神經纖維瘤病 (The Neurofibromatoses)
PART 21
代謝、遺傳與系統性疾病 (Metabolic, Genetic, and Systemic Diseases)
重點一覽 (AT-A-GLANCE)
■ 體染色體顯性 (autosomal dominant) 遺傳疾病,發生率為每 3000 名活產嬰兒中 1 例。
■ 若出現 2 項主要特徵即可在臨床上診斷(見表 135-1)。
■ 皮膚神經纖維瘤 (cutaneous neurofibromas):
■ 比周圍結締組織柔軟,略突出於皮膚表面,或位於皮膚正下方而其上方呈紫紅色 (violaceous) 色澤。
■ 皮下神經纖維瘤 (subcutaneous neurofibromas):
■ 源自周邊神經,可位於皮膚下方及深部內臟。
■ 一般質地硬得多。
■ 叢狀神經纖維瘤 (plexiform neurofibromas):
■ 一般在出生時即已存在,或在出生後最初數年內顯現。
■ 可能導致毀容、失明(繼發於弱視 amblyopia、青光眼 glaucoma 或眼球突出 proptosis)、肢體功能喪失,或因壓迫重要結構而造成器官功能障礙。
■ 鑲嵌型第一型神經纖維瘤病(mosaic neurofibromatosis Type 1,又稱節段型 NF-1,segmental NF-1):
■ NF-1 的表現,通常侷限於身體的某一區域。
■ 因 NF1 基因在受孕後 (postconceptional) 發生突變而造成體細胞鑲嵌現象 (somatic mosaicism)。
臨床特徵 (CLINICAL FEATURES)
美國國家衛生研究院 (National Institutes of Health) 於 1987 年召開了一場共識發展會議,以建立診斷標準,促進對第一型神經纖維瘤病 (neurofibromatosis Type 1, NF-1) 病人的更佳臨床研究與照護。¹
該會議所認定的 7 項診斷特徵(表 135-1),以及「在確立 NF-1 診斷前須具備這 7 項特徵中的 2 項或以上」的建議,已證實極為實用,並在 30 多年後仍持續未經修改地沿用。或許唯一需要注意的一點是,後來辨識出一種獨立的體染色體顯性症候群,其由編碼 sprouty 相關 EVH1 結構域含蛋白 1(sprouty-related EVH1 domain-containing protein 1, SPRED1)的基因發生失活突變所致,會導致咖啡牛奶斑 (café-au-lait spots)、間擦部位雀斑 (intertriginous freckling) 與巨頭畸形 (macrocephaly),但不會出現 NF-1 的其他任何表現(即 Legius 症候群)。²
咖啡牛奶斑 (Café-au-Lait Spots)
咖啡牛奶斑 (café-au-lait spots) 是平坦的色素性斑點 (pigmented macules),常為 NF-1 最先出現的表現(圖 135-1)。它們常於出生時即已存在,隨著嬰兒成長而數目增多;新的斑點可能在出生後第一個十年期間持續出現。一旦被注意到,它們往往會隨著孩童整體生長而按比例增大。雖然臉部不常見,但身體任何部位都可能出現。咖啡牛奶斑的大小、形狀與輪廓並無診斷意義,且常被引用的「邊緣平滑的咖啡牛奶斑較典型於 NF-1 而非 McCune-Albright 症候群」此一說法是不正確的。當咖啡牛奶斑彼此重疊時,重疊區域的顏色可能比任一單獨斑點更深。當其出現在石板灰色斑 (slate grey macules) 之中時,典型上會被一圈色素較淡的暈環所環繞。擁有大量咖啡牛奶斑的個體並不代表罹患「較嚴重」的 NF-1,且斑點的位置完全無法預測日後神經纖維瘤的位置。咖啡牛奶斑代表表皮中源自神經嵴 (neural crest) 的高度色素化黑色素細胞 (melanocytes) 的聚集。雖然這些細胞可能含有數目增多的「巨大型」黑色素小體 (giant melanosomes) 或黑色素巨球 (melanin macroglobules),但這些並非 NF-1 所特有,且其在切片中的有無對診斷並無幫助。
間擦部位雀斑 (Intertriginous Freckling)
小於 5 mm 的咖啡牛奶斑被稱為雀斑 (freckles),常見於腋窩 (axillae)、腹股溝區 (inguinal region) 與乳房下方(圖 135-2)。與這些部位的一般雀斑不同,這些病灶與日曬無關,且被認為實質上是 NF-1 的特異性表徵(Crowe 徵象,Crowe sign)。在所有最終被診斷為 NF-1 的孩童中,81% 會在 6 歲前出現間擦部位雀斑。³
個別神經纖維瘤 (Discrete Neurofibromas)
神經纖維瘤 (neurofibromas) 由許旺細胞 (Schwann cells)、肥大細胞 (mast cells)、纖維母細胞 (fibroblasts) 與神經周膜細胞 (perineural cells) 組成,是源自周邊神經的良性神經鞘瘤 (benign nerve sheath tumors),呈現為個別的腫塊。⁴ 皮膚神經纖維瘤略突出於皮膚表面,或位於皮膚正下方而其上方呈紫紅色色澤(圖 135-3)。它們比周圍結締組織柔軟,當以手指輕輕在其表面搓揉時,常會產生一種「鈕扣孔 (buttonholing)」的感覺(圖 135-4)。源自周邊神經、位於皮膚下方及深部內臟的皮下神經纖維瘤一般質地硬得多(圖 135-5)。若它們源自背根神經節 (dorsal root ganglia),可能會穿過神經孔 (neural foramina) 生長,壓迫脊髓,形成「啞鈴 (dumbbell)」狀外觀。頸部的皮下神經纖維瘤摸起來可能像「串珠項鍊 (beaded necklace)」,常被誤認為淋巴結。雖然不到 20% 的 10 歲以下孩童有皮膚神經纖維瘤,但神經纖維瘤一般在青春期後開始出現,並隨病人年齡增長而數目增多。罹患 NF-1 的女性常會提到皮膚神經纖維瘤在懷孕期間出現。大多數成年 NF-1 病人很可能有大量侵犯背根與其他較大神經的無症狀深部神經纖維瘤。偶爾,神經纖維瘤相關的搔癢 (pruritus) 可能嚴重到需要以抗組織胺 (antihistamines) 治療。
叢狀神經纖維瘤 (Plexiform Neurofibromas)
叢狀神經纖維瘤 (plexiform neurofibromas) 在組織學上與個別神經纖維瘤相似,是侵犯單條或多條神經束 (nerve fascicles) 的良性周邊神經鞘瘤,常源自主要神經的分支。⁵ 觸診時可能引發一種「蠕蟲狀 (wormy)」的感覺,因為觸者感受到多條增厚的神經束。其上方常有色素過度沉著(「巨大型咖啡牛奶斑」)或多毛症 (hypertrichosis)(圖 135-6)。大多數叢狀神經纖維瘤在出生時即已存在,或在出生後最初數年內顯現。外觀可見的叢狀神經纖維瘤容易辨識,可能導致毀容、失明(繼發於弱視、青光眼或眼球突出)或肢體功能喪失(圖 135-7 至圖 135-9)。相對地,胸腔或腹腔內的叢狀神經纖維瘤可能沒有任何外部表現,但可能因侵犯或壓迫重要結構(如輸尿管 ureters、腸道 bowel、脊髓 spinal cord)而造成同樣具毀滅性的後果。叢狀神經纖維瘤的生長速率變異極大。快速生長期與長期靜止期交替出現的情形很常見。惡性周邊神經鞘瘤 (malignant peripheral nerve sheath tumors) 一般源自叢狀神經纖維瘤,可能在深部叢狀神經纖維瘤中無聲無息地發展,直到發生遠端轉移才出現症狀。儘管 NF1 的異合子性喪失 (loss of heterozygosity) 可能導致良性神經纖維瘤的形成,但良性叢狀神經纖維瘤發生惡性轉化的產生,可能是由 RAS 致癌基因 (RAS oncogene) 以外的細胞週期調節因子所引起。例如,同時帶有 Nf1 與 p53 基因無效突變 (null mutations) 的小鼠一致地會發展出惡性腫瘤。⁶ 照護 NF-1 個體的醫師應警覺惡性腫瘤的發展;若叢狀神經纖維瘤呈現快速生長,或造成顯著疼痛或局部神經功能障礙,應予以切片。整合電腦斷層的正子斷層造影(以 2-deoxy-2-[fluorine-18]fluoro-d-glucose 為示蹤劑,¹⁸F-FDG PET/CT)對於辨識既有叢狀神經纖維瘤內惡性周邊神經鞘瘤的發展極為有用。⁷ ⁸
視神經路徑腫瘤 (Optic Pathway Tumors)
約 15% 的 NF-1 孩童會發展出視神經路徑腫瘤(optic pathway tumors, OPTs);然而這些病人中不到一半會出現症狀,因此有症狀 OPT 的整體發生率為 7%。⁹ ¹⁰ 近期關於 NF-1 相關 OPTs 的系列研究發現病人約有 2:1 的女性優勢,以及在非裔美籍孩童中發生率較低。這些觀察結果提示,荷爾蒙因素或修飾基因 (modifying genes) 可能影響 OPTs 的發展與預後。¹¹ ¹²
在 NF-1 中,發展出有症狀 OPTs 的風險最高的時期是在出生後最初 6 年;6 歲後才發展出有症狀腫瘤的情形極為罕見。¹³ ¹⁴ 因此,照護 NF-1 孩童的醫師應對此一年幼族群中 OPTs 的徵象與症狀保持敏感。約 30% 有症狀 OPTs 的孩童會以急速發作的眼球突出 (proptosis) 表現,患側眼有中度至重度視力喪失(圖 135-10)。另有 30% 有症狀 OPTs 的孩童會有異常的眼科檢查結果,但無任何視覺症狀,因而導致腫瘤被發現。由於年幼孩童即使視力喪失嚴重也很少抱怨,因此對所有年幼的 NF-1 孩童進行徹底的年度眼科檢查至關重要。當出現時,眼科徵象可能包括傳入性瞳孔缺損 (afferent papillary defect)、視神經萎縮 (optic nerve atrophy)、視乳頭水腫 (papilledema)、斜視 (strabismus) 或色覺缺損。最後,多達 40% 有視交叉腫瘤 (chiasmal tumors) 的孩童會發展出性早熟 (precocious puberty)。¹⁵
加速的線性生長 (accelerated linear growth) 將是性早熟的首個徵象,這凸顯了所有 NF-1 孩童都需要使用標準生長曲線圖進行年度生長評估。有視交叉腫瘤的孩童常無眼科症狀或徵象。性早熟的早期偵測很重要,因為加速的線性生長與第二性徵 (secondary sexual characteristics) 的發展均可藉由使用長效型黃體生成素釋放激素促效劑 (long-acting luteinizing hormone-releasing hormone agonist) 而被中止。
Lisch 結節 (Lisch Nodules)
Lisch 結節 (Lisch nodules) 是虹膜 (iris) 上略隆起、界限清楚的黑色素細胞性錯構瘤 (melanocytic hamartomas),被認為實質上是 NF-1 的特異性表徵(圖 135-11)。最佳的觀察方式是使用裂隙燈 (slitlamp),這對於將其與較常見、與 NF-1 無關的平坦虹膜痣 (flat iris nevi) 區分開來是必要的。它們不會造成任何視覺功能的損害。其被發現的頻率隨年齡增長而增加;雖然超過 90% 的成年 NF-1 病人有 Lisch 結節,但 6 歲以下的 NF-1 孩童中只有 30% 有此結節。³
特殊的骨性病變 (Distinctive Osseous Lesions)
有 2 種骨性病變具有足夠的特殊性,因而被納入 NF-1 的診斷標準。第一種是蝶骨翼 (wing of the sphenoid bone) 的發育不良,會導致眼眶 (orbit) 壁與/或底部的形成不良(圖 135-12)。這種先天性中胚層發育不良 (congenital mesodermal dysplasia) 可能、但並非總是在臨床上明顯可見,會導致眼球突出(因腦膜或腦組織疝入眼眶所致)或眼球內陷 (enophthalmos)。長骨 (long bone) 的發育不良以先天性變薄與彎曲為特徵,影響約 2% 的 NF-1 孩童(圖 135-13)。雖然脛骨 (tibia) 最常受影響,但股骨 (femur)、肱骨 (humerus) 與其他長骨也可能受侵犯。即使骨頭完整,變薄與彎曲也會產生可見的畸形,且骨頭力學性質的減弱會使其易於骨折,特別是在承重骨。骨折後初級癒合 (primary union) 失敗會導致「假關節 (false joint)」,即假關節病 (pseudarthrosis)。¹⁶
病因與發病機轉 (Etiology and Pathogenesis)
流行病學 (Epidemiology)
神經纖維瘤病的「現代時期」始於 1981 年,當時 Riccardi 對 von Recklinghausen 病的特徵與自然史做了詳細的臨床描述,隨後又有「合併雙側聽神經瘤的中樞型神經纖維瘤病 (central neurofibromatosis with bilateral acoustic neuroma)」之描述。¹⁷
隨著基因為基礎的診斷問世,已辨識出數種不同的臨床症候群:(a) NF-1,又稱 von Recklinghausen 病;(b) NF-2,其主要特徵為雙側前庭神經鞘瘤 (bilateral vestibular schwannomas) 的發展;(c) 節段型或鑲嵌型 NF-1;(d) Legius 症候群(SPRED1 突變),導致咖啡牛奶斑、間擦部位雀斑與巨頭畸形的體染色體顯性遺傳;以及 (e) 神經鞘瘤病 (schwannomatosis)。NF-1 與 NF-2 在所有族群中發生率相同。NF-1 的發生率為每 3000 名活產嬰兒中 1 例,而 NF-2 則少見得多,估計發生率為每 40,000 名活產嬰兒中 1 例。
遺傳學 (Genetics)
NF-1 以體染色體顯性方式遺傳。雖然 NF-1 的表現度 (expressivity) 變異相當大,即使在同一家族中基因型完全相同的個體之間亦然,但此疾病被認為具有 100% 的外顯率 (penetrance)。因此,NF-1 在世代間表面上的跳脫 (skipping),可能是誤診、非親生 (nonpaternity) 或孫輩發生新突變的結果。雖然已描述了大量的 NF1 突變,但僅注意到 2 種基因型–表現型相關性 (genotype–phenotype correlations)。整個基因發生缺失 (deletions) 的個體,發生顏面畸形 (facial dysmorphism)、智能障礙 (intellectual disability)、神經纖維瘤早期出現及叢狀神經纖維瘤存在的風險增加。¹⁸ 此外,在 NF1 第 17 外顯子 (exon 17) 有特定 3 鹼基對符合讀框 (3 base pair in-frame) 缺失的個體,可能有咖啡牛奶斑、間擦部位雀斑與 Lisch 結節,但不會發展出皮膚、皮下或叢狀神經纖維瘤。¹⁹ 有證據顯示,當 NF-1 從母親而非父親遺傳時,病情較為嚴重。NF1 位於第 17 號染色體的長臂,編碼一種稱為神經纖維瘤蛋白 (neurofibromin) 的蛋白質。尚未有 NF1 突變對偶基因同合子 (homozygosity) 的報告,可能是因為它是一種致死性的狀況。NF1 的突變率異常地高,估計為每世代每配子 2.4 × 10⁻⁵ 至 10⁻⁴,是已知遺傳疾病中最高者之一。這可能反映了該基因的龐大,其橫跨 350 kb 的基因組 DNA,並含有編碼一個含逾 2800 個胺基酸之多肽的 59 個外顯子 (exons)。²⁰ 另一種假說是該基因有某種結構特性使其特別易於發生突變。新突變約佔病例的 50%,且通常位於父系遺傳的對偶基因上。與許多其他顯性疾病相反,新突變的頻率似乎不會隨父親年齡的增長而增加。¹¹
分子生物學 (Molecular Biology)
大多數 NF1 突變導致 NF1 基因產物——神經纖維瘤蛋白——的細胞內含量降低;這似乎足以引起該疾病的許多臨床表現。腫瘤生成 (tumorigenesis),包括良性真皮神經纖維瘤的發展,似乎取決於體細胞中正常 NF1 對偶基因的失活,此一過程稱為異合子性喪失 (loss of heterozygosity)。²¹ 異合子性喪失是許多遺傳性癌症易感症候群(如視網膜母細胞瘤 retinoblastoma、Li-Fraumeni 症候群)中腫瘤生成的關鍵步驟,而所涉及的基因稱為腫瘤抑制基因 (tumor-suppressor genes)。當基因的兩套複本都停止正常運作時,腫瘤生成即被啟動——第一套因遺傳性突變所致,第二套則因第二次體細胞「打擊 (hit)」干擾了先前正常對偶基因的功能所致。神經纖維瘤蛋白存在於多種細胞類型中,包括神經元 (neurons)、寡突膠質細胞 (oligodendrocytes) 與非髓鞘形成性許旺細胞 (nonmyelinating Schwann cells)。已累積了相當多的證據支持神經纖維瘤蛋白作為 Ras 之負向調節因子 (negative regulator) 的角色。²⁰ RAS 基因家族編碼與膜結合、結合鳥嘌呤核苷酸 (guanine nucleotide-binding) 的蛋白質,這些蛋白質參與細胞增殖 (proliferation)、分化 (differentiation) 與學習的調節。Ras 存在於活化態(Ras-GTP [guanosine triphosphate],三磷酸鳥苷)與非活化態(Ras-GDP [guanosine diphosphate],二磷酸鳥苷)。神經纖維瘤蛋白藉由促進 Ras 從活化態轉變為非活化態,下調 Ras 的下游效應,這些效應包括促進學習、記憶、突觸可塑性 (synaptic plasticity) 以及細胞生長與增殖。Ras 下游效應的媒介物包括絲裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)、磷脂醯肌醇 3′-激酶 (phosphatidylinositol 3′-kinase)、蛋白激酶 B (protein kinase B) 與哺乳動物雷帕黴素標靶激酶 (mammalian target of rapamycin kinase)。²² SPRED1 也會抑制 MAPK 路徑的活化。²²
在腦中,有 3 種主要的 Ras 異構體 (isoforms);K-ras 是神經纖維瘤蛋白活性的主要標靶。²³ 神經纖維瘤蛋白對 Ras 的活化會刺激 G 蛋白 (G-protein) 活性,導致腺苷酸環化酶 (adenylyl cyclase) 的活化。環腺苷單磷酸 (cyclic adenosine monophosphate) 與其他下游中間產物似乎在細胞生長、學習與記憶中扮演角色。神經纖維瘤蛋白也與微管 (microtubules) 相結合,並在調節海馬迴 (hippocampus) 中 γ-胺基丁酸 (γ-aminobutyric acid)-能抑制性神經元活動中扮演角色。²⁴
Ras 是一種膜結合蛋白,其活性需要法尼基化 (farnesylation)(在接近 C 端 [C-terminus] 的半胱胺酸殘基上添加一個 C15 類異戊二烯 [isoprenoid]),此過程由法尼基轉移酶 (farnesyltransferase) 催化。抑制法尼基轉移酶的藥劑會造成 Ras 路徑的整體抑制,其作用與神經纖維瘤蛋白頗為相似。法尼基轉移酶抑制劑能逆轉 Nf1 缺損小鼠細胞的增殖表現型,並能藉由降低 Ras 含量,逆轉 NF-1 小鼠模型中的空間學習障礙。²⁵ MAPK 路徑下游激酶的抑制劑,如 MAPK 激酶(MAPK kinase, MEK),已被證實能在小鼠模型中使叢狀神經纖維瘤縮小,為目前進行中的臨床試驗開啟了道路。²⁶
許旺細胞中 NF1 的異合子性喪失已被證實是個別與叢狀神經纖維瘤發展的必要事件。²⁷
叢狀神經纖維瘤由許旺細胞、巨噬細胞 (macrophages)、肥大細胞、神經元與纖維母細胞組成。有趣的是,神經纖維瘤蛋白缺損的許旺細胞會分泌一種刺激肥大細胞遷移的物質,而後者反過來又刺激細胞外基質 (extracellular matrix) 的產生與血管新生 (angiogenesis)。⁵ 缺乏 NF1 表現的星狀膠質細胞 (astrocytes) 無法自行形成視神經膠質瘤 (optic nerve gliomas);這些腫瘤的發展需要一個對 NF1 為異合子的腦部環境。微膠細胞 (microglia),即中樞神經系統 (CNS) 的免疫監視細胞,參與視神經膠質瘤的形成與維持。²⁸
診斷 (Diagnosis)
儘管 NF1 基因已被辨識並完成完整定序,NF-1 的診斷仍主要是臨床診斷。雖然在特定情況下實驗室確認診斷極為有用,但在大多數病例中是不必要的。由於尚未辨識出常見的突變,分子診斷只能基於針對整個基因篩檢突變的策略。此一檢測可用於症狀前 (presymptomatic) 個體,並可應用於 NF-1 的產前診斷——當受影響的父母一方已被辨識出突變時。經驗不足的臨床醫師常會對皮膚腫塊進行切片,試圖在具有多個咖啡牛奶斑或其他 NF-1 表徵的個體中確認 NF-1 的診斷。此類處置一律是不必要的,因為 NF-1 的臨床診斷通常相當直接明確。叢狀神經纖維瘤的切片應保留給醫師希望排除惡性腫瘤可能性的情況。
鑑別診斷 (Differential Diagnosis)
有許多症候群可能出現咖啡牛奶斑(表 135-2)。然而,有數種症候群與 NF-1 有重疊的表現型,過去曾造成混淆。
最可能 (Most Likely)
■ 第一型神經纖維瘤病 (Neurofibromatosis Type 1)
■ 第二型神經纖維瘤病 (Neurofibromatosis Type 2)
■ 家族性咖啡牛奶斑(考慮 Legius 症候群)
■ LEOPARD 症候群(lentigines 雀斑樣痣、electrocardiographic abnormalities 心電圖異常、ocular hypertelorism 眼距過寬、pulmonary stenosis 肺動脈狹窄、abnormalities of genitalia 生殖器異常、retardation of growth 生長遲緩、deafness 耳聾)
較不可能 (Less Likely)
■ 結節性硬化症 (Tuberous sclerosis)
■ Fanconi 貧血 (Fanconi anemia)
■ 第 2B 型多發性內分泌腫瘤 (Multiple endocrine neoplasia Type 2B)
■ Bannayan-Riley-Ruvalcaba 症候群
■ McCune-Albright 症候群(多發性骨纖維化發育不良 polyostotic fibrous dysplasia)
■ Bloom 症候群
■ 共濟失調微血管擴張症 (Ataxia-telangiectasia)
第二型神經纖維瘤病 (Neurofibromatosis Type 2)
重點一覽 (AT-A-GLANCE)
■ 體染色體顯性遺傳疾病,發生率為每 40,000 名活產嬰兒中 1 例。
■ 標誌性特徵為雙側前庭神經鞘瘤 (bilateral vestibular schwannomas) 的存在。
■ 有在整個神經軸 (neural axis) 發展出多發性腦膜瘤 (meningiomas)、神經鞘瘤 (schwannomas)、神經膠質瘤 (gliomas) 與神經纖維瘤的風險。
NF-2 是一種體染色體顯性狀況,以雙側前庭神經鞘瘤、腦膜瘤(顱內、椎管內與視神經鞘)、神經鞘瘤(脊髓背根、周邊神經與顱神經)、CNS 的室管膜瘤 (ependymomas) 與神經膠質瘤,以及青少年型後囊下白內障 (juvenile posterior subcapsular cataracts) 為特徵。²⁹ 估計出生發生率為每 40,000 名活產嬰兒中 1 例。儘管它比 NF-1 少見得多,但其致病率 (morbidity) 卻大得多,病人常變得癱瘓與耳聾。最新的臨床診斷標準提高了確立診斷的敏感度,特別是在那些無陽性家族史的病例中(表 135-3)。約 60% 的病人在成年期以聽力喪失、耳鳴 (tinnitus) 或平衡喪失表現。發病年齡愈年輕,疾病最終的嚴重度愈大。孩童較易以非第八對顱神經腫瘤表現,例如視神經鞘腦膜瘤 (optic nerve sheath meningioma)。眼科發現包括青少年型後囊下白內障(60% 至 80% 的病人)、視網膜錯構瘤 (retinal hamartomas)、視網膜與視網膜色素上皮的合併錯構瘤 (combined hamartomas of the retina and retinal pigment epithelium),以及視神經鞘腦膜瘤。³⁰ ³¹
NF-2 個體可能有數個咖啡牛奶斑,但很少超過 6 個;不會出現間擦部位雀斑。NF-2 的特徵性皮膚病灶是皮膚神經鞘瘤 (cutaneous schwannoma)(圖 135-14)。它是一種斑塊狀、略隆起的病灶,帶有淡淡的紫紅色色澤,偶有毛髮。較不常見的是,可能發現與 NF-1 所見無法區分的皮膚神經纖維瘤。NF-2 是由第 22 號染色體上一個編碼膜相關蛋白 merlin 的基因發生突變所引起。如同 NF-1,NF2 基因是一種下調細胞生長的腫瘤抑制基因。此一控制所藉以發揮作用的確切細胞路徑仍有待闡明。
鑲嵌型第一型神經纖維瘤病 (Mosaic Neurofibromatosis Type 1)
節段型神經纖維瘤病 (segmental neurofibromatosis) 或鑲嵌型 NF-1 (mosaic NF-1) 一詞,是指那些具有 NF-1 表現(通常為咖啡牛奶斑與神經纖維瘤)、且侷限於身體某一區域的個體。³² Ruggieri 與 Huson 提議對這些個體使用「鑲嵌局部型 NF-1 (mosaic localized NF-1)」一詞,以承認此狀況的發病機轉是 NF1 在受孕後發生突變,導致體細胞鑲嵌現象。³³ 在一名病人身上證實了體細胞鑲嵌現象,其突變的 NF1 對偶基因以鑲嵌型態存在於培養自咖啡牛奶斑的纖維母細胞中,而在來自正常皮膚的纖維母細胞中則不存在。³⁴
絕大多數節段型 NF-1 病人有侷限於身體某一區域的咖啡牛奶斑或間擦部位雀斑(圖 135-15)。這些個體有在受影響區域發展出 NF-1 併發症的風險,最常見者為神經纖維瘤。NF-1 除咖啡牛奶斑、雀斑或皮膚神經纖維瘤以外的局部表現,也可能代表體細胞鑲嵌現象的例子。已有許多孤立性叢狀神經纖維瘤的報告,包括 1 例證實了腫瘤中許旺細胞發生 NF1 異合子性喪失的病例。由於只有 50% 的脛骨假關節病 (tibial pseudarthrosis) 病例發生在 NF-1 的背景下,其餘的可能實際上代表鑲嵌型 NF-1 的病例。鑲嵌型 NF-1 的其他例子包括一名孩童有單側視神經路徑膠質瘤,其生物學行為如同 NF-1 相關腫瘤,以及具有孤立性蝶骨發育不良的個體。對節段型 NF-1 病人的遺傳諮詢具有難度。這些病人中有許多被誤診為 NF-1,導致不必要的焦慮與不適當的遺傳諮詢。此外,已證實 NF-1 的性腺鑲嵌現象 (gonadal mosaicism);患有節段型 NF-1 的病人曾生下患有完全型 NF-1 的後代。
第一型神經纖維瘤病–Noonan 症候群 (Neurofibromatosis Type 1–Noonan Syndrome)
已認識到有些個體符合 NF-1 的診斷標準,卻又具有 Noonan 症候群的許多特徵;Noonan 症候群以眼距過寬 (hypertelorism)、眼瞼下垂 (ptosis)、瞼裂下斜 (downslanting palpebral fissures)、低位且向後旋轉的耳朵、頸蹼 (webbed neck)、胸廓畸形 (pectus deformities) 與身材矮小為特徵。超過 50% 的 Noonan 症候群孩童有心血管疾病,最常見者為肺動脈瓣狹窄 (pulmonary valve stenosis)。Noonan 症候群在 50% 的病例中是由 PTPN11 基因突變所引起。多個其他基因(KRAS、SOS1、BRAF、MEK1、MEK2、RIT1、HRAS 與 RAF1)的突變也與此表現型有關。基因檢測顯示,大多數 LEOPARD(lentigines 雀斑樣痣、electrocardiographic abnormalities 心電圖異常、ocular hypertelorism 眼距過寬、pulmonary stenosis 肺動脈狹窄、abnormalities of genitalia 生殖器異常、retardation of growth 生長遲緩、deafness 耳聾)症候群的個體也有 PTPN11 突變。³⁵
研究記錄了在 17 名臨床上具有 NF-1–Noonan 表現型的無血緣關係受試者中,有 16 名帶有 NF1 基因突變;未發現 PTPN11 突變。³⁶ 因此,絕大多數 NF-1–Noonan 症候群的病例似乎是由 NF1 突變所引起。
Legius 症候群 (Legius Syndrome)
已辨識出一些家族,其有咖啡牛奶斑、間擦部位雀斑與巨頭畸形的體染色體顯性遺傳,但無 NF-1 的任何其他表現(包括神經纖維瘤),且未偵測到 NF1 突變。對這些個體進一步的遺傳研究發現 SPRED1 突變,SPRED1 是一個負向調節 MAPK 路徑的基因。² 這些個體均無個別神經纖維瘤、叢狀神經纖維瘤、Lisch 結節、OPTs 或 NF-1 特有的骨性異常。一項針對 15 個 Legius 症候群家族的研究記錄到,受影響個體的全量表智商 (full scale IQ) 正常,但與未受影響的家族成員相比,作業智商 (performance IQ) 顯著較低。³⁷ 對於懷疑患有 Legius 症候群、且 NF1 定序未能偵測到突變的個體,可進行 SPRED1 突變的基因檢測。
神經鞘瘤病 (Schwannomatosis)
長久以來已注意到,有些病人會發展出多發性神經鞘瘤,卻不會發展出 NF-2 的其他表現,特別是前庭神經鞘瘤。患有此狀況(現稱為神經鞘瘤病,schwannomatosis)的病人,會在周邊神經與脊椎旁神經根 (paraspinal nerve roots) 內發展出多發性、疼痛的神經鞘瘤。³⁸ 這些腫瘤最常於人生的第二與第三個十年開始出現。為治療頑固性疼痛,常需對個別病灶進行手術。與 NF-2 相反,神經鞘瘤病個體有正常的壽命。在做出神經鞘瘤病的診斷之前,藉由基因檢測並施行 MRI 掃描以尋找前庭神經鞘瘤、以排除 NF-2 的可能性,是極為重要的。目前對於 30 歲或以上個體確切診斷神經鞘瘤病的診斷標準包括:(a) 2 個或以上的非皮內 (nonintradermal) 神經鞘瘤,至少 1 個有組織學確認,且 (b) 不符合 NF-2 的診斷標準,且 (c) MRI 上無前庭神經鞘瘤的證據,且 (d) 無罹患 NF-2 的一等親 (first-degree relative),且 (e) 無生殖細胞系 (germline) NF2 突變。³⁹ 腫瘤抑制基因 SMARCB1/INI1(先前被認為與嬰兒及年幼孩童橫紋肌樣瘤 [rhabdoid tumors] 的發展有關)的生殖細胞系突變,以及 LTZR1 的突變,已被辨識為大量家族性神經鞘瘤病病例的病因。然而,大多數病例為偶發性 (sporadic),由尚未辨識的基因突變所引起。⁴⁰
臨床病程、預後與處置 (Clinical Course, Prognosis, and Management)
放射學評估 (Radiographic Evaluation)
關於 NF-1 病人是否應「篩檢」隱藏的內部叢狀神經纖維瘤,存在爭議;較新的使用全身 MRI 的研究已證實其發生率高達 55%。⁴¹ ⁴² 若目前的化學治療試驗證明能成功治療快速生長的腫瘤,則使用此一技術在早期辨識它們可能會變得更為重要。
骨骼併發症 (Skeletal Complications)
所有 NF-1 孩童都應從幼兒早期開始定期篩檢脊柱側彎 (scoliosis)。脊柱側彎是 NF-1 最常見的骨骼表現,影響 10% 至 30% 的病人。脊柱側彎分為發育不良型 (dystrophic) 與非發育不良型 (nondystrophic)。發育不良型脊柱側彎是原發性骨發育不良的結果,可能在兒童期極早期就出現,典型上會導致橫跨相對少數椎體的急遽成角彎曲(圖 135-16)。它常伴隨極度的旋轉、椎體後緣的扇形改變 (scalloping)、椎體楔形變 (vertebral wedging)、椎弓根 (pedicles) 缺損,以及神經孔與脊椎管的擴大。彎曲可能快速進展,需要手術,而手術可能特別複雜,因為其彎曲複雜、多平面,骨質不佳,有神經根與脊髓損傷的潛在風險,且有椎管內與椎管外神經纖維瘤的存在。非發育不良型脊柱側彎較常見,與青少年的特發性脊柱側彎 (idiopathic scoliosis) 相似。許多孩童可以採取觀察等待或以支架 (bracing) 處置。⁴³
NF-1 孩童也可能出現局部區域的骨性過度生長 (bony overgrowth),且常位於無神經纖維瘤的區域。過度生長可能僅影響單一指(趾),或影響較大區域(如一隻手或一個肢體),但真正的半側肥大 (hemihypertrophy) 並不常見。骨性過度生長也可能發生在受叢狀神經纖維瘤影響的區域。
癌症 (Cancer)
NF-1 使個體易於罹患某些癌症的風險增加,這點幾乎毋庸置疑。在一項以族群為基礎的芬蘭研究中,估計終生癌症風險為 59.6%。研究發現 NF-1 個體罹患傳統上不被認為屬於 NF-1 疾病譜系的癌症的風險增加,最值得注意的是乳癌——與一般族群女性相比,40 歲以下 NF-1 女性的標準化發生率比 (standardized incidence ratio) 為 11.1。⁴⁴
惡性周邊神經鞘瘤 (Malignant Peripheral Nerve Sheath Tumors)
惡性周邊神經鞘瘤幾乎全都源自既有的叢狀神經纖維瘤。對於發展出此腫瘤的終生風險估計各有不同,但可能高達 13%。⁴⁵
由於治療結果不佳,醫師應勤於警覺這些腫瘤可歸因的早期症狀,包括快速生長的腫塊或無法解釋的疼痛或功能障礙。雖然傳統影像無法辨識良性叢狀神經纖維瘤內的惡性組織,但較新的影像方式(如 ¹⁸F-FDG PET 結合 CT 或 MRI)已被證實具有極佳的敏感度與特異度。⁷ ⁸
嗜鉻細胞瘤 (Pheochromocytoma)
嗜鉻細胞瘤明確與 NF-1 有關,發生率可能高達 1.4%。在一項全面性回顧中,平均發病年齡為 42 歲;84% 為單發性腎上腺腫瘤,10% 為雙側腎上腺腫瘤,6% 為位於腹部交感神經鏈 (abdominal sympathetic chain)、Zuckerkandl 器 (organ of Zuckerkandl) 與膀胱的異位性腫瘤。⁴⁶ 兒茶酚胺 (catecholamine) 相關症狀與高血壓各出現於 61% 的病人;22% 的病人兩者皆無。在 11.5% 的病人中發現惡性嗜鉻細胞瘤,常於發病時即有遠端轉移。NF1 的異合子性喪失已在 NF-1 相關與偶發性嗜鉻細胞瘤中均獲證實。⁴⁷
白血病 (Leukemia)
NF-1 孩童罹患青少年型慢性骨髓單核球性白血病 (juvenile chronic myelomonocytic leukemia) 與急性淋巴芽細胞性白血病 (acute lymphoblastic leukemia) 的風險增加。已在患有 NF-1 與惡性骨髓性疾病的孩童骨髓細胞中辨識出 NF1 的同合子失活 (homozygous inactivation),提示神經纖維瘤蛋白在下調未成熟骨髓性細胞 (immature myeloid cells) 的生長中扮演角色。⁴⁸ 此外,一項在英國以族群為基礎的研究辨識出,NF-1 個體發展出急性淋巴芽細胞性白血病的相對風險為一般族群的 5.4 倍。⁴⁹
其他腫瘤 (Other Tumors)
NF-1 孩童可能發展出幼年型黃色肉芽腫 (juvenile xanthogranulomas),這是直徑小於 1 cm 的黃色丘疹,通常見於頭部或軀幹,且可能為多發性(圖 135-17)。已有報告指出幼年型黃色肉芽腫、NF-1 與骨髓性白血病 (myeloid leukemias) 發展之間存在不尋常的關聯。⁵⁰ 雖然發病機轉不明,但此一關聯也曾在獨立於 NF-1 的情況下被觀察到。有些作者建議對同時患有幼年型黃色肉芽腫與 NF-1 的個體進行血球計數「篩檢」。鑑於此風險的確切大小未知,但肯定極小,此一建議只會增加父母的焦慮,而未提供任何有用資訊,且不為本文作者所支持。橫紋肌肉瘤 (rhabdomyosarcoma) 也較常發生於 NF-1 個體。⁵¹ 其他似乎較常發生於成年 NF-1 個體的腫瘤,包括十二指腸 (duodenum) 與 Vater 壺腹 (ampulla of Vater) 的體抑素瘤 (somatostatinomas),以及胃腸道間質瘤 (GI stromal tumors)。與非 NF-1 個體所見的腫瘤相比,NF-1 中的胃腸道間質瘤很少起源於胰臟,體積較小,且一般不伴隨荷爾蒙症狀(即糖尿病 diabetes、脂肪瀉 steatorrhea 與膽囊疾病 gallbladder disease)。⁵²
血管病變 (Vasculopathy)
NF-1 病人可能有影響任何動脈血管的血管病變。臨床表現可能包括腎動脈狹窄 (renal artery stenosis) 合併高血壓、腦梗塞 (cerebral infarcts)、出血性動脈瘤 (bleeding aneurysms),以及肢體的間歇性跛行 (intermittent claudication)。此血管病變是一種發育性問題,與神經纖維瘤對小動脈的壓迫無關,且似乎是出生後才獲得的,因為已有新病灶出現及既有病灶進展的描述。已在血管壁的所有層次中描述了特徵性的病理變化,最終導致動脈管腔的狹窄。NF-1 病人中最常被辨識出的血管病灶位於腎動脈,導致腎血管性高血壓 (renovascular hypertension)。⁵³ 對於任何使用單一降血壓藥物不易控制高血壓的成年 NF-1 病人,都應考慮腎動脈血管病變;所有 NF-1 孩童都應評估此一併發症。應使用主動脈攝影 (aortography) 合併腎動脈的選擇性血管攝影 (selective angiography) 來確認診斷。若使用口服降血壓藥物無法控制血壓,可施行經皮腔內血管成形術 (percutaneous transluminal angioplasty),若最初未成功可重複施行。腦動脈血管病變可能導致狹窄後 (poststenotic) 的小微血管增生,稱為moyamoya(煙霧病),可能反覆出血導致中風。
不明亮點 (Unidentified Bright Objects)
NF-1 孩童的腦部 MRI 常在 T2 加權影像上顯示訊號強度增加的區域,稱為「不明亮點 (unidentified bright objects)」或 UBOs(圖 135-18)。UBOs 可能見於內囊 (internal capsule)、基底核 (basal ganglia)、皮質 (cortex)、小腦半球 (cerebellar hemispheres)、視束 (optic tract) 或腦幹 (brainstem)。它們不會增強顯影,也不伴隨周圍組織的壓迫,這使它們得以與腫瘤區分。UBOs 出現於約 60% 的 NF-1 孩童,但會隨年齡消失,在成人中不常見。它們不伴隨局部神經徵象,其意義尚不確定。過去的組織病理學研究顯示,UBOs 對應於髓鞘空泡化 (myelin vacuolization)、含水量增加的區域。此外,近期一項使用擴散張量影像 (diffusion tensor imaging) 的研究提供了進一步的證據,顯示 UBOs 是由髓鞘內水池 (intramyelin water pool) 的變化所引起,並無去髓鞘 (demyelination) 或軸突損傷。⁵⁴
智力與學習障礙 (Intelligence and Learning Disabilities)
與較早文獻中嚴重失實的陳述相反,NF-1 病人的平均智商僅比一般族群與未受影響的手足低 5 至 10 分。然而,學習障礙(定義為能力 [智力] 與表現之間的差距)相當常見,估計盛行率為 30% 至 60%。⁵⁵ 雖然較早的報告提示 NF-1 中可能存在一種特定的「認知表現型 (cognitive phenotype)」,以視覺/知覺障礙過多為特徵,但較新的研究證實,在 NF-1 個體中言語缺損(如閱讀)至少與非言語障礙一樣常見。這些學習障礙是終生的,可能影響成年期的功能。許多 NF-1 孩童也有注意力與衝動控制不佳的問題,可能被診斷為注意力不足過動症 (attention deficit hyperactivity disorder),顯著干擾學業表現與學習。興奮劑藥物 (stimulant medications) 可能對這些個體在學業與職業上取得成功的能力有深遠的助益。⁵⁶ ⁵⁷
第一型神經纖維瘤病的處置 (Management of Neurofibromatosis Type 1)
NF-1 個體最好在一個能取得廣泛次專科醫師資源的多專科聯合門診 (multidisciplinary clinic) 中接受照護。門診的確切醫師組成不如能迅速取得次專科會診的能力來得重要。所有一等親都應接受 NF-1 皮膚表現的檢查,並應在首次就診時接受裂隙燈檢查以確認 Lisch 結節的存在。每年的就診讓醫師得以早期辨識 NF-1 的併發症,同時提供諮詢並傳播關於 NF-1 的資訊。個人與家庭可從 2 個全國性支持團體的網站獲得進一步資訊:Children’s Tumor Foundation(www.ctf.org)與 Neurofibromatosis Network(www.nfnetwork.org)。所有 10 歲或以下的 NF-1 孩童每年都應接受完整的眼科檢查,尋找 OPT 的徵象。這些檢查應包括視力 (visual acuity)、色覺、視野 (visual fields)、眼底鏡檢查 (funduscopy) 與裂隙燈檢查的評估。由於幾乎所有 OPTs 都發生在此一年齡層的孩童,10 歲以上孩童的眼科檢查頻率可以降低。每年應將體重與身高測量值繪製於標準化生長曲線圖上,因為性早熟最早期的指標可能是加速的線性生長。每次就診都應測量血壓,以尋找腎血管性高血壓的徵象。此外,每年應檢查脊柱以尋找脊柱側彎的早期徵象。
咖啡牛奶斑 (Café-au-Lait Spots)
NF-1 個體臉部的咖啡牛奶斑並不常見。偶爾確實會發生,受影響的個體可能會尋求改善外觀。嘗試以雷射治療去除咖啡牛奶斑的結果相當參差。銅蒸氣雷射 (copper vapor laser) 治療在 16 名接受治療的病人中有 15 名提供了良好至極佳的結果。⁵⁸ 使用 Q 開關 755-nm 紫翠玉雷射 (Q-switched 755-nm alexandrite laser) 在 56% 接受治療的病人中產生良好至極佳的結果,在 17% 中產生不佳的結果。⁵⁹ 使用低能量密度 (low-fluence) 1064-nm Q 開關釹:釔鋁石榴石雷射(neodymium:yttrium-aluminum-garnet, Nd:YAG)治療,在 75% 接受治療的咖啡牛奶斑中誘發了超過 50% 的清除。⁶⁰
個別皮膚神經纖維瘤 (Discrete Cutaneous Neurofibromas)
個別皮膚神經纖維瘤可藉由手術切除,以改善外觀或預防局部刺激,例如位於髮際線而在梳頭時受刺激,或位於足部而與鞋子摩擦。較深部的神經纖維瘤在壓迫重要結構時可能需要手術切除,例如浸潤神經孔並壓迫脊髓的背根神經纖維瘤。手術的併發症包括原腫瘤的再生長與神經損傷。對於因大量皮膚神經纖維瘤負荷而有嚴重搔癢的個體,抗組織胺可能提供症狀緩解。未對照的軼事性病例報告提示,ketotifen(一種抗組織胺與肥大細胞穩定劑)能緩解搔癢與疼痛,並預防新神經纖維瘤的快速生長。⁶¹ 在全身麻醉下使用二氧化碳雷射 (carbon dioxide laser) 去除數百個皮膚神經纖維瘤,使病人的生活品質顯著提升,疼痛與搔癢減少。⁶² 大多數病人認為遺留下平坦光滑的去色素瘢痕 (depigmented scar) 並不構成接受此類手術的阻礙。在另一項研究中,與電燒手術 (electrosurgery) 及二氧化碳雷射消融相比,鉺:釔鋁石榴石雷射(erbium:yttrium aluminium garnet, Er:YAG)消融較為優越。⁶³
叢狀神經纖維瘤 (Plexiform Neurofibromas)
直到最近,叢狀神經纖維瘤的治療仍侷限於手術減積 (surgical debulking),以改善外觀或預防功能喪失(如上呼吸道阻塞、失明)。此類手術的療效高度受限;鑑於這些腫瘤的高度浸潤性,完全切除是不可能的,且腫瘤再生長很常見。因此,開發用以對抗這些腫瘤的非傳統化學治療一直備受關注。不令人意外地,測試此類藥劑療效的研究設計最初充滿困難。叢狀神經纖維瘤在生物學上與較傳統的實體腫瘤頗為不同。治療後缺乏生長可能是腫瘤自然史的一部分,而非真正的治療反應。此外,腫瘤負荷可能難以在放射學上量化;叢狀神經纖維瘤可能沿神經根「擴散」,向外發出多個指狀突起,與單一實體腫瘤腫塊頗為不同。這些困難大致已藉由美國國家癌症研究所 (National Cancer Institute) 所收集的資料而被克服。體積測量 MRI (volumetric MRI) 使研究人員得以準確測量複雜腫瘤在三維空間中的生長變化,並顯示腫瘤體積是臨床試驗中可使用的有意義終點 (end point)。⁶⁴ 他們證實年紀較輕的病人其叢狀腫瘤生長最快,而新的叢狀腫瘤在出生後最初數年之後極不可能出現。⁶⁵ 最後,青春期的荷爾蒙變化似乎並不會加速這些腫瘤的生長。⁶⁶
掌握了這些資訊後,進行了數項研究以評估生物製劑 (biologics) 對叢狀神經纖維瘤生長的影響。使用 pirfenidone(一種抗纖維化藥劑,能減少纖維母細胞的增殖與膠原基質的合成)與一種下調 Ras 的法尼基轉移酶抑制劑的研究,均未產生有希望的結果。⁶⁷ ⁶⁸ Sirolimus(一種哺乳動物雷帕黴素標靶的抑制劑)在進展性叢狀神經纖維瘤病人中僅將進展時間延長了 4 個月。⁶⁹
基於對肥大細胞 c-Kit 受體訊息傳遞重要角色的體外研究,對一名患有無法切除的呼吸道叢狀神經纖維瘤的 3 歲孩童給予 imatinib,在 3 個月的治療期間使其體積縮小了 70%。⁷⁰ 此外,在一項後續研究中,17% 接受 imatinib 的病人其腫瘤體積減少了 20% 或以上。⁷¹ 若未來的研究證明其潛在療效,則篩檢性影像以偵測「隱藏」叢狀神經纖維瘤的角色將需要重新探討。
視神經路徑腫瘤 (Optic Pathway Tumors)
關於篩檢性神經影像 (screening neuroimaging) 在無症狀 NF-1 孩童照護中的角色,存在相當大的爭議。若例行「篩檢」能導致 OPTs 的早期偵測,並及早啟動可預防視力惡化的治療,則它將很重要。一項針對 NF-1 孩童的縱貫性研究未能辨識出任何因早期偵測而改變病人臨床病程的腫瘤。⁹ 此外,對極年幼 NF-1 孩童(OPTs 發展的高風險族群)進行針對性篩檢也告失敗,因為有 3 名孩童在 MRI 掃描結果正常後不久即發展出有症狀的腫瘤。⁷² NF-1 孩童與成人的腦幹腫瘤一般比非 NF-1 個體的對應腫瘤更為惰性 (indolent),提示例行篩檢並無益處。⁷³ 因此,National Neurofibromatosis Foundation 視神經路徑工作小組 (Optic Pathway Task Force) 建議反對對所有 NF-1 孩童進行例行篩檢性神經影像。¹⁴ 雖然關於成人進行頭部與脊髓「例行」MRI 掃描表現的可比縱貫性資料並不存在,但似乎合理的做法是僅對有提示 CNS 病理之症狀或徵象的個體進行此類檢查。OPT 一旦被辨識,應以系列 MRI 與眼科檢查追蹤。傳統上,治療僅在證實明確的放射學進展或視力惡化後才啟動。然而,近期一項來自 10 家機構、針對 115 名因 OPTs 接受治療之孩童的多中心回溯性研究證實,放射學結果無法預測視力結果。⁷⁴ 由於治療的目標應是在使治療副作用最小化的情況下保存視力,因此腫瘤生長而無視力下降,未必就先驗地 (a priori) 構成啟動治療的可接受理由。當有必要時,以 carboplatin 與 vincristine 治療已證實對這些腫瘤的處置有效。⁷⁵ 放射治療雖是非 NF-1 相關進展性 CNS 腫瘤治療的主要支柱,但並不適用於 NF-1 相關 OPTs,因為在極年幼的孩童中有血管病變、第二惡性腫瘤 (second malignancies) 以及有害的神經認知與內分泌副作用的風險。
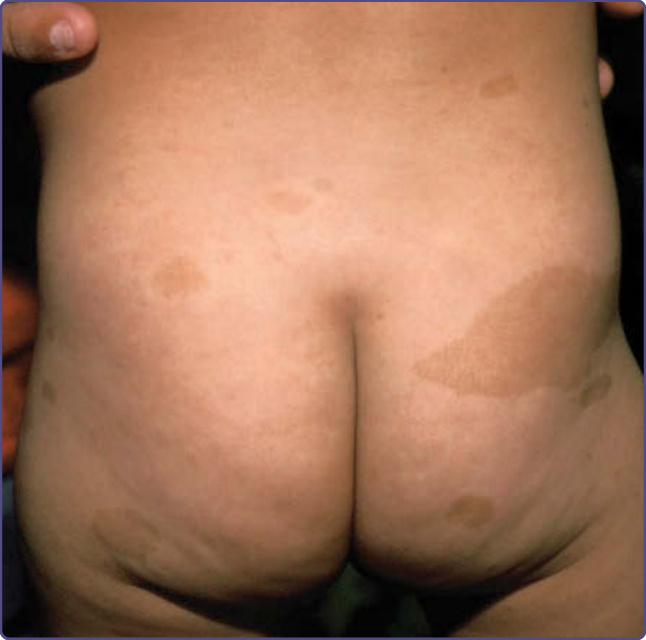
圖 135-1:咖啡牛奶斑 (café-au-lait spots)。
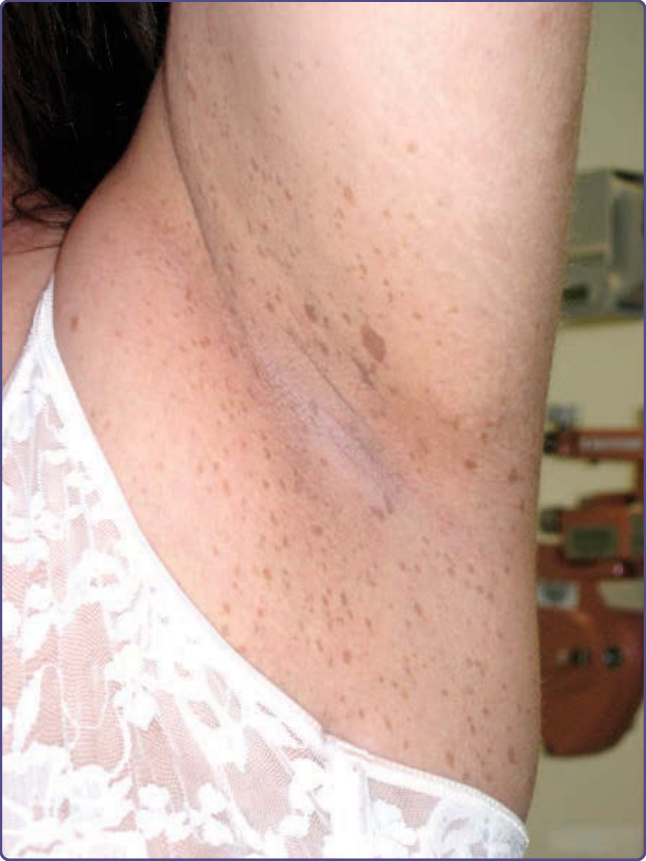
圖 135-2:腋窩雀斑 (axillary freckling)。
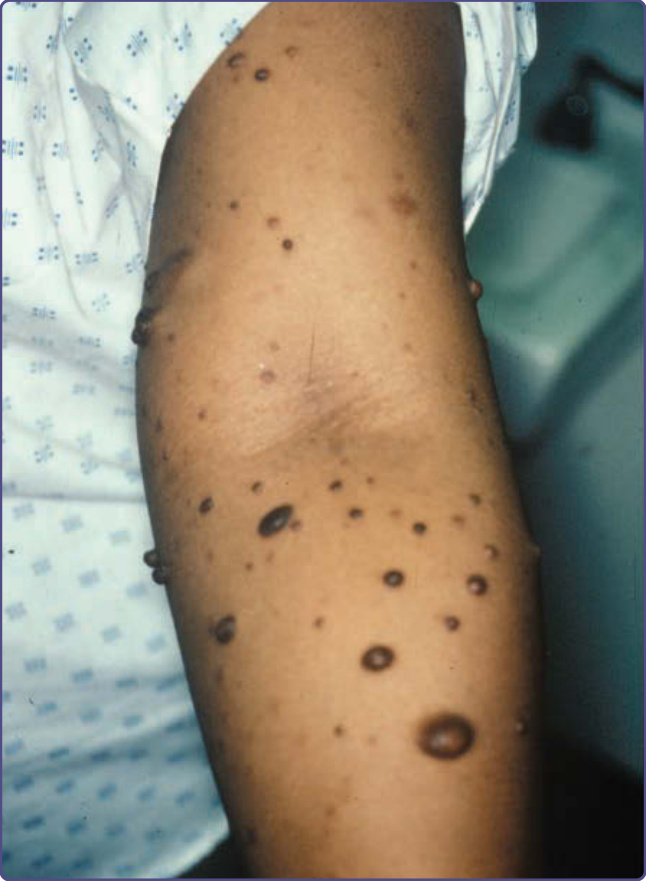
圖 135-3:皮膚神經纖維瘤 (cutaneous neurofibromas),其上方有色素過度沉著。
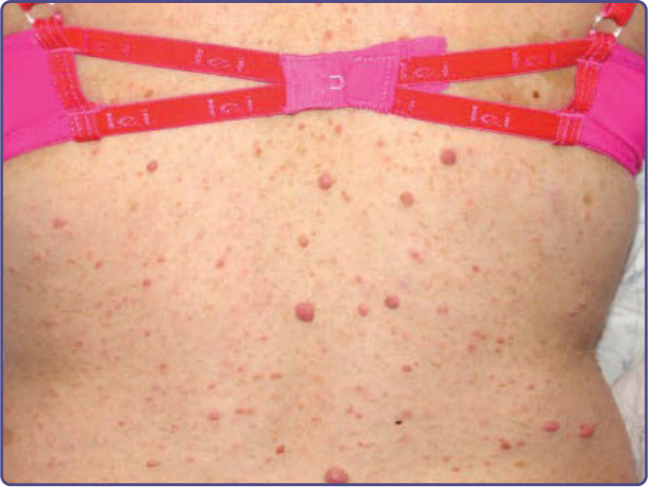
圖 135-4:多發性皮膚神經纖維瘤 (cutaneous neurofibromas)。
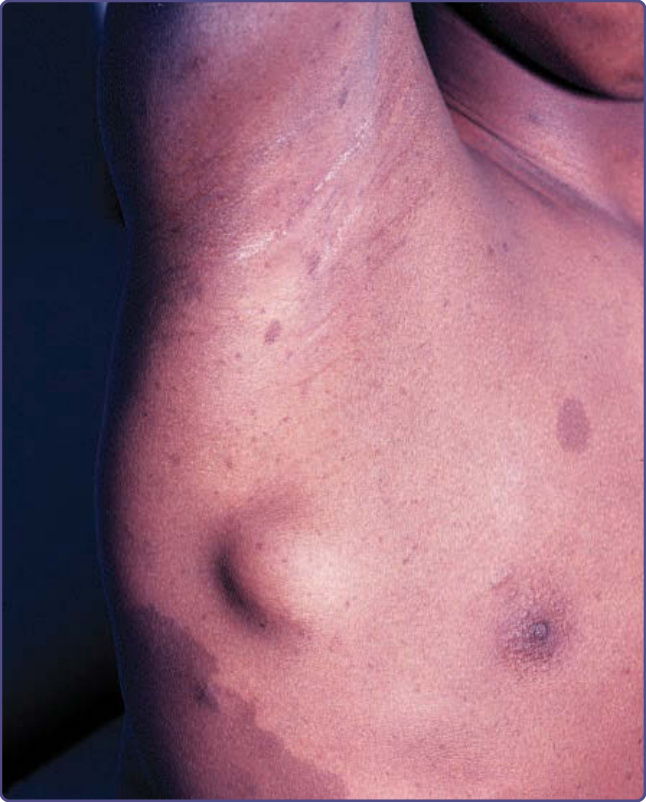
圖 135-5:皮下神經纖維瘤 (subcutaneous neurofibroma)。
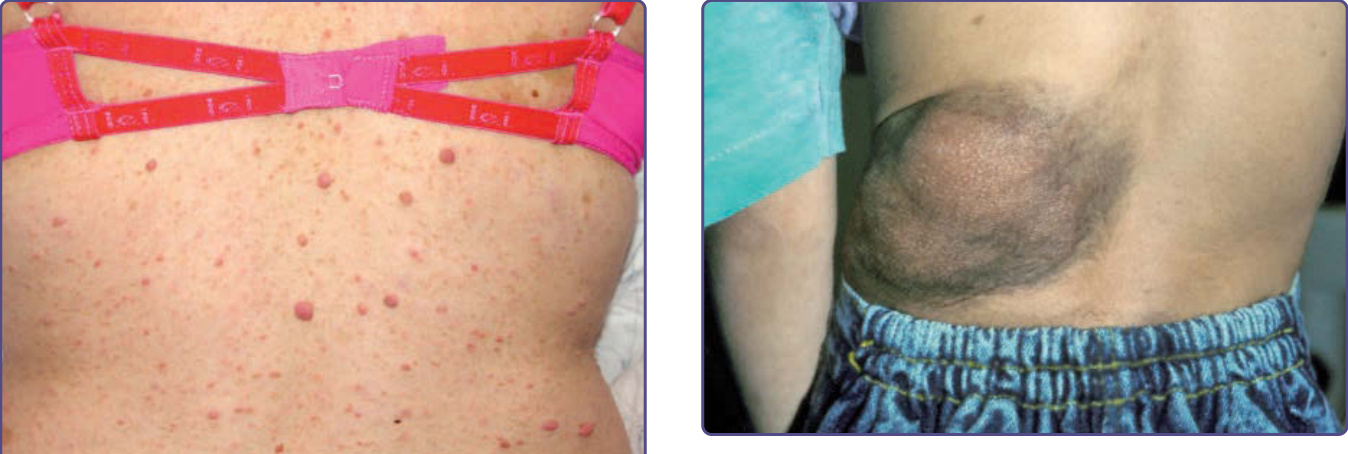
圖 135-6:叢狀神經纖維瘤 (plexiform neurofibroma),其上方有色素過度沉著與多毛症 (hypertrichosis)。
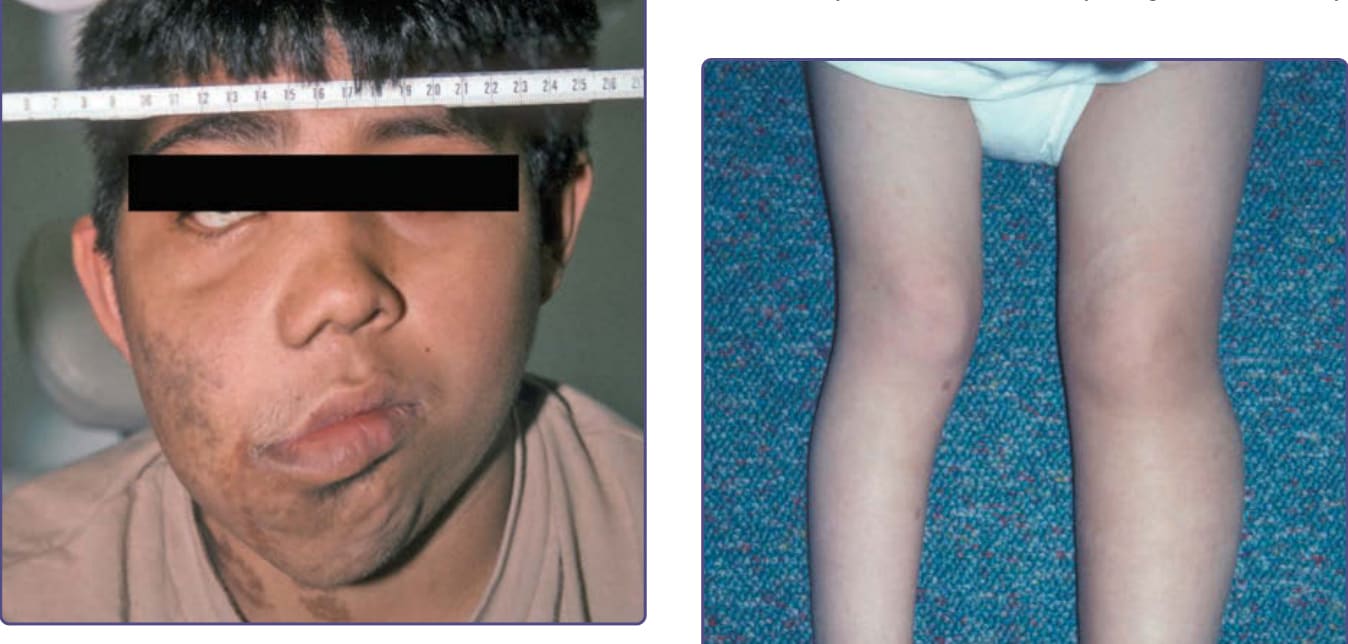
圖 135-7:顏面叢狀神經纖維瘤 (plexiform neurofibroma) 的進行性生長。
圖 135-8:左側第一趾的叢狀神經纖維瘤 (plexiform neurofibroma),導致孤立性巨趾症 (macrodactyly)。
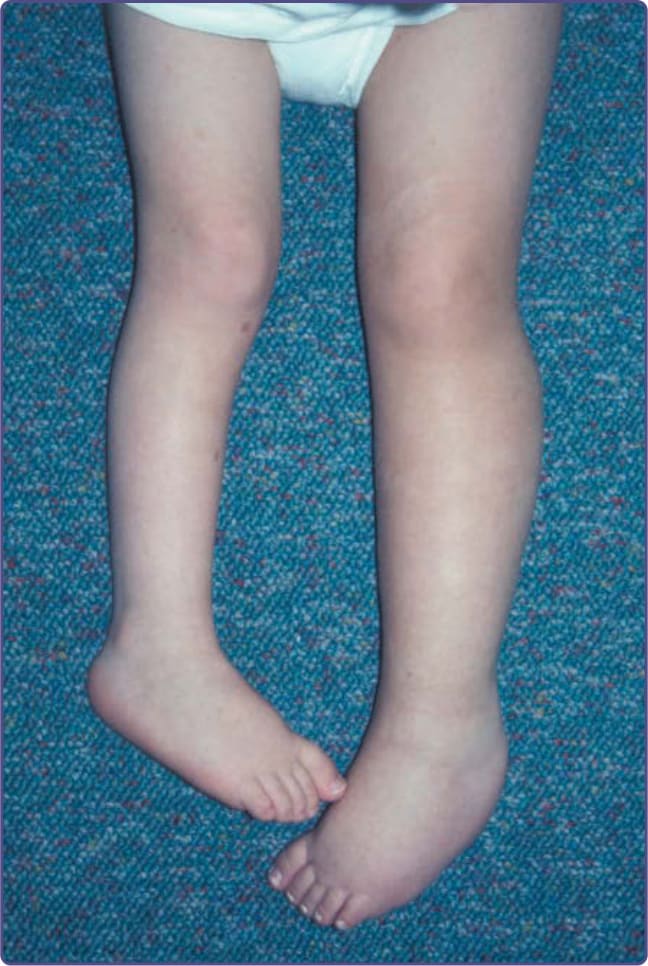
圖 135-9:左下肢的叢狀神經纖維瘤 (plexiform neurofibroma),導致雙腿長度不一 (leg-length discrepancy)。
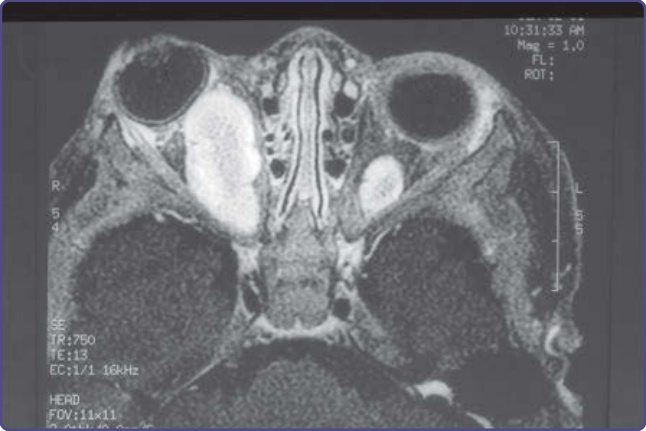
圖 135-10:眼眶內視神經腫瘤 (intraorbital optic nerve tumor) 造成眼球突出 (proptosis)。
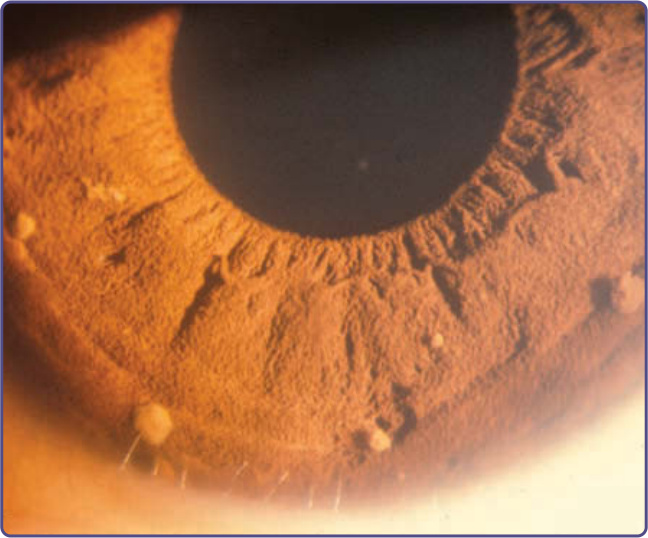
圖 135-11:Lisch 結節 (Lisch nodules)。
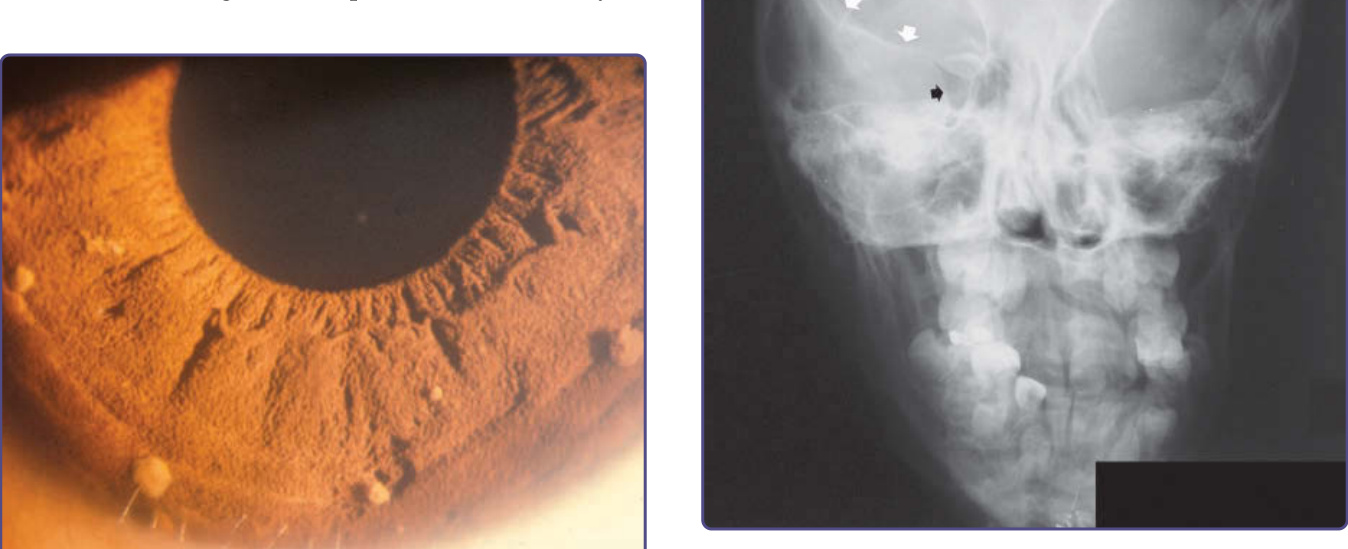
圖 135-12:蝶骨翼發育不良 (sphenoid wing dysplasia)。箭頭標示出右側眼眶壁中正常的蝶骨翼。對側的蝶骨翼則發育不良。
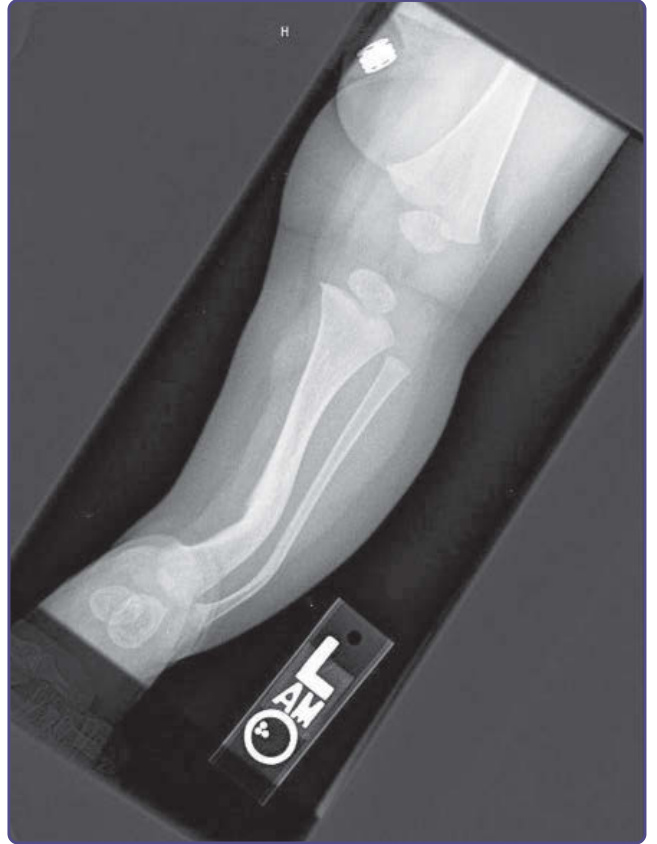
圖 135-13:脛骨與腓骨發育不良 (tibial and fibular dysplasia)。
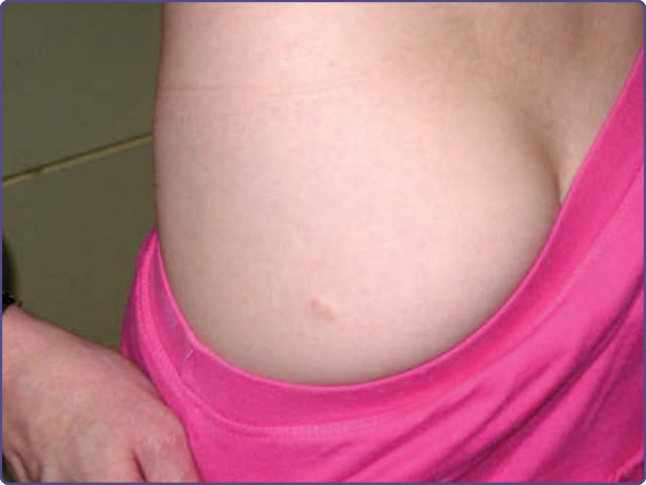
圖 135-14:皮膚神經鞘瘤 (cutaneous schwannoma)。
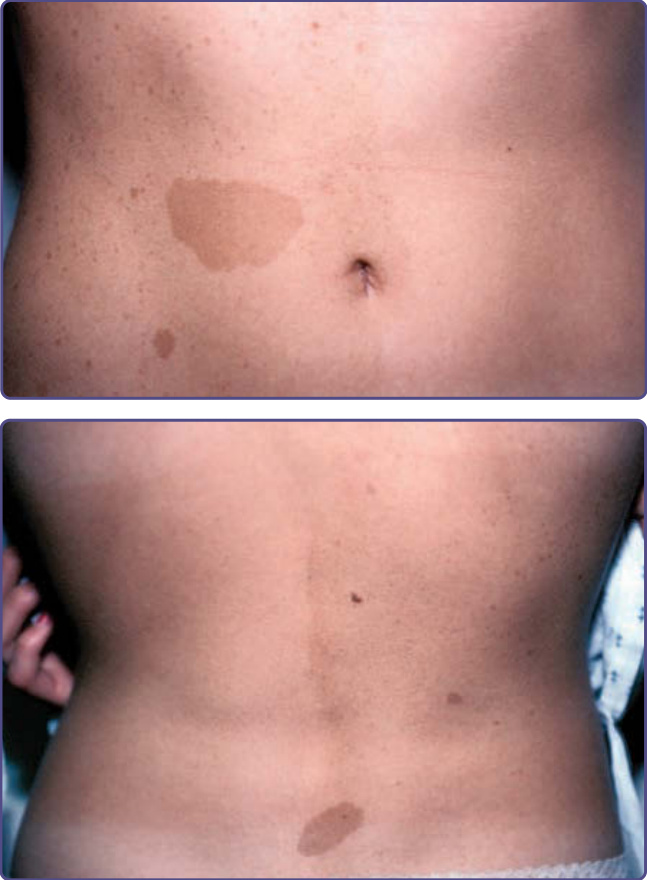
圖 135-15:鑲嵌型第一型神經纖維瘤病 (mosaic neurofibromatosis Type 1)。注意咖啡牛奶斑與雀斑侷限於身體的一側。
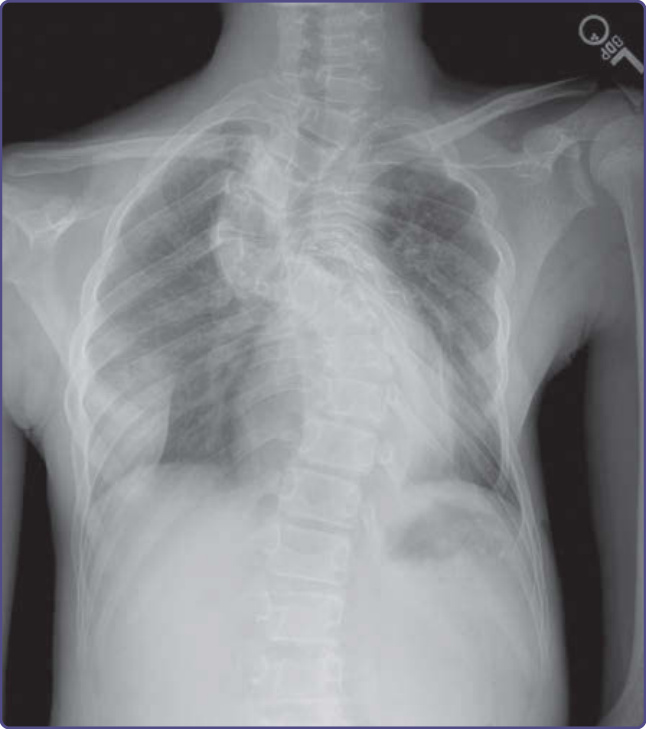
圖 135-16:發育不良型脊柱側彎 (dystrophic scoliosis)。
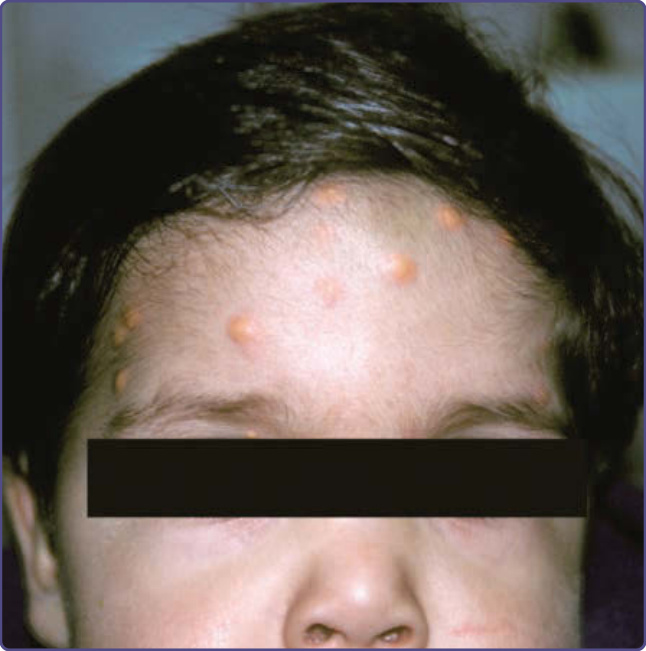
圖 135-17:前額上的多發性幼年型黃色肉芽腫 (juvenile xanthogranulomas)。
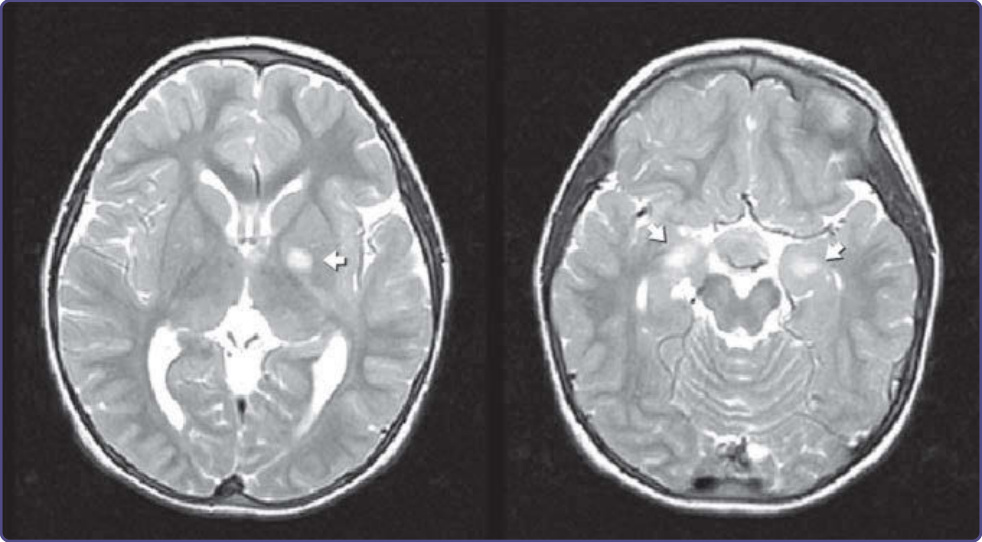
圖 135-18:T2 加權 MRI 的高訊號 (hyperintensity),稱為「不明亮點 (unidentified bright object)」。

表 135-1:第一型神經纖維瘤病 (Neurofibromatosis Type 1) 的診斷標準ᵃ
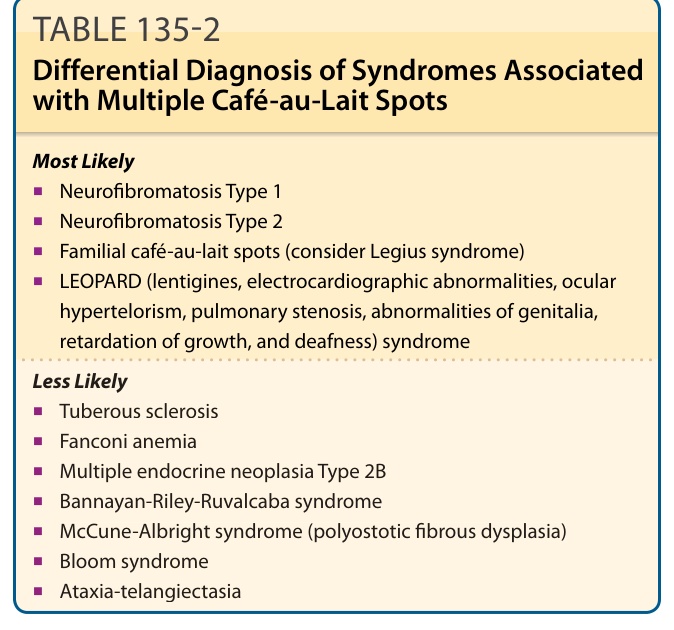
表 135-2:合併多發性咖啡牛奶斑 (Café-au-Lait Spots) 之症候群的鑑別診斷
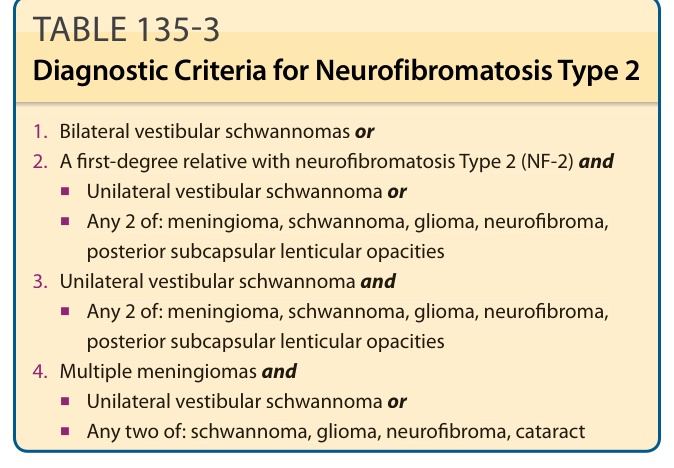
表 135-3:第二型神經纖維瘤病 (Neurofibromatosis Type 2) 的診斷標準