Systemic Necrotizing Arteritis
22
AT-A-GLANCE
■ Cutaneous vasculitis can be a presenting feature of a systemic disease affecting other organ systems. Thus, all patients with cutaneous vasculitis need to be evaluated for possible systemic vasculitis including all forms of small- and medium-vessel vasculitis.
■ Additional testing for vasculitis is guided by findings from medical history and physical examination, but should usually include routine laboratory testing of renal function, evaluating for hepatitis B and C viral infections, serologic testing for ANA and ANCA, selected radiographic imaging, and other tests as indicated by presentation.
■ Skin biopsy is nearly always essential to establish a diagnosis of vasculitis and biopsies can help determine the type of vasculitis.
■ Mimics of idiopathic vasculitis need to be considered, especially including infection, malignancy, thrombosis, or embolic disease.
■ Drug-induced vasculitis frequently presents with skin lesions. However, all patients with suspected drug-induced disease should still be evaluated for other possible causes or types of vasculitis.
■ Treatment of vasculitis that involves the skin is guided by the disease and the organ systems involved. For the systemic necrotizing vasculitides, glucocorticoids are always used, initially at high doses for severe disease. Additional immunosuppressive medications depend on the type of vasculitis, extent of disease, and comorbidities of the patient.
■ Treatment of suspected drug-induced vasculitis may include discontinuing the suspected etiologic agent and careful observation but could also involve treatment with glucocorticoids or other drugs.
■ Use of immunosuppressive medications should be overseen by physicians experienced with the use of this class of drugs.
■ Comprehensive clinical followup of all patients with vasculitis is an essential aspect of management.
INTRODUCTION
The term vasculitis can be defined broadly to mean inflammation of blood vessels. Although vasculitis sometimes affects a single organ, particularly the skin,
to most clinicians vasculitis connotes a group of diseases in which inflammation of the blood vessels is the major, but not the only, pathologic process. The systemic vasculitides are a wide-ranging set of diseases that are mostly idiopathic, rare, and multisystemic. These diseases involve such variety of clinical presentations and pathologies that all clinicians in every medical and surgical specialty will encounter such patients. Vasculitis of the skin is fairly common, and skin disease is a frequent manifestation in many forms of vasculitis, especially small- and medium-vessel arteritis where skin lesions may be the presenting symptom of a systemic illness. This chapter focuses on skin disease in the systemic vasculitides. Isolated forms of skin vasculitis and some of the systemic vasculitides are covered in Chap. 138. In addition to outlining the skin manifestations of vasculitis in general and for specific types of vasculitis, this chapter will provide an approach to patients with skin disease in which vasculitis is a diagnostic consideration. Multiple systems for classifying vasculitis exist, a situation that reflects a lack of clear understanding of the underlying pathophysiology and the overlap of clinical features among many types of vasculitis.1-6 The most commonly accepted approach is to sort them by the size(s) of the predominant vessel involved (small, medium, or large) and then subdivide or group diseases, as appropriate (Fig. 139-1). Classification criteria and definitions have been developed for many, but not all, specific types of vasculitis. These systems were designed for use in clinical research to create fairly homogenous study cohorts and were not meant to be used as “diagnostic” criteria.2 Nonetheless, clinicians will find these criteria helpful in their approach to the field. The most widely used criteria for vasculitis are from the classification criteria of the American College of Rheumatology4,5 and the disease definitions of the Chapel Hill Consensus Conference.6 Furthermore, some classes are no longer advised for use (eg, hypersensitivity vasculitis is a term that has lost specific meaning). It should be emphasized that the term leukocytoclastic vasculitis does not refer to a specific disease but is a pathologic description that often, but not always, applies to vasculitis in the skin or other organs. There are also some separate sets of criteria for pediatric patients.7
An important extension of the Chapel Hill Consensus Conference process is the recently completed Addendum that provides a comprehensive set of definitions for vasculitis of the skin.8
A new international initiative is under way to reconsider the classification of the vasculitides and take into consideration data regarding the clinical and pathophysiologic aspects of vasculitis not available when the
22
Classification of primary vasculitides
Predominantly small vessel
Predominantly medium vessel
Immune complex mediated:
Kawasaki PAN Other
Cryoglobulinemia HSP
Limited Skin
ANCA-associated MPA CSS GPA
Other
Predominantly large vessel
GCA TAK Isolated aortitis Other
No predominant size
Behçet Primary CNS Relapsing polychondiritis Cogan Other
prior systems were created; such new elements include testing for antineutrophil cytoplasmic autoantibodies (ANCA) and greater availability of advanced imaging techniques for large arterial disease.2
EPIDEMIOLOGY
With the probable exception of drug/toxin-induced vasculitis, all forms of idiopathic vasculitis are considered rare, “orphan” diseases in the United States (prevalences of less than 200,000 people); similar designations exist in Europe and elsewhere. Vasculitis occurs in people of both sexes, all ages, and all major racial/ethnic groups. However, some forms are more common in certain groups. For example, Takayasu arteritis is substantially more common in women than men, Kawasaki disease is almost exclusively a disease of young children, and giant cell arteritis is limited to older adults. Granulomatosis with polyangiitis (Wegener) mostly occurs in whites and Behçet disease is markedly more common in countries in the Eastern Mediterranean as well as Japan and Korea. The demographic differences among the vasculitides are of scientific interest as clues to etiology and can be helpful in developing a differential diagnosis.9 Not surprisingly, vasculitides in which there is strong regional variation have genetic risk factors in the HLA region.10-13 However, the epidemiologic tendencies, including genetics,
2540
are not so strong as to fully exclude the diagnosis of a specific form of vasculitis in any one person, and exceptions to the typical epidemiology occur regularly.
CLINICAL FEATURES
CUTANEOUS FINDINGS
CUTANEOUS FINDINGS
The appearance of cutaneous vasculitis reflects the size and depth of the involved vessels, the severity of inflammation and red blood cell extravasation, and the density of damaged vessels and resulting tissue destruction. For example, vasculitis caused by immune complexes in the dermal microvasculature often first appears as a tiny, bright red circle due to red blood cell extravasation, which becomes palpable once it reaches about 2 mm in diameter. A high density of such lesions, however, may produce areas of confluent purpura and/or central necrosis or ulceration. Vasculitis of small arteries (also referred to confusingly as “medium-vessel”), which lie deeper in the skin and subcutaneous tissues, produces nodules that are often erythematous but not as bright red as purpura, or livedo racemosa if the involved vessels are predominantly parallel to the surface. Ulceration and digit ischemia are more readily produced in medium-vessel than in small-vessel vasculitis.
The classic presentation of small-vessel vasculitis in the skin is palpable purpura, but other presentations are common. These include nonpalpable purpura that can be either round or have angular borders (retiform purpura), papules, urticarial lesions, nodules of different sizes and at different depths, bullous lesions, and ulcers (Figs. 139-3, 139-4, 139-6 to 139-12). Edema in and near visibly affected areas is common. Symptoms attributable to cutaneous vasculitis vary widely. Purpura or livedo may be asymptomatic or produce pain that is often described as stinging or burning but sometimes as itching. Nodules and ulcers are usually painful. Severe pain in a region not affected by nodules, ulcers, or confluent purpura should raise suspicion for neuropathy or inflammatory arthritis.
NONCUTANEOUS FINDINGS
NONCUTANEOUS FINDINGS
It is the presence or absence of particular features in the history and physical examination that give the first clue whether a patient has a disease involving multiple organ systems. A list of important signs and symptoms in different forms of vasculitis is shown in Table 139-1.
ETIOLOGY AND PATHOGENESIS
With the exception of infection-related vasculitis (eg, hepatitis B virus in many cases of polyarteritis nodosa and hepatitis C virus in most cases of cryoglobulinemic vasculitis) and drug-induced vasculitis, the etiology of most types of vasculitis is unknown. Similarly, the pathogenesis of the vasculitides remains an area of active investigation. This section summarizes the current thinking on causes and mechanisms of vasculitis with the caveat that although much of the evidence outlined in this section is strong, the pathogenesis of these diseases is almost certainly even more complex. Broadly speaking, the small-vessel vasculitides follow one of 2 pathways: (1) immune complex deposition or (2) non–immune complex mediated pathology that likely involves ANCA. Immune complexes consist of immunoglobulins, the antigens they are bound to, and complement components, and can form in a wide range of immune responses to microbes, autoantigens, or drugs. In a small percentage of antibody responses, the concentrations and properties of antibody and antigen favor deposition in the microvasculature, which may lead to activation of neutrophils and necrotizing vasculitis with destruction of endothelial cells. ANCA are directed against the neutrophil proteins myeloperoxidase (MPO) or proteinase-3 (PR3), which are primarily contained in intracellular granules but are also translocated to the cell surface or released via degranulation or excretion of neutrophil extracellular traps (NETs), allowing access to ANCA, activation of neutrophils, involvement of complement components, and necrotizing vasculitis. Genetics appear to contribute modestly to
22
ANCA-associated vasculitis, with the genes identified so far being associated with the specificity of the antibody to PR3 or MPO.10,11 Although the humoral immune system plays key roles in the pathogenesis of small-vessel vasculitis, there is evidence for the additional importance of cellular immune responses in these diseases. Large-vessel vasculitides, in contrast, are thought to be mediated by T cells activating macrophages in the walls of large arteries. In giant cell arteritis (GCA), both IL-17-secreting (Th17) and interferon-γ–secreting (Th1) cells are prominent. T cells enter the artery wall by the vasa vasorum, so the endothelium remains intact. Multiple studies implicating different infectious agents in GCA have not been reproducible, so it continues to be regarded as an autoimmune disease, as is Takayasu arteritis. Although these diseases have similar microscopic pathology, they have quite different epidemiology, moderately different vessel distributions, and different genetics. Genetics contributes modestly to both diseases via the HLA locus, but that is in the class II region in GCA and class I in Takayasu arteritis, with little overlap otherwise.12,13
The etiology and pathogenesis of polyarteritis nodosa (PAN) that is not associated with hepatitis B virus infection are unclear, including uncertainty about whether it is mediated by antibodies and whether it is a syndrome with many rare causes or has 1 or a few major causes. No genetic studies have been published; an HLA association would support the existence of a predominant target antigen but would not identify the target as an autoantigen or a microbe. Individual lesions may clearly show neutrophils with necrosis and destruction of the endothelium or may show a more mixed infiltrate with an intact endothelium, but it is uncertain whether the latter finding represents a different disease process or healing from a recent necrotizing lesion. This uncertainty is reflected in application of new nomenclature such as “macular arteritis” for the latter situation when it is limited to the skin, leading to the counterproposal that it be considered a subtype of “cutaneous PAN.”
DIAGNOSIS
DIAGNOSTIC ALGORITHM
DIAGNOSTIC ALGORITHM
When a patient presents with skin lesions that are concerning for possible vasculitis, answers to 3 questions should be sought quickly:
- Is the lesion due to vasculitis?
- Are other organ systems involved in the illness?
- Are there additional findings on medical interview, physical examination, laboratory testing, or radiographic imaging that can help establish a specific diagnosis?
If a diagnosis of vasculitis is obtained, then it is imperative to ask 2 more questions:
- Is it possible to make a diagnosis of a specific type of vasculitis for the patient?
- Does the patient need immediate treatment and/or hospitalization?
2541
22
ORGAN SYSTEM SIGNS AND SYMPTOMS DISEASE PROCESS TYPE OF VASCULITIS
General
Fever Systemic inflammation Many of the systemic vasculitides
Fatigue/malaise Systemic inflammation Most of the systemic vasculitides
Weight loss Systemic inflammation Most of the systemic vasculitides
Eye
Red eye Episcleritis, scleritis, uveitis, conjunctivitis GPA, MPA, RPC, BD, others
Acute visual loss or amaurosis fugax Arterial insufficiency GCA, TAK
Proptosis Orbital granuloma (“pseudotumor”) GPA
Tearing Dacryocystitis with lacrimal duct occlusion GPA, EGPA
Ear
Hearing loss Sensorineural hearing loss Conductive hearing loss GPA, MPA, EGPA, GCA GPA, EGPA, RPC
Ear pain and fullness Mastoiditis and/or other inflammation of upper airway, auditory tube, middle ear GPA, EGPA
External ear redness, tenderness, swelling Chondritis RPC, GPA, EGPA
Nose and Sinuses
Epistaxis, nasal crusting and discharge Nasal mucosal inflammation GPA, EGPA
Nasal bridge collapse (saddle nose deformity) Nasal cartilage inflammation GPA, RPC
Nasal polyps Eosinophilic nasal inflammation EGPA
Facial pain/tooth pain Sinusitis GPA, EGPA
Anosmia Olfactory epithelium/cells damage GPA, EGPA
Oral Cavity
Painful oral ulcers Aphthous ulcers BD, GPA
Gingival pain and swelling Gingival inflammation GPA
Jaw claudication Arterial insufficiency to muscles of mastication GCA
Pulmonary
Hemoptysis Alveolar hemorrhage Pulmonary artery rupture Pulmonary embolus Pulmonary nodules
GPA, MPA BD GPA, MPA, EGPA, BD GPA, EGPA
Dyspnea/cough (See causes of hemoptysis above) Pulmonary infiltrates Bronchitis/large airway collapse Pleuritis
GPA, MPA, EGPA GPA, RPC GPA, EGPA
Stridor/dyspnea Subglottic stenosis GPA, RPC
Wheezing Asthma Large airway collapse EGPA GPA, RPC
Cardiovascular
Angina Coronary arteritis Aortic root/valvular disease TAK TAK, GCA, BD, RPC
Congestive heart failure Myocarditis Aortic valve insufficiency EGPA, GPA TAK, GCA
Limb claudication Large artery stenosis TAK, GCA
GI
Abdominal ischemic pain Arterial insufficiency PAN, IGAV, GCA, TAK
Lower GI bleeding Mucosal ulcers or infarction IGAV, EGPA, GPA, MPA, PAN
2542
(Continued )
22
(Continued)
ORGAN SYSTEM SIGNS AND SYMPTOMS DISEASE PROCESS TYPE OF VASCULITIS
Renal
Gross hematuria Renal infarction Glomerulonephritis (rare cause of gross hematuria) PAN GPA, MPA, EGPA, IGAV, Cryo
CNS
Headache, scalp tenderness Cranial arteritis GCA, TAK
Lightheadedness/syncope Arterial insufficiency to brain GCA, TAK
Cranial neuropathy Inflammation of nerves; rarely mass lesion GCA, GPA, MPA
Peripheral Nervous System
Sensory/motor dysfunction Inflammation of nerves; rarely mass lesion EGPA, GPA, MPA, PAN, Cryo
Musculoskeletal
Polyarthralgia Polyarthritis GPA, GCA, TAK, Cryo, IGAV, BD
Shoulder and hip girdle pain Polymyalgia rheumatica GCA
Muscle weakness Myositis EGPA
Skin
Purpura Small-vessel vasculitis GPA, MPA, EGPA, PAN, IGAV, Cryo
Painful nodules, deep ulcers Medium-vessel vasculitis PAN, GPA, MPA, Cryo
Digital ischemia/gangrene Medium-large artery stenosis GCA, TAK, PAN, GPA, MPA, Cryo
Superficial nodules Granulomas GPA, EGPA
Papules, acnelike lesions Papulopustular lesions BD
Painful, red nodules Erythema nodosum BD, TAK
Peripheral edema Deep vein thrombosis GPA, MPA, EGPA, BD
Peripheral edema Deep vein thrombosis GPA, MPA, EGPA, BD
aThis list is not inclusive of all manifestations for all diseases. BD, Behçet disease; Cryo, cryoglobulinemic vasculitis; EGPA, eosinophilic granulomatosis with polyangiitis; GCA, giant cell arteritis; GPA, granulomatosis with polyangiitis; IgAV, IgA vasculitis (Henoch–Schönlein); MPA, microscopic polyangiitis; PAN, polyarteritis nodosa; RPC, relapsing polychondritis; TAK, Takayasu arteritis. RPC, an autoimmune disease defined by destruction of cartilage, is included in this table because several of its features overlap with those of GPA. As with BD, about 30% of patients with RPC also have vasculitis, with a wide range of vessel sizes involved.
A suggested approach to a patients with skin lesions suspected of being due to vasculitis is shown in Fig. 139-2. The answer to the first question is often obtained by skin biopsy, a procedure indicated in many cases of palpable purpura or other lesions when a diagnosis of vasculitis is not otherwise easily established. The second question is addressed by a thorough review of systems and physical examination and routine laboratory testing that can usually be completed rapidly. The third question is addressed by more specialized laboratory tests for which results typically take several days to return. It is important to quickly identify organ system involvement and how “sick” the patient is (or might soon be), because some causes of cutaneous vasculitis require no treatment but others require immediate hospitalization for initiation of immune-suppressive and supportive therapy. Given the broad range of entities that fall under the category “vasculitis,” and the even larger number of diseases that are also reasonably considered when evaluating a patient suspected of having vasculitis, a vast number and range of diagnostic tests are often considered in such cases. However, obtaining a thorough medical history and conducting a detailed physical examination should enable the clinician to limit the types of vasculitis under consideration and prioritize
the ordering of diagnostic tests. Not all tests need to be ordered for all patients suspected of having vasculitis. This approach must of course be balanced by a desire to be open-minded to atypical presentations of vasculitis as well as a range of infections, malignancies, and other diseases in the differential diagnosis of such patients. Evaluation for possible vasculitis usually occurs in parallel to evaluation for other processes.
REVIEW OF SYSTEMS
REVIEW OF SYSTEMS
A full review of systems with assessment of the overall severity of illness is the single most important component of the early evaluation of a patient suspected of having vasculitis. Together, the diseases that cause cutaneous vasculitis can affect all organ systems, and in most cases that involvement will cause symptoms, renal disease being a prominent exception. Although some symptoms are clearly more concerning than others (hemoptysis vs dry cough, painful red eye vs mild arthralgias), even relatively mild symptoms can be a clue that disease is not limited to the skin. A list of important signs and symptoms in different forms of vasculitis is shown in Table 139-1.
2543
22
Approach to patient with suspected vasculitis and skin lesions
Possible Cutaneous Small-Vessel Vasculitis High clinical suspicion: palpable purpura Moderate-low clinical suspicion: ulcers, nodules, vesicles, bullae
Assess severity/extent of disease Complete review of systems Review drug/toxin exposure Detailed physical examination Routine initial tests
- Complete blood count/differential
- Creatinine—urinalysis
- Liver function
- Chest imaging Consider alternative diagnoses
Diagnosis other than vasculitis established
Specific type of vasculitis established
Vasculitis not established but still a consideration
Skin biopsy
Obtain tissue for: Routine histopathology Immunofluorescence
Diagnosis of vasculitis established
Determine type and extent of vasculitis
Additional testing to determine type and extent of vasculitis (not all patients need all tests) Serologic tests
- ANCA
- Cryoglobulins
- SPEP (IFE), UPEP (UFE)
- Tests for hepatitis B and C viruses
- ANA, anti-Ro (SSA)
- Complement components (C3, C4)
Other diagnostic tests
- Chest CT, Sinus CT, angiogram audiogram, electromyogram Biopsies of other tissues
- Lung, kidney, sinus, other
Initiate treatment and close clinical followup
Speciatly consultation
- Dermatology, nephrology, neurology ophthalmology, otolaryngology, pulmonology, rheumatology
2544
MEDICAL HISTORY, MEDICATION USE, AND EXPOSURES TO TOXINS OR INFECTIOUS DISEASES
MEDICAL HISTORY,
MEDICATION USE, AND
EXPOSURES TO TOXINS OR
INFECTIOUS DISEASES
It is critical to know the full medical history of any patient suspected of having vasculitis. Other diseases may either have vasculitis as a component of the illness (eg, lupus) or may cause skin lesions that mimic vasculitis. Drug-induced vasculitis is common, and skin lesions, usually but not always purpura, are the most common manifestation of drug-induced vasculitis.14
The list of drugs reported to cause vasculitis is enormous, with almost every class of medication implicated in possible cases of drug-induced vasculitis. It is useful to ask about prescription, nonprescription, and “alternative” or herbal mediation use in the prior 6 to 12 months since the effect of some mediations may persist after usage ends. Patients should also be asked about use of illegal or recreational drugs because several such agents, including methamphetamines, cocaine, and others have been implicated in cases of vasculitis. Occupational or other exposure to nondrug toxins should be asked about. The patient should be asked about not only usual signs and symptoms of infection but also recent travel, sick contacts, and risks for sexually transmitted diseases.
PHYSICAL EXAMINATION
PHYSICAL EXAMINATION
Beyond a careful and full assessment of the skin, a multisystem examination is useful to determine whether symptoms are associated with objective abnormalities, or whether there are findings that a patient has not noticed. Vital signs are essential, but a patient with normal blood pressure can still have severe glomerulonephritis. The eyes should be inspected for redness and proptosis. The anterior nasal cavity can be easily visualized with an otoscope. Evidence of lymphadenopathy should be sought. Cardiac, lung, and abdominal examinations can give clues to underlying disease, but normal examinations do not rule out pathology. Similarly, absent pulses, asymmetric blood pressure readings, and bruits are helpful but imperfect measures to screen for large-vessel vasculitis. A complete joint examination is important and any findings suggestive of synovitis (joint swelling, warmth, redness) must be further investigated; however, many patients with vasculitis will have arthralgias without joint effusions. A full neurologic examination is one of the most valuable components of the evaluation and triage of a patient suspected of having vasculitis; subtle sensory and even motor abnormalities are often missed on initial evaluation. The more detailed and expert examinations that can be performed by ophthalmologists and
22
otolaryngologists are often extremely helpful in evaluating patients suspected of having vasculitis. Urgent referral is often indicated in patients with concerning symptoms such as new visual impairment, painful or red eyes, hoarseness or stridor, or hearing loss.
SUPPORTIVE STUDIES
SUPPORTIVE STUDIES
SKIN BIOPSY
The method for diagnosing vasculitis depends on the type of vasculitis suspected, which is often based on size of vessel involved. Vasculitides affecting the skin usually involve small and medium-sized vessels, and these vessels are amenable to biopsy (Fig. 139-3). Given the ease and low risk of skin biopsies, they play an important role in diagnosing vasculitis, and an equally important role in establishing a diagnosis other than vasculitis. A standard punch biopsy is sufficient to diagnose small-vessel vasculitis, but a deeper and wider excision may be necessary to capture information on medium-sized vessels.15,16 Lesions that should be approached with a deeper biopsy include subcutaneous nodules, livedo racemosa, or deep ulcers (Fig. 139-4). Many types of small-vessel vasculitis may also involve medium-sized skin vessels. It is important to realize that the difference between “small” and “medium” vessels is somewhat subjective, and skin pathologists often make such distinctions more than other pathologists who see larger biopsy specimens. Sometimes the presence of a typical clinical syndrome makes biopsy unnecessary. For example, IgA vasculitis (Henoch-Schönlein) in children is often diagnosed on clinical grounds alone and some cases of ANCA-associated or cryoglobulinemic vasculitis can be diagnosed confidently by combining clinical features with specific serologic tests. Behçet disease and Kawasaki disease are diagnosed based on the clinical syndromes; biopsy is usually not performed on the skin lesions that are common in these diseases, and such biopsies are often nondiagnostic. It is generally recommended to biopsy a skin lesion that has been clinically apparent for less than 48 hours, if possible, to maximize the chance of finding the typical features of acute neutrophilic vasculitis, including fibrinoid necrosis, extravasation of erythrocytes, extravasation of neutrophils with release of nuclear debris (leukocytoclasia), and the presence of immune deposits.15,16 Processing of tissue is different for conventional histopathology or immunofluorescence testing; if immunofluorescence is desired, then either 2 biopsies need to be performed or a single biopsy needs to be divided before processing. The latter approach may damage the tissue.15
As discussed throughout this chapter, the histologic finding of leukocytoclastic vasculitis is helpful in confirming the diagnosis of vasculitis but does nothing to establish an etiology from among the broad number of possibilities. Microscopy sometimes reveals features that are suggestive but not diagnostic of vasculitis,
2545
22
A B
C
such as leukocytoclasia without fibrinoid necrosis. The finding of a perivascular infiltrate, particularly if it consists predominantly of mononuclear cells but even if it is neutrophilic, is also nonspecific. Certain features, when seen in addition to leukocytoclastic vasculitis, are strongly suggestive of particular diseases, such as extravascular granulomas with geographic necrosis
A B
(granulomatosis with polyangiitis), or eosinophil-rich extravascular granulomas (eosinophilic granulomatosis with polyangiitis),17 but these features are seen in a minority of biopsies in these diseases. A predominance of IgA over IgG/IgM by immunofluorescence is suggestive but not diagnostic of IgA vasculitis (Henoch–Schönlein). The presence of
2546
A B
22
deposits of IgG, IgM, and/or complement is suggestive of one of several immune complex–mediated etiologies, including drug hypersensitivity, postinfectious vasculitis, cryoglobulinemia, and vasculitis secondary to systemic lupus erythematosus, Sjögren syndrome, or rheumatoid arthritis.17
OTHER BIOPSIES
Vasculitis is often diagnosed by biopsy of other organs, such as kidney, lung, muscle, or peripheral nerve or even from surgical specimens (Fig. 139-5). Kidney or lung biopsies are more likely than skin biopsies to show pathology diagnostic of a particular disease. Nonetheless, a skin biopsy establishing the diagnosis of vasculitis may preclude the need for more invasive biopsies.
LABORATORY TESTING
LABORATORY TESTING
Although individual laboratory tests on their own are almost never diagnostic for vasculitis, such tests are essential in the evaluation of a patient in whom cutaneous vasculitis is being considered. Laboratory testing may identify organ systems involved in the disease process, especially renal disease. Furthermore, in the proper setting, selected serologic tests may establish an etiology for vasculitis. However, serologic tests usually complement rather than substitute for biopsy, particularly in a patient with skin lesions that can be readily biopsied.
TESTS OF RENAL FUNCTION
Tests for renal disease are the most important to order in evaluating a patient suspected of having vasculitis because renal disease is common in many vasculitides and is rarely accompanied by signs or symptoms until end-stage renal failure occurs. Urinalysis, including both dipstick and microscopic examinations, should
be performed on all patients in whom vasculitis is suspected and repeated in patients in whom vasculitis of small or medium-sized vessels is established in another organ system. The presence of any blood on the routine dipstick tests needs to be followed by an examination for red blood cell casts by someone specifically trained to look for casts (many nephrologists, some rheumatologists, but few laboratory technicians in North America). Measurement of serum creatinine is critical to estimate the glomerular filtration rate (GFR). Small changes in creatinine, even within the normal range, may be early evidence of decline in GFR. Although small-vessel vasculitis affecting the glomeruli is expected to produce hematuria, usually accompanied by red blood cell casts and proteinuria, vasculitis affecting only medium-sized vessels (eg, polyarteritis nodosa) typically produces either isolated hematuria or a normal urinalysis. Urinalysis and serum creatinine are equally important tests and are complementary; neither alone is sufficient to exclude renal disease in vasculitis.
TESTS OF LIVER FUNCTION
Vasculitis, particularly polyarteritis nodosa, can involve the liver, but significant hepatic dysfunction is rare. Liver function tests are thus of limited value in diagnosing vasculitis, but they do provide a baseline against which future values can be compared if, as is often the case, potentially hepatotoxic drugs are to be used for treatment. Liver function tests can also provide an early hint at infection with hepatitis B or C viruses, both of which are associated with vasculitis, but do not substitute for serologic testing for these infections. Normal liver function tests do not rule out infectious hepatitis.
COMPLETE BLOOD COUNT
A complete blood count should be ordered on all patients suspected of having vasculitis. Many patients with active vasculitis have anemia and/or
2547
22
thrombocytosis, but the same is true of a wide range of inflammatory diseases. Severe anemia can be a clue to serious GI involvement from various forms of vasculitis. The white blood cell count and differential also can be clues to the presence of infection or hematologic malignancy. However, leukocytosis is usually nonspecific and is also commonly caused by use of glucocorticoids. An elevated absolute eosinophil is found in most untreated patients with eosinophilic granulomatosis with polyangiitis (Churg–Strauss), and a count greater than 1000 cells/µL helps differentiate this disease from asthma and atopy.
ACUTE PHASE REACTANTS
The erythrocyte sedimentation rate (ESR) and levels of C reactive protein (CRP) are elevated in many patients with vasculitis, but the diagnostic sensitivity and specificity of these tests are not particularly high. Thus, these tests are not particularly helpful in either establishing or excluding a diagnosis of vasculitis. Furthermore, the levels of ESR and CRP do not correlate well with stage or severity of disease. ESR and CRP are often elevated in conditions that mimic vasculitis in the skin, as well as in many serious systemic diseases, including infections and malignancies. Patients with active vasculitis can have normal ESR and CRP values, and patients may remain in clinical remission despite persistent elevation of these markers after treatment.
AUTOIMMUNE SEROLOGIES
Testing for autoantibodies is often a critical component of establishing the type of vasculitis present, but it is important to recognize that serologic testing on its own is never diagnostic and should never substitute for clinical judgment. Testing for ANCA and anti–glomerular basement membrane (GBM) antibodies, as well as antinuclear antibodies (ANA) to address the alternative possibility of systemic lupus erythematosus, is advised for any patient presenting with pulmonary hemorrhage and/ or acute renal insufficiency with an active urinary sediment. ANCA-associated vasculitis (AAV) and lupus can present with vasculitis of the skin, but anti-GBM disease does not, so the latter topic will not be discussed further.
ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES (ANCA)
Approximately 90% of patients with microscopic polyangiitis, 75% of patients with granulomatosis with polyangiitis, and 40% of patients with eosinophilic granulomatosis with polyangiitis will test positive for ANCA.18-21 Modern ANCA testing includes both immunofluorescence staining of neutrophils for the cytoplasmic (c-ANCA) or perinuclear (p-ANCA) patterns and enzyme-linked immunosorbent assays (ELISAs) for specific autoantigens (PR3 and MPO).21-23 Specificity of
2548
positive testing for anti-PR3 and anti-MPO antibodies for AAV is quite high,22-24 but specificity of p-ANCA staining in the absence of anti-MPO antibodies is low. Thus, positive tests for ANCA by ELISA are essential to consider ANCA testing positive for purposes of diagnosing vasculitis. The predictive value of positive ANCA testing depends on the setting. In cases of biopsy-proven vasculitis or clinical “surrogates” of a biopsy of vasculitis, such as diffuse alveolar hemorrhage or acute renal failure with an “active” urinary sediment, positive testing for anti-PR3/MPO ANCA is highly specific. In the setting of nonspecific constitutional and musculoskeletal symptoms, the positive predictive value of ANCA testing is lower.
ANTINUCLEAR ANTIBODIES (ANA)
Testing for ANA and related autoantibodies is useful when there is suspicion of systemic lupus or Sjögren syndrome. ANA testing is extremely sensitive (>95%) but not specific for the diagnosis of lupus. With the exception of anti-Ro (SSA) antibodies, additional tests for specific nuclear antigens, including doublestranded DNA, Smith, RNP, and La (SSB), should only be ordered if the ANA is positive and lupus is still under consideration. Only 80% of patients with Sjögren syndrome test positive for either rheumatoid factor, anti-Ro (SSA), or anti-La (SSB) antibodies, so negative tests do not rule out this diagnosis.
RHEUMATOID FACTOR (RF)
Testing for rheumatoid factor is rarely useful in establishing either the diagnosis or specific type of vasculitis. The sensitivity and specificity of rheumatoid factor for Sjögren syndrome or cryoglobulinemic vasculitis are low. Although at least 70% of patients with rheumatoid arthritis test positive for rheumatoid factor, the test is positive in more than 95% of patients with rheumatoid vasculitis.25 However, because rheumatoid vasculitis typically occurs in patients with longstanding, severe rheumatoid arthritis, such testing has little additive value.
PARAPROTEINS (ABNORMAL IMMUNOGLOBULINS, INCLUDING CRYOGLOBULINS)
Cryoglobulins are immune complexes (immunoglobulins and their target antigens) that precipitate in the cold and are associated with clinical syndromes in which vasculitis is a prominent component (see Chap. 144). Cryoglobulinemia most commonly results from chronic infection with hepatitis C virus, but rheumatoid arthritis, systemic lupus erythematosus, Sjögren syndrome, and hematologic malignancies are also all associated with cryoglobulinemia. Testing for cryoglobulins requires careful attention to specimen handling and processing since
incorrect practice at any one of several steps results in a high false-negative rate. Similarly, standard serum protein electrophoresis testing may not pick up some immunoglobulin clones, and immunofixation electrophoresis is a more comprehensive screen for clonal immunoglobulins. Vasculitis also has been associated with monoclonal gammopathies (myeloma, plasmacytoma, or lymphoma) in the absence of cryoglobulinemia.26
COMPLEMENT
Total hemolytic complement is measured using the CH50, but since this assay is cumbersome and suffers from variability between laboratories, measurement in serum of the complement proteins C3 and C4 is usually sufficient, and is useful for assessing patients with cutaneous vasculitis in several settings. In patients with cryoglobulinemic vasculitis, C4 levels are usually severely depleted whereas C3 levels are less depleted or even normal.27,28 One or both of these components are low in 70% of patients with rheumatoid vasculitis,25
which is useful because rheumatoid arthritis is generally not associated with low circulating complement. In contrast, because systemic lupus erythematosus is commonly associated with low complement in a variety of settings, low complement helps to raise the suspicion for lupus but is not specific for vasculitis in SLE. A subset of patients whose cutaneous vasculitis presents as urticaria (hence the term urticarial vasculitis), but who cannot be diagnosed with lupus or another underlying disease, have depletion of complement (Chap. 138).
SELECTED TESTING FOR INFECTIOUS DISEASES
Many infections can cause skin lesions that either include vasculitis or mimic vasculitis. Chronic infection with hepatitis C virus is strongly associated with cryoglobulinemic vasculitis, and it also can be associated with polyarteritis nodosa in the absence of cryoglobulins.29 Chronic hepatitis B virus infection was the cause of many cases of polyarteritis nodosa before the widespread adoption of vaccination programs.30
Thus, patients with known or suspected vasculitis affecting small or medium-sized arteries should be screened for hepatitis B and C infections. Endocarditis can cause both true vasculitis, presumably through deposition of immune complexes, and lesions that mimic vasculitis, through septic emboli. Blood cultures are appropriate for some patients suspected of small-vessel vasculitis. Interestingly, bacteremia can be a cause of a positive test for ANCA that is not associated with vasculitis.31
Numerous and diverse infections have been implicated in causing secondary vasculitis, usually of small vessels and limited to the skin. Testing for specific organisms should therefore be based on a history of exposure or a suspicious clinical syndrome (eg, sore throat or acute diarrhea).
22
Several uncommon infections directly infect and damage vascular endothelial cells and thus produce lesions that can either be regarded as vasculitis or as mimics of vasculitis; numerous organisms have been implicated, mostly in the form of case reports.
SCREENING FOR DRUGS OF ABUSE
SCREENING FOR DRUGS OF
ABUSE
Toxicology screens for commonly used drugs of abuse may be appropriate for some clinical situations where vasculitis is suspected. In particular, both cocaine and methamphetamines have been associated with cutaneous vasculitis and/or arterial vasospasm, and nasal inhalation of cocaine can produce destructive nasal disease as severe as that seen in ANCA-associated vasculitis, although certain clinical features may help distinguish the 2 causes.32,33 There is a now wellrecognized form of destructive vasculopathy/vasculitis associated with exposure to levamisole, an antihelminthic and immunomodulatory agent that is an adulterant of illegal cocaine, especially in North America. Levamisole can lead to characteristic gangrenous lesions as well as positive tests for both anti-MPO and anti-PR3 ANCA.34 Levamisole use can be confirmed by testing in urine.
IMAGING
IMAGING
CHEST IMAGING
A chest radiograph is an appropriate screening test for any patient suspected of having vasculitis. For a patient with pulmonary symptoms, computed tomography (CT) is usually indicated, because plain radiographs will frequently not detect small nodules or subtle but significant infiltrates. In patients diagnosed with granulomatosis with polyangiitis, microscopic polyangiitis, or eosinophilic granulomatosis with polyangiitis (Churg–Strauss), a screening CT is indicated for staging purposes and to establish a baseline, even in asymptomatic patients. If subglottic stenosis is suspected, CT of the neck/ trachea can be a helpful adjunct to direct laryngoscopy.
SINUS IMAGING
Sinus involvement is extremely common in granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, and the ability to evaluate the sinuses on physical examination is limited even for an otolaryngologist. CT of the sinuses can help assess the possibility of granulomatosis with polyangiitis or eosinophilic granulomatosis with polyangiitis and is useful in staging and restaging disease once one of those diagnoses is made and treatment is initiated. However, the CT appearance of sinus inflammation
2549
22
in these diseases does not allow discrimination from other causes of sinusitis, and patients with prior damage from vasculitis often have persistent abnormalities. Nasal inflammation is better assessed by physical examination than by CT.
ANGIOGRAPHY
Angiography has a central role in the diagnosis and management of large- and medium-vessel vasculitis. Conventional catheter-based dye angiography has the highest resolution but is an invasive procedure and still does not allow visualization of most small vessels. Angiography based on CT and magnetic resonance (MR) are increasingly replacing the use of catheterbased angiography.35
The role of angiography in the diagnosis of vasculitis of the skin is limited to either establishing the underlying type of vasculitis (eg, abdominal angiography demonstrating multiple aneurysms and stenoses in polyarteritis nodosa) or evaluating the arterial supply in patients with gangrene.
OTHER DIAGNOSTIC STUDIES
OTHER DIAGNOSTIC
STUDIES
NERVE CONDUCTION STUDIES AND ELECTROMYOGRAPHY
Nerve conduction studies should never replace a full neurologic examination, and most patients with neurologic manifestations of vasculitis do not need such testing. Thus, nerve conduction testing is not recommended for screening asymptomatic patients, but can be useful for providing objective evidence of neuropathy and for distinguishing between compressive (ie, mechanical) and non-compressive neuropathy, with the latter type including neuropathy due to vasculitis and many other medical causes. Electromyography (EMG) can establish the presence of myopathy, but not the cause. Nerve conduction studies are painful and require expertise not always readily available.
AUDIOLOGY TESTING
An audiogram is critical in diagnosing and distinguishing between conductive and/or sensorineural hearing loss. Hearing loss is a commonly missed manifestation of small-vessel vasculitis, including among elderly patients.36 Sensorineural hearing loss is a cranial neuropathy and may rapidly lead to irreversible hearing loss. Although an audiogram is not generally indicated for screening an asymptomatic patient, a baseline audiogram is advised for all patients with an established diagnosis of ANCA-associated vasculitis (granulomatosis with polyangiitis, microscopic polyangiitis, or eosinophilic granulomatosis with polyangiitis).
2550
SUMMARIES OF THE VASCULITIDES
MICROSCOPIC POLYANGIITIS
MICROSCOPIC POLYANGIITIS
Microscopic polyangiitis (MPA) is a multisystem vasculitis of small and also sometimes medium-sized vessels.37,38 Pauci-immune glomerulonephritis develops in the majority of patients, and pulmonary hemorrhage, peripheral and cranial neuropathy, musculoskeletal, and constitutional symptoms are also common; cardiac and GI involvement are less common. Most patients with microscopic polyangiitis are positive for ANCA, usually with specificity for antibodies to myeloperoxidase (MPO).39 Although rapidly progressive glomerulonephritis and/or diffuse alveolar hemorrhage commonly lead to diagnosis, microscopic polyangiitis often features a prolonged prodrome limited to musculoskeletal and constitutional symptoms.40
The skin is frequently involved in microscopic polyangiitis.20 The most common cutaneous lesion is palpable purpura and the pathology is indistinguishable from other types of leukocytoclastic vasculitis17,41
(Fig. 139-6). Vasculitis of medium-sized vessels, leading to digital ischemia, subcutaneous nodules, livedo reticularis, and deep ulcers, can rarely occur in microscopic polyangiitis.
GRANULOMATOSIS WITH POLYANGIITIS
GRANULOMATOSIS WITH
POLYANGIITIS
Granulomatosis with polyangiitis encompasses all the features of microscopic polyangiitis but also many additional manifestations caused by necrotizing granulomatous inflammation, and the 2 syndromes are currently considered distinct entities. Chronic inflammation of the upper airway (nasal cavity, sinuses, auditory tube, and middle ear) is present in about
90% of patients, often but not always as the initial manifestation.18,42,43 Cavitary pulmonary nodules, orbital pseudotumor, and subglottic stenosis are also common and important features. Vasculitis of the eye (scleritis and episcleritis) is also much more common in granulomatosis with polyangiitis than in microscopic polyangiitis.18,37,42-44 Most patients with granulomatosis with polyangiitis who have involvement of multiple organ systems will test positive for ANCA.44,45 The majority ANCA type in this disease is C-ANCA/anti- PR3; however, P-ANCA/anti-MPO is also not uncommon. Patients with disease seemingly restricted to the upper airway are only ANCA-positive in about 70% of cases, which can make diagnosis more challenging.21,44
In addition to palpable purpura and other presentations typical of small-vessel vasculitis, additional skin lesions occur in granulomatosis with polyangiitis and reflect a combination of vasculitis and necrotizing granulomatous disease, including neutrophilic and granulomatous dermatitis with papules (particularly on the extensor surfaces of the elbows), subcutaneous nodules, and ulcers17,46-49 (Figs. 139-7 and 139-8). Vasculitis and extravascular granulomatous disease are sometimes seen in the same biopsy, facilitating
A
22
diagnosis. As with MPA, manifestations attributable to involvement of medium-sized vessels also can be seen (Fig. 139-9).
EOSINOPHILIC GRANULOMATOSIS WITH POLYANGIITIS
EOSINOPHILIC
GRANULOMATOSIS WITH
POLYANGIITIS
Eosinophilic granulomatosis with polyangiitis is often included among the ANCA-associated vasculitides since approximately 40% of patients test positive for ANCA,50 and there is considerable overlap in features of eosinophilic granulomatosis with polyangiitis with both granulomatosis with polyangiitis and microscopic polyangiitis. However, eosinophilic granulomatosis with polyangiitis has unique features, the most prominent being a history of asthma (often severe or poorly controlled), and blood eosinophilia. Nasal polyps, constitutional symptoms, and rashes, all typical of atopy, are also common. The presence of pulmonary infiltrates on chest imaging (eosinophilic pneumonia) provides an important distinction from asthma. The most common presentation of severe vasculitis in eosinophilic granulomatosis with polyangiitis is acute peripheral neuropathy with involvement of the heart, GI tract, brain, and eyes less commonly. Glomerulonephritis and pulmonary hemorrhage occur in about 10% and less than 5%, respectively, of patients with eosinophilic granulomatosis with polyangiitis.19,51,52
Cutaneous disease is more common in eosinophilic granulomatosis with polyangiitis than in systemic granulomatous vasculitis or microscopic polyangiitis.19,51,52 Palpable and nonpalpable purpura, reflecting vasculitis histologically, comprise about 50% of cutaneous lesions.17 Eosinophilic dermatitis and granulomatous dermatitis rich in both neutrophils and eosinophils are also common and produce erythematous macules, papules, and nodules. As with granulomatosis with polyangiitis, small-vessel vasculitis and extravascular eosinophilic and/or granulomatous
B
2551
22
A B
disease are sometimes seen in the same biopsy. While the skin lesions of eosinophilic granulomatosis with polyangiitis often contain eosinophils, with or without vasculitis, eosinophil-rich skin biopsies are not unique to this disease and can be seen in other types of vasculitis.
CRYOGLOBULINEMIC VASCULITIS
CRYOGLOBULINEMIC
VASCULITIS
See Chap. 144 “Cryoglobulinemia and Cryofibrinogenemia.”
IGA VASCULITIS (HENOCH–SCHÖNLEIN)
IGA VASCULITIS
(HENOCH–SCHÖNLEIN)
See Chap. 138 “Cutaneous Necrotizing Venulitis.”
VASCULITIS ASSOCIATED WITH OTHER AUTOIMMUNE DISEASES
VASCULITIS ASSOCIATED
WITH OTHER AUTOIMMUNE
DISEASES
Vasculitis is seen relatively commonly in patients with systemic lupus (10% to 36%)53,54 and Sjögren syndrome (10%)55 but is now extremely uncommon in rheumatoid arthritis.56,57 Vasculitis in these diseases affects mostly small vessels, with some medium-vessel disease. The vasculitis may cause peripheral neuropathy, GI ischemia, and CNS disease, with other visceral involvement being rare. Inflammatory bowel disease and relapsing polychondritis58 also have been associated with vasculitis involving small, medium, or large vessels.
2552
MALIGNANCY-ASSOCIATED VASCULITIS
MALIGNANCY-ASSOCIATED
VASCULITIS
Vasculitis temporally associated with a solid malignancy has been described in many case reports and a few larger series.59 Diverse malignancies have been implicated. Small-vessel vasculitis of the skin appears to be most common, and reports of remission of vasculitis after surgical resection of the tumor (and no other therapy) are suggestive of a causal relationship. Hematologic malignancies with or without associated paraproteinemias can result in vasculitis.26 On the other hand, the comprehensive medical testing and imaging associated with evaluating a patient with possible vasculitis can lead to discovery of a malignancy in a patient with vasculitis, and the 2 diagnoses may be unrelated.
CUTANEOUS LEUKOCYTOCLASTIC ANGIITIS
CUTANEOUS
LEUKOCYTOCLASTIC
ANGIITIS
All of the diseases and syndromes described to this point are defined by a combination of clinical and laboratory parameters. Cutaneous leukocytoclastic angiitis, in contrast, is a histologic term defined by the absence of evidence of systemic disease. The term undoubtedly encompasses many etiologies, and appears in this list only to serve as a reminder that cutaneous vasculitis is not always a marker of systemic disease. If, and only if, a patient has biopsyproven vasculitis, has no evidence of involvement of other organ systems by vasculitis, and has no clinical of laboratory evidence to support a specific form of vasculitis or a coexisting autoimmune inflammatory disease, should the diagnosis of cutaneous
leukocytoclastic angiitis be tentatively made. This term appears in the nomenclature of the Chapel Hill Consensus Conference of 1994 to acknowledge that vasculitis limited to the skin is relatively common.7 It has been estimated that about 20% of such cases follow a wide range of infections, and another 20% are associated with drug exposure.17 However, it is quite problematic to label skin-only vasculitis as a separate disease entity, and all such patients should be followed carefully for the possible evolution to a more systemic form of vasculitis.
DRUG-INDUCED VASCULITIS
DRUG-INDUCED
VASCULITIS
Reactions to drugs have been implicated in about 20% of cases of cutaneous small-vessel vasculitis,15
but the exact frequency of drug-induced disease is hard to establish due to incomplete reporting of cases and difficulty in firmly establishing causality of any one agent. Most categories of drugs have been implicated as causing vasculitis, but the number of reports for a given drug may represent reporting bias rather than relative risk.14,60 Cutaneous small-vessel disease is the norm, but medium-vessel vasculitis and visceral involvement have been reported. The literature is undoubtedly biased toward severe cases, but there are numerous reports of serious or fatal internal organ involvement (particularly renal, pulmonary, and hepatic). There is also a now well-accepted subset of drug-induced ANCA-associated vasculitis (antibodies usually directed against myeloperoxidase), especially involving propylthiouracil and related agents, hydralazine, but also other drugs.61,62
The cutaneous presentation of drug-induced vasculitis is indistinguishable from other causes of smallvessel vasculitis (ie, usually purpura but sometimes with other lesions) (Fig. 139-10). It is essential that a comprehensive review of all prescription, nonprescription, illegal, and “alternative”
22
drugs and supplements be undertaken for all patients suspected of having vasculitis. Establishing a diagnosis of drug-induced vasculitis may lead to avoidance of treatment with glucocorticoids and immunosuppressive drugs in favor of clinical followup after drug discontinuation. However, 2 important caveats must be kept in mind in such cases: (1) drug discontinuation alone may not resolve the disease and severe disease may be present necessitating treatment, and (2) the diagnosis of drug-induced vasculitis may be wrong and the patient really has another form of vasculitis; thus, careful and prolonged followup of all patients is required.
BEHÇET DISEASE
BEHÇET DISEASE
See Chap. 141 “Adamantiades-Behçet Disease.”
POLYARTERITIS NODOSA
POLYARTERITIS NODOSA
Idiopathic vasculitis of medium-sized vessels (ie, small or medium-sized muscular arteries) can present in one or multiple organ systems. “Classic” polyarteritis nodosa involves multiple organ systems and presents with some combination of skin disease, myalgia, hypertension (from renal artery involvement), abdominal pain, neuropathy, and/or testicular pain. However, many patients do not present with the full set of manifestations, and disease limited to muscle and nerve, single internal organs, or to the skin are well-described variants. When the disease is limited to the skin, it is sometimes referred to as cutaneous polyarteritis nodosa; however, some of these patients will later manifest disease in other organs. Interpreting the literature on polyarteritis nodosa is problematic since the term was formerly used to describe several forms of vasculitis now considered to be distinct diseases (particularly microscopic polyangiitis).63,64 Historically, many cases of polyarteritis nodosa were associated with chronic infection with hepatitis B, but that association has declined markedly in countries in which vaccination against hepatitis B has become routine.64 Whether the remaining, rare entity still known as polyarteritis nodosa represents one, a few, or many etiologies, or even whether there is a unified fundamental pathophysiology (eg, immune complex disease), is unclear. The most common cutaneous features of polyarteritis nodosa are livedo reticularis/racemosa (a lacy pattern of cutaneous blood vessels on the legs, and not always easy to distinguish from a benign consequence of vasoconstriction of more superficial vessels) (Fig. 139-11), painful cutaneous nodules or ulcers (Fig. 139-12), and digital ischemia.65,66 These manifestations reflect vasculitis of “medium-sized” arteries in the subcutis, which are frequently too deep to be sampled in a routine punch biopsy.15
2553
22
KAWASAKI DISEASE
KAWASAKI DISEASE
See Chap. 142 “Kawasaki Disease.”
PRIMARY VASCULITIS OF THE CNS
PRIMARY VASCULITIS OF
THE CNS
This rare disease is limited to the CNS (hence, no cutaneous findings) and presents with symptoms of
2554
encephalopathy, multiple small strokes, and often headache.67,68
GIANT CELL ARTERITIS
GIANT CELL ARTERITIS
Giant cell arteritis (temporal arteritis) is currently considered strictly a disease of adults older than 50 years that is markedly more common with advancing age. It is mostly a disease of people of Northern European ancestry. Cranial arteritis is a common feature with this term indicating stenosis or occlusion of 1 or more branches of the carotid artery to produce headache (70% to 80%), jaw claudication (50%), and monocular (and rarely binocular) blindness (15%). Polymyalgia rheumatica, which includes pain and stiffness of the shoulder and hip girdles, is seen in at least 30% to 40% of patients and may occur without cranial disease. Constitutional symptoms are common, including fever, malaise, and weight loss. Involvement of the aorta and its major branches produces symptoms similar to Takayasu arteritis and is present in 15% to 20% of patients. Diagnosis of GCA is usually confirmed by temporal artery biopsy (Fig. 139-13). Palpable nodularity of the temporal artery is present in 30% to 40% of cases and is the only cutaneous manifestation of GCA apparent on examination. Scalp necrosis and digital ischemia are rare complications.69,70
TAKAYASU ARTERITIS
TAKAYASU ARTERITIS
Takayasu is a rare form of vasculitis (prevalence <1:100,000) involving the aorta and its major branches.71-76 Many patients are diagnosed as young adults, and 90% are female. The typical presentation of Takayasu is limb claudication. Dizziness, constitutional symptoms, and severe hypertension (from renal artery stenosis) are also common. Cerebral infarction can occur. Coronary occlusions with angina or infarction and bowel ischemia are less common but lifethreatening complications. Absent pulse(s), abnormal blood pressure readings, and arterial bruits are common but not universal findings. Diagnosis is made by angiography. Lesions resembling erythema nodosum or pyoderma gangrenosum77 have been described repeatedly in Takayasu, with pathology of nodular lesions often, but not always, showing vasculitis and thereby differing from typical erythema nodosum.78,79 Although complete occlusion of subclavian arteries is common, digital ischemia is rare.
CLINICAL COURSE AND PROGNOSIS
The severity of vasculitis varies widely, but all of the named diseases have the potential to cause permanent damage to vital organs. All forms of vasculitis
22
A B
are treatable, and the goal is to make the diagnosis and start treatment before damage has occurred. After successful treatment with immune-suppressive drugs, some forms of vasculitis are more likely to recur than others. Granulomatosis with polyangiitis with anti- PR3 antibodies recurs in well over 50% of cases in the absence of long-term treatment, whereas microscopic polyangiitis with anti-MPO antibodies recurs in fewer than 50%. Polyarteritis nodosa and eosinophilic granulomatosis with polyangiitis also recur in fewer than 50% of patients, although in eosinophilic granulomatosis with polyangiitis, chronic asthma and/or sinonasal disease recur in most patients and require long-term treatment. IgA vasculitis is usually monophasic or resolves after a few episodes in children but is more likely to be chronic in adults. Cryoglobulinemic vasculitis secondary to hepatitis C virus is usually, but not always, cured by eradication of the virus, whereas it usually recurs in the absence of eradication. Vasculitis associated with connective tissue diseases can be monophasic, episodic, or chronic and varies widely in severity. Treatment is also a source of morbidity. Glucocorticoids are used in almost all forms of severe vasculitis and carry numerous risks at high doses or with longterm use even at moderate or low doses. Almost all other medications used to treat vasculitis increase risk of infection but are considered preferable to glucocorticoids on those grounds and others. Mortality in the first 3 to 6 months of severe vasculitis affecting vital organs is around 10%, because of either complications of vasculitis (eg, pulmonary hemorrhage, bowel perforation, or cardiomyopathy) or to infection. After that, as treatment is reduced and vasculitis usually remains in control, mortality is reduced
dramatically but appears to remain somewhat higher than expected.80 Other than advanced age, the predictors of higher risk of mortality reflect prior organ damage: kidney damage in the case of granulomatosis with polyangiitis and microscopic polyangiitis,81 and cardiac damage in the case of eosinophilic granulomatosis with polyangiitis.82
MANAGEMENT
GENERAL APPROACH TO TREATMENT OF SYSTEMIC VASCULITIS
GENERAL APPROACH TO
TREATMENT OF SYSTEMIC
VASCULITIS
When establishing a treatment regimen for a patient with vasculitis, consideration must be given to both the severity of the current presentation and the likelihood of disease progression and recurrence. An overview is presented in Fig. 139-14. For an increasing number of vasculitides, treatment is guided by results of relatively large randomized controlled trials. However, for many situations, clinicians still rely upon either extrapolation from trials in other diseases or empiric treatment based on small case series or personal experience. For vasculitides expected to have extended courses and/or to include severe manifestations, the general approach is to plan for 2 phases of treatment: remission induction and remission maintenance.83,84 Remission induction usually involves use of high-dose glucocorticoids with steady dose taper combined with a short course (3-6 months) of a fairly rapid-acting and potent immunosuppressive agent. For example,
2555
22
General guidelines for treatment of small- and medium-vessel vasculitis
Diagnosis of vasculitis
Yes
Discontinue drug or treat infection
Drug exposure or active infection?
No
Assessment of severity
Nonsevere Severe
Nonsevere Cryo, CTD-V, EGPA, IgAV, PAN (often self-limited), or persistent drug-induced vasculitis Observe or treat based on symptom relief
Nonsevere GPA, MPA (but likely to become severe if not treated)
Remission Induction
Severe CTD-V, EGPA, IgAV, PAN, Severe Cryo, GPA, MPA
GC + MTX GC + CYC GC + [CYC or RTX] +/– PLEX
GC
Remission Maintenance Careful observation alone +/– GC (often low-dose)
AZA, MMF, MTX, or RTX +/– GC (often low-dose)
cyclophosphamide, rituximab, and methotrexate all have roles in remission induction for ANCAassociated vasculitis. Remission maintenance usually involves prolonged use of a non–cyclophosphamidebased regimen to allow for glucocorticoids to be either fully discontinued or maintained at a low dose (eg, ≤10 mg prednisone daily). However, although the above approach is usual for the ANCA-associated vasculitides, polyarteritis nodosa, and some other forms of severe systemic disease for which there are data from clinical trials to guide treatment, it is less commonly used for other vasculitides in which glucocorticoids alone may suffice as therapy.
2556
Clinicians must always remain alert to the possibilities of (1) a different diagnosis (either a different form of vasculitis or an entirely different disease); (2) development of additional manifestations of vasculitis; and (3) treatment-related side effects, some of which can mimic vasculitis (infections, skin reactions, etc). An important aspect of caring for patients with vasculitis is determining who and when not to treat. “Watchful waiting” may be a reasonable management approach when (1) a diagnosis is not clear and no major organ system appears threatened; (2) there is an obvious cause or etiology for the vasculitis that is either reversible (toxin/drug) or self-limited
(some infections); (3) uncertainly exists as to whether the patient’s symptoms are due to a flare of disease or the result of either chronic damage or another process. A critical component to the care of patients with vasculitis is close, regular clinical followup. Vasculitis often progresses rapidly, and most forms of vasculitis have high rates of relapse. Depending on the form of vasculitis, regular office visits should be accompanied by laboratory and radiographic monitoring. Close followup should occur not only at the start of the disease process, but also for years following diagnosis.
TREATMENTS FOR SYSTEMIC VASCULITIS
TREATMENTS FOR
SYSTEMIC VASCULITIS
A comprehensive list of drugs used in the treatment of the various systemic vasculitides and details of treatment regimens is beyond the scope of this chapter (additional information about specific agents is available in Chaps. 184, “Glucocorticoids,” 190, “Cytotoxic and Antimetabolic Agents,” 192, “Immunosuppressive and Immunomodulatory Drugs,” and 193, “Immunobiologicals: Targeted Therapy Against Cytokines, Cytokine Receptors, and Growth Factors in Dermatology”).
GLUCOCORTICOIDS
GLUCOCORTICOIDS
Glucocorticoids remain the mainstay of therapy for vasculitis given the rapidity of action, reliability of response, and physicians’ familiarity with dosing and side effects. Glucocorticoids are usually the initial drug used to treat vasculitis and may be the only agent used for some forms of the disease. However, additional agents are often prescribed because either the dose of glucocorticoids needed to maintain disease control is unacceptably high or disease control is not attained by glucocorticoids alone. The acute and chronic toxicities are often underappreciated and the potential cumulative damage from chronic or recurrent use of glucocorticoids may be substantial.
OTHER IMMUNOSUPPRESSIVE AGENTS
OTHER
IMMUNOSUPPRESSIVE
AGENTS
A wide variety of additional immunosuppressive agents are used for treatment of the vasculitides. The proven effectiveness of the alkylating agent cyclophosphamide for the ANCA-associated vasculitides and, to a lesser extent, other forms of vasculitis, have helped establish this drug as the standard of care for initial treatment of severe forms of vasculitis.42,85-87
Other alkylating agents are now rarely prescribed for
22
vasculitis. Although effective in many, but certainly not all, cases, cyclophosphamide is also associated with serious toxicities, many of which are related to total cumulative dose (eg, female and male infertility, bladder cancer). Therefore, the past 3 decades have seen the emergence of “cyclophosphamide-sparing” regimens that usually have patients transition from an initial course of treatment with cyclophosphamide to a more prolonged course of a less toxic immunosuppressive agent, especially either methotrexate88,89
or azathioprine.86,90 Mycophenolate, cyclosporine A, and other agents also have been used for maintenance therapy. Apremilast has now been demonstrated to have efficacy in the treatment of mucocutaneous manifestations of Behçet disease.91,92
BIOLOGIC AGENTS
BIOLOGIC AGENTS
More recently, “biologic” agents, drugs created using recombinant DNA techniques to target specific components of the immune system, have been studied for use in treating vasculitis, often with a goal of being either cyclophosphamide-sparing or glucocorticoidsparing. The recent demonstration that rituximab, a B cell–depleting therapy, is as effective as cyclophosphamide for induction of remission in AAV is considered a major advance for the field.93-95 Mepolizumab, a monoclonal antibody to anti-interleukin 5, has been demonstrated to have efficacy in the treatment of eosinophilic granulomatosis with polyangiitis.96 Two trials have demonstrated the efficacy of tocilizumab, a monoclonal antibody to interleukin 6, for the treatment of giant cell arteritis.97,98 Promising data on the use of abatacept, a CTLA-4 immunoglobin, to treat giant cell arteritis also has been published.99 However, studies examining the efficacy of anti-TNF agents for either granulomatosis with polyangiitis or giant cell arteritis have been highly disappointing.100,101
There are many new biologic agents under consideration for use in the treatment of vasculitis, and clinical trials are needed to properly evaluate these new treatments.
OTHER TREATMENTS
OTHER TREATMENTS
Although a variety of other drug classes, including colchicine, antibiotics (dapsone and others), and “alternative” therapies have been promoted for treatment of various forms of vasculitis, good evidence is generally lacking for the efficacy of these agents. The role of plasma exchange in the treatment of vasculitis remains controversial. There is some evidence for efficacy of plasma exchange in patients with AAV and severe renal disease102 and possibly forms of cryoglobulinemic vasculitis. A large international multicenter, but randomized, trial of plasma exchange for AAV is nearing completion.
2557
22
COMMON ERRORS IN THE DIAGNOSIS AND TREATMENT OF SYSTEMIC VASCULITIS
COMMON ERRORS IN
THE DIAGNOSIS AND
TREATMENT OF SYSTEMIC
VASCULITIS
Given the huge spectrum of disease manifestations in vasculitis, the potential toxicities of treatments for vasculitis, and the serious ramifications of missing alternative diagnoses, especially infections, diagnostic misclassification is a major concern. Establishing a firm diagnosis of vasculitis based on physical examination and laboratory findings alone is a common problem. This is especially true for skin disease since not all purpura is due to vasculitis and not all skin disease in vasculitis is purpuric. Clinicians are cautioned against making a diagnosis of vasculitis in the skin without a biopsy; the exception to this guideline is if the patient has a clearly established diagnosis of vasculitis based on other evidence, but even then, it may be necessary to know what is causing a skin lesion. Many types of skin lesions improve with treatment with glucocorticoids so response to empiric use of this treatment is not diagnostically useful. Undertreatment or delayed initiation of immunosuppressive therapy is a common problem for patients with systemic vasculitis. Undertreatment may take the form of failure to recognize multiorgan system disease, delay or reluctance to initiate immunosuppressive medications other than glucocorticoids for those forms of vasculitis for which such therapy has been shown to be useful, or underdosing of immunosuppressive agents. Another common mistake is extending the course of treatment with medium-high doses of glucocorticoids beyond what is necessary to control an acute flare of vasculitis or the more serious manifestations.
MEDICAL EMERGENCIES RELATED TO THE PRIMARY VASCULITIDES
MEDICAL EMERGENCIES
RELATED TO THE PRIMARY
VASCULITIDES
The systemic vasculitides vary greatly in their rate of progression and level of medical severity. However, it is imperative that all clinicians caring for patients with vasculitis understand that these diseases are often organ- and life-threatening and that progression to emergency situations may be rapid. Furthermore, vasculitis can accelerate rapidly even after a long period of slowly changing or even indolent disease. Table 139-2 outlines several of the most common situations in which patients require emergency care but this list is incomplete and clinicians must be vigilant about considering these and other potentially rapidly progressive problems.
2558
EMERGENCY MANIFESTATIONS OF VASCULITIS TYPE OF VASCULITIS
Ophthalmologic
Threatened vision; monocular blindness
GCA, TAK
■Scleritis
GPA, MPA, RPC BD
■Uveitis
Pulmonary
GPA, RPC GPA, MPA, EGPA GPA, MPA, EGPA GPA, MPA, EGPA, BD
Subglottic stenosis
■Alveolar hemorrhage
■Diffuse inflammatory infiltrates
■Pulmonary embolus
Cardiovascular
TAK, GCA, PAN
Malignant hypertension—Renal arterial stenoses
■Rapid-onset cardiomyopathy/ heart failure
EGPA
■Angina/myocardial infarction
TAK, GCA GCA, TAK, PAN, GPA, MPA PAN, BD, GCA, TAK
■Gangrene—digital ischemia
■Aneurysm dissection (aorta or branches)
GI
Mesenteric ischemia PAN, IgAV, GCA, TAK
Renal
Glomerulonephritis/rising creatinine/renal failure GPA, MPA, EGPA, IgAV, PAN, Cryo
Neurologic
GCA GCA, TAK GPA, MPA, EGPA, BD, GCA, TAK GPA, MPA
Headache
■Syncope
■Stroke
■Sensorineural hearing loss or other cranial neuropathies
■Mononeuritis multiplex
EGPA, PAN, GPA, MPA
Other Organ-Threatening Disease Any systemic vasculitis
Other Organ-Threatening Disease Any systemic vasculitis
BD, Behçet disease; Cryo, cryoglobulinemic vasculitis; EGPA, eosinophilic granulomatosis with polyangiitis; GCA, giant cell arteritis; GPA, granulomatosis with polyangiitis; IgAV, IgA vasculitis (Henoch–Schönlein); MPA, microscopic polyangiitis; PAN, polyarteritis nodosa; RPC, relapsing polychondritis; TAK, Takayasu arteritis.
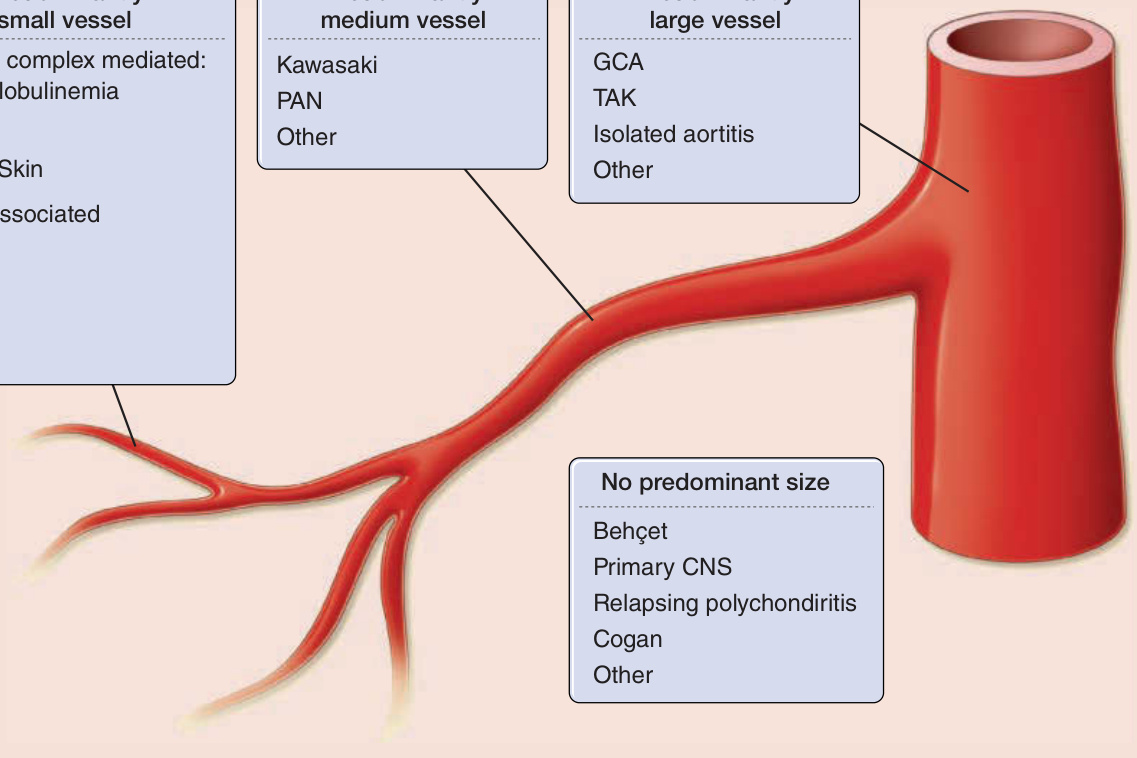
Figure 139-1 Classification of the primary vasculitides. ANCA, antineutrophil cytoplasmic antibodies; CSS, Churg–Strauss syndrome; GCA, giant cell arteritis; GPA, granulomatosis with polyangiitis (Wegener); HSP, Henoch–Schönlein purpura; MPA, microscopic polyangiitis; PAN, polyarteritis nodosa; TAK, Takayasu arteritis. (Redrawn from Watts RA et al: Systemic vasculitis—Is it time to reclassify? Rheumatology (Oxford). 2011;50(4):643-645.)
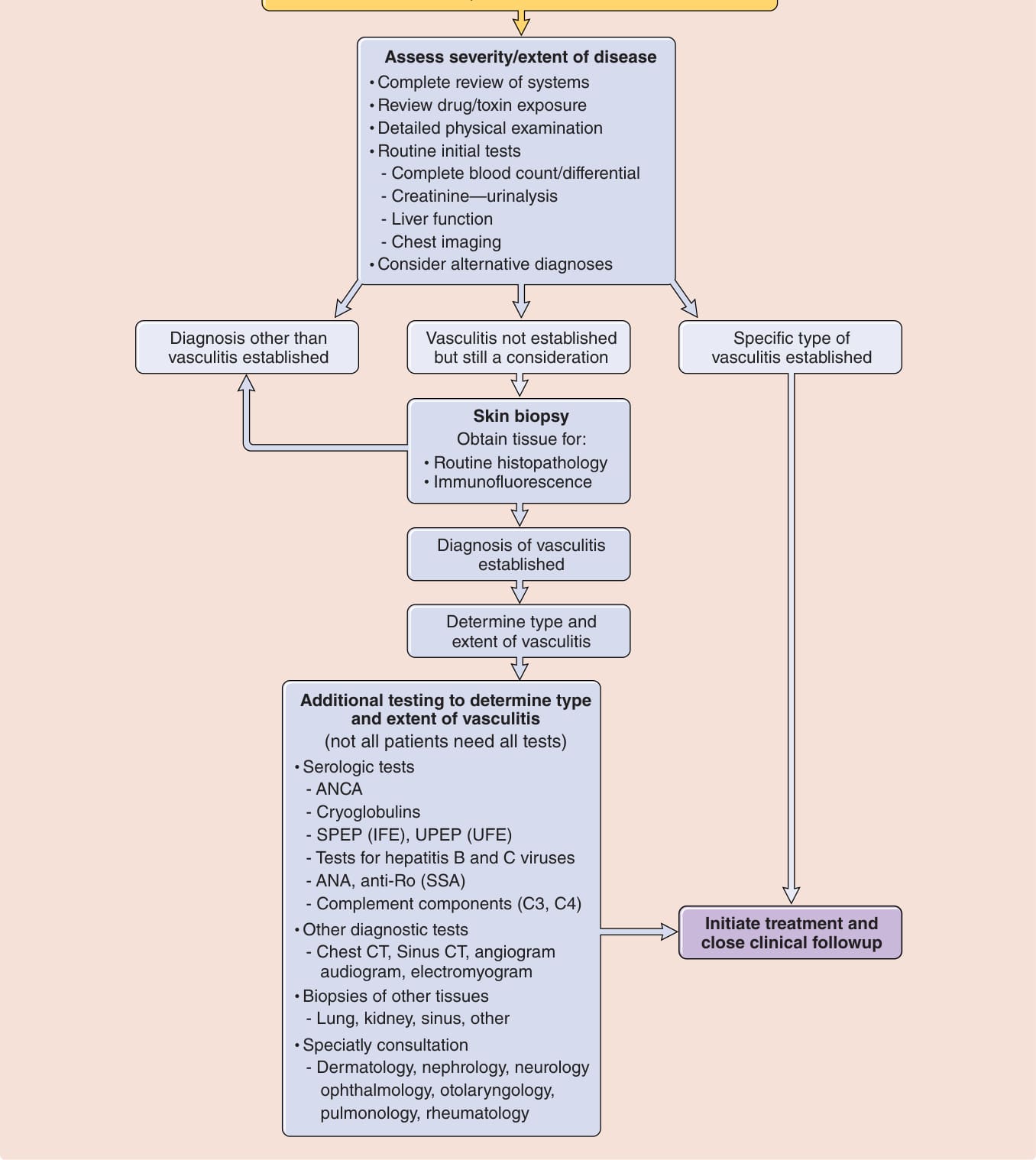
Figure 139-2 Approach to the patient with suspected vasculitis and skin lesions. ANCA, antineutrophil cytoplasmic antibodies; ANA, antinuclear antibodies; CT, computed tomography; IFE, immunofixation electrophoresis; SPEP, serum protein electrophoresis; UPEP, urine protein electrophoresis.
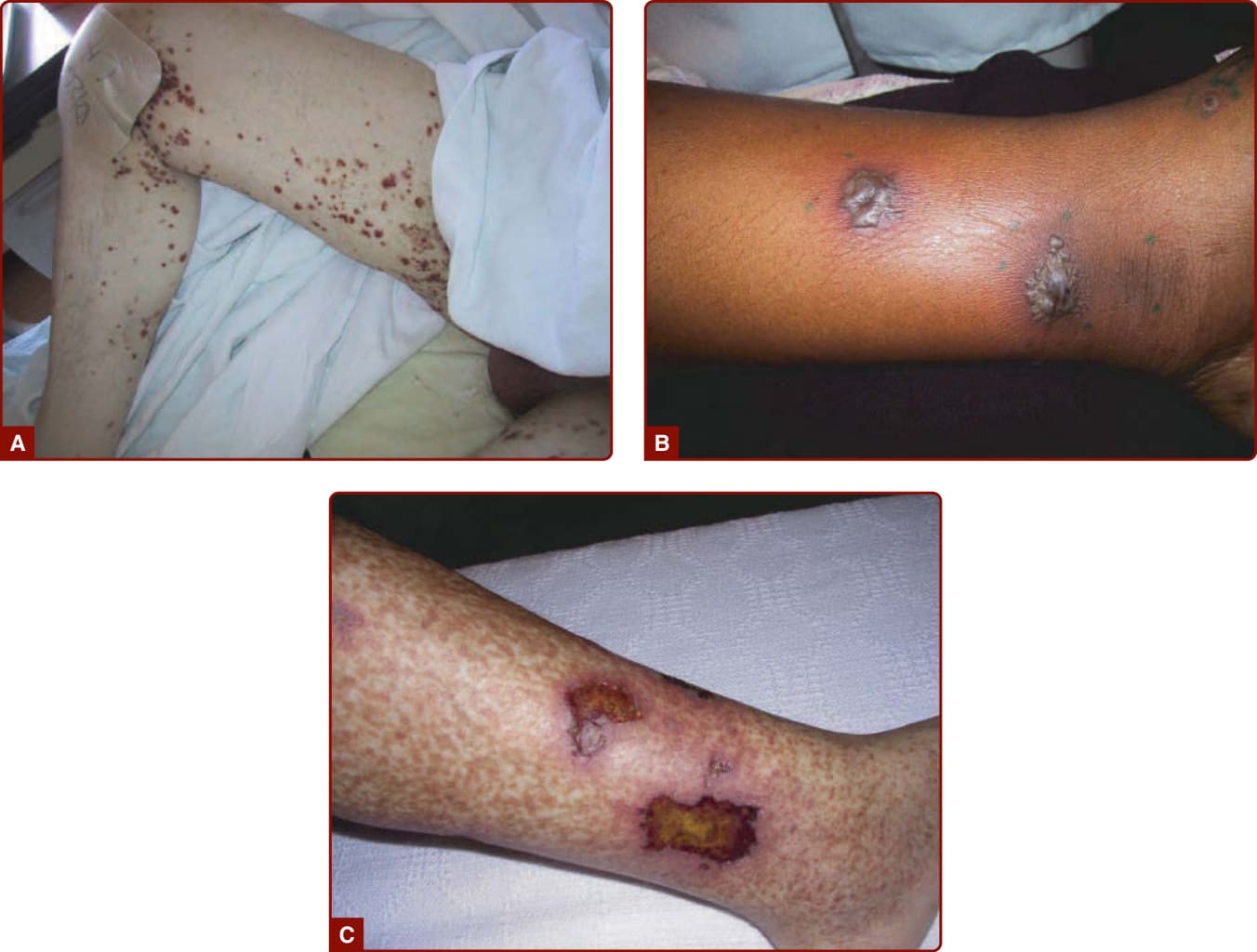
Figure 139-3 Different skin manifestations of systemic vasculitis. A, Purpura. B, Bullae. C, Ulcer.
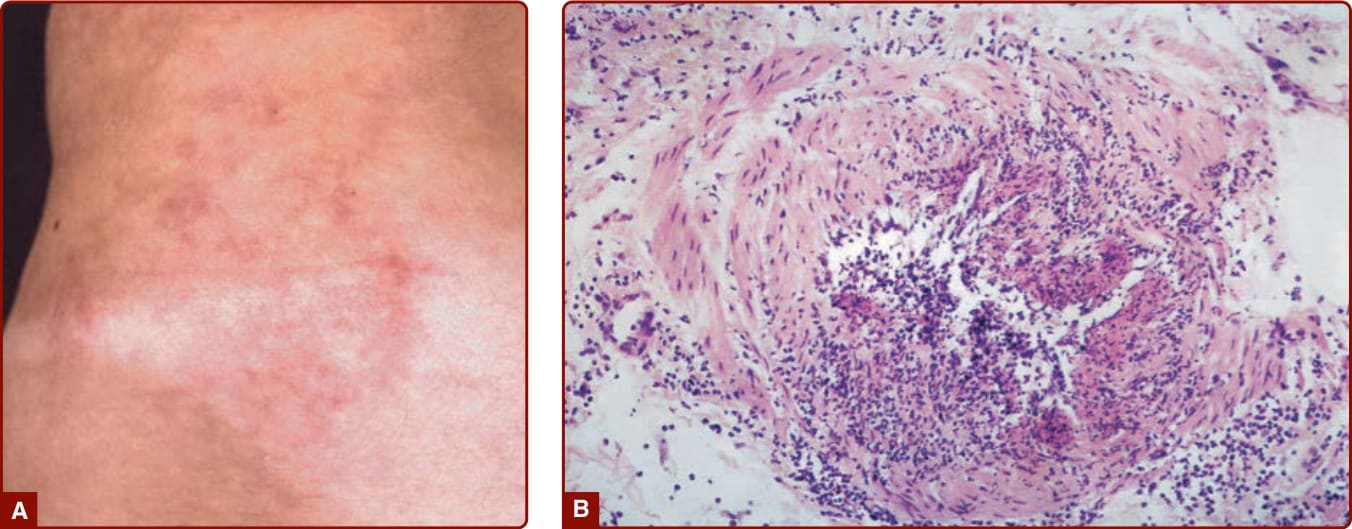
Figure 139-4 A, A patient with polyarteritis nodosa with “starburst” livedo made up of a cluster of nodular lesions. B, Histopathology of skin lesions in polyarteritis nodosa showing segmental necrotizing arteritis.
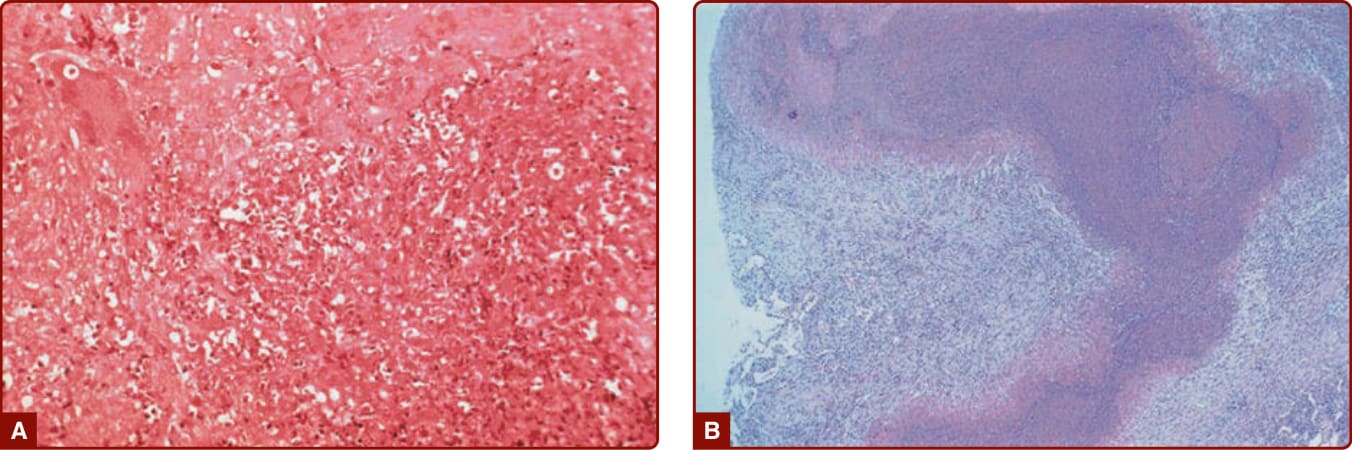
Figure 139-5 A, Lung histopathology from a patient with granulomatosis with polyangiitis (Wegener) demonstrating necrosis, giant cells, and mixed cellular inflammation. B, “Geographic necrosis” in a low-power view of an open lung biopsy specimen from a patient with granulomatosis with polyangiitis (Wegener).
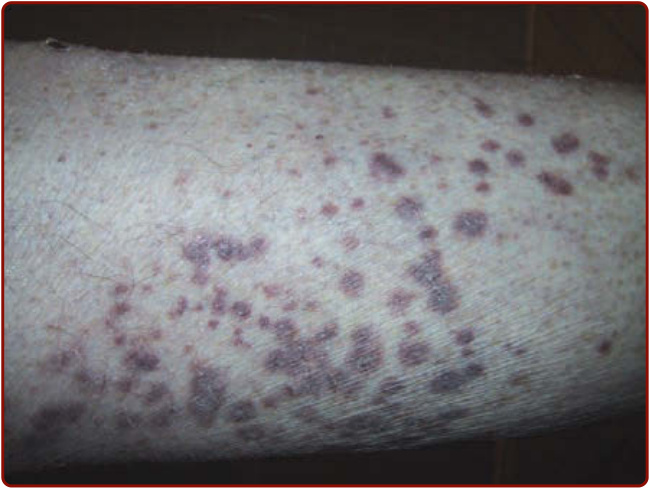
Figure 139-6 Purpura in a patient with microscopic polyangiitis.
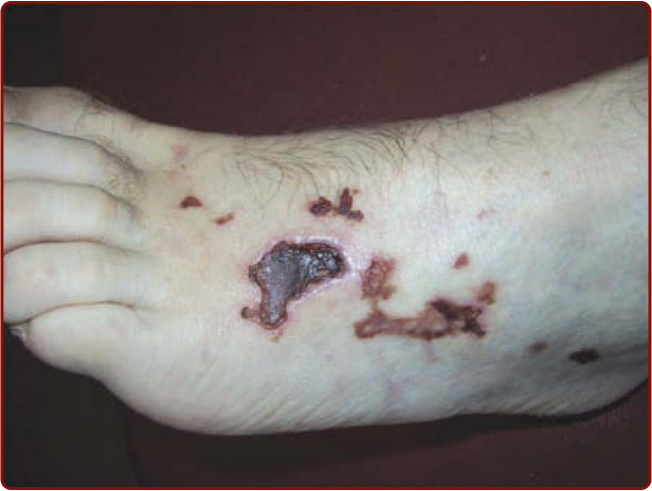
Figure 139-7 Skin ulcer in a patient with granulomatosis with polyangiitis (Wegener).
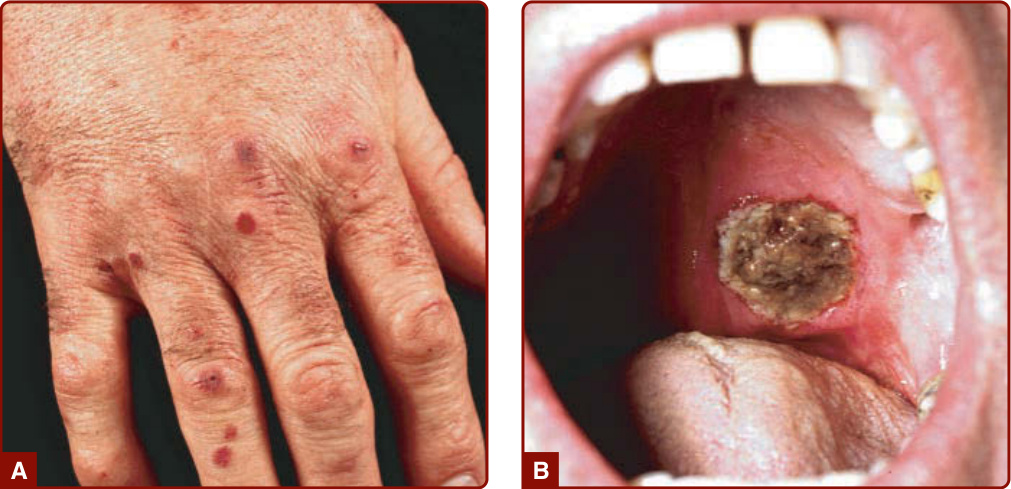
Figure 139-8 Granulomatosis with polyangiitis (Wegener). A, Palpable purpura B, Deep ulcer on the soft palate.
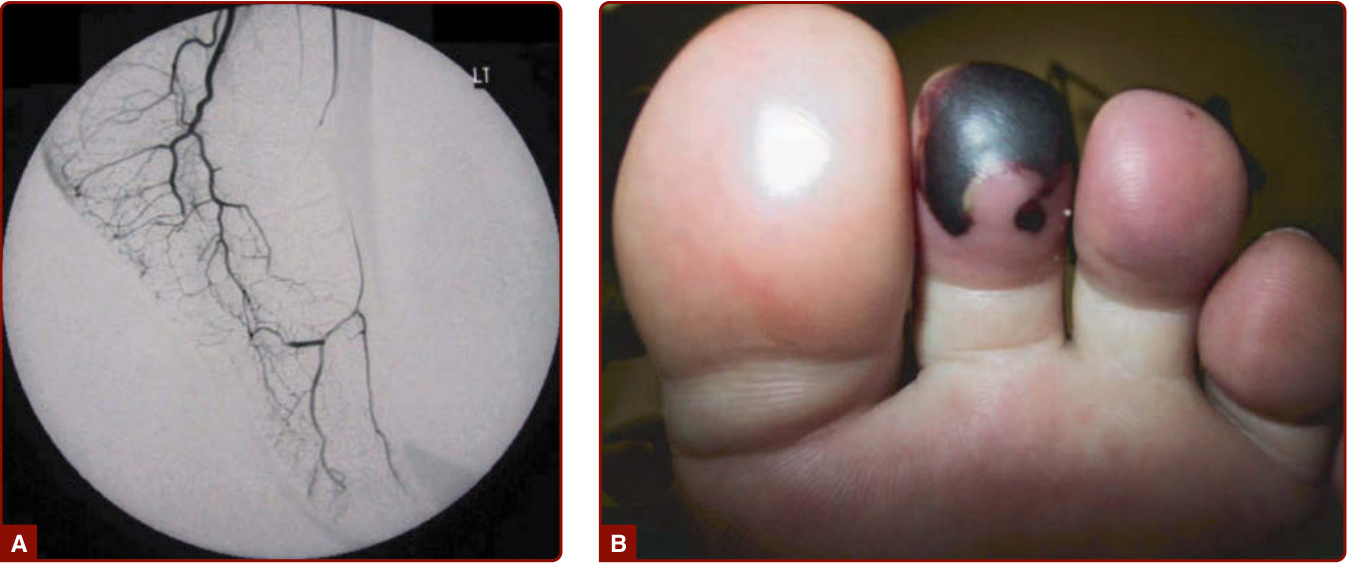
Figure 139-9 Granulomatosis with polyangiitis (Wegener). A, Vasculitis and occlusion of the dorsal pedal artery. B, Gangrenous toe secondary to the occlusion in panel A.
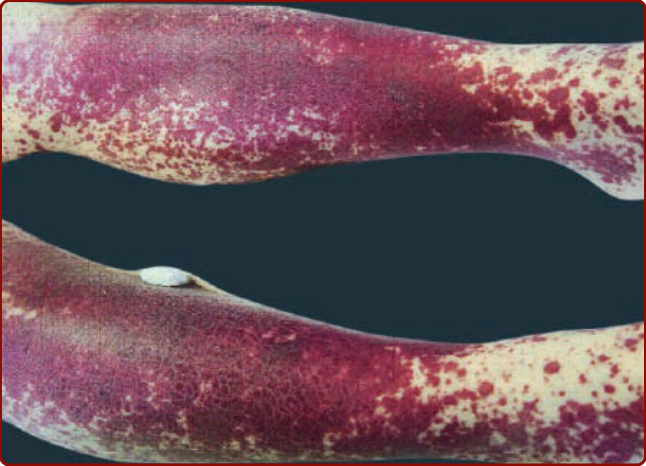
Figure 139-10 Leukocytoclastic vasculitis after administration of granulocyte macrophage colony-stimulating factor for aplastic anemia.
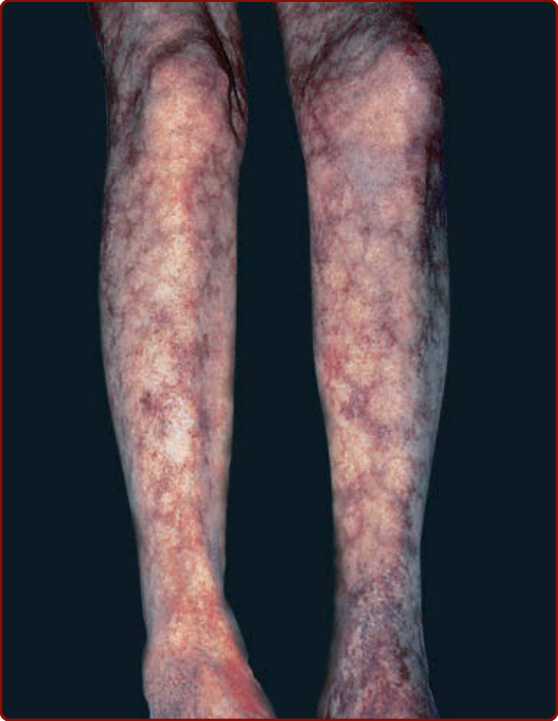
Figure 139-11 Livedo reticularis.
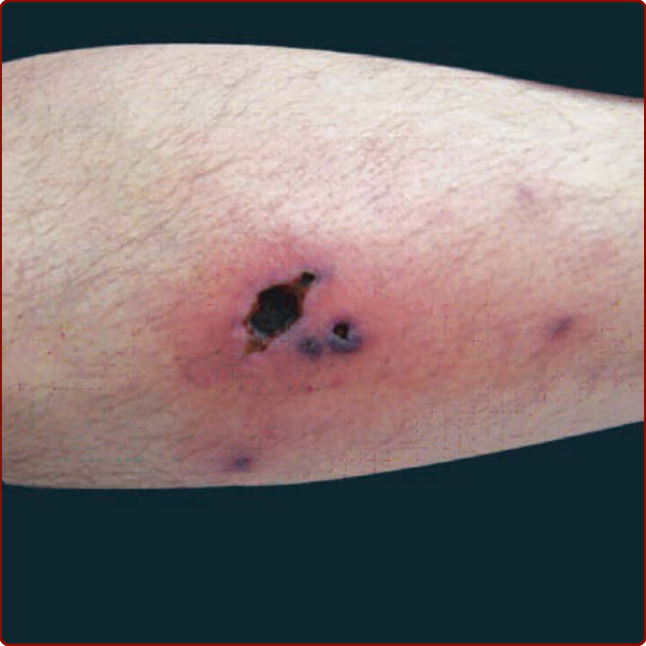
Figure 139-12 Leg ulcer in a patient with polyarteritis nodosa.
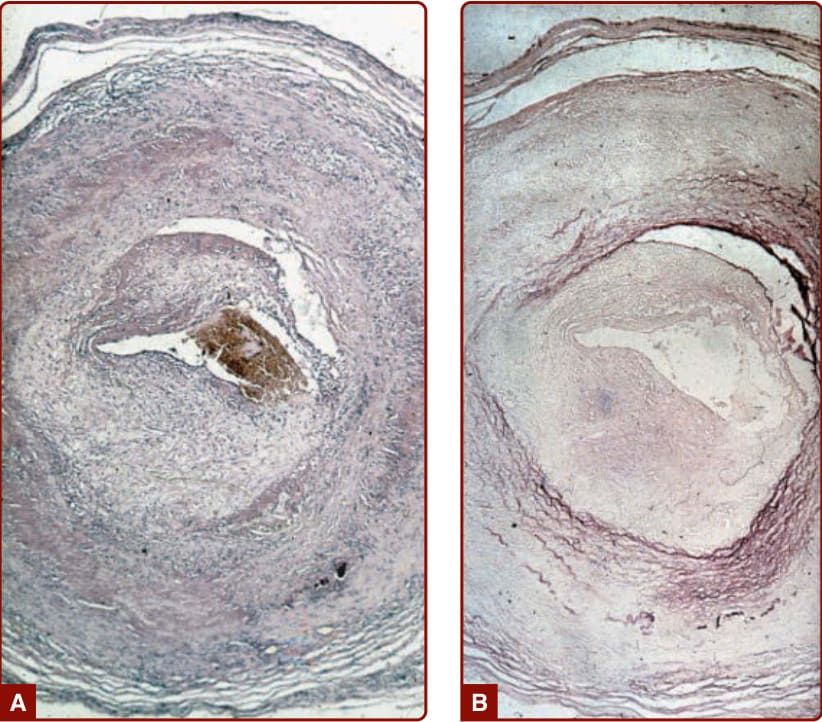
Figure 139-13 A, Histopathology of the temporal artery from a patient with giant cell arteritis shows necrosis of the media, inflammatory infiltrates consisting of lymphocytes, and giant cells. There is also subintimal proliferation of fibroblasts and fibrosis. B, Elastic tissue stain reveals destruction of the lamina interna and externa.
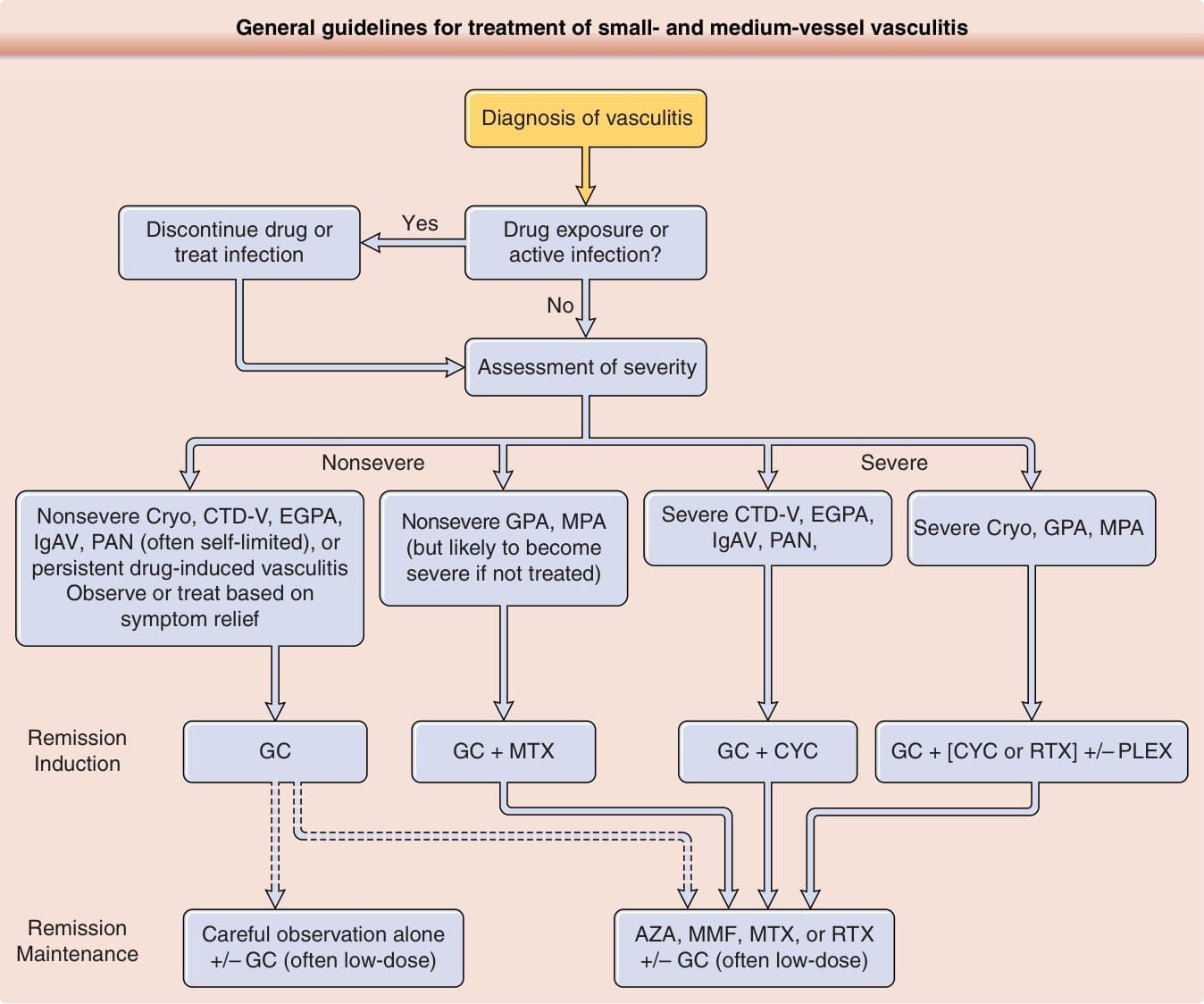
Figure 139-14 General guidelines for treatment of small- and medium-vessel vasculitis. It is important to recognize that approaches will vary substantially based on severity and comorbidity, and high-quality data are not available to guide treatment of many forms of vasculitis. Once the diagnosis is established and treatable causes are identified or excluded, severity is assessed by the organ systems involved and the severity of disease in that organ system. For example: CNS and cardiac involvement are always considered severe; GI, renal, and peripheral nerve disease usually severe; skin manifestations usually not severe; musculoskeletal not severe. Dashed lines indicate where multiple options exist. CTD-V, vasculitis associated with a systemic connective tissue disease (eg, systemic lupus erythematosus, Sjögren syndrome, rheumatoid arthritis); Cryo, cryoglobulinemic vasculitis; EGPA, eosinophilic granulomatosis with polyangiitis (Churg–Strauss); GPA, granulomatosis with polyangiitis (Wegener); IgAV, IgA vasculitis (Henoch–Schönlein); MPA, microscopic polyangiitis; PAN, polyarteritis nodosa; GC, glucocorticoids; AZA, azathioprine; CYC, cyclophosphamide; MMF, mycophenolate; MTX, methotrexate; PLEX, plasma exchange; RTX, rituximab. Treatment of giant cell arteritis and Takayasu arteritis includes GC, but the options for glucocorticoid-sparing drugs differ from the algorithm shown here. The approach to treatment for Behçet disease is complex and does not fit readily into algorithms with other vasculitides.
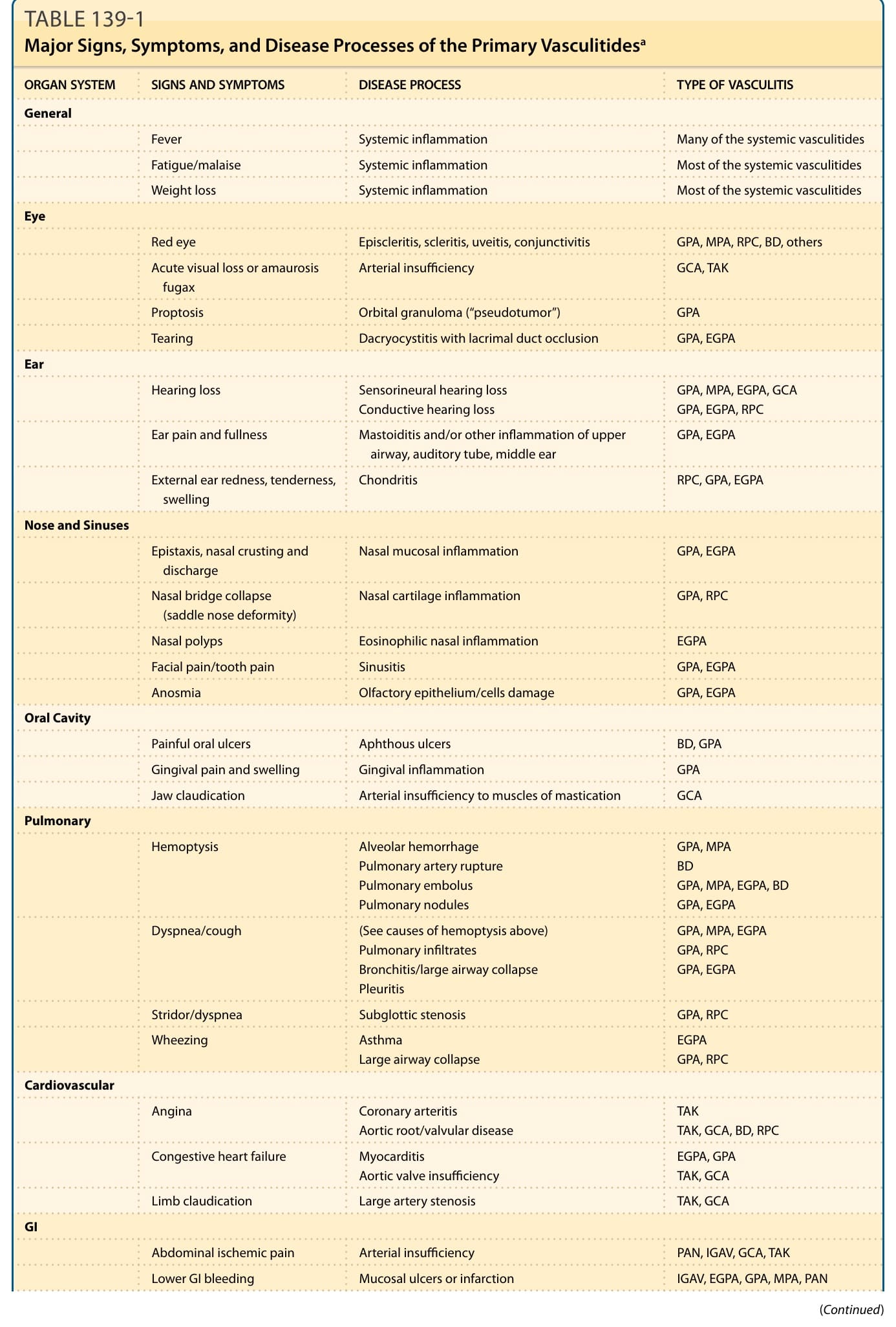
TABLE 139-1 Major Signs, Symptoms, and Disease Processes of the Primary Vasculitidesa
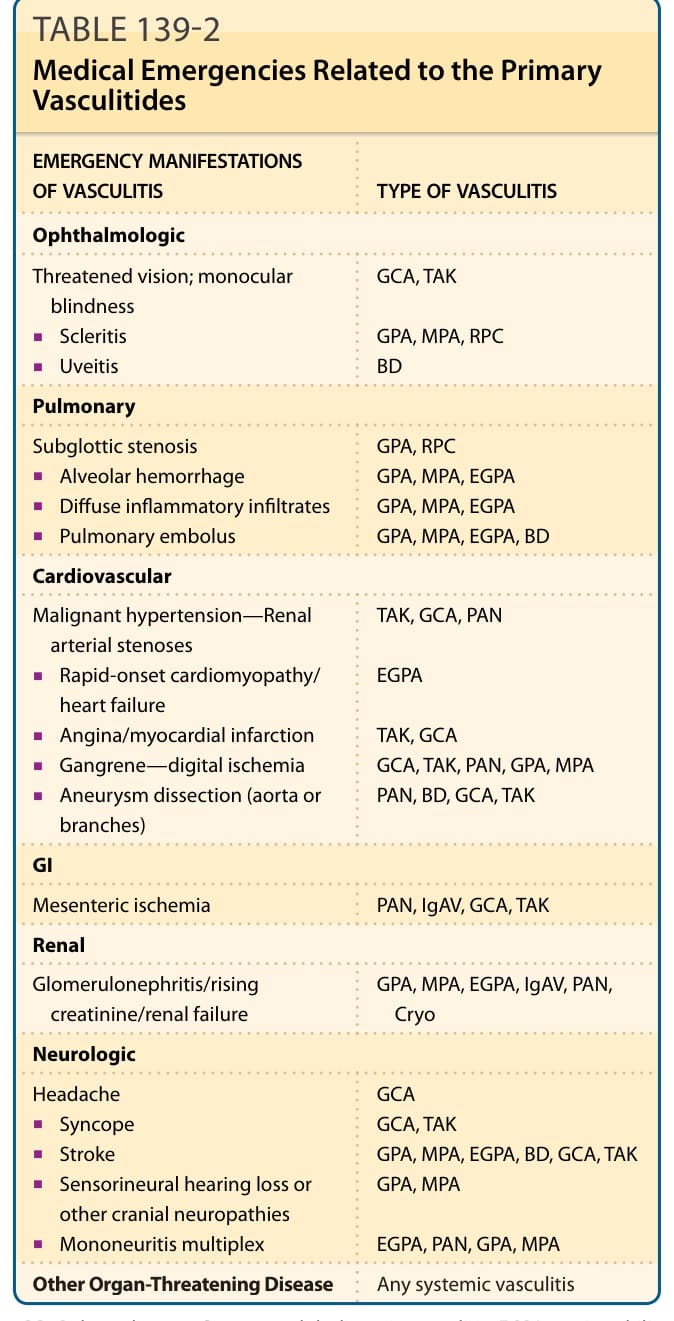
TABLE 139-2 Medical Emergencies Related to the Primary Vasculitides