Vascular Malformations
22
VASCULAR MALFORMATIONS
AT-A-GLANCE
■ The worldwide prevalence is roughly 0.3%, mostly accounted for by capillary malformations.
■ Congenital, localized (although sometimes extensive or multifocal), and well-demarcated lesions of malformed vessels of various types: capillary, venous, lymphatic, arteriovenous, and combined
■ Histologically consist of enlarged, tortuous vessels of various types.
■ Caused by inherited or somatic mutations in various gene
■ Can be isolated, combined, or part of a syndrome
■ Management: multidisciplinary approach
INTRODUCTION
INTRODUCTION
DEFINITION
Vascular malformations are believed to arise because of errors in the development of vessels that occur during the 4th to 10th weeks of intrauterine life. Most vascular malformations are sporadic, although several families with inherited forms have been identified. They are very heterogeneous.1
CLASSIFICATION
For many years, vascular anomalies were grouped under the term angioma, hampering precise classification and leading to incorrect diagnosis and improper management. For example, the term hemangioma has been used both for vascular malformations, often venous (cavernous hemangioma), as well as for vascular tumors (strawberry hemangioma). This nomenclature changed in 1982 with the development of a biologic classification by Mulliken and Glowacki.2,3
This classification system organized vascular anomalies based on clinical, hemodynamic, radiologic, and histologic features. It divided vascular anomalies into two major categories: (1) vascular tumors (with cellular proliferation, hemangioma being the most
common; see Chap. 118), and (2) vascular malformations (structural anomalies of blood vessels) that are subsequently subdivided, depending on the affected vessel type, into arterial, capillary, lymphatic, or venous malformations (VMs). In 1996, this classification was adopted and further developed by the International Society for the Study of Vascular Anomalies (ISSVA; Table 147-1).4
Vascular malformations mostly affect a single vessel type (Fig. 147-1), yet combined malformations also exist. They are named according to the affected vessel types, capillary-venous or venolymphatic malformation, for instance. In addition to isolated forms, vascular malformations occur in syndromes such as Klippel-Trenaunay syndrome (KTS, capillarylymphatic-VM with limb hypertrophy), Maffucci syndrome (multiple enchondromas associated with multiple venous anomalies and high incidence of malignancy), CLOVES (congenital lipomatous overgrowth with vascular malformations, epidermal nevi, and spinal or skeletal anomalies or scoliosis) syndrome, or Parkes Weber syndrome (high-flow vascular malformation of the extremity with soft tissue hypertrophy).4
EPIDEMIOLOGY
EPIDEMIOLOGY
Vascular malformations affect about 0.3% of the population with most of these being capillary malformations (CMs). Vascular malformations are mostly congenital, even though they may be diagnosed later in life.5
CLINICAL FEATURES
CLINICAL FEATURES
Vascular malformations grow proportionately with the patient. Usually they do not regress. Most frequently, they are well-demarcated and localized. They can affect any part of the body, including the viscera. In rare instances, they can be the stigmata of deep lesions or the first sign of a syndrome. Vascular malformations are rheologically divided into slow flow (capillary, lymphatic, venous, and combined) and fast flow (arterial, arteriovenous, and combined) (Table 147-2).4 They can lead to esthetic or functional impairment or even threaten life in rare instances.6
VASCULAR TUMORS VASCULAR MALFORMATIONS
Hemangiomas Capillary
Infantile hemangioma Capillary malformation (CM, also called port-wine stain)
Congenital hemangioma Telangiectasia (hereditary benign telangiectasia, essential telangiectasia)
Rapidly-involuting congenital hemangioma (RICH)
Hereditary hemorrhagic telangiectasia (HHT)
Noninvoluting congenital hemangioma (NICH) Capillary malformation–arteriovenous malformation (CM-AVM)
Sturge-Weber syndrome
Hemangioendotheliomas Venous
Kaposiform hemangioendothelioma Venous malformation (VM)
Tufted angioma
Familial form: cutaneo-mucosal venous malformation (VMCM)
Glomuvenous malformation (GVM)
Blue rubber bleb nevus or Bean syndrome (BRBN)
Angiosarcoma
Lymphatic
Lymphatic malformation (LM)
Primary Lymphedemas
Arterial
Arteriovenous malformation (AVM)
Capillary malformation–arteriovenous malformation (CM-AVM)
Arteriovenous fistula (AVF)
Syndromic Malformations
Slow flow
Klippel-Trenaunay syndrome (capillary– lymphatic–venous malformation with limb hypertrophy)
Maffucci syndrome CLOVES (congenital lipomatous overgrowth with vascular malformations, epidermal nevi, and spinal or skeletal anomalies or scoliosis) syndrome
Fast flow
Parkes Weber syndrome
Parkes Weber syndrome
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Several genes have been identified to be mutated in the inherited forms of vascular malformations (Table 147-3). Somatic genetic mutations have also been unraveled as the cause of many common sporadic lesions.7-15
22
DIAGNOSIS
DIAGNOSIS
The diagnosis is usually based on clinical features in 90% of superficial malformations.
SUPPORTIVE STUDIES Pathology: Histologically, vascular malformations consist of enlarged, tortuous vessels with quiescent endothelium. In contrast to hemangioma, there is neither a parenchymal mass nor overt cellular proliferation.
Imaging: Radiologic investigation is needed to delineate the extent of a malformation but rarely for diagnosis unless the malformation is deeply located. Doppler ultrasonography is a very useful noninvasive radiologic examination that provides clues for differentiating the various types. Magnetic resonance imaging (MRI) details the extension and precise location of the lesion.16
Genetic Testing: Because a germline or somatic genetic cause is known for a large number of vascular anomalies, genetic testing can be used to confirm or help make the precise diagnosis. For somatic mutations, a biopsy of the affected tissue is needed. For inherited mutations, a blood test is sufficient to make the diagnosis.
MANAGEMENT
MANAGEMENT
Treatment of vascular malformations depends on the affected vessel type, the location of the lesion, the age of the patient, and the symptoms. Because many lesions are extensive, patients should be aware that a complete cure is often not possible and that recurrence occurs. Treatment can be difficult and with severe complications. Extensive or complex lesions should always be managed by a multidisciplinary team.
CAPILLARY MALFORMATIONS
AT-A-GLANCE
■ Worldwide occurrence. Prevalence is roughly 0.3%.
■ Congenital, slow-flow malformations of the capillary bed
■ Pinkish-red to purple in color; tend to darken and thicken with time
■ Can be part of a sporadic syndrome, such as Sturge-Weber syndrome or KTS
■ Can be part of the inherited capillary malformationarteriovenous malformation phenotype
■ Pathology: capillaries that are increased in size and number with abnormal innervation.
2637
22
A
B
D
C E
Capillary malformations mainly occur sporadically, although there are well-documented pedigrees showing autosomal dominant inheritance.17 When inherited, they are usually multiple and part of the capillary malformation–arteriovenous malformation (CM-AVM) phenotype, which associates atypical CMs with arteriovenous malformation (AVM, see Arteriovenous Malformations later).18-23
EPIDEMIOLOGY
EPIDEMIOLOGY
CMs, commonly called port-wine stain, is a slow-flow vascular malformation with a prevalence of 0.3%.5
2638
There is no sex preponderance.
Although most often an isolated finding, in rare instances, CM can be the cutaneous hallmark of occult spinal dysraphism, especially if located in the lumbosacral area.24 Other blanchable, pink patches, known as stork bite, angel’s kiss, salmon patch, nevus simplex, or nevus flammeus neonatorum, are often confused with CM. They are located on the nape of the neck (81%), the eyelids (45%), or the glabella (33%).25 When located on the occiput, the moniker Unna nevus may be used (Fig. 147-2). These patches have a much higher incidence (42%) in white infants than in black infants (31%).26 They are also present in various syndromes, such as Beckwith-Wiedemann and Rubinstein-Taybi syndromes. These lesions (with the exception of the Unna nevus) disappear spontaneously around the age of 1 to 4 years. In contrast, true CMs persist lifelong.
22
CAPILLARY VENOUS LYMPHATIC ARTERIOVENOUS
Skin color Red Normal-bluish-purple Normal to yellowish-purple Normal to red
Aspect Flat Flat to raised Raised to vesicular Flat to raised
Temperature Normal Normal Normal Warm
Palpation Normal Compressible Firm, noncompressible Thrill, bruit
Associations — Phleboliths, deformation — Ulceration, deformation
Radiology No flow Slow flow Slow flow, cyst Fast flow
Histology Dilated capillaries D2-40 negative Thin-walled venous channels D2-40 negative Cystic lymphatic channels D2-40 positive Arterialized veins D2-40 negative
Etiology See Table 147-3
Treatment Pulsed-dye laser Sclerotherapy, surgery,
Surgery, sclerotherapy,
Embolization followed by surgical resection
Treatment Pulsed-dye laser Sclerotherapy, surgery, sirolimus medication Surgery, sclerotherapy, sirolimus medication Embolization followed by surgical resection of nidus
sirolimus medication
SYNDROMIC DISORDERS
SYNDROMIC DISORDERS
CMs can be part of a syndrome, such as phakomatosis pigmentovascularis (PPV), Sturge-Weber syndrome (SWS), KTS, Parkes Weber syndrome, CLOVES syndrome, PTEN (phosphatase and tensin homolog) Hamartoma tumor syndrome, diffuse capillary malformation with overgrowth (DCMO), and macrocephaly–capillary malformation (M-CM, also called megalencephaly–capillary malformation– polymicrogyria syndrome). None of these syndromes is inherited.
PHAKOMATOSIS PIGMENTOVASCULARIS
PPV is thought to be an embryogenic anomaly affecting the vasomotor nerves and the melanocytes, both derived from neural crest. It manifests as a large, metameric CMs, usually located on the trunk or the extremities, in association with pigmented cutaneous lesions, such as a pigmented nevus, a nevus spilus, a café- au-lait patch, or an atypical Mongolian spot (see Chap. 77) that is not located on the sacrum (Fig. 147-3).27
Nevus anemicus can also be seen in the vicinity as a twin spot.28
STURGE-WEBER SYNDROME
When located in the frontopalpebral area, CM can be part of SWS (Fig. 147-4A). This neuro-oculo-cutaneous syndrome associates a cutaneous CM of the ophthalmic branch of the trigeminal nerve (V1) with a homolateral leptomeningeal capillary-venous malformation (CVM) and a choroid CVM. Glaucoma is often present (Fig. 147-4B). SWS is associated with a high risk of epilepsy and mental retardation because of anomalies of the venous drainage of the encephalon, as well as with glaucoma, buphthalmos, and sometimes retinal detachment.29
sirolimus medication
of nidus
DIFFUSE CAPILLARY MALFORMATION WITH OVERGROWTH
Patients with DCMO have hemihypertrophy, which can be total, regional, or contralateral. CM can be diffuse over the entire body. This entity is characterized by a reticulated, ill-defined CM.30 The lesions do not follow the lines of Blaschko (Fig. 147-5A).
MACROCEPHALY–CAPILLARY MALFORMATION
A well-delineated, dark CM of the vermillion border, the tip of the nose, or both associated with macrocephaly is often pathognomonic of M-CM. These patients have megalencephaly and are at risk of mental retardation.31 There are two reports of M-CM associated with Wilms tumor in the literature.32
CLINICAL FEATURES
CLINICAL FEATURES
CUTANEOUS FINDINGS
CM is a red, homogenous, congenital lesion that is often unilateral, sometimes bilateral, but usually not median. CMs involve skin and subcutis and sometimes mucosa (see Fig. 147-5). Their color varies from pinkish-red to deep purple with a geographic contour or a dermatomal distribution. Lesions are flat and painless, do not bleed spontaneously, and are never warm on palpation in contrast to AVM (see later). Acquired CM can be seen after trauma.33
Fifty percent of CMs are located on the face, where they follow the distribution of the trigeminal nerve: ophthalmic branch V1 (front and upper eyelid), maxillary branch V2 (lower eyelid, cheek, and upper lip) (see Figs. 147-4A and 147-5B), or mandibular V3
2639
22
SPORADIC MALFORMATIONS INHERITED MALFORMATIONS
DIAGNOSIS MUTATED GENE DIAGNOSIS MUTATED GENE
Capillary Malformations (CMs)
CM GNAQ CM-AVM1 RASA1
Sturge-Weber syndrome
CM-AVM2 EPHB4
Phakomatosis pigmentovascularis (PPV) GNAQ or GNA11
Macrocephaly–capillary malformation (M-CM) AKT3, PIK3CA, PIK3R2
Venous Malformations (VMs)
VM TIE2 (60%) PIK3CA (20%) Mucocutaneous venous malformations (VMCM) TIE2
Blue rubber bleb nevus (BRBN) TIE2 Glomuvenous malformation (GVM) GLOMULIN
Multifocal venous malformation (MVM)
Verrucous venous malformation (VVM) MAP3K3
Maffucci syndrome IDH1, IDH2
Lymphatic Malformations (LMs)
LM PIK3CA Nonne-Milroy lymphedema VEGFR3
Nonne-Milroy–like lymphedema VEGF-C
CLOVES (congenital lipomatous overgrowth with vascular malformations, epidermal nevi, and spinal or skeletal anomalies or scoliosis)
Klippel-Trenaunay syndrome (KTS)
Lymphedema distichiasis FOXC2
Microcephaly, chorioretinopathy, lymphedema, mental retardation KIF11
Hypotrichosis–lymphedema–telangiectasia SOX18
Hennekam lymphedema–lymphangiectasia 1 CCBE1
Hennekam lymphedema–lymphangiectasia 2 FAT4
Hennekam lymphedema–lymphangiectasia 3 ADAMTS3
Emberger syndrome GATA2
Fetal chylothorax ITGA9
Lymphedema–choanal atresia PTPN14
Four-limb lymphedema GJC2 (CX47)
Arteriovenous Malformations (AVMs)
Extracranial AVM MAP2K1 Hereditary hemorrhagic telangiectasia (HHT)1 ENG
Brain AVM
KRAS HHT2 ACVRL1 (ALK1)
HHT3 SMAD4, GDF2
PTEN
Phosphatase and tensin (PTEN) homolog
Phosphatase and tensin (PTEN) homolog (protein encoded by PTEN gene) PTEN
(lower lip, chin, and mandible). CM can be diffuse over the entire body, often with associated hemihypertrophy (see Fig. 147-5A). This entity is called diffuse capillary malformation with overgrowth (see earlier discussion). When inherited, CMs are usually small and multifocal, such as in CM-AVM1 and 2. These small CMs vary in size from a couple mm to several centimeters in size. They are often round-to-oval, pink, red, or brown in color and are often surrounded with a pale halo (Fig. 147-6).18,34 The lesions are well-circumscribed and blanch on pressure with a diascopy. Some of them are present at birth, but others appear later. They grow
2640
(protein encoded by PTEN gene)
with the child and are asymptomatic. Bier spots, white cutaneous spots surrounded by a pale halo of redness, are seen in both entities. In contract to CM- AVM1, CMs in CM-AVM2 can be more telangiectatic in appearance, especially periorally and on the upper thorax.23 CM-AVM1 is characterized by the presence of CMs in 97%, AVMs or arteriovenous fistulas (AVFs) in 24% (10% intra–central nervous system [CNS], 13% extra-CNS), and Parkes Weber phenotype in 8% of patients.21,35 In CM-AVM2, the risk of a fast-flow lesion seems to be less frequent because the respective penetrances are 93% (CM), 19% (AVM), and 7% (Parkes Weber).
A B
22
NONCUTANEOUS FINDINGS
In rare instances, CM can be the stigmata of an underlying anomaly such as lumbar dysraphism underneath a sacral CM. Cutaneous lesions of PPV can be associated with systemic, visceral (hypoplasia of the larynx, intestinal polyposis), muscular (scoliosis), or neurologic (mental retardation, epilepsy, intracranial) signs and symptoms in 60% of cases.36
SWS is associated with a high risk of epilepsy and mental retardation because of anomalies of the venous drainage of the encephalon, as well as with glaucoma, buphthalmos, and sometimes retinal detachment.29
Patients with DCMO have hemihypertrophy, which can be total, regional, or contralateral. They have also associated toe anomalies (in 30%) and prominent veins.30 In CM-AVM1 or 2, there is a 20% to 30% risk of an associated AVM located on the skin, the brain, or the spine.21,35 A well-delineated dark CM of the vermillion border or the tip of the nose associated with macrocephaly is often pathognomonic of M-CM, and these patients have megalencephaly and are at risk of mental retardation.31
COMPLICATIONS
The major concern for a patient with a CM is cosmetic because of the visible discoloration. Hypertrophy of soft (often lips and gums) and underlying hard tissues can occur with time, especially when the CM affects the V2 and V3 dermatomes or the extremities (Fig. 147-7D and 147-7). Overgrowth of the maxilla or mandible leads to skeletal asymmetry, occlusal tilt, and open-bite deformity. When the child has atopic dermatitis, psoriasis, or acne, lesions are worse in the area of CM, a finding known as the Meyerson phenomenon.37
Evenly thickened skin, purple nodules, and pyogenic granulomas can develop by adolescence (Fig. 147-7B and 147-7C).38,39
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Facial CM is thought to be caused by clonal expansion of abnormal cells originating from the neural crest.40
2641
22
B
A
Somatic activating mutations in GNAQ have been identified as the cause of facial CM with hypertrophy as well as of SWS.13 GNAG is a q class of G-protein α subunits that mediates signals between G protein– coupled receptors and their downstream effectors. Mutations in GNAQ and GNA11 have also been identified in pigmented lesions such as blue nevi, nevus of Ota, uveal melanoma, and PPV.41,42
The inherited CM-AVM1 is caused by inactivating mutations in RASA1.21 This gene encodes a GTPaseactivating protein, which negatively regulates Ras activity. CM-AVM2 is caused by inactivating mutations in EPHB4.23 EPHB4 is usually expressed in venous endothelial cells and its ligand EPHINB2 on arterial endothelial cells. They regulate arteriovenous identity. EPHB4 interacts with RASA1 to regulate RAS- MAPK signaling, and loss of EPHB4 or RASA1 in CM- AVM likely activates this signaling pathway. M-CM is caused by activating mutations in AKT3, PIK3CA, and PIK3R2.43
DIAGNOSIS
DIAGNOSIS
SUPPORTIVE STUDIES Pathology: Histologically, CM is characterized by dilated capillaries of the papillary and upper reticular dermis combined with areas of increased number of normal-looking capillaries (Fig. 147-8).44 Endothelial cells are flat. Factor VIII, fibronectin, and basement membrane protein are normal, but S100 staining shows abnormal innervation.45,46
Imaging: No imaging studies are mandatory for a CM except in rare situations. If a so-called “CM” is painful, warm, or spontaneously bleeds, Doppler ultrasound is indicated to exclude the diagnosis of a fast-flow malformation, such as an AVM, Parkes Weber syndrome, or a proliferating hemangioma.
B A C
2642
A B
C D
22
CM located in the frontopalpebral area, especially if the inner part of the upper eyelid is involved, can be part of SWS. Therefore, for these CMs, an ophthalmologic and neurologic examination is mandatory. They need to be done during the first months of life and repeated once a year until puberty, even if normal at younger age.47 A brain MRI should also be done to evaluate the occurrence of associated leptomeningeal CVM. In patients with PPV, ophthalmologic, neurologic, and orthopedic follow-up is mandatory because of the common association of CM with systemic lesions. In patients with extensive CMs of the lower extremity, such as in DCMO, a scaniometry study (leg length films) is needed to evaluate possible progressive growth discrepancy around the age of 4 to 6 years and needs to be repeated until the end of growth to allow the orthopedist to correct the discrepancy at the adequate time. Serial renal ultrasound examinations are also indicated to rule out the appearance of a Wilms tumor. MRI of the spinal cord is indicated in the presence of a lumbosacral CM. Brain and spinal MRI should be considered in patients with CM-AVM.
Genetic Testing: Genetic testing is indicated, especially in patients with multifocal lesions (CM- AVM1 and 2) because of increased risk for intracerebral fast-flow lesions. These patients should be screened for
RASA1 and EPHB4. Pale, uncharacteristic, and inconspicuous CMs can be sign of PHTS, especially if these vascular lesions are associated with macrocephaly (see AVM later). Because of increased cancer risk, such patients should be screened for PTEN mutations. Identification of a germline mutation would enable precise counseling and surveillance.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
See Table 147-4.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE
AND PROGNOSIS
CM is present at birth and never regresses spontaneously. During the first weeks of life, they can slightly fade, as the hemoglobin level of the newborn decreases. Subsequently, the red hue stabilizes, and the lesion grows in proportion to the rest of the body. Around puberty, as well as later in life, CM slowly thickens and darkens with time. It often becomes raised and nodular (see Fig. 147-7).38 Pyogenic
2643
22
B A C
D E
granuloma can occur with time, as well as soft tissue or bony hypertrophy.39
MANAGEMENT
MANAGEMENT
The goal of management is to monitor for associated complications and minimize the impact of the CM on self-esteem and quality of life. Camouflage using a foundation such as Covermark is still an effectively approach (Fig. 147-9)
INTERVENTIONS Orthopedic Considerations: In the presence of leg-length discrepancy, an early-adapted shoe lift is needed if leg-length discrepancy is over 1.5 cm to prevent compensatory tilting of the pelvis. Epiphysiodesis is sometimes necessary when the child is approximately 11 to 13 years old.
2644
Procedures: Laser treatment is the gold standard therapy for most CM. Pulsed-dye laser with its specific wavelength (585 or 595 nm) and a short pulse duration (400–1500 ms) currently gives the best results in infants and children by lightening the red color of the CM. There are few complications. Multiple sessions (6–12) are needed, and general anesthesia may be necessary as the procedure is painful. Laser treatment is more efficient on the cervicofacial and trunk area than on the extremities. Early treatment during childhood does not reduce the number of laser sessions.48 The use of a dynamic cooling system to avoid heating the epidermis allows higher laser fluencies, resulting in optimal lightening of the lesion. Recurrence can occur after cessation of therapy.49 Laser has no impact on associated hypertrophy.6,50
Contour resection is considered to treat complications such as pyogenic granuloma or lip hypertrophy.6
Counseling: Genetic and psychological counseling for the patients and their families are part of multidisciplinary management. Patient organizations dedicated to vascular anomalies also have an important role in supporting patients and their families.
A B
22
PREVENTION
PREVENTION
In patients with SWS, ophthalmologic follow-up, started immediately after birth is essential because
A B
reducing visual impairment will depend on the promptness of treatment. Prophylactic antiepileptic medication to prevent neural cell death has also been advocated, although randomized prospective studies to support efficacy are lacking at this time.51,52
2645
22
Based on Morphology Most Likely Faint pink median patch
■Unna nevus
■Nevus flammeus neonatorum (nevus simplex) Multiple, inherited
■CM of CM-AVM Telangiectatic patch
■CMTC
■HHT
■CM-AVM2
■Essential telangiectasia
■Unilateral nevoid telangiectasia Warm on palpation
■Proliferating hemangioma
■AVM
■AVF Ulcerated or painful
■AVM
■AVF Rule Out Small, multiple
■Mastocytosis
■CM of CM-AVM
Based on Location Most Likely Occiput
Based on Location Most Likely Occiput
■Unna nevus Glabella, upper eyelid, nape of neck
■Unna nevus Glabella, upper eyelid, nape of neck
■Stork bite, angel’s kiss, salmon patch Consider Extremity
■Stork bite, angel’s kiss, salmon patch Consider Extremity
■CMTC
■CMTC
■Klippel-Trenaunay syndrome
■Klippel-Trenaunay syndrome
■Parkes Weber syndrome Faint on trunk
■Parkes Weber syndrome Faint on trunk
■M-CM Rule Out Scalp
■M-CM Rule Out Scalp
■Adams-Oliver syndrome
■Adams-Oliver syndrome
■Multiple telangiectasia near canthus
■Multiple telangiectasia near canthus
■Ataxia telangiectasia
■Ataxia telangiectasia
AVF, arteriovenous fistula; AVM, arteriovenous malformation; CM, capillary malformation; CM-AVM, capillary malformation-arteriovenous malformation; CMTC, cutis marmorata telangiectatica congenita; HHT, hereditary hemorrhagic telangiectasia; M-CM, macrocephaly capillary malformation.
VENOUS ANOMALIES
AT-A-GLANCE
■ Most common referral to specialized centers for vascular anomalies; incidence unknown but is substantially lower than for CM and estimated at 1 in 10,000
2646
■ Congenital slow-flow malformation of the venous bed
■ Bluish in color, localized or extensive, solitary or multifocal
■ Compressible on palpation; presence of phleboliths
■ Associated with consumptive coagulation abnormalities in about 40% of cases
■ Histologically consists of ectatic venous-like channels with anomalies in mural cells
■ Mainly sporadic but can be inherited as an autosomal dominant trait
■ Inherited venous anomalies: cutaneomucosal venous malformation (1%), glomuvenous malformation (>5%)
■ Syndromic venous malformation: KTS, blue rubber bleb nevus, Maffucci syndrome.
INTRODUCTION
INTRODUCTION
A VM is a congenital lesion made up of venous-type vessels of the skin or mucosa but can involve any structure (subcutis, muscles, bones, and nerves) and any organ (CNS, gastrointestinal [GI] tract). Fifty percent of VMs are located in the cervicofacial area, and 37% are on the extremities. VMs are mainly isolated but can be part of complex vascular disorders, such as the KTS, Maffucci, and blue rubber bleb nevus (BRBN) syndromes.53,54
EPIDEMIOLOGY
EPIDEMIOLOGY
Venous anomalies are the most common VMs referred to specialized center with an overall incidence of 1 in 10,000 in the population.54 They mainly occur sporadically, although 1% are inherited mucocutaneous venous malformations (VMCMs) and 5% are inherited glomuvenous malformations (GVMs). No sex preponderance is reported.55 Both VMCM and GVM have an age-dependant variation in penetrance, which reaches its maximum by 20 years of age (87% for VMCM and 92.7% for GVM).56 Large VMCMs and GVMs are present at birth. However, 17% of affected individuals develop new small lesions over time.55 BRBN syndrome is rare and occurs sporadically with about 200 cases reported in the literature.
SYNDROMIC DISORDERS
SYNDROMIC DISORDERS
Syndromes with a venous anomaly include BRBN syndrome, KTS (see Lymphatic Malformations later), and Maffucci syndrome.
BLUE RUBBER BLEB NEVUS SYNDROME
BRBN is characterized by numerous malformations of the venous system that involve the skin and visceral organs. It is commonly characterized by one large “dominant” VM lesion associated with multiple small, dark blue, nipple-like lesions, the latter being typically located on the palm and soles (Fig. 147-10).15 These cutaneous lesions are associated with multiple small GI VMs, often responsible for chronic anemia.15,57
MAFFUCCI SYNDROME
Maffucci syndrome is a rare disorder characterized by multiple enchondromas associated with subcutaneous VMs of the distal extremities (Fig. 147-11). The disease starts during childhood with the development of enchondromas of the bones of hands and feet, as well as of the long bones. Deformities and shortening of extremities often occur. Subcutaneous vascular nodules appear later, around puberty, on the fingers and the toes. Phleboliths may become present.58
B A
22
CLINICAL FEATURES
CLINICAL FEATURES
The features of venous anomalies are summarized in Table 147-5.
CUTANEOUS FINDINGS
VMs are usually solitary but can be multifocal, the latter suggesting an inheritable disorder in most instances.54 They may be relatively localized or extensive within an anatomic region. The most common location is the head and neck area (Fig. 147-12A and 147-12D–F). VMs can affect any tissue or organ (Figs. 147-12 to 147-14). VMs are congenital, light to dark bluish lesions. Deep VM has a normal overlying skin color and is often diagnosed only at puberty or later in life with the onset of pain. Size varies from small spongy blebs to large lesions of several centimeters in diameter. Skin temperature is normal. There is no thrill or bruit. VMs are larger when in a dependent position and can easily be emptied by compression or facilitating venous drainage. Palpation is not painful unless thrombosis occurs.54,59
VM lesions in VMCM are small and multifocal. They are more reminiscent of small sporadically occurring VMs than BRBNs. They can enlarge with time and are often asymptomatic. The VMs are generally small (<5 cm), and more than 80% of patients have more than two lesions. Some are present at birth; most lesions appear by puberty. Mucosal lesions involve the lips, tongue, and buccal mucosa.60,61
In rare instances, VM can be sporadic yet multiple. These multifocal venous malformations (MVMs) are very similar to the VMCM lesions, yet there is no family history of the same findings.15
GVM is a bluish to purple, raised venous anomaly characterized by multifocality, hyperkeratosis, and nodularity with a cobblestone surface (see Fig. 147-13C). In rare cases (especially in newborns), the lesions may be flat and purple in color, mimicking CM62; this plaquelike GVM usually darkens with time. GVMs are usually present at birth and slowly expand during childhood.
2647
22
VENOUS MALFORMATION VENOUS MALFORMATION CUTANEOMUCOSAL GLOMUVENOUS MALFORMATION BLUE RUBBER BLEB NEVUS SYNDROME
Location 50% head and neck Extremities, mucosa 70% extremities Especially palms and soles
Skin color Normal to bluish-purple Bluish Bluish-red-purple Bluish-dark-blue
Aspect Flat to raised Raised, dome shaped Raised, cobblestone Plaquelike, hyperkeratotic Dome shaped, nipple-like
Single, large Multiple, small Multiple Multiple, small, often + 1 large lesion
Palpation Phleboliths Compressible Noncompressible, painful Firm, rubbery, spongy
Associated Coagulopathy — — Visceral venous malformation, coagulopathy
Radiologic Phleboliths Venous channels No phlebolith Venous channels
Tissue involved Often deep location Cutis, subcutis Cutis, subcutis Often deep component
Histology Thin-walled venous channels Thin-walled venous channels Glomus cells Thin-walled venous channels
Inheritance Sporadic Autosomal dominant Autosomal dominant Sporadic
Inheritance Sporadic Autosomal dominant Autosomal dominant Sporadic
New small lesions appear with time. In contrast to VM, GVM is often painful on palpation and cannot be completely emptied by compression. GVMs are usually multifocal and located on the extremities, involving the skin and subcutis. They are rarely encountered in mucosae,
and intestinal hemorrhage is not present. Patients with GVMs have normal mental and physical development. Diagnosis of GVM is based on clinical and histologic evaluation of the cutaneous lesions. Genetic testing can differentiate small multifocal GVMs from VMCMs.55
A B C
D E F
2648
A
C
22
B
D
Verrucous Venous Malformation: Verrucous venous malformation (VVM), previously known as verrucous hemangioma, is a rare congenital
vascular anomaly. Although VVM expresses an immunohistochemical profile similar to vascular neoplasms (Wilms tumor 1 and GLUT-1 positive), it is distinct from infantile hemangioma because it is usually noted at birth and never spontaneously involutes. Mainly located on the legs, VVM can also be found on the head and the trunk. It initially appears as a bluish macule that later becomes erythematousviolaceous in color and after trauma and secondary infections often evolves into a verrucous nodule (see Fig. 147-14). It never involves muscles or deep tissues.63
NONCUTANEOUS FINDINGS
Patients with VMCM, like those with sporadically occurring MVM, seldom have VMs located in internal
2649
22
organs, such as the lungs, kidneys, brain, or GI tract.15 Extracutaneous manifestations of BRBN include GI tract involvement. The small bowel is the predominant region; however, VMs can occur in any site from the oral cavity to the anal mucosa. In rare instances, VMs can also be seen in brain, lungs, and kidneys, as well as other organs.57
COMPLICATIONS
VM is usually unilateral, causing asymmetry and progressive, slowly worsening distortion (see Figs. 147-12 and 147-13). Depending on their size and location, VMs can cause pain, particularly in the morning on awakening, and anatomic distortion and can even threaten life because of bleeding, expansion, or obstruction of vital structures. Migraine is a common feature when VM is located in the temporal muscle. Whereas oropharyngeal VM can impair speech and cause difficulties in swallowing, pharyngeal or laryngeal lesions can compromise the airway and cause snoring and sleeping apnea.54
On the extremities, VMs often involve muscles and joints. They cause muscle weakness, hypotrophy, and sometimes hypertrophy, resulting in leg-length discrepancy, which is less severe than in KTS. Intraarticular bleeding, if the malformation is located in the knee joint, leads to early-onset arthrosis.64 Genital VM often occurs with limb VM and can cause dyspareunia. GI VM may lead to chronic anemia. Local thrombosis is usually responsible for acute pain that lasts for several days and resolves as phleboliths, which can be identified by palpation or by radiography. VM rarely causes pulmonary embolism, although this complication can occur when large draining veins exist.65,66
Chronic localized intravascular coagulopathy (LIC) is associated with solitary or syndromic spongy VMs. It is characterized by elevated D-dimer level (>500 ng/mL) in 40% of patients. Severe LIC with low fibrinogen levels is common in large VMs of the extremities and rare in the cervicofacial area. A number of events, such as surgery, sclerotherapy, and hormonal influences, can trigger the conversion of LIC to disseminated intravascular coagulopathy (DIC).67
In contrast to skin lesions, GI lesions, as seen in patients with BRBN, have a tendency to bleed. They may spontaneously rupture, causing acute hemorrhage and even death. However, most of the bleeding tends to progress slowly, resulting in chronic and occult blood loss that can lead to iron-deficiency anemia. Other complications include intussusception, volvulus, and bowel infarction, which are to be considered in patients with BRBN and abdominal pain. Morbidity depends on the extent of GI involvement.57 VVM often ulcerates.63
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
VMCM is inherited as an autosomal dominant disorder caused by germline mutations in the TEK gene,
2650
which encodes the angiopoietin receptor, TIE2.68,69 The inherited mutations cause hyperphosphorylation of the receptor even in the absence of a ligand. Somatic second hits have been found in VMCM tissues of two patients: one caused loss of membranous expression of the second, wild-type, allele; two others occurred on the allele with the inherited mutation, causing an increase in hyperphosphorylation.15,70 These mutations activate the PI3K/AKT signaling pathway in endothelial cells.71,72
GVM is caused by dominant, loss-of-function mutations in the glomulin gene.73 In addition, a somatic second hit is required for lesions to form, leading to complete loss of function of glomulin. The most common second hit is acquired uniparental isodisomy, which leads to homozygosity of the inherited mutation.73,74 This explains the variable expressivity regarding penetrance, the extent of the lesions, and the number of lesions within family members.75 So far, a mutation in glomulin has been found in almost all GVM families tested, demonstrating locus homogeneity.75-77 Glomulin seems to be expressed in vascular smooth muscle cells, but its exact function remains unknown. Sporadic VMs are also caused by genetic mutations. Sixty percent are caused by activating somatic mutations in TIE2.70 The mutations differ from the inherited ones seen in VMCM. The most common mutation in VMs (L914F) has not been identified in VMCM, and the most common inherited mutation in VMCM (R849W) has not been identified in single somatic VM. This suggests that whereas the recurrent somatic mutations have effects too detrimental to be supported in the germline and probably cause lethality, the inherited ones have weaker effects that require additional changes (somatic second hits).70-72
Another 20% of sporadically occurring VMs are caused by somatic mutations in the PIK3CA gene.10
The same mutations are seen in cancers. They activate the PI3K/AKT signaling pathway, as do the mutations in TIE2, underscoring perturbations in this signaling activity to underlie pathogenesis of VMs.10,11
MVM is also caused by TIE2 mutations. Similar to VMCM, these patients often have two TIE2 mutations. Most frequently, a R915C mutation is present as a mosaic change, and a somatic Y897C mutation occurs on top of the R915C change on the same allele. This explains the lack of family history (the first mutation has appeared de novo in a mosaic fashion in the patient), multifocality (the mosaic mutation is a predisposing change, like the inherited mutations are in VMCM), and the clinical similarity with VMCM lesions (both have similar, albeit not the same, double cis mutations).15 VVM is caused by somatic MAP3K3 mutations.78 BRBN syndrome, like VMCM, MVM, and 60% of VMs, is also caused by mutations in TIE2. BRBN mutations are not detectable in the blood; they are somatic.15 Yet in distinct, distally located lesions of the same patient with BRBN, the same double mutations can be identified with equal allele frequencies. The most common is a T1105N–T1106P double mutation. This suggests that the separate and even newly appearing lesions are formed by endothelial cells
that originate form a single common site, such as the dominant lesion or bone marrow. The BRBN-causing TIE2 mutations cause strong hyperphosphorylation of the receptor, and the endothelial cells acquire increased colony-forming capacity, survival, and faster migration.15
Maffucci syndrome is caused by somatic mutations in IDH1 and 2.79-81
DIAGNOSIS
DIAGNOSIS
SUPPORTIVE STUDIES Laboratory Testing: A coagulation profile with platelet count, fibrinogen, and D-dimer level should be done in any patients with a pure VM or complex malformation with a venous component because the D-dimer level is elevated in about 40% of VMs. This blood test, done at the first consultation, should be repeated around puberty and whenever the malformation becomes painful. This is never the case in a patient with GVM or other simple vascular anomalies. Associated low fibrinogen is pathognomonic of severe LIC. Severe LIC can easily decompensate to DIC with severe bleeding during interventional or surgical procedures.67,82-84
Pathology: VMs are histologically composed of ectatic vascular channels of venous type with flat endothelium. The vascular walls are thin and lined by a discontinuous layer of mural smooth muscle cells positive for smooth muscle-α-actin.85 Basement membranes are also thin. In contrast, GVM is histologically characterized by distended venous channels surrounded by mural “glomus cells,” which are aberrantly differentiated smooth muscle cells. Glomus cells are round or polygonal, instead of being elongated, like the normal vascular smooth muscle cells.86 By immunohistochemistry, glomus cells stain positively for smooth muscle-α-actin and vimentin, but they are negative for desmin, von Willebrand factor, and S100.87
By in situ hybridization, they are also negative for glomulin.76
VVM is characterized by hyperkeratotic epidermis on top of small blood vessels with a multilaminated membrane located in the dermis and the subcutis. Focal positive staining for GLUT-1 is seen.63 Histopathologic examination of patients with Maffucci syndrome shows features of spindle cell hemangioendothelioma.
Imaging: Plain radiography can identify the calcification of phleboliths, which are pathognomonic of LIC in a VM. It can also show the presence of multiple enchondromas in patients with Maffucci syndrome (see Fig. 147-11). Doppler ultrasonography is noninvasive and can confirm the slow-flow nature of the lesion. In contrast to lymphatic malformations (LMs), the venous channels are compressible with the probe.88
MRI imaging with spin-echo T1- and T2-weighted and fat-saturation sequences depicts the exact anatomic
22
location of the lesion (see Fig. 147-12).89 This will highlight the more cellular component, as well as the more superficial location of GVM compared with VM. Atypical MRI findings require a biopsy to rule out a sarcoma or neurofibroma. In the presence of palmar-plantar multifocal lesions typical of BRBN (see Fig. 147-10), an endoscopy, colonoscopy, or wireless capsule endoscopy should be performed to detect GI lesions. These radiologic examinations are mandatory for pretherapeutic evaluation of any venous anomaly.
Genetic Testing: Consider genetic testing for TIE2 and glomulin mutations in patients with multifocal lesions. This differentiates the conditions and enables genetic counseling.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
See Table 147-6.
Based on Morphology Most Likely Bluish subcutaneous mass, noncompressible
■Infantile hemangioma (deep)
■Lymphatic malformation With multiple enchondromas
■Maffucci syndrome Consider Hyperkeratotic
■GVM, if on extremity
■Cutaneous lesions of CCM
■VVM Rule Out Superficial, collateral normal veins
■Deep venous insufficiency, iatrogenic stenosis, or congenital agenesis Bluish nonvascular lesion
Based on Location Consider Base of the nose
Based on Location Consider Base of the nose
■Normal prominent veins Scalp or forehead
■Normal prominent veins Scalp or forehead
■Sinus pericranii
■Sinus pericranii
■Underlying Galen malformation Subungual
■Underlying Galen malformation Subungual
■Solitary glomus tumor Cheek
■Solitary glomus tumor Cheek
■Blue nevus Brain
■Blue nevus Brain
■Developmental venous anomaly
■Developmental venous anomaly
■CCM Rule Out Neck; deep mass
■CCM Rule Out Neck; deep mass
■Thyroglossal duct
■Thyroglossal duct
■Bronchogenic cyst
■Bronchogenic cyst
2651
CCM, cerebral cavernous malformation; GVM, glomuvenous malformation; VVM, verrucous venous malformation
22
An unusual pathway of cerebral drainage, a cerebral developmental venous anomaly that consists of dilated intramedullary veins converging into a larger draining vein, can be seen in 0.5% of the population. In contrast, it is present in 20% of patients with extensive head and neck VMs. Although usually asymptomatic, this anomaly can cause headache.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE
AND PROGNOSIS
VMs grow proportionately with the patient. They never regress spontaneously. Initially, asymptomatic lesions often become painful in response to trauma or in altered hormonal states (puberty or pregnancy). They may also expand following thrombosis. No oncogenic transformation of this condition has been described. The major complications are life-threatening GI hemorrhage and airway compression. Patients with inherited venous anomalies, as well as BRBN and MVM, tend to develop additional small lesions with time.15 Patients with Maffucci syndrome have a high incidence of malignancies (40%), mainly chondrosarcoma but also glioma, fibrosarcoma, and angiosarcoma.58
MANAGEMENT
MANAGEMENT
Treatment of venous anomalies is warranted when lesions are symptomatic or for cosmetic purposes. The most common indications for treatment are pain and functional impairment.54 Their management is often multidisciplinary, involving hematologists, surgeons, and interventional radiologists.
INTERVENTIONS Medical Management: Compression garments can be helpful to decrease swelling and pain in VMs of the extremity by decreasing venous pressure. In contrast, compression increases pain in GVM patients and is not indicated in this situation. Low-molecular-weight heparin (LMWH; LMWH, 100 IU anti-Xa/kg/day) is usually given to patients with signs of LIC 24 hours before and for another 5 days after surgical procedures to minimize hemorrhagic risk. The same treatment is given to patients with painful episodes of local thrombosis. In this circumstance, the duration of treatment is usually 2 weeks.54,67,84
Bleeding from GI BRBN lesions can be managed conservatively with iron supplementation and blood transfusions. With the understanding of the underlying pathophysiologic mechanisms (activation of the PI3K-AKT signaling), targeted molecular therapies are becoming possible. Rapamycin (sirolimus) was promising in
2652
a preclinical VM-mouse model trial.90 A phase II clinical trial for difficult-to-treat extensive VMs or complex slow-flow vascular anomalies not amenable to conventional management also gave encouraging results.90,91
Patients experienced almost complete relief of pain and symptoms, reduced coagulopathy when present, improved function, and increased self-perceived quality of life. Bleeding in sporadic VM as well as in BRBN stopped within 24 hours. Side effects were minor and included mucositis, mild headache, fatigue, and diarrhea. Moreover, a statistically significant reduction in volume was observed with MRIs in most patients that reached 1-year follow-up. A multicentric European Study (VASE; NCT 02638389) is ongoing to determine which subtypes of VMs are best suited for rapamycin treatment and for how long the treatment should be continued. Rapamycin should not be considered as treatment for small, localized, and asymptomatic slowflow vascular malformations that respond to standard of care.92
Procedures: Percutaneous intralesional sclerotherapy is the primary treatment for VM, in contrast to GVM. Absolute ethanol is the most efficient sclerosing agent.93 Local compression or intralesional coils are used to prevent dissemination of ethanol into the systemic circulation. Local complications include inflammation, edema, blistering, necrosis, chronic drainage, and temporary or permanent nerve deficit. Systemic complications, such as renal or pulmonary toxicity, myocardial depression, and cardiac arrest, have been reported. Multiple sclerotherapy sessions are often needed because VMs have a propensity to recanalize and recur. Alternatives to ethanol sclerotherapy are sodium tetradecyl-sulphate foam or lauromacrogol that are effective for small VMs and cause fewer local adverse effects. Positive response was seen in 49.5% (for pain) and 52.7% (for mass reduction), within the 86 patients (91 VMs) involved in one study.94 Detergent sclerosants have been used as microfoams, using air bubbles or carbon dioxide to increase volume and surface contact with endothelium. However, neurologic complications have been described in 2% of cases. A modified radiopaque ethanol sclerosing agent was therefore developed. It traps ethanol within a mesh of ethylcellulose to increase viscosity.95,96
Surgical resection is often performed after sclerotherapy.53 In patients with BRBN, endoscopic coagulation with Nd:YAG (neodymium-doped yttrium aluminum garnet)laser, bipolar or argon plasma coagulation, and band-ligation are useful. An open surgery allowing surgical resection of all GI lesions can eradicate the need for blood transfusion as well as the common intermittent abdominal pain caused by intussuception.97,98 In patients with GVM, skin graft is often needed if excision is undertaken. VVM is also best treated with surgical resection. Closure often necessitates a skin graft.
Counseling: In patients with VMCM or GVM, genetic counseling should be provided for affected families, informing patients of a 50% risk of inheriting
the disease-causing mutation and of the variability in clinical expression. Psychosocial counseling and patient organizations dedicated to vascular anomalies are also an important asset for patients and their families.
LYMPHATIC ANOMALIES
AT-A-GLANCE
■ Worldwide occurrence of LM is unknown, but it is less frequent than that of VM.
■ Congenital, sporadic, slow-flow malformations of lymphatic vessels
■ LMs consist of micro- or macrocysts filled with lymphatic fluid.
■ Histologically consist of dilated lymphatic channels with flat endothelium that expresses D2-40.
■ Primary lymphedema can be inherited as an autosomal trait.
■ Infection is the most common complication that can lead to septicemia.
INTRODUCTION
INTRODUCTION
LMs are localized morphogenic errors of the lymphatic vessels. They consist of small vesicles or large cysts filled with lymphatic fluid. Macrocystic LM can be diagnosed in utero as early as the first trimester of pregnancy.99 However, most LMs are diagnosed during infancy, before the age of 2 years. Some can manifest only at puberty or during adulthood. They can be isolated, combined with other vascular anomalies, or be part of a syndrome. Primary lymphedema is not a localized lesion but a more generalized condition, in which the lymph fluid accumulates in the interstitial tissue. Lower extremities are most commonly affected. It can be uni- or bilateral. Primary lymphedema can be an isolated condition or part of a syndrome, and it can occur sporadically or as an inherited disorder.
EPIDEMIOLOGY
EPIDEMIOLOGY
LM is a congenital disorder of unknown incidence. It occurs sporadically in contrast to primary lymphedema, which can be inherited, usually as an autosomal dominant trait, in up to 20% of cases. Primary lymphedema is divided into congenital (Milroy disease, Online Mendelian Inheritance in Man [OMIM] #153100) and late onset (presenting at puberty, Meige disease, OMIM #153200). It can be isolated or
22
part of a syndrome, such as in Turner and Noonan syndromes.100
SYNDROMIC DISORDERS
SYNDROMIC DISORDERS
KLIPPEL-TRENAUNAY SYNDROME
Klippel-Trenaunay syndrome is a combined capillary– lymphatic–venous malformation associated with hypertrophy of the affected limb. Lower limbs are affected in 70% of cases. It is characterized by a geographic, widespread CM associated with lymphatic vesicles. The persistence of a persistent embryonic vein located on the lateral side of the thigh is pathognomonic.101,102
CLOVES SYNDROME
CLOVES syndrome is an eponym for congenital lipomatous overgrowth with vascular malformations, epidermal nevi, and skeletal anomalies.103 This nonhereditary disorder is characterized by progressive asymmetric hypertrophy, multiple truncal lipomatous masses with paraspinal fast-flow or slow-flow vascular anomalies (or both), epidermal nevus or nevi, acral lesions, and skeletal or spinal anomalies (Fig. 147-15).
2653
22
The most important differential diagnoses are other overgrowth syndromes, such as the Proteus syndrome and Proteus-like syndrome.104
GENERALIZED LYMPHATIC ANOMALY
Generalized lymphatic anomaly (GLA) is a rare condition in which LMs can invade several organs such as the mediastinum, lungs, pleura, GI tract, bones, and soft tissue.105
GORHAM-STOUT SYNDROME
Gorham-Stout syndrome, or “vanishing bone disease,” is an aggressive rare lymphatic disorder characterized by progressive demineralization and destruction of bones, which are replaced by lymphatic vessels and capillaries.106
CLINICAL FEATURES
CLINICAL FEATURES
CUTANEOUS FINDINGS
LMs are composed of microcysts (Figs. 147-16 and 147-17), previously termed lymphangioma circumscriptum) or macrocysts (>1 cm in diameter, previously known as cystic hygroma) that grow proportionally with the child (see Figs. 147-1B, 147-16, and 147-17). They are filled with clear or serosanguineous fluid (see Fig. 147-17B). Whereas macrocystic LM manifests as a soft, multilobulated, well-defined mass, microcystic LMs are ill-defined and often invade adjacent structures. Dermal LMs can manifest as small, millimeter-sized vesicles, clear (see Figs. 147-16D and 147-17A) or dark red in color (when there is intracystic bleeding), that can bleed and become purple and nodular.
A B C
D
2654
22
A B C
Angiokeratoma Circumscriptum: Angiokeratoma circumscriptum, or capillary-lymphatic malformation (CLM), is a combined, well-demarcated lesion often located on an extremity. The lesion is pink to bluish-red in color, slightly raised, and usually hyperkeratotic. Clinically, angiokeratoma circumscriptum, angiokeratoma of Mibelli (circumscribed, darkred, hyperkeratotic plaques on distal extremities), and angiokeratoma of Fordyce (very common hyperkeratotic, blue-black papules on the scrotum of elderly men) are recognized. They are to be differentiated from angiokeratomas of Fabry disease (see Chap. 127) and from VVM (previously known as verrucous hemangioma; see earlier VM section).
Lymphedema: Lymphedema is characterized by swelling of the affected body part, usually a lower extremity, and caused by accumulation of lymphatic fluid into the extracellular space caused by intrinsic lymphatic dysfunction (Fig. 147-18). Various phenotypes exist depending on the age of onset, location, and associated anomalies.107,108
Congenital Lymphedema: Milroy disease is suspected in the presence of swelling of the dorsum of the feet with a family history of lymphedema. Lymphedema is present at birth, often bilateral, and affects the lower limbs below the knees.109 Other features can be associated with congenital lymphedema, such as hydrocele (37% of males), prominent veins (23%), upslanting toenails (14%), papillomatosis (10%), or urethral abnormalities in males (4%).
NONCUTANEOUS FINDINGS
Pulmonary embolism can occur in patients with KTS or CLOVES syndrome.110 Depending on the affected organ, symptoms such as pleural effusion, ascites, and malabsorption can occur in patients with GLA.105 Painful pathological fractures are common in patients with Gorham-Stout syndrome.106
COMPLICATIONS
LM can suddenly enlarge in response to cough, inflammation, fever, viral or bacterial infection, or intralesional bleeding (Fig. 147-19). Recurrent cellulitis is the major complication, especially in patients with KTS, and it can evolve into septicemia if not promptly treated. Local redness and warmth appear, and the lesion becomes painful. Facial asymmetry, especially of the mandible, is commonly associated with microcystic LM (see Fig. 147-16). Intraorbital LM is responsible for ocular dystopia and exophthalmia, as well as orbital enlargement. LM on the tongue impairs speech and produces oozing and halitosis. Airway obstruction is common when the base of the tongue or the cervicofacial area is affected.111 Extensive limb LM can cause elephantiasis. Extension into the pleura can occur and cause chylothorax. Visceral LM can cause protein-losing enteropathy and hypoalbuminemia. Patients with KTS as well as patients with CLOVES are at high risk of pulmonary embolism because of the persistent embryonic vein and the presence of ectatic thoracic veins, respectively.110
Lymphedema is complicated by cellulitis in 20% of cases. More rarely, pleural effusion, even in utero; hydrops fetalis; and chylous ascites can be observed.108,112,113
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
LMs, as well as KTS and CLOVES syndrome, are caused by mosaic or somatic mutations in PIK3CA that activate the PI3K/AKT/mTor signaling pathway.11,12,114,115
More than 20 genes have been identified to be mutated in various forms primary lymphedema.108
Milroy disease is caused by inherited loss-of-function mutations in vascular endothelial growth factor receptor 3 (VEGFR3).116-118 Mutations in the same gene
2655
22
A B
C D
are also responsible for sporadic hydrops fetalis and generalized subcutaneous edema.119 Lymphedemadistichiasis is caused by loss-of-function mutations
A B
in the FOXC2 transcription factor.120 Lymphedema associated with microcephaly, with or without chorioretinopathy or developmental delay, is caused
2656
by dominantly inherited mutations in KIF11.121,122
Hypotrichosis–lymphedema–telangiectasia, which has an autosomal-dominant or -recessive pattern of inheritance, is caused by mutations in SOX18.118 Hennekam syndrome (OMIM #235510) is an autosomal recessive generalized lymphatic dysplasia characterized by intestinal lymphangiectasia with severe and progressive lymphedema of the limbs, genitalia, and face, as well as severe mental retardation.123 Mutations have been identified in CCBE1 (collagen and calciumbinding EGF domains 1).107,124 Lymphedema is associated with hematologic malignancies in Emberger syndrome caused by GATA2 mutations.125 In about 35% to 40% of patients with familial primary lymphedema, a germline mutation can be identified.108,126
DIAGNOSIS
DIAGNOSIS
SUPPORTIVE STUDIES Pathology: LMs are characterized by dilated, flatendothelium-lined channels of variable wall thickness (see Fig. 147-8C). No blood cells are seen in these spaces, except after intracystic bleeding or in the presence of a combined lymphatic-venous malformation. Macrocystic LM consists of a single or multiple lymphatic cysts surrounded by a thick fibrous membrane. The cysts do not communicate with each other. Endothelial cells express specific lymphatic markers, such as podoplanin, D2-40, and VEGFR-3.44,127 Lymphedema is characterized by abnormalities in the primary peripheral lymphatic capillaries, collecting lymphatic vessels, or lymphatic valves and lymphovenous valves. This is pronounced in the affected limb but is sometimes also seen on the contralateral side. Extensive dermal fibrosis is often present.44
Imaging: Doppler ultrasound shows microcysts, macrocysts, or both separated by thin septa. In contrast to VMs, they are not compressible by the probe. MRI also demonstrates the cystic nature of the lesion, with often discernable fluid-fluid levels.128,129 Computed tomography (CT) is the best study to show bony involvement. Doppler ultrasonography of the deep venous system of the affected lower limb and the pelvic area is needed in patients with KTS and CLOVES. Moreover, because CLOVES syndrome is characterized by progressive overgrowth during infancy, careful followup every 6 months is recommended until the end of puberty. Follow-up consists of clinical examination, scaniometry (leg length films), and abdominal ultrasonography because of the increased risk of cryptorchidism, hydrocele, renal atrophy, and Wilms tumor.130 Additional investigations should be done according to symptoms.
Genetic Testing: Genetic panel–based testing is available in accredited diagnostic laboratories. Such screens help identify germline mutations and can aid
22
Most Likely
■Infantile hemangioma
Consider
■Venous malformation
■Teratoma
■Fibrosarcoma or rhabdomyosarcoma
Rule Out Vulvar
Rule Out Vulvar
■Acquired lymphangiectasia caused by radiotherapy
■Acquired lymphangiectasia caused by radiotherapy
■Acquired lymphangiectasia caused by Crohn disease
■Acquired lymphangiectasia caused by Crohn disease
management. For example, identification of a germline GATA2 mutation indicates specific cancer surveillance programs, and a KIF11 mutation implies a need for detailed ophthalmologic and developmental examination.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
See Table 147-7.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE
AND PROGNOSIS
LM usually grows with the child. It can cause asymmetry with bony overgrowth. Increases in size can be seen during infection or intralesional bleeding. Episodic reports of spontaneous involution exist. Because regression is commonly seen after local infection, it may be caused by postinflammatory autosclerosis. Patients with Gorham-Stout syndrome have a poor prognosis; it is lethal in 16% of cases.
MANAGEMENT
MANAGEMENT
INTERVENTIONS Medical Management: Systemic antibiotics should be used to treat intralesional bacterial infection as well as early cellulitis in both isolated and syndromic LM. Antiinflammatory drugs may be required to alleviate pain. Patients with CLOVES or KTS syndrome need to receive LMWH at prophylactic dosages of 100 anti-Xa/kg/day before any surgical procedure; this should be continued for another 10 to 20 days postoperatively to avoid perioperative decompensation of their coagulation abnormalities, and more importantly, to reduce the risk of life-threatening postoperative pulmonary embolism.110 Rapamycin has been effectively
2657
22
B A
used for extensive LMs resistant to standard treatment, as well as for patients with KTS and GLA. Although none had complete resolution, several patients experienced cessation of lymphatic leakage and infection and reduction of volume of the malformation. Side effects were minor.131,132
Lymphedema is best treated with elastic stockings, massage, and pneumatic compression devices. Sildenafil was suggested as effective in the treatment of LM,133 but further studies have not confirmed its efficacy and highlighted potential side effects, such as infections and bleeding.134,135
Procedures: Nd:YAG laser or carbon dioxide laser photocoagulation has been used to treat dermal LM vesicles that cause oozing (Fig. 147-20). Macrocystic LM is treated with fluid aspiration followed by percutaneous, intralesional injection of sclerosing agents performed by an interventional radiologist. Sclerosing agents include sodium tetradecyl sulfate, pure ethanol, OK432 (extract from a killed strain of group A Streptococcus pyogenes: picibanil), doxycycline, and bleomycin. Side effects vary from fever and erythema to edema.136,137
Surgical resection is another alternative for LM, CLOVES, and KTS syndrome.138,139 Results depend on the anatomical site and extension of the lesion. Recurrence is frequent because it is difficult to differentiate microcystic LM from adjacent normal tissue. This leads to either incomplete resection or unnecessary sacrifice of normal structures.
Counseling: Family and patient education regarding the cause and treatment is necessary for primary lymphedema because it can be inherited and associated with other important medical
2658
problems, necessitating inclusion in special surveillance programs. Psychosocial counseling and patient organizations dedicated to vascular anomalies are also an important help for patients and their families.
ARTERIOVENOUS MALFORMATIONS
AT-A-GLANCE
■ Worldwide occurrence; rare but exact frequency unknown
■ Congenital, fast-flow malformations that can be occult until puberty
■ Histologically consist of direct communications between arteries and veins
■ Usually sporadic but can have a genetic predisposition as in CM-AVM, hereditary hemorrhagic telangiectasia, or PHTS
■ Most difficult vascular malformation to treat; need a multidisciplinary approach
INTRODUCTION
INTRODUCTION
Fast-flow vascular malformations, namely AVM and AVF, are the most severe and devastating malformations. AVF is usually the result of trauma. AVM is characterized by the presence of a “nidus,” the epicenter of the lesion that is composed of direct communications between multiple feeding arteries and draining veins
without an intervening normal capillary bed. AVMs are present at birth, although they are not always visible. They can be localized or extensive and evolve with age.
EPIDEMIOLOGY
EPIDEMIOLOGY
The incidence of AVM is unknown. No sex preponderance has been identified. It is a rare, usually sporadic, fast-flow vascular malformation. Hereditary hemorrhagic telangiectasia (HHT) is an autosomal inherited disorder with a prevalence of 1 in 5000 individuals.140
CM-AVM is another familial form with autosomal dominant inheritance and a similar prevalence estimate.19-23
SYNDROMIC DISORDERS
SYNDROMIC DISORDERS
BONNET-DECHAUME-BLANC OR WYBURN-MASON SYNDROME
Bonnet-Dechaume-Blanc or Wyburn-Mason syndrome is a sporadic, syndromic AVM located in the centrofacial or hemifacial area (or both), with oculo-orbital and cerebral involvement.141 It rarely follows a trigeminal distribution like SWS. Intracerebral AVMs are common in Bonnet-Dechaume syndrome and can cause epistaxis, exophthalmos, and hemianopia. Mental retardation can also occur.
COBB SYNDROME
Cobb syndrome is another sporadic, syndromic AVM that associates cutaneous and spinal cord AVMs of the same metamere.142 The cutaneous lesion masquerades as a CM, although it is warm on palpation. Cobb syndrome manifests in childhood with a sudden onset of back or lower extremity pain associated with sensory disturbance. Other neurologic complications (pain, sensory and motor disturbances, and neurogenic bladder) can occur depending on the location and extension of the AVM.
HEREDITARY HEMORRHAGIC TELANGIECTASIA
Individuals with HHT demonstrate combinations of the following triad: multiple cutaneous and mucosal telangiectasias, often located on the mucosal lip; epistaxis; and a positive family history.143 The diagnosis is clinical, in accordance with the Curaçao criteria. The estimated frequencies of manifestations in HHT patients are spontaneous, recurrent epistaxis, 90%; skin telangiectasia, 75%; hepatic or pulmonary AVMs, 30%; and GI bleeding, 15%.144 In 30% of patients with HHT, a hepatic, pulmonary, or cerebral AVM (or a
22
combination of any of these AVMs) can be seen.144 In contrast, in 23% of patients with CM-AVM1 and 13% of patients with CM-AVM2, an intracerebral or intraspinal AVM is present.18,19,22,23,35], but there is no risk for visceral AVM as in HHT. Vein of Galen aneurysmal malformation is also part of the phenotype. Recurrent epistaxis is typically the initial manifestation of HHT. The nosebleeds can be severe enough to require blood transfusions. Pulmonary AVMs affect approximately 30% of patients with HHT and are particularly common in HHT1. These patients are at higher risk of stroke and brain abscess than in the healthy population because the normal filtering function of the lung is lost. Migraine headaches occur in 13% to 50% of cases. Recurrent painless GI bleeding occurs in 10% to 40% of patients. Symptoms may include abdominal pain, jaundice, symptoms of high-output cardiac failure, and bleeding from esophageal varices.145
PARKES WEBER SYNDROME
Parkes Weber syndrome is characterized by a large, congenital, cutaneous, red vascular stain on an extremity in association with soft tissue and skeletal hypertrophy of the affected limb and underlying multiple arteriolar-venular microfistulas.146 The affected extremity, often the lower one, is longer and larger than the contralateral one. Although often sporadic, it can be part of CM-AVM. In these cases, there are small, multifocal CMs located on other parts of the body. The signs and symptoms worsen with age. Affected patients can develop congestive heart failure.
PHOSPHATASE AND TENSIN HOMOLOG HAMARTOMA TUMOR SYNDROME
PHTS is an autosomal-dominant disorder that includes patients with Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome because 60% and 81% of them, respectively, have a mutation in PTEN.147 These patients typically have macrocephaly, penile freckling, multiple developmental venous anomalies in the brain, fastflow VMs (54%), and an increased risk of malignancy. The VMs are often multifocal (57%) and musculoskeletal and associated with ectopic fat deposition and disruption of the normal tissue architecture.147,148
CLINICAL FEATURES
CLINICAL FEATURES
CUTANEOUS FINDINGS
AVMs usually manifest as cutaneous, faint, red to purple, ill-defined masses with a thrill, a bruit, or a pulsation of increased amplitude (Figs. 147-21 to 147-23; see also Figs. 147-1D and 147-1E). About one third of AVMs are present at birth, another -third appear during childhood or at puberty, and the rest
2659
22
A B
manifest in adulthood because hormonal changes and trauma usually trigger the growth of an AVM. AVMs can affect any tissue and organ. Seventy percent of AVMs are located on the head and neck. They never regress spontaneously and get worse with time.149
In 1985, Schobinger staged AVMs according to their severity. Schobinger stage I is a red stain with bruit and pulses of increased amplitude. With age, puberty, or trauma, AVM can worsen, and veins become prominent and tortuous (Schobinger stage II; Fig. 147-22) and subsequently darker and painful, and they ulcerate and bleed (Schobinger stage III; see Fig. 147-23). The final stage of a large AVM is cardiac failure (Schobinger stage IV; see Fig. 147-23).150 The patient often reports hearing his or her cardiac pulse.
CM-AVM (see earlier discussion of CM-AVM) manifests as multiple atypical CMs that are randomly distributed (see Fig. 147-6) and associated with fast-flow lesions—AVM, AVF, or Parkes Weber syndrome—in 30% (CM-AVM1) or 20% (CM-AVM2) of patients. The expressivity is highly variable within families, from small, asymptomatic CMs to life-threatening AVMs. The fast-flow lesions (AVM or AVF) are either cutaneous or subcutaneous, with or without intramuscular and intraosseous involvement. About 10% to 15% of patients with CM-AVM have a Parkes Weber phenotype affecting the lower (two thirds of such cases) or the upper extremity. The AVMs can be asymptomatic, but complications occur depending on the location and size.19-21
A B C
2660
A B
22
NONCUTANEOUS FINDINGS
In 30% of patients with HHT, a hepatic, pulmonary, or cerebral AVM (or a combination of any of these AVMs).144 In 23% of patients CM-AVM1 and 13% of patients with CM-AVM2, an intracerebral or intraspinal AVM (or both) is present.18,19,22,23,35
COMPLICATIONS
COMPLICATIONS
AVMs involving the face can as well destroy the bone structure underneath and can cause gingival bleeding or epistaxis as well as hypertrophic asymmetry. Bony hypertrophy is a common feature of facial AVM, which causes asymmetry. AVM of an extremity often causes peripheral ischemia due to blood flow steel phenomenon. Cardiac failure is rare (Schobinger stage IV), especially during childhood. Puberty and trauma as well as hormonal influence can trigger appearance of AVMs. Intracerebral AVM such as in Bonnet-Dechaume syndrome, can cause epistaxis, exophthalmos, and hemianopia. Mental retardation can also occur. Cobb syndrome manifests in childhood with a sudden onset of back or lower extremity pain associated with sensory disturbance. Other neurologic complications (pain, sensory and motor disturbances, and neurogenic bladder) can occur depending on the location and extension of the AVM. Recurrent epistaxis is typically the initial manifestation of HHT. The nosebleeds can be severe enough to require blood transfusions. The prevalence of brain AVM is 1000-fold higher in patients with HHT1 than in the general population (1 in 10,000) and 100-fold
higher in patients with HHT2. A similar increased risk is seen in CM-AVM1 and 2. Patients with HHT with pulmonary AVMs and telangiectasia of the GI tract are at risk for life-threatening hemorrhage. Other sites of bleeding may include sites in the kidney, spleen, bladder, liver, meninges, and brain. Strokes may be either hemorrhagic or ischemic. These patients are at higher risk of stroke and brain abscess than in the healthy population because the normal filtering function of the lung is lost. Migraine headaches occur in 13% to 50% of the cases. Recurrent painless GI bleeding occurs in 10% to 40% of patients. Symptoms may include abdominal pain, jaundice, symptoms of high-output cardiac failure, and bleeding from esophageal varices.145
Patients with CM-AVM can exhibit intrauterine lifethreatening intracerebral bleeding caused by vein of Galen aneurysmal malformation.22,151 The brain and medullary AVMs can also bleed postnatally.22,35,151
ETIOLOGY AND PATHOGENESIS
ETIOLOGY AND
PATHOGENESIS
Underlying genetic defects have been identified for the fast-flow vascular malformations of CM-AVM (OMIM #608354), HHT (OMIM #187300), and PHTS (OMIM #153480 and 158350). These disorders are inherited as an autosomal-dominant pattern with a penetrance of about 95% for the cutaneous CMs or telangiectasias.18,19,22,151
HHT is caused by alterations in the transforming growth factor-β signaling pathway. The genes implicated
2661
22
in HHT are ENG, encoding endoglin (HHT type 1), and ACVRL1 (formerly called ALK1, HHT type 2); SMAD4, and GDF2 are less frequently involved.152-155
Endoglin and ACVL1 are type III and type I transforming growth factor-β receptors, and both are well-expressed on vascular endothelial cells. Lossof-function of this signaling concomitantly increases PI3K-AKT signaling.156
CM-AVM1 is caused by loss-of-function mutations in RASA1.18,20 RASA1 encodes P120RASGAP, a GTPase regulating RAS activity. It converts the active GTP-bound RAS into its inactive GDP-bound form.61,62,157 The high intrafamilial phenotypic variability is explained by the necessity of a somatic second-hit mutation to occur.21,158 In 30% of patients with CM-AVM1, the mutation is de novo. CM-AVM2 is caused by loss-of-function mutation in EPHB4, which encodes an endothelial cell receptor in venous vessels. The ligand EPHRINB2, in contrast to EPHB4, is expressed in arterial endothelial cells. This bidirectional ligand–receptor system is important for arteriovenous identity and separation. EPHB4 signals using p120RASGAP, and the loss of function of either gene causes increased RAS-MAPK signaling.18,159-162
AVM can also be caused by loss-of-function mutation in PTEN, a tumor suppressor gene, as seen in patients with PHTS.163 PTEN regulates PI3K-AKT activity, and this seems to be regulated at least in part by the HHT receptor complex (see earlier). Loss of function of one of the partners of this complex or of PTEN itself leads to activation of PI3K. Sporadic extracranial AVMs are caused by somatic activating MAP2K1 (MEK) mutations.14 Brain AVM are caused by another activating mutation in KRAS, which is also involved in the RAS-MAPK signaling pathway.164
DIAGNOSIS
DIAGNOSIS
SUPPORTIVE STUDIES Pathology: Histologically, AVMs are poorly demarcated and consist of distorted arteries and veins with thickened muscle walls caused by arteriovenous shunting and fibrosis (see Fig. 147-22C).
Imaging: Ultrasonography and color Doppler show no mass but an aggregation of high-velocity arterial and pulsatile venous flow with low resistance. Vessels are tortuous. On the extremities, noninvasive follow-up is performed by comparing the arterial outflow of the affected limb with the unaffected one. MRI is preferred over CT to delineate the extent of an AVM and differentiate AVM from hemangiomas and other VMs. Flow voids, corresponding to fast-flow vessels, are pathognomonic of AVM. Arteriography is needed before any treatment to determine feeding arteries and the nidus (see Figs. 147-1E and 147-22B).
Genetic Testing: Genetic testing is indicated in patients with an AVM that is associated with multifocal
2662
cutaneous CMs (CM-AVM) or telangiectasias (HHT). Molecular diagnosis helps genetic counseling and guide further imaging for eventual undetected malformations. Patients with AVM with associated macrocephaly should be screened for PTEN mutations. Identification of a germline mutation would enable precise counseling and inclusion in cancer surveillance programs.
DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
See Table 147-8. During childhood, AVM is often mistaken for a CM or hemangioma because of its presentation as a faint, ill-defined, macular red stain. However, disproportionate warmth on palpation is a clue to its fast-flow component. A pulse, thrill, and bruit are other signs supporting AVM. Doppler ultrasonography can help differentiate a slow-flow CM from a fast-flow AVM or hemangioma. Hemangiomas have equatorial feeding arteries, peripheral veins, variable echogenicity, and fast flow but no true arteriovenous shunting. Other dermatologic lesions can mimic AVM. Epithelioid hemangioendothelioma occurs in the extremity as a purple and locally aggressive lesion. Tumid lupus erythematosus, sarcoidosis of the face, and Melkersson-Rosenthal syndrome of the lip can mimic AVM. Dabska tumor can simulate an AVM on a child when it is located in the ear.
CLINICAL COURSE AND PROGNOSIS
CLINICAL COURSE
AND PROGNOSIS
AVMs tend to worsen with time, causing local destruction or life-threatening bleeding. Puberty and trauma trigger growth of the lesion. Improper management, often because of misdiagnosis, such as ligation of feeding arteries, or partial resection of the nidus, can have
Most Likely At birth
■Congenital hemangioma
■Infantile fibrosarcoma
■Other sarcoma Rapid growth after birth
■Infantile hemangioma
Rule Out Ear, nose
Rule Out Ear, nose
■Lupus erythematosus tumidus
■Lupus erythematosus tumidus
■Sarcoidosis
■Sarcoidosis
■Dabska tumor Lip
■Dabska tumor Lip
■Melkersson-Rosenthal syndrome
■Melkersson-Rosenthal syndrome
dramatic consequences and expansion of the AVM (see Fig. 147-23). The CMs in CM-AVM are harmless and usually of only cosmetic importance, although flow characteristics are commonly abnormal.
MANAGEMENT
MANAGEMENT
This fast-flow VM is the most complex and difficult vascular anomaly to treat. Its management is multidisciplinary. AVMs should be followed by periodic evaluation. The goal of cure of an AVM is based on obliteration (by embolization) and complete removal of the nidus. Partial resection and proximal ligation of the feeding arteries lead to severe complications, recurrence, and reexpansion over time. Furthermore, ligature of the proximal arteries prevents access to the nidus for embolization, stimulates recruitment of new feeding arteries, and therefore expands the malformation. Mismanagement of AVM can lead to amputation.19
INTERVENTIONS
Medications: In a limb AVM or multiple AVFs, elastic stockings can stabilize the lesion and protect the skin, especially in the early stage. Because AVM is difficult to eradicate, the focus of management is often to control the evolution of the malformation rather than to cure the patient.40,45 A pharmacologic approach with Marimastat, a synthetic matrix metalloproteinase inhibitor, has been used to treat extensive AVMs with good results. It reduced extension of the malformation and decreased pain and bruit.46 Unfortunately, the drug is no longer available. Thalidomide was successfully used to reduce the frequency and duration of nosebleeds in patients with HHT.165 It has also been used in unresectable stage 3 AVMs for which standard treatment had not been successful. For some patients, it was used in combination with embolization to improve efficacy.
Procedures: Superselective embolization: In contrast to AVF, superselective arterial embolization alone is only palliative and is done only in unresectable, complicated AVM. The embolized particles need to reach the epicenter of the lesion to avoid refilling of the nidus through new collaterals.166 Direct puncture of the nidus is another possibility in patients with previous arterial ligation or embolization.167 The most common materials are liquids (ethanol, isobutylcyanoacrylate), particles (Ivalon), foam, and implantable devices (coils, microspheres). Embolization is rarely curative unless performed for an arteriovenous fistula. Embolization can be used alone (palliative approach) or followed by complete surgical excision (curative approach). The site of injection must be as close as possible to the nidus to permit a superselective approach. Proximal occlusion is responsible of secondary refilling of the nidus through collateral arteries.24
Radical excision: Surgical resection is usually done only after embolization to minimize perioperative
22
bleeding.168 Excision should be complete and radical with carcinologic margins when possible. It is preceded with superselective embolization to occlude the nidus and reduce intraoperative bleeding.19,26 The overlying skin can be saved if normal; otherwise, it is widely excised. Coverage options include tissue expansion or a local or free flap.29 Cutaneous expansion can be done before embolization or surgery but may act as a trigger to stimulate growth of the malformation. Early intervention for “quiescent AVM” (Schobinger stage I) is debatable and should be considered only if complete resection is possible.169 Follow-up for at least 5 years with annual Doppler ultrasonography or MRI (or both) is mandatory after any treatment. For patients with Parkes Weber syndrome, treatment should be as conservative as possible. Epiphysiodesis may be necessary to control leg-length discrepancy. This procedure can sometimes aggravate the AVM.146
For patients with HHT, multiple treatments have been tried to reduce bleeding, including topical application of antiinflammatory drugs, laser, or surgery. Because of the extensiveness of the lesions, they all give limited symptom-free intervals.
Counseling: Genetic counseling is mandatory in CM-AVM, HHT, and PHTS.
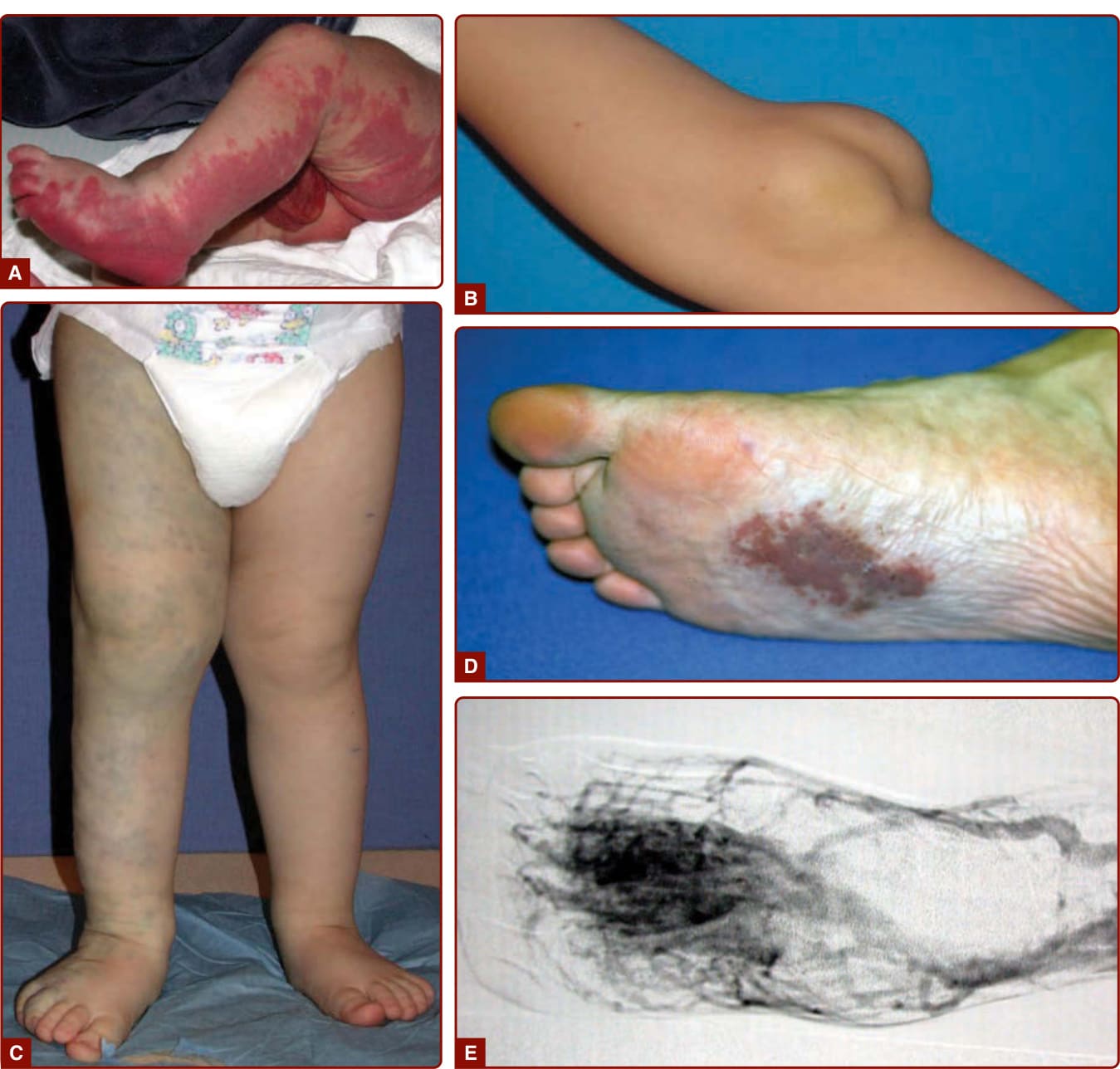
Figure 147-1 Vascular malformations of various vessel types. A, Capillary malformation of left lower extremity and genitalia. B, Subcutaneous lymphatic malformation invading the cubital nerve. C, Extensive venous malformation of right lower extremity, causing pain, swelling, and coagulopathy. D, Arteriovenous malformation of the sole. E, Nidus of the lesion on arteriography.
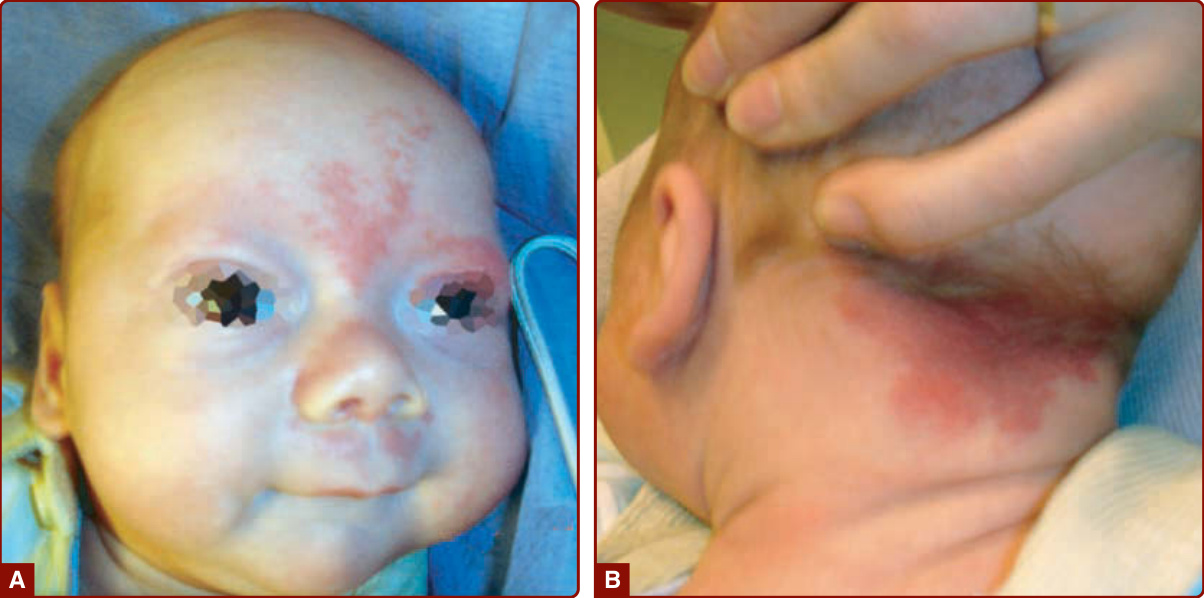
Figure 147-2 Nevus flammeus neonatorum (also called nevus simplex) on the glabella, upper eyelids, and upper lip (A) of a 6-week-old neonate who also has an Unna nevus on the occiput (B).
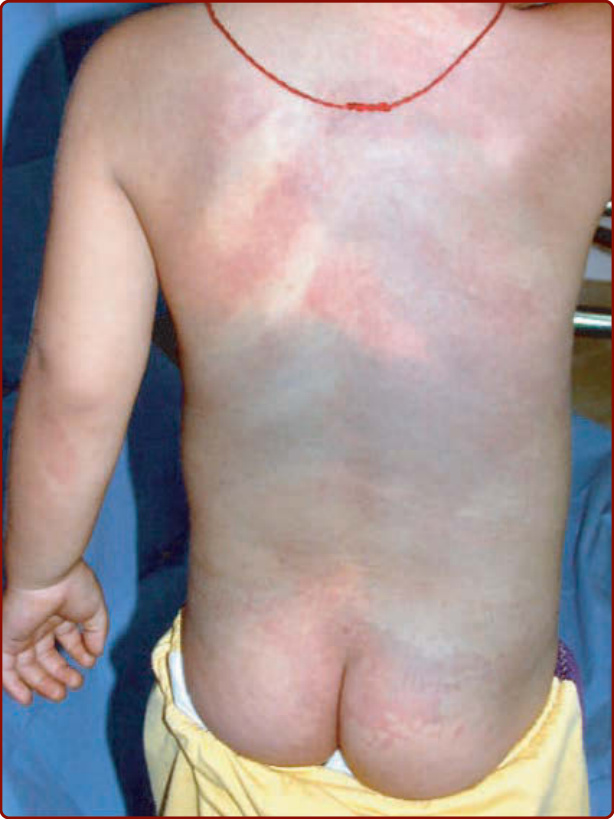
Figure 147-3 Clinical characteristics of phakomatosis pigmentovascularis. A large metameric capillary malformation with atypical extensive dermal melanocytosis located on the back.
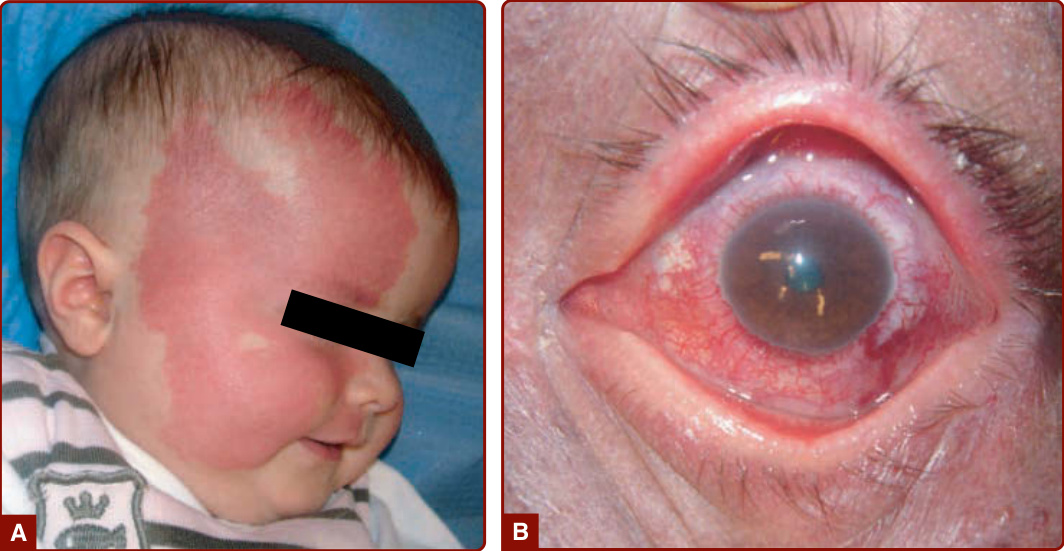
Figure 147-4 Extensive right facial capillary malformation. A, Involving the ophthalmic and maxillary branches of the trigeminal nerve in a 6-month-old girl with Sturge-Weber syndrome. B, Choroid capillary–venous malformation causing vision loss in a 50-year-old man with Sturge-Weber syndrome.
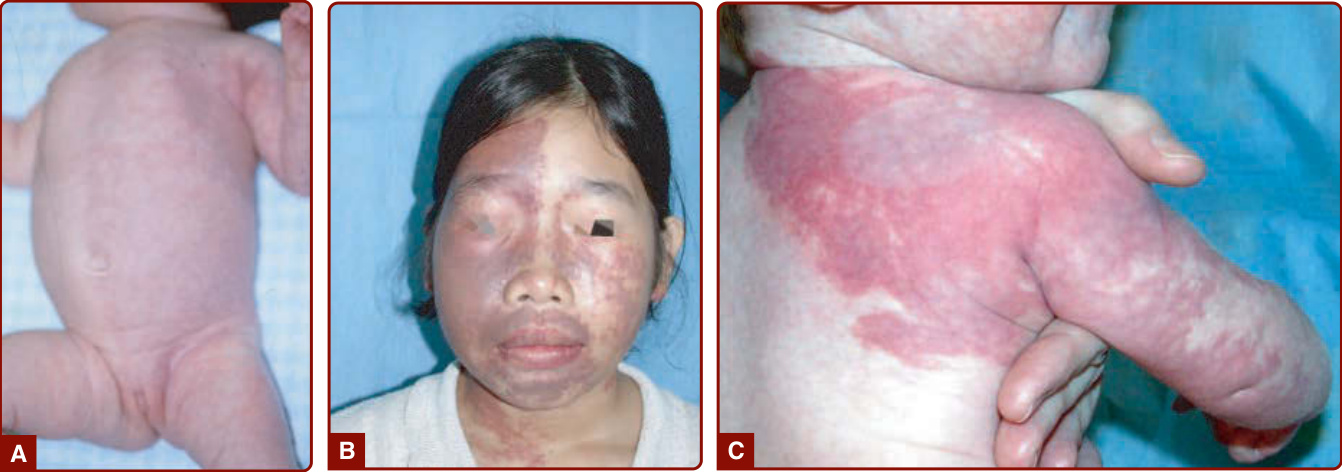
Figure 147-5 Several aspects of capillary malformations. A, Light red lesions involving half of the body. B, Dark red lesions on the face with mucosal involvement and soft tissue hypertrophy. C, More localized on the shoulder and arm.
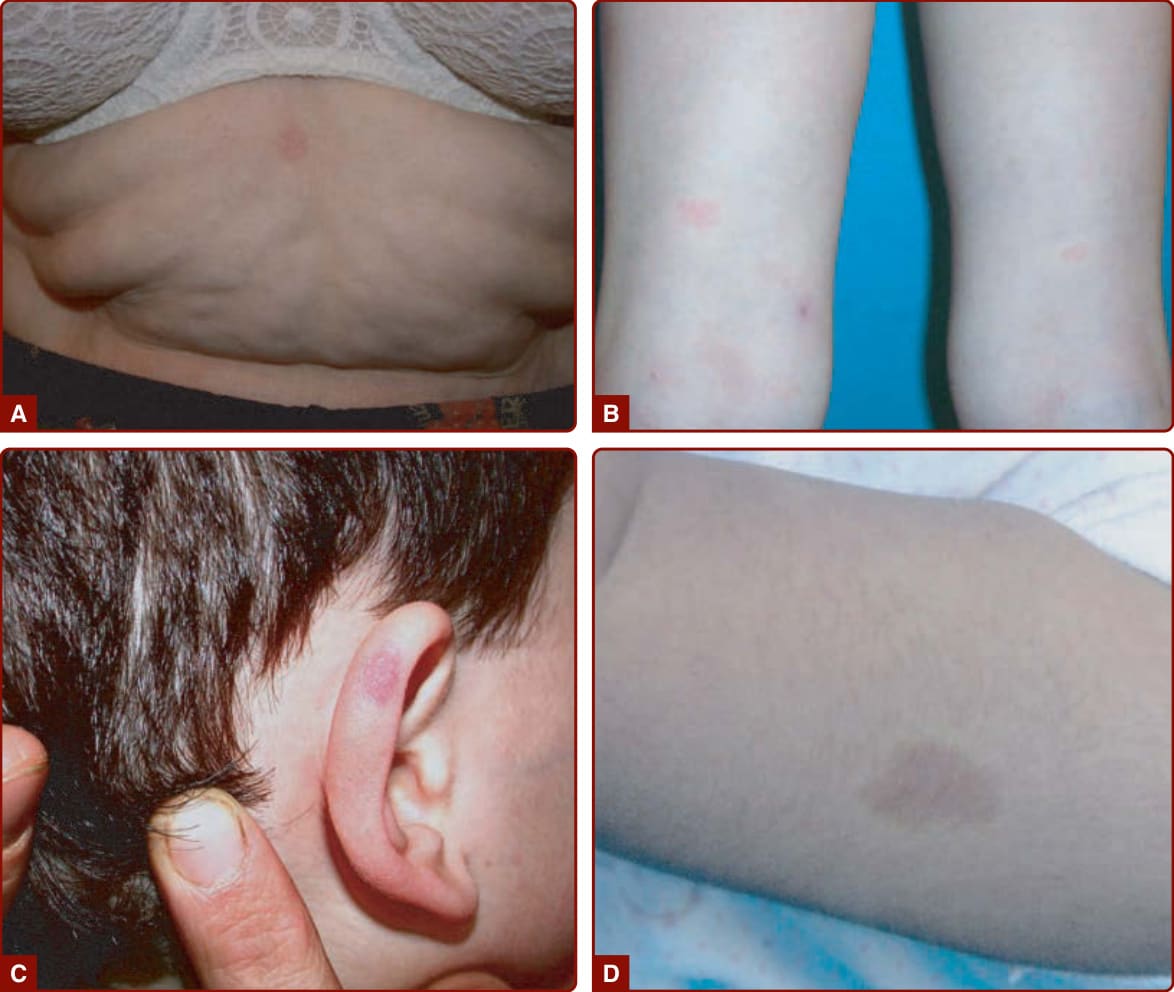
Figure 147-6 A–D, Examples of small atypical capillary malformations which belong to CM-AVM (capillary malformationarteriovenous malformation). D, Note the pale halo surrounding the lesion.
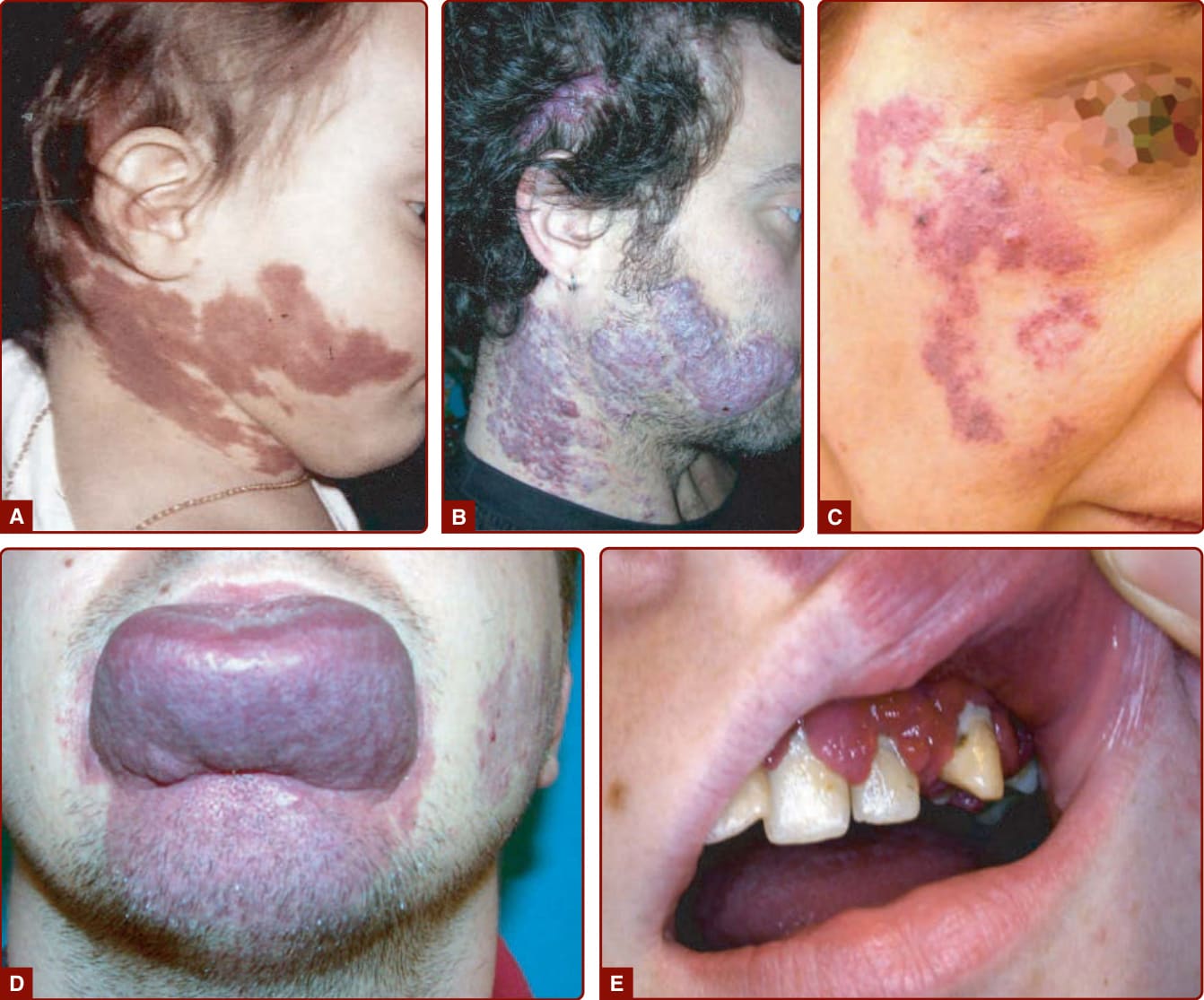
Figure 147-7 Clinical evolution of capillary malformation: darkening and thickening with time. A, At age 6 months. B, At age 33 years. C, Development of pyogenic granuloma. D, Soft tissue hypertrophy. E, Mucosal hypertrophy.
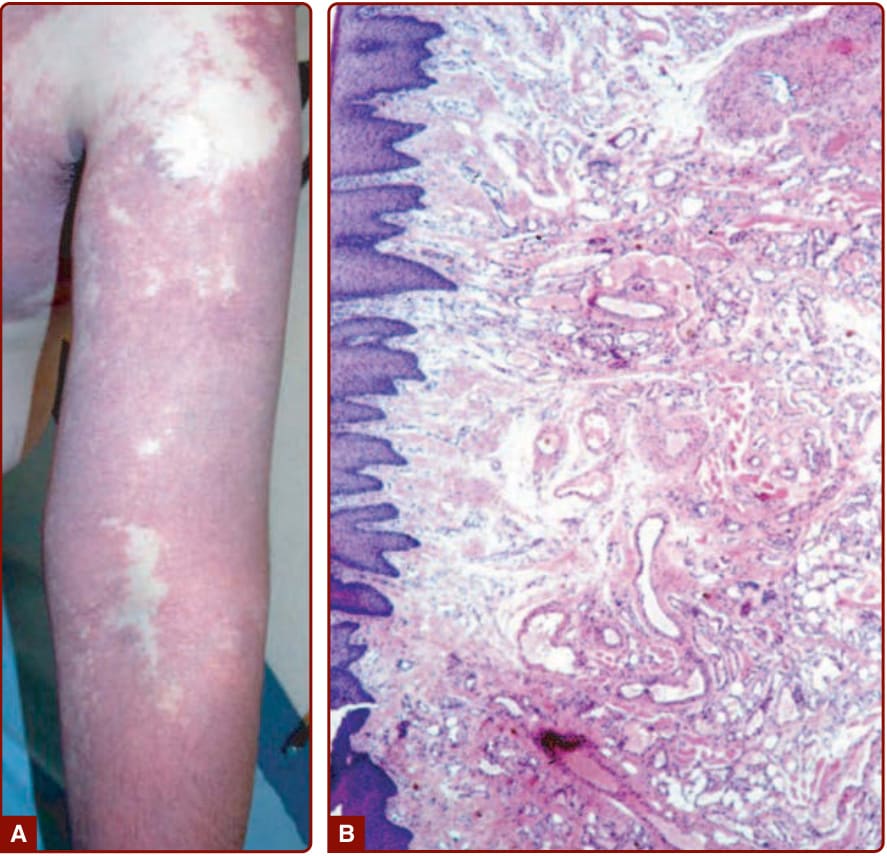
Figure 147-8 A, Extensive capillary malformation of upper extremity. B, Histologically, capillary malformation is characterized by dilated capillaries of normal number in the papillary and upper reticular dermis in combination with areas of increased number of normal-looking capillaries (hematoxylin and eosin staining).
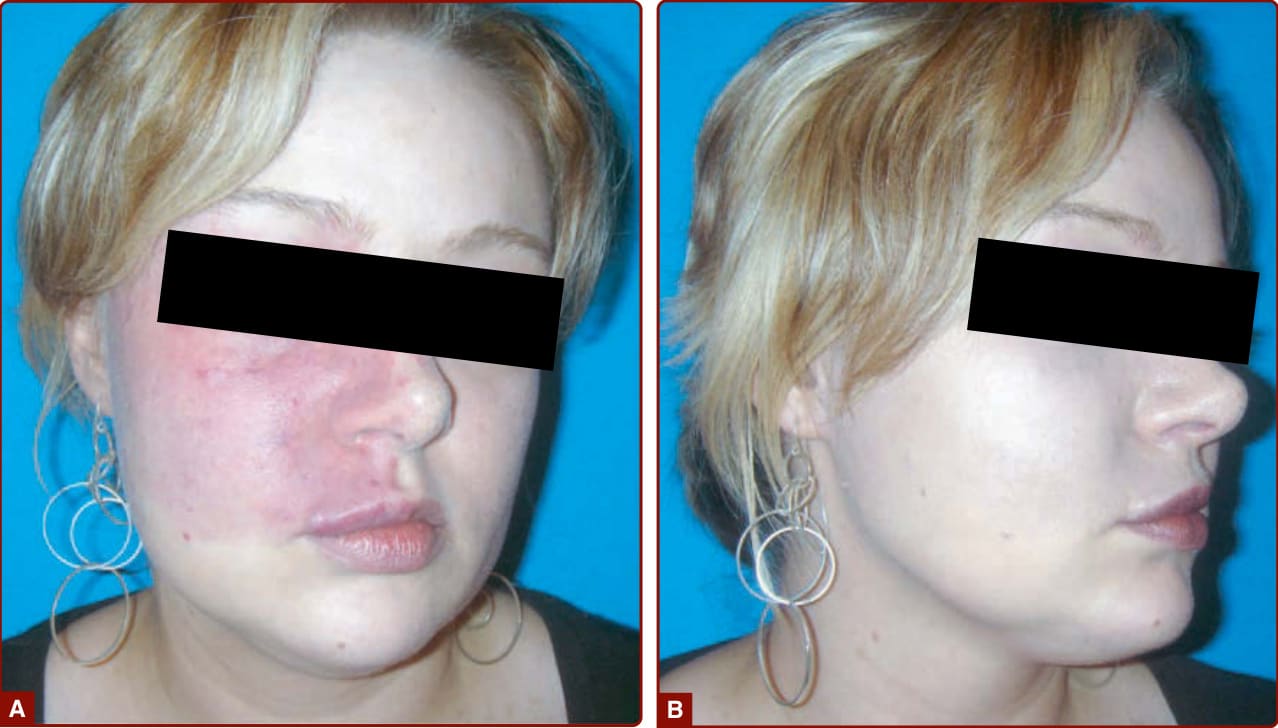
Figure 147-9 Efficient noninvasive management of facial capillary malformation using cosmetic makeup.
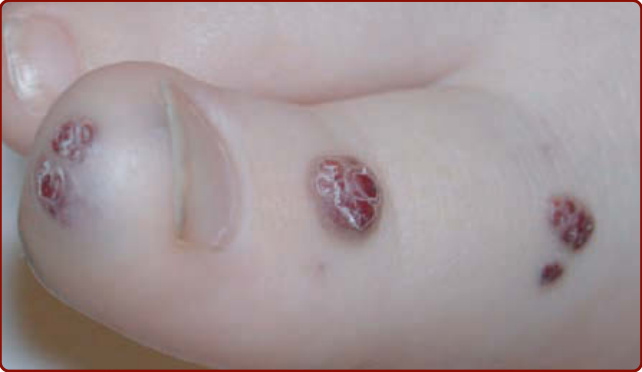
Figure 147-10 Blue rubber bleb nevus syndrome. Pathognomonic multiple small, dark blue, nipple-like lesions on the toe.
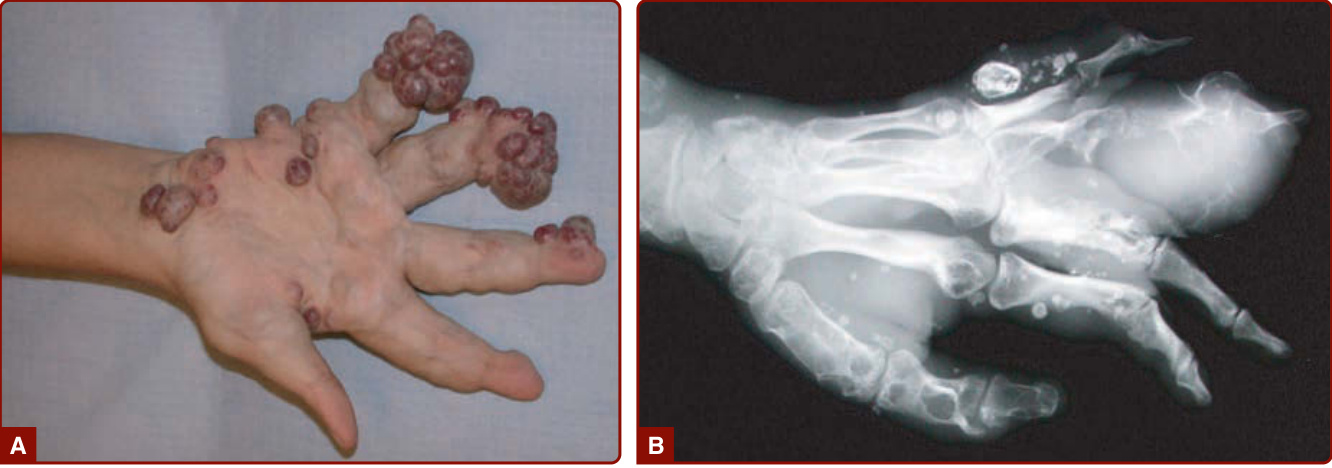
Figure 147-11 Clinical (A) and radiologic (B) aspects of Maffucci syndrome. Multiple venous malformations and enchondromas deforming the hand and fingers.
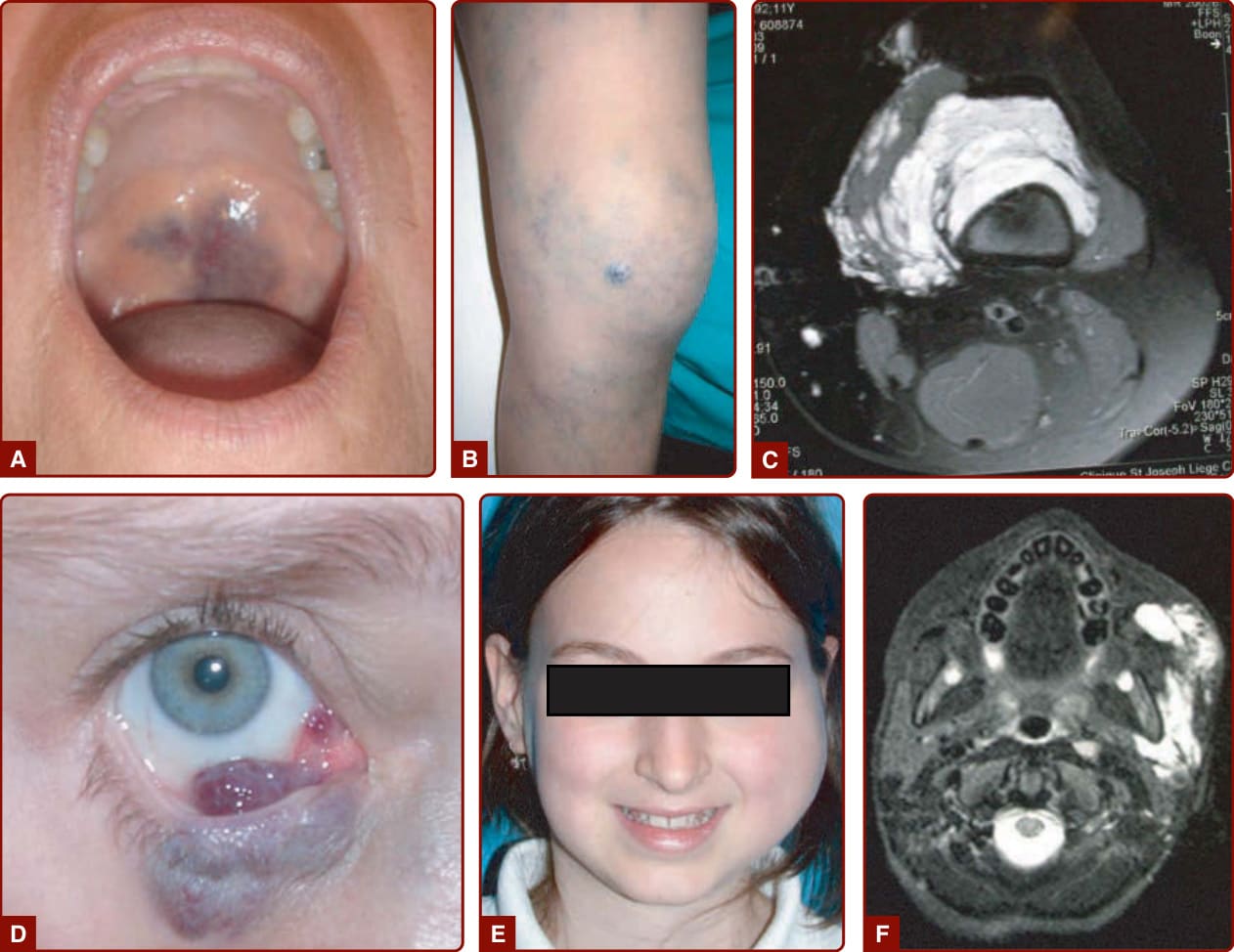
Figure 147-12 Various venous malformations. A. Of the soft palate, causing sleeping apnea. B and C, Of the knee, causing hemarthroses; confirmed by T2-weighted images. D. Of the mucosal eyelid. E and F, Of the cheek, causing facial asymmetry and pain.
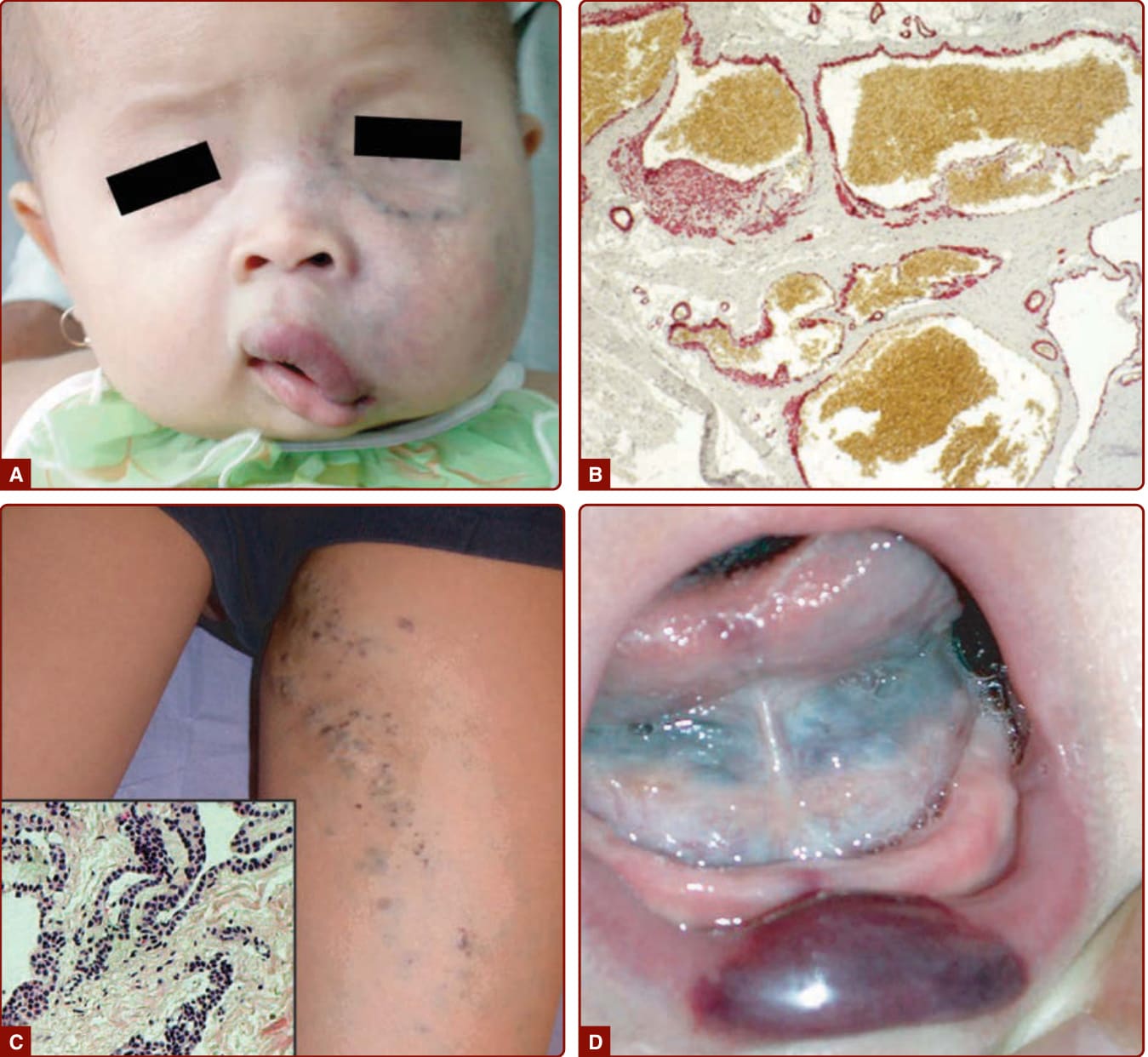
Figure 147-13 Venous malformations (VMs). A, Extensive hemifacial VM causing soft tissue hypertrophy and lip deformation in a 6-month-old boy. B, Histologically, VM consists of ectatic venous-like channels with anomalies in mural cells (hematoxylin and eosin). C, Superficial glomuvenous malformation of the inner thigh. Note the presence of mural glomus cells on histology (hematoxylin and eosin). D, Mucosal VM involving the lower lip and floor of the mouth.
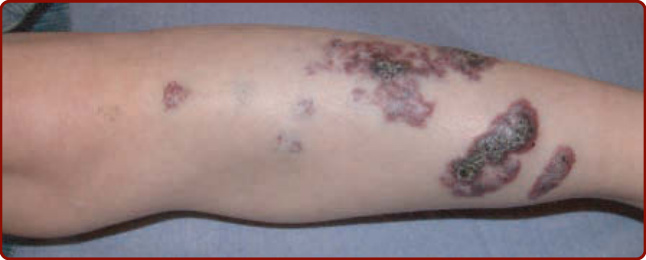
Figure 147-14 Verrucous venous malformation (previously known as verrucous hemangioma) on the lower extremity, causing oozing. It is purple-red in color and slightly raised, with hyperkeratosis.
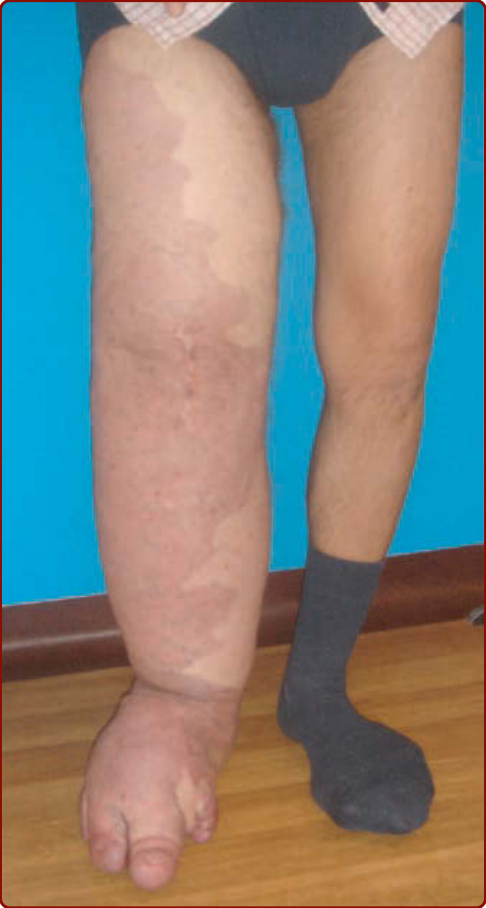
Figure 147-15 Capillary–lymphatic–venous malformation of right lower extremity with soft tissue hypertrophy (CLOVES syndrome).
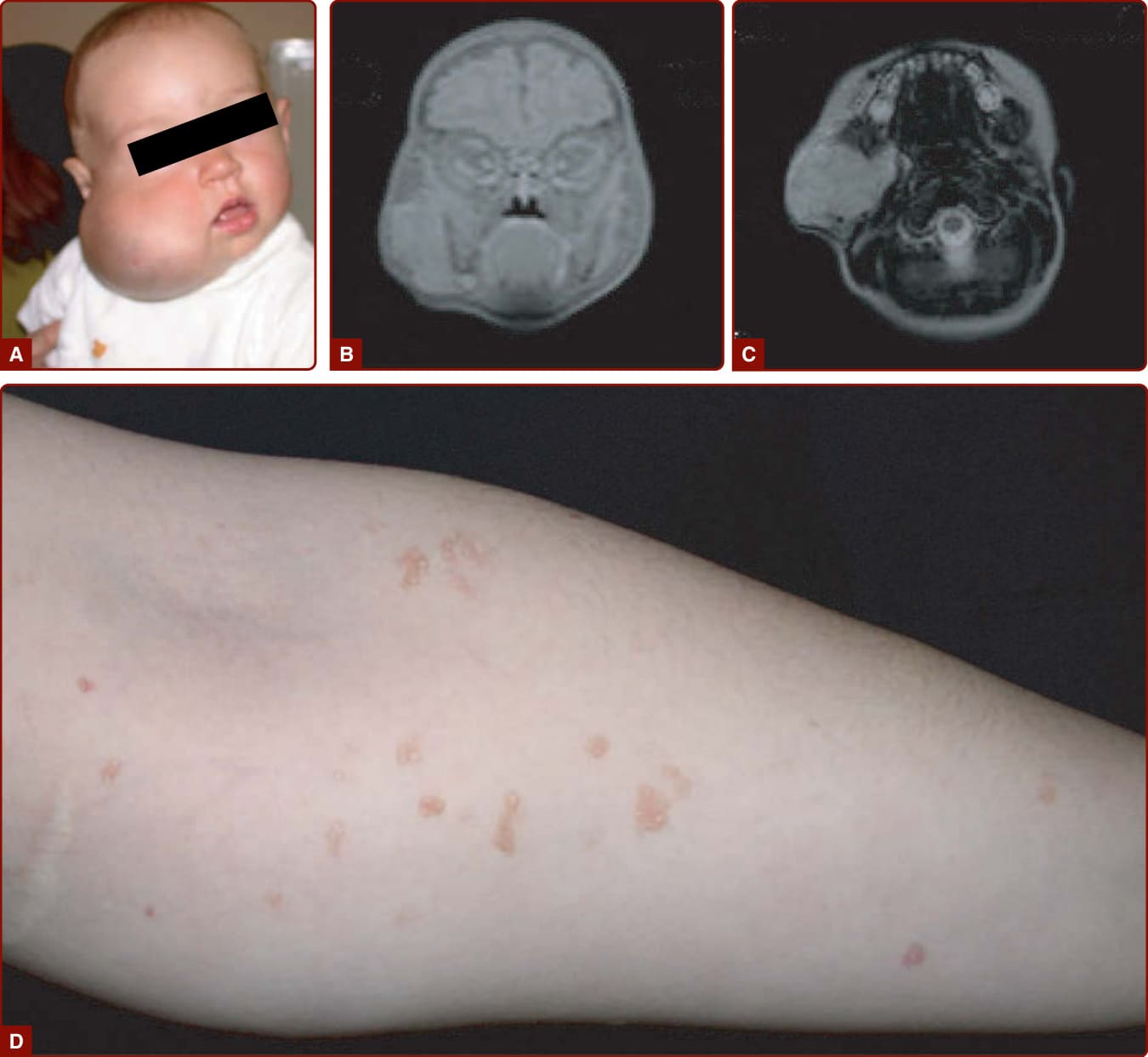
Figure 147-16 Various aspects of lymphatic malformation (LM). Bluish discoloration (A) of extensive LM of cheek, as seen on computed tomography scan (B and C). Small clear vesicles of dermal LM (D).
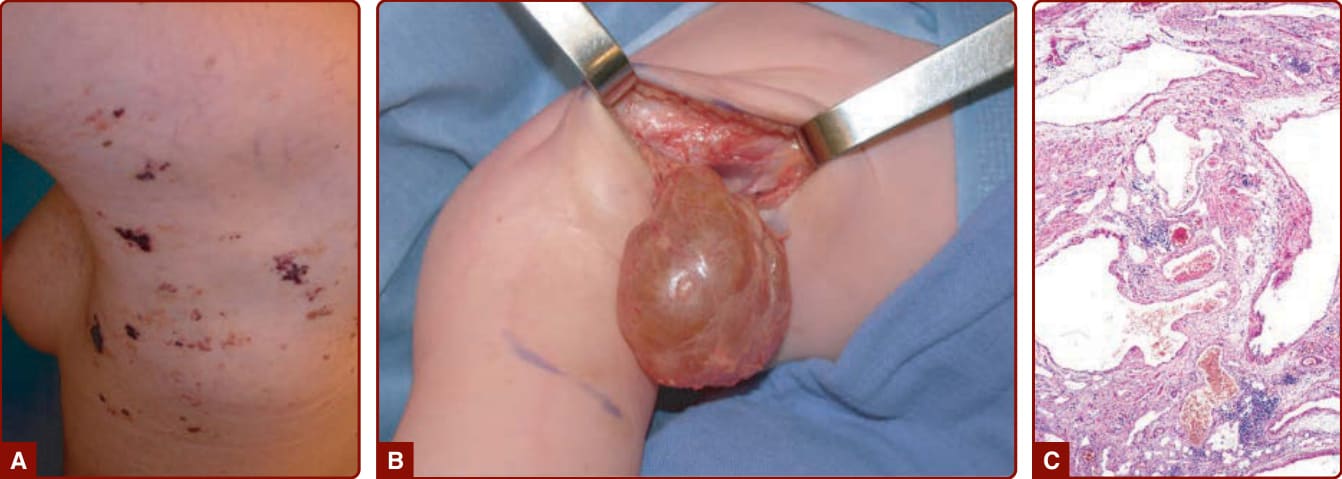
Figure 147-17 A, Microcystic, dermal lymphatic malformation (LM) consisting of clear and dark-red vesicles. B, Intraoperative view of a macrocystic LM of axilla showing a well-delineated cyst filled with clear lymph fluid. C, Histologically, LM consists of dilated lymphatic channels with flat endothelium (hematoxylin and eosin).
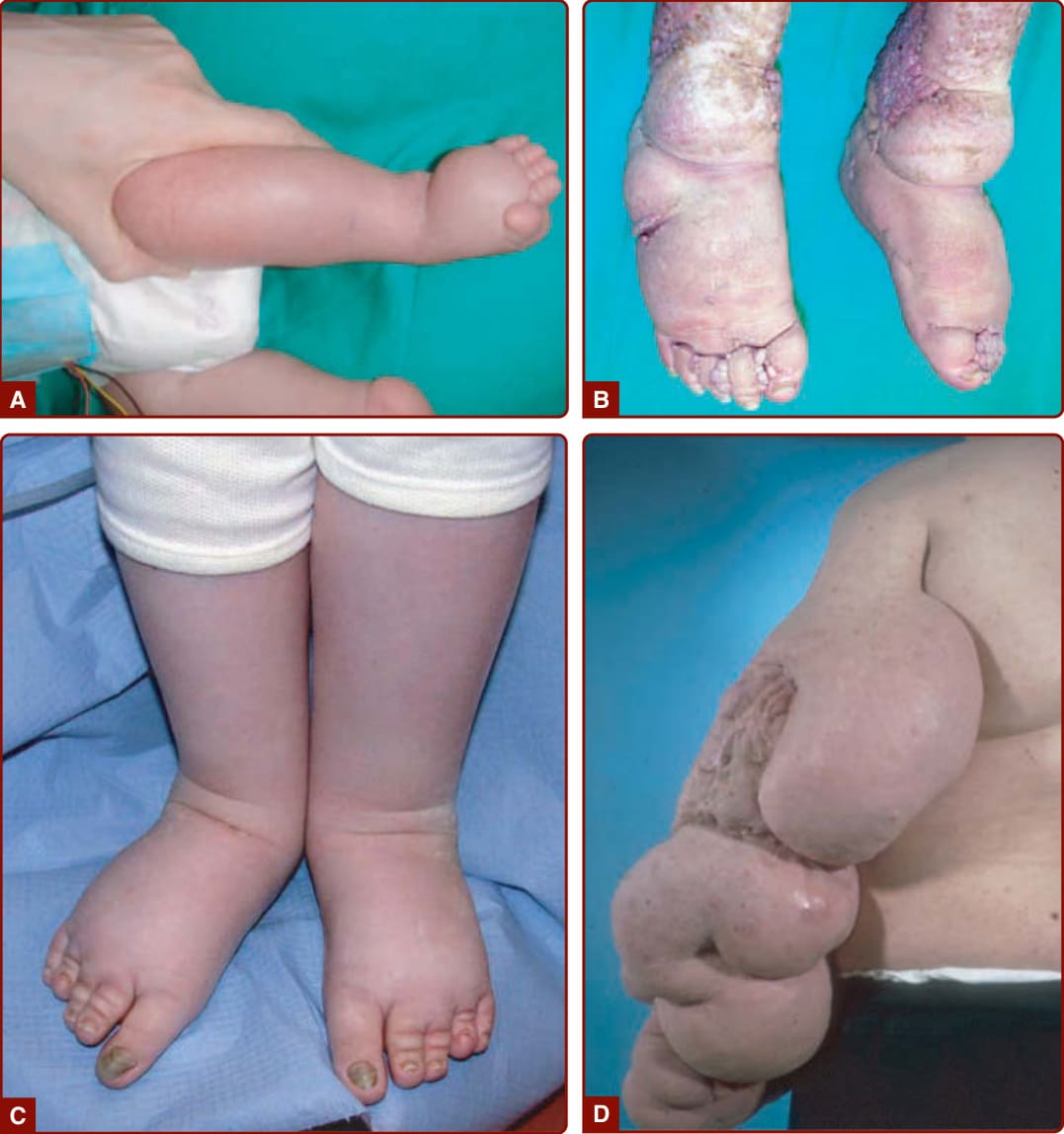
Figure 147-18 Various types of congenital primary lymphedema characterized by lymphedema of lower limb, below the knees (A); with papillomatosis (B); with upslanting toenails (C); that can evolve into elephantiasis (D).
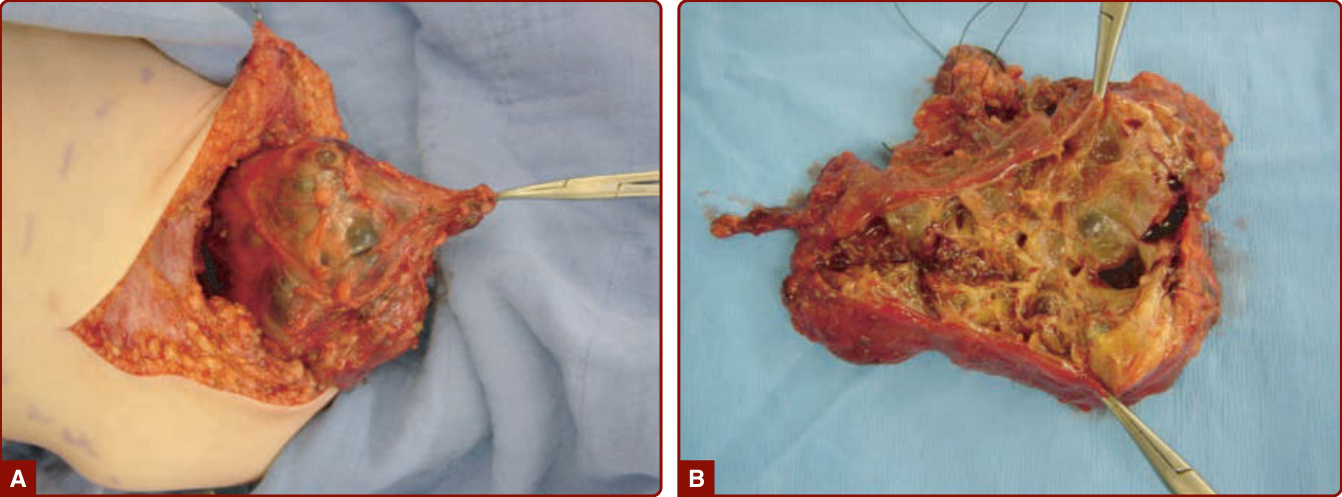
Figure 147-19 Intraoperative view of an axillary lymphatic malformation that had suddenly increased in size because of intracystic bleeding.
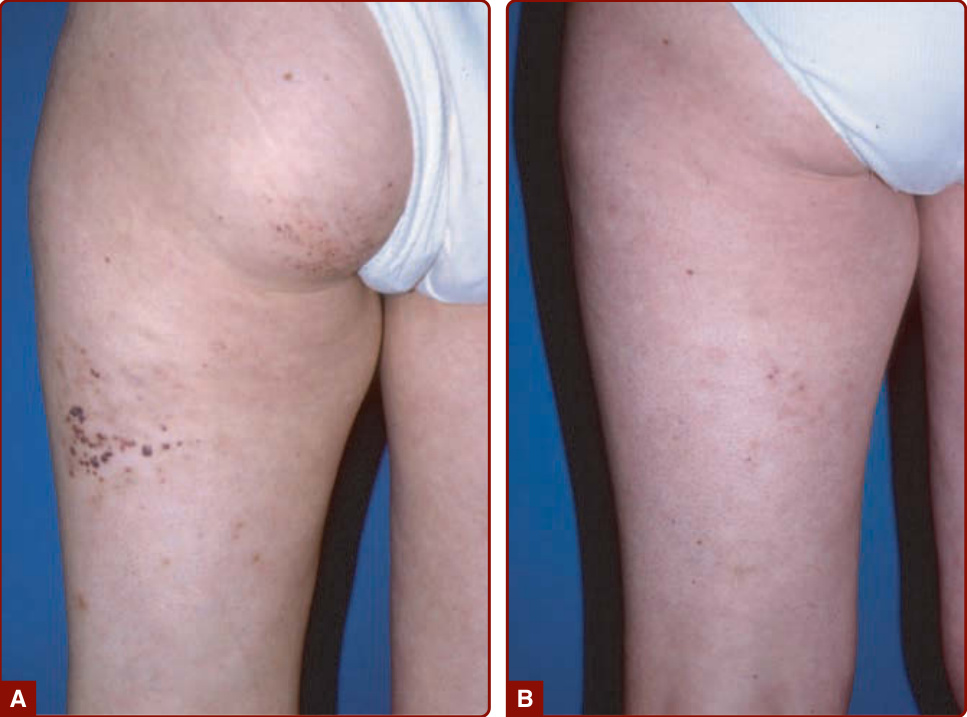
Figure 147-20 Dermal lymphatic malformation (A) efficiently treated with carbon dioxide laser (B).
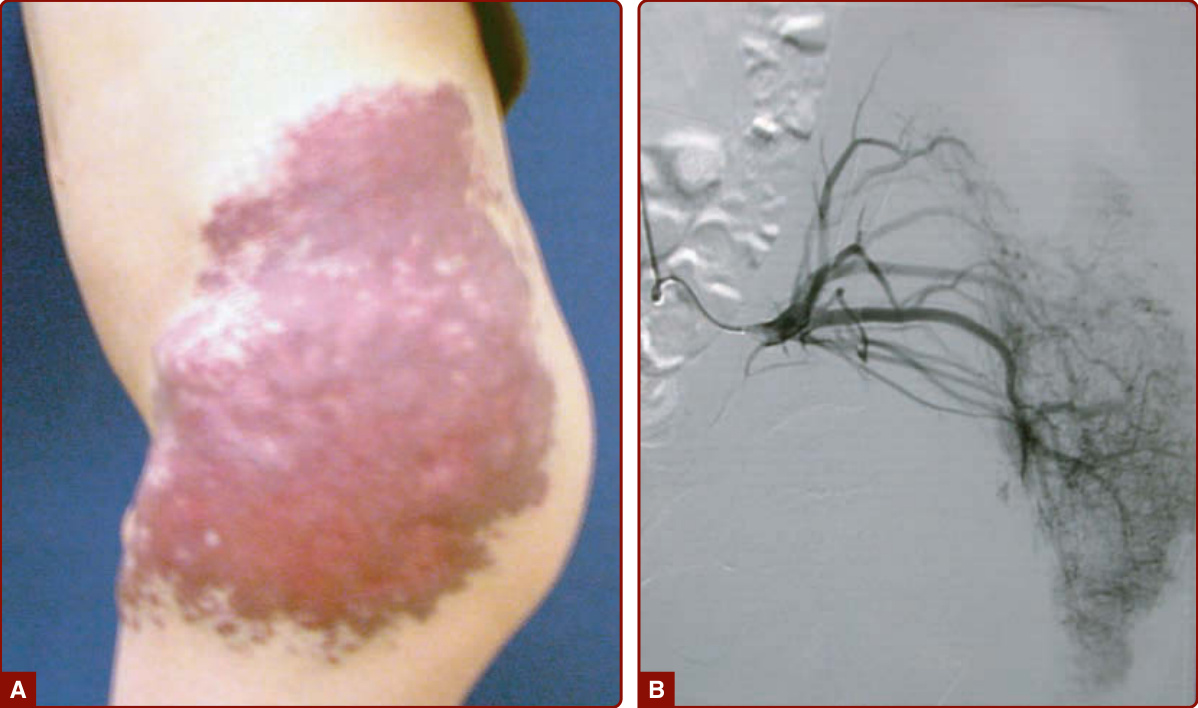
Figure 147-21 Stage 2 arteriovenous malformation of the left thigh mimicking an infantile hemangioma that did not disappear at 8 years of age (A). Arteriography showing the nidus confirmed the diagnosis (B).
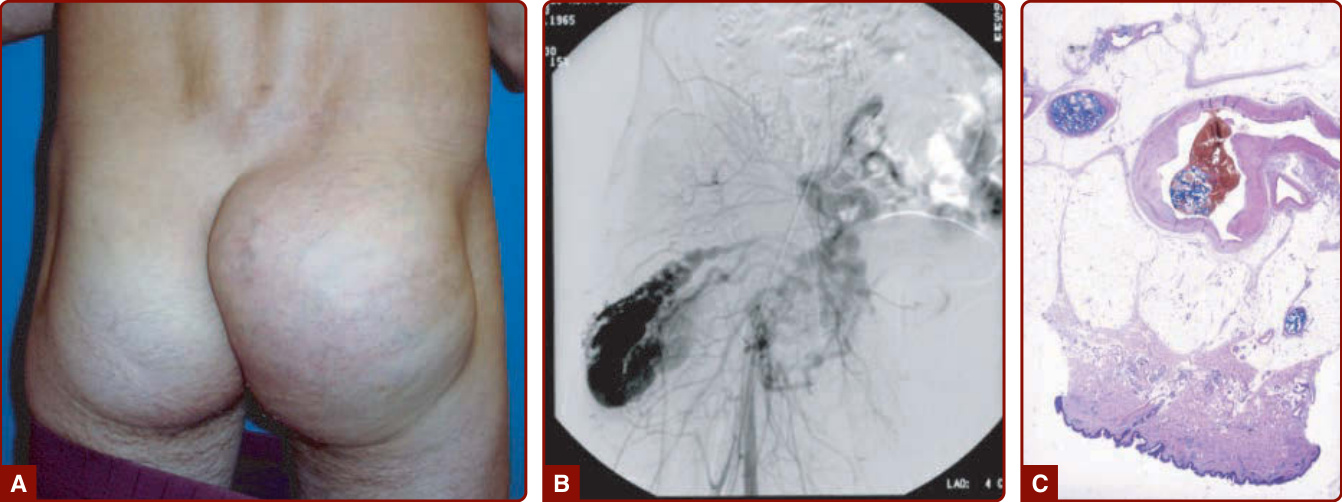
Figure 147-22 A, Extensive arteriovenous malformation deforming the right buttock. B, Arteriography showing nidus of the malformation. C, Histology shows direct connection between artery and vein, with thrombotic material secondary to embolization procedure.
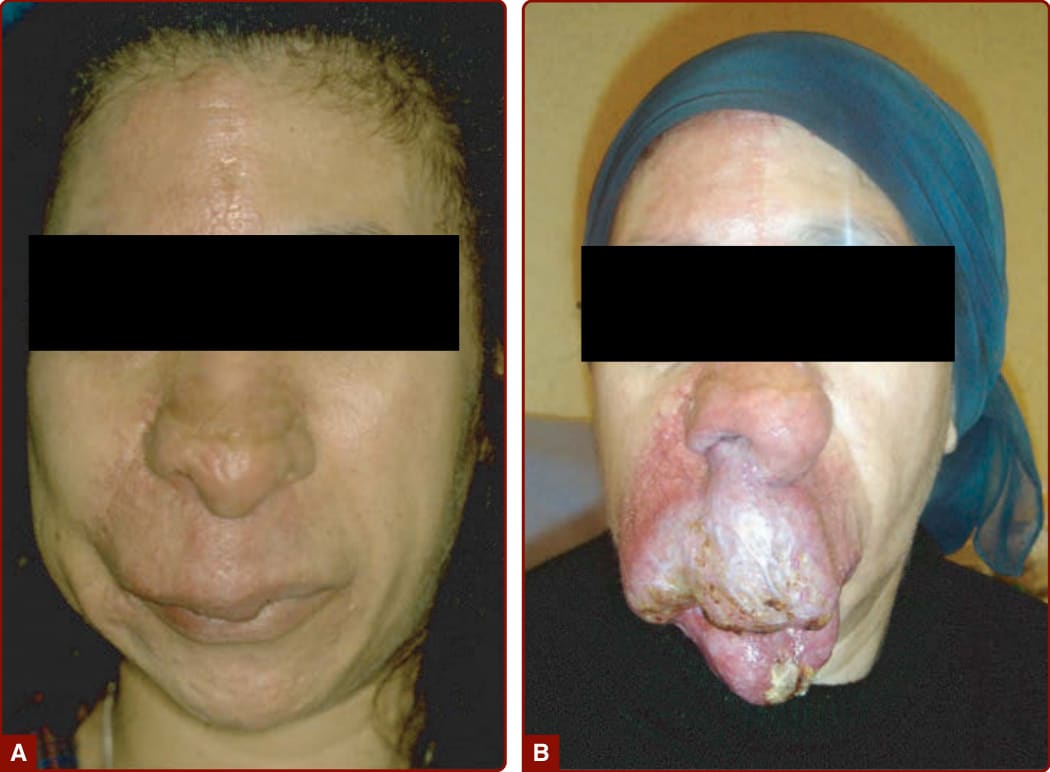
Figure 147-23 Stage 2 arteriovenous malformation (AVM; A) evolving into stage 4 AVM (B).
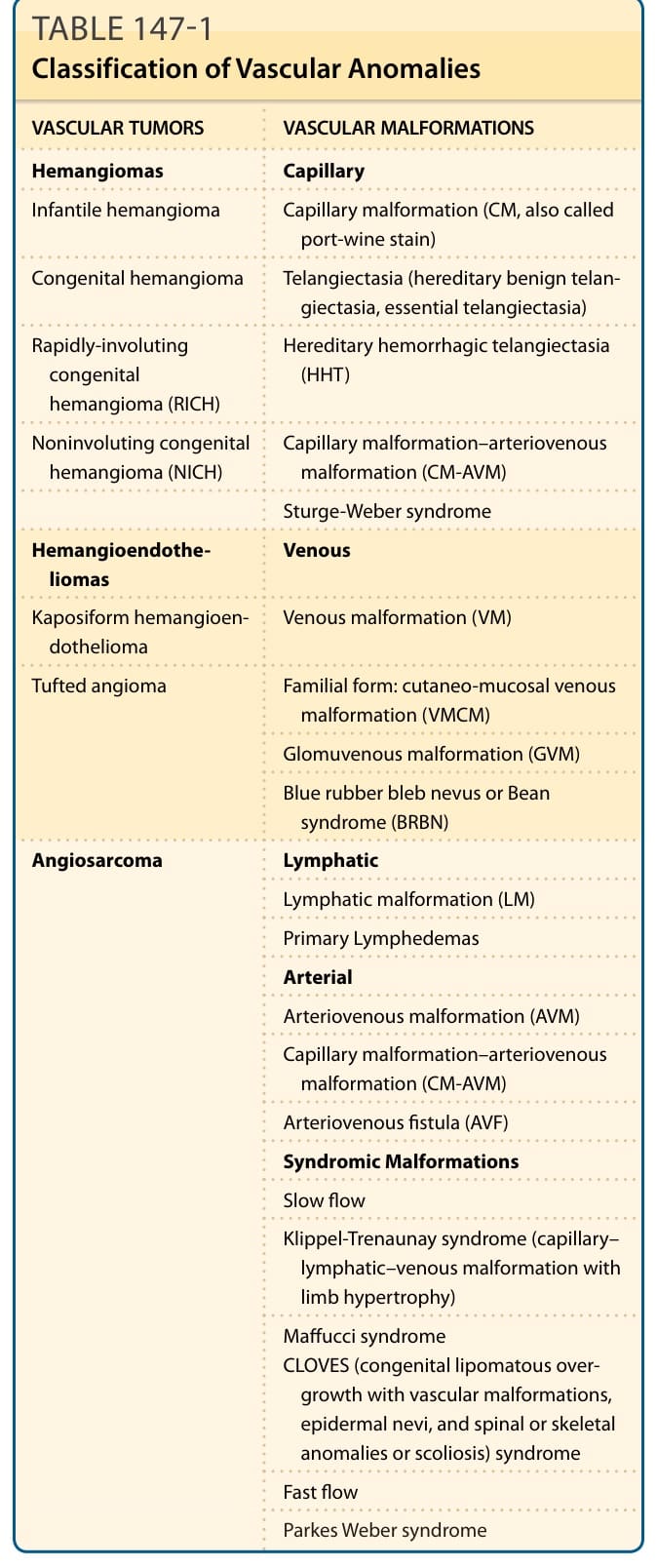
TABLE 147-1 Classification of Vascular Anomalies
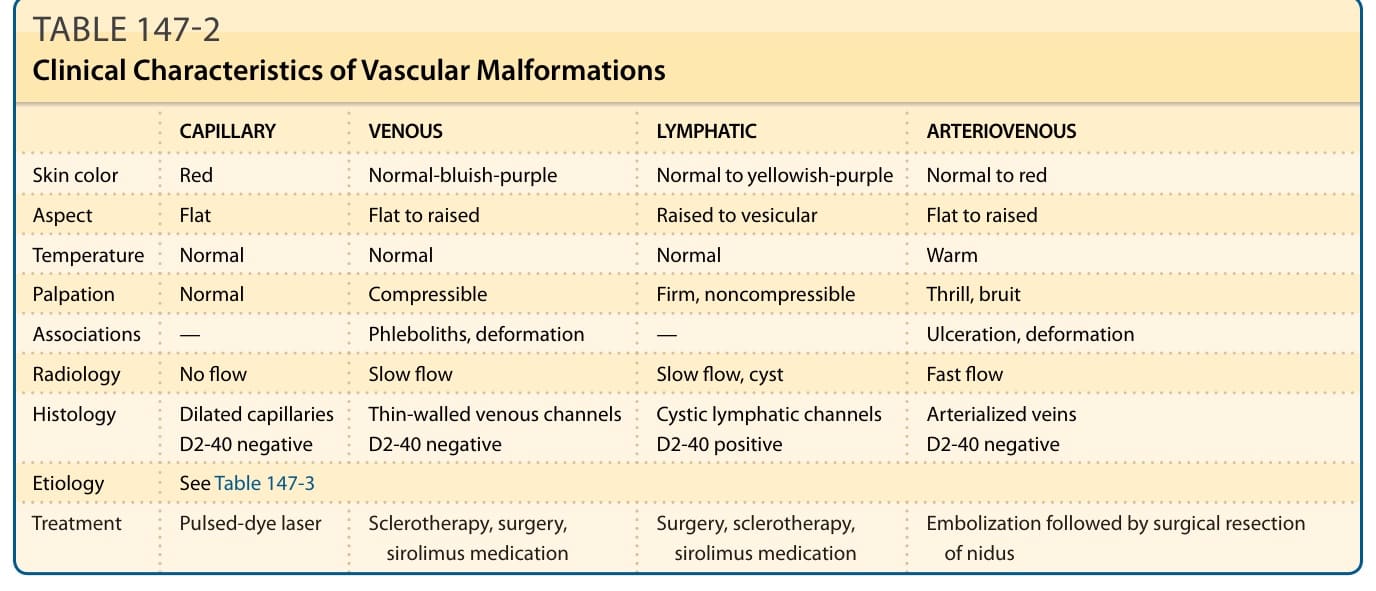
TABLE 147-2 Clinical Characteristics of Vascular Malformations
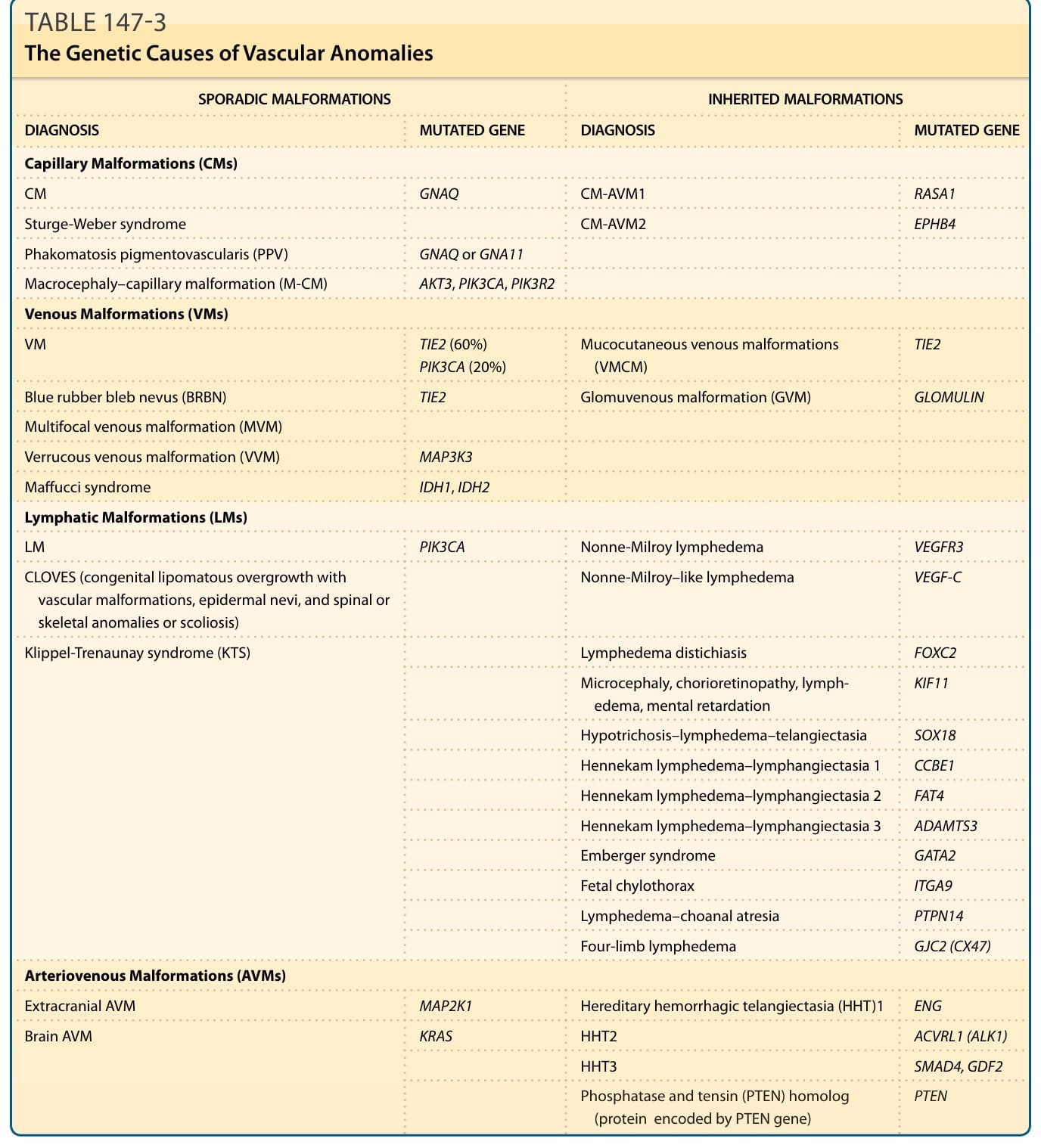
TABLE 147-3 The Genetic Causes of Vascular Anomalies

TABLE 147-4 Differential Diagnosis of Capillary Malformation
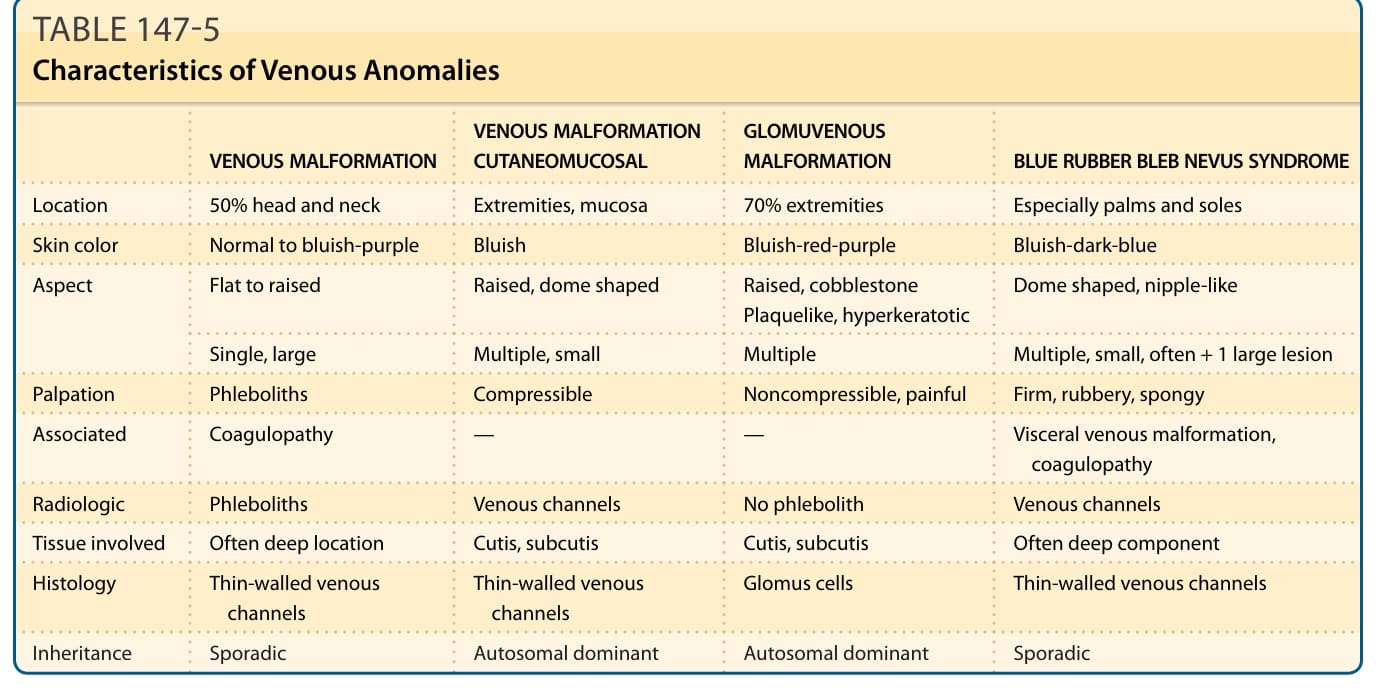
TABLE 147-5 Characteristics of Venous Anomalies
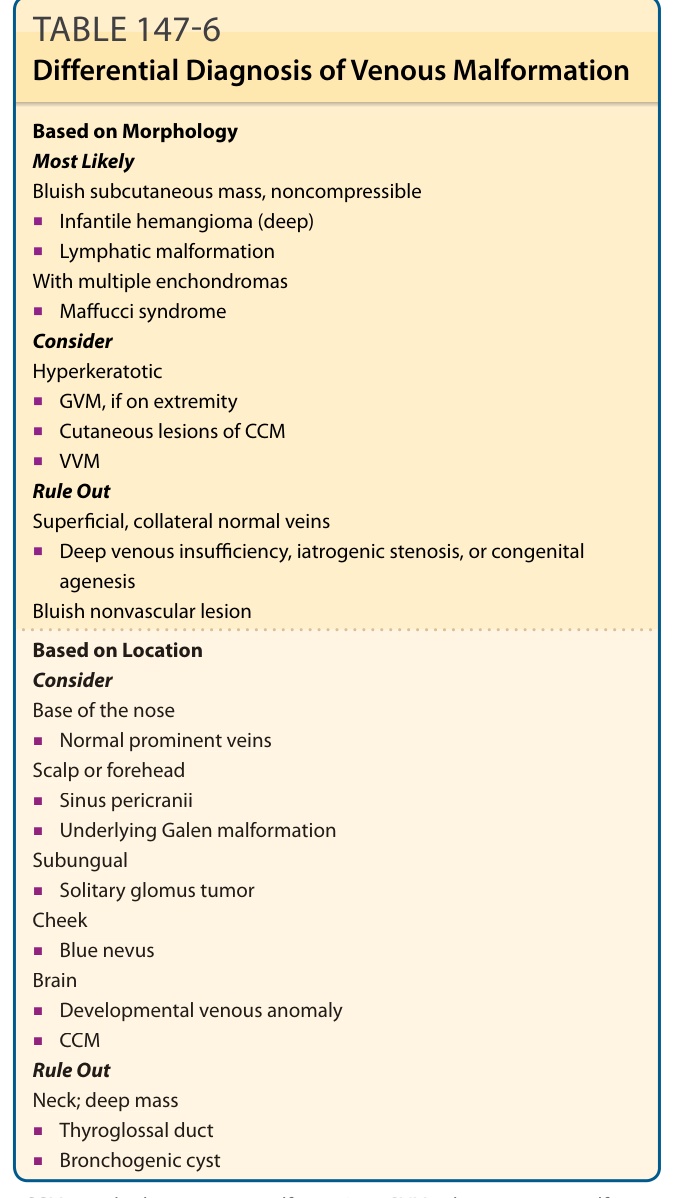
TABLE 147-6 Differential Diagnosis of Venous Malformation

TABLE 147-7 Differential Diagnosis of Lymphatic Malformation
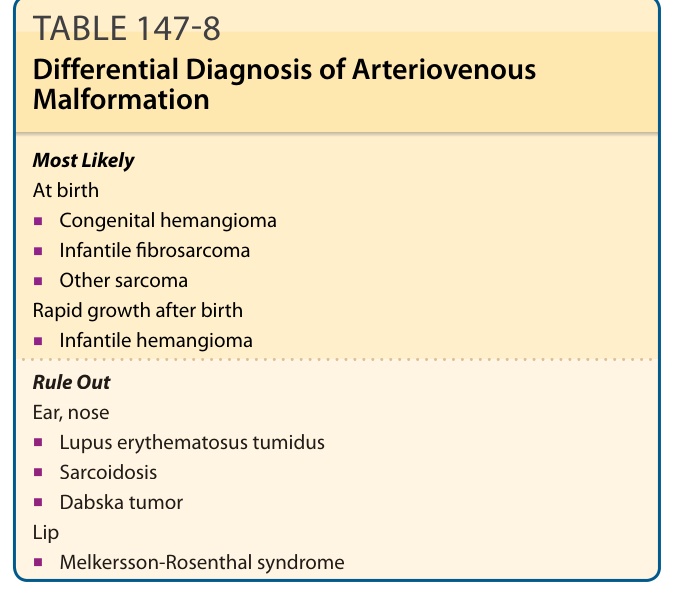
TABLE 147-8 Differential Diagnosis of Arteriovenous Malformation