The Neurofibromatoses
21
AT-A-GLANCE
■ Autosomal dominant condition with incidence of 1 in 3000 live births.
■ Diagnosed clinically if 2 major features are present (see Table 135-1).
■ Cutaneous neurofibromas:
■ Softer than the surrounding connective tissue and protrude just above the skin surface or lie just under the skin with an overlying violaceous hue.
■ Subcutaneous neurofibromas:
■ Arise from peripheral nerves, both under the skin and deep in the viscera.
■ Generally much harder.
■ Plexiform neurofibromas:
■ Generally present at birth or apparent during the first several years of life.
■ May lead to disfigurement, blindness (secondary to amblyopia, glaucoma, or proptosis), loss of limb function, or organ dysfunction by compression of vital structures.
■ Mosaic neurofibromatosis Type 1 (segmental NF-1):
■ Manifestations of NF-1, usually limited to one area of the body.
■ Occurs as result of a postconceptional mutation in the NF1 gene, leading to somatic mosaicism.
CLINICAL FEATURES
A consensus development conference was held by the National Institutes of Health in 1987 to establish diagnostic criteria to promote better clinical research and care of patients with neurofibromatosis Type 1 (NF-1).1
The 7 diagnostic features recognized at this conference (Table 135-1), and the recommendation that 2 or more of these 7 features be present before a diagnosis of NF-1 is established, have proven extremely useful and continue to be used without modification more than 30 years later. Perhaps the one caveat is the identification of a separate autosomal dominant syndrome caused by an inactivating mutation of the gene encoding sprouty-related EVH1 domain-containing protein 1 (SPRED1), which leads to the development of café-au-lait spots, intertriginous freckling, and macrocephaly, but none of the other manifestations of NF-1 (Legius syndrome).2
CAFÉ-AU-LAIT SPOTS
CAFÉ-AU-LAIT SPOTS
Café-au-lait spots, which are flat, pigmented macules, are often the first manifestation of NF-1 to appear (Fig. 135-1). Frequently present at birth, they become more numerous as the infant grows; new ones may continue to appear throughout the first decade of life. Once noticed, they tend to grow in size in proportion to the overall growth of the child. Although infrequently found on the face, they may be noted anywhere on the body. The size, shape, and contour of café-au-lait spots are of no diagnostic significance, and the oft-quoted adage about smooth-edged café-au-lait spots being more typical of NF-1 rather than McCune-Albright syndrome is incorrect. When café-au-lait spots overlap each other, the area of overlap may be darker than either individual spot. When found within slate grey macules, they are typically surrounded by a more lightly pigmented halo. Individuals with large numbers of café-au-lait spots do not have “more-severe” NF-1, and the location of the macules in no way predicts the location of subsequent neurofibromas. Café-au-lait spots represent collections of heavily pigmented melanocytes of neural crest origin in the epidermis. Although the cells may contain increased numbers of “giant” melanosomes, or melanin macroglobules, these are not unique to NF-1, and their presence or absence in biopsies is not helpful diagnostically.
INTERTRIGINOUS FRECKLING
INTERTRIGINOUS
FRECKLING
Café-au-lait spots smaller than 5 mm are referred to as freckles, and are commonly present in the axillae, inguinal region, and under the breasts (Fig. 135-2). Unlike ordinary freckles in these locations, these lesions are not related to sun exposure, and are considered virtually pathognomonic of NF-1 (Crowe sign). Of all children ultimately diagnosed with NF-1, 81% will have intertriginous freckling by age 6 years.3
DISCRETE NEUROFIBROMAS
DISCRETE
NEUROFIBROMAS
Neurofibromas, which consist of Schwann cells, mast cells, fibroblasts, and perineural cells, are benign nerve sheath tumors that appear as discrete masses
21
■Six or more café-au-lait macules larger than 5 mm in greatest diameter in prepubertal individuals, and larger than 15 mm in greatest diameter in postpubertal individuals.
■Six or more café-au-lait macules larger than 5 mm in greatest
diameter in prepubertal individuals, and larger than 15 mm in greatest diameter in postpubertal individuals.
■Two or more neurofibromas of any type or 1 plexiform neurofibroma.
■Two or more neurofibromas of any type or 1 plexiform r r neurofibroma.
■Freckling in the axillary or inguinal regions.
■Freckling in the axillary or inguinal regions.
■Optic glioma.
■Optic glioma.
■Two or more iris Lisch nodules.
■Two or more iris Lisch nodules.
■A distinctive osseous lesion such as sphenoid dysplasia or thinning of long bone cortex with or without pseudarthrosis.
■A distinctive osseous lesion such as sphenoid dysplasia or thinning
of long bone cortex with or without pseudarthrosis.
■A first-degree relative (parent, sibling, or offspring) with neurofibromatosis Type 1 by the above criteria.
■A first-degree relative (parent, sibling, or offspring) with neurofibro-
matosis Type 1 by the above criteria.
aPatient must meet 2 or more of the listed criteria for diagnosis.
arising from peripheral nerves.4 Cutaneous neurofibromas protrude just above the skin surface or lie just under the skin with an overlying violaceous hue (Fig. 135-3). They are softer than the surrounding connective tissue, often creating a “buttonholing” sensation when a finger is rubbed gently over the surface (Fig. 135-4). Subcutaneous neurofibromas that arise from peripheral nerves, both under the skin and deep in the viscera, are generally much harder (Fig. 135-5). If they arise from the dorsal root ganglia, they may grow through neural foramina, compressing the spinal cord, creating a “dumbbell” appearance. Subcutaneous neurofibromas in the neck may feel like a “beaded necklace,” often being confused with lymph nodes. While fewer than 20% of children younger than 10 years of age have cutaneous neurofibromas, neurofibromas generally start appearing after puberty and increase in number as the patient grows older. Women who have NF-1 often comment on the appearance of cutaneous neurofibromas during pregnancy. The majority of adult patients with NF-1 probably have numerous
2466
asymptomatic deep neurofibromas involving the dorsal roots and other larger nerves. On occasion, neurofibroma-associated pruritus may be severe enough to require treatment with antihistamines.
PLEXIFORM NEUROFIBROMAS
PLEXIFORM
NEUROFIBROMAS
Plexiform neurofibromas, histologically similar to discrete neurofibromas, are benign peripheral nerve sheath tumors that involve single or multiple nerve fascicles, often arising from branches of major nerves.5 They may elicit a “wormy” sensation on palpation, as a person
21
feels multiple thickened nerve fascicles. Often there is overlying hyperpigmentation (“giant café-au-lait spot”) or hypertrichosis (Fig. 135-6). Most plexiform neurofibromas are present at birth or become apparent during the first several years of life. Externally visible plexiform neurofibromas are easily identified and may lead to disfigurement, blindness (secondary to amblyopia, glaucoma, or proptosis), or loss of limb function (Figs. 135-7 to 135-9). In contrast, thoracic or abdominal plexiform neurofibromas may have no external manifestations but may have equally devastating consequences as a consequence of invasion or compression of vital structures (eg, ureters, bowel, spinal cord). The growth rate of plexiform neurofibromas is highly variable. Periods of rapid growth alternating with long periods of quiescence are common. Malignant peripheral nerve sheath tumors, which generally arise from plexiform neurofibromas, may develop silently in deep plexiform neurofibromas and not give rise to symptoms until distant metastases have occurred. Even though loss of heterozygosity of NF1 may lead to the formation of benign neurofibromas, the generation of malignant transformation of a benign plexiform neurofibroma may be caused by cell-cycle regulators beyond the RAS oncogene. For example, mice that have null mutations in both the Nf1 and p53 genes uniformly develop malignant tumors.6 Physicians caring for individuals with NF-1 should be alert to development of a malignancy; plexiform neurofibromas should be biopsied if they exhibit rapid growth or cause significant pain or focal neurologic dysfunction. Positron emission tomography with 2-deoxy-2-[fluorine-18]fluoro-d-glucose integrated with computed tomography (18F-FDG PET/ CT) can be extremely useful in identifying the development of malignant peripheral nerve sheath tumor within a preexisting plexiform neurofibroma.7,8
OPTIC PATHWAY TUMORS
OPTIC PATHWAY TUMORS
Approximately 15% of children with NF-1 will develop optic pathway tumors (OPTs); however, less than half of these patients will ever develop symptoms, giving an
2467
21
overall incidence of symptomatic OPT of 7%.9,10 Recent series of NF-1–associated OPTs have identified an approximately 2:1 female predominance of patients, as well as a lower incidence in African American children. These observations suggest the possibility that either hormonal factors or modifying genes may influence the development and prognosis of OPTs.11,12
The period of greatest risk for the development of symptomatic OPTs in NF-1 is during the first 6 years of life; the development of a symptomatic tumor after
2468
age 6 years is extremely unusual.13,14 Thus, physicians caring for children with NF-1 should be sensitive to the signs and symptoms of OPTs in this young age group. Approximately 30% of children with symptomatic OPTs will present with the rapid onset of proptosis, with moderate to severe visual loss in the affected eye (Fig. 135-10). Another 30% of children who have symptomatic OPTs will have abnormal ophthalmologic examinations, without any visual symptoms, leading to discovery of the tumors. As young children rarely
complain of visual loss, even when severe, thorough annual eye examinations are imperative in all young children with NF-1. When present, ophthalmologic signs may include an afferent papillary defect, optic nerve atrophy, papilledema, strabismus, or defects in color vision. Finally, as many as 40% of children who have chiasmal tumors develop precocious puberty.15
Accelerated linear growth will be the first sign of precocious puberty, underscoring the need for all children with NF-1 to have annual assessments of growth using standard growth charts. Children with chiasmal tumors often have no ophthalmologic symptoms or signs. Early detection of precocious puberty is important as both the accelerated linear growth and the development of secondary sexual characteristics can be can be aborted with the use of a long-acting luteinizing hormone-releasing hormone agonist.
LISCH NODULES
LISCH NODULES
Lisch nodules are slightly raised, well-circumscribed melanocytic hamartomas of the iris thought to be virtually pathognomonic of NF-1 (Fig. 135-11). They are best seen using a slitlamp, which is necessary to
21
distinguish them from the more commonly seen flat iris nevi, which are not associated with NF-1. They do not cause any functional impairment of vision. The frequency with which they are found increases with age; although Lisch nodules are found in more than 90% of adults with NF-1, only 30% of children with NF-1 who are younger than 6 years of age have them.3
DISTINCTIVE OSSEOUS LESIONS
DISTINCTIVE OSSEOUS
LESIONS
There are 2 bony lesions distinctive enough to be included in the diagnostic criteria for NF-1. The first, dysplasia of the wing of the sphenoid bone, results in poor formation of the wall and/or floor of the orbit (Fig. 135-12). This congenital mesodermal dysplasia may be, but is not always, clinically apparent, leading to proptosis (from herniation of meninges or brain into the orbit) or enophthalmos. Dysplasia of a long bone, characterized by congenital thinning and bowing, affects approximately 2% of children with NF-1 (Fig. 135-13). Although the tibia is most commonly affected, the femur, humerus and other long bones also may be involved. Even when the bone is intact, thinning and bowing produce a visible deformity, and the weakened mechanical properties of the bone predispose to fracture, particularly in the weightbearing bones. Failure of primary union following a fracture results in a “false joint,” or pseudarthrosis.16
2469
21
ETIOLOGY AND PATHOGENESIS
EPIDEMIOLOGY
EPIDEMIOLOGY
The “modern age” of neurofibromatosis began in 1981, with Riccardi’s detailed clinical descriptions of the features and natural history of von Recklinghausen disease and the subsequent description of “central neurofibromatosis with bilateral acoustic neuroma.”17
With the availability of gene-based diagnosis, several distinct clinical syndromes have been identified: (a) NF-1, also known as von Recklinghausen disease; (b) NF-2, whose cardinal feature is the development of bilateral vestibular schwannomas; (c) segmental or mosaic NF-1; (d) Legius syndrome (SPRED1 mutation) leading to autosomal dominant transmission of café-au-lait spots, intertriginous freckling, and macrocephaly; and (e) schwannomatosis. Both NF-1 and NF-2 occur equally in all ethnic groups. NF-1 occurs at an incidence of 1 in 3000 live births, whereas NF-2 is much less common with an estimated incidence of 1 in 40,000 live births.
GENETICS
GENETICS
NF-1 is inherited in an autosomal dominant fashion. Although the expressivity of NF-1 varies considerably, even among individuals in the same family who are
2470
genotypically identical, the disorder is considered to be 100% penetrant. Thus, apparent skipping of NF-1 between generations may be the result of misdiagnosis, nonpaternity, or the occurrence of a new mutation in the grandchild. Although a large number of NF1 mutations have been described, only 2 genotype– phenotype correlations have been noted. Individuals who have deletions of the entire gene have an increased risk of facial dysmorphism, intellectual disability, early appearance of neurofibromas, and the presence of plexiform neurofibromas.18 In addition, individuals who have a specific 3 base pair in-frame deletion in exon 17 of NF1 may have café-au-lait spots, intertriginous freckling and Lisch nodules, but do not develop cutaneous, subcutaneous, or plexiform neurofibromas.19 There is evidence that NF-1 is more severe when inherited from the mother rather than from the father. NF1 is located on the long arm of chromosome 17, and encodes a protein called neurofibromin. Homozygosity for mutant NF1 alleles has not been reported, probably because it is a lethal condition. NF1 has an unusually high mutation rate, estimated at 2.4 × 10−5
to 10−4 gametes per generation, one of the highest of known inherited disorders. This may reflect the large size of the gene, which spans 350 kb of genomic DNA, and contains 59 exons that encode a peptide containing more than 2800 amino acids.20 An alternative hypothesis is that there is some structural property of the gene that renders it particularly susceptible to mutation. New mutations account for approximately 50% of cases, and are usually on the paternally inherited allele. In contrast to many other dominant disorders, the frequency of new mutations does not appear to increase with advancing paternal age.11
MOLECULAR BIOLOGY
MOLECULAR BIOLOGY
Most NF1 mutations result in reduced intracellular levels of the NF1 gene product, neurofibromin; this appears to be sufficient to cause many of the clinical manifestations of the disease. Tumorigenesis, including the development of benign dermal neurofibromas, appears to be dependent on inactivation of the normal NF1 allele in somatic cells, a process referred to as loss of heterozygosity.21 Loss of heterozygosity is a critical step in tumorigenesis in many inherited cancer predisposition syndromes (eg, retinoblastoma, Li-Fraumeni syndrome), and the genes involved are called tumorsuppressor genes. Tumorigenesis is initiated when both copies of the gene cease functioning normally—the first as a result of an inherited mutation, and the second as the result of a second somatic “hit” interfering with the function of the previously normal allele. Neurofibromin is found in a variety of cell types, including neurons, oligodendrocytes, and nonmyelinating Schwann cells. Considerable evidence has been developed for the role of neurofibromin as a negative regulator of Ras.20 The RAS gene family encodes membrane-associated, guanine nucleotide-binding proteins that are involved in the regulation of cellular
proliferation, differentiation, and learning. Ras exists in an active (Ras-GTP [guanosine triphosphate]) and inactive (Ras-GDP [guanosine diphosphate]) state. By favoring conversion of Ras from its active state to its inactive state, neurofibromin downregulates the downstream effects of Ras, which include promoting learning, memory, synaptic plasticity, and cell growth and proliferation. Mediators of the downstream effects of Ras include mitogen-activated protein kinase (MAPK), phosphatidylinositol 3′-kinase, protein kinase B, and mammalian target of rapamycin kinase.22 SPRED1 also inhibits activation of the MAPK pathway.22
In the brain, there are 3 major isoforms of Ras; K-ras is the primary target of neurofibromin activity.23 Activation of Ras by neurofibromin stimulates G-protein activity, resulting in activation of adenylyl cyclase. Cyclic adenosine monophosphate and other downstream intermediates appear to play a role in cell growth, learning, and memory. Neurofibromin also associates with microtubules and plays a role in regulating γ-aminobutyric acid-ergic inhibitory neuronal activity in the hippocampus.24
Ras is a membrane-bound protein, and requires farnesylation (addition of a C15 isoprenoid to a cysteine residue near the C-terminus) for activity, a process that is catalyzed by farnesyltransferase. Agents that inhibit farnesyltransferase cause general inhibition of the Ras pathway, much as neurofibromin does. Farnesyltransferase inhibitors reverse the proliferative phenotype in Nf1-deficient mouse cells, and can reverse spatial learning impairments in a mouse model of NF-1, by decreasing Ras levels.25 Inhibitors of downstream kinases of the MAPK pathway, such as MAPK kinase (MEK), have been shown to shrink plexiform neurofibromas in mouse models, leading the way for clinical trials that are ongoing.26
Loss of heterozygosity of NF1 in Schwann cells has been shown to be a necessary event for the development of both discrete and plexiform neurofibromas.27
Plexiform neurofibromas are comprised of Schwann cells, macrophages, mast cells, neurons and fibroblasts. Interestingly, neurofibromin-deficient Schwann cells secrete a substance that stimulates mast cell migration, which, in turn, stimulates production of extracellular matrix and angiogenesis.5 Astrocytes lacking NF1 expression cannot form optic nerve gliomas on their own; a brain environment heterozygous for NF1 is necessary for the development of these tumors. Microglia, CNS immune surveillance cells, are involved in both optic nerve glioma formation and maintenance.28
DIAGNOSIS
Despite the identification of the NF1 gene and its complete sequencing, the diagnosis of NF-1 remains primarily a clinical one. Although laboratory confirmation of the diagnosis is extremely useful in specific circumstances, in most cases it is unnecessary. As no common mutations have been identified, molecular diagnosis can only be based on strategies that screen
21
Most Likely
■Neurofibromatosis Type 1
■Neurofibromatosis Type 2
■Familial café-au-lait spots (consider Legius syndrome)
■LEOPARD (lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness) syndrome
Less Likely
Less Likely
■Tuberous sclerosis
■Tuberous sclerosis
■Fanconi anemia
■Fanconi anemia
■Multiple endocrine neoplasia Type 2B
■Multiple endocrine neoplasia Type 2B
■Bannayan-Riley-Ruvalcaba syndrome
■Bannayan-Riley-Ruvalcaba syndrome
■McCune-Albright syndrome (polyostotic fibrous dysplasia)
■McCune-Albright syndrome (polyostotic fibrous dysplasia)
■Bloom syndrome
■Bloom syndrome
■Ataxia-telangiectasia
■Ataxia-telangiectasia
the entire gene for mutations. This testing is available for both presymptomatic individuals and can be applied to prenatal diagnosis of NF-1 when a mutation has been identified in an affected parent. Inexperienced clinicians often biopsy cutaneous masses in an effort to confirm the diagnosis of NF-1 in an individual with multiple café-au-lait spots or other stigmata of NF-1. Such procedures are invariably unnecessary as the clinical diagnosis of NF-1 is usually quite straightforward. Biopsy of a plexiform neurofibroma should be reserved for those situations in which the physician wishes to exclude the possibility of a malignancy.
DIFFERENTIAL DIAGNOSIS
There are numerous syndromes in which café-au-lait spots may be seen (Table 135-2). However, there are several syndromes that have overlapping phenotypes with NF-1 that have caused confusion in the past.
NEUROFIBROMATOSIS TYPE 2
NEUROFIBROMATOSIS
TYPE 2
AT-A-GLANCE
■ Autosomal dominant disorder with incidence of 1 in 40,000 live births.
■ Hallmark is the presence of bilateral vestibular schwannomas.
■ At risk for development of multiple meningiomas, schwannomas, gliomas, and neurofibromas throughout neural axis.
2471
21
-
Bilateral vestibular schwannomas or
-
A first-degree relative with neurofibromatosis Type 2 (NF-2) and
-
Bilateral vestibular schwannomas or
-
A first-degree relative with neurofibromatosis Type 2 (NF-2) and
■Unilateral vestibular schwannoma or
■Unilateral vestibular schwannoma or
■Any 2 of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
3. Unilateral vestibular schwannoma and
■Any 2 of: meningioma, schwannoma, glioma, neurofibroma,
posterior subcapsular lenticular opacities
3. Unilateral vestibular schwannoma and
■Any 2 of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
4. Multiple meningiomas and
■Any 2 of: meningioma, schwannoma, glioma, neurofibroma,
posterior subcapsular lenticular opacities
4. Multiple meningiomas and
■Unilateral vestibular schwannoma or
■Unilateral vestibular schwannoma or
■Any two of: schwannoma, glioma, neurofibroma, cataract
■Any two of: schwannoma, glioma, neurofibroma, cataract
NF-2 is an autosomal dominant condition characterized by bilateral vestibular schwannomas, meningiomas (intracranial, intraspinal, and optic nerve sheath), schwannomas (dorsal roots of the spinal cord, peripheral nerves, and cranial nerves), ependymomas and gliomas of the CNS, and juvenile posterior subcapsular cataracts.29 The estimated birth incidence is 1 in 40,000 live births. Even though it is much less common than NF-1, its morbidity is much greater, with patients frequently becoming paralyzed and deaf. The most recent clinical diagnostic criteria have led to increased sensitivity in establishing the diagnosis, particularly in those cases without a positive family history (Table 135-3). Approximately 60% of patients present in adulthood with hearing loss, tinnitus, or loss of balance. The younger the age at presentation, the greater the ultimate severity of the disease. Children are more apt to present with a non–eighth nerve tumor, such as an optic nerve sheath meningioma. Ophthalmologic findings include juvenile posterior subcapsular cataracts (60% to 80% of patients), retinal hamartomas, combined hamartomas of the retina and retinal pigment epithelium, and optic nerve sheath meningiomas.30,31
Individuals with NF-2 may have several café-au-lait spots, but rarely have more than 6; intertriginous freckling is not seen. The characteristic cutaneous lesion of NF-2 is the cutaneous schwannoma (Fig. 135-14). It is
2472
a plaque-like, slightly raised lesion with a faint violaceous hue, occasionally with hair. Less commonly, cutaneous neurofibromas, indistinguishable from those seen in NF-1, may be found. NF-2 is caused by mutations in a gene on chromosome 22 which encodes the membrane-related protein merlin. As in NF-1, the NF2 gene is a tumor-suppressor gene that downregulates cellular growth. The exact cellular pathways by which this control is exerted have yet to be delineated.
MOSAIC NEUROFIBROMATOSIS TYPE 1
MOSAIC
NEUROFIBROMATOSIS
TYPE 1
The term segmental neurofibromatosis or mosaic NF-1 refers to individuals who have manifestations of NF-1, usually café-au-lait macules and neurofibromas, limited to one area of the body.32 Ruggieri and Huson have proposed use of the term mosaic localized NF-1 for these individuals, as an acknowledgment that the pathogenesis of this condition is a postconceptional mutation in NF1 leading to somatic mosaicism.33 Somatic mosaicism was confirmed in a patient in whom the mutant NF1 allele was present in a mosaic pattern in cultured fibroblasts from a café-au-lait macule and absent in fibroblasts from normal skin.34
The vast majority of patients with segmental NF-1 have café-au-lait macules or intertriginous freckling limited to one area of the body (Fig. 135-15). These individuals are at risk for developing complications of NF-1 in the affected area, most commonly neurofibromas. Localized manifestations of NF-1 other than café-au-lait macules, freckling, or cutaneous neurofibromas may also represent examples of somatic mosaicism. There have been numerous reports of isolated plexiform neurofibromas, including 1 case in which loss of heterozygosity of NF1 in Schwann cells from the tumor was demonstrated. As only 50% of cases of tibial pseudarthrosis occur in the context of NF-1, the others may actually represent cases of mosaic NF-1. Other examples of mosaic NF-1 include a child who had a unilateral optic pathway glioma that acted that acted biologically like an NF-1–associated tumor and individuals with isolated sphenoid bone dysplasia. Genetic counseling of patients with segmental NF-1 is problematic. Many of these patients are misdiagnosed as having NF-1, leading to unnecessary anxiety and inappropriate genetic counseling. In addition, gonadal mosaicism for NF-1 has been demonstrated; patients with segmental NF-1 have had offspring with complete NF-1.
NEUROFIBROMATOSIS TYPE 1–NOONAN SYNDROME
NEUROFIBROMATOSIS
TYPE 1–NOONAN SYNDROME
It has been recognized that there are individuals who meet the diagnostic criteria for NF-1, yet have many
features of Noonan syndrome, which is characterized by hypertelorism, ptosis, downslanting palpebral fissures, low-set, posteriorly rotated ears, webbed neck, pectus deformities, and short stature. More than 50% of children with Noonan syndrome have cardiovascular disease, most commonly pulmonary valve stenosis. Noonan syndrome is caused by mutations in the gene PTPN11 in 50% of cases. Mutations in multiple other genes (KRAS, SOS1, BRAF, MEK1, MEK2, RIT1, HRAS, and RAF1) also are associated with this phenotype. Genetic testing shows that most individuals with LEOPARD (lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness) syndrome also have mutations in PTPN11.35
Studies have documented mutations in the NF1 gene in 16 of 17 unrelated subjects who clinically had the NF-1–Noonan phenotype; no mutations in PTPN11 were found.36 Thus, it appears that the vast majority of cases of NF-1–Noonan syndrome are caused by mutations in NF1.
LEGIUS SYNDROME
LEGIUS SYNDROME
Families have been identified who have autosomal dominant transmission of café-au-lait spots, intertriginous freckling, and macrocephaly without any other manifestations of NF-1, including neurofibromas,
21
in whom no NF1 mutation was detected. Further genetic studies of these individuals revealed mutations in SPRED1, a gene that negatively regulates the MAPK pathway.2 None of these individuals has discrete neurofibromas, plexiform neurofibromas, Lisch nodules, OPTs, or NF-1–specific bony abnormalities. A study of 15 families with Legius syndrome documented a normal full scale IQ in affected individuals but a significantly lower performance IQ when compared to unaffected family members.37 Genetic testing for SPRED1 mutations is available in individuals suspected of having Legius syndrome in whom sequencing of NF1 has failed to detect a mutation.
SCHWANNOMATOSIS
SCHWANNOMATOSIS
It had been noted for some time that there are patients who develop multiple schwannomas but who fail to develop other manifestations of NF-2, particularly vestibular schwannomas. Patients with this condition, now called schwannomatosis, develop multiple, painful schwannomas within peripheral nerves and paraspinal nerve roots.38 The tumors most often start appearing during the second and third decades of life. Surgery of individual lesions is often necessary to treat intractable pain. In contrast to NF-2, individuals with schwannomatosis have a normal life span. Before making a diagnosis of schwannomatosis, it is extremely important to exclude the possibility of NF-2 by both genetic testing and performing an MRI scan to look for vestibular schwannomas. Current diagnostic criteria for a definite diagnosis of schwannomatosis in individuals 30 years of age or older include: (a) 2 or more nonintradermal schwannomas, at least 1 with histologic confirmation, and (b) diagnostic criteria for NF-2 not fulfilled, and (c) no evidence of vestibular schwannoma on MRI, and (d) no first-degree relative with NF-2, and (e) no germline NF2 mutation.39 Germline mutations in the tumor-suppressor genes SMARCB1/INI1, previously implicated in the development of rhabdoid tumors in infants and young children, as well as mutations in LTZR1 have been identified as the cause of a large number of the familial cases of schwannomatosis. However, most cases are sporadic and caused by mutations in as-yet unidentified genes.40
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
RADIOGRAPHIC EVALUATION
RADIOGRAPHIC EVALUATION
There is debate as to whether patients with NF-1 should be “screened” for the presence of hidden, internal plexiform neurofibromas; newer studies using whole-body MRI have demonstrated an incidence as high as 55%.41,42 If current chemotherapeutic trials
2473
21
prove successful in the treatment of rapidly growing tumors, their identification at an early age may become more important using this technique.
SKELETAL COMPLICATIONS
SKELETAL COMPLICATIONS
All children with NF-1 should be regularly screened for scoliosis, beginning in early childhood. Scoliosis is the most common skeletal manifestation of NF-1, affecting 10% to 30% of patients. Scoliosis is divided into dystrophic and nondystrophic types. Dystrophic scoliosis is the result of primary bone dysplasia, and may present very early in childhood, typically resulting in a sharply angulated curve spanning relatively few vertebral bodies (Fig. 135-16). It is often accompanied by extreme rotation, scalloping of the posterior margins of the vertebral bodies, vertebral wedging, defective pedicles, and enlargement of the neural foramina and spinal canal. The curvature may progress rapidly, necessitating surgery that may be particularly complicated because of the complex, multiplanar curves, poor quality of bone, potential for nerve root and spinal cord injury, and the presence of intraspinal and extraspinal neurofibromas. The nondystrophic type of scoliosis is more common, and is similar to idiopathic scoliosis in adolescents. Many children can be managed expectantly or with bracing.43
Localized regions of bony overgrowth may also occur in children with NF-1 and are often in areas free of neurofibromas. The overgrowth may affect only a single digit or a larger region such as a hand or an extremity, but true hemihypertrophy is unusual. Bony overgrowth may also occur in a region affected by a plexiform neurofibroma.
2474
CANCER
CANCER
There is little doubt that NF-1 predisposes an individual to an increased risk for certain cancers. In a populationbased Finnish study, an estimated lifetime cancer risk was 59.6%. Individuals with NF-1 were found to be at increased risk for cancers not traditionally thought to be part of the NF-1 spectrum of disease, most notably breast cancer which had a standardized incidence ratio of 11.1 for NF-1 women younger than 40 years of age when compared to women in the general population.44
MALIGNANT PERIPHERAL NERVE SHEATH TUMORS
Malignant peripheral nerve sheath tumors almost exclusively arise from preexisting plexiform neurofibromas. Estimates of the lifetime risk for the development of this tumor vary but may be as high as 13%.45
As the outcome of therapy is poor, physicians should be diligently aware of the early symptoms referable to these tumors, including a rapidly growing mass or unexplained pain or dysfunction. Although conventional imaging cannot identify malignant tissue within a benign plexiform neurofibroma, newer modalities, such as 18F-FDG PET coupled with either CT or MRI, have been shown to have excellent sensitivity and specificity.7,8
PHEOCHROMOCYTOMA
Pheochromocytoma clearly is associated with NF-1, with an incidence potentially as high as 1.4%. In a comprehensive review, the mean age at presentation was 42 years; 84% had solitary adrenal tumors, 10% had bilateral adrenal tumors, and 6% had ectopic tumors in the abdominal sympathetic chain, organ of Zuckerkandl, and the bladder.46 Catecholamineassociated symptoms and hypertension each were present in 61% of the patients; 22% of the patients had neither of these findings. Malignant pheochromocytomas were found in 11.5% of patients, often with distant metastases at presentation. Loss of heterozygosity of the NF1 has been demonstrated both in NF-1– associated and sporadic pheochromocytomas.47
LEUKEMIA
There is an increased risk of juvenile chronic myelomonocytic leukemia and acute lymphoblastic leukemia in children with NF-1. Homozygous inactivation of NF1 in the bone marrow cells children with NF-1 and malignant myeloid disorders has been identified, suggesting that neurofibromin plays a role in downregulating the growth of immature myeloid cells.48 In addition, a population-based study in the United Kingdom identified that NF-1 individuals had a relative risk of developing acute lymphoblastic leukemia of 5.4 times the general population.49
OTHER TUMORS
Children with NF-1 may develop juvenile xanthogranulomas, which are yellowish papules less than 1 cm in diameter that are usually found on the head or trunk and may be multiple (Fig. 135-17). An unusual association between juvenile xanthogranulomas, NF-1, and the development of myeloid leukemias has been reported.50 Although the pathogenesis is obscure, this association also has been observed independent of NF-1. Some authors have recommended “screening” blood counts for individuals who have juvenile xanthogranulomas and NF-1. Given that the precise magnitude of the risk is unknown, but certainly extremely small, this recommendation only serves to increase parental anxiety without providing any useful information and is not endorsed by the authors. Rhabdomyosarcoma also occurs more frequently in individuals with NF-1.51 Other tumors that appear to occur more frequently in adults with NF-1 are somatostatinomas of the duodenum and the ampulla of Vater, and GI stromal tumors. Compared to those tumors seen in non–NF-1 individuals, GI stromal tumors in NF-1 rarely arise in the pancreas, are smaller, and generally are not associated with hormonal symptoms (ie, diabetes, steatorrhea, and gallbladder disease).52
VASCULOPATHY
VASCULOPATHY
Patients with NF-1 may have a vasculopathy affecting essentially any arterial vessel. Clinical manifestations may include renal artery stenosis with hypertension, cerebral infarcts, bleeding aneurysms, and intermittent claudication of an extremity. This vasculopathy is a developmental problem not related to compression
21
of an arteriole by a neurofibroma, and appears to be acquired after birth, as the appearance of new lesions and the progression of preexisting ones have been described. Characteristic pathologic changes have been described in all layers of the vascular wall which ultimately leads to narrowing of the arterial lumen. The most commonly identified vascular lesion in patients with NF-1 is in the renal artery, leading to renovascular hypertension.53 Renal artery vasculopathy should be considered in any adult NF-1 patient with hypertension that is not easily controlled with a single antihypertensive medication; all NF-1 children should be evaluated for this complication. Aortography with selective angiography of the renal arteries should be used to confirm the diagnosis. If the blood pressure is uncontrollable using oral antihypertensives, percutaneous transluminal angioplasty can be performed and repeated if initially unsuccessful. Cerebral artery vasculopathy may lead to poststenotic proliferation of small capillaries, termed, moyamoya, which can recurrently bleed leading to strokes.
UNIDENTIFIED BRIGHT OBJECTS
UNIDENTIFIED BRIGHT
OBJECTS
Brain MRI of children with NF-1 frequently demonstrates regions of increased signal intensity on T2-weighted images, referred to as “unidentified bright objects” or UBOs (Fig. 135-18). UBOs may be found in the internal capsule, basal ganglia, cortex, cerebellar hemispheres, optic tract, or brainstem. They do not enhance and are not associated with compression of surrounding tissue, which distinguish them from neoplasms. UBOs are present in approximately 60% of children with NF-1, but disappear with age and are uncommon in adults. They are not associated with focal neurologic signs and are of uncertain significance. Past histopathologic studies have shown that the UBOs corresponded to areas of myelin vacuolization with increased water content. In addition, a recent study using diffusion tensor imaging provided further evidence that UBOs were caused by changes in the intramyelin water pool without demyelination or axonal damage.54
INTELLIGENCE AND LEARNING DISABILITIES
INTELLIGENCE AND
LEARNING DISABILITIES
Contrary to gross misstatements in the older literature, the mean IQ of patients with NF-1 is only 5 to 10 points lower than the general population and unaffected siblings. However, learning disabilities, defined as discrepancies between ability (intellect) and performance, are quite common with an estimated prevalence of 30% to 60%.55 Although earlier reports suggested there might be a specific “cognitive phenotype” in NF-1
2475
21
characterized by an excess of visual/perceptual disabilities, newer studies demonstrate that verbal deficits (eg, reading) are at least as common as nonverbal disabilities in individuals with NF-1. These learning disabilities are lifelong and can affect adult functioning. Many children with NF-1 also have poor attention and impulse control, and may be diagnosed with attention deficit hyperactivity disorder, significantly interfering with school performance and learning. Stimulant medications may have a profound beneficial effect on the ability of these individuals to succeed academically and professionally.56,57
MANAGEMENT OF NEUROFIBROMATOSIS TYPE 1
MANAGEMENT OF
NEUROFIBROMATOSIS
TYPE 1
Individuals with NF-1 are best cared for within a multidisciplinary clinic that has access to a wide range of subspecialists. The exact physician composition of the clinic is less important than the ability to obtain expeditious subspecialty consultation. All first-degree relatives should be examined for the cutaneous manifestations of NF-1 and should undergo slitlamp examination at the first visit to ascertain the presence of Lisch nodules. Yearly visits allow the physician to identify NF-1 complications early, while providing counseling and dissemination of information regarding NF-1. Individuals and families can obtain further information from the websites of the 2 national support groups: the Children’s Tumor Foundation (www.ctf.org) and the Neurofibromatosis Network (www.nfnetwork.org). All children with NF-1 who are 10 years old or younger should have yearly complete ophthalmologic examinations looking for signs of an OPT. These should include assessment of visual acuity, color vision, visual fields, funduscopy, and slitlamp examination. As almost all OPTs arise in children in this age
2476
group, the frequency of ophthalmologic examinations can be reduced in children older than 10 years of age. Yearly measurements of weight and height should be plotted on standardized growth charts, as the earliest indication of precocious puberty may be accelerated linear growth. Blood pressure measurements should be obtained at each visit to look for signs of renovascular hypertension. In addition, the spine should be examined each year for early signs of scoliosis.
CAFÉ-AU-LAIT SPOTS
Café-au-lait spots on the face are unusual in individuals with NF-1. On occasion, they do occur and affected individuals may seek to improve cosmesis. Attempts at removing café-au-lait spots with laser therapy have provided very mixed results. Copper vapor laser therapy provided good to excellent results in 15 of 16 treated patients.58 Use of the Q-switched 755-nm alexandrite laser produced good to excellent results in 56% of treated patients and poor results in 17%.59 Use of low-fluence 1064-nm Q-switched neodymium:yttriumaluminum-garnet (Nd:YAG) laser therapy induced greater than 50% clearance in 75% of the café-au-lait spots treated.60
DISCRETE CUTANEOUS NEUROFIBROMAS
Discrete cutaneous neurofibromas may be removed surgically to improve cosmesis or to prevent local irritation, for example, in the hairline while brushing or on the foot rubbing against the shoe. Deeper neurofibromas may require surgical removal when pushing on vital structures, such as a dorsal root neurofibroma that infiltrates the neural foramen and compresses the spinal cord. Complications of surgery include regrowth of the original tumor and nerve damage. In individuals who have severe pruritus from a large burden of cutaneous neurofibromas, antihistamines
may provide symptomatic relief. Uncontrolled anecdotal case reports suggest that ketotifen, an antihistamine and mast cell stabilizer, has provided relief from pruritus and pain and prevented the rapid growth of new neurofibromas.61 Use of the carbon dioxide laser under general anesthesia to remove hundreds of cutaneous neurofibromas has resulted in markedly increased quality of life and decreased pain and pruritus for patients.62 The residua of a flat smooth depigmented scar was not thought to be an impediment to such surgery by most patients. In another study, when compared to electrosurgery and carbon dioxide laser ablation, erbium:yttrium aluminium garnet (Er:YAG) laser ablation was superior.63
PLEXIFORM NEUROFIBROMAS
Until recently, the treatment of plexiform neurofibromas was limited to surgical debulking either to improve cosmesis or to prevent loss of function (eg, upper airway obstruction, blindness). Such surgery was highly limited in its efficacy; complete resection was impossible given the highly infiltrative nature of these tumors and tumor regrowth was common. Thus, there has been considerable interest in the development of nontraditional chemotherapy for use against these tumors. Unsurprisingly, the design of studies to test the efficacy of such agents was initially fraught with complications. Plexiform neurofibromas are quite different biologically from more conventional solid neoplasms. Lack of growth following treatment may be part of the natural history of the tumor rather than a true therapeutic response. In addition, tumor burden may be difficult to quantify radiographically; plexiform neurofibromas may “spread” along nerve roots sending out multiple finger-like projections, quite different than a single, solid tumor mass. These difficulties largely have been overcome by data collected at the National Cancer Institute. Volumetric MRI has enabled researchers to accurately measure changes in growth of complex tumors in 3 dimensions and to show that tumor volume was a meaningful end point to use in clinical trials.64 They demonstrated that younger patients had the most rapid growth of plexiform tumors that that new plexiform tumors were extremely unlikely to appear after the first few years of life.65 Finally, the hormonal changes in puberty did not seem to accelerate the growth of these tumors.66
Armed with this information, several studies were performed to assess the impact of biologics on the growth of plexiform neurofibromas. Studies using pirfenidone, an antifibrotic agent that decreases proliferation of fibroblasts and collagen matrix synthesis, and a farnesyltransferase inhibitor that downregulates Ras, did not yield promising results.67,68 Sirolimus, an inhibitor of the mammalian target of rapamycin, prolonged the time to progression by only 4 months in patients with progressive plexiform neurofibromas.69
Based on in vitro studies of the important role of mast cell c-Kit receptor signaling, administration of imatinib to a 3-year-old with an unresectable airway plexiform
21
neurofibroma led to 70% diminution in size during 3 months of therapy.70 In addition, in a subsequent study, 17% of patients receiving imatinib had a 20% or more decrease in tumor volume.71 If future studies demonstrate potential efficacy, the role of screening imaging to detect “hidden” plexiform neurofibromas will need to be readdressed.
OPTIC PATHWAY TUMORS
There has been considerable debate regarding the role of screening neuroimaging in the care of asymptomatic children with NF-1. Routine “screening” would be important if it led to early detection of OPTs and early initiation of therapy that prevented visual deterioration. A longitudinal study of children with NF-1 failed to identify any tumors in which early detection altered the patient’s clinical course.9 Moreover, targeted screening of very young children with NF-1, the high-risk group for the development of OPTs, failed, as 3 children developed symptomatic tumors shortly after having had normal MRI scans.72 Brainstem tumors in both children and adults with NF-1 are generally more indolent than their counterparts in non–NF-1 individuals, suggesting that routine screening would not be beneficial.73 Thus, the National Neurofibromatosis Foundation Optic Pathway Task Force has recommended against routine screening neuroimaging of all children with NF-1.14 Although there are no comparable longitudinal data for adults regarding the performance of “routine” MRI scans of the head and spinal cord, it seems reasonable to perform such testing only in individuals who have symptoms or signs suggestive of CNS pathology. Once identified, OPT should be followed by serial MRI and ophthalmologic examinations. Traditionally, treatment had been initiated only after demonstration of clear radiographic progression or deterioration of vision. However, a recent multicenter retrospective study from 10 institutions of 115 children treated for OPTs demonstrated that radiographic outcomes did not predict visual acuity outcomes.74 Because the goal of treatment should be preservation of vision with minimization of side effects of therapy, tumor growth without visual decline might not be a priori an acceptable reason to institute therapy. When necessary, treatment with carboplatin and vincristine has proven effective in the management of these tumors.75 Radiation therapy, while a mainstay of treatment of progressive CNS neoplasms not associated with NF-1, is not appropriate for NF-1– associated OPTs because of the risk of vasculopathy, second malignancies, and detrimental neurocognitive and endocrinologic side effects in very young children.
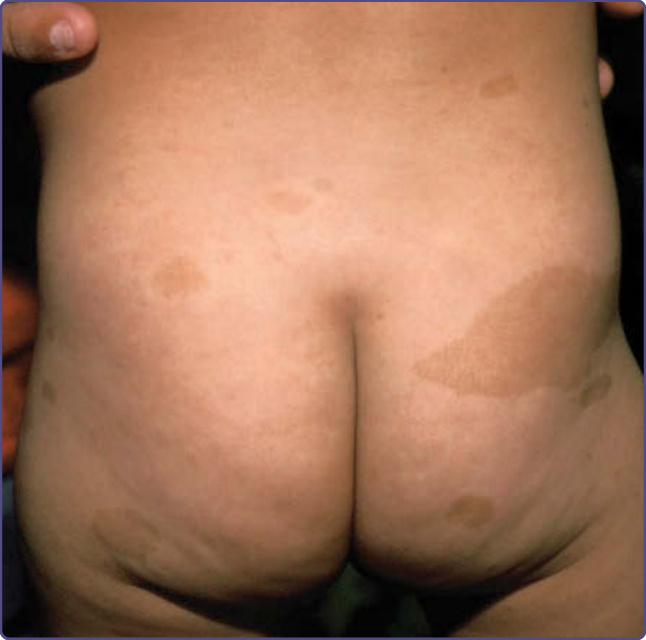
Figure 135-1 Café-au-lait spots.
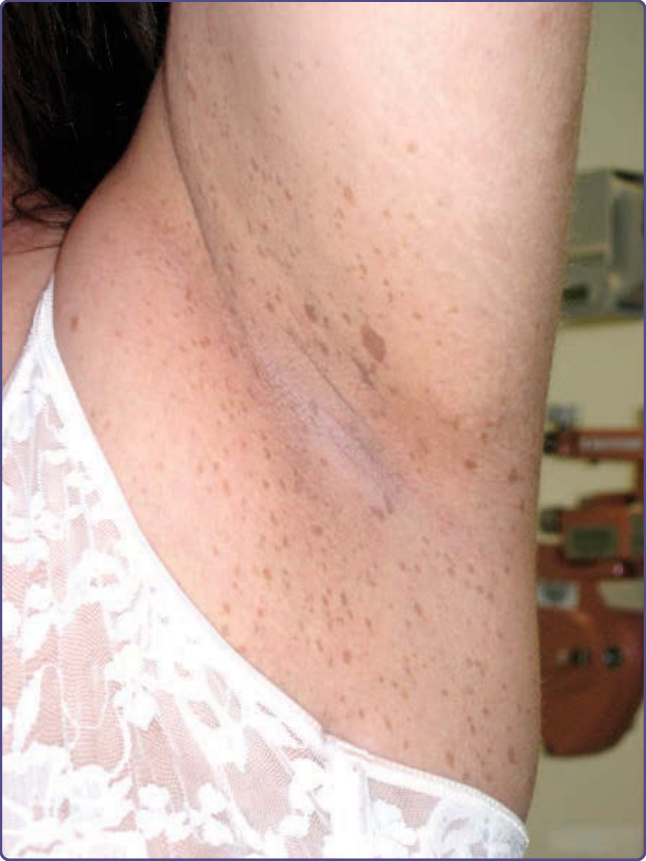
Figure 135-2 Axillary freckling.
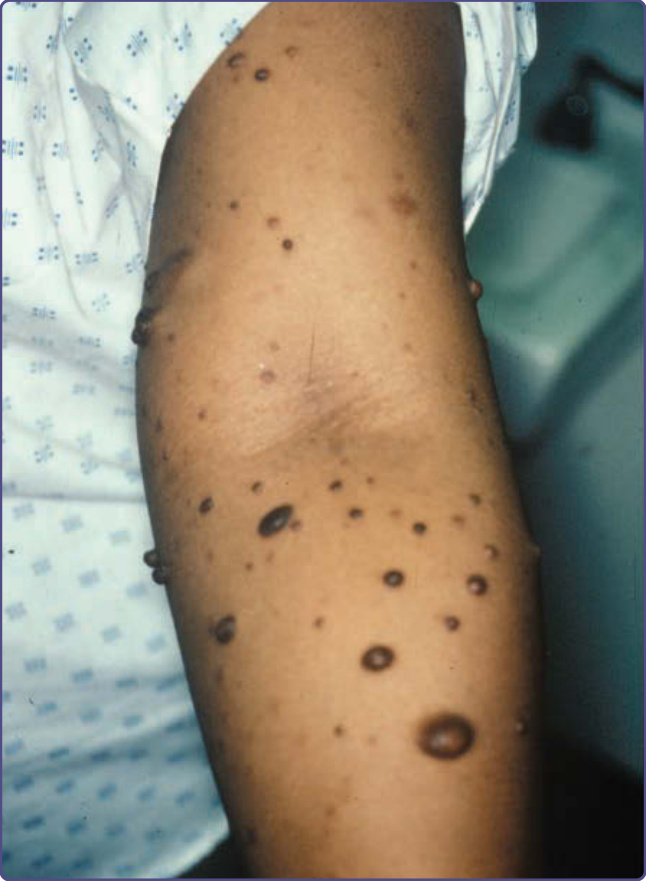
Figure 135-3 Cutaneous neurofibromas with overlying hyperpigmentation.
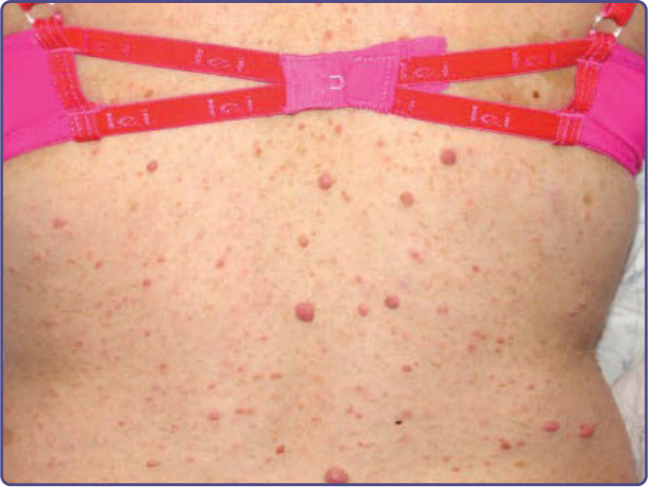
Figure 135-4 Multiple cutaneous neurofibromas.
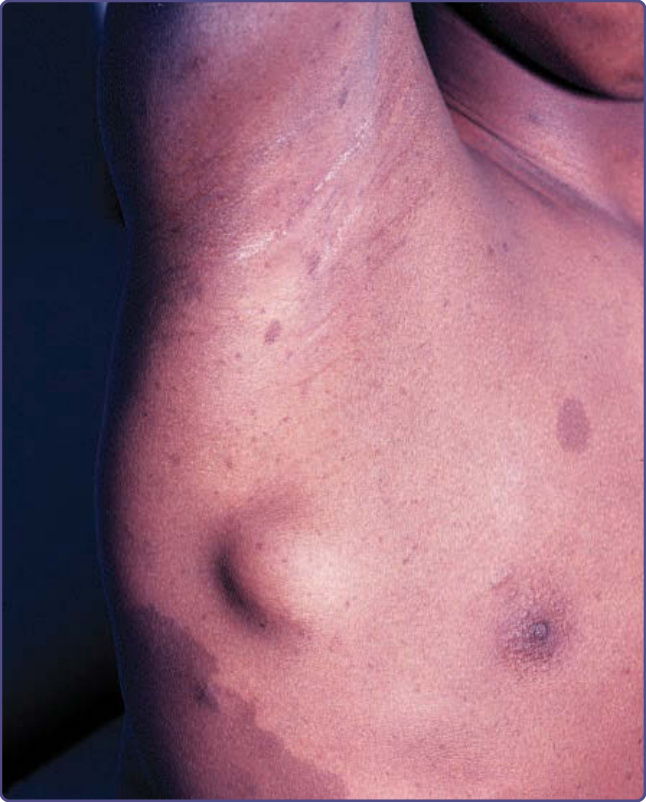
Figure 135-5 Subcutaneous neurofibroma.
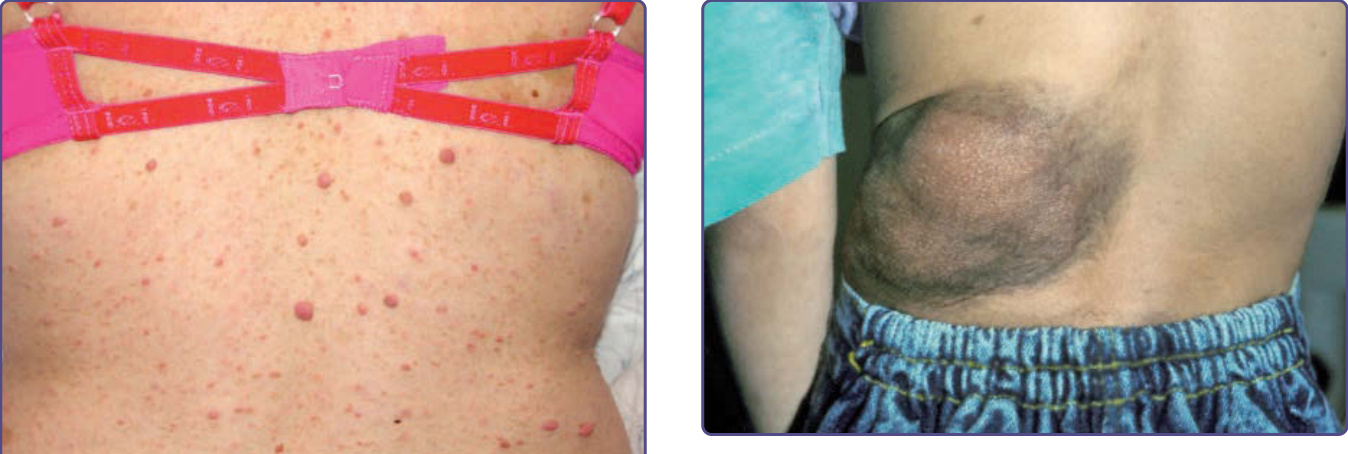
Figure 135-6 Plexiform neurofibroma with overlying hyperpigmentation and hypertrichosis.
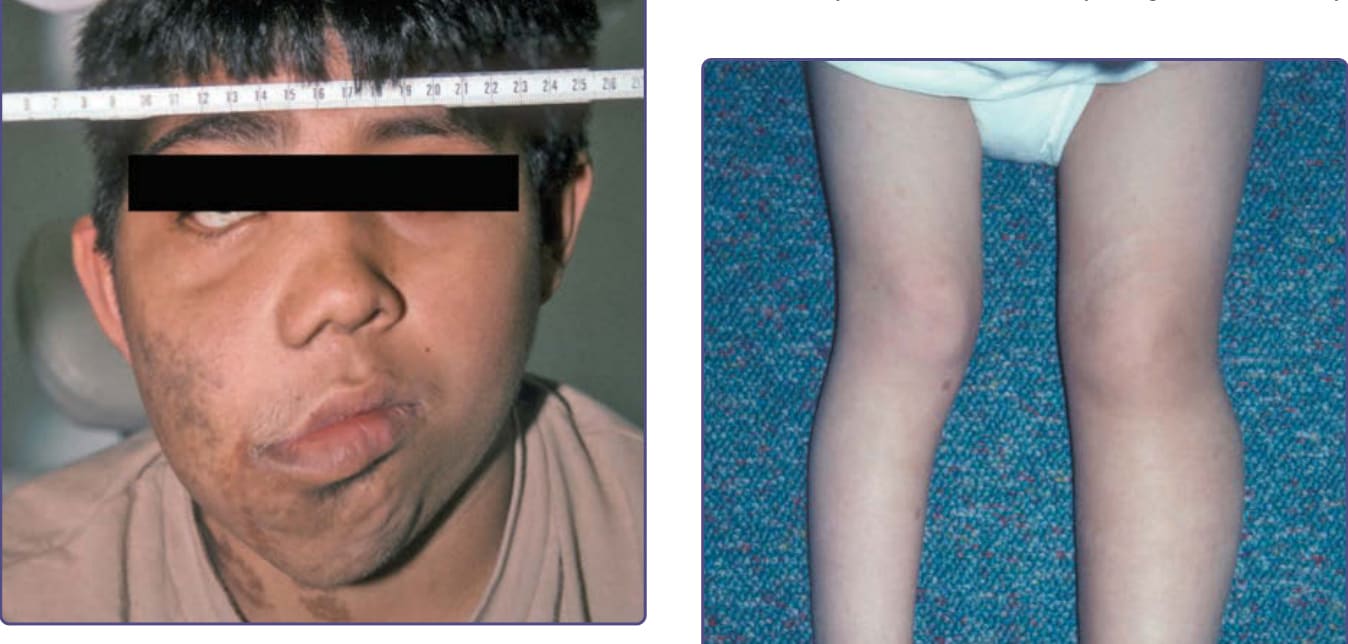
Figure 135-7 Progressive growth of facial plexiform neurofibroma.
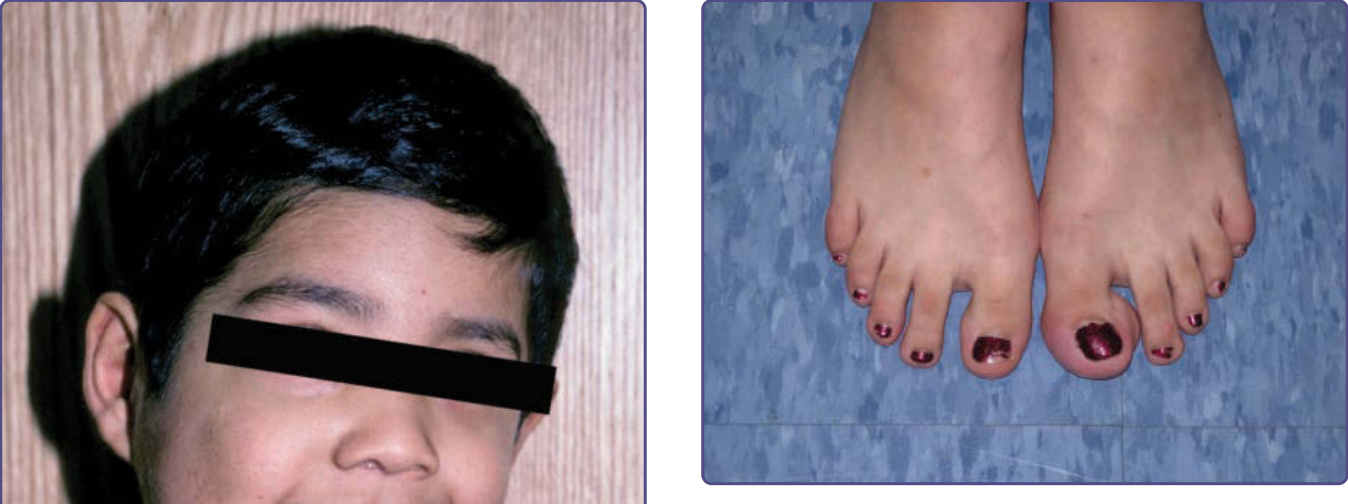
Figure 135-8 Plexiform neurofibroma of left first toe leading to isolated macrodactyly.
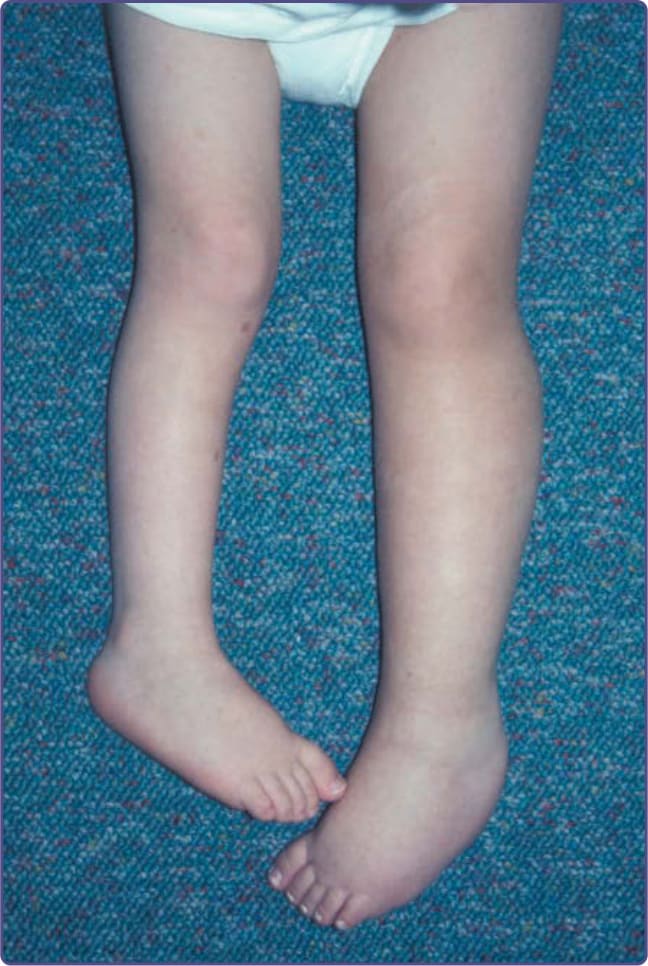
Figure 135-9 Plexiform neurofibroma of left lower extremity leading to leg-length discrepancy.
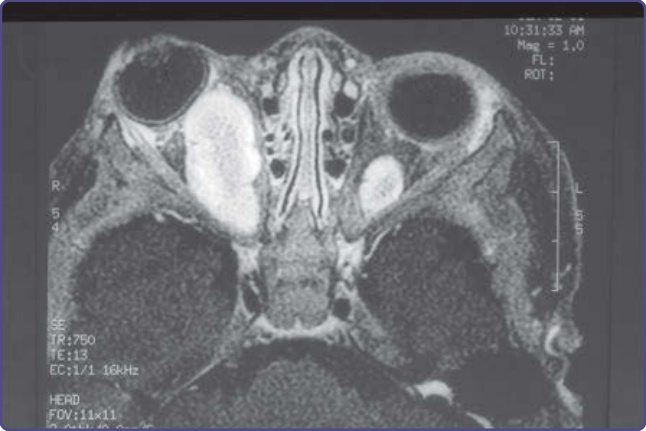
Figure 135-10 Intraorbital optic nerve tumor causing proptosis.
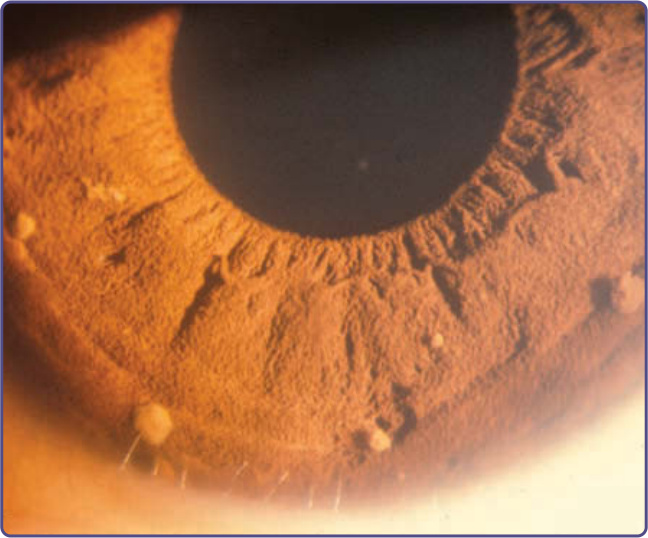
Figure 135-11 Lisch nodules.
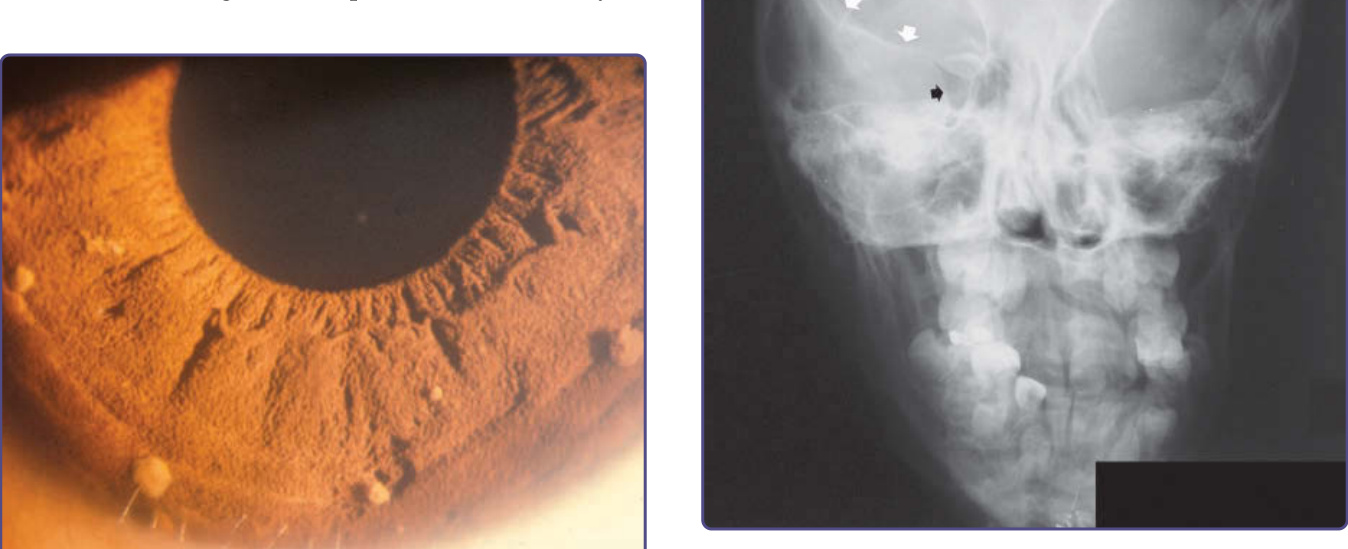
Figure 135-12 Sphenoid wing dysplasia. Arrows outline normal sphenoid wing in wall of right orbit. Contralateral sphenoid wing is dysplastic.
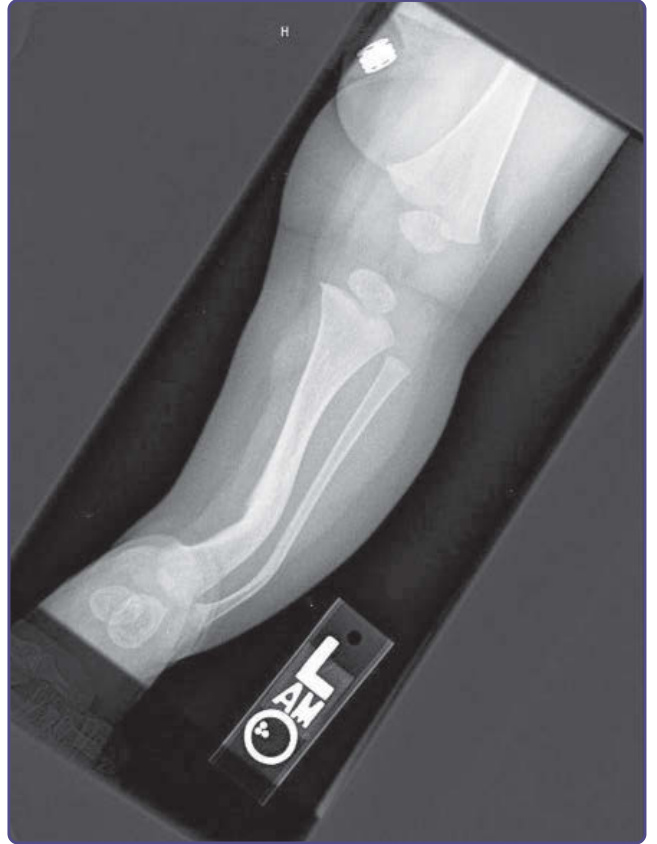
Figure 135-13 Tibial and fibular dysplasia.
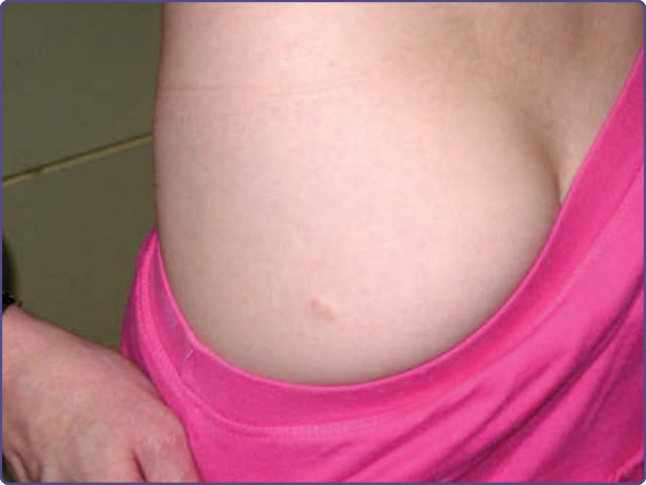
Figure 135-14 Cutaneous schwannoma.
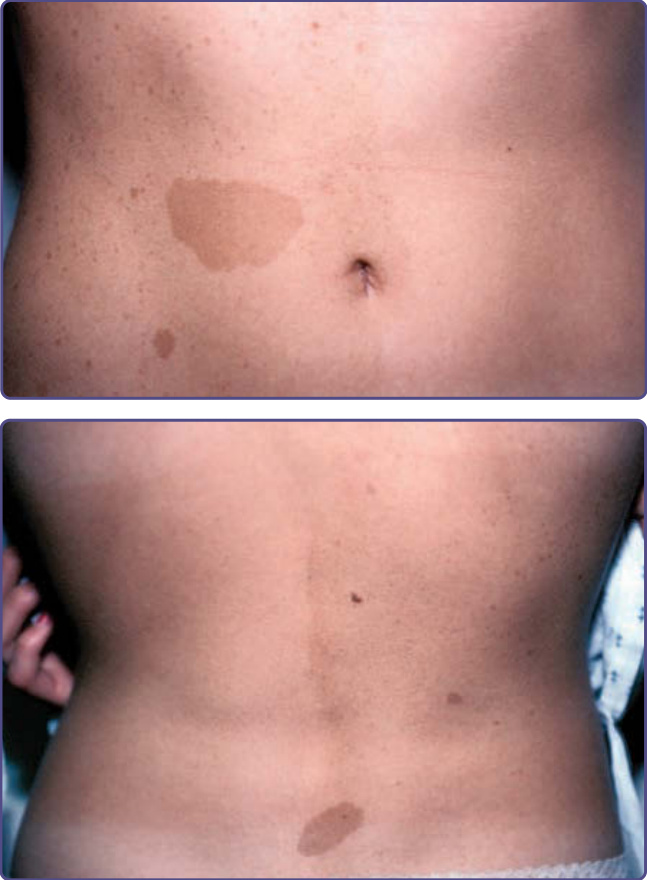
Figure 135-15 Mosaic neurofibromatosis Type 1. Note that the café-au-lait spots and freckling are limited to one side of body.
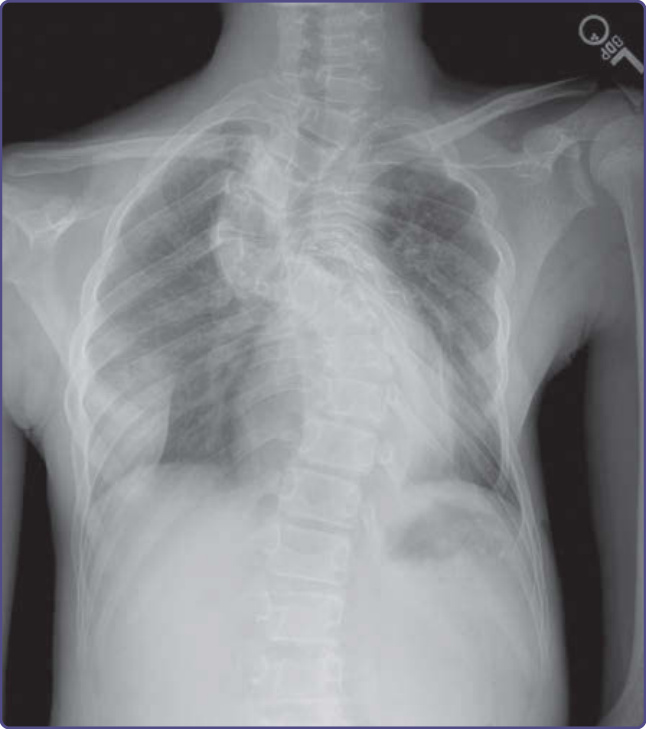
Figure 135-16 Dystrophic scoliosis.
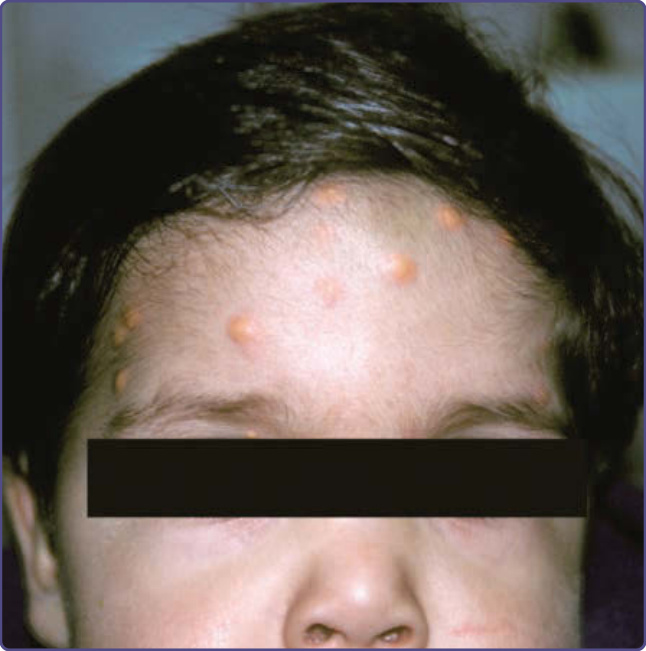
Figure 135-17 Multiple juvenile xanthogranulomas on the forehead.
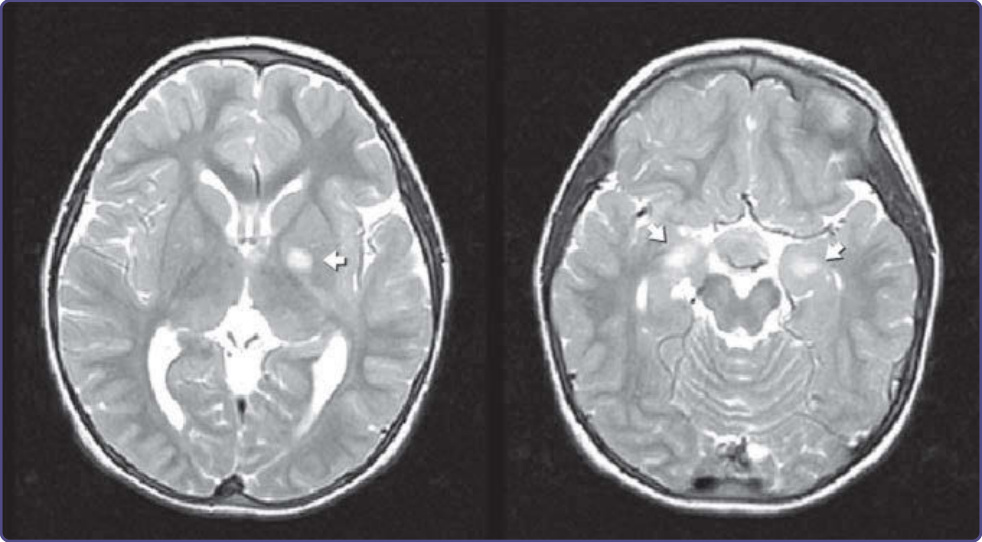
Figure 135-18 T2- weighted MRI hyperintensity, termed “unidentified bright object.”

TABLE 135-1 Diagnostic Criteria for Neurofibromatosis Type 1a
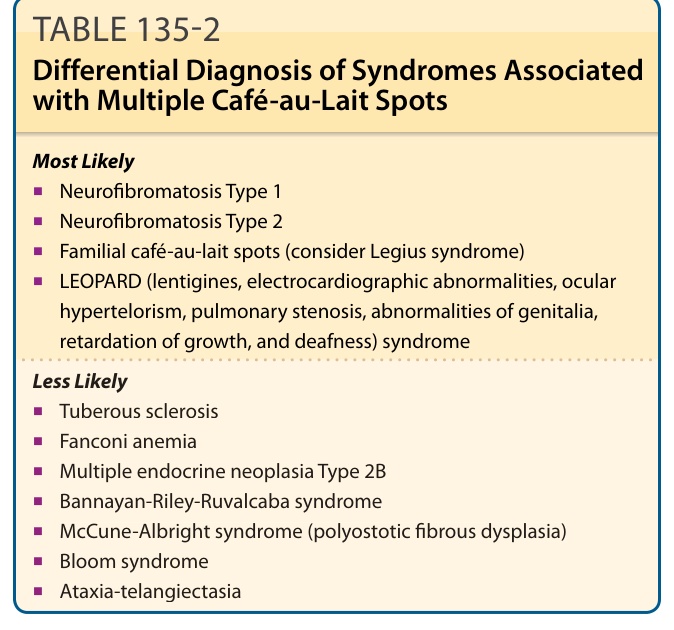
TABLE 135-2 Differential Diagnosis of Syndromes Associated with Multiple Café-au-Lait Spots
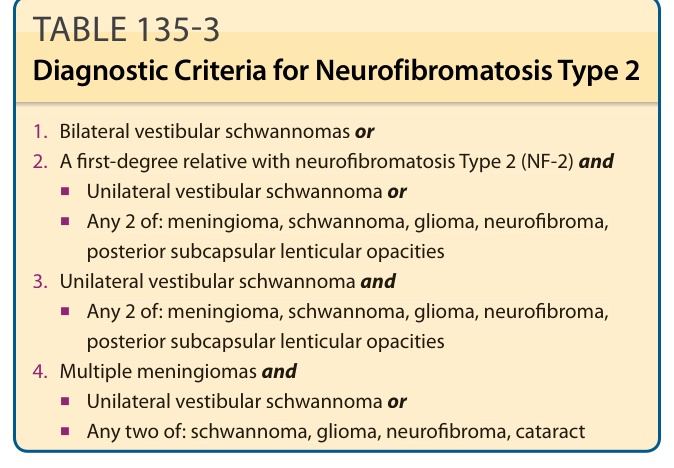
TABLE 135-3 Diagnostic Criteria for Neurofibromatosis Type 2