Genetic Immunodeficiency Diseases
21
OVERVIEW OF GENETIC IMMUNODEFICIENCY DISEASES
Primary immunodeficiency diseases are inherited disorders of the immune system that result in an increased susceptibility to infection and an increased morbidity and mortality.1 Many of these genetic immunodeficiency diseases are associated with a variety of cutaneous abnormalities, and recognition of these clinical features may allow an early diagnosis of primary immunodeficiency. Cutaneous abnormalities may include cutaneous infections, atopic-like or seborrheic-like dermatitis, macular erythemas, alopecia, poor wound healing, purpura, petechiae, telangiectasias, pigmentary dilution, cutaneous granulomas, extensive warts, angioedema, and lupus-like changes (Table 132-1). Other clinical features often include failure to thrive, visceral infection, autoimmune disorders, connective tissue/rheumatologic diseases, allergic reactions, and neoplasias.
CLINICAL FEATURES
CLINICAL FEATURES
Immunodeficiency should be suspected when patients have recurrent infections of increased duration or severity, particularly with unusual organisms. Incomplete clearing of infections, unexpected or severe complications of infection, or poor response to antibiotics may be associated.2 Affected infants often grow poorly (failure to thrive). The most common noncutaneous abnormalities are infections, diarrhea, vomiting, hepatosplenomegaly, arthritis, adenopathy or paucity of lymph nodes/tonsils, and hematologic abnormalities. The classification of genetic immunodeficiency disorders includes: (a) antibody deficiencies, (b) cellular deficiencies, (c) combined antibody and cellular deficiencies, (d) disorders of phagocytosis and cell killing, and (e) complement defects. The characteristic clinical signs of each group suggest that proper classification and laboratory tests may be used to confirm the diagnosis. The laboratory testing and clinical patterns of illness associated with each group of immunodeficiency disorders that allow their differential diagnosis are outlined in Table 132-2 and Fig. 132-1. The most important disorder in the differential diagnosis of all genetic immunodeficiency disorders is HIV infection. In addition to the lack of HIV antigen as detected by polymerase chain reaction in patients with genetic immunodeficiency, other features help to differentiate the disorders. Patients with HIV infection tend to show
an inverted CD4-to-CD8 ratio and hypergammaglobulinemia, in contrast to the hypogammaglobulinemia of many patients with genetic immunodeficiency.
ANTIBODY DEFICIENCY DISORDERS
AGAMMAGLOBULINEMIA
AGAMMAGLOBULINEMIA
AT-A-GLANCE
■ Synonym: Bruton disease.
■ Early-onset, recurrent bacterial infections.
■ Absent or barely detectable tonsillar and cervical lymph node tissue.
■ Profound hypogammaglobulinemia and decreased or absent peripheral B cells.
CLINICAL FEATURES
Agammaglobulinemia is characterized by recurrent pyogenic infections that often begin after 6 months of age, concurrent with the waning of maternal immunoglobulins. These patients have absent or barely detectable tonsils and cervical lymph nodes.3 Skin infections, especially furunculosis and impetigo, are common and often surround body orifices. An atopic-like eczematous eruption that fails to improve with immunoglobulin (Ig) therapy has been described in many affected children. Pyoderma gangrenosum and noninfectious cutaneous granulomas have been reported. Childhood exanthematous disorders are handled appropriately, but the infections may recur, owing to a failure to develop specific antibodies. Recurrent otitis media, sinusitis, bronchitis, and pneumonia are the earliest infectious manifestations and usually are caused by pneumococci, staphylococci, or Haemophilus. Untreated pulmonary infections may lead to progressive bronchiectasis and chronic pulmonary disease.4 Patients can also suffer from chronic enteroviral infections and hearing loss from repeated otitis and sinusitis infections. Other common bacterial infections include conjunctivitis, osteomyelitis, septic arthritis, and meningitis. Protracted diarrhea may be caused by infection, particularly with Giardia, Salmonella, Campylobacter, or Cryptosporidium spp. Three virus groups cause problems: enterovirus, hepatitis B virus, and rotavirus. Patients have developed
21
CUTANEOUS MANIFESTATIONS ASSOCIATED IMMUNODEFICIENCY CUTANEOUS MANIFESTATIONS ASSOCIATED IMMUNODEFICIENCY
Angioedema Hereditary angioedema Lupus-like cutaneous change IgA deficiency Chronic granulomatous disease, autosomal recessive Chronic granulomatous disease, X-linked carrier Common variable immunodeficiency Complement deficiency, early components Elevated IgM with hypogammaglobulinemia
Angioedema Hereditary angioedema Lupus-like cutaneous
Atopic-like dermatitis IgA deficiency/IgM deficiency Chronic granulomatous disease Common variable immunodeficiency Elevated IgM with
Atopic-like dermatitis IgA deficiency/IgM deficiency Chronic granulomatous disease Common variable immunodeficiency Elevated IgM with hypogammaglobulinemia Ectodermal dysplasia with immunodeficiency Hyperimmunoglobulinemia E syndrome Severe combined immunodeficiency Wiskott-Aldrich syndrome X-linked agammaglobulinemia
hypogammaglobulinemia Ectodermal dysplasia with
immunodeficiency Hyperimmunoglobulinemia
E syndrome Severe combined immunodeficiency Wiskott-Aldrich syndrome X-linked agammaglobulinemia
Cutaneous
Chronic granulomatous disease Hyperimmunoglobulinemia
Cutaneous abscesses Chronic granulomatous disease Hyperimmunoglobulinemia E syndrome Leukocyte adhesion deficiency
abscesses
E syndrome Leukocyte adhesion deficiency
Cutaneous
Ataxia-telangiectasia Chronic granulomatous disease Chronic mucocutaneous candidiasis Common variable immunodeficiency Severe combined immunodeficiency,
Cutaneous granulomas Ataxia-telangiectasia Chronic granulomatous disease Chronic mucocutaneous candidiasis Common variable immunodeficiency Severe combined immunodeficiency, especially TAP2 deficiency X-linked hypogammaglobulinemia
granulomas
especially TAP2 deficiency X-linked hypogammaglobulinemia
Cutaneous candidal
Chronic mucocutaneous candidiasis DiGeorge syndrome Hyperimmunoglobulinemia
Cutaneous candidal infection Chronic mucocutaneous candidiasis DiGeorge syndrome Hyperimmunoglobulinemia E syndrome Severe combined immunodeficiency especially TAP2 deficiency
infection
E syndrome Severe combined immunodeficiency
especially TAP2 deficiency
Eosinophilic folliculitis,
Hyperimmunoglobulinemia E syndrome
Eosinophilic folliculitis, infantile Hyperimmunoglobulinemia E syndrome
infantile
Graft-versus-host
DiGeorge syndrome Severe combined immunodeficiency
Graft-versus-host disease DiGeorge syndrome Severe combined immunodeficiency
disease
IgA deficiency Chronic granulomatous disease,
change
autosomal recessive Chronic granulomatous disease,
X-linked carrier Common variable
immunodeficiency Complement deficiency, early
components Elevated IgM with
hypogammaglobulinemia
Mucocutaneous
Ataxia-telangiectasia
Mucocutaneous telangiectasias Ataxia-telangiectasia
telangiectasias
Petechiae and/or purpura Chédiak-Higashi syndrome
Petechiae and/or purpura Chédiak-Higashi syndrome
Pigmentary dilution/silvery
Griscelli syndrome
Pigmentary dilution/silvery hair Griscelli syndrome
hair
Pyoderma gangrenosum-like
Wiskott-Aldrich syndrome Chédiak-Higashi syndrome Griscelli syndrome IgA deficiency Chédiak-Higashi syndrome Chronic granulomatous disease Hyperimmunoglobulinemia
Pyoderma gangrenosum-like ulcerations Wiskott-Aldrich syndrome Chédiak-Higashi syndrome Griscelli syndrome IgA deficiency Chédiak-Higashi syndrome Chronic granulomatous disease Hyperimmunoglobulinemia E syndrome Leukocyte adhesion deficiency
ulcerations
E syndrome Leukocyte adhesion
deficiency
Seborrheic-like or exfoliative
X-linked
Seborrheic-like or exfoliative dermatitis X-linked hypogammaglobulinemia Ataxia-telangiectasia “Leiner” phenotype: complement deficiency or dysfunction
dermatitis
hypogammaglobulinemia Ataxia-telangiectasia “Leiner” phenotype: complement
deficiency or dysfunction
Warts, extensive Severe combined
Warts, extensive Severe combined immunodeficiency X-linked agammaglobulinemia Common variable immunodeficiency Elevated IgM with hypogammaglobulinemia Epidermodysplasia verruciformis (see Chap. 166) IgM deficiency Severe combined immunode- ficiency after transplantation (mutations in IL2RG or JAK3) WHIM syndrome
immunodeficiency X-linked agammaglobulinemia Common variable
immunodeficiency Elevated IgM with
hypogammaglobulinemia Epidermodysplasia verruciformis
(see Chap. 166) IgM deficiency Severe combined immunode-
ficiency after transplantation (mutations in IL2RG or JAK3) WHIM syndrome
Ig, immunoglobulin; TAP, transporter associated with antigen processing or transporter, ATP-binding cassette; WHIM, warts, hypogammaglobulinemia, infections, myelokathexis.
2395
21
DISORDER INFECTION OTHER
Antibody Sinopulmonary (pyogenic bacteria) Autoimmune disease (autoantibodies, inflammatory bowel disease)
GI (enterovirus, Giardia) Minimal growth retardation
Normal handling of fungal and viral infections (exception is enterovirus) Paucity of lymphoid tissue
Cellular Low-grade or opportunistic infections Growth retardation
Pneumonia (pyogenic bacteria, Pneumocystis, viruses) Graft-versus-host disease
GI (viruses) Fatal infections from live vaccines
Skin, mucous membranes (fungi) Malignancy
Phagocytosis and cell killing Skin, reticuloendothelial system (Staphylococcus, enteric bacteria, Aspergillus, Mycobacteria) Ulcerative stomatitis
Complement Alternative, late components Early components
Sepsis/blood-borne (streptococci, pneumococci, Neisseria) Autoimmune disease (systemic lupus erythematosus, glomerulonephritis)
paralysis after administration of the live polio vaccine. A rheumatoid-like arthritis, characterized by chronic inflammation and swelling of the large joints, may develop in as many as one-third to one-half of boys with X-linked agammaglobulinemia (XLA) and is often caused by mycoplasmal infection (Ureaplasma urealyticum). Disseminated echovirus infection has caused meningoencephalitis and a dermatomyositislike disorder with brawny edema, induration of the muscles with accompanying weakness, muscle contractures, and poikiloderma.
ETIOLOGY AND PATHOGENESIS
Agammaglobulinemia results from failure of development of B cells, most commonly from gene defects that prevent the assembly of a full B-cell antigen receptor. XLA is the most common cause of agammaglobulinemia and results from defects in a cytoplasmic tyrosine kinase, Bruton tyrosine kinase. XLA is inherited in an X-linked fashion, and approximately 50% of affected boys have a family history of the disorder.5 Autosomal recessive agammaglobulinemia affects males and females equally and results from defects in the genes that encode for components of the pre–B-cell and B-cell receptors or in BLNK (B-cell linker), a scaffold protein that assembles signaling molecules associated with the pre–B-cell and B-cell receptor.
DIAGNOSIS
The diagnosis of agammaglobulinemia is made by serum concentrations of IgG, IgA, and IgM that are far below the 95% confidence limits for appropriate controls (usually less than 100 mg/dL total Ig) and by the virtual absence of B cells in the peripheral circulation
2396
C1 esterase inhibitor deficiency
Angioedema
Angioedema
(<1% of normal). The absence of the Bruton tyrosine kinase protein in patients with XLA can be detected by flow cytometry. Identification of a defect in one of the known genetic causes of agammaglobulinemia confirms the diagnosis and allows for genetic counseling and prenatal diagnosis.
CLINICAL COURSE, PROGNOSIS, AND MANAGEMENT
Early Ig replacement, intravenously or subcutaneously, and antibiotic use markedly reduces the risk of infections, although it may not be helpful in diminishing the risk and morbidity of chronic lung disease or chronic enterovirus infection.
COMMON VARIABLE IMMUNODEFICIENCY
COMMON VARIABLE
IMMUNODEFICIENCY
AT-A-GLANCE
■ Heterogeneous group of disorders in which both antibody deficiency and abnormalities of T cells may be found.
■ Most common underlying gene defect is in the transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) gene.
■ May manifest during childhood, with peak onset in the second and third decades of life.
(Continued)
21
Algorithm for immunodeficiency disorders in neonates and infants
Diagnosis of 1° immunodeficiency
History, physical examination, height & weight, head circumference
Suspected 1° immunodeficiency?
Recurrent sinopulmonary bacterial infections? (otitis media, sinusitis, pneumonia)
Recurrent viral and/or fungal infections? -disseminated infections? -opportunistic infections?
Bacteremia/meningitis with encapsulated bacteria? (including Neisseria, streptococci, pneumococci)
Recurrent skin abscesses and/or fungal infections?
Screen humoral immunity Screen cellular immunity Screen for phagocyte defect Screen for complement deficiency
Humoral immunity screen
Quantitative immunoglobulin levels
Normal Abnormal
Screen cellular immunity (see panel below)
Diphtheria and tetanus titer (specific antibody production)
Abnormal
Abnormal Normal Normal
Consider
- X-linked agammaglobulinemia (↓ IgG, IgA, and IgM)
- isolated IgA or IgM deficiency
- hyper-IgM (normal or ↑ IgM, ↓ IgG, IgA, IgE)
Consider diagnosis of combined defect (CVID) -↓ IgG, IgA, and/or IgM
Consider complement, phagocyte defect, other conditions
Cellular immunity screen
CBC/differential + chest radiography - *if no thymus consider DiGeorge syndrome; AT, SCID (especially absolute lymphocyte count)
T-cell panel
Normal
Abnormal
Abnormal consider SCID
Carry out molecular genetic testing or testing for specific enzyme defect
-
Functional testing -mitogen stimulation -antigen stimulation
-
B cell/NK cell marker studies
*SCID do DNA testing or test for specific enzyme defect *Complete DiGeorge syndrome (no T cells, NL B cells, NL NK cells)
↓
- HIV testing
Abnormal
Phagocyte defect screen
CBC/differential
Leukocytosis
Consider leukocyte adhesion deficiency (LAD)
Normal
Dihydroadamine test
Is oxidase function normal?
Abnormal Normal
Consider chronic granulomatous disease (CGD)
Chédiak-Higashi syndrome (large cytoplasmic granules) or specific defect
Complement deficiency screen
Suspected complement deficiency
- Measure CH50 (total complement levels) and AH50
CH50 Abnormal AH50 Abnormal
CH50 Abnormal AH50 Normal
CH50 Normal AH50 Abnormal
Measure C3, C4 levels Measure C3, C4 levels
Deficiency of component of alternative pathway
Absent to near absent C4
Normal or high
NL or high levels
Measure C1 components, C2
Moderately low levels = complement consumption and NOT a complement deficiency
Low
Defect in classical pathway Defects in C5-C9 (terminal pathway)
2397
21
AT-A-GLANCE (Continued)
Continued
AT A GLANCE (
)
■ Diagnosis requires the presence of low levels of serum immunoglobulin (Ig) G with either low IgM and/or low IgA, defective antibody response to immunization, especially with pneumococcal antigens, with recurrent infections and/or typical autoimmune complications.
■ Variable severity of autoimmune and infectious complications.
CLINICAL FEATURES
Common variable immunodeficiency (CVID) usually presents in young adults, but approximately 20% of cases are diagnosed before the age of 21 years.6
Patients have infections similar to those in patients with XLA, particularly sinopulmonary infections, but are less susceptible to enteroviral infections and more susceptible to Giardia infections. Many patients with CVID have liver disease and GI disease, causing malabsorption syndromes. Noncaseating granulomas of skin (Fig. 132-2), lungs, liver, and spleen have been reported. Caseating granulomas of the skin and viscera, although rare, also have been described.7,8
Extensive warts can be a major problem in individuals with CVID (Fig. 132-3). A comparison of skin manifestations in patients with CVID compared to IgA deficiency found that there was an increased incidence of atopic dermatitis without an elevated
2398
IgE level in patients with IgA deficiency, while there was an increased prevalence of psoriasis, skin infections, acne, alopecia, vitiligo, and aphthous ulcers in patients with CVID.9 Lymphoid tissues often are enlarged, and splenomegaly with hypersplenism is also found, with 8.2% of CVID patients undergoing splenectomy.6 Autoimmune disorders are especially frequent (28.6%), particularly autoimmune thrombocytopenia, autoimmune hemolytic anemia, rheumatoid arthritis, sicca syndrome, and pernicious anemia. Alopecia areata and lupus also have been described. In 10% to 20% of CVID patients, at least 1 family member is also immunodeficient, particularly with CVID or IgA deficiency.10 The incidence of lymphoreticular malignancy and gastric carcinoma are markedly increased, particularly in the fifth and sixth decades of life.
SELECTIVE IMMUNOGLOBULIN DISORDERS
SELECTIVE
IMMUNOGLOBULIN
DISORDERS
AT-A-GLANCE
■ Immunoglobulin A deficiency is usually asymptomatic; only 10% to 15% of affected individuals demonstrate clinical manifestations, including bacterial sinopulmonary infections and autoimmune disorders.
CELLULAR DEFICIENCIES
CHRONIC MUCOCUTANEOUS CANDIDIASIS
CHRONIC MUCOCUTANEOUS
CANDIDIASIS
AT-A-GLANCE
■ Heterogeneous group of disorders with altered immune responses selective to Candida.
■ Recurrent, progressive candidal infections of the skin, nails, and mucous membranes.
■ May be associated with the later development of endocrinopathy (APECED [autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy]).
Several clinical subtypes of chronic mucocutaneous candidiasis (CMC) have been defined (Table 132-3). They have varied clinical manifestations, variable immunodeficiency, and different forms of genetic inheritance. Patients with CMC may have childhood or mature onset, and familial or sporadic occurrence. In addition, CMC may be present with or without endocrinopathy. Patients with autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED or autoimmune polyendocrine syndrome, Type 1) often have affected siblings. APECED and other familial forms of CMC are autosomal recessive. Autosomal dominant inheritance is seen in patients with associated keratitis.
CLINICAL FEATURES
Patients with CMC have recurrent, progressive infections of the skin, nails, and mucous membranes most commonly caused by Candida albicans. Depending on the subtype, the clinical presentation ranges from recurrent, recalcitrant thrush (Fig. 132-4) to mild erythematous scaling plaques (Fig. 132-5) with a few dystrophic nails to severe generalized, crusted granulomatous plaques (Fig. 132-6). The cutaneous plaques occur most commonly in intertriginous areas, periorificial sites, and the scalp, but they may be generalized. The nails are thickened, brittle, and discolored, and the paronychial areas are often erythematous, swollen, and tender. Scalp infections may lead to scarring and alopecia. Although the oral mucosa is the most frequent site of mucosal alteration, esophageal, genital, and laryngeal mucosae may be affected. Strictures may be formed by candidal infection at these mucosal sites. Scrapings and cultures from cutaneous or mucosal lesions demonstrate candidal organisms. Patients with CMC rarely develop systemic candidiasis, but 50% may develop recurrent or severe infections caused by other organisms. In one study, 81%
21
of patients with early-onset CMC also had infections with bacteria, fungi, and parasites, including bacterial septicemia.11 Concomitant dermatophyte infections may occur. In patients with APECED, the candidal infections tend to begin by 5 years of age, although the endocrinologic dysfunction may not be apparent until 12 to 13 years of age (see Fig. 132-6). The most commonly associated endocrinopathies are hypoparathyroidism (88%) and hypoadrenocorticism (60%). One-third of patients have candidiasis, hypoparathyroidism, and defective adrenal function. Other associated endocrinopathies or autoimmune disorders include gonadal insufficiency (45%), alopecia areata (20%), pernicious anemia (16%), thyroid abnormalities (12%), chronic active hepatitis or juvenile cirrhosis (9%), vitiligo, diabetes mellitus, and hypopituitarism. Chronic diarrhea and malabsorption have been reported in 25% of patients and usually are associated with hypoparathyroidism. Some affected patients also have pulmonary fibrosis, dental enamel hypoplasia, and keratoconjunctivitis. The “ectodermal dysplasia” features are likely to be secondary to the candidal infections or autoimmunity. Patients with APECED often have autoimmune antibodies, including antithyroglobulin, antimicrosomal, antiadrenal, and antimelanocyte antibodies, and rheumatoid factor. Autoantibodies also have been found in patients with CMC who do not have clinical endocrinologic disease.
ETIOLOGY AND PATHOGENESIS
T-helper 17 (Th17) cells play a critical role in immune defense against Candida and are impaired in their responsiveness in both APECED and non-APECED CMC patients.12 Heterozygous mutations that activate the signal transducer and activator of transcription 1 gene (STAT1) are the most common cause,13,14 and repress expression of interleukin (IL)-17 while promoting interferon signaling (see Table 132-3).15-18 Mutations in isoforms of IL-17, IL-17 receptors, or TRAF3IP2, which regulates responses to IL-17, also have been described. APECED, an autosomal recessive form, results from loss-of-function mutations in the autoimmune regulator gene (AIRE). AIRE regulates the expression of self-antigens in thymic medullary epithelial cells, promoting immune tolerance through negative selection of autoreactive T cells and generation of antigen-specific regulatory T cells.19 The retention of peripheral tissue-specific autoreactive T cells in APECED leads to the production of neutralizing autoantibodies, including against Th17- associated cytokines, leading to Candida infections and autoimmune disease.20,21 Predisposition to Candida and dermatophyte infections is also a feature of mutations in the caspase recruitment domain family member-9 gene (CARD9)22 and in the gene encoding dectin-1.23
DIAGNOSIS
Scrapings and cultures from lesions show candidal organisms. If a biopsy is done, the C. albicans are seen
2399
21
CHRONIC MUCOCUTANEOUS CANDIDIASIS TYPE INHERITANCE/GENE DEFECT CLINICAL FEATURES ASSOCIATED FEATURES
Familial chronic mucocutaneous candidiasis
Autosomal dominant gain-offunction mutations in STAT1 Candidiasis of the oral mucosa, nails and skin often begin in infancy; occasionally dermatophyte infections; squamous cell carcinoma, dental enamel defects, arterial aneurysms have been described
Autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED) syndrome
Autosomal recessive Mutations in the AIRE (autoimmune regulator) gene More common in the Finnish population, Iranian Jews, and Sardinians
Others Autosomal recessive IL-17RA, IL-17RC (interleukin-17 receptors A and C) TRAF3IP2 (TRAF3-interacting protein 2) RORC (RAR-related orphan receptor C) CARD9 deficiency (caspase recruitment domain family member 9) Dectin-1 deficiency
Other genetic disorders with increased risk of chronic candida infections:
Other genetic disorders with increased risk of chronic candida infections:
■Acrodermatitis enteropathica
■Acrodermatitis enteropathica
■DiGeorge syndrome
■DiGeorge syndrome
■Ectodermal dysplasia-ectrodactyly-clefting
■Ectodermal dysplasia-ectrodactyly-clefting
Most common are thyroid disease, vitiligo and alopecia areata, but other immune-mediated disorders occur with increased frequency May have bacterial (especially skin and lungs), and viral infections; invasive fungal and mycobacterial infections are uncommon
Oral and diaper area candidiasis > skin and nail begin before 5 years of age; up to 60% have urticarial eruptions
Often does not develop until adolescence or adulthood Hypoparathyroidism (90%) Hypoadrenalism (80%) Chronic diarrhea (up to 80%) Dental enamel hypoplasia (70%) Hypogonadism (up to 50%) Hepatitis (up to 40%) Pneumonitis (up to 40%) Sjögren syndrome (up to 40%) Alopecia areata (30%) Vitiligo (20%) Thyroid disease (20%) Diabetes (Type 1) (20%) Pernicious anemia (20%) Keratoconjunctivitis (20%) Hypopituitarism (15%) Hyposplenism (15%) Hypertension (15%) Nephritis (10%) Mucosal squamous cell carcinoma (10% of adults) Almost 100% have anti-Type I interferon antibodies Other autoantibodies common
All with mucosal candidiasis, dermatophytes
Mycobacterial infections (RORC)
Invasive fungal infections (especially of brain) (CARD9)
■Hypohidrotic ectodermal dysplasia with immunodeficiency (NFκB [nuclear factor kappa B] IA)
■Hypohidrotic ectodermal dysplasia with immunodeficiency (NFκB [nuclear factor kappa B] IA)
■Hyper-IgE syndrome
■Hyper-IgE syndrome
■Interleukin-12B/ IL-12RB1 deficiency
■Interleukin-12B/ IL-12RB1 deficiency
■Multiple carboxylase deficiency
■Multiple carboxylase deficiency
■Severe combined immunodeficiency
■Severe combined immunodeficiency
2400
only in the stratum corneum. Evidence of an immunologic defect, including skin test anergy and deficient in vitro lymphoproliferation or cytokine release in response to Candida antigens, is found in 75% of CMC patients. However, the variability reflects the underlying heterogeneity of CMC. Autoantibodies against Type I interferons are a sensitive and specific marker for APECED.24
DIFFERENTIAL DIAGNOSIS
Frequent Candida infections in infants, especially thrush, commonly accompany recurrent use of antibiotics, such as for otitis; without a reason for the recurrent Candida infections, HIV infection and CMC should be considered. Autoimmune disorders and
21
endocrinopathies are a feature of IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome,25 but infections tend to be bacterial. However, fungal, bacterial, and viral infections may accompany autoimmune endocrinopathies in IL-2 receptor α chain (CD25) deficiency.26
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
Candidal lesions in patients with CMC generally respond to long-term systemically administered azole antifungal agents (itraconazole, fluconazole) or terbinafine.27 Patients who are resistant usually respond to voriconazole (with its risk of phototoxicity), posaconazole, echinocandins, and/or amphotericin B with or without flucytosine. Cutaneous granulomas often are less responsive despite clearance of infection. Other therapies are granulocyte colony-stimulating factor (may increase IL-17 production),28 nail avulsion, drainage of abscesses, and debridement of thick-crusted cutaneous plaques. All patients with CMC should have an annual endocrine evaluation and patients with documented endocrinopathy or a family history of APECED should be monitored more closely. Patients who have a history of infections other than candidal should have further evaluation of their immune status.
2401
21
CARTILAGE–HAIR HYPOPLASIA SYNDROME
CARTILAGE–HAIR
HYPOPLASIA SYNDROME
AT-A-GLANCE
■ Synonym: metaphyseal chondrodysplasia McKusick type.
■ Autosomal recessive disorder, common in Amish and Finnish populations.
■ Mutations in the RNA component of a ribonucleoprotein endoribonuclease, leading to defective cell-mediated and humoral immunity.
■ Characterized by fine, sparse, hypopigmented hair, and short-limbed dwarfism.
■ Supportive treatment with appropriate antibiotic use; bone marrow transplantation corrects immune deficiency but not dermis or cartilage.
COMBINED ANTIBODY AND T-CELL DEFICIENCY
HYPERIMMUNOGLOBULIN M SYNDROME
HYPERIMMUNOGLOBULIN
M SYNDROME
AT-A-GLANCE
■ Most cases of hyperimmunoglobulin M syndrome have the X-linked recessive form with deficiency of CD40 ligand; common clinical features are recurrent sinopulmonary and GI tract infections, oral ulcerations, and verrucae.
■ Autosomal recessive hyperimmunoglobulin M syndrome, except CD40 deficiency, does not have a susceptibility to opportunistic infections and lymphoid hyperplasia.
WISKOTT-ALDRICH
WISKOTT-ALDRICH SYNDROME
SYNDROME
AT-A-GLANCE
■ X-linked recessive.
■ Mutations in WASP (Wiskott-Aldrich syndrome protein).
2402
■ Recalcitrant dermatitis.
■ Recurrent pyogenic infections.
■ Hemorrhage caused by thrombocytopenia and platelet dysfunction.
■ Therapy: bone marrow stem cell transplantation.
CLINICAL FEATURES
Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disorder with an incidence of approximately 4 per 1 million male births.29,30 The classic triad of WAS is (a) hemorrhage caused by thrombocytopenia and platelet dysfunction, (b) recurrent pyogenic infections, and (c) recalcitrant dermatitis, but this triad appears in only 25% of patients. The bleeding diathesis, which is the most common manifestation of mutations in WAS, being present in 84% of patients, often manifests initially during the first weeks or months of life with bloody diarrhea. Epistaxis, hematemesis, hematuria, mucocutaneous petechiae, and intracranial hemorrhage also may occur. Recurrent bacterial infections begin in infancy as levels of placentally transmitted maternal antibodies diminish. These infections include furunculosis, conjunctivitis, otitis media and otitis externa, pansinusitis, pneumonia, meningitis, and septicemia. Infections with encapsulated bacteria such as Pneumococcus, Haemophilus influenzae, and Neisseria meningitidis predominate. Patients are also susceptible to infections caused by herpes and other viruses and to Pneumocystis jiroveci. The atopic dermatitis associated with WAS, which occurs in approximately 80% of patients, usually develops during the first few months of life and may be quite severe. The face, scalp, and flexural areas are the most severely involved, although patients commonly have widespread involvement with progressive lichenification. The eruption may be more exfoliative than that of atopic dermatitis in individuals without WAS, and excoriated areas frequently have serosanguineous crusts (Fig. 132-7). Secondary bacterial infection of eczematous lesions is common, as are eczema herpeticum (Fig. 132-8) and molluscum contagiosum. IgE-mediated allergic problems, such as urticaria, food allergies, and asthma, are seen in addition to the atopic dermatitis. Up to 40% of patients with WAS develop an autoimmune disorder.31 The most common are vasculitis (particularly involving the skin, GI tract, brain, and heart) in 20% of patients, autoimmune hemolytic anemia in 14% of patients, and IgA nephropathy in up to 10% of patients.32 Other immune-mediated cutaneous manifestations are angioedema, dermatomyositis, pyoderma gangrenosum, and erythema nodosum. Hepatosplenomegaly is common, and lymphadenopathy, transient arthritis, and joint effusions are present occasionally.
ETIOLOGY AND PATHOGENESIS
The defective gene is WASP, mapped to Xp11.22- 11.23, which encodes WASP, a hematopoietic specific
21
cytoplasmic protein that functions in signaling and cytoskeletal organization. WASP couples the signals arising at the cell membrane with reorganization of the cellular cytoskeleton, resulting in cellular activation and promotion of cell motility.33 Mutations in WASP affect organization of the immunologic synapse and T-cell activation, T-lymphocyte and B-lymphocyte migration, and initiation of the primary antibody response. There is a strong phenotype– genotype correlation; classic WAS occurs when WASP is absent or truncated, whereas X-linked thrombocytopenia occurs when mutated WASP is expressed. The atopic dermatitis is likely associated with the observed skewing of CD4+ T-cell differentiation toward Th2 cells with suppression of Th1 and regulatory T cells differentiation.34 Alteration in homeostasis of peripheral B cells and reduced activation of regulatory T cells is thought to promote the development of the autoimmune manifestations of WAS.35-37
Given the expression of WASP on epidermal Langerhans cells, abnormal interactions of Langerhans cells with T cells and the ability of Langerhans cells to move to the lymph node after antigen stimulation may be involved as well. WAS patients have decreased function and number of both T and B lymphocytes, beginning in the first years of life. The lymphocytes of patients with WAS lack microvilli formed by actin bundles, resulting in defective chemotaxis and, in some patients, there is decreased expression of sialoglycoproteins (eg, CD43 and others) on lymphocytes and platelets. Defects in humoral immunity include abnormal serum Ig and decreased antibody response to polysaccharide antigens. WAS patients also have defects in natural killer (NK) cell cytotoxicity, dendritic cell migration, and activation, and impaired macrophage chemotaxis.
DIAGNOSIS
The diagnosis is suspected on the basis of the platelet abnormalities and associated atopic dermatitis, and confirmed by laboratory testing, particularly genotyping to identify the WASP mutation. The thrombocytopenia of WAS is persistent, and platelet counts may range from 1000 to 80,000 platelets/µL. A platelet count of <70,000/µL is required for formal diagnostic criteria. The platelets are small, and platelet aggregation is defective. Levels of IgM and, sometimes, IgG are low, and isohemagglutinins are absent. IgA, IgE, and IgD levels usually are elevated. Eosinophilia, leukopenia, and lymphopenia are also seen. Delayed hypersensitivity skin-test reactivity is diminished, and patients fail to respond to polysaccharide antigens. WASP can be detected by flow cytometry using intracellular staining with an antibody to WASP and sequencing of the WASP gene can confirm the diagnosis of WAS or X-linked thrombocytopenia, which results also from mutations in the WASP gene that do not affect immune function. Mutation analysis allows for prenatal diagnosis. Female carriers of WAS can be
2403
21
detected by their selective inactivation of the abnormal X chromosome in lymphocytes and platelets.38
DIFFERENTIAL DIAGNOSIS
Other immunodeficiencies are characterized by eczematous dermatitis and increased susceptibility to infections, but WAS can usually be distinguished by the bleeding tendency and laboratory evidence of microthrombocytopenia.
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
Therapeutic interventions allow some patients with WAS to survive into adulthood; however, a significant proportion die before the age of 10 years from infections secondary to hemorrhage, malignancies, or the complications of transplantation.29,39
Thirteen percent of patients with WAS develop lymphoreticular malignancies, especially non-Hodgkin lymphoma (especially diffuse large B-cell lymphomas), with a predominance of extranodal and brain involvement. Development of autoimmune hemolytic anemia is a poor prognostic factor and associated with the development of lymphoid malignancies; overall, 25% of patients with autoimmunity develop a malignancy.32 10% of patients die from these malignancies, usually as adolescents or young adults. Appropriate antibiotics, immunizations, and transfusions of platelets and plasma decrease the risk of fatal infections and hemorrhage. Immunoglobulin replacement therapy is useful in some patients. Splenectomy has been advocated to ameliorate the bleeding abnormality in patients with recurrent severe hemorrhage, but this procedure increases the risk of infection from encapsulated bacterial organisms. Topical glucocorticoid preparations and Ig replacement may improve the dermatitis, and chronic administration of oral acyclovir is appropriate for patients with recurrent eczema herpeticum. Bone marrow or stem cell transplantation is the treatment of choice for patients with recurrent problems, especially significant autoimmunity.29,39 Full engraftment results in normal platelet number and function, normal immunologic status, and clearance of the dermatitis (T-lymphocyte engraftment). The 5-year survival rate for children younger than 5 years of age with human leukocyte antigen (HLA)- matched sibling donors is 87%; older patients and those with mismatched donors have survival rates of approximately 50%. Lentivirally transduced, WAS-reconstituted, autologous CD34+ cells have been administered in gene therapy trials, resulting in sustained clinical benefit in the majority of treated patients; dermatitis is improved or cleared and the risk of infection and autoimmune manifestations is reduced.37,40,41 To avoid the risk of insertional oncogenesis from viral vectors, ongoing trials use selfinactivating lentiviral vectors and, more recently,
2404
zinc finger nucleases as a nonviral approach to the correction of WAS cells.42
SEVERE COMBINED IMMUNODEFICIENCY
SEVERE COMBINED
IMMUNODEFICIENCY
AT-A-GLANCE
■ Heterogeneous group of X-linked and autosomal recessive disorders with deficient cell-mediated and humoral immunity.
■ Failure to thrive in early infancy; diarrhea; recurrent mucocutaneous candidiasis, bacterial, and viral infections; risk of graft-versus-host disease.
■ Hematopoietic stem cell transplantation may provide a long-term cure.
CLINICAL FEATURES
Infants with severe combined immunodeficiency (SCID) usually fail to gain weight by 3 to 6 months of age, following the onset of recurrent sinopulmonary and skin infections.43,44 Persistent mucocutaneous candidiasis is often present at the time of diagnosis, and systemic candidal infections occur occasionally. Patients with SCID also may have chronic diarrhea and malabsorption caused by viral infections. P. jiroveci (carinii) pneumonia is often a presenting feature. Although bacterial infections usually respond to systemic antibiotics, viral infections tend to be fatal. Infants with SCID lack palpable lymphoid tissue despite recurrent infections. In addition to cutaneous bacterial and candidal infections, the most common cutaneous eruptions are morbilliform or resemble seborrheic dermatitis. In some infants with SCID, biopsy sections show graft-versus-host disease (GVHD) (see Chap. 129). GVHD may result from in utero exposure to maternal lymphocytes, which is usually nonfatal, from transfusion with nonirradiated blood products, which is usually fatal, or may follow stem cell transplantation. Patients with GVHD most commonly present acutely with morbilliform erythema, papular dermatitis, or diffuse erythroderma. The face, neck, palms, and soles are usually affected initially, before the eruption becomes generalized. Severe cases are complicated by diffuse bullae or toxic epidermal necrolysis. The clinical presentation of GVHD from maternal engraftment (without transplantation) is
21
LYMPHOCYTE PROFILE DISORDER GENE GENE PRODUCT FUNCTION
ADA
T−/B−/NK− ADA deficiencya,b
PNP
PNP deficiency
RAG1/RAG2
T−/B−/NK+ RAG 1 or 2c
Artemis
Artemis deficiencyd,e
LIG4
LIG4 syndromee,93
IL2RG
T−/B+/NK− X-linked severe combined immunodeficiencya
JAK3
Janus protein kinase deficiency
Enzyme necessary for detoxification of metabolic products of the purine salvage pathway that cause lymphocytes to undergo apoptosis Enzyme necessary for detoxification of metabolic products of the purine salvage pathway that cause lymphocytes to undergo apoptosis: located downstream from ADA
Required for somatic V, D, and J rearrangements during Band T-cell development Required for DNA repair during somatic V, D, and J rearrangements during B-cell and T-cell development Ligase required for ATP-dependent repair of double-stranded DNA
Common γ chain of IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21
Tyrosine kinase; primary signal transducer from common γ chain
α Chain of IL-7 receptor Facilitates expression of T-cell receptor/CD3 complex; block at pre–T-cell receptor stage of development Phosphatase that regulates immune cell signaling Defect in thymic development. Human homolog of nude mouse with total alopecia and onychodystrophy; critical transcription factor for thymic development
IL7R CD3δ and CD3ε
T−/B+/NK+ IL-7 receptor (IL-7R) deficiency T-cell receptor/CD3 complex deficiency CD45 deficiency92
PTPRC WHN
Winged helix nude deficiency
Other Defects in T-Cell Development
CD8A
CD4+ CD8− /B+/NK+ CD8 deficiency94
ZAP-70 TAP1 TAP2
ZAP-70 deficiency MHC class I deficiency/TAP deficiency (bare lymphocyte syndrome I)
CD4low CD8+/B+/NK+ MHC class II deficiency (bare lymphocyte syndrome II) CIITA RFXB RFX5 RFXAP
Promotes survival and differentiation of activated T cells to memory CD8+ T cells Signaling tyrosine kinase Transporter associated with antigen processing, transports peptides to assemble the class I molecule, dysregulated NK cells
Defects in the transactivating factors of MHC II molecules, nonfunctional CD4 T cells if present
CD3γ Chain facilitates expression of T-cell receptor/CD3 complex
T+/B+/NK+ T-cell receptor/CD3 complex deficiency CD3γ Chain facilitates expression of T-cell receptor/CD3 complex
T+/B+/NK+ T-cell receptor/CD3 complex
deficiency
aMost common severe combined immunodeficiency defects. X-linked severe combined immunodeficiency accounts for approximately 50% of patients. Adenosine deaminase deficiency accounts for approximately 20% of patients.
bEighty-five percent to 90% of patients present in infancy with severe immunodeficiency; one-half have skeletal deformities, neurologic symptoms, behavior problems, and decreased IQ.
cDeletions/frameshift mutations of RAG1 /RAG2 genes result in severe combined immunodeficiency phenotype. Less-severe mutations result in Omenn syndrome, which is clinically distinct from other forms of severe combined immunodeficiency. Mutations in other genes may lead to the Omenn phenotype and immunodeficiency.
dCommon genetic defect seen in Athabascan-speaking Native Americans, the defect is called SCIDA in this population. Gene frequency may be as high as 2%. These patients have a higher incidence of oral/genital ulcers.
eBoth Artemis and LIG4 mutations lead to hypersensitivity to irradiation. ADA, adenosine deaminase; ATP, adenosine triphosphate; IL, interleukin; MHC, major histocompatibility complex; NK, natural killer cell; PNP, purine nucleoside phosphorylase; RAG, recombination activating gene; TAP, transporter associated with antigen processing.
indistinguishable from the clinical presentation of GVHD from transplantation, but histopathologic features differ.45 Biopsy sections from GVHD secondary to maternal engraftment show psoriasiform hyperplasia with parakeratosis and variable spongiosis in contrast to the vacuolar interface pattern observed in conventional GVHD. Overall, 83% of SCID patients
with maternal engraftment develop skin manifestations. Oral and genital ulcerations are also seen in SCID, particularly in Athabascan-speaking Native American children with a defect in the Artemis gene.46
Patients with Omenn syndrome show erythroderma, hepatosplenomegaly and lymphadenopathy with eosinophilia and increased IgE production.47 B cells
2405
21
tend to be undetectable, and T cells, although often increased in number, are nonfunctional. Omenn syndrome has been shown to be a phenotype with several underlying genetic bases. Most patients have RAG1/ RAG2 mutations, but the phenotype of Omenn syndrome has been described in patients with mutations in Artemis and IL-7R. Two individuals with cartilagehair hypoplasia and another with complete DiGeorge syndrome also showed features of Omenn syndrome. The natural history of SCID is changing with the introduction of newborn screening for SCID and other T-cell lymphopenia.48 Except for a few causes of SCID that may not present with T-cell lymphopenia at birth, patients are identified by newborn screening soon after birth and the diagnosis is established before serious infections develop. Early stem cell transplantation provides an excellent outcome irrespective of the source of the stem cells or the preparation regimen.49
ECTODERMAL DYSPLASIA WITH IMMUNODEFICIENCY
ECTODERMAL DYSPLASIA
WITH IMMUNODEFICIENCY
AT-A-GLANCE
■ Mutations in nuclear factor κB (NF-κB) essential modulator, leading to abnormal NF-κB signaling.
■ Characteristic facies of hypohidrotic ectodermal dysplasia.
■ Often severe immunodeficiency; bacterial, atypical mycobacterial, and viral infections are common.
■ Therapy: stem cell transplantation can correct the immunodeficiency but not the other features of the disease.
EPIDEMIOLOGY
X-linked recessive transmission is most common with an estimated incidence of 1:250,000 live male births.50
An autosomal dominant form of ectodermal dysplasia with immunodeficiency also has been described.51
ETIOLOGY AND PATHOGENESIS
Several genetic disorders result from gene mutations in the nuclear factor κB (NF-κB) essential modulator (NEMO) and lead to abnormal NF-κB signaling (see Chaps. 75 and 131). One of these, hypohidrotic ectodermal dysplasia with immunodeficiency, results from hypomorphic mutations in the NEMO gene (also called ectodermal dysplasia, anhidrotic, with immunodeficiency or Hyperimmunoglobulinemia M, immunodeficiency, X-linked with ectodermal dysplasia). NF-κB is a DNA-binding transcription factor, and its ability to transcribe genes is
2406
regulated by a cytoplasmic inhibitor, IKB (inhibitor of nuclear factor κB). NF-κB signaling affects inflammation, apoptosis, development, and immunity. NF-κB activation requires phosphorylation of IKB by IKB kinase (IKK). IKK is composed of 2 catalytic components (IKKα and IKKβ) and a regulatory subunit, IKKγ or NEMO. NEMO has no catalytic function, but is the structural scaffold that supports the IKK complex. When NEMO is absent or defective, no functional IKK complex is formed and, as a result, NF-κB cannot translocate to the nucleus and activate gene transcription. The hypomorphic mutations that cause anhidrotic ectodermal dysplasia with immunodeficiency allow early survival in males, in contrast to the large deletions in NEMO (usually of exons 4 to 10) that lead to incontinentia pigmenti in carrier females and fetal death in affected males (see Chap. 75). Female carriers of hypomorphic NEMO mutations often show mild features of incontinentia pigmenti, but may also express an incontinentia pigmenti phenotype with transient immunodeficiency.52 In boys with hypomorphic mutations of NEMO, the ectodermal dysplasia phenotype results from impaired NF-κB signaling.53 Immunodeficiency results from impaired NF-κB activation in response to signaling via antigen receptors, Toll-like receptors, IL-1 receptor, and the tumor necrosis factor (TNF) receptor family.54 Gain-of-function mutations in IKBα, the component of the inhibitor of NF-κB that is phosphorylated by IKK, lead to an autosomal dominant form of ectodermal dysplasia with a distinct immunologic phenotype, characterized by a profound T-cell deficiency, but normal NK cytotoxicity and responses to Mycobacteria. Mutations in NEMO that cause immunodeficiency without the ectodermal dysplasia phenotype also have been reported.
CLINICAL FEATURES
Affected patients present with classic characteristics of hypohidrotic ectodermal dysplasia (Fig. 132-9). These include the characteristic facies, hypotrichosis or atrichia, hypohidrosis (leading to heat intolerance), hypodontia or anodontia with conical incisors, and associated dermatitis (see Chap. 131). A subset of patients has associated osteopetrosis and lymphedema in association with severe immunodeficiency. Boys with immunodeficiency from NEMO mutations without the features of hypohidrotic ectodermal dysplasia also have been described. In an analysis of 72 patients with NEMO mutations, only 77% had ectodermal dysplasia.50 Bacterial infections early in infancy are common, particularly sepsis, pneumonia, otitis, sinusitis, and lymphadenitis. Recurrent pneumonias may lead to bronchiectasis. Common pathogens include Streptococcus pneumoniae, H. influenzae, Klebsiella, Salmonella, and Pseudomonas. Infections with atypical Mycobacteria and viruses, including cytomegalovirus (systemic and GI involvement), herpes simplex virus, molluscum contagiosum, human papillomavirus, and Pneumocystis carinii, are also reported. The immunodeficiency is often severe, but its characteristics are variable with an apparent genotype–phenotype correlation that can be further
identified by in vitro reconstitution of the mutations. Immune dysfunction includes impaired B-cell Ig class switching with hypogammaglobulinemia (and often increased levels of IgM and/or IgA), impaired specific antibody production (particularly to polysaccharide antigens), deficient NK cell cytotoxicity, poor cytokine production in response to CD40 signaling, and autoinflammation, especially of the gut. Biopsy of skin may show evidence of keratinocyte apoptosis, reflecting the dysfunction in NF-κB signaling, and must be differentiated from GVHD.
TREATMENT AND PROGNOSIS
There is an increased mortality, with 36% of patients dying at a mean age of 6.4 years.50 Therapy is guided by the patient’s clinical and immunologic phenotype. Ectodermal dysplasia is treated supportively (see Chap. 131). Patients with impaired antibody production may benefit from Ig replacement. All infections (bacterial and viral) should be treated aggressively with the appropriate antibiotics/antivirals. Prophylaxis for both P. jiroveci and Mycobacterium aviumintracellulare should be considered. Stem cell transplantation may lead to immune reconstitution but may not correct the other manifestations of the disease.55 The mothers and sisters of these patients should be offered genetic testing for the NEMO mutation and counseling.
21
ATAXIA-TELANGIECTASIA
ATAXIA-TELANGIECTASIA
AT-A-GLANCE
■ Autosomal recessive disorder with mutations in ataxia-telangiectasia mutated (ATM).
■ Early onset of ataxia with progressive neurologic deterioration; conjunctival telangiectasia first appears in preschool years in most patients.
■ Sinopulmonary infections, lymphoreticular neoplasia.
■ Deficiency of IgA, IgE, IgG2, IgG4; variable manifestations of T-cell deficiency; high levels α-fetoprotein; chromosomal breaks; sensitive to irradiation.
EPIDEMIOLOGY
Ataxia-telangiectasia (AT; Online Mendelian Inheritance in Man [OMIM] #208900), also called Louis-Bar syndrome, is an autosomal recessive disorder with an estimated incidence of up to 1:40,000 and a carrier rate of up to 1%. Carriers have an increased risk of breast cancer, hematologic malignancies,56,57 and ischemic heart disease, with a reduced life expectancy of approximately 8 years.58 These heterozygotes show an increased risk of chromosomal breaks after exposure to irradiation in vitro, suggesting that mammograms in known carriers of AT are contraindicated.
CLINICAL FEATURES
Characteristic oculocutaneous telangiectasias begin near the ocular canthi and progress across the bulbar conjunctivae (Fig. 132-10). These telangiectasias usually appear when patients are 3 to 6 years of age; rarely have they been described at earlier ages. Cutaneous telangiectasias subsequently may develop on the malar prominences, ears, eyelids, anterior chest, and popliteal and antecubital fossae, and the dorsal aspects of the hands and feet (Fig. 132-11). The telangiectasias may be subtle and resemble fine petechiae, especially in the flexural areas. The development of telangiectasias may be related to sun exposure, because ocular,
2407
21
but not cutaneous, telangiectasias develop in affected dark-skinned children. The development of telangiectasias may relate, at least in part, to the sensitivity of some AT strains to ultraviolet light.59
Progeric changes of the skin, including xerosis and gray hair, occur in 90% of patients.60 During adolescence, the facial skin may become progressively more atrophic and sclerotic, causing a mask-like appearance. Occasionally, the ears, arms, and hands also become sclerodermatous. The hair may be diffusely gray by adolescence, and subcutaneous fat is generally lost in childhood. Recurrent severe impetigo often develops. Seborrheic dermatitis occurs in many patients, and the associated blepharitis may lead to a diagnosis of blepharoconjunctivitis rather than ocular telangiectasia. Mottled hyperpigmentation and hypopigmentation commonly occur and, together with the telangiectasias and atrophy, can resemble the poikiloderma of radiodermatitis, actinic damage, or scleroderma. Other pigmentary changes include café-au-lait spots that may be found in a dermatomal distribution, multiple ephelides, and vitiligo. Hypertrichosis of the arms and legs, alopecia areata, multiple warts, atopic dermatitis, keratosis pilaris, nummular eczema, and acanthosis nigricans also have been described in association with AT. Among the most common cutaneous manifestations are noninfectious cutaneous granulomas (Fig. 132-12).61 These persistent, atrophic, and often ulcerative lesions are often mistaken for other granulomatous processes, including sarcoidosis, necrobiosis lipoidica diabeticorum, granuloma annulare, and granulomatous dermatitis.
2408
Usually, the progressive cerebellar ataxia first becomes apparent during infancy (median age: 1.2 years) with swaying of the head and trunk and apraxia of eye movements, often years before skin or conjunctival abnormalities develop. In childhood, dysarthric speech, drooling, choreoathetosis, and myoclonic jerks become prominent. The diagnosis of AT is usually made at a median age of 7 years, after appearance of the mucocutaneous telangiectasia. Patients usually require a wheelchair by their teenage years. Recurrent bacterial and viral sinopulmonary infections occur in up to 80% of patients; these are the most common cause of death, which is usually from bronchiectasis and respiratory failure. Approximately 75% of patients with AT may have growth retardation and endocrine disorders, especially ovarian agenesis or testicular hypoplasia and insulin-resistant diabetes. Neoplasia occurs in 40% of surviving adolescents or young adults, although lymphoid malignancy has been described as the presenting sign during infancy. Most common are lymphomas (especially B-cell lymphoma; 200-fold increased risk) and leukemia (especially T-cell chronic lymphocytic leukemia; 70-fold increased risk). Basal cell carcinomas have been reported in young adults. Patients with AT tend to have both humoral and cellular immunologic abnormalities. Serum IgA and IgE are absent or deficient in 70% and up to 80% of patients, respectively. Circulating anti-IgA antibodies are common in AT patients with IgA deficiency. Approximately 60% of patients have selective IgG2 and IgG4 deficiencies. Defective cell-mediated immunity is found in 70% of patients, particularly lymphopenia and deficient in vitro responses to antigens and
mitogens; T cells bearing γ/δ receptors are increased in number, while CD4+ T cells tend to be reduced. Virtually all patients have elevated levels of α-fetoprotein (which is particularly significant after 2 years of age), and many have detectable carcinoembryonic antigen. Patients with AT often have elevated hepatic transaminases (40% to 50%), and glucose intolerance. DNA is exquisitely sensitive to X-irradiation, and patients also show an increased rate of telomere shortening. Spontaneous chromosomal abnormalities (fragments, breaks, gaps, and translocations) occur 2 to 18 times more frequently in patients with AT than in normal individuals and mainly involve chromosomes 2, 7, and 14. Rearrangements of chromosomes 7 and 14, and especially 14:14 translocations, seem to predict the development of lymphoreticular malignancy, including leukemia. The thymus is absent or hypoplastic, and the spleen may be reduced in size. Among techniques to confirm diagnosis are an elevated serum α-fetoprotein level in children older than 8 months of age, analysis of radioresistant DNA synthesis (which demonstrates an abnormal S-phase checkpoint), radiosensitivity testing with the colony survival assay, immunoblotting for the ATM (ataxia-telangiectasia mutated) protein, assessment of ATM kinase activity, and molecular genetic testing.
PROGNOSIS, CLINICAL COURSE, AND TREATMENT
Therapy for AT is supportive and includes administration of antibiotics for infection, physiotherapy for pulmonary bronchiectasis, physical therapy to prevent contractures in patients with neurologic dysfunction, and sunscreens and sun avoidance to diminish actinic-like changes. Patients should be aggressively screened for malignancy, especially after the first decade of life. Intralesional injections of triamcinolone have helped promote healing of the painful, associated ulcerations, although the lesions do not clear completely with treatment. Autopsy findings indicate that approximately 50% of the patients die from pulmonary disease, the most common cause of death. Lymphoreticular malignancies, including lymphoid leukemia, are the second most common cause of death (15% of patients with AT). The remaining patients tend to die of both pulmonary disease and malignancy. Therapeutic radiation and radiomimetic chemotherapeutic agents, especially bleomycin, may lead to extensive tissue necrosis. The administration of small doses of other chemotherapeutic drugs and low-dose, fractionated radiation is the least-harmful means of managing these malignancies. In a small subset of patients with milder AT, treatment with aminoglycosides increased ATM gene function.62 Death usually occurs by late childhood or early adolescence; the oldest surviving patient died at the age of 50 years. Prenatal diagnosis is currently best achieved by DNA analysis.
21
DISORDERS OF PHAGOCYTOSIS AND CELL KILLING
Patients with defects in phagocyte function or cell killing typically present during infancy or childhood with recurrent, unusual, and/or difficult-to-clear bacterial infections. Infections commonly seen include those of skin or mucosa, lung, lymph nodes, deeptissue abscesses, or childhood periodontitis. The most distinctive disorders of phagocytosis and cell killing are chronic granulomatous disease (CGD), leukocyte adhesion deficiencies, Hyperimmunoglobulinemia E syndrome (HIES), and the silvery hair syndromes with immunodeficiency.63 However, several disorders with neutropenia, neutrophil dysfunction, or cytokine dysfunction also lead to a decreased ability to engulf and/or kill organisms. These are reviewed in Table 132-5.
CHRONIC GRANULOMATOUS DISEASE
CHRONIC
GRANULOMATOUS DISEASE
AT-A-GLANCE
■ Group of disorders in which defective production of reactive oxygen intermediates impairs intracellular killing of microorganisms.
■ X-linked or autosomal recessive.
■ Mutations in genes that encode components of nicotinamide adenine dinucleotide phosphate oxidase system.
■ Pneumonias, lymphadenopathy, hepatosplenomegaly, and skin infections.
■ Granulomas, most commonly of lungs and liver.
■ Lupus-like inflammation in carriers (X-linked) and patients.
CGD is a heterogeneous group of X-linked and autosomal recessive disorders.64,65 Ninety percent of patients with the disorder are male, and the overall incidence is 1 in 200,000 to 250,000 persons. X-linked disease with deficiency of gp91phox occurs in 70% of affected individuals with signs and symptoms manifesting in the first year of life. Individuals with autosomal recessive disease are more likely to have some nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, tend to present later in life (mean age of onset: 8 years), have milder signs and symptoms, and have longer survival.66,67
2409
21
DISORDER GENETICS GENE FUNCTIONAL CONSEQUENCE CLINICAL FINDINGS
Neutropenia
Cyclic neutropenia AD ELA2 Defects in neutrophil elastase Cycles of neutropenia and monocytopenia, approximately every 3 weeks; oral ulcers and fever when low
Severe congenital neutropenia AD ELA1 Defects in neutrophil elastase lead to abnormal neutrophil trafficking Neutropenia, myelodysplastic syndrome, and acute myelogenous leukemia; neutropenia and lymphopenia; myeloid progenitors in circulation
AD GF11 Repressor of elastase transcription leads to accumulation of elastase
AR SBDS SBDS protein participates in metabolism of ribosomal RNA Pancytopenia with myelodysplastic syndrome and acute myelogenous leukemia; pancreatic insuffi- ciency; chondrodysplasia
Shwachman- Diamond syndrome (SBDS)
WHIM128 AD CXCR4 Receptor regulates development and migration of neutrophils; affects viral entry into cells
Cytokine Defects
Chronic cutaneous and cervical warts, antibioticresponsive bacterial sinopulmonary infections; often low Ig levels, peripheral neutropenia in the face of bone marrow hypercellularity (myelokathexis), and distinctive neutrophil morphology
AR IL12B Absence of component of IL-12 and IL-23 that stimulates IFN-γ production
Severe mycobacterial infections. Disseminated bacille Calmette-Guérin after vaccination; severe Salmonella infections
AR IL12RB1 β1-Chain of IL-12 and IL-23 receptors
AR, AD IFNGR1 Affects ligand binding to IFN-γ receptor
AR IFNR2 Affects IFN-γ receptor signaling
AR, AD STAT1 Affects transcription in IFN receptor signaling Also severe viral infections
AR STAT5B As in Stat1, but also growth hormone receptor signaling Also growth hormone insensitivity
Defects of Killing
Myeloperoxidase deficiency AR MPO Myeloperoxidase required for bacterial killing Especially Staphylococcus aureus and candidal infections; may be asymptomatic
AR C/EBPE C/EBPε transcription factor required
Specific granule deficiency AR C/EBPE C/EBPε transcription factor required for granulocyte differentiation Recurrent bacterial infections; neutrophils with 2 lobes
Specific granule
deficiency
Recurrent bacterial infections; neutrophils with 2 lobes
for granulocyte differentiation
AD, autosomal dominant; AR, autosomal recessive; IFN, interferon; IL, interleukin; WHIM, warts, hypoimmunoglobulinemia, infections, and myelokathexis.
CLINICAL FEATURES
Pyodermas with associated regional lymphadenopathy/adenitis and dermatitis tend to affect body sites that are exposed to organisms, especially on the skin and lungs. Staphylococcal abscesses are found in 40% of patients, particularly of the perianal area, nares, and ears. Purulent inflammatory reactions may develop at sites of lymph node drainage or minor cutaneous trauma and heal slowly with scarring. Ecthyma gangrenosum may be the presentation in a neonate.68 Staphylococcal infections are the most common, but other bacterial and fungal organisms that are killed by generation of reactive oxygen species may cause infection (Aspergillus spp., Burkholderia cepacia, Candida spp., Klebsiella spp., mycobacteria (including severe/disseminated bacille
2410
Calmette-Guérin infection),69 Nocardia spp., and Serratia marcescens (especially abscesses with ulceration and osteomyelitis).70
Patients with CGD often develop chronic inflammatory granulomas, most commonly of the lungs and liver. Cutaneous granulomas are nodular and often necrotic. Granulomas can occlude vital structures, especially of the GI and genitourinary systems. Intraoral ulcerations resembling aphthous stomatitis, chronic gingivitis, perioral ulcers, scalp folliculitis, and seborrheic dermatitis also have been described in many patients. Female carriers for X-linked CGD do not have the increased risk of infections but may have cutaneous lesions of discoid or systemic lupus erythematosus, Jessner lymphocytic infiltration of the skin, aphthous stomatitis,
granulomatous cheilitis, photosensitivity, and/or Raynaud phenomenon.71,72
The lymph nodes, lungs, liver, spleen, and GI tract are the most frequent areas of noncutaneous involvement. Suppurative lymphadenitis with abscess and fistula formation usually affects cervical nodes. Pneumonia occurs in almost all affected children and may lead to abscess formation, cavitation, and empyema. Hepatosplenomegaly has been reported in 80% to 90% of patients; more than 30% of patients develop hepatic abscesses, and staphylococcal liver abscesses are almost pathognomonic to CGD. Other excessive inflammatory responses can include hemophagocytic lymphohistiocytosis73 or features that resemble inflammatory bowel disease (anal fistulae, diarrhea), IgA nephropathy, sarcoidosis, and rheumatoid arthritis.74,75
Patients with bacterial and Nocardia spp. infections tend to be symptomatic and frequently have leukocytosis, anemia, and elevated sedimentation rate. In contrast, lack of fever, normal erythrocyte sedimentation rate, and few symptoms are more common in Aspergillus spp. infections. Thus, laboratory values within normal limits do not rule out infection in a CGD patient. Patients show an increased erythrocyte sedimentation rate, hypergammaglobulinemia, leukocytosis, and mild anemia; other immune function tests are otherwise usually normal.
ETIOLOGY AND PATHOGENESIS
CGD is caused by defects in the reduced NADPH oxidase, the enzyme complex responsible for the generation of superoxide. Normal bactericidal activity after phagocytosis requires the NADPH oxidase system, which consists of NADPH, an unusual phagocyte cytochrome b (b558), and cytosolic proteins. In patients with CGD, this membrane-associated NADPH oxidase system fails to produce superoxide and other toxic oxygen metabolites. The oxidative molecules have direct microbicidal activity, but also act as intracellular signaling molecules, activating the release of primary granule proteins neutrophil elastase and cathepsin G inside the phagocytic vacuole that, in turn, are necessary to kill microbes. Mouse models of CGD show decreased regulatory T-cell activity, unrestrained γ/δ T-cell activity, and increased production of IL-1β, IL-8, and IL-17.76 NADPH oxidase also modulates major histocompatibility complex Class II antigen presentation by B cells.77
In X-linked kindreds (approximately 70%), the CYBB gene encoding the gp91phox (phagocyte oxidase) subunit of cytochrome b558 is mutated. Patients with autosomal recessive CGD are deficient in NADPH oxidase cytosolic factors (p47phox—encoded by NCF1—in approximately 20% of patients; p67phox—encoded by NCF2—in ≤5%; and p40phox—encoded by NCF4—very rare); occasionally, the p22phox (γ subunit of cytochrome b558), which contains a docking site for p47phox, is deficient (mutation in CYBA; ≤5% of patients). It is thought that cytochrome b558 is the membrane attachment site for these cytosolic
21
factors that translocate from the cytosol to the plasma membrane, assembling oxidase components to allow activation of the NADPH oxidase. The types of microbial organisms that cause infections in patients with CGD require intracellular killing and are usually catalase-positive. Only 5 organisms are responsible for the overwhelming majority of infections in CGD in North America and Europe: Staphylococcus aureus, S. marcescens, B. cepacia, Nocardia spp., and Aspergillus spp. The intense humoral and granulomatous responses of CGD are thought to be compensatory in the robust but ineffectual response to organisms.
DIAGNOSIS
The diagnosis of CGD is made on the basis of assays that rely on superoxide production. The dihydrorhodamine flow cytometry-based test (DHR 123 assay) is currently favored and can readily identify patients or carriers of X-linked disease78; the ferricytochrome c reduction assay is another quantitative measure of the respiratory burst. The screening test for CGD is the nitroblue tetrazolium (NBT) reduction assay, in which the yellow NBT becomes insoluble, oxidized form (blue formazan) when precipitated with normal oxidative metabolism. Quantitative NBT tests and chemiluminescence assays also may be performed. Immunoblot analysis confirms the absence of the glycoprotein 91phox
(gp91phox) component; because deficiency of one component of cytochrome b558 leads to absence of the other, sequencing of the gp91phox or p22phox gene is necessary if absence is noted by immunoblot analysis. Immunoblots that show the absence of p47phox or p67phox can be diagnostic. Biopsy of cutaneous granulomas shows histiocytic infiltrates associated with foreign-body giant cells and accumulation of neutrophils with necrosis. Although the histopathologic features of lupus erythematosuslike skin lesions in CGD patients and carriers may resemble those of lupus patients, immunofluorescence examination of lesional skin is usually negative.
DIFFERENTIAL DIAGNOSIS
Although the tests to measure respiratory burst are an easy screening test, other disorders of phagocytosis may be confused with CGD. Among these are the leukocyte adhesion defects (see section “Leukocyte Adhesion Deficiencies”), Shwachman-Diamond syndrome, myeloperoxidase deficiency, and defects in Toll receptor, interferon, or IL-12 signaling.
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
Patients with X-linked CGD, p22phox CGD, and p67phox CGD tend to have a more-severe clinical course compared to patients with p47phox CGD. The mean age of
2411
21
TREATMENT INDICATION DOSE
Trimethoprim-sulfamethoxazolea (dicloxacillin if allergic) Bacterial infections 5 mg/kg/day divided twice daily
Itraconazolea Fungal infections 100 mg/day if patient weighs <50 kg; 200 mg/day if patient weighs >50 kg
Interferon-γa Infections 50 mcg/m2 subcutaneously 3 × /week
Leukocyte transfusion Life-threatening infections
Bone marrow transplantation Life-threatening infections
Systemic glucocorticoids Granulomas, particularly if obstructive
Debridement/drainage/surgical intervention Abscesses (particularly liver)
Debridement/drainage/surgical intervention Abscesses (particularly liver)
aRegardless of genetic subgroup, the current recommendation is to use prophylaxis with trimethoprim-sulfamethoxazole, itraconazole, and interferon-γ.136
diagnosis of X-linked CGD is 3 years of age, and of autosomal recessive forms, 8 years of age. More than 90% of patients with non-p47phox CGD have undetectable levels of superoxide production. Some patients develop severe infections as early as infancy whereas others unexpectedly develop a serious infection typical of CGD later during childhood. Small foci of localized inflammation may not be associated with fever and may be difficult to detect without vigorous investigation of the lungs, liver, and bones by routine screening radiographs, scans, or ultrasonography. Cultures should be performed to identify the infectious agent, and invasive procedures may be necessary to obtain adequate tissue samples. Patients with evidence of infection should be treated empirically with broad-spectrum parenteral antibiotics that cover S. aureus as well as Gram-negative organisms.79 Intravenous therapy should be continued for at least 10 to 14 days, followed by a several-week course of oral antibiotics. Surgical interventions (drainage, debridement) may be required for deeper infections (Table 132-6). Trimethoprim-sulfamethoxazole therapy decreases the incidence of bacterial infection without increasing the incidence of fungal infection. Itraconazole is an effective agent for prophylaxis for fungal infections.80 Prophylactic interferon (IFN)-γ decreases the number and severity of infections without increasing the incidence of chronic inflammatory complications in both X-linked and autosomal recessive CGD.81 Use of IFN-γ is not accompanied by any measurable improvement in NADPH oxidase activity; its clinical benefit is related to enhanced phagocyte function and killing by nonoxidative mechanisms. Granulocyte transfusions have been used for rapidly progressive, life-threatening infections. The prophylactic administration of antibiotics and IFN-γ has reduced the mortality of CGD to approximately 2% per patient-year for autosomal CGD and 5% per year for X-linked CGD.65 The most common causes of death are pneumonia and/or sepsis caused by Aspergillus or B. cepacia. Systemic glucocorticoids have been helpful for patients with obstructive visceral granulomas.
2412
Other therapies that may be of benefit for the inflammatory manifestations include anakinra, azathioprine, hydroxychloroquine, pioglitazone,82 sirolimus, and thalidomide83; TNF inhibitors improve the colitis, but increase the risk of infection. Stem cell transplantation may cure CGD and its use has increased.84 Younger patients without infection at transplantation have the best outcome (survival >95%), but reduced-intensity conditioning regimens have been used for older individuals (including adults) and patients with recalcitrant infections or inflammation,85 and should be considered if recurrent serious infections or corticosteroid-dependent inflammatory disease before irreversible organ damage occurs.86
Gene therapy has been performed for the p47phoxdeficient and X-linked forms of CGD. The use of retroviruses, however, may lead to insertional oncogenesis and myelodysplasia, necessitating safer approaches such as self-inactivating lentiviral vectors or nonviral techniques.86-90
LEUKOCYTE ADHESION DEFICIENCIES
LEUKOCYTE ADHESION
DEFICIENCIES
AT-A-GLANCE
■ Group of 4 disorders: 3 autosomal recessive (mutations in ITGB2, SLC35C1, or FERMT3) and 1 autosomal dominant (mutations in Rac2).
■ Gingivitis and periodontitis.
■ Poor wound healing; delayed separation of the umbilical stump and development of pyoderma gangrenosum-like necrotic ulcerations after wounding.
■ Life-threatening bacterial and fungal infections.
HYPERIMMUNOGLOBU- LINEMIA E SYNDROME
HYPERIMMUNOGLOBU
LINEMIA E SYNDROME
AT-A-GLANCE
■ Synonyms: Job syndrome, Buckley syndrome, hyperimmunoglobulin E recurrent infection syndrome.
■ Most cases are autosomal dominant, but recessive forms with some different features have been described.
■ Classic triad: (a) recurrent staphylococcal skin abscesses, (b) pneumonia with pneumatocele formation, and (c) high serum levels of IgE.
■ Atopic dermatitis is a common manifestation.
■ Scoliosis, fractures, and dental abnormalities are uniquely features of the more prevalent, autosomal dominant form.
■ Autosomal recessive cases have severe viral infections and can develop severe neurologic complications.
HIES is rare (incidence 1 in 106).91 It is found equally in males and females. Most cases are sporadic or consistent with an autosomal dominant inheritance. Autosomal recessive inheritance also has been described, but patients show different associated features.
CLINICAL FEATURES
The clinical presentation of individual patients with HIES may vary greatly.91-93 Table 132-7 summarizes
INCIDENCE (%)
Immune System
■Dermatitis
100 87 87
■Skin abscesses
■Recurrent pneumonia (3 or more, proved by radiography)
■Pneumatoceles
77 83
■Mucocutaneous candidiasis
Skeletal System
■Characteristic fades
83 65 72 68 57 63
■Wide nose
■Failure of dental exfoliation (>3 teeth)
■Hyperextensibility
■Recurrent pathologic fractures
■Scoliosis (>10°)
Laboratory Values
Laboratory Values
■Immunoglobulin E >2000 IU/mL or >10 times the age-specific norm
■Immunoglobulin E >2000 IU/mL or >10 times
97
97
the age-specific norm
■Eosinophilia (>2 SD above the norm)
■Eosinophilia (>2 SD above the norm)
93
93
21
the most common clinical features and laboratory values of patients with sporadic or autosomal dominant HIES. The neonatal or infantile rash of HIES is often a papulopustular eruption with prominent crusting distributed on the scalp, face, neck, axillae, and diaper area.94,95 The most consistent finding on skin biopsy is an eosinophilic spongiotic dermatitis, sometimes centered in the dermal follicles.94 Early candidal infections of the skin, mucosae, and nails and/or infantile atopic dermatitis are other presentations; in patients with the eosinophilic papulopustules, the atopic dermatitis commonly follows later in infancy. Superinfection of the dermatitis with S. aureus is very common,95 and patients show high levels of antistaphylococcal IgE antibodies. Staphylococcal skin infections include impetigo, furunculosis, paronychia, cellulitis, and characteristic “cold” abscesses (Fig. 132-13) that do not demonstrate the anticipated degree of erythema, warmth, and purulence. The abscesses occur most commonly on the head and neck and in intertriginous areas. Recurrent streptococcal pyoderma may also develop. Pulmonary bacterial pneumonia, abscesses, and empyema are the most frequent systemic infections and may result in pneumatoceles that serve as the nidus for bacterial (often Pseudomonas aeruginosa) or fungal (especially Aspergillus) infection. The most common infecting organisms are S. aureus and H. influenzae. Other than pneumonias, deep-seated infections, bacteremia, and sepsis are rare.96
Facial and skeletal abnormalities are common. Patients develop progressive coarsening of facial features (see Fig. 132-13), probably reflecting the skeletal defects, but the recurrent facial pustulation and lichenification from dermatitis may contribute. Distinctive facial features, including prominent forehead, a broad nasal bridge, and wide nasal tip are universally present
2413
21
by the age of 16 years. Dental anomalies include retention of primary teeth and lack of eruption of secondary teeth.97 Osteopenia is common, and 57% of patients have had at least 3 pathologic fractures, especially of the long bones, ribs, and pelvis.98,99 Scoliosis occurs in 63% of adult patients and hyperextensibility of joints in 68% of patients.96 Milder phenotypes may reflect mosaicism.100
Autosomal recessive disease shares with autosomal recessive HIES the high serum IgE levels, peripheral eosinophilia, chronic eczematous dermatitis, and recurrent skin (including cold abscesses) and respiratory tract staphylococcal infections. However, patients with autosomal recessive HIES show no tendency to form pneumatoceles and have no skeletal or dental abnormalities.101 Instead, autosomal recessive HIES is characterized by recurrent viral infections (eg, molluscum contagiosum, warts, herpes simplex and varicella-zoster), opportunistic infections, autoimmunity and devastating neurologic complications (especially vasculitis) that are often fatal in childhood (Fig. 132-14).102-107 Of patients with autosomal recessive HIES, up to 24% may have the neonatal the neonatal papulopustular eruption and food allergies/asthma are more common than in autosomal dominant HIES. Patients with autosomal recessive HIES have an increased risk of mucocutaneous squamous cell carcinoma and lymphomas.
ETIOLOGY AND PATHOGENESIS
Three different gene defects have been identified in HIES. The most common is a dominant negative mutation in signal transducer and activator of transcription 3 (STAT3),108,109 and is associated with a severe reduction of Th17 cells.110 STAT3 signals downstream of IL-6, which, together with transforming growth factor–β, is important for the development of Th17 cells. The Th17 cell deficiency accounts for the increased susceptibility to Candida infection and, as Th17 activation is key, for innate immunity at epithelial surfaces
2414
through the development of antimicrobial peptides. The decreased cytokine signaling may account for the “cold” abscesses and destructive inflammation from increased expression of other proinflammatory cytokines. The osteopenia of HIES is thought to result from loss of STAT3-dependent downregulation of osteoclast differentiation. Autosomal recessive HIES most often results from biallelic mutations in the dedicator of cytokinesis 8 (DOCK8), which encodes for a protein that is implicated in the regulation of the actin cytoskeleton that is critical for T-cell expansion and antibody responses.102,103
Mutations in TYK2 have been found in a single patient with HIES,111 although other patients with TYK2 deficiency have increased viral and mycobacterial infections, but not HIES, suggesting that HIES may not be an important feature. Biallelic mutations in phosphoglucomutase 3, a glycosylation enzyme, also have been found to underlie an HIES-like autosomal recessive disorder with elevated IgE levels, atopy, autoimmunity, and neurocognitive abnormalities.112-114
DIAGNOSIS
By definition, serum IgE levels must be elevated, but normal levels of IgE increase markedly during childhood. Diagnostic criteria for autosomal dominant HIES have been established that include IgE levels greater than 1000 IU/mL and a weighted score of 5 clinical features (typical newborn rash, recurrent pneumonia, pathologic bone fractures, characteristic facies, and a high palate)115; these features and Th17 cell deficiency lead to a “probably” diagnosis, but are definitive with a heterozygous STAT3 mutation. It should be recognized, however, that normal levels of IgE are age-dependent. Levels greater than 2000 IU/mL are seen in adolescents and adults; considerably lower levels are seen during infancy, given that the upper limit of normal in infants younger than age 1 year is 12.7 IU/mL and by 5 years of age is 47.1 IU/mL; an IgE level 10-fold above the 95th percentile for age is required for diagnosis. Eosinophilia of at least 2 SD above normal (usually above 700 cells/µL) is also a clinical manifestation. The total white blood cell count is typically normal and often fails to elevate in the setting of acute infection. In the autosomal recessive forms, eosinophilia tends to be more severe (eg, 17,500/µL).116 These patients have a global defect in T-cell activation (CD4>CD8 cells), which may explain their susceptibility to viral infections (including herpes and molluscum) in addition to bacterial and fungal infections. In contrast to autosomal dominant HIES, autosomal recessive HIES is associated with Th2 cell skewing. Deficiency of DOCK8 expression can be detected by flow cytometry.117
DIFFERENTIAL DIAGNOSIS
HIES is often considered in young pediatric patients with atopic dermatitis or WAS because of the high levels of IgE, eosinophilia, dermatitis, and recurrent staphylococcal infections. The coarse facial features, osteopenia,
recurrent pneumonia, and cold abscesses of autosomal dominant HIES help to distinguish it; the presence of platelet abnormalities also helps to distinguish WAS. In a study of 70 pediatric patients with a serum IgE level higher than 2000 IU/mL, 54 (77%) had atopic dermatitis and only 6 (8%) had HIES; the IgE level did not correlate with the diagnosis of HIES.118 Other immunodeficiency syndromes can be confused with HIES (eg, Omenn, DiGeorge, IPEX, and IRAK-4 [IL-1R–associated kinase 4] deficiency), as well as Netherton syndrome and GVHD. Early candidal infections may lead to consideration of mucocutaneous candidiasis (and with mosaic STAT3 mutations, candida infections may be present without detectable Th17 cell deficiency.100
CLINICAL COURSE, PROGNOSIS, AND TREATMENT
Given the lack of correlation between serum levels of IgE, eosinophilia, and the susceptibility to severe infections, the clinical course is difficult to predict. Patients with autosomal recessive HIES have a more severe course related to the neurologic complications. Antistaphylococcal antibiotics are effective for most cutaneous infections in patients with HIES, and oral triazole antifungals treat the mucocutaneous candidiasis. The prophylactic use of antistaphylococcal antibiotics markedly reduces the incidence of skin abscesses and pneumonia. The cutaneous and pulmonary abscesses often require incision and drainage and may require partial lung resections. Intravenous immunoglobulin therapy has been used successfully. Intravenous immunoglobulin may influence IgE levels as a consequence of an increased Ig catabolism or neutralization of the IgE. Ascorbic acid and cimetidine have decreased the number of infections and the chemotactic defect in some patients. Isotretinoin has been reported to eliminate the recurrent staphylococcal abscesses in an isolated patient without any change in immunologic status. Cyclosporine also has been used with good clinical and laboratory response. IFN-γ has shown inconsistent effects on IgE levels and infection susceptibility. Alendronate has been administered for the osteopenia.99 There are anecdotal reports of improvement of the eczematous dermatitis of HIES with use of omalizumab, a monoclonal antibody directed against IgE.119 INF-α has effectively treated the numerous warts120 and severe herpetic infections121,122
in DOCK8-deficient HIES. Stem cell transplantation is generally reserved for DOCK8-deficient HIES,123-125 but has been successful in severe cases of STAT3-deficient HIES.126
WHIM SYNDROME
WHIM SYNDROME
WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome is an immunodeficiency that is characterized by neutropenia,
21
hypogammaglobulinemia, and a susceptibility to infection by human papillomavirus. The disorder results from defects in the chemokine receptor gene CXCR4,127,128 and thus mature neutrophils are not able to exit the bone marrow (myelokathexis). The susceptibility to human papillomavirus infections suggests an important role for CXCR4 in immunity to this virus. Plerixafor, a CXCR4 antagonist, increases circulating leukocytes, reduces infections and, in combination with imiquimod, markedly reduces associated warts that had previously been resistant to treatment.129
SILVERY HAIR SYNDROMES: CHÉDIAK-HIGASHI AND GRISCELLI SYNDROMES
SILVERY HAIR SYNDROMES:
CHÉDIAK-HIGASHI AND
GRISCELLI SYNDROMES
The silvery hair syndromes comprise a group of autosomal recessive disorders in which a peculiar metallic sheen to hair, and often to skin, is noted (see also Chap. 75). Of these, Chédiak-Higashi and Griscelli (Type 2) syndromes have associated immunodeficiency (Table 132-8). Another syndrome with silvery hair, Elejalde syndrome, does not have associated immunodeficiency and may be allelic with the myosin 5a-deficient form of Griscelli syndrome (GS), with severe associated CNS dysfunction.
CHÉDIAK-HIGASHI SYNDROME
CHÉDIAK-HIGASHI
SYNDROME
AT-A-GLANCE
■ Autosomal recessive disorder of vesicle trafficking that results in giant organelles, including melanosomes, leukocyte granules, and plateletdense granules.
■ Mutations in LYST.
■ Silvery hair; pigment speckling and often hyperpigmentation on nose and ears.
■ Variable degree of photophobia and nystagmus.
■ Diagnosis can be confirmed by pigment clumping in hair shaft and leukocyte granules in blood smears.
■ Recurrent pyogenic infections and a mild bleeding diathesis.
■ Life-threatening “accelerated phase” with pancytopenia and organomegaly as a result of lymphohistiocytic infiltration.
■ Progressive neurologic deterioration.
2415
■ Therapy: transplantation.
21
GRISCELLI (GS1) GRISCELLI (GS2) GRISCELLI (GS3) CHÉDIAK-HIGASHI
Gene defect/protein MYO5A/myosin5a RAB27A/Rab27a a. Slac2-a/mlph b. MYO5A F-exon deletion LYST/lysosomal transport protein
Functional consequence Intracellular organelle transport and neurotransmitter secretion; interacts with Slac2-a/ mlph
GTPase for organelle movement and transfer. Essential for T- and NK-lymphocyte cytotoxic granule release and target cell death; interacts with Slac2-a/ mlph
Interacts with both myosin 5a and Rab27 to form a protein complex essential for melanosome movement to melanocytes
Protein required for final steps of membrane fusion and vesicle trafficking
Tissue expression Brain; not in cytotoxic cells Cytotoxic cells; not in brain Slac2-a/mlph is not in cytotoxic cells
Clinical features Silvery metallic hair; mild skin pigment dilution; onset of neurologic disorder in infancy
Silvery metallic hair; mild skin pigment dilution; severe immune disorder with hemophagocytic syndrome; neurologic symptoms are associated with hemophagocytic syndrome and CNS infiltration
Silvery metallic hair Silvery metallic hair and skin; recurrent infections; bleeding tendencies; progressive neurologic defects; immune disorder with hemophagocytic syndrome
Immune defects None T- and NK-cell function; hypogammaglobulinemia None T- and NK-cell function, diminished neutrophil chemotaxis and bactericidal activity
Hair mount Large, uneven clusters of pigment Same as GS1 Same as GS1 Granular, evenly distributed pigment
Skin in light microscopy Pigmentary dilution in keratinocytes, accumulation of melanosomes in melanocytes
Same as GS1 Same as GS1 Pigmentary dilution in both keratinocytes and melanocytes
Skin in electron microscopy Mature melanosomes filling epidermal melanocytes
Same as GS1 Same as GS1 Reduced numbers of giant melanosomes
Leukocyte granules None None None Large, cytoplasmic
Leukocyte granules None None None Large, cytoplasmic
GTPase, guanosine triphosphatase; mlph, melanophilin; NK, natural killer.
EPIDEMIOLOGY
Chédiak-Higashi syndrome (CHS) is a rare autosomal recessive disorder.130,131 Parental consanguinity is often reported, and there are approximately 300 reported cases worldwide.
CLINICAL FINDINGS
Patients usually first show manifestations during infancy or early childhood. Pigment abnormalities occur in 75% of patients, particularly the silver sheen to the hair (Fig. 132-15A). Ocular hypopigmentation may cause photophobia, and strabismus and nystagmus are common. Visual acuity does not tend to be reduced. The skin is typically fair compared to other family members, but dark, slate-colored areas of pigmentation and diffuse, speckled hypopigmentation may be seen in the sun-exposed skin of dark-skinned individuals.132-134
2416
Infections can be observed in the newborn period and continue throughout the lifetime of the patient. Infections most commonly involve the skin, lungs, and respiratory tract where microbes such as S. aureus, Streptococcus pyogenes, and Pneumococcus are prevalent. Deep ulcerations resembling pyoderma gangrenosum have been described. Neurologic manifestations include muscle weakness, cranial and peripheral neuropathy, and progressive neurologic deterioration. CHS patients also exhibit a mild coagulation defect. Patients bruise easily, manifest petechiae, and have some mucosal bleeding, although platelet numbers remain normal.
ETIOLOGY AND PATHOGENESIS
CHS is caused by mutations in the LYST gene, located on chromosome lq42, which encodes a protein required for the final steps of vesicle trafficking and
A
B
C
secretion.135 The LYST gene is expressed in lysosomes and other secretory organelles (melanosomes, cytolytic granules, and platelet dense granules). Characteristic giant granules are thought to result from altered granule maturation and fusion. Enlarged melanosomes are unable to be transferred to keratinocytes. NK cells and cytotoxic T lymphocytes do not release proteolytic enzymes necessary for target cell killing. Cytotoxic T-lymphocyte–associated antigen is trapped in abnormally large vesicles rather than on the cell surface and thus may be unable to regulate T-cell activation, increasing the risk of lymphoproliferative disease. Platelet-dense granules are delayed in secretion of storage pools required for normal coagulation. Diminished chemotaxis of neutrophils and
21
monocytes and delayed intracellular microorganism killing are often associated.
DIAGNOSIS
Diagnosis is usually suspected clinically because of the silvery sheen to the hair. The finding of large cytoplasmic granules in blood leukocytes is highly diagnostic. Hair shafts have evenly distributed small pigment granules (see Fig. 132-15B), in contrast to the irregular, large pigmentary clumping of GS (see Fig. 132-15C). Although not performed for diagnostic purposes, skin biopsy shows pigmentary dilution in both melanocytes and keratinocytes, and electron microscopy reveals giant melanosomes.
DIFFERENTIAL DIAGNOSIS
The silvery hair sheen is also seen in GS, but an unusual metallic hair sheen and skin hypopigmentation is a component as well of Hermansky-Pudlak syndrome (see Chap. 75). Silvery hair also has been described in a neonate with hypoproteinemia from congenital hydrops fetalis; the hair spontaneously repigmented136 with clinical improvement. Pseudo- CHS granules are seen on smears in some cases of acute leukemia.137
The hemophagocytic syndrome associated with lymphohistiocytic proliferation of silvery hair syndromes needs to be distinguished from massive lymphoproliferation in other immunodeficiency disorders, particularly X-linked lymphoproliferative disorder (see “Antibody Deficiency Disorders” above), familial hemophagocytic lymphohistiocytosis, autoimmune lymphoproliferative syndrome, and immunodeficiency caused by mutations in the IL-2R. Familial hemophagocytic lymphohistiocytosis is a group of autosomal recessive disorders with hemophagocytosis and absence of NK-cell cytotoxicity that tends to be fatal without early stem cell transplantation.138,139 The condition can result from mutations in either PRF1, which encodes perforin; UNC13D, which encodes Munc 13-4140; STX11, encoding syntaxin 11; or STXBP2, encoding Munc 18-2. All of these proteins are involved in the exocytosis and function of cytotoxic intracellular granules NK cells and cytotoxic T lymphocytes. Mutations in several genes can lead to autosomal recessive or dominant autoimmune lymphoproliferative syndrome, also known as Canale-Smith syndrome.141,142 These include the genes that encode Fas or CD95 (TNFRSF6), the cell-surface apoptosis receptor, and its ligand (TNFSF6), and the caspase proteins 8 and 10, proteases in the cascade leading to apoptosis. This group of disorders features autoimmune disorders (cytopenias, leukocytoclastic vasculitis, lupus erythematosus) and an increased risk of lymphoma, and is associated with increased numbers of circulating CD4−/CD8− α/β T cells. Immunodeficiency with extensive lymphocytic infiltration
2417
21
of viscera is also a manifestation of autosomal recessive mutations in the α chain of the IL-2 receptor.143
Patients demonstrate bacterial, viral, and fungal infections because of early apoptosis of the developing thymic T lymphocytes.
PROGNOSIS, CLINICAL COURSE, AND TREATMENT
The “accelerated phase” of hemophagocytic lymphohistiocytosis occurs in 85% of CHS patients and is usually triggered by Epstein-Barr virus infection. Viscera become infiltrated with lymphoid and histiocytic cells, which are sometimes atypical in appearance. Hepatosplenomegaly with jaundice, lymphadenopathy, pancytopenia, and a leukemia-like gingivitis with pseudomembranous sloughing of the buccal mucosa are associated. In this phase, petechiae, bruising and gingival bleeding are commonly seen, resulting from the thrombocytopenia and decreased hepatic synthesis of coagulation factors. Overwhelming infection or hemorrhage during the accelerated phase often lead to death. CHS may present with the accelerated phase.144
The mean age of death for untransplanted patients with CHS is 6 years, usually from overwhelming infection or hemorrhage during the lymphoma-like accelerated phase. Prenatal diagnosis has been achieved by examination of hair from fetal scalp biopsies and of leukocytes from fetal blood samples. The treatment of choice for patients with CHS is early transplantation,145-148 which corrects the immunologic status but does not affect the pigment abnormality nor inhibit the development of neurologic disorders that grow increasingly worse with age. Reduced intensity conditioning has been used with success, especially in patients transplanted before the accelerated phase occurs. Absence of cytotoxic T-cell function has been touted as predictive of the later occurrence of hemophagocytic lymphohistiocytosis, and as a biomarker for early transplantation. Management of CHS is otherwise largely supportive, with use of prophylactic antibiotics to avoid recurrent infections. Acyclovir, high-dose intravenous γ-globulin, vincristine, cyclosporine, and prednisone have been used to control the accelerated phase, but the accelerated phase is usually fatal.
GRISCELLI SYNDROME
GRISCELLI SYNDROME
AT-A-GLANCE
■ Autosomal recessive group of disorders.
■ Mutations in MYO5A (more neurologic), RAB27A (hemophagocytosis), or Slac-2a.
2418
■ Silvery hair is a hallmark.
■ Diagnosis can be confirmed by pigment clumping in hair shaft, but smears show no leukocyte granules.
■ Hemophagocytosis often precipitated by Epstein- Barr virus infection.
■ Treatment: transplantation.
EPIDEMIOLOGY
The majority of affected patients with this autosomal recessive disorder are of Mediterranean or Middle Eastern origin and born into consanguineous families.
CLINICAL FINDINGS
The pigmentary dilution of GS is often limited to the hair, characterized by a silver-gray sheen with a few cases of skin involvement. The hair shaft shows large uneven clusters of pigment (see Fig. 132-15C) and the skin shows pigmentary dilution in keratinocytes but accumulation of melanosomes in melanocytes. Electron microscopy reveals numerous stage IV melanosomes in epidermal melanocytes. In contrast to CHS, smears of leukocytes do not show giant granules. The additional clinical features present in each subtype are dependent on the function and tissue expression of the defective protein. Patients with GS1 have a primary neurologic disorder that presents in infancy with hypotonia and developmental delay. Severe immune disorder characterized by hemophagocytic syndrome or an “accelerated phase of the disease” is seen in GS2. Hemophagocytic syndrome is characterized by infiltration of various organ systems by activated lymphocytes and macrophages. It develops at mean age of 36 months, and often is precipitated by a virus, particularly Epstein-Barr virus. Patients have fever, hepatosplenomegaly, neurologic impairment, coagulopathy, and pancytopenia. Patients with GS3 present solely with the pigmentary abnormalities.149,150
ETIOLOGY AND PATHOGENESIS
Mutations in 3 genes may underlie GS. GS1 is caused by a mutation in the MYO5A gene that encodes myosin-5a, a motor protein responsible for intracellular organelle transport. MYO5A is abundantly expressed in brain tissue and is important for neurotransmitter secretion. It is not expressed in cytotoxic cells. GS2 is caused by a mutation in the RAB27A gene, which encodes Rab27a, a guanosine triphosphatase protein involved in the function of the intracellular-regulated secretory pathway. It is essential for T-lymphocyte and NK-lymphocyte cytotoxic granule release and cell death. This defective cytotoxic activity most likely
Protein interactions required for melanosome transfer
Myosin-5a
Slac2-a/ melanophilin
Globular tail
Medial tail
Head
GTP-Rab27a
F-axon
Proximal tail
Neck
Melanosome
Actin
leads to the uncontrolled activation of lymphocytes and macrophages in the hemophagocytic syndrome. Both MYO5A and RAB27A have been mapped to chromosome 15q21.1. The third type, GS3, is the result of a homozygous missense mutation in Slac2-a/melanophilin or a deletion of the MYO5A F-exon.151 Melanophilin (also not expressed in cytotoxic cells) encodes a member of the Rab effector family and interacts with both myosin-5a (through the F-exon) and RAB27A to form a triprotein complex essential for the capture and local transport of melanosomes to the melanocytes (Fig. 132-16). All 3 genetic subtypes share the inability to construct this complex for melanosome transport, thus leading to the characteristic pigmentary abnormality of GS.
PROGNOSIS, CLINICAL COURSE, AND TREATMENT
GS has been uniformly fatal but now can be reversed by successful hematopoietic stem cell transplantation. Of patients with GS2, 85% have mutations in the RAB27A gene that lead to early protein truncation. They have the most-severe form of the disease, with early development of hemophagocytic syndrome and rapid progression. The median survival from the onset of the accelerated phase to death in these patients is 5 months. Most patients die by 5 years of age secondary to progressive CNS
21
disease or recurrent infections. Missense mutations in the RAB27A gene show a milder phenotype with later age of hemophagocytic syndrome onset and a good response to chemotherapy treatment. Patients with GS1 do not develop hemophagocytic syndrome. Prenatal diagnosis has been achieved by examination of hair from fetal scalp biopsies and DNA analysis.
COMPLEMENT DEFICIENCY DISORDERS
AT-A-GLANCE
■ Deficiency or dysfunction of early complement components is associated with autoimmune disorders, particularly systemic lupus erythematosus.
■ Deficiency or dysfunction of early complement components leads to risk of infections caused by encapsulated bacteria.
■ Deficiency of late complement components markedly increases susceptibility to neisserial infections.
HEREDITARY ANGIOEDEMA
HEREDITARY ANGIOEDEMA
Hereditary angioedema (HAE) is almost always inherited in an autosomal dominant fashion with a spontaneous mutation rate of 25% (for clinical features and management, see Chap. 41).152 It occurs in 1 in 150,000 persons and has no racial or ethnic predilection. Seventy-five percent of patients report a positive family history. There are 3 types of HAE. Type I is characterized by low antigenic levels of C1 inhibitor (C1 INH) and affects 85% of patients, whereas the remainder of patients have Type II, with low functional levels but normal/high antigenic levels of C1 INH. A third type of HAE does not show a deficiency of C1 INH. This type of HAE was previously thought to only affect women, although eventually, a family was described in which three male family members were affected.153 One family in which the disease is transmitted as an autosomal recessive trait because of a mutation in the promoter region of the gene has been identified.154
HAE causes significant morbidity, especially if attacks are frequent and laryngeal edema is a
2419
21
MANIFESTATION COMPONENT PATHWAY ASSOCIATIONS
Infection C1q, C1r, C1s, C4, C2, C3 Classical Autoimmune disorders, especially SLE
Increased encapsulated bacterial infections
Mannan-binding lectin Lectin Pyogenic infections in neonates and children (SLE)
Factors H, I Inhibitors, alternative Recurrent pyogenic infections (C3 depletion)
Properdin Stabilizer, alternative Fulminant neisserial infections
CR3 Receptor for iC3b LAD (see LAD Type 1)
C5 to C9 Terminal (membrane attack complex) Recurrent neisserial infections; milder with C9 deficiency
Autoimmune disorders C1, C4, C2 Classical Autoimmune disorders, especially SLE
Increased encapsulated bacterial infections
SLE
Exfoliative erythroderma C3, C5 Shared, terminal Failure to thrive, diarrhea, infections
Angioedema C1 esterase inhibitor Inhibitor, classical Angioedema
Renal disease and/or hemolysis Factor H Inhibitor, alternative Glomerulonephritis, atypical hemolytic uremic syndrome
Decay accelerating factor (CD59) Inhibitor, alternative Hemolysis, thrombosis
Membrane cofactor protein (CD46) Inhibitor, alternative Atypical hemolytic uremic syndrome
C3 Shared Membranoproliferative glomerulonephritis, severe bacterial infections
C3 Shared Membranoproliferative glomerulonephritis, severe
LAD, leukocyte adhesion deficiency; SLE, systemic lupus erythematosus.
significant risk for mortality. There are several treatment modalities available including the use of attenuated androgens, antifibrinolytic agents, plasma-derived C1 INH, a kallikrein inhibitor and a bradykinin receptor antagonist.152 Plasma-derived C1 INH can be used acutely to treat angioedema attacks or preventatively if attacks are frequent or before known procedures.
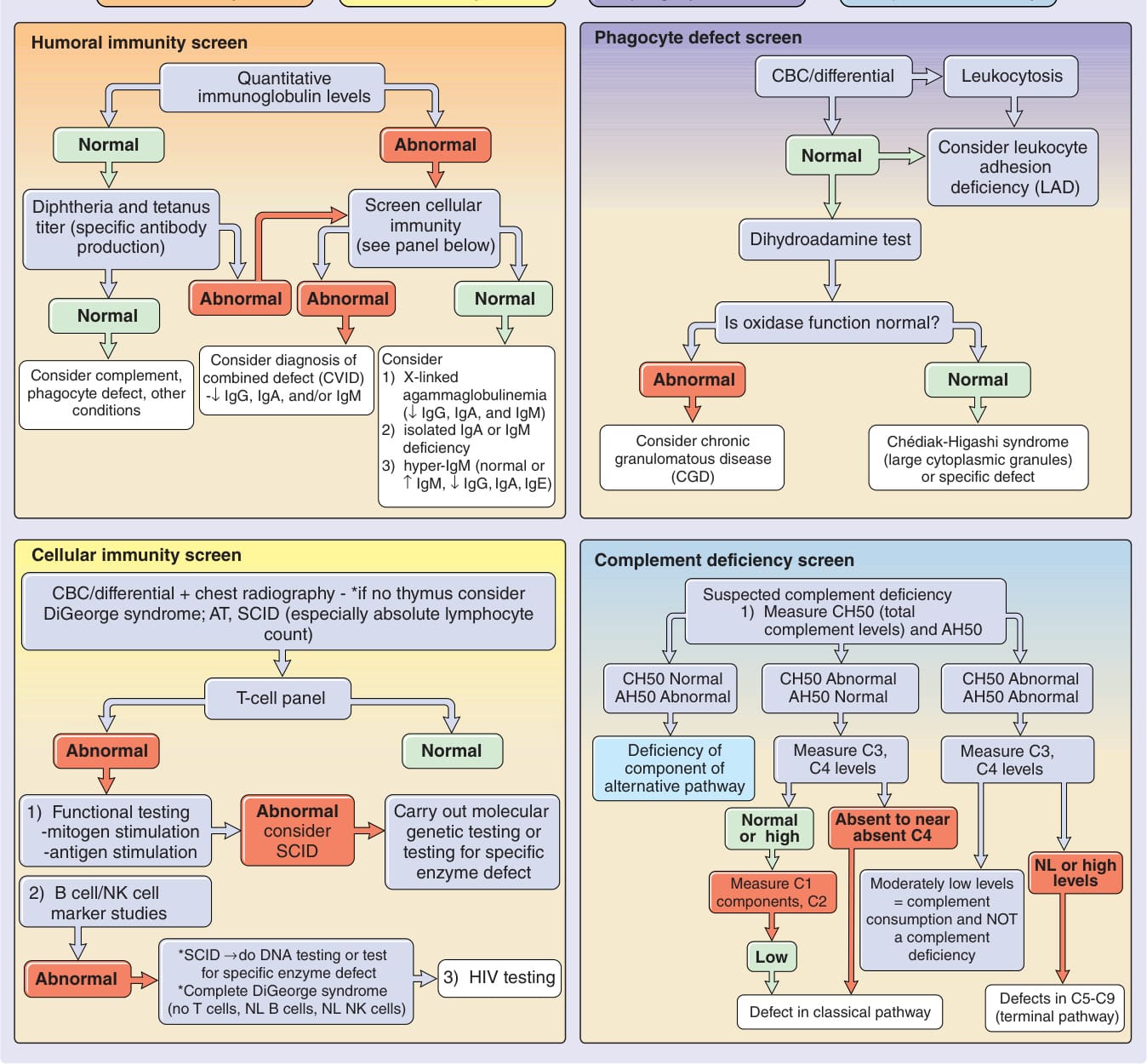
Figure 132-1 Algorithm for immunodeficiency disorders in neonates and infants. If the clinical presentation is concerning for severe combined immunodeficiency (SCID), this is a medical emergency and the patient needs immediate referral to an immunologist. AH50, alternative pathway 50% hemolytic activity; AT, ataxia-telangiectasia; CBC, complete blood cell count; CH50, 50% hemolytic complement; CVID, common variable immunodeficiency; Ig, immunoglobulin; NK cell, natural killer cell; NL, normal; ↓, decreased; ↑, increased.
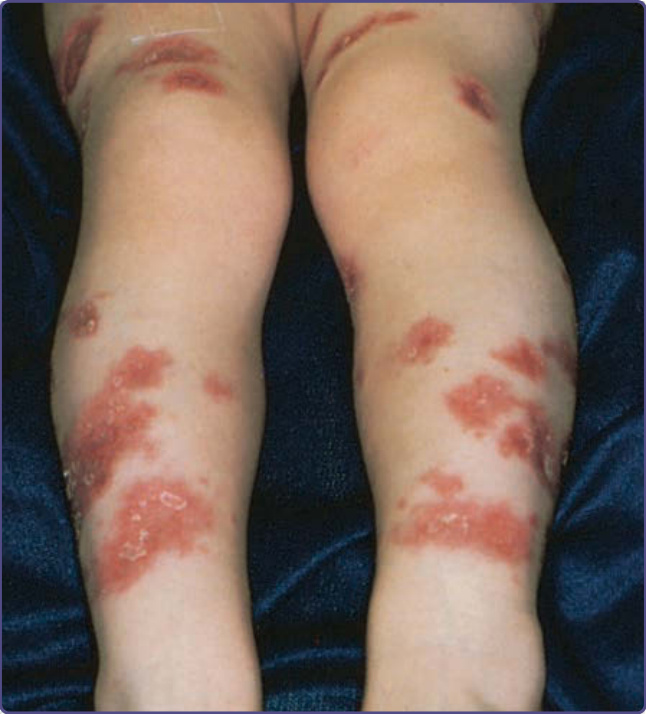
Figure 132-2 Noncaseating granulomas on the legs of a child with common variable immunodeficiency. Cultures and special stains showed no organisms.
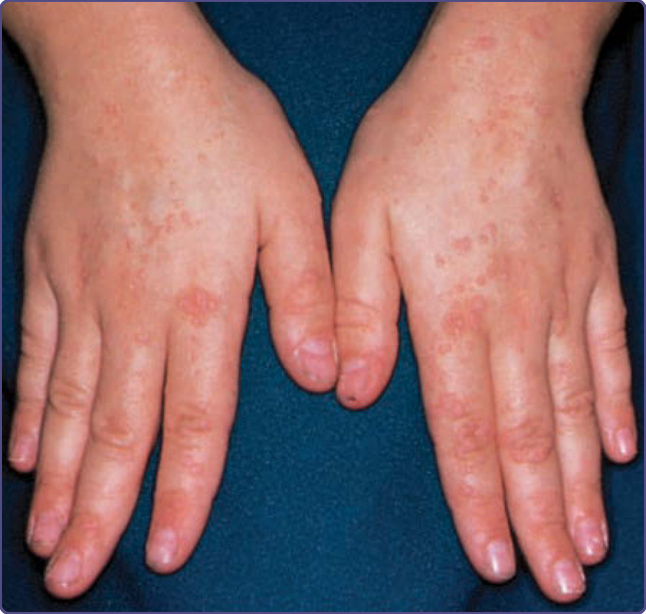
Figure 132-3 The dorsal aspect of the hands were covered with recalcitrant verrucae vulgaris in this girl with common variable immunodeficiency.
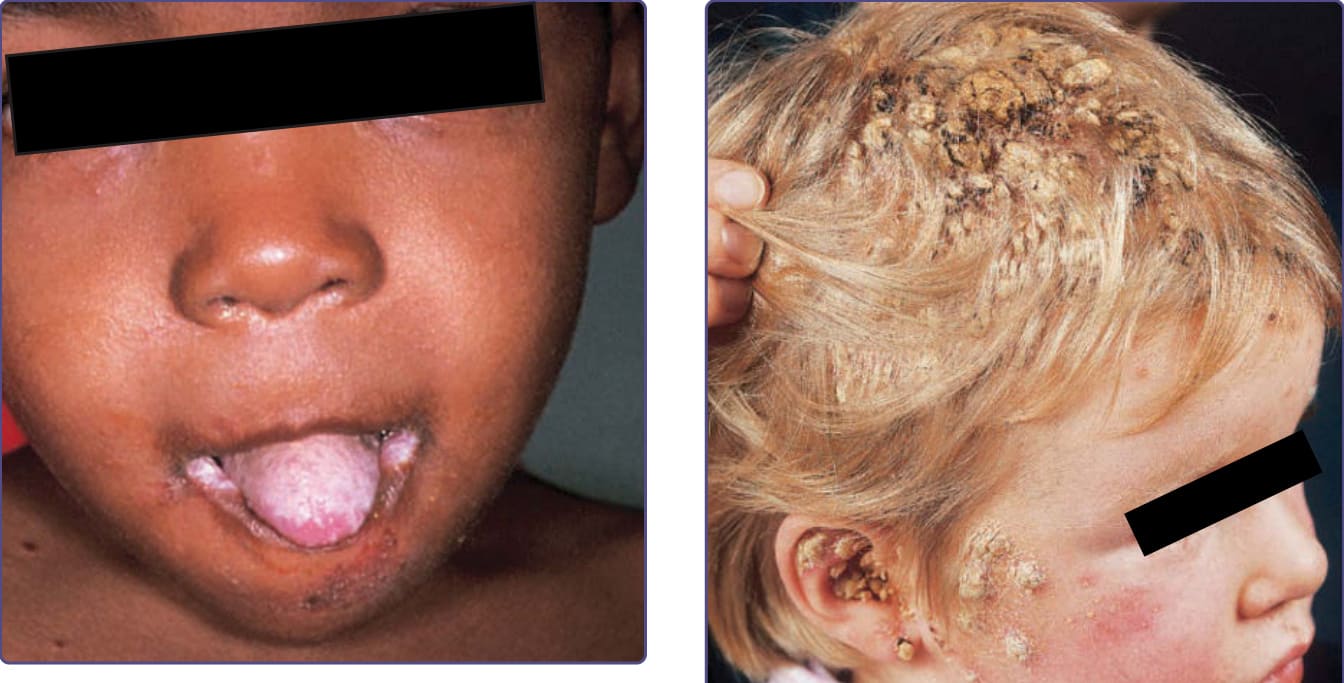
Figure 132-4 Recurrent thrush and candidal cheilitis in a boy with mucocutaneous candidiasis.
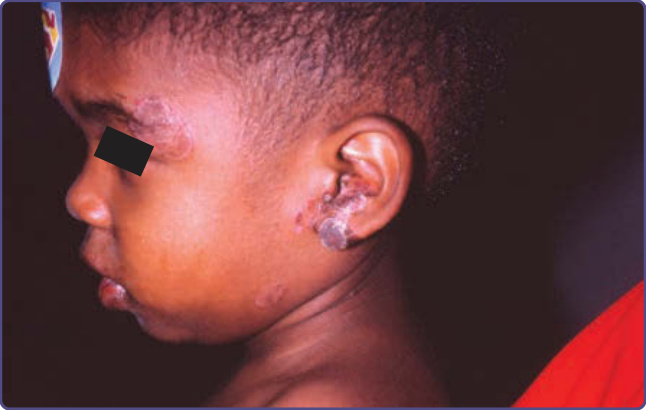
Figure 132-5 Cutaneous candidal infections with hyperkeratotic candidal granulomas. The child responded to oral azole antifungal medications, but not to topical medications.
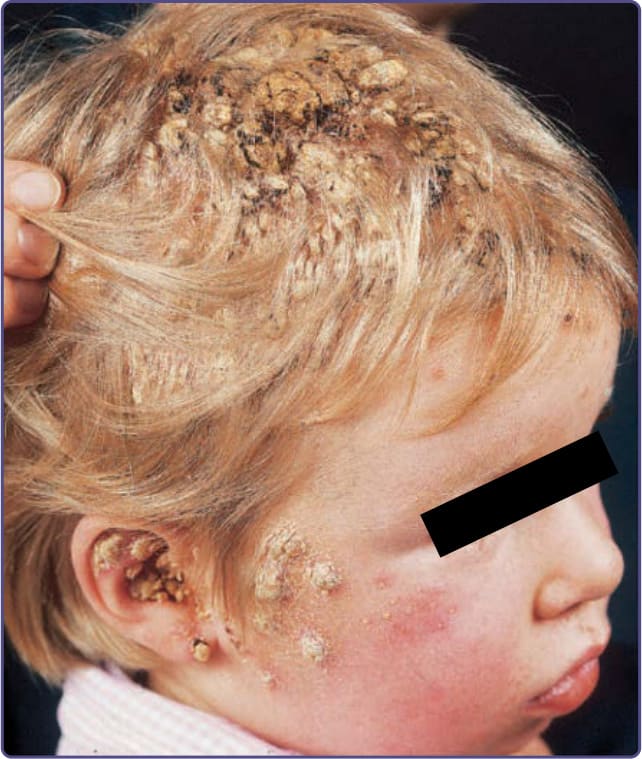
Figure 132-6 This 3-year-old child with hypothyroidism had oral thrush, intertriginous candidiasis, verrucous crusting on the scalp and face, and candidal onychomycosis. The warty growths shown in the photograph consisted of dried pus and serum, and grew only Candida albicans.
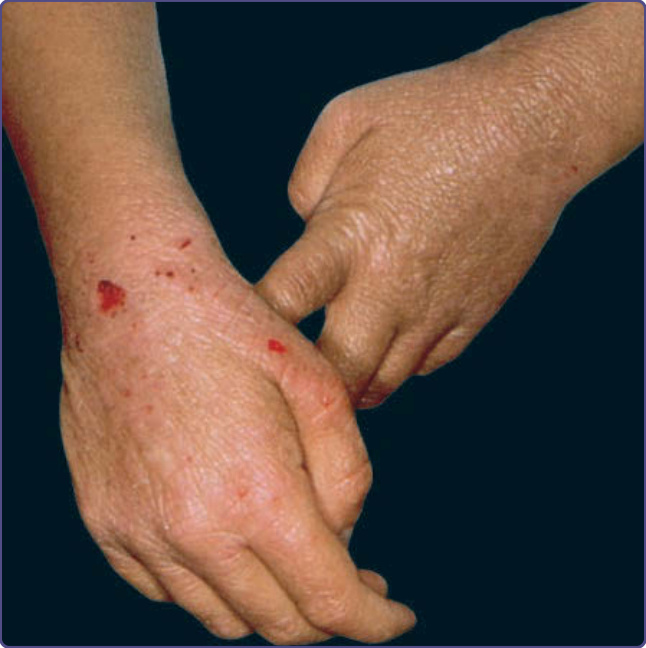
Figure 132-7 Severe atopic dermatitis in a boy with Wiskott- Aldrich syndrome. Note the serosanguineous crusting.
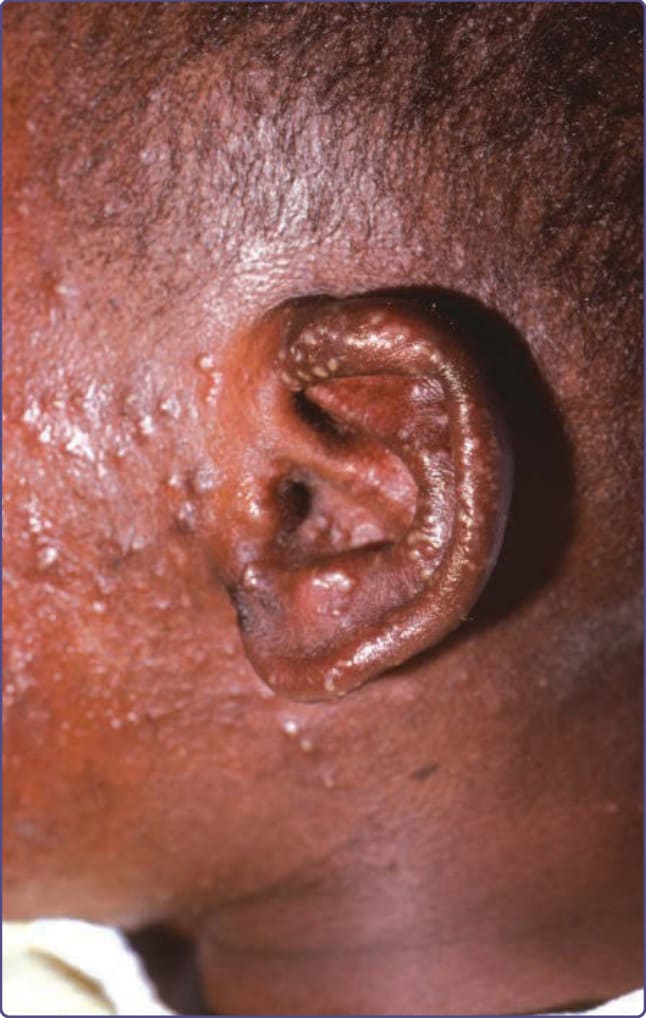
Figure 132-8 Herpetic infection with pustules on the ear of a teenager with Wiskott-Aldrich syndrome. The patient was blind in this left eye owing to a previous ocular infection. After the pictured infection, the patient was administered prophylactic acyclovir and had no subsequent herpetic infections for a decade. Eventually, he became resistant to acyclovir and succumbed to his immunodeficiency.
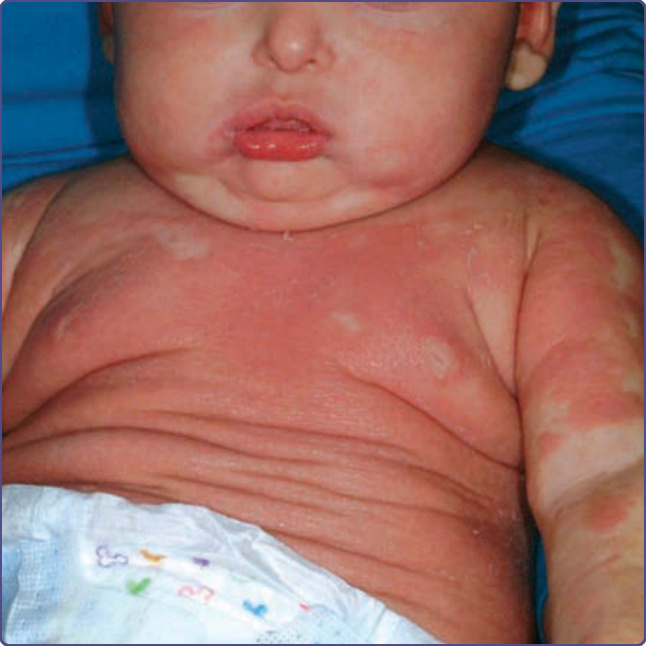
Figure 132-9 Ectodermal dysplasia with immunodeficiency gene mutation. This boy with a hypomorphic mutation in NEMO (nuclear factor κB essential modulator) showed failure to thrive, slightly exfoliative dermatitis, and the typical facial features of hypohidrotic ectodermal dysplasia. Note the small chin, the “pouty” lower lip, thin upper lip, and small, pinched nose with malar hypoplasia. His mother had a pigmented streak following a line of Blaschko, typical of that seen in incontinentia pigmenti. (Used with permission from Dr. Anthony Mancini.)
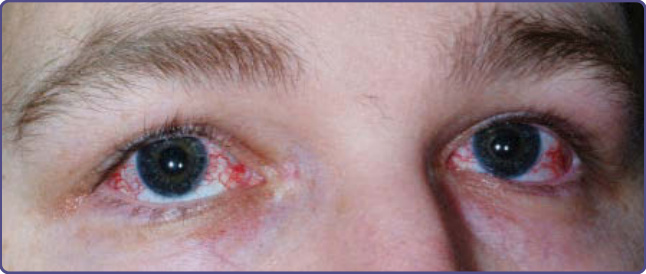
Figure 132-10 Bulbar conjunctival telangiectasias in a patient with ataxia-telangiectasia.
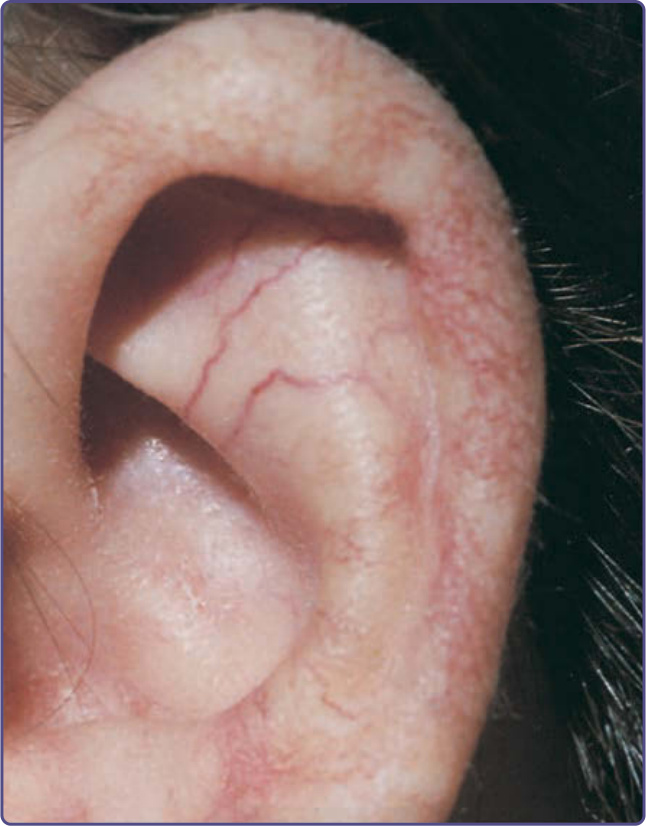
Figure 132-11 Ataxia-telangiectasia. Telangiectasias inside and on the helix.
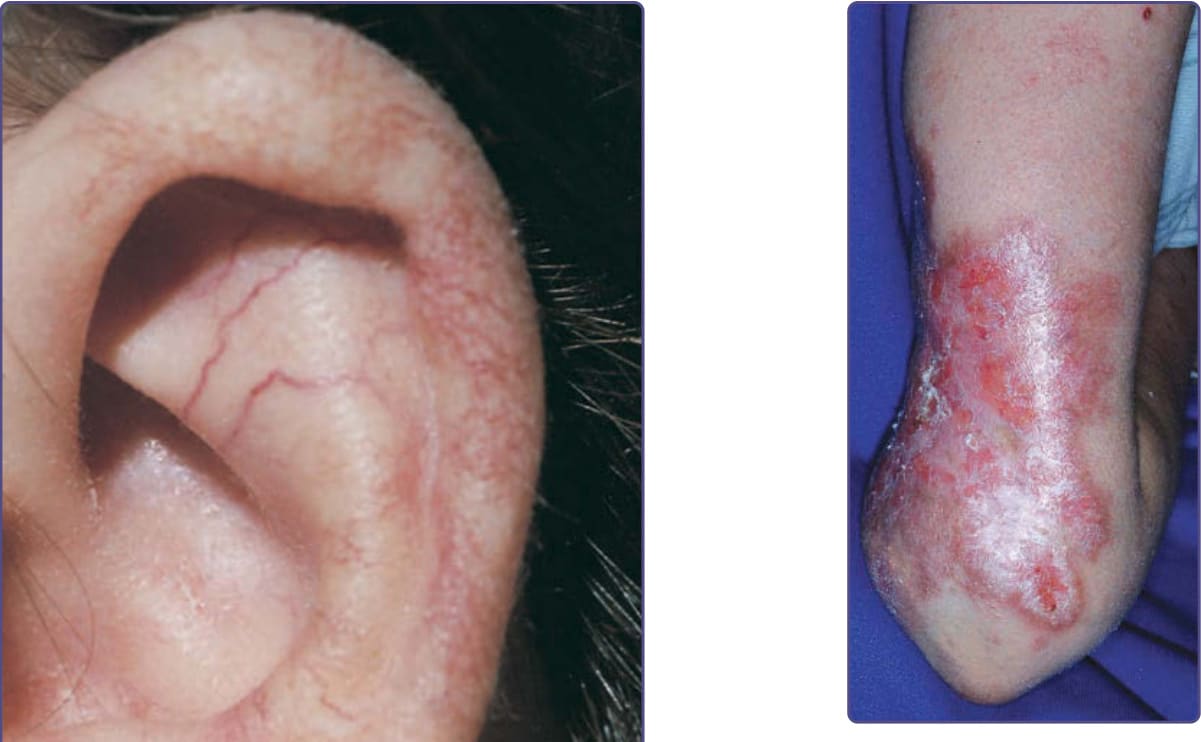
Figure 132-12 Noninfectious granulomatous dermatitis of a patient with ataxia-telangiectasia. These persistent lesions tend to ulcerate, but often respond to injections of triamcinolone acetonide.
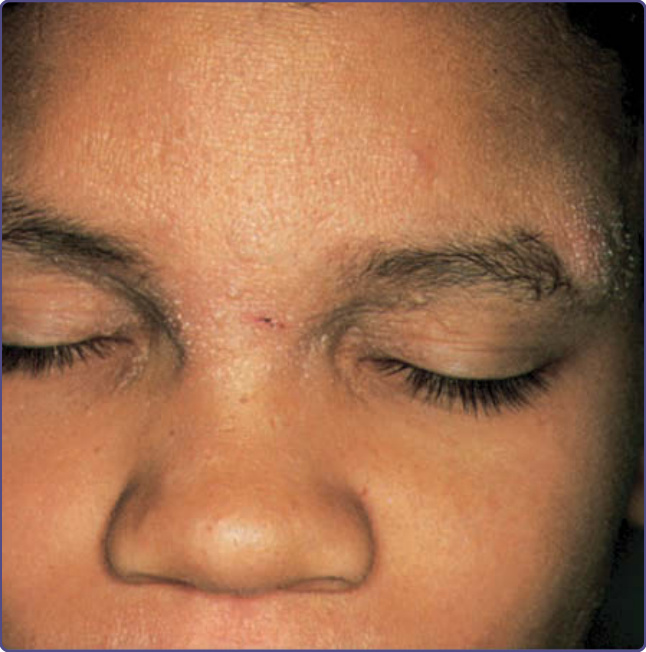
Figure 132-13 Coarse facial features, atopic dermatitis furuncles, and a cold abscess at the glabella in a boy with Hyperimmunoglobulinemia E syndrome.
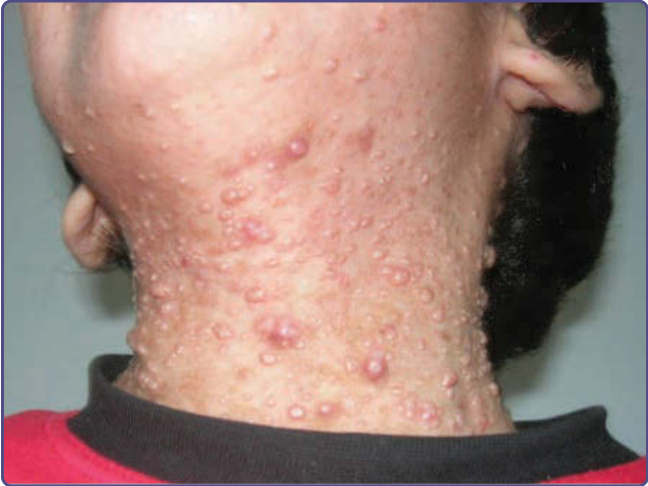
Figure 132-14 This boy with autosomal recessive HIES caused by a DOCK8 (dedicator of cytokinesis 8) mutation shows extensive molluscum contagiosum infection on the neck. (Used with permission from Dr. I. Barlan.)
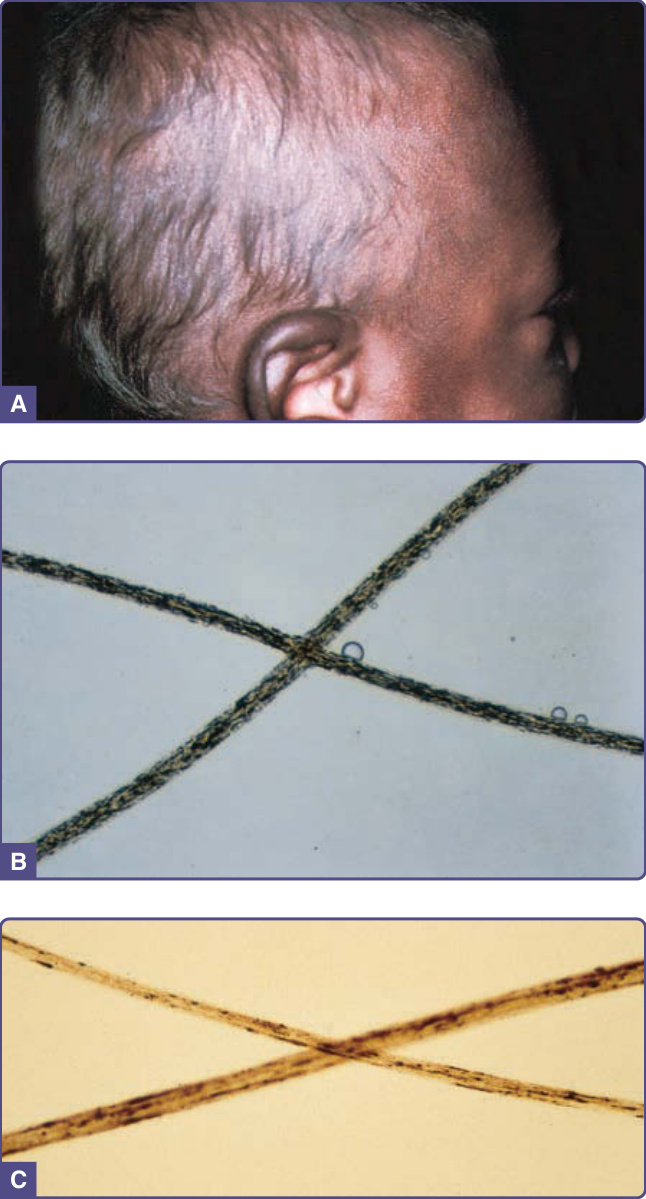
Figure 132-15 Silvery hair syndromes. A, Silvery sheen to the hair in a black infant; the patient had darkly pigmented eyes and skin. Note the accentuation of pigmentation on the ear helix. B, Small, evenly clumped pigment granules in the hairs of Chédiak-Higashi syndrome. C, Larger aggregates of irregularly spaced pigmentation in the hairs of a patient with Griscelli syndrome.
Figure 132-16 Protein interactions required for melanosome transfer. GTP, guanosine triphosphate. (Adapted with permission of ASCI: Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456. Permission conveyed through Copyright Clearance Center, Inc.)
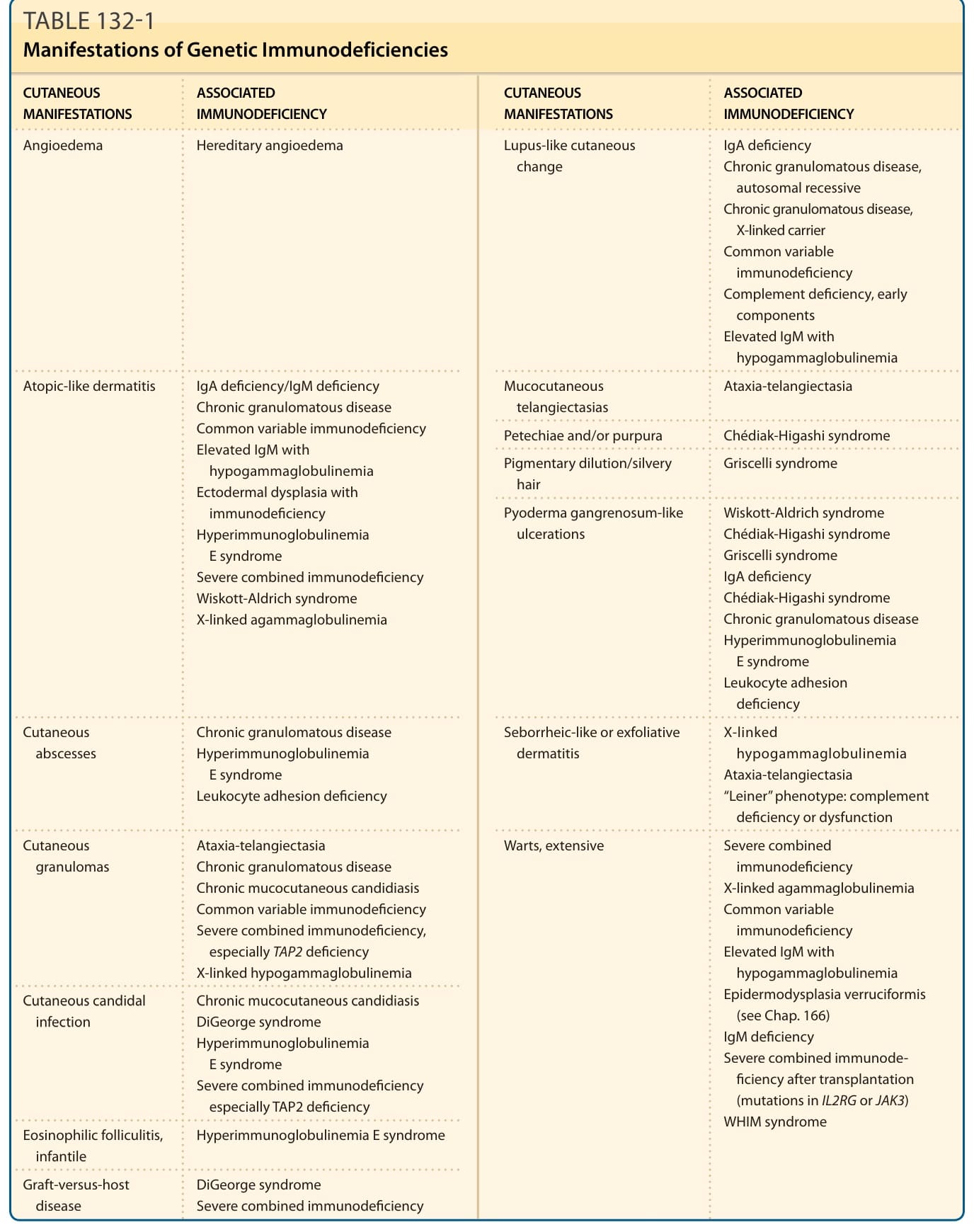
TABLE 132-1 Manifestations of Genetic Immunodeficiencies
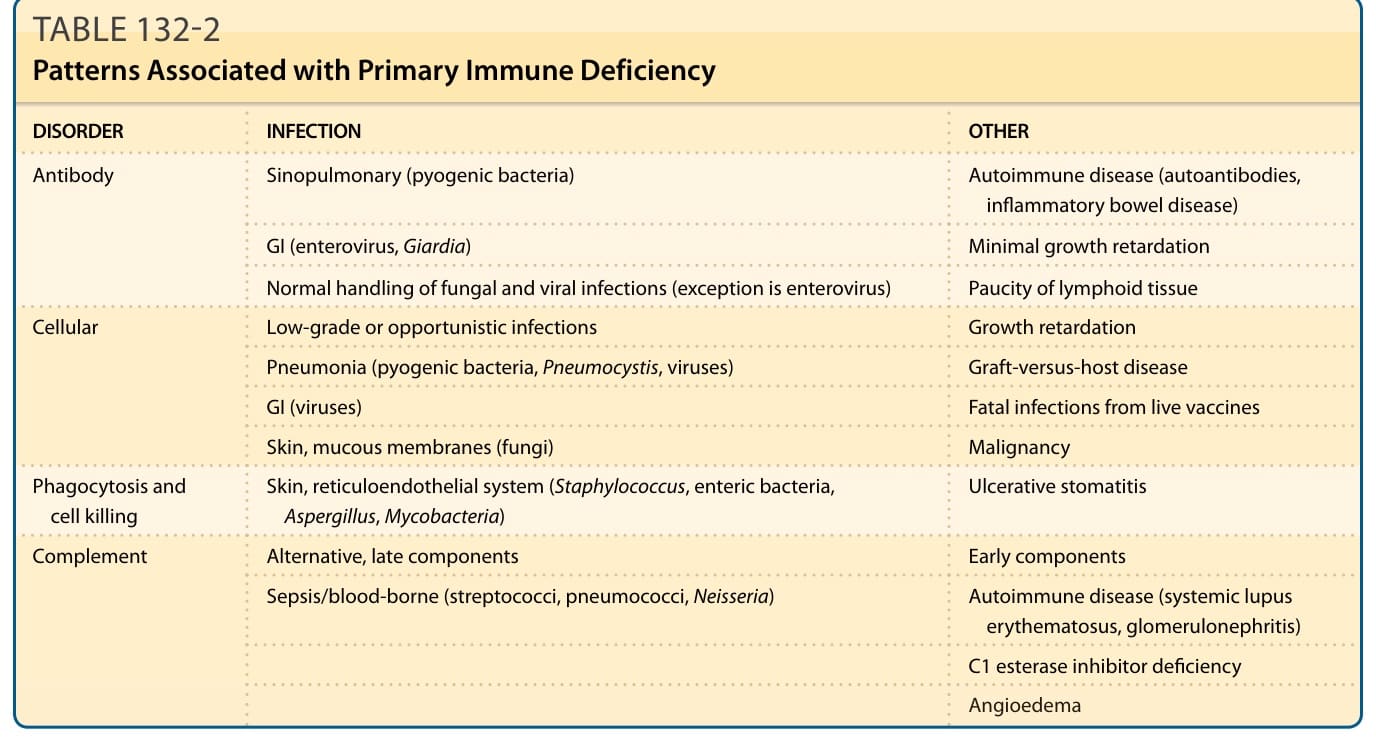
TABLE 132-2 Patterns Associated with Primary Immune Deficiency
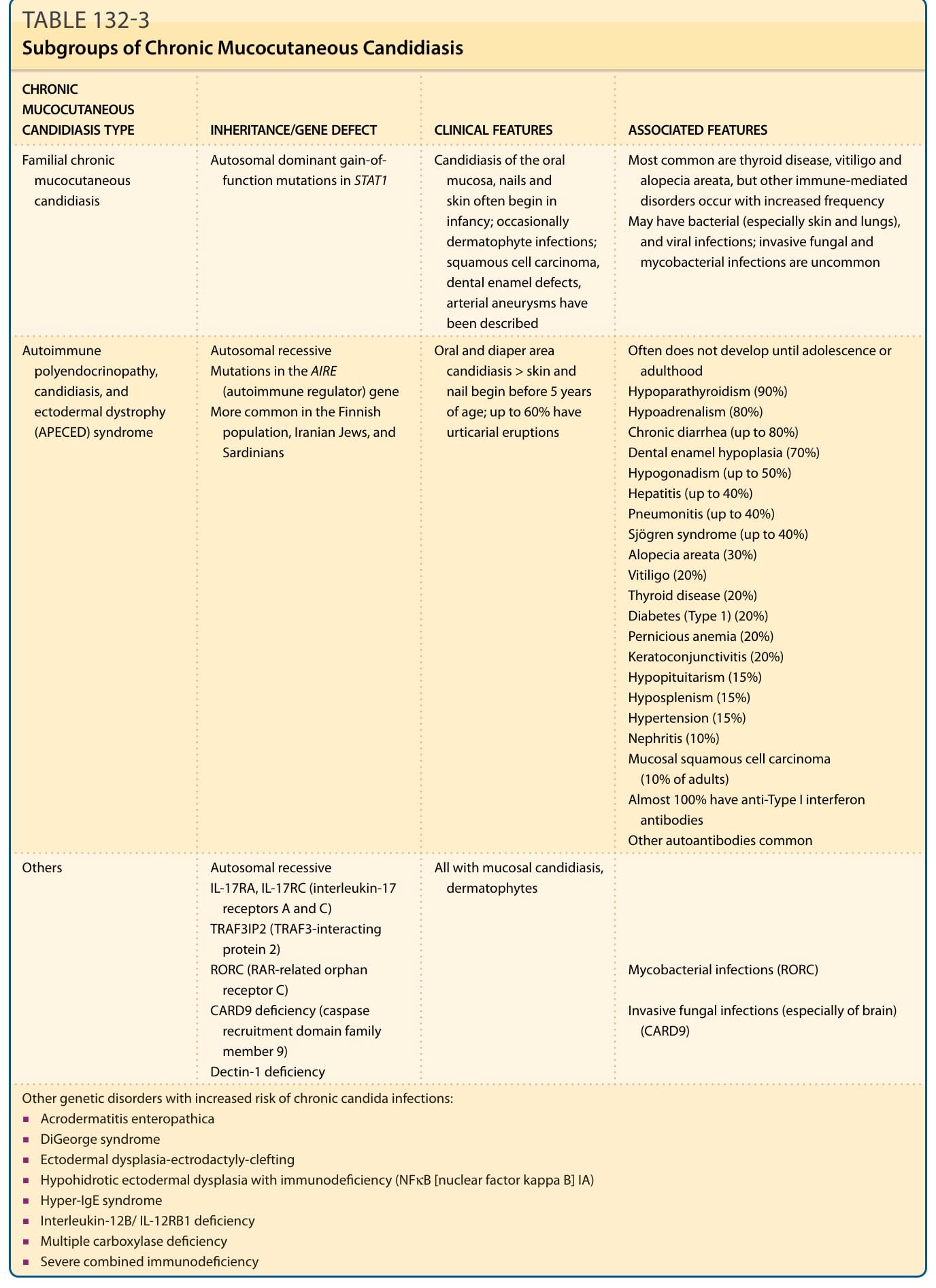
TABLE 132-3 Subgroups of Chronic Mucocutaneous Candidiasis
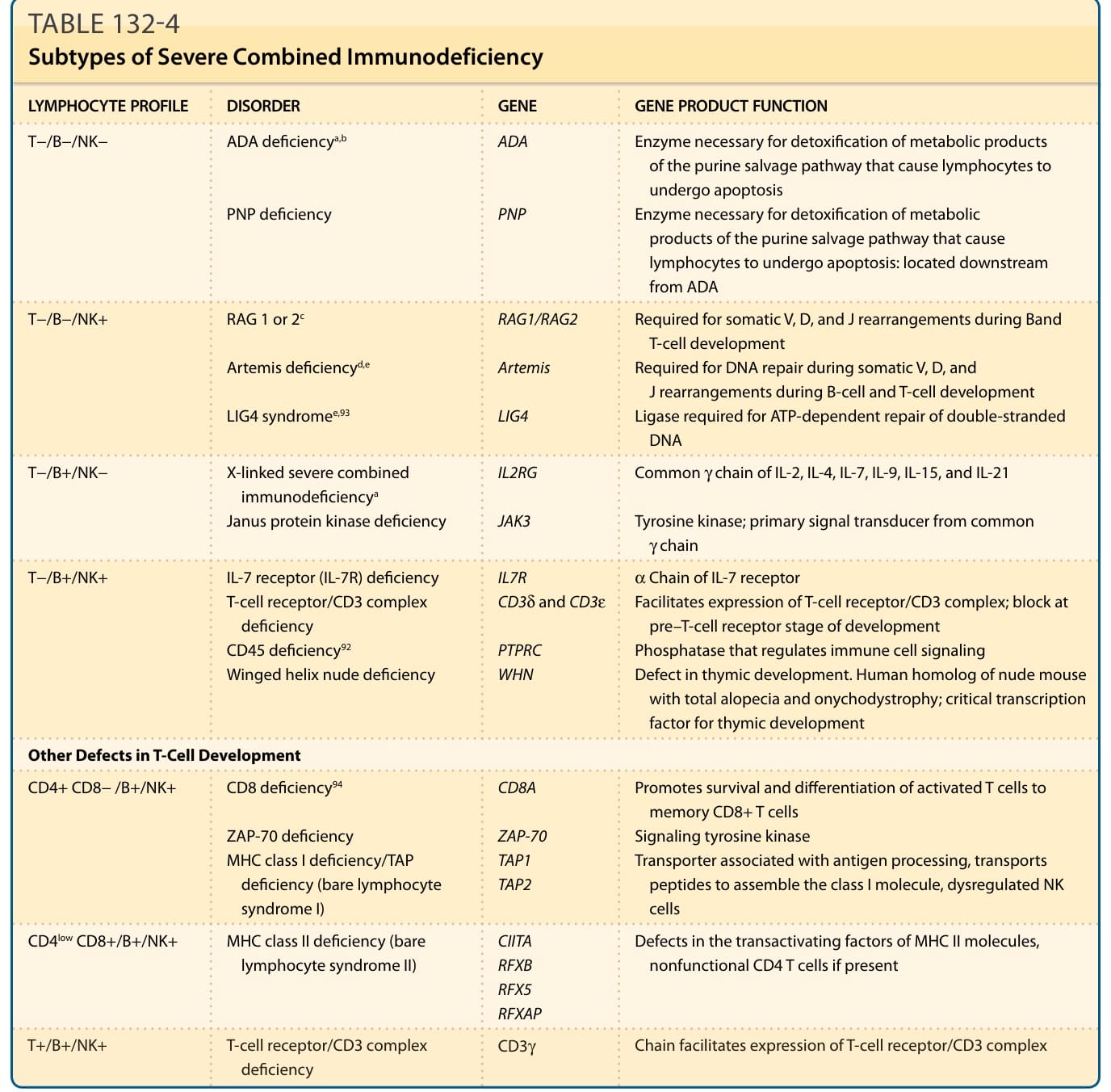
Table 132-4 outlines the subtypes of severe combined immunodeficiency.
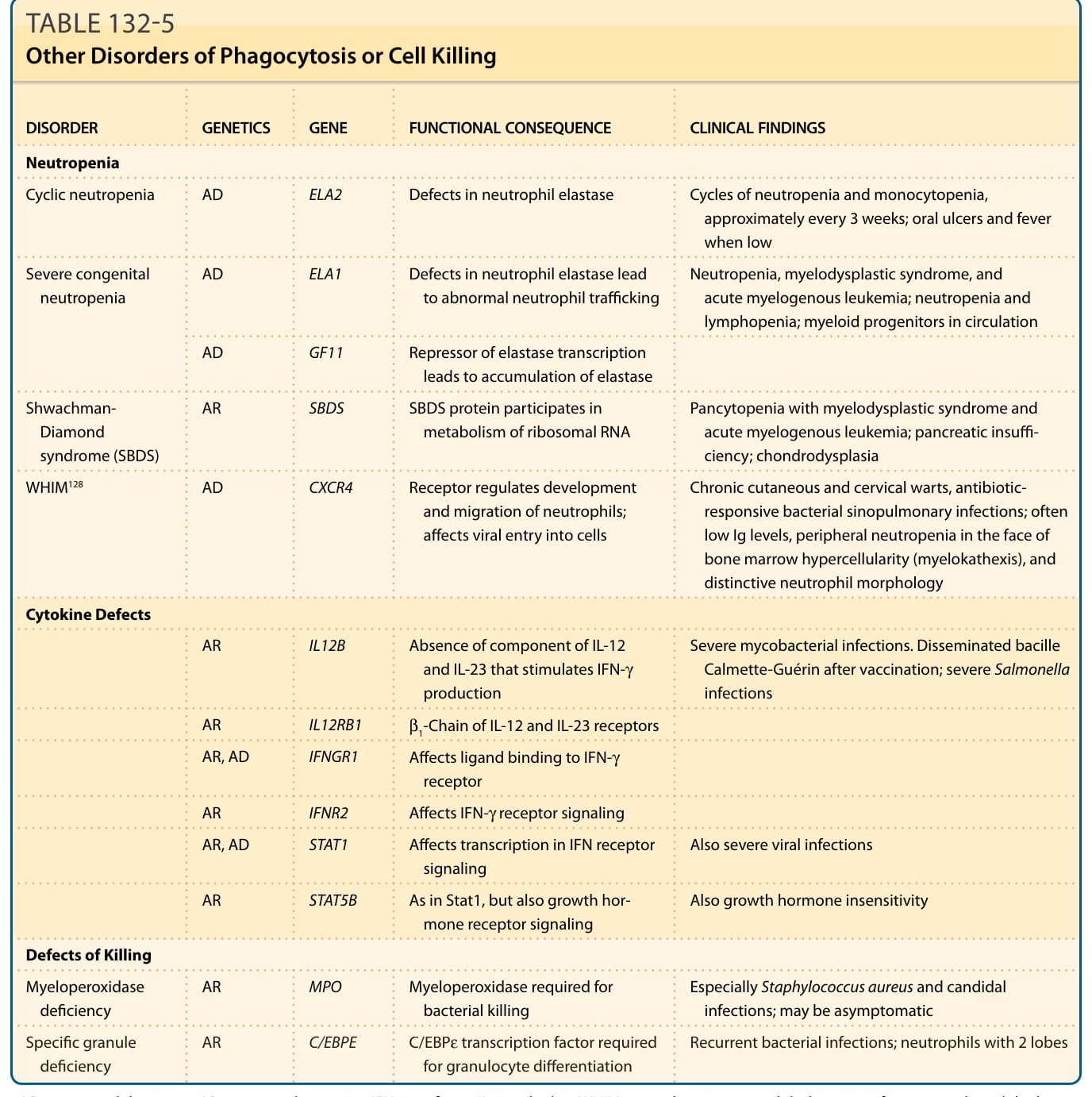
TABLE 132-5 Other Disorders of Phagocytosis or Cell Killing
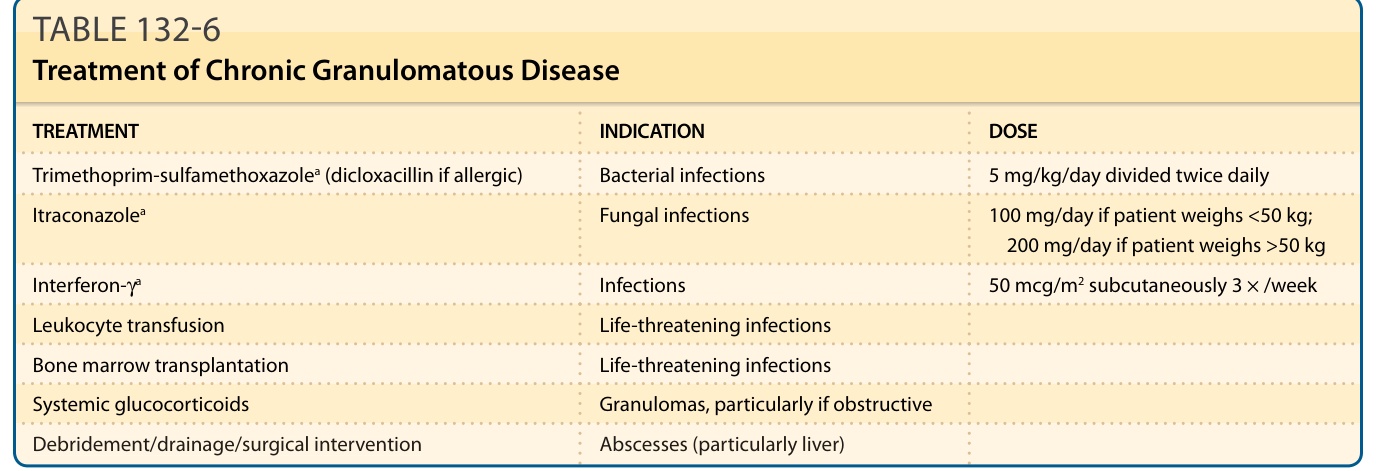
TABLE 132-6 Treatment of Chronic Granulomatous Disease
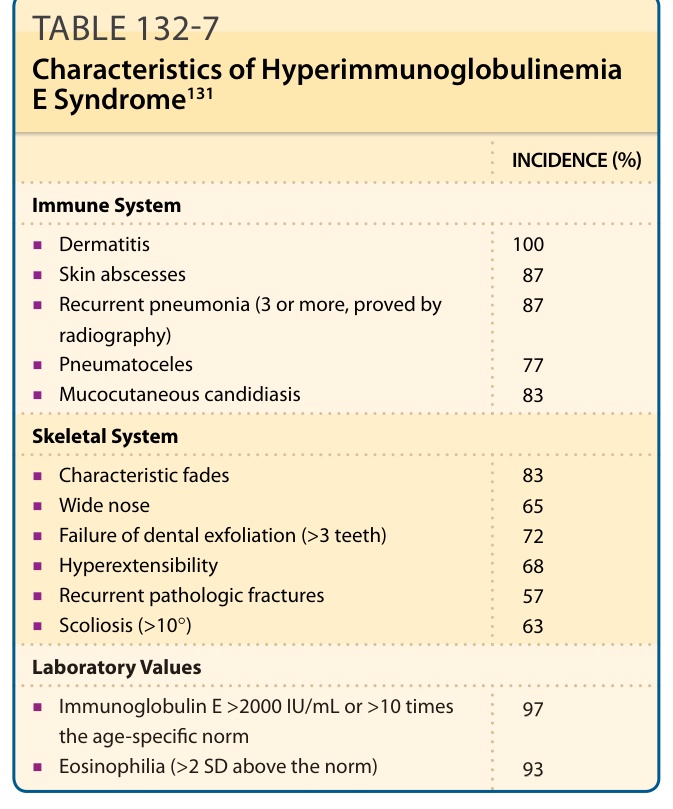
TABLE 132-7 Characteristics of Hyperimmunoglobulinemia E Syndrome131
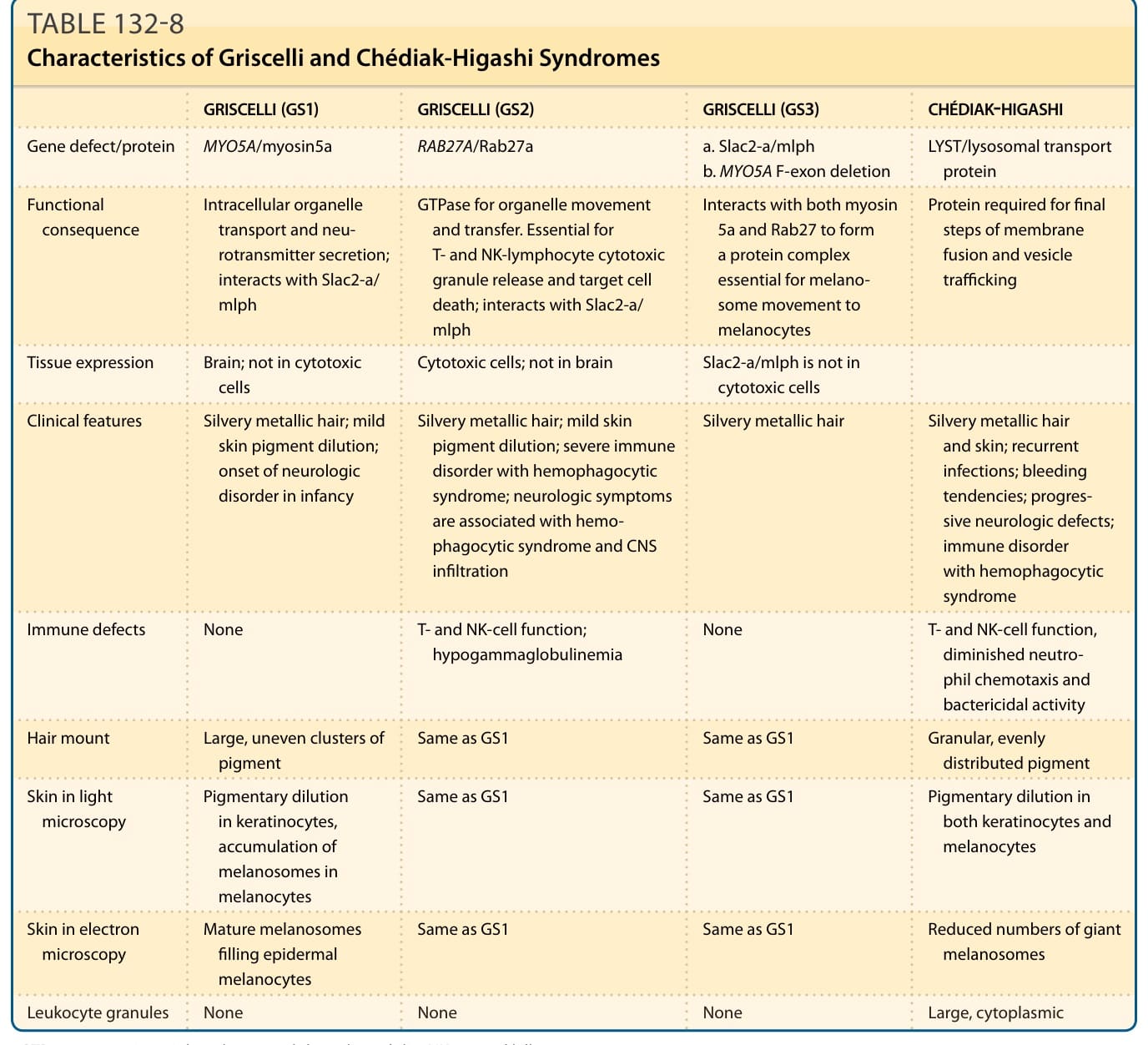
TABLE 132-8 Characteristics of Griscelli and Chédiak-Higashi Syndromes
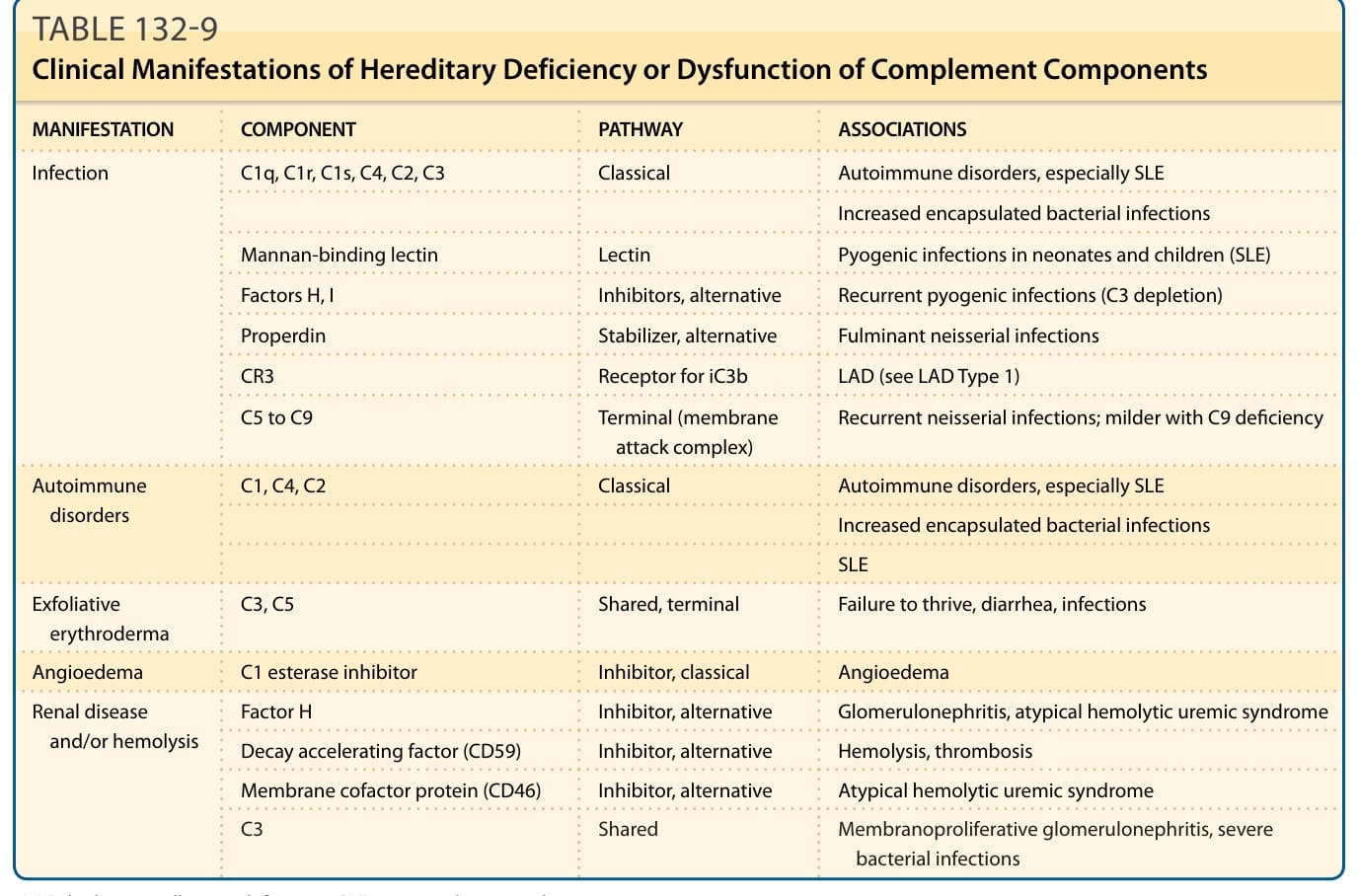
Table 132-9 outlines the clinical manifestations of hereditary deficiency or dysfunction of complement components.