系統性壞死性動脈炎 (Systemic Necrotizing Arteritis)
PART 22
血管疾病 (Vascular Diseases)
重點一覽 (AT-A-GLANCE)
■ 皮膚血管炎 (cutaneous vasculitis) 可能是侵犯其他器官系統的系統性疾病的初發表現。因此,所有皮膚血管炎病人都需要評估是否可能有系統性血管炎 (systemic vasculitis),包括所有型態的小血管與中血管血管炎。
■ 血管炎的進一步檢查應依據病史與理學檢查的發現來引導,但通常應包括腎功能的常規實驗室檢查、評估 B 型與 C 型肝炎病毒感染、ANA 與 ANCA 的血清學檢查、選擇性的影像學檢查,以及其他依臨床表現所需的檢查。
■ 皮膚切片 (skin biopsy) 幾乎總是診斷血管炎所必需的,切片也有助於判定血管炎的類型。
■ 必須考慮特發性血管炎 (idiopathic vasculitis) 的擬似疾病 (mimics),尤其包括感染、惡性腫瘤、血栓 (thrombosis) 或栓塞性疾病 (embolic disease)。
■ 藥物誘發性血管炎 (drug-induced vasculitis) 常以皮膚病灶表現。然而,所有疑似藥物誘發性疾病的病人仍應評估其他可能的病因或血管炎類型。
■ 侵犯皮膚的血管炎治療,應依據疾病與所侵犯的器官系統來引導。對於系統性壞死性血管炎,一律使用糖皮質素 (glucocorticoids),重症初期以高劑量使用。額外的免疫抑制藥物則取決於血管炎類型、疾病範圍與病人的共病 (comorbidities)。
■ 疑似藥物誘發性血管炎的治療可能包括停用疑似致病藥物並仔細觀察,但也可能涉及以糖皮質素或其他藥物治療。
■ 免疫抑制藥物的使用應由具有此類藥物使用經驗的醫師監督。
■ 對所有血管炎病人進行全面的臨床追蹤,是治療管理上不可或缺的一環。
前言 (INTRODUCTION)
血管炎 (vasculitis) 一詞可廣義地定義為血管的發炎。雖然血管炎有時只侵犯單一器官,尤其是皮膚,但對多數臨床醫師而言,血管炎意指一群以血管發炎為主要(但非唯一)病理過程的疾病。系統性血管炎 (systemic vasculitides) 是一組範圍廣泛的疾病,多數為特發性、罕見且多系統侵犯。這些疾病涉及極為多樣的臨床表現與病理變化,使得每一個內、外科次專科的臨床醫師都會遇到此類病人。皮膚血管炎相當常見,而皮膚疾病是許多型態血管炎的常見表現,尤其是小血管與中血管動脈炎,其皮膚病灶可能是系統性疾病的初發症狀。本章聚焦於系統性血管炎中的皮膚疾病。孤立型 (isolated) 的皮膚血管炎與部分系統性血管炎則涵蓋於第 138 章。除了概述一般性與特定型態血管炎的皮膚表現外,本章也將提供一套處理皮膚疾病中需考慮血管炎作為鑑別診斷之病人的方法。血管炎的分類系統有多種並存,這種情況反映出我們對其潛在病理生理學缺乏清楚的理解,以及許多血管炎類型之間臨床特徵的重疊。1-6 最被廣泛接受的方法是依主要受侵犯血管的大小(小、中或大)來分類,然後依適當情況再細分或歸類疾病 (Fig. 139-1)。許多(但非全部)特定型態的血管炎已制定出分類標準與定義。這些系統是為了臨床研究而設計,用以建立相當同質的研究世代 (study cohorts),並非作為「診斷」標準使用。2 儘管如此,臨床醫師仍會發現這些標準對其面對此領域時有所助益。最被廣泛使用的血管炎標準來自美國風濕病學院 (American College of Rheumatology) 的分類標準4,5 與 Chapel Hill 共識會議 (Chapel Hill Consensus Conference) 的疾病定義。6 此外,有些分類已不再建議使用(例如過敏性血管炎 hypersensitivity vasculitis 已失去明確的意義)。應強調的是,白血球碎裂性血管炎 (leukocytoclastic vasculitis) 一詞並非指特定疾病,而是一種病理描述,常(但非總是)適用於皮膚或其他器官的血管炎。也有一些針對兒童病人的另外的標準。7
Chapel Hill 共識會議過程的一項重要延伸,是近期完成的增補文件 (Addendum),提供了一套針對皮膚血管炎的完整定義。8
目前正進行一項新的國際性倡議,重新檢討血管炎的分類,並納入先前系統建立時尚無法取得的血管炎臨床與病理生理學面向的相關資料;此類新元素包括抗嗜中性球細胞質自體抗體 (antineutrophil cytoplasmic autoantibodies, ANCA) 的檢測,以及針對大動脈疾病更普及的進階影像技術。2
原發性血管炎的分類 (Classification of primary vasculitides)
以小血管為主 (Predominantly small vessel)
- 免疫複合體媒介型 (Immune complex mediated):冷凝球蛋白血症 (Cryoglobulinemia)、HSP、局限性皮膚型 (Limited Skin)
- ANCA 相關型 (ANCA-associated):MPA、CSS、GPA
- 其他 (Other)
以中血管為主 (Predominantly medium vessel)
- 川崎病 (Kawasaki)、PAN、其他 (Other)
以大血管為主 (Predominantly large vessel)
- GCA、TAK、孤立性主動脈炎 (Isolated aortitis)、其他 (Other)
無主要血管大小之分 (No predominant size)
- 貝賽特氏病 (Behçet)、原發性中樞神經系統血管炎 (Primary CNS)、復發性多發軟骨炎 (Relapsing polychondritis)、Cogan 症候群、其他 (Other)
流行病學 (EPIDEMIOLOGY)
除了藥物/毒素誘發性血管炎可能例外之外,所有型態的特發性血管炎在美國均被視為罕見的「孤兒」疾病(盛行率低於 200,000 人);在歐洲與其他地區亦有類似的歸類。血管炎發生於兩性、所有年齡層與所有主要種族/族裔群體。然而,某些型態在特定群體中較為常見。例如,高安動脈炎 (Takayasu arteritis) 在女性中遠較男性常見,川崎病 (Kawasaki disease) 幾乎完全是幼兒的疾病,而巨細胞動脈炎 (giant cell arteritis) 則局限於年長成人。肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener) 大多發生於白人,而貝賽特氏病 (Behçet disease) 在東地中海國家以及日本與韓國則明顯較為常見。血管炎之間的人口學差異具有科學上的意義,可作為病因的線索,並有助於建立鑑別診斷。9 不令人意外的是,具有強烈地區性差異的血管炎在 HLA 區域具有遺傳危險因子。10-13 然而,包括遺傳學在內的流行病學傾向,其強度尚不足以完全排除任何個別病人罹患某特定型態血管炎的診斷,而典型流行病學的例外情況也經常發生。
臨床特徵 (CLINICAL FEATURES)
皮膚發現 (CUTANEOUS FINDINGS)
皮膚血管炎的外觀反映出受侵犯血管的大小與深度、發炎與紅血球外滲的嚴重度,以及受損血管的密度與由此造成的組織破壞。例如,由真皮微血管系統 (dermal microvasculature) 中免疫複合體所引起的血管炎,常最初表現為一個微小、鮮紅色的圓圈,係因紅血球外滲所致,當其直徑達到約 2 mm 時即變得可觸知 (palpable)。然而,高密度的此類病灶可能產生融合性紫斑 (confluent purpura) 與/或中央壞死或潰瘍的區域。小動脈血管炎(也令人混淆地被稱為「中血管」),其血管位於皮膚與皮下組織較深處,會產生常呈紅斑性但不如紫斑那麼鮮紅的結節 (nodules),或當受侵犯的血管主要平行於體表時產生網狀青斑 (livedo racemosa)。中血管血管炎較小血管血管炎更易產生潰瘍與指(趾)缺血 (digit ischemia)。
皮膚小血管血管炎的典型表現是可觸性紫斑 (palpable purpura),但其他表現也很常見。這些包括可為圓形或具有角狀邊界的非可觸性紫斑(網狀紫斑 retiform purpura)、丘疹 (papules)、蕁麻疹樣病灶 (urticarial lesions)、大小不一且深度不一的結節、水疱性病灶 (bullous lesions) 與潰瘍 (Figs. 139-3, 139-4, 139-6 to 139-12)。受侵犯區域之內與其鄰近處的水腫 (edema) 也很常見。可歸因於皮膚血管炎的症狀差異很大。紫斑或青斑可能無症狀,或產生常被描述為刺痛或灼熱、但有時為搔癢的疼痛。結節與潰瘍通常會疼痛。若在未受結節、潰瘍或融合性紫斑侵犯的區域出現劇烈疼痛,應提高對神經病變 (neuropathy) 或發炎性關節炎 (inflammatory arthritis) 的懷疑。
非皮膚發現 (NONCUTANEOUS FINDINGS)
正是病史與理學檢查中特定特徵的有無,提供了病人是否罹患侵犯多重器官系統之疾病的第一個線索。不同型態血管炎的重要徵象與症狀清單列於 Table 139-1。
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
除了感染相關血管炎(例如許多結節性多動脈炎 polyarteritis nodosa 病例中的 B 型肝炎病毒,以及多數冷凝球蛋白血症性血管炎 cryoglobulinemic vasculitis 病例中的 C 型肝炎病毒)與藥物誘發性血管炎之外,多數型態血管炎的病因仍屬未知。同樣地,血管炎的發病機轉仍是個積極研究中的領域。本節摘述目前對血管炎病因與機轉的看法,但需注意:雖然本節所概述的許多證據都相當有力,這些疾病的發病機轉幾乎可確定遠較此更為複雜。廣義而言,小血管血管炎遵循兩條途徑之一:(1) 免疫複合體沉積 (immune complex deposition),或 (2) 非免疫複合體媒介、可能涉及 ANCA 的病理過程。免疫複合體由免疫球蛋白、其所結合的抗原,以及補體成分組成,可在針對微生物、自體抗原或藥物的廣泛免疫反應中形成。在一小部分的抗體反應中,抗體與抗原的濃度與性質有利於在微血管系統中沉積,這可能導致嗜中性球的活化與壞死性血管炎,並破壞內皮細胞。ANCA 針對的是嗜中性球的蛋白質髓過氧化酶 (myeloperoxidase, MPO) 或蛋白酶-3 (proteinase-3, PR3),這些蛋白主要存在於細胞內顆粒中,但也會被轉位至細胞表面,或經由脫顆粒 (degranulation) 或嗜中性球胞外陷阱 (neutrophil extracellular traps, NETs) 的排出而釋放,使 ANCA 得以接觸、活化嗜中性球、牽涉補體成分,並導致壞死性血管炎。遺傳學似乎對 ANCA 相關血管炎有中度的貢獻,目前所辨識出的基因與抗體對 PR3 或 MPO 的專一性有關。10,11 雖然體液免疫系統在小血管血管炎的發病機轉中扮演關鍵角色,但也有證據顯示細胞免疫反應在這些疾病中具有額外的重要性。相對地,大血管血管炎被認為是由 T 細胞在大動脈管壁中活化巨噬細胞所媒介。在巨細胞動脈炎 (giant cell arteritis, GCA) 中,分泌 IL-17 的細胞 (Th17) 與分泌干擾素-γ 的細胞 (Th1) 都很顯著。T 細胞經由血管的滋養血管 (vasa vasorum) 進入動脈壁,因此內皮維持完整。多項牽涉不同感染原於 GCA 的研究均無法被重現,因此它持續被視為一種自體免疫疾病,高安動脈炎 (Takayasu arteritis) 亦然。雖然這些疾病具有相似的顯微病理,但它們的流行病學相當不同、血管分布有中度差異,且遺傳學也不同。遺傳學經由 HLA 基因座 (locus) 對兩種疾病都有中度貢獻,但在 GCA 是位於第 II 類 (class II) 區域,在高安動脈炎則是第 I 類 (class I),兩者間幾無其他重疊。12,13
未與 B 型肝炎病毒感染相關的結節性多動脈炎 (polyarteritis nodosa, PAN) 的病因與發病機轉尚不清楚,包括它是否由抗體媒介、它究竟是一個具有許多罕見病因的症候群,抑或具有一個或少數幾個主要病因等不確定性。尚無遺傳學研究發表;若有 HLA 相關性,將支持有一個主要標的抗原 (target antigen) 存在,但無法辨識該標的究竟是自體抗原或微生物。個別病灶可能清楚顯示嗜中性球伴隨壞死與內皮的破壞,或可能顯示伴隨完整內皮的較混合性浸潤,但後者的發現究竟代表不同的疾病過程,抑或是近期壞死性病灶的癒合,仍不確定。這種不確定性反映在新命名法的應用上,例如當後者情況局限於皮膚時稱為「斑狀動脈炎」(macular arteritis),並由此衍生出反提案 (counterproposal),主張將其視為「皮膚型 PAN」(cutaneous PAN) 的一個亞型。
診斷 (DIAGNOSIS)
診斷流程 (DIAGNOSTIC ALGORITHM)
當病人以令人擔憂可能為血管炎的皮膚病灶就診時,應迅速尋求以下 3 個問題的答案:
- 此病灶是否由血管炎所引起?
- 此疾病是否侵犯其他器官系統?
- 在病史詢問、理學檢查、實驗室檢查或影像學檢查中,是否有其他有助於建立特定診斷的發現?
若已取得血管炎的診斷,則必須再詢問 2 個問題:
- 是否可能為病人做出特定型態血管炎的診斷?
- 病人是否需要立即治療與/或住院?
Table 139-1:原發性血管炎的主要徵象、症狀與疾病過程
| 器官系統 | 徵象與症狀 | 疾病過程 | 血管炎類型 |
|---|---|---|---|
| 全身性 (General) | 發燒 | 系統性發炎 | 許多系統性血管炎 |
| 疲倦/倦怠 | 系統性發炎 | 多數系統性血管炎 | |
| 體重減輕 | 系統性發炎 | 多數系統性血管炎 | |
| 眼 (Eye) | 紅眼 | 表層鞏膜炎、鞏膜炎、葡萄膜炎、結膜炎 | GPA, MPA, RPC, BD, 其他 |
| 急性視力喪失或一過性黑矇 (amaurosis fugax) | 動脈供血不足 | GCA, TAK | |
| 眼球突出 (Proptosis) | 眼窩肉芽腫(「假性腫瘤」) | GPA | |
| 流淚 | 淚囊炎合併淚管阻塞 | GPA, EGPA | |
| 耳 (Ear) | 聽力喪失 | 感音神經性聽力喪失;傳導性聽力喪失 | GPA, MPA, EGPA, GCA;GPA, EGPA, RPC |
| 耳痛與耳脹 | 乳突炎與/或上呼吸道、耳咽管、中耳的其他發炎 | GPA, EGPA | |
| 外耳發紅、壓痛、腫脹 | 軟骨炎 (Chondritis) | RPC, GPA, EGPA | |
| 鼻與鼻竇 (Nose and Sinuses) | 鼻出血、鼻結痂與分泌物 | 鼻黏膜發炎 | GPA, EGPA |
| 鼻樑塌陷(鞍鼻畸形 saddle nose deformity) | 鼻軟骨發炎 | GPA, RPC | |
| 鼻息肉 | 嗜酸性球性鼻部發炎 | EGPA | |
| 顏面痛/牙痛 | 鼻竇炎 (Sinusitis) | GPA, EGPA | |
| 嗅覺喪失 (Anosmia) | 嗅覺上皮/細胞損傷 | GPA, EGPA | |
| 口腔 (Oral Cavity) | 疼痛性口腔潰瘍 | 阿弗他潰瘍 (Aphthous ulcers) | BD, GPA |
| 牙齦疼痛與腫脹 | 牙齦發炎 | GPA | |
| 顎跛行 (Jaw claudication) | 咀嚼肌動脈供血不足 | GCA | |
| 肺 (Pulmonary) | 咳血 (Hemoptysis) | 肺泡出血;肺動脈破裂;肺栓塞;肺結節 | GPA, MPA;BD;GPA, MPA, EGPA, BD;GPA, EGPA |
| 呼吸困難/咳嗽 | (見上述咳血原因);肺浸潤;支氣管炎/大氣道塌陷;胸膜炎 | GPA, MPA, EGPA;GPA, RPC;GPA, EGPA | |
| 喘鳴/呼吸困難 | 聲門下狹窄 (Subglottic stenosis) | GPA, RPC | |
| 哮鳴 (Wheezing) | 氣喘;大氣道塌陷 | EGPA;GPA, RPC | |
| 心血管 (Cardiovascular) | 心絞痛 | 冠狀動脈炎;主動脈根部/瓣膜疾病 | TAK;TAK, GCA, BD, RPC |
| 充血性心衰竭 | 心肌炎;主動脈瓣閉鎖不全 | EGPA, GPA;TAK, GCA | |
| 肢體跛行 | 大動脈狹窄 | TAK, GCA | |
| 腸胃 (GI) | 腹部缺血性疼痛 | 動脈供血不足 | PAN, IGAV, GCA, TAK |
| 下消化道出血 | 黏膜潰瘍或梗塞 | IGAV, EGPA, GPA, MPA, PAN | |
| 腎 (Renal) | 肉眼血尿 | 腎梗塞;腎絲球腎炎(肉眼血尿的罕見原因) | PAN;GPA, MPA, EGPA, IGAV, Cryo |
| 中樞神經系統 (CNS) | 頭痛、頭皮壓痛 | 顱動脈炎 | GCA, TAK |
| 頭暈/昏厥 | 腦部動脈供血不足 | GCA, TAK | |
| 顱神經病變 | 神經發炎;罕見為腫塊病灶 | GCA, GPA, MPA | |
| 周邊神經系統 (Peripheral Nervous System) | 感覺/運動功能障礙 | 神經發炎;罕見為腫塊病灶 | EGPA, GPA, MPA, PAN, Cryo |
| 肌肉骨骼 (Musculoskeletal) | 多關節痛 | 多關節炎 | GPA, GCA, TAK, Cryo, IGAV, BD |
| 肩部與髖部帶狀區疼痛 | 風濕性多肌痛 (Polymyalgia rheumatica) | GCA | |
| 肌肉無力 | 肌炎 (Myositis) | EGPA | |
| 皮膚 (Skin) | 紫斑 | 小血管血管炎 | GPA, MPA, EGPA, PAN, IGAV, Cryo |
| 疼痛性結節、深部潰瘍 | 中血管血管炎 | PAN, GPA, MPA, Cryo | |
| 指(趾)缺血/壞疽 | 中至大動脈狹窄 | GCA, TAK, PAN, GPA, MPA, Cryo | |
| 表淺結節 | 肉芽腫 (Granulomas) | GPA, EGPA | |
| 丘疹、痤瘡樣病灶 | 丘疹膿疱性病灶 | BD | |
| 疼痛性紅色結節 | 結節性紅斑 (Erythema nodosum) | BD, TAK | |
| 周邊水腫 | 深部靜脈血栓 | GPA, MPA, EGPA, BD |
表 139-1:原發性血管炎的主要徵象、症狀與疾病過程。a此清單並未涵蓋所有疾病的所有表現。BD, 貝賽特氏病 (Behçet disease);Cryo, 冷凝球蛋白血症性血管炎 (cryoglobulinemic vasculitis);EGPA, 嗜酸性球性肉芽腫性多血管炎 (eosinophilic granulomatosis with polyangiitis);GCA, 巨細胞動脈炎 (giant cell arteritis);GPA, 肉芽腫性多血管炎 (granulomatosis with polyangiitis);IgAV, IgA 血管炎(Henoch–Schönlein);MPA, 顯微鏡下多血管炎 (microscopic polyangiitis);PAN, 結節性多動脈炎 (polyarteritis nodosa);RPC, 復發性多發軟骨炎 (relapsing polychondritis);TAK, 高安動脈炎 (Takayasu arteritis)。RPC 是一種以軟骨破壞為定義的自體免疫疾病,因其數項特徵與 GPA 重疊而納入本表。與 BD 相同,約 30% 的 RPC 病人也合併血管炎,且所侵犯的血管大小範圍廣泛。
處理疑似因血管炎所致皮膚病灶之病人的建議方法,如 Fig. 139-2 所示。第一個問題的答案常經由皮膚切片取得,此程序適用於許多可觸性紫斑或其他病灶、且血管炎診斷無法以其他方式輕易建立的病例。第二個問題藉由徹底的系統性回顧 (review of systems)、理學檢查與常規實驗室檢查來處理,這些通常可迅速完成。第三個問題則藉由更專門的實驗室檢查來處理,其結果通常需數天才能回報。重要的是要迅速辨識器官系統的侵犯情況,以及病人「病得多重」(或可能很快病得多重),因為某些皮膚血管炎的病因不需治療,但其他病因則需要立即住院以開始免疫抑制與支持性治療。鑑於歸類於「血管炎」此類別之下的疾病範圍廣泛,加上評估疑似血管炎病人時也合理需考慮的疾病數目更為龐大,此類病例常需考慮為數眾多且範圍廣泛的診斷檢查。然而,取得詳盡的病史並進行詳細的理學檢查,應能使臨床醫師限縮所考慮的血管炎類型,並排定診斷檢查的優先順序。並非所有疑似血管炎的病人都需要進行所有檢查。此方法當然必須與一種開放心態取得平衡,即對血管炎的非典型表現以及此類病人鑑別診斷中的一系列感染、惡性腫瘤與其他疾病保持開放態度。對可能血管炎的評估通常與對其他病程的評估同時進行。
系統性回顧 (REVIEW OF SYSTEMS)
進行完整的系統性回顧並評估整體疾病嚴重度,是疑似血管炎病人早期評估中最重要的單一環節。引起皮膚血管炎的這些疾病合起來可侵犯所有器官系統,且多數情況下該侵犯會造成症狀,腎臟疾病則是一個顯著的例外。雖然某些症狀顯然比其他症狀更令人擔憂(咳血 vs 乾咳、疼痛性紅眼 vs 輕度關節痛),但即使是相對輕微的症狀,也可能是疾病不局限於皮膚的線索。不同型態血管炎的重要徵象與症狀清單列於 Table 139-1。
Fig. 139-2 內容:對疑似血管炎且有皮膚病灶之病人的處理方法
可能的皮膚小血管血管炎 (Possible Cutaneous Small-Vessel Vasculitis)
- 高度臨床懷疑:可觸性紫斑
- 中至低度臨床懷疑:潰瘍、結節、水疱、大疱
評估疾病的嚴重度/範圍
- 完整的系統性回顧
- 檢視藥物/毒素暴露
- 詳細的理學檢查
- 常規初始檢查:全血球計數/分類、肌酸酐—尿液分析、肝功能、胸部影像
- 考慮其他鑑別診斷
→ 可能結果:已建立血管炎以外的診斷/已建立特定型態血管炎/血管炎尚未確立但仍須考慮
皮膚切片 (Skin biopsy):取得組織進行常規組織病理學與免疫螢光 (Immunofluorescence)
→ 已建立血管炎診斷 → 判定血管炎的類型與範圍
進一步檢查以判定血管炎的類型與範圍(並非所有病人都需要所有檢查)
- 血清學檢查:ANCA、冷凝球蛋白、SPEP (IFE)、UPEP (UFE)、B 型與 C 型肝炎病毒檢測、ANA、anti-Ro (SSA)、補體成分 (C3, C4)
- 其他診斷檢查:胸部 CT、鼻竇 CT、血管攝影、聽力圖、肌電圖;其他組織切片(肺、腎、鼻竇、其他)
開始治療並密切臨床追蹤
專科會診:皮膚科、腎臟科、神經科、眼科、耳鼻喉科、胸腔科、風濕免疫科
病史、用藥史與毒素或感染性疾病暴露 (MEDICAL HISTORY, MEDICATION USE, AND EXPOSURES TO TOXINS OR INFECTIOUS DISEASES)
了解任何疑似血管炎病人的完整病史至關重要。其他疾病可能將血管炎作為疾病的一部分(例如紅斑性狼瘡 lupus),或可能造成擬似血管炎的皮膚病灶。藥物誘發性血管炎很常見,而皮膚病灶(通常但非總是紫斑)是藥物誘發性血管炎最常見的表現。14
被報告會引起血管炎的藥物清單極為龐大,幾乎每一類藥物都曾被牽涉於可能的藥物誘發性血管炎病例中。詢問先前 6 至 12 個月內的處方藥、非處方藥及「另類」或草藥使用情形是有用的,因為某些藥物的效應在停用後可能仍持續。也應詢問病人是否使用非法或娛樂性藥物,因為包括甲基安非他命 (methamphetamines)、古柯鹼 (cocaine) 及其他在內的數種此類藥物都曾被牽涉於血管炎病例中。也應詢問職業性或其他非藥物毒素的暴露。除了詢問一般的感染徵象與症狀外,也應詢問病人近期旅遊、接觸生病者,以及性傳染病的風險。
理學檢查 (PHYSICAL EXAMINATION)
除了仔細且完整地評估皮膚之外,多系統的檢查有助於判定症狀是否伴隨客觀的異常,或是否有病人未注意到的發現。生命徵象至關重要,但血壓正常的病人仍可能有嚴重的腎絲球腎炎。應檢視眼睛是否有發紅與眼球突出。前鼻腔 (anterior nasal cavity) 可輕易以耳鏡 (otoscope) 視診。應尋找淋巴結病變 (lymphadenopathy) 的證據。心臟、肺與腹部檢查可提供潛在疾病的線索,但檢查正常並不能排除病變。同樣地,脈搏消失、血壓讀數不對稱與雜音 (bruits) 是有助於篩檢大血管血管炎、但不完美的指標。完整的關節檢查很重要,任何提示滑膜炎 (synovitis) 的發現(關節腫脹、發熱、發紅)都必須進一步檢查;然而,許多血管炎病人會有關節痛但無關節積液。完整的神經學檢查是評估與分流疑似血管炎病人最有價值的環節之一;細微的感覺甚至運動異常常在初次評估時被遺漏。眼科醫師與耳鼻喉科醫師所能執行的更詳細、更專門的檢查,常對評估疑似血管炎的病人極有幫助。對於有令人擔憂症狀(如新發視力障礙、疼痛或發紅的眼睛、聲音嘶啞或喘鳴、或聽力喪失)的病人,常需緊急轉介。
輔助性檢查 (SUPPORTIVE STUDIES)
皮膚切片 (SKIN BIOPSY)
診斷血管炎的方法取決於所懷疑的血管炎類型,而這常基於所侵犯血管的大小。侵犯皮膚的血管炎通常涉及小型與中型血管,而這些血管適合切片 (Fig. 139-3)。鑑於皮膚切片的簡便性與低風險,切片在診斷血管炎上扮演重要角色,在建立血管炎以外的診斷上也扮演同等重要的角色。標準的穿孔切片 (punch biopsy) 足以診斷小血管血管炎,但要取得中型血管的資訊則可能需要較深且較寬的切除 (excision)。15,16 應以較深切片處理的病灶包括皮下結節、網狀青斑 (livedo racemosa) 或深部潰瘍 (Fig. 139-4)。許多型態的小血管血管炎也可能侵犯中型皮膚血管。重要的是要認識到「小型」與「中型」血管之間的差異有些主觀,而皮膚病理學家較常做出此類區分,多於那些看到較大切片檢體的其他病理學家。有時典型臨床症候群的存在使切片變得不必要。例如,兒童的 IgA 血管炎(Henoch-Schönlein)常僅以臨床基礎即可診斷,而某些 ANCA 相關或冷凝球蛋白血症性血管炎病例可藉由結合臨床特徵與特定血清學檢查而有信心地診斷。貝賽特氏病 (Behçet disease) 與川崎病 (Kawasaki disease) 是依臨床症候群來診斷;通常不對這些疾病常見的皮膚病灶進行切片,且此類切片常無診斷意義。一般建議若可能的話,對臨床上出現未滿 48 小時的皮膚病灶進行切片,以最大化找到急性嗜中性球性血管炎典型特徵的機會,包括纖維素樣壞死 (fibrinoid necrosis)、紅血球外滲、嗜中性球外滲伴隨核碎屑釋放(白血球碎裂 leukocytoclasia),以及免疫沉積物的存在。15,16 為常規組織病理學或免疫螢光檢查所做的組織處理方式不同;若需要免疫螢光,則需執行 2 次切片,或將單一切片在處理前加以分割。後者方法可能損壞組織。15
如本章通篇所討論,白血球碎裂性血管炎的組織學發現有助於確認血管炎的診斷,但對於從廣泛的可能性中建立病因毫無助益。顯微鏡檢查有時會顯示提示但非診斷性的血管炎特徵,例如有白血球碎裂但無纖維素樣壞死。血管周圍浸潤 (perivascular infiltrate) 的發現也是非特異性的,尤其是當其主要由單核細胞組成時,即使是嗜中性球性浸潤也是如此。某些特徵在白血球碎裂性血管炎之外若同時被觀察到,則強烈提示特定疾病,例如血管外肉芽腫伴隨地圖狀壞死 (geographic necrosis)(肉芽腫性多血管炎 granulomatosis with polyangiitis),或富含嗜酸性球的血管外肉芽腫(嗜酸性球性肉芽腫性多血管炎 eosinophilic granulomatosis with polyangiitis),17 但這些特徵僅見於這些疾病少數的切片中。免疫螢光上 IgA 優勢多於 IgG/IgM 是提示但非診斷性的 IgA 血管炎(Henoch–Schönlein)。IgG、IgM 與/或補體沉積物的存在,提示數種免疫複合體媒介病因之一,包括藥物過敏、感染後血管炎、冷凝球蛋白血症,以及繼發於系統性紅斑性狼瘡、修格蘭氏症候群 (Sjögren syndrome) 或類風濕性關節炎的血管炎。17
其他切片 (OTHER BIOPSIES)
血管炎常藉由其他器官的切片來診斷,例如腎、肺、肌肉或周邊神經,甚至從手術檢體 (Fig. 139-5)。腎或肺切片較皮膚切片更可能顯示某特定疾病的診斷性病理。儘管如此,能建立血管炎診斷的皮膚切片,可能免去更具侵入性切片的需要。
實驗室檢查 (LABORATORY TESTING)
雖然個別實驗室檢查本身幾乎從不能診斷血管炎,但此類檢查在評估考慮皮膚血管炎的病人時不可或缺。實驗室檢查可辨識疾病過程中所侵犯的器官系統,尤其是腎臟疾病。此外,在適當的情境下,選擇性的血清學檢查可能為血管炎建立病因。然而,血清學檢查通常補充而非取代切片,尤其是在皮膚病灶可輕易切片的病人。
腎功能檢查 (TESTS OF RENAL FUNCTION)
在評估疑似血管炎病人時,腎臟疾病檢查是最重要的,因為腎臟疾病在許多血管炎中很常見,且在末期腎衰竭發生之前很少伴隨徵象或症狀。所有疑似血管炎的病人都應進行尿液分析,包括試紙 (dipstick) 與顯微鏡檢查,且對在另一器官系統已確立小型或中型血管血管炎的病人應重複進行。常規試紙檢查上任何血液的存在,都需要由受過特定訓練尋找紅血球柱 (red blood cell casts) 的人員進一步檢查(在北美,許多腎臟科醫師、部分風濕免疫科醫師會找,但很少有實驗室技術員會找)。測量血清肌酸酐 (serum creatinine) 對估計腎絲球過濾率 (glomerular filtration rate, GFR) 至關重要。肌酸酐的微小變化,即使在正常範圍內,也可能是 GFR 下降的早期證據。雖然侵犯腎絲球的小血管血管炎預期會產生血尿,通常伴隨紅血球柱與蛋白尿,但僅侵犯中型血管的血管炎(例如結節性多動脈炎 polyarteritis nodosa)通常產生孤立性血尿或正常的尿液分析。尿液分析與血清肌酸酐是同等重要且互補的檢查;單憑任一項都不足以排除血管炎的腎臟疾病。
肝功能檢查 (TESTS OF LIVER FUNCTION)
血管炎,尤其是結節性多動脈炎,可侵犯肝臟,但顯著的肝功能障礙很罕見。因此肝功能檢查在診斷血管炎上的價值有限,但如常見情況般若要使用具潛在肝毒性的藥物治療時,它們確實提供了一個可供未來數值比較的基準。肝功能檢查也可提供 B 型或 C 型肝炎病毒感染的早期提示,這兩者都與血管炎相關,但不能取代這些感染的血清學檢查。肝功能檢查正常並不能排除感染性肝炎。
全血球計數 (COMPLETE BLOOD COUNT)
所有疑似血管炎的病人都應進行全血球計數。許多活動性血管炎的病人有貧血與/或血小板增多 (thrombocytosis),但廣泛的發炎性疾病也是如此。嚴重貧血可能是各型血管炎嚴重腸胃道侵犯的線索。白血球計數與分類也可能是感染或血液惡性腫瘤存在的線索。然而,白血球增多通常是非特異性的,也常由使用糖皮質素所引起。在多數未治療的嗜酸性球性肉芽腫性多血管炎(Churg–Strauss)病人中可發現絕對嗜酸性球升高,而大於 1000 cells/µL 的計數有助於區分此疾病與氣喘及異位性體質 (atopy)。
急性期反應物 (ACUTE PHASE REACTANTS)
紅血球沉降速率 (erythrocyte sedimentation rate, ESR) 與 C 反應蛋白 (C reactive protein, CRP) 濃度在許多血管炎病人中升高,但這些檢查的診斷敏感度與特異度都不特別高。因此,這些檢查在建立或排除血管炎診斷上都不特別有幫助。此外,ESR 與 CRP 的濃度與疾病的分期或嚴重度並無良好相關。ESR 與 CRP 在皮膚擬似血管炎的情況,以及許多嚴重的系統性疾病(包括感染與惡性腫瘤)中也常升高。活動性血管炎的病人可能有正常的 ESR 與 CRP 數值,而病人也可能在治療後這些標記持續升高的情況下仍維持臨床緩解。
自體免疫血清學 (AUTOIMMUNE SEROLOGIES)
檢測自體抗體常是建立血管炎類型的關鍵環節,但重要的是要認識到血清學檢查本身從不具診斷性,且絕不應取代臨床判斷。對於任何以肺出血與/或伴隨活動性尿沉渣 (active urinary sediment) 之急性腎功能不全就診的病人,建議檢測 ANCA 與抗腎絲球基底膜 (anti–glomerular basement membrane, GBM) 抗體,以及抗核抗體 (antinuclear antibodies, ANA) 以處理系統性紅斑性狼瘡的鑑別可能性。ANCA 相關血管炎 (ANCA-associated vasculitis, AAV) 與狼瘡可表現為皮膚血管炎,但抗 GBM 疾病不會,因此後者主題不再進一步討論。
抗嗜中性球細胞質抗體 (ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES, ANCA)
約 90% 的顯微鏡下多血管炎病人、75% 的肉芽腫性多血管炎病人,以及 40% 的嗜酸性球性肉芽腫性多血管炎病人 ANCA 檢測呈陽性。18-21 現代的 ANCA 檢測包括對嗜中性球進行免疫螢光染色以辨識細胞質型 (c-ANCA) 或核周型 (p-ANCA) 圖樣,以及針對特定自體抗原(PR3 與 MPO)的酵素連結免疫吸附分析 (enzyme-linked immunosorbent assays, ELISAs)。21-23 anti-PR3 與 anti-MPO 抗體陽性檢測對 AAV 的特異度相當高,22-24 但在無 anti-MPO 抗體情況下 p-ANCA 染色的特異度則很低。因此,為診斷血管炎之目的,以 ELISA 檢測 ANCA 呈陽性才是判定 ANCA 陽性的關鍵。ANCA 陽性檢測的預測值取決於情境。在切片證實的血管炎或血管炎切片的臨床「替代指標」(如瀰漫性肺泡出血或伴隨「活動性」尿沉渣的急性腎衰竭)病例中,anti-PR3/MPO ANCA 陽性檢測具高度特異性。在非特異性的體質性與肌肉骨骼症狀情境中,ANCA 檢測的陽性預測值則較低。
抗核抗體 (ANTINUCLEAR ANTIBODIES, ANA)
當懷疑系統性紅斑性狼瘡或修格蘭氏症候群時,檢測 ANA 與相關自體抗體是有用的。ANA 檢測對狼瘡的診斷極為敏感(>95%)但不具特異性。除了 anti-Ro (SSA) 抗體之外,針對特定核抗原的其他檢測,包括雙股 DNA、Smith、RNP 與 La (SSB),僅應在 ANA 呈陽性且仍考慮狼瘡時才進行。只有 80% 的修格蘭氏症候群病人對類風濕因子、anti-Ro (SSA) 或 anti-La (SSB) 抗體中任一項檢測呈陽性,因此陰性檢測並不能排除此診斷。
類風濕因子 (RHEUMATOID FACTOR, RF)
檢測類風濕因子在建立血管炎的診斷或特定類型上很少有用。類風濕因子對修格蘭氏症候群或冷凝球蛋白血症性血管炎的敏感度與特異度都很低。雖然至少 70% 的類風濕性關節炎病人類風濕因子檢測呈陽性,但在超過 95% 的類風濕性血管炎 (rheumatoid vasculitis) 病人中此檢查呈陽性。25 然而,由於類風濕性血管炎通常發生於患有長期、嚴重類風濕性關節炎的病人,此類檢測的附加價值不大。
副蛋白(異常免疫球蛋白,包括冷凝球蛋白)(PARAPROTEINS, ABNORMAL IMMUNOGLOBULINS, INCLUDING CRYOGLOBULINS)
冷凝球蛋白 (cryoglobulins) 是在低溫下沉澱的免疫複合體(免疫球蛋白及其標的抗原),與血管炎為顯著成分的臨床症候群相關(見第 144 章)。冷凝球蛋白血症最常源於 C 型肝炎病毒的慢性感染,但類風濕性關節炎、系統性紅斑性狼瘡、修格蘭氏症候群與血液惡性腫瘤也都與冷凝球蛋白血症相關。檢測冷凝球蛋白需要對檢體的處理與處置給予仔細的注意,因為數個步驟中任一步驟操作不當都會導致高偽陰性率。同樣地,標準的血清蛋白電泳檢測可能無法偵測某些免疫球蛋白株 (clones),而免疫固定電泳 (immunofixation electrophoresis) 是篩檢株性免疫球蛋白較全面的方法。血管炎也曾與在無冷凝球蛋白血症情況下的單株丙種球蛋白病 (monoclonal gammopathies)(骨髓瘤、漿細胞瘤或淋巴瘤)相關。26
補體 (COMPLEMENT)
總溶血補體以 CH50 測量,但由於此分析法繁瑣且實驗室間變異性大,通常測量血清中的補體蛋白 C3 與 C4 即已足夠,且在數種情境下對評估皮膚血管炎病人很有用。在冷凝球蛋白血症性血管炎病人中,C4 濃度通常嚴重耗竭,而 C3 濃度則較不耗竭或甚至正常。27,28 70% 的類風濕性血管炎病人這些成分之一或二者偏低,25 這很有用,因為類風濕性關節炎一般不與低循環補體相關。相對地,由於系統性紅斑性狼瘡在多種情境下常與低補體相關,低補體有助於提高對狼瘡的懷疑,但對 SLE 的血管炎並不具特異性。一部分皮膚血管炎表現為蕁麻疹(因而稱為蕁麻疹性血管炎 urticarial vasculitis)、但無法被診斷為狼瘡或其他潛在疾病的病人,會有補體耗竭(第 138 章)。
感染性疾病的選擇性檢測 (SELECTED TESTING FOR INFECTIOUS DISEASES)
許多感染可造成包含血管炎或擬似血管炎的皮膚病灶。C 型肝炎病毒的慢性感染與冷凝球蛋白血症性血管炎強烈相關,它也可能在無冷凝球蛋白情況下與結節性多動脈炎相關。29 在廣泛採用疫苗接種計畫之前,慢性 B 型肝炎病毒感染是許多結節性多動脈炎病例的病因。30
因此,患有已知或疑似侵犯小型或中型動脈血管炎的病人,應篩檢 B 型與 C 型肝炎感染。心內膜炎 (Endocarditis) 可同時造成真正的血管炎(推測是經由免疫複合體沉積)以及經由敗血性栓子 (septic emboli) 擬似血管炎的病灶。對某些疑似小血管血管炎的病人,血液培養是適當的。有趣的是,菌血症 (bacteremia) 可能是 ANCA 檢測陽性而與血管炎無關的一個原因。31
許多種類多樣的感染都曾被牽涉於引起繼發性血管炎,通常為小血管且局限於皮膚。因此,針對特定微生物的檢測應基於暴露史或可疑的臨床症候群(例如喉嚨痛或急性腹瀉)。
數種不常見的感染會直接感染並損害血管內皮細胞,因而產生可被視為血管炎或血管炎擬似病灶的病灶;許多微生物都曾被牽涉,大多以病例報告的形式呈現。
濫用藥物篩檢 (SCREENING FOR DRUGS OF ABUSE)
在某些懷疑血管炎的臨床情況下,針對常用濫用藥物的毒物學篩檢可能是適當的。特別是,古柯鹼與甲基安非他命都曾與皮膚血管炎與/或動脈血管痙攣相關,而經鼻吸入古柯鹼可產生如 ANCA 相關血管炎所見一樣嚴重的破壞性鼻部疾病,雖然某些臨床特徵可能有助於區分這 2 種病因。32,33 現在有一種廣為人知的破壞性血管病變/血管炎,與暴露於左旋咪唑 (levamisole) 相關,這是一種驅蟲與免疫調節藥物,是非法古柯鹼的摻雜物,尤其在北美。左旋咪唑可導致特徵性的壞疽性病灶以及 anti-MPO 與 anti-PR3 ANCA 二者皆陽性的檢測結果。34 左旋咪唑的使用可藉由尿液檢測確認。
影像學 (IMAGING)
胸部影像 (CHEST IMAGING)
胸部 X 光攝影是任何疑似血管炎病人的適當篩檢檢查。對於有肺部症狀的病人,通常需要電腦斷層 (computed tomography, CT),因為平片常無法偵測小結節或細微但顯著的浸潤。在已診斷肉芽腫性多血管炎、顯微鏡下多血管炎或嗜酸性球性肉芽腫性多血管炎(Churg–Strauss)的病人中,即使是無症狀病人,篩檢性 CT 也適用於分期目的並建立基準。若懷疑聲門下狹窄,頸部/氣管的 CT 可作為直接喉鏡檢查的有用輔助。
鼻竇影像 (SINUS IMAGING)
鼻竇侵犯在肉芽腫性多血管炎與嗜酸性球性肉芽腫性多血管炎中極為常見,且即使對耳鼻喉科醫師而言,以理學檢查評估鼻竇的能力也很有限。鼻竇 CT 可協助評估肉芽腫性多血管炎或嗜酸性球性肉芽腫性多血管炎的可能性,並在做出其中一種診斷且開始治療後,對疾病的分期與再分期有用。然而,這些疾病中鼻竇發炎的 CT 表現無法與其他鼻竇炎病因區分,且先前曾因血管炎受損的病人常有持續性的異常。鼻部發炎以理學檢查評估較 CT 更佳。
血管攝影 (ANGIOGRAPHY)
血管攝影在大血管與中血管血管炎的診斷與處置中扮演核心角色。傳統的導管式顯影劑血管攝影具有最高解析度,但屬侵入性程序,且仍無法顯影多數的小血管。基於 CT 與磁振 (magnetic resonance, MR) 的血管攝影正日益取代導管式血管攝影的使用。35
血管攝影在皮膚血管炎診斷中的角色,僅限於建立潛在的血管炎類型(例如腹部血管攝影顯示結節性多動脈炎中的多發性動脈瘤與狹窄),或評估有壞疽病人的動脈供應。
其他診斷檢查 (OTHER DIAGNOSTIC STUDIES)
神經傳導檢查與肌電圖 (NERVE CONDUCTION STUDIES AND ELECTROMYOGRAPHY)
神經傳導檢查絕不應取代完整的神經學檢查,且多數有血管炎神經學表現的病人並不需要此類檢查。因此,不建議以神經傳導檢查篩檢無症狀病人,但它有助於提供神經病變的客觀證據,並區分壓迫性(即機械性)與非壓迫性神經病變,後者類型包括血管炎所致以及許多其他內科病因所致的神經病變。肌電圖 (electromyography, EMG) 可確立肌病變 (myopathy) 的存在,但無法確立其病因。神經傳導檢查會疼痛,且需要不一定隨時可得的專業技術。
聽力檢查 (AUDIOLOGY TESTING)
聽力圖 (audiogram) 對診斷並區分傳導性與/或感音神經性聽力喪失至關重要。聽力喪失是小血管血管炎常被遺漏的表現,包括在年長病人中。36 感音神經性聽力喪失是一種顱神經病變,可能迅速導致不可逆的聽力喪失。雖然聽力圖一般不適用於篩檢無症狀病人,但建議為所有已確立 ANCA 相關血管炎(肉芽腫性多血管炎、顯微鏡下多血管炎或嗜酸性球性肉芽腫性多血管炎)診斷的病人進行基準聽力圖。
各種血管炎概述 (SUMMARIES OF THE VASCULITIDES)
顯微鏡下多血管炎 (MICROSCOPIC POLYANGIITIS)
顯微鏡下多血管炎 (microscopic polyangiitis, MPA) 是一種侵犯小型、有時也侵犯中型血管的多系統血管炎。37,38 多數病人會發展出寡免疫性腎絲球腎炎 (pauci-immune glomerulonephritis),而肺出血、周邊與顱神經病變、肌肉骨骼與體質性症狀也很常見;心臟與腸胃道侵犯則較不常見。多數顯微鏡下多血管炎病人 ANCA 呈陽性,通常對抗髓過氧化酶 (MPO) 的抗體具專一性。39 雖然快速進展性腎絲球腎炎與/或瀰漫性肺泡出血常導致診斷,但顯微鏡下多血管炎常以局限於肌肉骨骼與體質性症狀的延長前驅期 (prodrome) 為特徵。40
皮膚在顯微鏡下多血管炎中常受侵犯。20 最常見的皮膚病灶是可觸性紫斑,其病理與其他類型的白血球碎裂性血管炎無法區分17,41 (Fig. 139-6)。中型血管的血管炎導致指(趾)缺血、皮下結節、網狀青斑 (livedo reticularis) 與深部潰瘍,在顯微鏡下多血管炎中可罕見發生。
肉芽腫性多血管炎 (GRANULOMATOSIS WITH POLYANGIITIS)
肉芽腫性多血管炎涵蓋顯微鏡下多血管炎的所有特徵,但也具有許多由壞死性肉芽腫性發炎所造成的額外表現,而這 2 種症候群目前被視為不同的疾病實體。約 90% 的病人有上呼吸道(鼻腔、鼻竇、耳咽管與中耳)的慢性發炎,常(但非總是)作為初始表現。18,42,43 空洞性肺結節 (cavitary pulmonary nodules)、眼窩假性腫瘤 (orbital pseudotumor) 與聲門下狹窄也是常見且重要的特徵。眼睛的血管炎(鞏膜炎與表層鞏膜炎)在肉芽腫性多血管炎中也較顯微鏡下多血管炎遠為常見。18,37,42-44 多數有多重器官系統侵犯的肉芽腫性多血管炎病人 ANCA 檢測呈陽性。44,45 此疾病中多數的 ANCA 類型是 C-ANCA/anti-PR3;然而 P-ANCA/anti-MPO 也不少見。看似局限於上呼吸道的病人僅約 70% 的病例 ANCA 呈陽性,這可能使診斷更具挑戰性。21,44
除了可觸性紫斑與其他小血管血管炎典型表現之外,肉芽腫性多血管炎也出現額外的皮膚病灶,反映出血管炎與壞死性肉芽腫疾病的組合,包括伴隨丘疹的嗜中性球性與肉芽腫性皮膚炎(尤其在肘部伸側表面)、皮下結節與潰瘍17,46-49 (Figs. 139-7 and 139-8)。血管炎與血管外肉芽腫疾病有時可見於同一切片中,有助於診斷。如同 MPA,也可見到可歸因於中型血管侵犯的表現 (Fig. 139-9)。
嗜酸性球性肉芽腫性多血管炎 (EOSINOPHILIC GRANULOMATOSIS WITH POLYANGIITIS)
嗜酸性球性肉芽腫性多血管炎常被歸入 ANCA 相關血管炎,因為約 40% 的病人 ANCA 檢測呈陽性,50 且嗜酸性球性肉芽腫性多血管炎與肉芽腫性多血管炎及顯微鏡下多血管炎二者的特徵都有相當的重疊。然而,嗜酸性球性肉芽腫性多血管炎具有獨特特徵,最顯著的是氣喘病史(常為嚴重或控制不良)與血液嗜酸性球增多。鼻息肉、體質性症狀與皮疹(皆為異位性體質的典型)也很常見。胸部影像上肺浸潤(嗜酸性球性肺炎 eosinophilic pneumonia)的存在,提供了與氣喘的重要區別。嗜酸性球性肉芽腫性多血管炎中重度血管炎最常見的表現是急性周邊神經病變,伴隨心臟、腸胃道、腦與眼睛的侵犯則較不常見。腎絲球腎炎與肺出血分別發生於約 10% 與少於 5% 的嗜酸性球性肉芽腫性多血管炎病人。19,51,52
皮膚疾病在嗜酸性球性肉芽腫性多血管炎中較系統性肉芽腫性血管炎或顯微鏡下多血管炎更為常見。19,51,52 可觸性與非可觸性紫斑(組織學上反映血管炎)約占皮膚病灶的 50%。17 嗜酸性球性皮膚炎與富含嗜中性球及嗜酸性球的肉芽腫性皮膚炎也很常見,並產生紅斑性斑、丘疹與結節。如同肉芽腫性多血管炎,小血管血管炎與血管外嗜酸性球性與/或肉芽腫性疾病有時可見於同一切片中。雖然嗜酸性球性肉芽腫性多血管炎的皮膚病灶常含有嗜酸性球(無論有無血管炎),但富含嗜酸性球的皮膚切片並非此疾病所獨有,也可見於其他類型的血管炎。
冷凝球蛋白血症性血管炎 (CRYOGLOBULINEMIC VASCULITIS)
見第 144 章「冷凝球蛋白血症與冷凝纖維蛋白原血症」(Cryoglobulinemia and Cryofibrinogenemia)。
IgA 血管炎 (Henoch–Schönlein) (IGA VASCULITIS [HENOCH–SCHÖNLEIN])
見第 138 章「皮膚壞死性小靜脈炎」(Cutaneous Necrotizing Venulitis)。
與其他自體免疫疾病相關的血管炎 (VASCULITIS ASSOCIATED WITH OTHER AUTOIMMUNE DISEASES)
血管炎在系統性紅斑性狼瘡病人(10% 至 36%)53,54 與修格蘭氏症候群病人(10%)55 中相對常見,但在類風濕性關節炎中現在已極為罕見。56,57 這些疾病中的血管炎主要侵犯小血管,伴隨部分中血管疾病。此血管炎可能造成周邊神經病變、腸胃道缺血與中樞神經系統疾病,其他內臟侵犯則很罕見。發炎性腸道疾病 (Inflammatory bowel disease) 與復發性多發軟骨炎58 也曾與侵犯小型、中型或大型血管的血管炎相關。
惡性腫瘤相關血管炎 (MALIGNANCY-ASSOCIATED VASCULITIS)
在時間上與實質固態惡性腫瘤相關的血管炎,已在許多病例報告與少數較大型系列中被描述。59 多種惡性腫瘤都曾被牽涉。皮膚的小血管血管炎似乎最為常見,而腫瘤手術切除後(無其他治療)血管炎緩解的報告,提示有因果關係。具有或不具相關副蛋白血症 (paraproteinemias) 的血液惡性腫瘤可導致血管炎。26 另一方面,評估可能血管炎病人所伴隨的全面醫學檢查與影像,可能導致在血管炎病人身上發現惡性腫瘤,而這 2 種診斷可能無關。
皮膚白血球碎裂性血管炎 (CUTANEOUS LEUKOCYTOCLASTIC ANGIITIS)
至此所描述的所有疾病與症候群,都是由臨床與實驗室參數的組合所定義。相對地,皮膚白血球碎裂性血管炎是一個組織學術語,以無系統性疾病證據來定義。此術語無疑涵蓋了許多病因,列於此清單中僅作為提醒:皮膚血管炎並非總是系統性疾病的標記。若且唯若病人有切片證實的血管炎、無其他器官系統受血管炎侵犯的證據,且無臨床或實驗室證據支持某特定型態血管炎或共存的自體免疫發炎性疾病,才應暫時做出皮膚白血球碎裂性血管炎的診斷。此術語出現於 1994 年 Chapel Hill 共識會議的命名法中,以承認局限於皮膚的血管炎相對常見。7 據估計,約 20% 的此類病例繼發於廣泛的感染,另有 20% 與藥物暴露相關。17 然而,將僅限於皮膚的血管炎標記為一個獨立的疾病實體相當有問題,且所有此類病人都應仔細追蹤,以防可能演變為更為系統性的血管炎型態。
藥物誘發性血管炎 (DRUG-INDUCED VASCULITIS)
藥物反應被牽涉於約 20% 的皮膚小血管血管炎病例中,15 但藥物誘發性疾病的確切頻率難以確立,這是由於病例通報不完整以及難以確切建立任一藥物的因果關係。多數類別的藥物都曾被牽涉為引起血管炎,但某一藥物的報告數目可能代表通報偏差,而非相對風險。14,60 皮膚小血管疾病是常態,但中血管血管炎與內臟侵犯也曾被報告。文獻無疑偏向嚴重病例,但有許多嚴重或致命內臟器官侵犯(尤其是腎、肺與肝)的報告。現在也有一個廣為接受的藥物誘發性 ANCA 相關血管炎亞群(抗體通常針對髓過氧化酶),尤其涉及丙基硫氧嘧啶 (propylthiouracil) 與相關藥物、聯胺嗪 (hydralazine),但也包括其他藥物。61,62
藥物誘發性血管炎的皮膚表現與其他小血管血管炎病因無法區分(即通常為紫斑,但有時伴隨其他病灶)(Fig. 139-10)。對所有疑似血管炎的病人,全面檢視所有處方藥、非處方藥、非法藥物與「另類」藥物及補充品至關重要。建立藥物誘發性血管炎的診斷可能促使避免以糖皮質素與免疫抑制藥物治療,而改以停藥後的臨床追蹤。然而,在此類病例中必須謹記 2 個重要的注意事項:(1) 單純停藥可能無法使疾病緩解,且可能存在需要治療的重症;(2) 藥物誘發性血管炎的診斷可能有誤,病人實際上罹患的是另一型態的血管炎;因此,對所有病人都需要仔細且延長的追蹤。
貝賽特氏病 (BEHÇET DISEASE)
見第 141 章「Adamantiades-Behçet 病」(Adamantiades-Behçet Disease)。
結節性多動脈炎 (POLYARTERITIS NODOSA)
中型血管(即小型或中型肌肉性動脈)的特發性血管炎可表現於一個或多個器官系統。「典型」結節性多動脈炎侵犯多重器官系統,並以皮膚疾病、肌痛、高血壓(源於腎動脈侵犯)、腹痛、神經病變與/或睪丸痛的某種組合表現。然而,許多病人並不以全套的表現就診,而局限於肌肉與神經、單一內臟器官或皮膚的疾病也都是有完整描述的變異型。當疾病局限於皮膚時,有時稱為皮膚型結節性多動脈炎 (cutaneous polyarteritis nodosa);然而,其中部分病人日後會在其他器官出現疾病。解讀結節性多動脈炎的文獻有問題,因為此術語過去曾被用來描述數種現在被視為不同疾病的血管炎型態(尤其是顯微鏡下多血管炎)。63,64 歷史上,許多結節性多動脈炎病例與慢性 B 型肝炎感染相關,但在已將 B 型肝炎疫苗接種列為常規的國家,這種相關性已明顯下降。64 至於剩下、仍被稱為結節性多動脈炎的這個罕見疾病實體究竟代表一個、少數幾個或許多病因,甚至是否有一個統一的基本病理生理學(例如免疫複合體疾病),仍不清楚。結節性多動脈炎最常見的皮膚特徵是網狀青斑 (livedo reticularis/racemosa)(腿部皮膚血管呈現的蕾絲狀圖樣,且不一定容易與較表淺血管血管收縮的良性後果區分)(Fig. 139-11)、疼痛性皮膚結節或潰瘍 (Fig. 139-12),以及指(趾)缺血。65,66 這些表現反映出皮下「中型」動脈的血管炎,這些動脈常太深而無法以常規穿孔切片取樣。15
川崎病 (KAWASAKI DISEASE)
見第 142 章「川崎病」(Kawasaki Disease)。
原發性中樞神經系統血管炎 (PRIMARY VASCULITIS OF THE CNS)
這種罕見疾病局限於中樞神經系統(因此無皮膚發現),並以腦病變、多發性小中風以及常見的頭痛症狀表現。67,68
巨細胞動脈炎 (GIANT CELL ARTERITIS)
巨細胞動脈炎(顳動脈炎 temporal arteritis)目前被視為嚴格屬於大於 50 歲成人的疾病,且隨年齡增長而明顯更為常見。它主要是北歐血統人士的疾病。顱動脈炎是常見特徵,此術語意指頸動脈一條或多條分支的狹窄或阻塞,以產生頭痛(70% 至 80%)、顎跛行(50%),以及單眼(罕見為雙眼)失明(15%)。風濕性多肌痛 (Polymyalgia rheumatica),包括肩部與髖部帶狀區的疼痛與僵硬,見於至少 30% 至 40% 的病人,且可在無顱部疾病情況下發生。體質性症狀很常見,包括發燒、倦怠與體重減輕。主動脈及其主要分支的侵犯產生類似高安動脈炎的症狀,見於 15% 至 20% 的病人。GCA 的診斷通常經由顳動脈切片確認 (Fig. 139-13)。顳動脈可觸性結節見於 30% 至 40% 的病例,且是檢查時可見的 GCA 唯一皮膚表現。頭皮壞死 (Scalp necrosis) 與指(趾)缺血是罕見的併發症。69,70
高安動脈炎 (TAKAYASU ARTERITIS)
高安動脈炎是一種罕見型態的血管炎(盛行率 <1:100,000),侵犯主動脈及其主要分支。71-76 許多病人在年輕成人時被診斷,且 90% 為女性。高安動脈炎的典型表現是肢體跛行。頭暈、體質性症狀與嚴重高血壓(源於腎動脈狹窄)也很常見。可發生腦梗塞。伴隨心絞痛或梗塞的冠狀動脈阻塞與腸缺血是較不常見但危及生命的併發症。脈搏消失、血壓讀數異常與動脈雜音是常見但非普遍的發現。診斷藉由血管攝影做出。類似結節性紅斑或壞疽性膿皮症 (pyoderma gangrenosum) 的病灶77 在高安動脈炎中曾被反覆描述,結節性病灶的病理常(但非總是)顯示血管炎,從而與典型的結節性紅斑不同。78,79 雖然鎖骨下動脈的完全阻塞很常見,但指(趾)缺血很罕見。
臨床病程與預後 (CLINICAL COURSE AND PROGNOSIS)
血管炎的嚴重度差異很大,但所有具名的疾病都有對重要器官造成永久性損害的潛力。所有型態的血管炎都是可治療的,目標是在損害發生之前做出診斷並開始治療。在以免疫抑制藥物成功治療後,某些型態的血管炎較其他型態更可能復發。具 anti-PR3 抗體的肉芽腫性多血管炎在無長期治療情況下復發率遠超過 50%,而具 anti-MPO 抗體的顯微鏡下多血管炎復發率則低於 50%。結節性多動脈炎與嗜酸性球性肉芽腫性多血管炎的復發率也低於 50%,雖然在嗜酸性球性肉芽腫性多血管炎中,慢性氣喘與/或鼻竇鼻部疾病在多數病人會復發並需要長期治療。IgA 血管炎在兒童通常為單相 (monophasic) 或在數次發作後緩解,但在成人較可能轉為慢性。繼發於 C 型肝炎病毒的冷凝球蛋白血症性血管炎通常(但非總是)可藉由根除病毒而治癒,而在未根除病毒情況下則通常復發。與結締組織疾病相關的血管炎可為單相、間歇性或慢性,嚴重度差異很大。治療本身也是發病率的來源。糖皮質素用於幾乎所有型態的重度血管炎,且在高劑量或長期使用(即使在中或低劑量)下都帶有許多風險。幾乎所有其他用於治療血管炎的藥物都會增加感染風險,但基於此及其他理由,仍被認為較糖皮質素更為可取。重度血管炎侵犯重要器官的最初 3 至 6 個月死亡率約為 10%,原因為血管炎的併發症(例如肺出血、腸穿孔或心肌病變)或感染。此後,隨著治療減少而血管炎通常維持在控制中,死亡率大幅降低,但似乎仍略高於預期。80 除了高齡之外,較高死亡風險的預測因子反映出先前的器官損害:在肉芽腫性多血管炎與顯微鏡下多血管炎是腎臟損害,81 在嗜酸性球性肉芽腫性多血管炎則是心臟損害。82
處置 (MANAGEMENT)
系統性血管炎治療的一般原則 (GENERAL APPROACH TO TREATMENT OF SYSTEMIC VASCULITIS)
在為血管炎病人建立治療方案時,必須同時考量當前表現的嚴重度,以及疾病進展與復發的可能性。概要呈現於 Fig. 139-14。對於數目日增的血管炎,治療由相對大型的隨機對照試驗結果所引導。然而,對許多情況而言,臨床醫師仍仰賴從其他疾病試驗的外推,或基於小型病例系列或個人經驗的經驗性治療。對於預期會有延長病程與/或包含嚴重表現的血管炎,一般方法是規劃 2 個治療階段:緩解誘導 (remission induction) 與緩解維持 (remission maintenance)。83,84 緩解誘導通常涉及使用高劑量糖皮質素並穩定遞減劑量,並結合一短療程(3-6 個月)相當速效且強效的免疫抑制藥物。例如,環磷醯胺 (cyclophosphamide)、利妥昔單抗 (rituximab) 與甲胺喋呤 (methotrexate) 在 ANCA 相關血管炎的緩解誘導中都有其角色。緩解維持通常涉及長期使用非以環磷醯胺為基礎的方案,以使糖皮質素得以完全停用或維持在低劑量(例如 ≤10 mg prednisone daily)。然而,雖然上述方法對於 ANCA 相關血管炎、結節性多動脈炎,以及部分有臨床試驗資料引導治療的其他型態重度系統性疾病是常用的,但對於糖皮質素單獨即可能足以作為治療的其他血管炎,則較不常使用。
Fig. 139-14 內容:小血管與中血管血管炎治療的一般原則
血管炎的診斷 (Diagnosis of vasculitis)
→ 藥物暴露或活動性感染?(Drug exposure or active infection?)
- 是 → 停藥或治療感染 (Discontinue drug or treat infection)
- 否 → 評估嚴重度 (Assessment of severity)
評估嚴重度:分為非重度 (Nonsevere) 與重度 (Severe)
- 非重度 Cryo、CTD-V、EGPA、IgAV、PAN(常為自限性)或持續性藥物誘發性血管炎 → 觀察或基於症狀緩解進行治療
- 非重度 GPA、MPA(但若不治療可能變為重度)→ 緩解誘導 (Remission Induction)
- 重度 CTD-V、EGPA、IgAV、PAN,重度 Cryo、GPA、MPA → 緩解誘導 (Remission Induction)
緩解誘導 (Remission Induction) 選項:
- GC(單獨)
- GC + MTX
- GC + CYC
- GC + [CYC 或 RTX] +/– PLEX
緩解維持 (Remission Maintenance):
- 單獨密切觀察 +/– GC(常為低劑量)
- AZA、MMF、MTX 或 RTX +/– GC(常為低劑量)
臨床醫師必須始終警覺以下可能性:(1) 不同的診斷(不同型態的血管炎,或完全不同的疾病);(2) 血管炎出現額外的表現;以及 (3) 治療相關的副作用,其中部分可能擬似血管炎(感染、皮膚反應等)。照護血管炎病人的一個重要面向是判定哪些人以及何時不予治療。「觀察等待」(Watchful waiting) 在以下情況可能是合理的處置方法:(1) 診斷不明確且無主要器官系統看似受威脅;(2) 血管炎有明顯的原因或病因,且該病因為可逆(毒素/藥物)或自限性(某些感染);(3) 對於病人的症狀究竟是疾病惡化、慢性損害的結果或另一病程所致,存在不確定性。照護血管炎病人的一個關鍵環節是密切、規律的臨床追蹤。血管炎常迅速進展,且多數型態的血管炎復發率高。依血管炎型態而定,規律的門診回診應伴隨實驗室與影像學監測。密切追蹤不僅應在疾病過程開始時進行,也應在診斷後持續數年。
系統性血管炎的治療藥物 (TREATMENTS FOR SYSTEMIC VASCULITIS)
用於治療各種系統性血管炎的藥物完整清單與治療方案細節已超出本章範圍(關於特定藥物的額外資訊可參見第 184 章「糖皮質素」(Glucocorticoids)、第 190 章「細胞毒性與抗代謝藥物」(Cytotoxic and Antimetabolic Agents)、第 192 章「免疫抑制與免疫調節藥物」(Immunosuppressive and Immunomodulatory Drugs),以及第 193 章「免疫生物製劑:針對皮膚科中細胞激素、細胞激素受體與生長因子的標靶治療」(Immunobiologicals: Targeted Therapy Against Cytokines, Cytokine Receptors, and Growth Factors in Dermatology))。
糖皮質素 (GLUCOCORTICOIDS)
鑑於糖皮質素的作用迅速、反應可靠,以及醫師對其劑量與副作用的熟悉,它仍是血管炎治療的主力。糖皮質素通常是用於治療血管炎的初始藥物,且對某些型態的疾病可能是唯一使用的藥物。然而,常需額外開立其他藥物,因為維持疾病控制所需的糖皮質素劑量高得無法接受,或單靠糖皮質素無法達到疾病控制。其急性與慢性毒性常被低估,而慢性或反覆使用糖皮質素的潛在累積損害可能相當可觀。
其他免疫抑制藥物 (OTHER IMMUNOSUPPRESSIVE AGENTS)
各式各樣的額外免疫抑制藥物用於治療血管炎。烷化劑 (alkylating agent) 環磷醯胺對 ANCA 相關血管炎、以及在較小程度上對其他型態血管炎已證實的有效性,已協助確立此藥作為重度型態血管炎初始治療的照護標準。42,85-87 其他烷化劑現在很少用於血管炎。雖然環磷醯胺在許多(但當然非全部)病例中有效,它也與嚴重毒性相關,其中許多與總累積劑量有關(例如女性與男性不孕、膀胱癌)。因此,過去 30 年來出現了「節省環磷醯胺」(cyclophosphamide-sparing) 的方案,通常讓病人從環磷醯胺的初始治療療程轉換至毒性較低的免疫抑制藥物的較長療程,尤其是甲胺喋呤88,89 或硫唑嘌呤 (azathioprine)。86,90 黴酚酸酯 (Mycophenolate)、環孢素 A (cyclosporine A) 與其他藥物也曾用於維持治療。阿普斯特 (Apremilast) 現已證實對貝賽特氏病黏膜皮膚表現的治療有效。91,92
生物製劑 (BIOLOGIC AGENTS)
較近期,「生物製劑」(biologic agents)(使用重組 DNA 技術製造、用以標靶免疫系統特定成分的藥物)已被研究用於治療血管炎,常以節省環磷醯胺或節省糖皮質素為目標。近期證實利妥昔單抗(一種 B 細胞耗竭療法)在 AAV 緩解誘導上與環磷醯胺一樣有效,這被視為此領域的一項重大進展。93-95 美泊利單抗 (Mepolizumab)(一種抗介白素 5 的單株抗體)已證實對嗜酸性球性肉芽腫性多血管炎的治療有效。96 兩項試驗證實托珠單抗 (tocilizumab)(一種抗介白素 6 的單株抗體)對巨細胞動脈炎的治療有效。97,98 關於使用阿巴西普 (abatacept)(一種 CTLA-4 免疫球蛋白)治療巨細胞動脈炎的可喜資料也已發表。99 然而,檢視抗 TNF 藥物對肉芽腫性多血管炎或巨細胞動脈炎二者之有效性的研究結果則極為令人失望。100,101
有許多新的生物製劑正被考慮用於血管炎的治療,需要臨床試驗來適當地評估這些新療法。
其他治療 (OTHER TREATMENTS)
雖然各種其他藥物類別,包括秋水仙素 (colchicine)、抗生素(dapsone 與其他)以及「另類」療法,都曾被提倡用於治療各種型態的血管炎,但這些藥物的有效性一般缺乏良好證據。血漿置換 (plasma exchange) 在血管炎治療中的角色仍有爭議。有一些證據顯示血漿置換對患有 AAV 與重度腎臟疾病的病人有效,102 並可能對某些型態的冷凝球蛋白血症性血管炎有效。一項大型國際多中心隨機試驗評估血漿置換對 AAV 的療效,已接近完成。
系統性血管炎診斷與治療的常見錯誤 (COMMON ERRORS IN THE DIAGNOSIS AND TREATMENT OF SYSTEMIC VASCULITIS)
鑑於血管炎疾病表現的巨大範圍、血管炎治療的潛在毒性,以及遺漏其他鑑別診斷(尤其是感染)的嚴重後果,診斷錯誤分類是一大隱憂。僅基於理學檢查與實驗室發現即確立血管炎的確切診斷是一個常見的問題。這對皮膚疾病尤其如此,因為並非所有紫斑都由血管炎所致,也並非血管炎的所有皮膚疾病都是紫斑性的。臨床醫師應避免在未做切片的情況下做出皮膚血管炎的診斷;此原則的例外是病人已基於其他證據明確確立血管炎的診斷,但即使如此,仍可能有必要知道是什麼造成某個皮膚病灶。許多類型的皮膚病灶在以糖皮質素治療後會改善,因此對此治療的經驗性使用之反應在診斷上並無用處。治療不足或延遲開始免疫抑制治療是系統性血管炎病人的常見問題。治療不足可能採取以下形式:未能辨識多重器官系統疾病、對於已證實此類療法有用的血管炎型態延遲或不願開始糖皮質素以外的免疫抑制藥物,或免疫抑制藥物劑量不足。另一個常見錯誤是將中至高劑量糖皮質素的治療療程延長超過控制血管炎急性惡化或較嚴重表現所必需的時間。
與原發性血管炎相關的醫療緊急狀況 (MEDICAL EMERGENCIES RELATED TO THE PRIMARY VASCULITIDES)
系統性血管炎在其進展速度與醫療嚴重度上差異極大。然而,所有照護血管炎病人的臨床醫師都必須理解這些疾病常危及器官與生命,且可能迅速進展為緊急狀況。此外,血管炎即使在一段長時間緩慢變化或甚至惰性的疾病之後,仍可能迅速加劇。Table 139-2 概述數種病人最常需要緊急照護的情況,但此清單並不完整,臨床醫師必須對考慮這些以及其他可能快速進展的問題保持警覺。
Table 139-2:與原發性血管炎相關的醫療緊急狀況
| 血管炎的緊急表現 | 血管炎類型 |
|---|---|
| 眼科 (Ophthalmologic) | |
| 視力受威脅;單眼失明 | GCA, TAK |
| 鞏膜炎 (Scleritis) | GPA, MPA, RPC |
| 葡萄膜炎 (Uveitis) | BD |
| 肺 (Pulmonary) | |
| 聲門下狹窄 (Subglottic stenosis) | GPA, RPC |
| 肺泡出血 (Alveolar hemorrhage) | GPA, MPA, EGPA |
| 瀰漫性發炎性浸潤 (Diffuse inflammatory infiltrates) | GPA, MPA, EGPA |
| 肺栓塞 (Pulmonary embolus) | GPA, MPA, EGPA, BD |
| 心血管 (Cardiovascular) | |
| 惡性高血壓—腎動脈狹窄 | TAK, GCA, PAN |
| 快速發作的心肌病變/心衰竭 | EGPA |
| 心絞痛/心肌梗塞 | TAK, GCA |
| 壞疽—指(趾)缺血 | GCA, TAK, PAN, GPA, MPA |
| 動脈瘤剝離(主動脈或其分支) | PAN, BD, GCA, TAK |
| 腸胃 (GI) | |
| 腸繫膜缺血 (Mesenteric ischemia) | PAN, IgAV, GCA, TAK |
| 腎 (Renal) | |
| 腎絲球腎炎/肌酸酐上升/腎衰竭 | GPA, MPA, EGPA, IgAV, PAN, Cryo |
| 神經學 (Neurologic) | |
| 頭痛 | GCA |
| 昏厥 (Syncope) | GCA, TAK |
| 中風 (Stroke) | GPA, MPA, EGPA, BD, GCA, TAK |
| 感音神經性聽力喪失或其他顱神經病變 | GPA, MPA |
| 多發性單神經炎 (Mononeuritis multiplex) | EGPA, PAN, GPA, MPA |
| 其他危及器官的疾病 (Other Organ-Threatening Disease) | 任何系統性血管炎 |
表 139-2:與原發性血管炎相關的醫療緊急狀況。BD, 貝賽特氏病 (Behçet disease);Cryo, 冷凝球蛋白血症性血管炎 (cryoglobulinemic vasculitis);EGPA, 嗜酸性球性肉芽腫性多血管炎 (eosinophilic granulomatosis with polyangiitis);GCA, 巨細胞動脈炎 (giant cell arteritis);GPA, 肉芽腫性多血管炎 (granulomatosis with polyangiitis);IgAV, IgA 血管炎(Henoch–Schönlein);MPA, 顯微鏡下多血管炎 (microscopic polyangiitis);PAN, 結節性多動脈炎 (polyarteritis nodosa);RPC, 復發性多發軟骨炎 (relapsing polychondritis);TAK, 高安動脈炎 (Takayasu arteritis)。
圖表 (FIGURES AND TABLES)
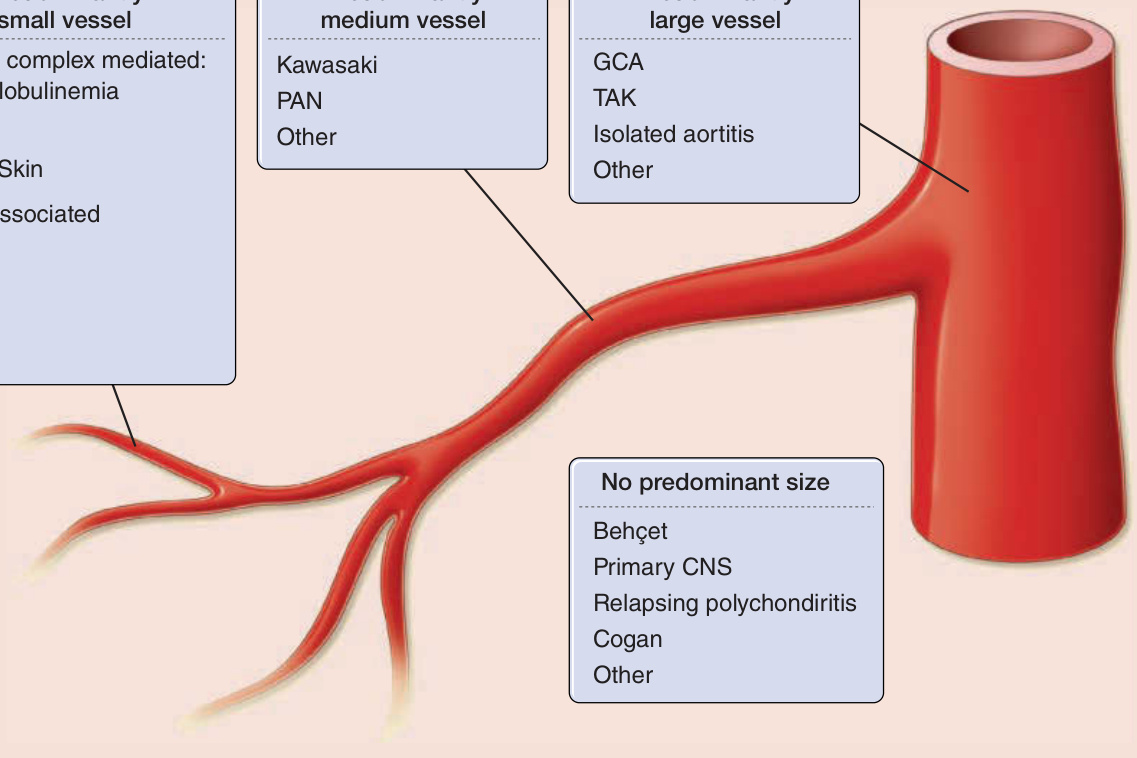
圖 139-1:原發性血管炎的分類。ANCA, 抗嗜中性球細胞質抗體 (antineutrophil cytoplasmic antibodies);CSS, Churg–Strauss 症候群;GCA, 巨細胞動脈炎 (giant cell arteritis);GPA, 肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener);HSP, Henoch–Schönlein 紫斑 (Henoch–Schönlein purpura);MPA, 顯微鏡下多血管炎 (microscopic polyangiitis);PAN, 結節性多動脈炎 (polyarteritis nodosa);TAK, 高安動脈炎 (Takayasu arteritis)。(Redrawn from Watts RA et al: Systemic vasculitis—Is it time to reclassify? Rheumatology (Oxford). 2011;50(4):643-645.)
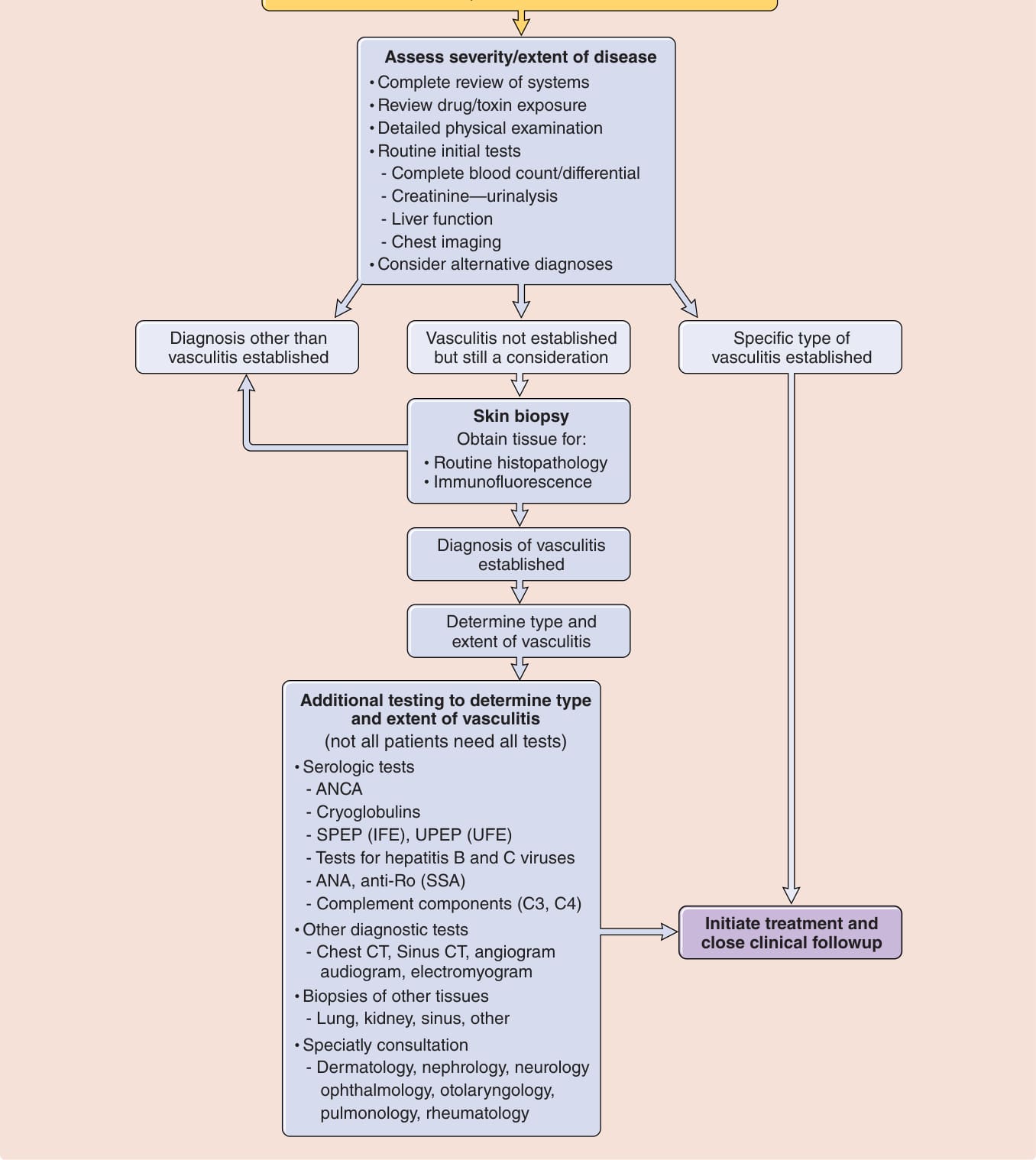
圖 139-2:對疑似血管炎且有皮膚病灶之病人的處理方法。ANCA, 抗嗜中性球細胞質抗體 (antineutrophil cytoplasmic antibodies);ANA, 抗核抗體 (antinuclear antibodies);CT, 電腦斷層 (computed tomography);IFE, 免疫固定電泳 (immunofixation electrophoresis);SPEP, 血清蛋白電泳 (serum protein electrophoresis);UPEP, 尿液蛋白電泳 (urine protein electrophoresis)。
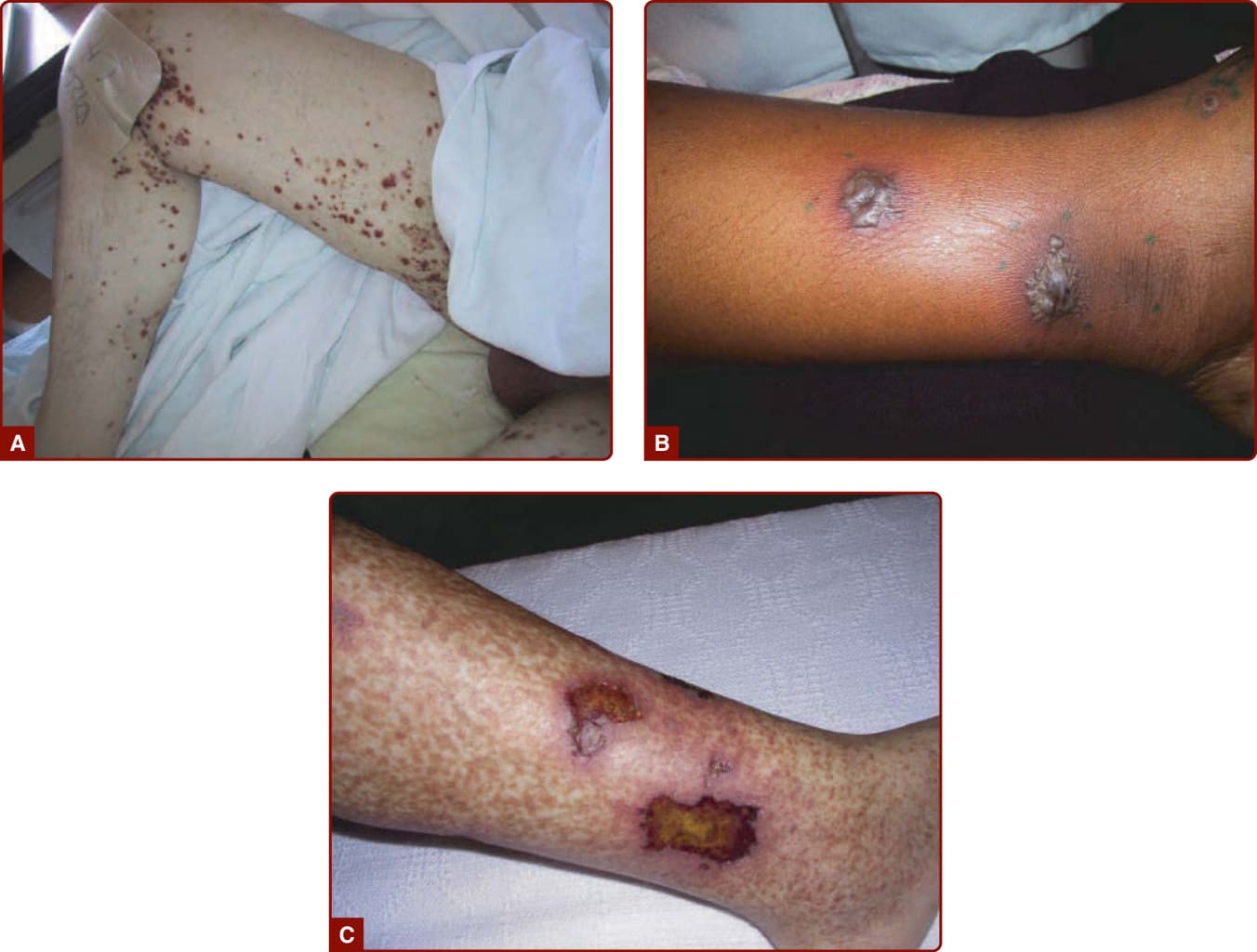
圖 139-3:系統性血管炎的不同皮膚表現。A,紫斑 (Purpura)。B,大疱 (Bullae)。C,潰瘍 (Ulcer)。
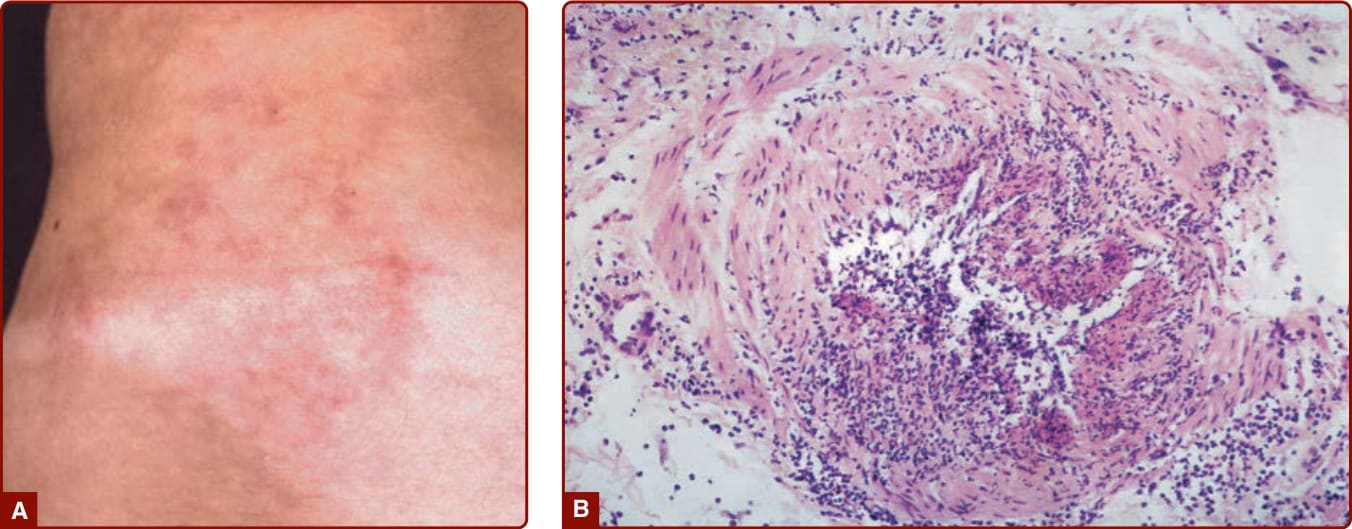
圖 139-4:A,一名結節性多動脈炎 (polyarteritis nodosa) 病人,具有由一群結節性病灶組成的「星爆狀」(starburst) 青斑。B,結節性多動脈炎皮膚病灶的組織病理,顯示節段性壞死性動脈炎 (segmental necrotizing arteritis)。
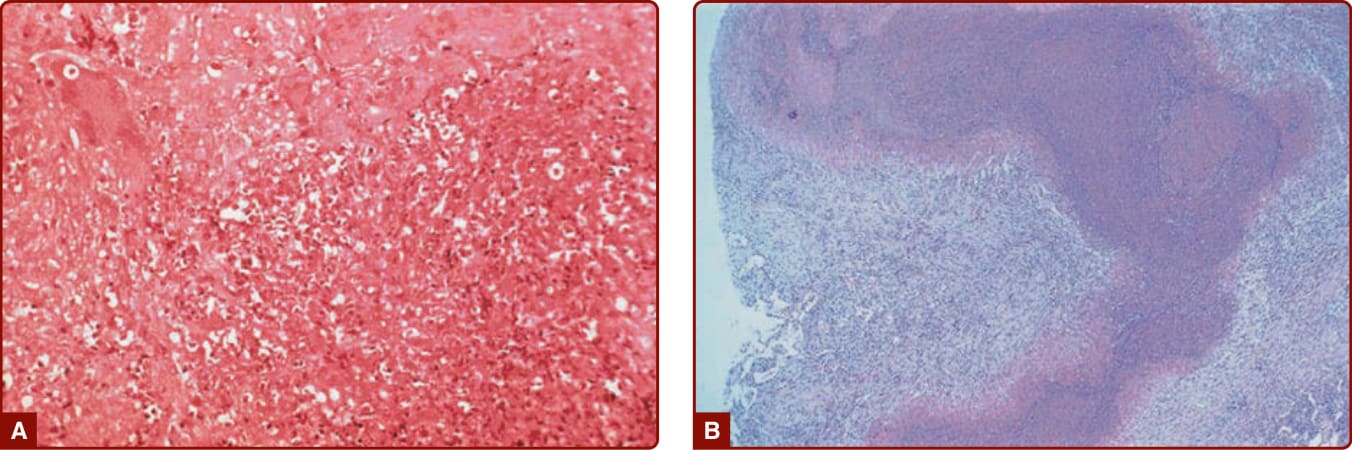
圖 139-5:A,一名肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener) 病人的肺組織病理,顯示壞死、巨細胞與混合性細胞發炎。B,一名肉芽腫性多血管炎 (Wegener) 病人開放性肺切片檢體的低倍視野中的「地圖狀壞死」(Geographic necrosis)。
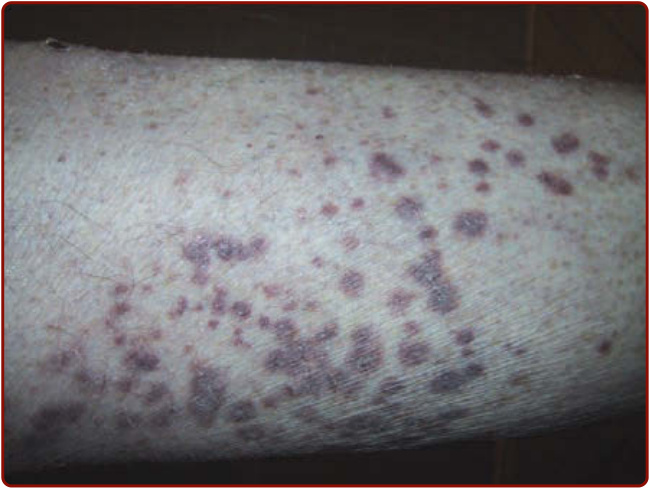
圖 139-6:一名顯微鏡下多血管炎 (microscopic polyangiitis) 病人的紫斑。
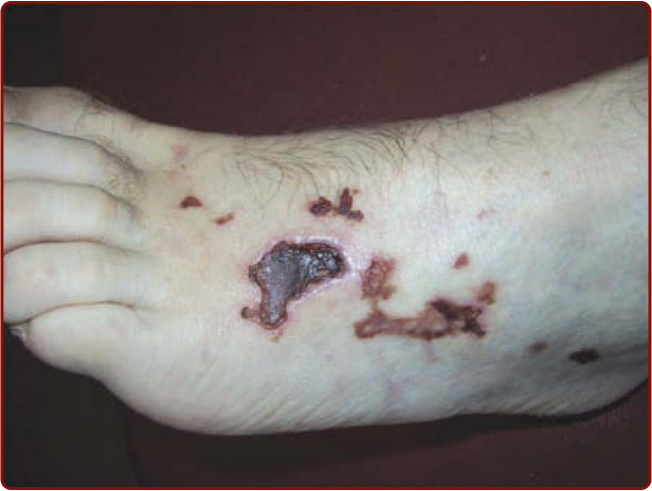
圖 139-7:一名肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener) 病人的皮膚潰瘍。
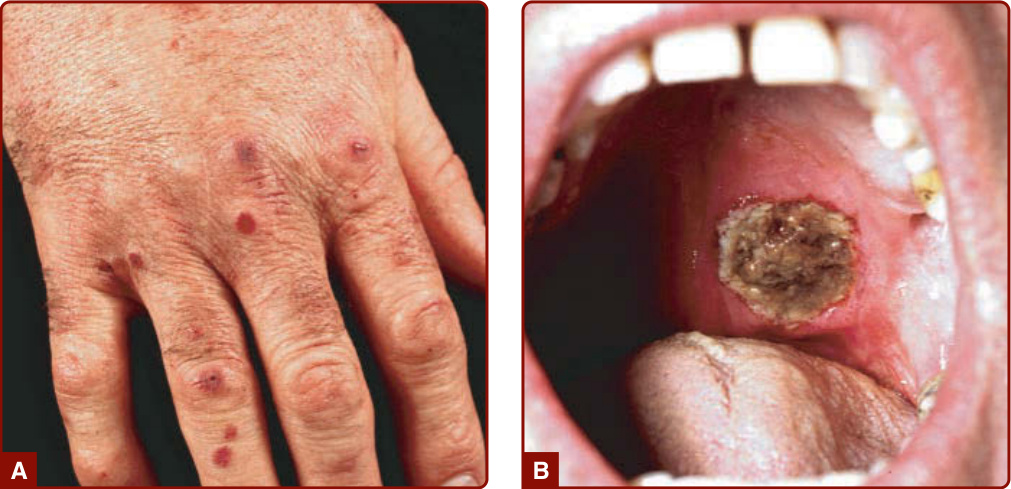
圖 139-8:肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener)。A,可觸性紫斑。B,軟顎上的深部潰瘍。
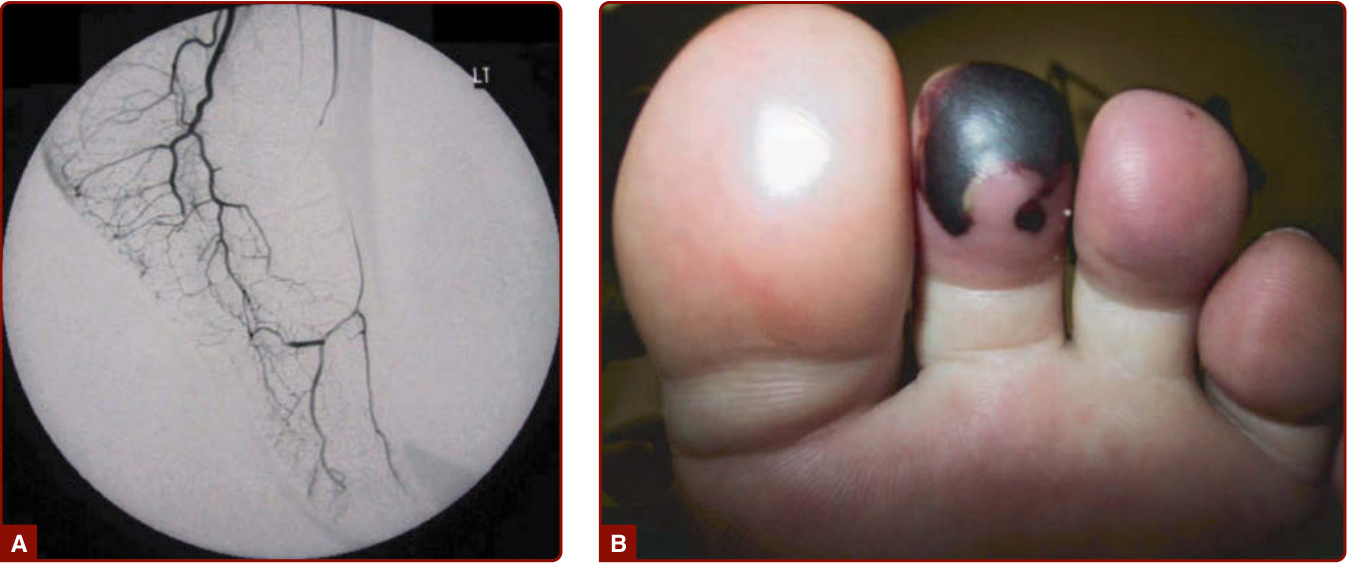
圖 139-9:肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener)。A,足背動脈 (dorsal pedal artery) 的血管炎與阻塞。B,繼發於 A 圖阻塞的壞疽性趾。
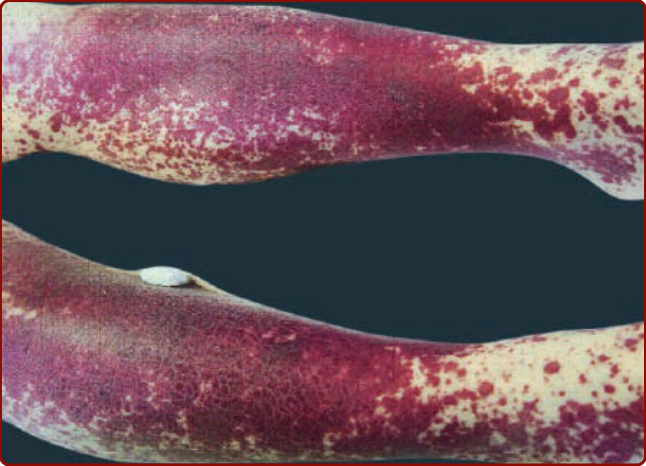
圖 139-10:因再生不良性貧血 (aplastic anemia) 投予顆粒球巨噬細胞群落刺激因子 (granulocyte macrophage colony-stimulating factor) 後的白血球碎裂性血管炎 (leukocytoclastic vasculitis)。
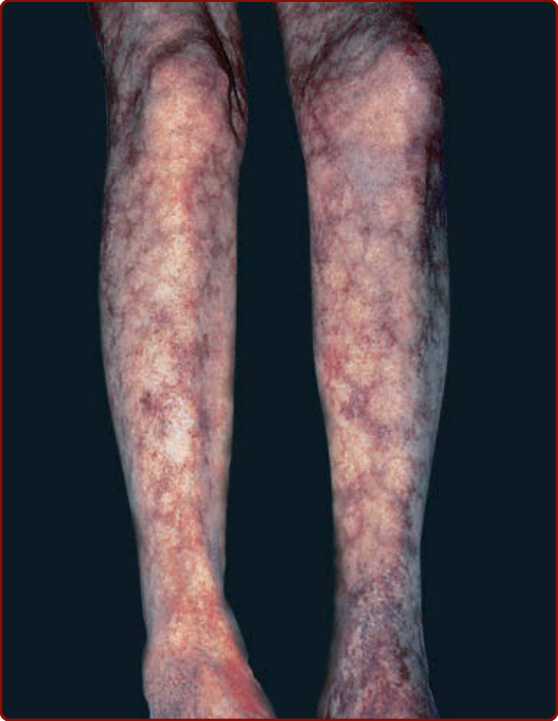
圖 139-11:網狀青斑 (Livedo reticularis)。
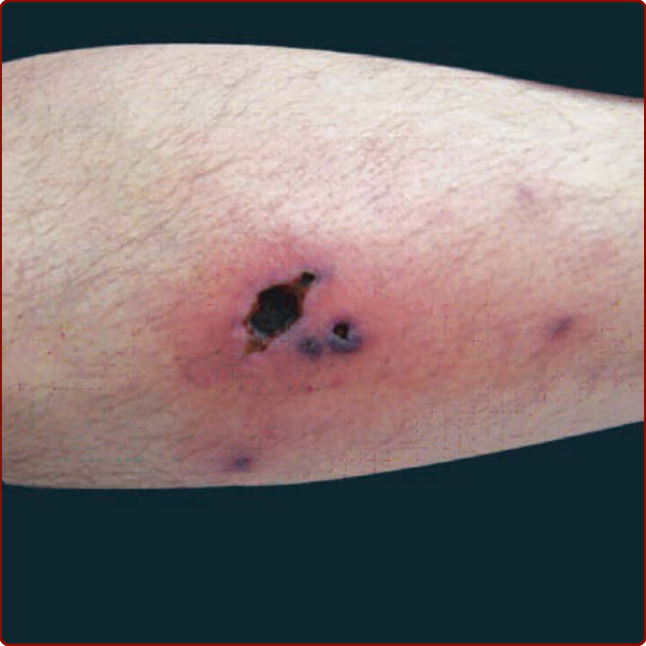
圖 139-12:一名結節性多動脈炎 (polyarteritis nodosa) 病人的腿部潰瘍。
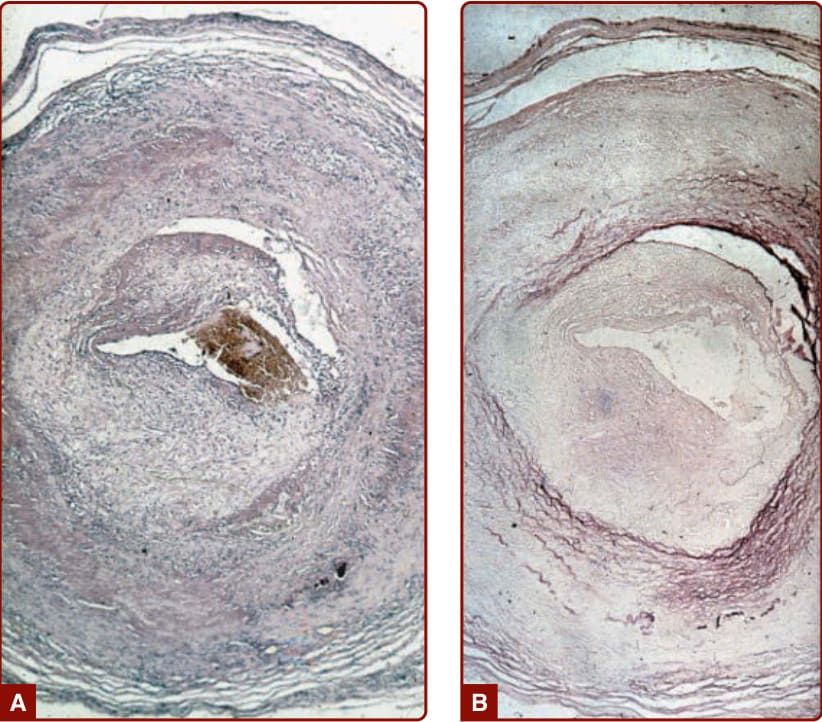
圖 139-13:A,一名巨細胞動脈炎 (giant cell arteritis) 病人的顳動脈組織病理,顯示中膜 (media) 壞死、由淋巴球組成的發炎性浸潤,以及巨細胞。亦有內膜下 (subintimal) 纖維母細胞增生與纖維化。B,彈性組織染色 (Elastic tissue stain) 顯示內彈性膜 (lamina interna) 與外彈性膜 (lamina externa) 的破壞。
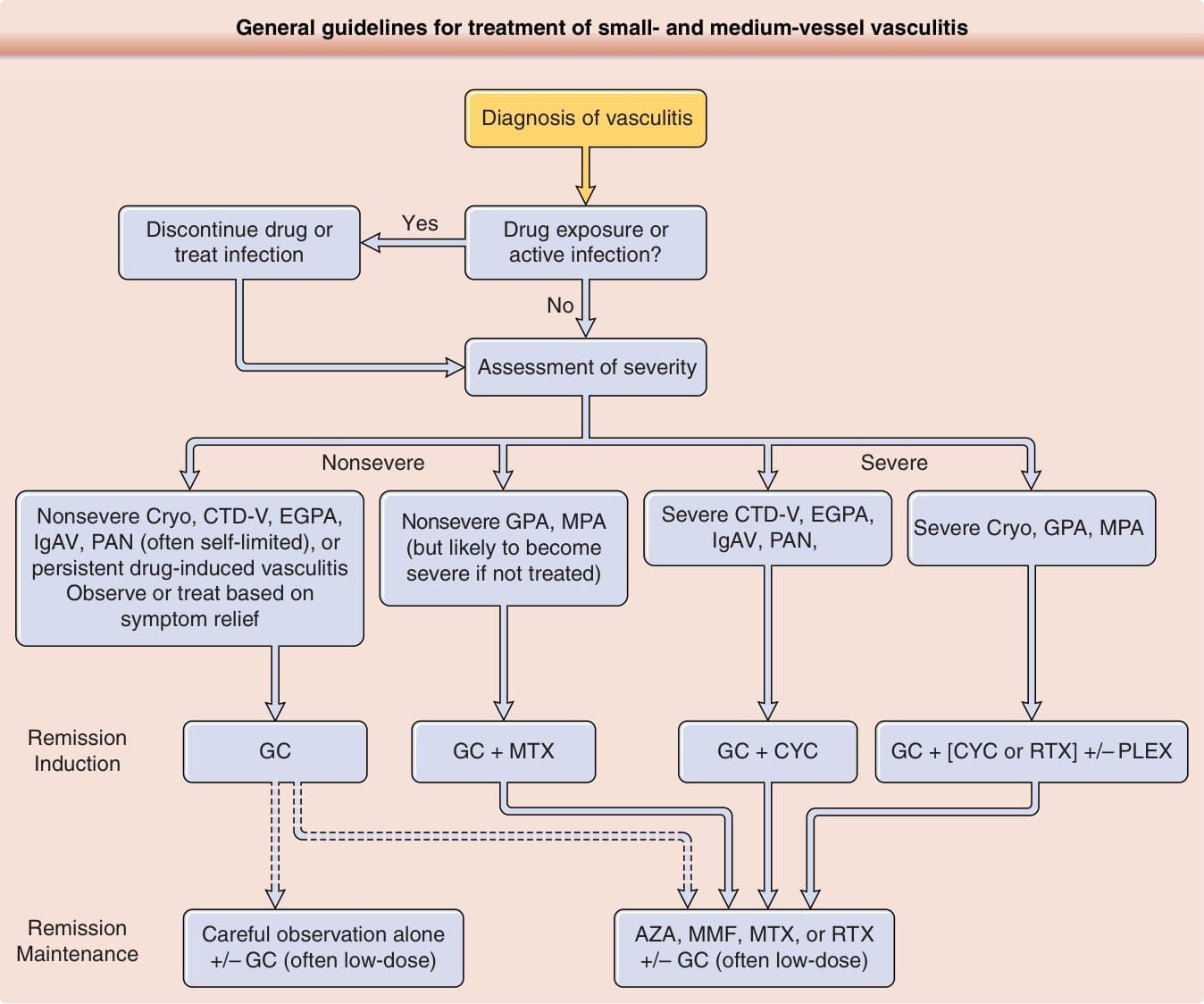
圖 139-14:小血管與中血管血管炎治療的一般原則。重要的是要認識到,方法會基於嚴重度與共病而有顯著差異,且許多型態血管炎的治療尚無高品質資料可引導。一旦診斷確立且可治療的病因被辨識或排除,便依所侵犯的器官系統與該器官系統的疾病嚴重度來評估嚴重度。例如:中樞神經系統與心臟侵犯一律視為重度;腸胃道、腎與周邊神經疾病通常為重度;皮膚表現通常非重度;肌肉骨骼非重度。虛線表示存在多種選項之處。CTD-V, 與系統性結締組織疾病相關的血管炎(例如系統性紅斑性狼瘡、修格蘭氏症候群、類風濕性關節炎);Cryo, 冷凝球蛋白血症性血管炎 (cryoglobulinemic vasculitis);EGPA, 嗜酸性球性肉芽腫性多血管炎 (eosinophilic granulomatosis with polyangiitis, Churg–Strauss);GPA, 肉芽腫性多血管炎 (granulomatosis with polyangiitis, Wegener);IgAV, IgA 血管炎(Henoch–Schönlein);MPA, 顯微鏡下多血管炎 (microscopic polyangiitis);PAN, 結節性多動脈炎 (polyarteritis nodosa);GC, 糖皮質素 (glucocorticoids);AZA, 硫唑嘌呤 (azathioprine);CYC, 環磷醯胺 (cyclophosphamide);MMF, 黴酚酸酯 (mycophenolate);MTX, 甲胺喋呤 (methotrexate);PLEX, 血漿置換 (plasma exchange);RTX, 利妥昔單抗 (rituximab)。巨細胞動脈炎與高安動脈炎的治療包括 GC,但節省糖皮質素藥物的選項與此處所示演算法不同。貝賽特氏病的治療方法複雜,且不易納入與其他血管炎共用的演算法中。
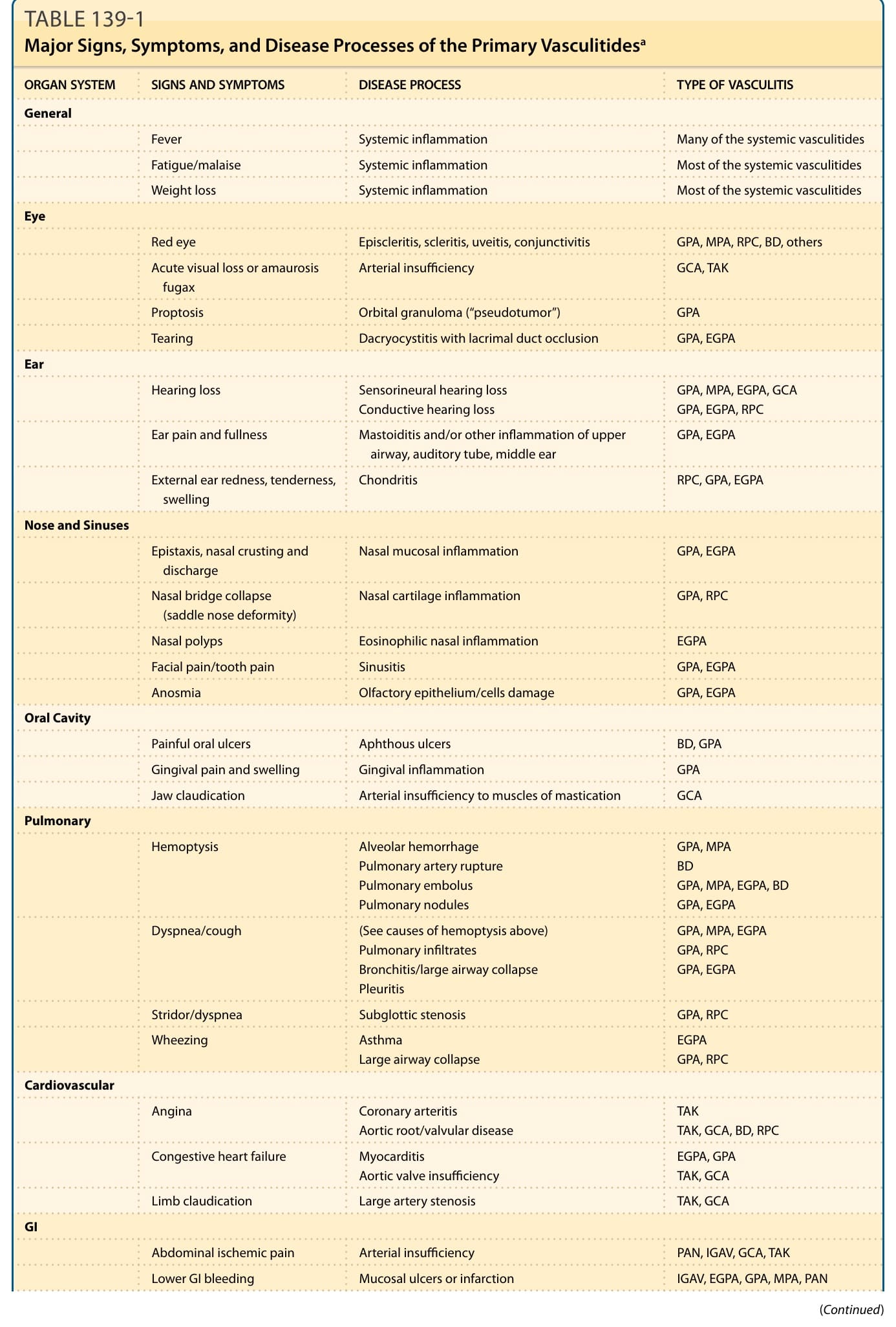
表 139-1:原發性血管炎的主要徵象、症狀與疾病過程 (Major Signs, Symptoms, and Disease Processes of the Primary Vasculitides)。
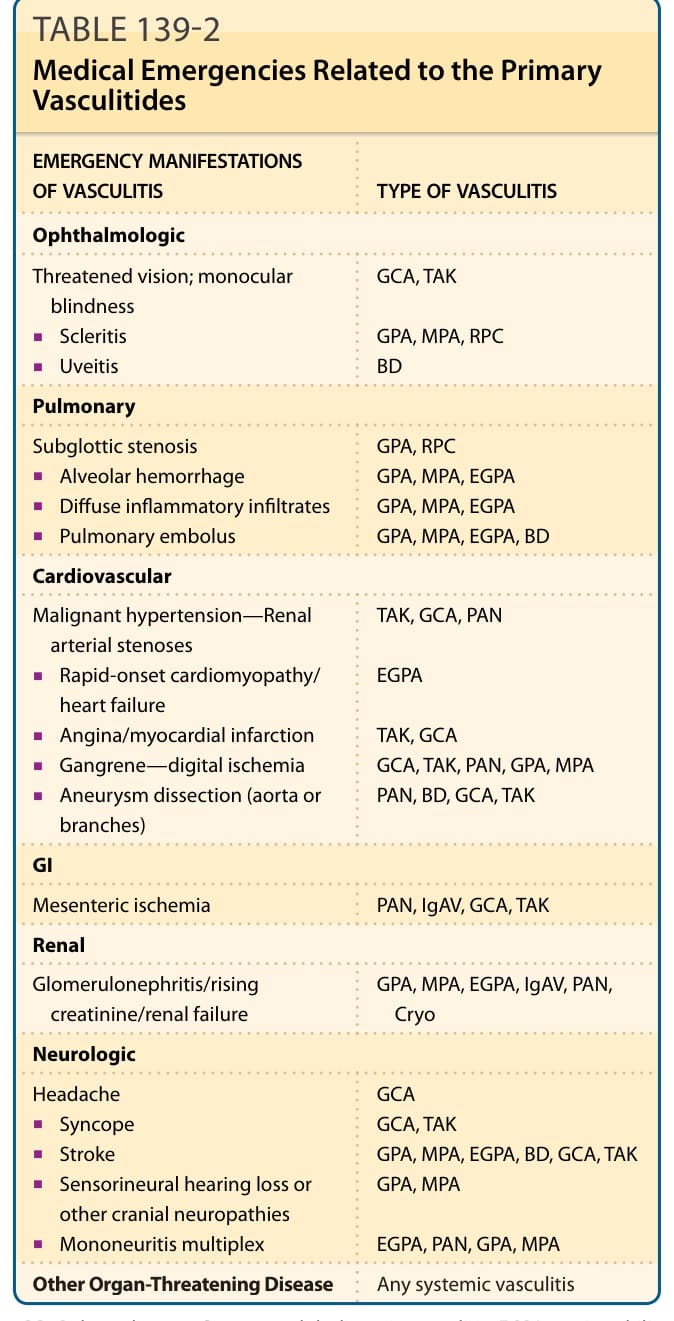
表 139-2:與原發性血管炎相關的醫療緊急狀況 (Medical Emergencies Related to the Primary Vasculitides)。