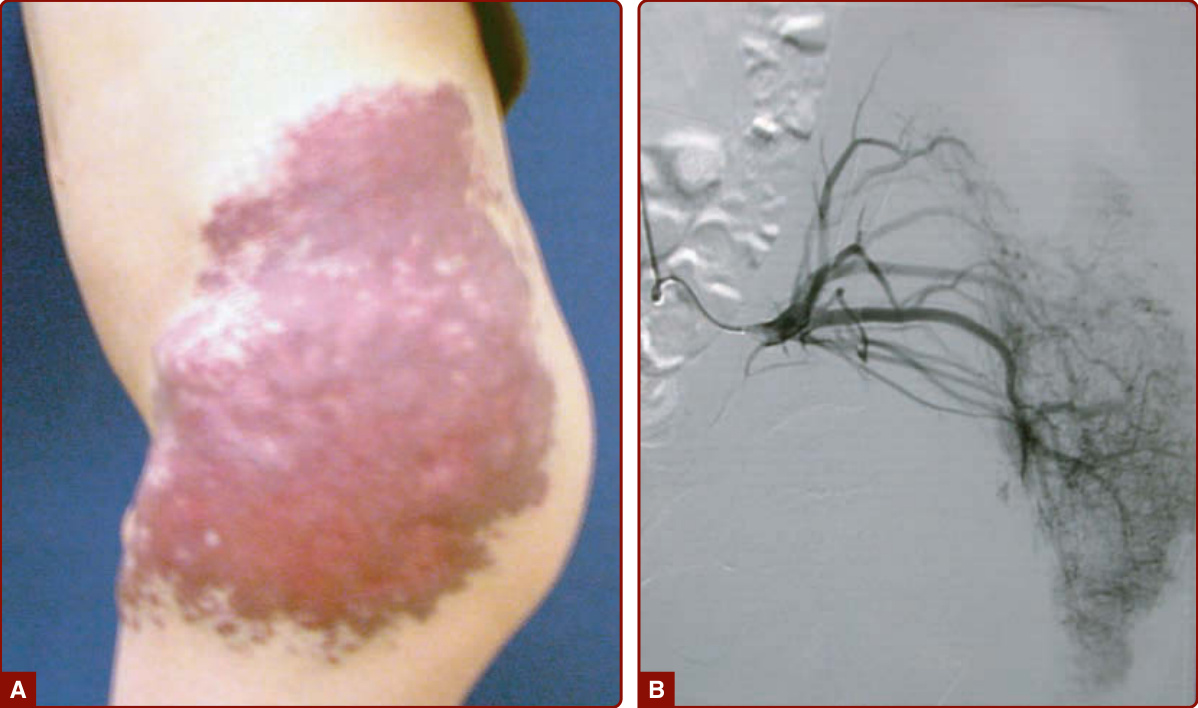
圖 147-21:左大腿第 2 期動靜脈畸形,模擬一個在 8 歲時仍未消失的嬰兒血管瘤 (A)。動脈攝影顯示病灶核心,確認了診斷 (B)。
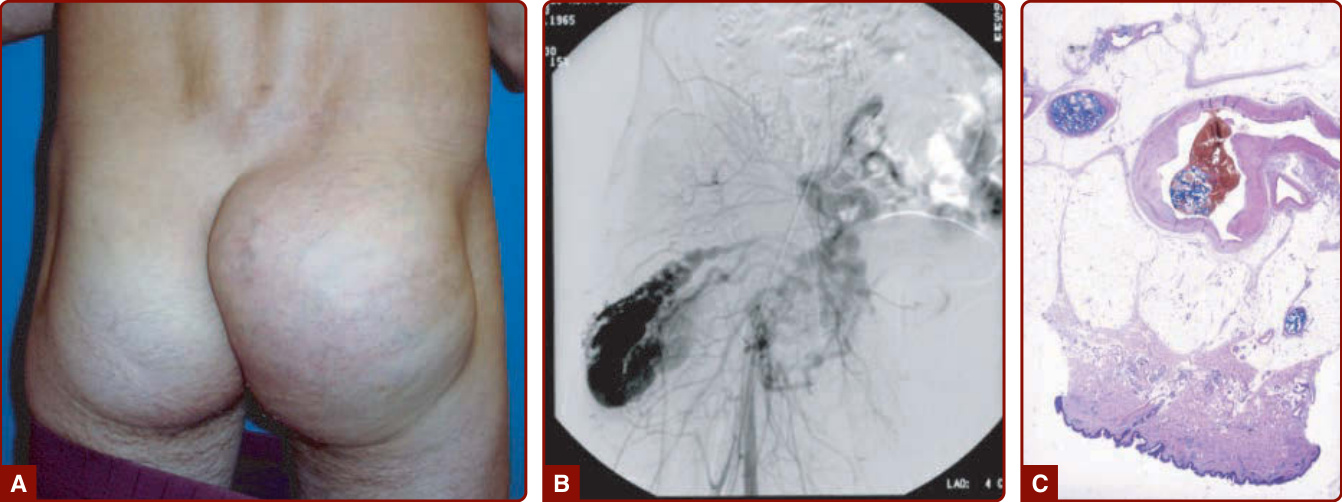
圖 147-22:A,使右臀部變形的廣泛動靜脈畸形。B,動脈攝影顯示畸形的病灶核心。C,組織學顯示動脈與靜脈之間的直接連接,並有繼發於栓塞程序的血栓物質。
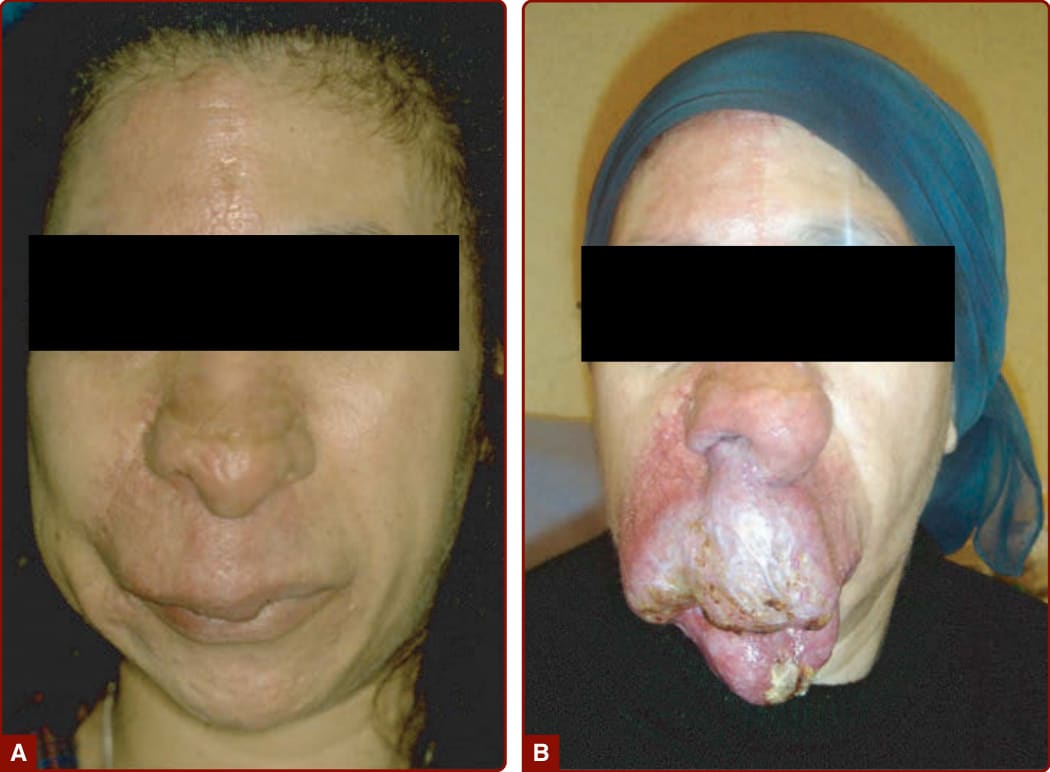
圖 147-23:第 2 期動靜脈畸形 (AVM;A) 演變為第 4 期 AVM (B)。