系統性壞死性動脈炎 (Systemic Necrotizing Arteritis)
概論與分類
- 血管炎 (vasculitis):血管的發炎;系統性血管炎 (systemic vasculitides) 多為特發性、罕見且多系統侵犯。
- 皮膚血管炎常見,皮膚病灶可能是系統性疾病的初發症狀,故所有皮膚血管炎病人都需評估是否有系統性血管炎。
- 分類以主要受侵犯血管的大小(小、中、大)為核心,再細分。常用標準:美國風濕病學院 (ACR) 分類標準與 Chapel Hill 共識會議疾病定義。
- 白血球碎裂性血管炎 (leukocytoclastic vasculitis) 並非特定疾病,而是病理描述。
- 原發性血管炎分類:
- 以小血管為主:免疫複合體媒介(冷凝球蛋白血症、HSP)、ANCA 相關(MPA、CSS/EGPA、GPA)。
- 以中血管為主:川崎病、PAN。
- 以大血管為主:GCA、TAK、孤立性主動脈炎。
- 無主要血管大小:貝賽特氏病、原發性 CNS 血管炎、復發性多發軟骨炎、Cogan 症候群。
流行病學
- 多數特發性血管炎為罕見「孤兒」疾病(盛行率 <200,000 人)。
- 族群傾向:高安動脈炎 (Takayasu arteritis) 女性遠多於男性;川崎病幾乎只見於幼兒;巨細胞動脈炎 (giant cell arteritis) 限於年長成人;GPA 多為白人;貝賽特氏病在東地中海、日韓較常見。具強烈地區性差異者多有 HLA 區域遺傳危險因子。
致病機轉 (pathogenesis)
- 多數血管炎病因未知;例外為感染相關(PAN 之 B 型肝炎、冷凝球蛋白血症性血管炎之 C 型肝炎)與藥物誘發性。
- 小血管血管炎走兩條途徑:(1) 免疫複合體沉積;(2) 非免疫複合體媒介、可能涉及 ANCA 的病理。
- ANCA 針對嗜中性球的髓過氧化酶 (myeloperoxidase, MPO) 或蛋白酶-3 (proteinase-3, PR3),活化嗜中性球並導致壞死性血管炎。
- 大血管血管炎由 T 細胞活化大動脈壁巨噬細胞媒介;GCA 中 Th17 與 Th1 細胞顯著,T 細胞經滋養血管 (vasa vasorum) 進入、內皮維持完整。GCA 與 TAK 皆視為自體免疫疾病(HLA:GCA 屬 class II,TAK 屬 class I)。
臨床表現(皮膚)
- 外觀反映受侵犯血管的大小、深度與密度。
- 小血管血管炎典型表現為可觸性紫斑 (palpable purpura)(直徑約 2 mm 即可觸知);高密度可融合成融合性紫斑與中央壞死/潰瘍。其他:網狀紫斑 (retiform purpura)、丘疹、蕁麻疹樣病灶、結節、水疱、潰瘍。
- 中血管血管炎位置較深,產生紅斑性結節或網狀青斑 (livedo racemosa),較易造成潰瘍與指(趾)缺血 (digit ischemia)。
- 紫斑可無症狀或刺痛/灼熱;結節與潰瘍通常疼痛;無病灶處劇痛應疑神經病變或發炎性關節炎。
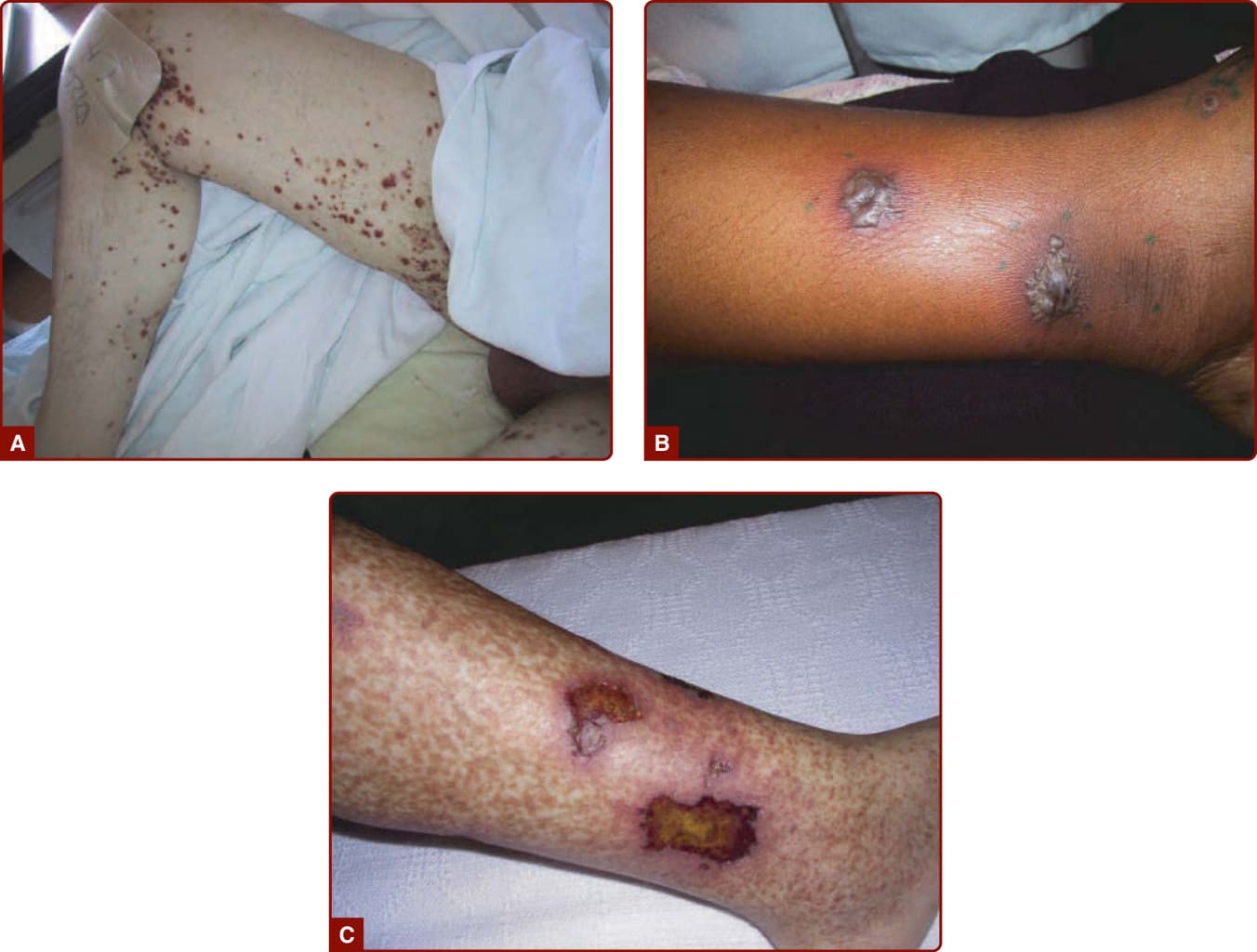
圖 139-3:系統性血管炎的不同皮膚表現(A 紫斑、B 大疱、C 潰瘍)。
診斷
- 就診時先問 3 問題:(1) 病灶是否由血管炎引起?(2) 是否侵犯其他器官系統?(3) 有無其他發現可建立特定診斷?已確立血管炎後再問:能否做特定型態診斷?是否需立即治療/住院?
- 皮膚切片 (skin biopsy):診斷血管炎幾乎總是必需。標準穿孔切片 (punch biopsy) 足以診斷小血管血管炎;皮下結節、網狀青斑、深部潰瘍需較深、較寬的切除 (excision)。建議切片臨床出現未滿 48 小時的病灶,以提高找到急性嗜中性球性血管炎特徵(纖維素樣壞死、紅血球外滲、白血球碎裂、免疫沉積物)的機會。需免疫螢光時需做 2 次切片或分割單一切片。
- 切片可確認血管炎但無法定病因。免疫螢光 IgA 優勢提示 IgA 血管炎;IgG/IgM/補體沉積提示免疫複合體媒介病因。
- 其他器官切片(腎、肺、肌肉、周邊神經)較皮膚切片更可能顯示診斷性病理。
實驗室與影像檢查
- 腎功能最重要:腎臟疾病常無症狀直到末期腎衰竭。所有疑似病人應做尿液分析(試紙+顯微鏡,找紅血球柱 red blood cell casts)與血清肌酸酐 (serum creatinine)。中血管血管炎(如 PAN)常為孤立性血尿或正常尿液分析。
- 全血球計數:常見貧血/血小板增多;EGPA 多數未治療病人絕對嗜酸性球升高,>1000 cells/µL 有助與氣喘/異位性體質區分。
- ANCA:約 90% MPA、75% GPA、40% EGPA 呈陽性。需以 ELISA 測 anti-PR3/anti-MPO(特異度高);單純 p-ANCA 染色而無 anti-MPO 特異度低。
- ANA:對狼瘡敏感(>95%)但不特異;ANA 陽性才續驗 dsDNA、Smith、RNP、La (SSB)。
- 補體 (C3, C4):冷凝球蛋白血症性血管炎 C4 嚴重耗竭、C3 較不耗竭或正常;類風濕性血管炎 70% 補體偏低;低補體提高對狼瘡懷疑。
- 感染篩檢:侵犯小/中型動脈者應篩 B、C 型肝炎;心內膜炎可造成真血管炎或敗血性栓子擬似病灶。
- 濫用藥物:古柯鹼 (cocaine)、甲基安非他命 (methamphetamines) 與皮膚血管炎/血管痙攣相關;左旋咪唑 (levamisole)(古柯鹼摻雜物)導致壞疽性病灶與 anti-MPO 及 anti-PR3 雙陽性,可尿液檢測確認。
- 影像:胸部 X 光為篩檢,有肺部症狀用 CT;確診 GPA/MPA/EGPA 即使無症狀也做篩檢 CT 分期。血管攝影對中、大血管血管炎(如 PAN 顯示多發性動脈瘤與狹窄)重要。
各型血管炎重點
- 顯微鏡下多血管炎 (MPA):侵犯小型、有時中型血管。多數有寡免疫性腎絲球腎炎 (pauci-immune glomerulonephritis)、肺出血、神經病變。多數 ANCA 陽性,多為 anti-MPO。最常見皮膚病灶為可觸性紫斑。
- 肉芽腫性多血管炎 (GPA):涵蓋 MPA 特徵加壞死性肉芽腫發炎。約 90% 有上呼吸道慢性發炎;空洞性肺結節、眼窩假性腫瘤、聲門下狹窄、眼血管炎(鞏膜炎)常見。多為 C-ANCA/anti-PR3。皮膚病灶含伸側丘疹、皮下結節、潰瘍。
- 嗜酸性球性肉芽腫性多血管炎 (EGPA):約 40% ANCA 陽性。獨特特徵為氣喘病史與血液嗜酸性球增多;鼻息肉常見;嗜酸性球性肺炎可與氣喘區分。重度血管炎最常表現為急性周邊神經病變。腎絲球腎炎、肺出血分別約 10%、<5%。皮膚病灶比 GPA/MPA 更常見,紫斑約占 50%。
- 與其他自體免疫疾病相關:狼瘡 10–36%、修格蘭氏症候群 10% 相對常見;類風濕性關節炎現已極罕見。
- 皮膚白血球碎裂性血管炎:組織學術語,以無系統性疾病證據定義;約 20% 繼發於感染、20% 與藥物暴露相關;應仔細追蹤以防演變為系統性。
- 藥物誘發性血管炎:約占皮膚小血管血管炎 20%;皮膚表現與其他病因無法區分。藥物誘發性 ANCA 相關血管炎(多為 anti-MPO)尤涉及丙基硫氧嘧啶 (propylthiouracil) 與聯胺嗪 (hydralazine)。
- 結節性多動脈炎 (PAN):中型(小/中肌肉性動脈)特發性血管炎。「典型」PAN 以皮膚疾病、肌痛、高血壓(腎動脈侵犯)、腹痛、神經病變、睪丸痛組合表現。歷史上多與慢性 B 型肝炎相關。最常見皮膚特徵:網狀青斑 (livedo reticularis/racemosa)、疼痛性結節或潰瘍、指(趾)缺血。
- 巨細胞動脈炎 (GCA,顳動脈炎):嚴格屬 >50 歲成人,多為北歐血統。顱動脈炎產生頭痛(70% 至 80%)、顎跛行(50%)、單眼失明(15%)。風濕性多肌痛見於至少 30% 至 40%。主動脈侵犯見於 15% 至 20%。顳動脈切片確診;顳動脈可觸性結節見於 30% 至 40%,為唯一可見皮膚表現。
- 高安動脈炎 (TAK):罕見(盛行率 <1:100,000),侵犯主動脈及主要分支;90% 為女性,多於年輕成人診斷。典型表現為肢體跛行;血管攝影診斷。
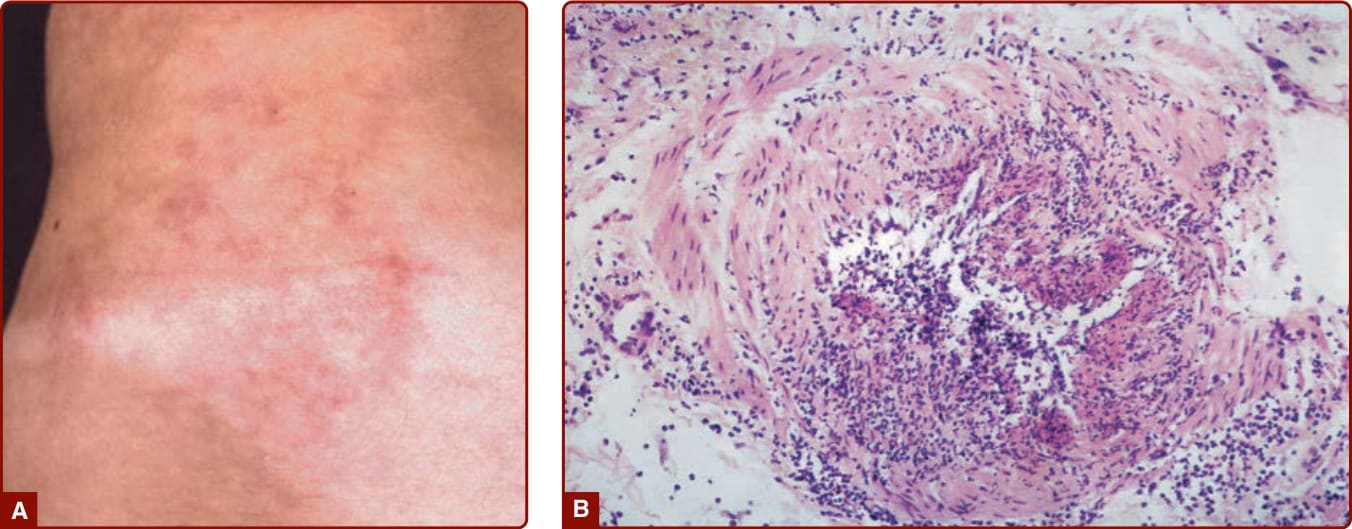
圖 139-4:結節性多動脈炎病人的「星爆狀」青斑;B 為節段性壞死性動脈炎組織病理。
臨床病程與預後 (prognosis)
- 所有具名疾病皆可能造成重要器官永久損害,但皆可治療,目標為損害發生前診斷與治療。
- 復發傾向:具 anti-PR3 的 GPA 無長期治療復發率遠超過 50%;具 anti-MPO 的 MPA、PAN、EGPA 皆低於 50%(EGPA 之慢性氣喘/鼻竇鼻部疾病多數會復發)。IgA 血管炎兒童多為單相,成人較易慢性。繼發於 C 型肝炎的冷凝球蛋白血症性血管炎通常隨病毒根除而治癒。
- 重度血管炎侵犯重要器官最初 3 至 6 個月死亡率約 10%(肺出血、腸穿孔、心肌病變或感染);之後大幅降低但仍略高於預期。死亡風險預測因子:高齡、先前器官損害(GPA/MPA 之腎損害、EGPA 之心臟損害)。
治療
- 分緩解誘導 (remission induction) 與緩解維持 (remission maintenance) 兩階段。誘導:高劑量糖皮質素並穩定遞減,加短療程(3-6 個月)速效強效免疫抑制藥物。維持:長期非環磷醯胺方案,使糖皮質素停用或維持低劑量(例如 ≤10 mg prednisone daily)。
- 評估嚴重度後處置:藥物暴露或活動性感染 → 停藥或治療感染;非重度 Cryo/CTD-V/EGPA/IgAV/PAN(常自限)→ 觀察或依症狀治療;非重度 GPA/MPA → 緩解誘導;重度 → 緩解誘導。
- 糖皮質素 (glucocorticoids):血管炎治療主力,作用迅速;部分疾病可單獨使用。急慢性毒性常被低估。
- 其他免疫抑制藥物:環磷醯胺 (cyclophosphamide) 為重度血管炎初始治療標準(毒性與總累積劑量相關:不孕、膀胱癌),故發展「節省環磷醯胺」方案轉換至甲胺喋呤 (methotrexate) 或硫唑嘌呤 (azathioprine);黴酚酸酯 (mycophenolate)、環孢素 A (cyclosporine A) 用於維持。阿普斯特 (apremilast) 對貝賽特氏病黏膜皮膚表現有效。
- 生物製劑:利妥昔單抗 (rituximab,B 細胞耗竭) 在 AAV 緩解誘導與環磷醯胺一樣有效;美泊利單抗 (mepolizumab,抗 IL-5) 對 EGPA 有效;托珠單抗 (tocilizumab,抗 IL-6) 與阿巴西普 (abatacept) 對 GCA 有效;抗 TNF 藥物對 GPA、GCA 效果令人失望。
- 其他:秋水仙素、dapsone 等證據不足;血漿置換 (plasma exchange) 角色仍有爭議,對 AAV 合併重度腎臟疾病可能有效。
- 常見錯誤:未切片即診斷皮膚血管炎;治療不足或延遲開始免疫抑制;中高劑量糖皮質素療程過長。
- 觀察等待 (watchful waiting) 合理時機:診斷不明且無主要器官受威脅;病因可逆(毒素/藥物)或自限(感染);症狀來源不確定。
- 緊急狀況(Table 139-2):威脅視力/單眼失明 (GCA, TAK)、肺泡出血、聲門下狹窄、惡性高血壓、心肌病變、壞疽、腸繫膜缺血、腎衰竭、中風、多發性單神經炎等。血管炎可在長期惰性後迅速加劇,需密切規律追蹤。