神經纖維瘤病 (The Neurofibromatoses) 精華筆記
概論與分類
- 神經纖維瘤病為體染色體顯性 (autosomal dominant) 遺傳病;隨基因診斷問世已辨識數種症候群:
- NF-1(von Recklinghausen 病):每 3000 名活產嬰兒 1 例。
- NF-2:標誌為雙側前庭神經鞘瘤 (bilateral vestibular schwannomas);每 40,000 名活產嬰兒 1 例。
- 節段型/鑲嵌型 NF-1 (segmental / mosaic NF-1)。
- Legius 症候群(SPRED1 突變):咖啡牛奶斑、間擦部位雀斑、巨頭畸形 (macrocephaly),無 NF-1 其他表現。
- 神經鞘瘤病 (schwannomatosis)。
- NF-1 與 NF-2 在各族群發生率相同。
第一型神經纖維瘤病 (NF-1)
診斷標準
- 1987 年 NIH 共識會議訂出 7 項診斷特徵,具備 2 項或以上即可臨床診斷(表 135-1),沿用逾 30 年未修改。
- NF-1 診斷主要為臨床診斷;基因檢測僅在症狀前診斷或產前診斷等特定情況下有用,多數病例不必要。對皮膚腫塊切片以「確認」NF-1 是不必要的。
主要臨床表現
- 咖啡牛奶斑 (café-au-lait spots):平坦色素性斑點,常為 NF-1 最先出現的表現;出生時常已存在,隨成長數目增多(第一個十年持續出現)。大小、形狀、輪廓無診斷意義;數目多寡不代表病情較重,位置也無法預測日後神經纖維瘤位置。為表皮中神經嵴源的高度色素化黑色素細胞 (melanocytes) 聚集。
- 間擦部位雀斑 (intertriginous freckling):小於 5 mm 的咖啡牛奶斑,見於腋窩、腹股溝、乳房下方;與日曬無關,被視為 NF-1 的特異性表徵(Crowe 徵象)。最終診斷 NF-1 的孩童中,百分之八十一會在 6 歲前出現。
- 個別神經纖維瘤 (discrete neurofibromas):源自周邊神經的良性神經鞘瘤,由許旺細胞 (Schwann cells)、肥大細胞、纖維母細胞與神經周膜細胞組成。
- 皮膚神經纖維瘤:較周圍組織柔軟,搓揉有「鈕扣孔 (buttonholing)」感。
- 皮下神經纖維瘤:源自周邊神經,質地硬得多;源自背根神經節者可呈「啞鈴 (dumbbell)」狀壓迫脊髓。
- 一般於青春期後開始出現、隨年齡增多;不到 20% 的 10 歲以下孩童有皮膚神經纖維瘤。嚴重搔癢可用抗組織胺 (antihistamines)。
- 叢狀神經纖維瘤 (plexiform neurofibromas):侵犯單條或多條神經束的良性神經鞘瘤;觸診呈「蠕蟲狀」感,上方常有色素過度沉著或多毛症。多數出生時即有或數年內顯現。可致毀容、失明(弱視、青光眼或眼球突出)、肢體功能喪失;胸腹腔者可無外觀表現卻壓迫重要結構。惡性周邊神經鞘瘤一般源自此瘤;若快速生長、明顯疼痛或局部神經功能障礙應切片,¹⁸F-FDG PET/CT 有助辨識惡性轉化。
- 視神經路徑腫瘤 (optic pathway tumors, OPTs):約 15% 孩童發生,但不到一半有症狀(有症狀整體發生率 7%);女性約 2:1 優勢。最高風險期為出生後最初 6 年,6 歲後極罕見。約 30% 以急速眼球突出 (proptosis) 加視力喪失表現;多達 40% 視交叉腫瘤孩童會性早熟(首徵常為加速的線性生長,可用長效黃體生成素釋放激素促效劑中止)。
- Lisch 結節 (Lisch nodules):虹膜上略隆起的黑色素細胞性錯構瘤,特異性高,裂隙燈最佳觀察;不影響視力。超過 90% 成人有,6 歲以下孩童僅 30% 有。
- 特殊骨性病變:(1) 蝶骨翼 (wing of sphenoid) 發育不良 → 眼眶壁/底形成不良;(2) 長骨發育不良(先天變薄彎曲,影響約 2% 孩童,脛骨最常見),易骨折,癒合失敗形成假關節病 (pseudarthrosis)。
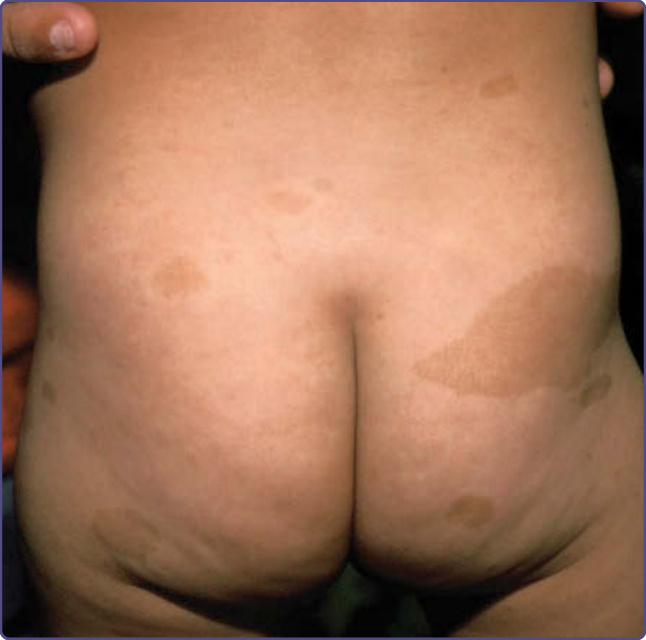
圖 135-1:咖啡牛奶斑 (café-au-lait spots)。
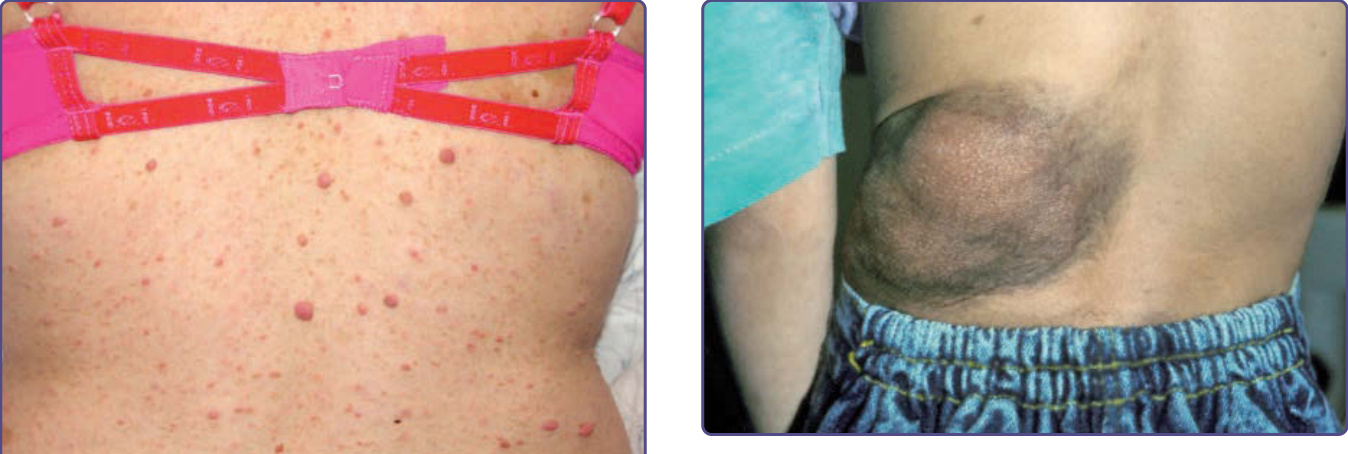
圖 135-6:叢狀神經纖維瘤 (plexiform neurofibroma),其上方有色素過度沉著與多毛症。
病因與發病機轉
- 遺傳學:體染色體顯性,表現度變異大但外顯率 (penetrance) 100%;世代間表面跳脫多因誤診、非親生或新突變。NF1 位於第 17 號染色體長臂,編碼神經纖維瘤蛋白 (neurofibromin),含 59 個外顯子、編碼逾 2800 個胺基酸的多肽。突變率極高(每世代每配子 2.4 × 10⁻⁵ 至 10⁻⁴);新突變約佔 50%,多位於父系對偶基因,且不隨父親年齡增加。母系遺傳病情較重。
- 基因型–表現型相關性僅 2 種:(1) 整個基因缺失 → 顏面畸形、智能障礙、神經纖維瘤早現、叢狀瘤;(2) 第 17 外顯子 3 鹼基對符合讀框缺失 → 有咖啡牛奶斑、雀斑、Lisch 結節但無神經纖維瘤。
- 分子生物學:神經纖維瘤蛋白為 Ras 的負向調節因子,促進 Ras 由活化態 (Ras-GTP) 轉為非活化態 (Ras-GDP),下調 MAPK、PI3K、蛋白激酶 B 與 mTOR 路徑;SPRED1 亦抑制 MAPK。腫瘤生成依賴正常 NF1 對偶基因失活(異合子性喪失 loss of heterozygosity,第二次體細胞「打擊」),故 NF1 屬腫瘤抑制基因 (tumor-suppressor gene)。法尼基轉移酶 (farnesyltransferase) 抑制劑與 MEK 抑制劑在小鼠模型可縮小叢狀瘤,已進入臨床試驗。
鑑別診斷
- 多種症候群可見咖啡牛奶斑(表 135-2)。最可能:NF-1、NF-2、家族性咖啡牛奶斑(Legius 症候群)、LEOPARD 症候群。較不可能:結節性硬化症、Fanconi 貧血、第 2B 型多發性內分泌腫瘤、Bannayan-Riley-Ruvalcaba 症候群、McCune-Albright 症候群、Bloom 症候群、共濟失調微血管擴張症。
第二型神經纖維瘤病 (NF-2)
- 體染色體顯性,每 40,000 名活產 1 例;以雙側前庭神經鞘瘤為標誌,並有多發性腦膜瘤、神經鞘瘤、神經膠質瘤、室管膜瘤與青少年型後囊下白內障。
- 雖較 NF-1 少見,但致病率更高(常癱瘓與耳聾)。約 60% 成年期以聽力喪失、耳鳴或平衡喪失表現;發病愈年輕、病情愈重。眼科:青少年型後囊下白內障(60% 至 80% 病人)、視網膜錯構瘤、視神經鞘腦膜瘤。
- 皮膚病灶為皮膚神經鞘瘤 (cutaneous schwannoma)(斑塊狀、略隆起、淡紫紅色,偶有毛髮);可有數個咖啡牛奶斑但很少超過 6 個,無間擦部位雀斑。
- 致病基因位於第 22 號染色體,編碼膜相關蛋白 merlin,亦為下調細胞生長的腫瘤抑制基因。
圖 135-14:皮膚神經鞘瘤 (cutaneous schwannoma)。
其他相關症候群
- 鑲嵌型 NF-1(節段型):NF-1 表現侷限身體某區域,因 NF1 受孕後突變造成體細胞鑲嵌;有在受影響區域發展併發症(最常為神經纖維瘤)的風險。已證實性腺鑲嵌現象 (gonadal mosaicism),節段型病人可生下完全型 NF-1 後代,故遺傳諮詢具難度。
- NF-1–Noonan 症候群:符合 NF-1 標準又具 Noonan 特徵(眼距過寬、眼瞼下垂、頸蹼、肺動脈瓣狹窄等);絕大多數由 NF1 突變引起(17 名中 16 名有 NF1 突變、無 PTPN11 突變)。
- Legius 症候群:SPRED1 突變致咖啡牛奶斑、間擦雀斑、巨頭畸形,無神經纖維瘤、Lisch 結節、OPTs 或 NF-1 骨性異常;NF1 定序陰性者可檢測 SPRED1。
- 神經鞘瘤病 (schwannomatosis):多發性疼痛性神經鞘瘤於周邊神經與脊椎旁神經根,多在第二、三個十年出現,無前庭神經鞘瘤,壽命正常。診斷前須以基因檢測與 MRI 排除 NF-2。部分家族性病例由 SMARCB1/INI1 或 LTZR1 生殖細胞系突變引起,但多數為偶發性。
臨床病程、預後與併發症
- 隱藏內部叢狀瘤:全身 MRI 顯示發生率高達 55%。
- 脊柱側彎 (scoliosis):NF-1 最常見骨骼表現,影響 10% 至 30%;分發育不良型(原發骨發育不良,急遽成角,可能需複雜手術)與非發育不良型(似特發性,可觀察或支架)。亦可見局部骨性過度生長。
- 癌症:終生癌症風險估計 59.6%(芬蘭族群研究);40 歲以下 NF-1 女性乳癌標準化發生率比達 11.1。
- 惡性周邊神經鞘瘤:幾乎全源自既有叢狀瘤,終生風險可能高達 13%;警覺快速生長腫塊、不明疼痛或功能障礙,¹⁸F-FDG PET 結合 CT/MRI 敏感度與特異度佳。
- 嗜鉻細胞瘤 (pheochromocytoma):發生率可達 1.4%,平均發病 42 歲;84% 單發腎上腺,11.5% 為惡性。
- 白血病:青少年型慢性骨髓單核球性白血病與急性淋巴芽細胞性白血病風險增加(英國研究示後者相對風險為一般族群 5.4 倍)。
- 其他腫瘤:幼年型黃色肉芽腫 (juvenile xanthogranulomas)、橫紋肌肉瘤、體抑素瘤、胃腸道間質瘤。
- 血管病變 (vasculopathy):可影響任何動脈,最常見腎動脈狹窄致腎血管性高血壓;高血壓難以單一藥物控制的成人應評估,無法控制時可行經皮腔內血管成形術。腦動脈病變可致 moyamoya(煙霧病)反覆出血。
- 不明亮點 (UBOs):T2 加權 MRI 高訊號區,約 60% 孩童有,隨年齡消失;不增強、不壓迫,對應髓鞘空泡化,意義不明。
- 智力與學習障礙:平均智商僅比常人低 5 至 10 分,但學習障礙常見(盛行率 30% 至 60%);注意力不足過動症常見,興奮劑藥物有助益。
治療與處置
- 整體照護:多專科聯合門診每年追蹤。所有 10 歲以下孩童每年完整眼科檢查(視力、色覺、視野、眼底鏡、裂隙燈)找 OPT;每年量身高體重畫生長曲線(偵測性早熟);每次量血壓(找腎血管性高血壓);每年檢查脊柱(找脊柱側彎)。
- 咖啡牛奶斑(顏面者偶求美容改善):雷射結果參差。銅蒸氣雷射 16 名中 15 名良好至極佳;Q 開關 755-nm 紫翠玉雷射 56% 良好至極佳、17% 不佳;低能量密度 1064-nm Q 開關 Nd:YAG 在 75% 病灶誘發超過 50% 清除。
- 個別皮膚神經纖維瘤:手術切除改善外觀或防刺激;嚴重搔癢可用抗組織胺,ketotifen(抗組織胺兼肥大細胞穩定劑)軼事報告可緩解搔癢、疼痛。全身麻醉下二氧化碳雷射可去除數百個瘤;另有研究示 Er:YAG 雷射消融優於電燒與二氧化碳雷射。
- 叢狀神經纖維瘤:傳統限於手術減積(無法完全切除、常再生長)。生物製劑:pirfenidone 與法尼基轉移酶抑制劑無希望結果;sirolimus(mTOR 抑制劑)僅延長進展時間 4 個月;imatinib 在一名 3 歲孩童 3 個月內使腫瘤縮小 70%,後續研究 17% 病人腫瘤體積減少 20% 或以上。
- 視神經路徑腫瘤:不建議對所有 NF-1 孩童例行篩檢性神經影像(早期偵測未改變病程);OPT 一旦辨識以系列 MRI 與眼科檢查追蹤。治療目標為保存視力;必要時 carboplatin 與 vincristine 有效。放射治療因血管病變、第二惡性腫瘤及神經認知/內分泌副作用風險,不適用於 NF-1 相關 OPTs。