遺傳性免疫缺乏疾病 (Genetic Immunodeficiency Diseases)
總覽與臨床特徵
- 原發性免疫缺乏疾病 (primary immunodeficiency diseases) 為免疫系統遺傳性疾患,導致感染易感性增加、罹病率與死亡率升高;常伴皮膚異常,辨識皮膚徵象有助早期診斷。
- 皮膚表現:皮膚感染、異位樣 (atopic-like) 或脂漏樣皮膚炎、斑狀紅斑、禿髮、傷口癒合不良、紫斑、瘀點、毛細血管擴張、色素稀釋、皮膚肉芽腫、廣泛性疣、血管性水腫、狼瘡樣變化。其他:生長遲滯、內臟感染、自體免疫、結締組織/風濕病、過敏、腫瘤。
- 懷疑時機:反覆感染且時間延長/嚴重度增加、不尋常微生物、清除不完全、對抗生素反應不佳。
- 分類:(a) 抗體缺乏、(b) 細胞性缺乏、(c) 合併抗體與細胞性缺乏、(d) 吞噬作用與細胞殺傷障礙、(e) 補體缺陷。
- 最重要鑑別診斷為 HIV 感染:HIV 經 PCR 偵測不到 HIV 抗原;HIV 傾向倒置的 CD4/CD8 比值與高γ球蛋白血症,與多數遺傳性免疫缺乏的低γ球蛋白血症形成對比。
圖 132-1:新生兒與嬰兒免疫缺乏疾患的診斷流程。
抗體缺乏疾患
無γ球蛋白血症 (Agammaglobulinemia)
- 同義詞 Bruton 病;早發、反覆化膿性感染,通常 6 個月大後(母源 Ig 消退時)開始;扁桃腺與頸部淋巴結缺如或幾乎無法偵測。
- 皮膚:癤病 (furunculosis)、膿痂疹 (impetigo)、異位樣濕疹疹(對 Ig 治療無反應)、壞疽性膿皮症、非感染性肉芽腫。
- 感染:中耳炎、鼻竇炎、支氣管炎、肺炎(肺炎球菌、葡萄球菌、嗜血桿菌);未治療可致支氣管擴張。一種類風濕性關節炎可見於 1/3–1/2 的 X 連鎖無γ球蛋白血症 (XLA) 男孩,常由 Ureaplasma urealyticum 引起。
- 機轉:B 細胞發育失敗。XLA 最常見,源於 Bruton 酪胺酸激酶缺陷,約 50% 有家族史;體染色體隱性型源於前 B 細胞/B 細胞受體組件或 BLNK 缺陷。
- 診斷:血清 IgG、IgA、IgM 遠低於對照(通常總 Ig <100 mg/dL),周邊 B 細胞 <正常值 1%。
- 治療:早期靜脈或皮下 Ig 替代+抗生素,顯著降低感染風險。
常見變異型免疫缺乏 (CVID)
- 異質性疾患,可同時有抗體缺乏與 T 細胞異常;最常見基因缺陷在 TACI 基因。多於年輕成人表現,約 20% 在 21 歲前診斷,發病高峰在第二、三個十年。
- 感染似 XLA(尤其鼻竇肺部),對腸病毒較不易感、對梨形鞭毛蟲較易感;常有肝病與胃腸道病致吸收不良。可見皮膚(圖 132-2)、肺、肝、脾的非乾酪性肉芽腫。
- 皮膚:廣泛性疣(圖 132-3)、乾癬、皮膚感染、痤瘡、禿髮、白斑、口瘡性潰瘍盛行率增加。
- 自體免疫常見(28.6%);淋巴網狀惡性腫瘤與胃癌發生率顯著增加(第五、六個十年)。
選擇性免疫球蛋白疾患
- IgA 缺乏通常無症狀;僅 10%–15% 有臨床表現(細菌性鼻竇肺部感染、自體免疫)。
細胞性缺乏
慢性皮膚黏膜念珠菌病 (CMC)
- 異質性疾患,對念珠菌 (Candida) 的免疫反應選擇性改變;皮膚、指甲、黏膜反覆進行性念珠菌感染(多為 C. albicans)。可伴內分泌病變(APECED)。
- 臨床範圍:難治鵝口瘡(圖 132-4)→輕度紅斑鱗屑斑塊(圖 132-5)→嚴重結痂性肉芽腫斑塊(圖 132-6)。好發間擦部位、開口周圍、頭皮。50% 可能發展由其他微生物引起的反覆或嚴重感染。
- APECED(第 1 型自體免疫多內分泌腺症候群):念珠菌感染 5 歲前開始,內分泌障礙常至 12–13 歲才明顯;最常為副甲狀腺低能症 (88%) 與腎上腺皮質低能症 (60%)。
- 機轉:Th17 細胞反應受損。STAT1 雜合性活化突變最常見(抑制 IL-17、促進干擾素訊息);APECED 為 AIRE 功能喪失突變(體染色體隱性)。CARD9 與 dectin-1 突變亦增加念珠菌與皮癬菌感染。
- 診斷:刮取與培養見念珠菌,切片見念珠菌僅存於角質層。針對第 I 型干擾素的自體抗體為 APECED 敏感且特異標記。
- 治療:長期全身性唑類 (itraconazole、fluconazole) 或 terbinafine;抗藥者用 voriconazole(光毒性風險)、posaconazole、棘白菌素類、合併或不合併 flucytosine 的 amphotericin B。皮膚肉芽腫反應較差。所有病人應每年內分泌評估。
軟骨毛髮發育不全症候群 (Cartilage–Hair Hypoplasia)
- 同義詞 McKusick 型幹骺端軟骨發育不良;體染色體隱性,常見於 Amish 與芬蘭族群;核糖核蛋白內切核糖核酸酶 RNA 組件突變,致細胞介導與體液免疫缺陷。
- 特徵:細、稀疏、色素減退毛髮,短肢侏儒症。支持性治療+抗生素;骨髓移植可矯正免疫缺陷,但無法矯正真皮或軟骨。
合併抗體與 T 細胞缺乏
高免疫球蛋白 M 症候群
- 多為 X 連鎖隱性、CD40 配體缺乏;常見反覆鼻竇肺部與胃腸道感染、口腔潰瘍、疣。
威斯科特-奧德里奇症候群 (WAS)
- X 連鎖隱性、WASP 基因突變;發生率約每百萬男嬰 4 例。
- 典型三聯徵(僅 25% 出現):(a) 血小板減少/功能障礙致出血、(b) 反覆化膿性感染、(c) 難治性皮膚炎。出血傾向最常見(84%),常以血便初現。
- 皮膚:異位性皮膚炎約 80%(顏面、頭皮、屈側最重,可剝脫性、血清血性結痂,圖 132-7),常繼發細菌感染、疱疹性濕疹(圖 132-8)、傳染性軟疣。多達 40% 發展自體免疫(血管炎 20%、自體免疫性溶血性貧血 14%、IgA 腎病變多達 10%)。
- 機轉:WASP(Xp11.22-11.23)編碼造血特異性細胞質蛋白,主管訊息傳遞與細胞骨架;影響免疫突觸、T/B 細胞遷移與初級抗體反應。WASP 缺如/截短→典型 WAS;突變但仍表現→X 連鎖血小板減少症。
- 診斷:持續性血小板減少(1000–80,000/µL;正式標準需 <70,000/µL)、血小板體積小、IgM(有時 IgG)低、IgA/IgE/IgD 升高。流式細胞儀偵測 WASP、基因定序確認。
- 治療:抗生素、免疫接種、血小板與血漿輸注;Ig 替代對某些病人有用;局部糖皮質類固醇+Ig 改善皮膚炎;反覆疱疹性濕疹用長期口服 acyclovir。骨髓或幹細胞移植為反覆問題(尤其顯著自體免疫)者首選;HLA 配對手足捐贈者、<5 歲兒童 5 年存活率 87%,年長/不相配者約 50%。百分之十三 (Thirteen percent) 發展淋巴網狀惡性腫瘤(尤其非何杰金氏淋巴瘤)。基因治療(慢病毒轉導 CD34+ 細胞)有持久益處。
嚴重複合型免疫缺乏 (SCID)
- 異質性 X 連鎖與體染色體隱性疾患,細胞介導與體液免疫均缺乏;嬰兒早期生長遲滯、腹瀉、反覆皮膚黏膜念珠菌病與細菌/病毒感染;有移植物對抗宿主疾病 (GVHD) 風險。
- 通常 3–6 個月大無法增重;常有持續性皮膚黏膜念珠菌病;肺囊蟲肺炎常為初發;病毒感染傾向致命;儘管反覆感染仍缺乏可觸及淋巴組織。皮疹多為麻疹樣或似脂漏性皮膚炎。
- GVHD 可源於子宮內母源淋巴球暴露(通常非致命)、未照射血品輸注(致命)或幹細胞移植;母源植入之 GVHD 切片見乾癬樣增生伴角化不全(與傳統 GVHD 空泡性界面型態不同)。
- Omenn 症候群:紅皮症、肝脾腫大、淋巴結腫大、嗜伊紅性白血球增多、IgE 增加;B 細胞傾向無法偵測、T 細胞增多但無功能;多為 RAG1/RAG2 突變。
- 治療:造血幹細胞移植可能提供長期治癒;新生兒篩檢引入後,早期移植結果極佳。
外胚層失養症合併免疫缺乏
- NEMO(NF-κB 必要調節因子)基因突變致 NF-κB 訊息傳遞異常;X 連鎖隱性最常見(每 250,000 活產男嬰 1 例)。
- 弱效突變 (hypomorphic) 允許男性早期存活;女性帶因者常顯示色素失調症 (incontinentia pigmenti) 輕微特徵。臨床:無汗性外胚層失養症典型面容(圖 132-9)、毛髮稀少、少汗(耐熱不良)、缺牙伴錐狀門齒;嬰兒期細菌感染(敗血症、肺炎、中耳炎),亦見非典型分枝桿菌與病毒感染。
- 治療:依表現型而定;外胚層失養症支持性治療;Ig 替代;積極抗感染;考慮對肺囊蟲與鳥型分枝桿菌複合體預防性投藥;幹細胞移植可重建免疫但不矯正其他表現。死亡率增加,36% 於平均 6.4 歲死亡。
運動失調-毛細血管擴張症 (AT)
- 體染色體隱性、ATM 基因突變(OMIM #208900;又稱 Louis-Bar 症候群);發生率可達 1:40,000,帶因率達 1%。帶因者乳癌、血液惡性腫瘤、缺血性心臟病風險增加;放射線致染色體斷裂風險增加(已知帶因者乳房 X 光攝影為禁忌)。
- 眼皮膚毛細血管擴張始於眼眥、橫越球結膜(圖 132-10),通常 3–6 歲出現,後及顴突、耳、眼瞼、屈側(圖 132-11)。早老性變化(乾燥症、灰髮)見於 90%。最常見皮膚表現之一為非感染性皮膚肉芽腫(圖 132-12),常潰瘍、易誤為其他肉芽腫病程。
- 進行性小腦運動失調通常嬰兒期首現(中位 1.2 歲),診斷中位年齡 7 歲;青少年期前多需輪椅。多達 80% 反覆鼻竇肺部感染(最常見死因)。腫瘤見於 40% 存活至青少年/年輕成人者,以淋巴瘤(B 細胞,風險增 200 倍)與白血病(T 細胞 CLL,風險增 70 倍)為主。
- 免疫:IgA 與 IgE 分別在 70%、達 80% 缺如/缺乏;約 60% 選擇性 IgG2、IgG4 缺乏;70% 細胞介導免疫缺陷。幾乎所有病人 α 胎兒蛋白升高(2 歲後具意義)。
- 治療:支持性(抗生素、物理治療、防曬);積極篩檢惡性腫瘤;triamcinolone 病灶內注射助癒疼痛性潰瘍。bleomycin 等類放射性化療藥可致廣泛組織壞死;用小劑量化療+低劑量分次放射。死亡多於兒童晚期或青春期早期,存活最久者 50 歲死亡。
吞噬作用與細胞殺傷障礙
- 典型嬰兒/兒童期反覆、不尋常、難清除的細菌感染(皮膚黏膜、肺、淋巴結、深部膿瘍、牙周炎)。
慢性肉芽腫病 (CGD)
- 活性氧中間產物產生缺陷,損害細胞內殺傷;X 連鎖或體染色體隱性;NADPH 氧化酶系統組件基因突變。90% 為男性,發生率約每 200,000–250,000 人 1 例。
- gp91phox 缺乏的 X 連鎖型佔 70%,第一年顯現;體染色體隱性型較晚發病(平均 8 歲)、徵象較輕、存活較長。
- 皮膚:膿皮症伴區域淋巴結腫大/淋巴結炎、皮膚炎,好發暴露部位(皮膚、肺);葡萄球菌膿瘍 40%(肛周、鼻孔、耳);壞疽性深膿瘡可為新生兒初發。皮膚肉芽腫呈結節狀且常壞死。X 連鎖女性帶因者可有圓盤狀/全身性紅斑性狼瘡病灶、光敏感、雷諾現象。
- 病原:金黃色葡萄球菌、黏質沙雷氏菌、洋蔥伯克氏菌、諾卡氏菌屬、麴菌屬(觸酶陽性微生物)。葡萄球菌肝膿瘍幾乎為 CGD 特異表現。肝脾腫大見於 80%–90%。
- 機轉:NADPH 氧化酶缺陷致無法產生超氧化物與毒性氧代謝產物。X 連鎖為 CYBB(gp91phox)突變;體染色體隱性缺乏 p47phox(NCF1,約 20%)、p67phox(NCF2,≤5%)等細胞質因子。
- 診斷:DHR 123 流式細胞儀檢測(較受青睞)、亞鐵細胞色素 c 還原;篩檢用 NBT 還原分析。
- 治療:有感染證據者經驗性用涵蓋金黃色葡萄球菌與革蘭氏陰性菌的廣效非經腸道抗生素,靜脈至少 10–14 天後接口服數週(表 132-6)。Trimethoprim-sulfamethoxazole 降低細菌感染;itraconazole 預防真菌;預防性 IFN-γ 降低感染數量與嚴重度。預防性抗生素+IFN-γ 已將死亡率降至體染色體型約每病人年 2%、X 連鎖約 5%。幹細胞移植可能治癒(無感染年輕病人存活率 >95%)。
白血球黏附缺乏症
- 4 種疾患(3 種體染色體隱性:ITGB2、SLC35C1、FERMT3;1 種體染色體顯性:Rac2);牙齦炎與牙周炎、傷口癒合不良、臍帶殘端分離延遲、壞疽性膿皮症樣壞死潰瘍、危及生命的細菌與真菌感染。
高免疫球蛋白 E 症候群 (HIES)
- 同義詞 Job/Buckley 症候群;多為體染色體顯性,亦有隱性型。典型三聯徵:(a) 反覆葡萄球菌皮膚膿瘍、(b) 伴肺氣囊腫的肺炎、(c) 高血清 IgE。發生率約 10⁶ 分之 1。
- 皮膚:新生兒/嬰兒期丘疹膿疱性疹伴結痂(頭皮、顏面、頸、腋、尿布區);切片最常見嗜伊紅性海綿水腫性皮膚炎。特徵性「冷」膿瘍(圖 132-13),不顯現預期紅斑、溫熱與化膿。
- 顯性型特有骨骼/牙齒異常:粗糙面容、突出前額、寬鼻、乳牙滯留、骨質減少(57% 至少 3 次病理性骨折)、脊柱側彎(成人 63%)。
- 隱性型(多為 DOCK8 雙等位基因突變):反覆病毒感染(軟疣、疣、單純疱疹、水痘-帶狀疱疹,圖 132-14)、伺機性感染、自體免疫、毀滅性神經併發症;皮膚黏膜鱗狀細胞癌與淋巴瘤風險增加。
- 機轉:最常見為 STAT3 顯性負向突變(Th17 嚴重減少);隱性型多為 DOCK8 突變。
- 診斷:IgE 升高(顯性型診斷標準含 IgE >1000 IU/mL 加 5 項臨床特徵加權分數,合併雜合性 STAT3 突變為確定診斷);嗜伊紅性白血球增多(通常 >700 cells/µL)。
- 治療:抗葡萄球菌抗生素、口服三唑類 (triazole) 抗真菌、預防性抗葡萄球菌抗生素;膿瘍切開引流。IFN-α 治療 DOCK8 缺乏型的多發性疣與嚴重疱疹。幹細胞移植一般保留給 DOCK8 缺乏型,嚴重 STAT3 缺乏型亦曾成功。
WHIM 症候群
- 疣、低γ球蛋白血症、感染、骨髓滯留 (myelokathexis);源於 CXCR4 缺陷,成熟嗜中性白血球無法離開骨髓。Plerixafor(CXCR4 拮抗劑)增加循環白血球、減少感染。
銀髮症候群:Chédiak-Higashi 與 Griscelli
- 一群體染色體隱性疾患,毛髮(常含皮膚)有金屬光澤;Chédiak-Higashi (CHS) 與 Griscelli(第 2 型)伴免疫缺乏。
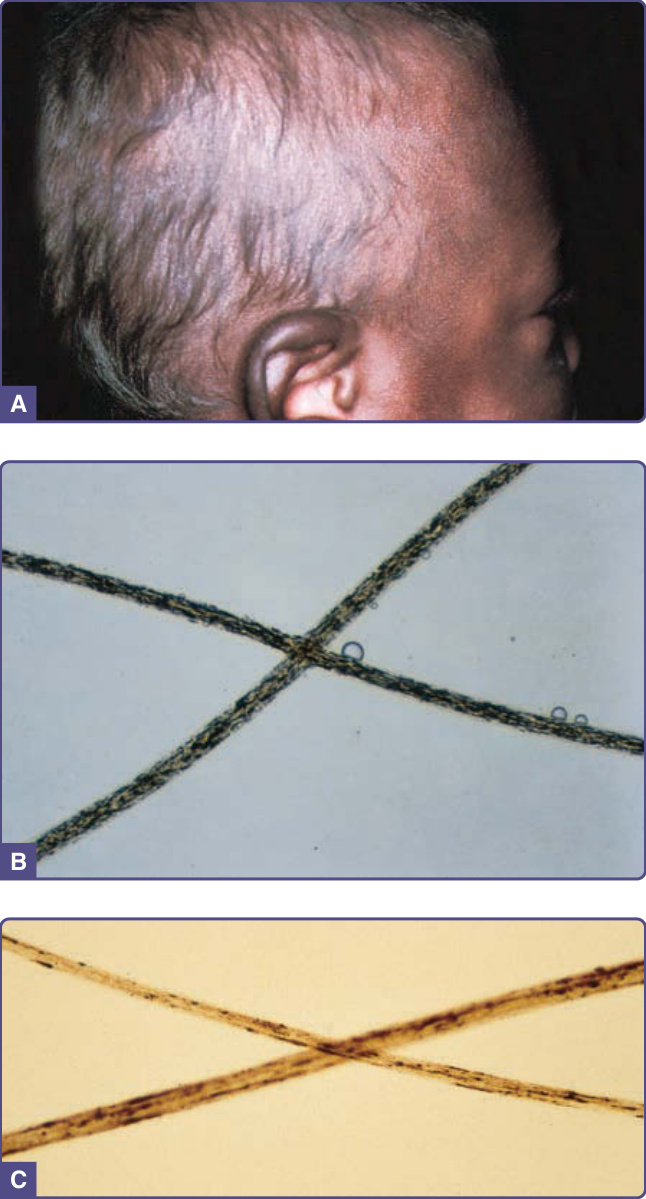
圖 132-15:銀髮症候群。A,毛髮銀色光澤;B,CHS 毛髮均勻聚集的小型色素顆粒;C,Griscelli 症候群不規則間隔的較大型色素聚集。
Chédiak-Higashi 症候群 (CHS)
- 囊泡運輸體染色體隱性疾患,LYST 基因突變(染色體 1q42),致巨大胞器(黑色素小體、白血球顆粒、血小板緻密顆粒);全球約 300 例。
- 銀髮(75% 色素異常)、眼部色素減退伴畏光與眼球震顫、反覆化膿性感染(葡萄球菌、化膿性鏈球菌、肺炎球菌)、輕度出血傾向(瘀青、瘀點,血小板數正常)。
- 診斷:血液白血球見大型細胞質顆粒;毛幹均勻分布小型色素顆粒(與 Griscelli 不規則大型聚集對比)。
- 加速期(噬血細胞性淋巴組織球增生症)見於 85%,常由 EB 病毒觸發,伴肝脾腫大、全血球減少,常致命。未移植平均死亡年齡 6 歲。首選早期移植(矯正免疫但不影響色素異常與神經退化)。
Griscelli 症候群 (GS)
- 體染色體隱性;MYO5A(偏神經性,GS1)、RAB27A(噬血細胞作用,GS2)或 Slac-2a(GS3)突變;銀髮為標誌;毛幹大型不均勻色素叢集,但抹片不顯示白血球顆粒。
- GS2 有嚴重免疫疾患/疾病加速期(噬血細胞症候群,常由 EB 病毒促發);GS1 有原發性神經疾患;GS3 僅色素異常。
- 過去一律致命,現可經造血幹細胞移植逆轉;GS2 中 85% 有早期截短突變、最嚴重,多於 5 歲前死亡。
補體缺乏疾患
- 早期補體組件缺乏/功能障礙與自體免疫(尤其全身性紅斑性狼瘡)及莢膜細菌感染相關;晚期組件缺乏顯著增加奈瑟氏菌感染易感性。
遺傳性血管性水腫 (HAE)
- 幾乎總為體染色體顯性,自發突變率 25%;每 150,000 人 1 例,75% 有陽性家族史。第 I 型 (85%) C1 抑制因子 (C1 INH) 抗原濃度低;第 II 型功能濃度低但抗原正常/高;第三型不顯示 C1 INH 缺乏。
- 喉部水腫為死亡重大風險。治療:衰減型雄激素、抗纖溶劑、血漿來源 C1 INH、激肽釋放酶抑制劑、緩激肽受體拮抗劑;C1 INH 可急性治療發作或預防性使用。